

Abstract
The activation of memory T cells is a very rapid and concerted cellular response that requires coordination between cellular processes in different compartments and on different time scales. In this study, we use ribosome profiling and deep RNA sequencing to define the acute mRNA translation changes in CD8 memory T cells following initial activation events. We find that initial translation enables subsequent events of human and mouse T cell activation and expansion. Briefly, early events in the activation of Ag-experienced CD8 T cells are insensitive to transcriptional blockade with actinomycin D, and instead depend on the translation of pre-existing mRNAs and are blocked by cycloheximide. Ribosome profiling identifies ∼92 mRNAs that are recruited into ribosomes following CD8 T cell stimulation. These mRNAs typically have structured GC and pyrimidine-rich 5′ untranslated regions and they encode key regulators of T cell activation and proliferation such as Notch1, Ifngr1, Il2rb, and serine metabolism enzymes Psat1 and Shmt2 (serine hydroxymethyltransferase 2), as well as translation factors eEF1a1 (eukaryotic elongation factor α1) and eEF2 (eukaryotic elongation factor 2). The increased production of receptors of IL-2 and IFN-γ precedes the activation of gene expression and augments cellular signals and T cell activation. Taken together, we identify an early RNA translation program that acts in a feed-forward manner to enable the rapid and dramatic process of CD8 memory T cell expansion and activation.
Visual Abstract

Key Points
mRNA translation shapes early activation of CD8 memory T cells.
Ribosome footprinting identifies a translational program in CD8 memory T cells.
RNA-GQ, GC, and PRTE motifs regulate the translation of key genes in T cells.
Introduction
Resting naive T cells are long-lived and metabolically quiescent cells that show little ongoing DNA, RNA, and protein synthesis (1–3). T cells are activated by encountering adequately presented Ag on MHC class I or II and accompanying B7 receptor binding that engages TCR and CD28 costimulatory signals, respectively (4, 5). The first signaling consequences of Ag encounter are detectable within minutes (6–10). They are followed during the next 24 h by metabolic reprogramming, rapid cell proliferation, and the acquisition of new cellular functions (11–18). Ag-experienced/memory T cells show much more dramatic responses to repeated Ag exposure (boost) than do naive T cells (15, 16), and this requires accurate coordination of transcription and translation that occur in the nucleus and cytoplasm, respectively, and proceed on vastly different time scales. Specifically, transcription is a slow process and requires several hours to yield new transcripts (19, 20). Accordingly, the epigenetic (21–23) and transcriptional influences on RNA abundance in T cell activation are best observed at 24 h or later (24–30). In contrast, existing mRNAs are translated into proteins as often as two to three times per minute following initial signaling events (3, 31, 32). Resting T cells are known to harbor large numbers of “idling” ribosomes, and untranslated RNAs and are poised for rapid activation (19, 20, 32–34). Previous polysome profiling studies have implicated mTOR as an activator of translation and driver of T cell exit from quiescence, differentiation, and growth (35–38). Technological advances in deep RNA sequencing (RNA-seq) and ribosome footprinting (also known as ribosome profiling) enabled more rapid assessments of translation at earlier time points and provides codon-level resolution of ribosome occupancy and precise measurements of the translation efficiency for every transcript across the transcriptome (39). These new techniques overcome the methodological shortcomings of polysome profiling that relied on manual separation of heavy and light ribosome fractions and semiquantitative measurements of RNAs in these fractions. In the current study, we use ribosome footprinting to examine the immediate translational response in memory CD8+ T cells exposed to Ag. We identify a concise program of critical mRNAs whose early translational activation triggers and augments subsequent steps leading to complete CD8 memory T cell activation.
Materials and Methods
Mice
Eight- to 12-wk-old OT-1 C57BL/6-Tg(TcraTcrb)1100Mjb/J and C57BL/6 mice were obtained from The Jackson Laboratory. All animal experiments were performed in accordance with regulations from the Memorial Sloan Kettering Cancer Center’s Institutional Animal Care and Use Committee.
In vitro culture and stimulation
Lymph node OT-1 T cells were cultured in RPMI 1640 (Sigma-Aldrich) supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% l-glutamine and 100 IU of mouse rIL-2 (PeproTech). For B cell culture, lymphocytes were isolated on a Ficoll gradient from spleens of female C57BL/6 mice. The OVA257–264 peptide (SIINFEKL) (GenScript) was loaded overnight on B cells (1 μg/ml). For T cell priming, OT-1 cells were cultured with irradiated (1500 rad) or mitomycin C–treated (Sigma-Aldrich), OVA peptide–pulsed C57BL/6 splenocytes. After stimulation, T cells were expanded for an additional 6 d. The T cell purity was ∼95%. T cells were restimulated with CD3/CD28 Dynabeads (Thermo Fisher Scientific) according to the manufacturer’s protocol. Where indicated, T cells were treated with actinomycin D (AMD; 1 μM) or cycloheximide (CHX; 50 μg/ml, Sigma-Aldrich)
Isolation of human primary CD8+ T cells
Human primary lymphocytes were isolated from total blood packs (NY Blood Center) by Ficoll-Paque density centrifugation followed by enrichment of CD8+ T cells by magnetic separation (130-096-495, Miltenyi Biotec) according to the manufacturer’s instruction. CD8+ human T cells were then activated with human-specific CD3/CD28 Dynabeads (Thermo Fisher Scientific) and cultured for indicated time intervals.
Isolation of mouse primary T cells from OT-1 mice
Mouse primary T cells were isolated from the spleen and lymph nodes of OT-1 mice using the EasySep mouse T cell isolation kit (STEMCELL Technologies, catalog no. 19851) and following the instructions provided with the kit. This kit isolates total untouched T cells using negative selection. After isolation, we tested the purity of the cells by CD3-FITC staining.
Flow cytometry analysis
All Abs were purchased from BioLegend and included CD69 (clone H1.2F3, FITC), CD44 (IM7, allophycocyanin), and CD62L (MEL-14, BV421). Live cells were stained for 20 min at room temperature in PBS + 1% BSA. The samples were acquired on a BD LSRFortessa flow cytometer and analyzed with FlowJo software (Tree Star) on singlet events.
De novo protein synthesis assay
Cultured OT-1 cells were labeled with Cy5-conjugated puromycin (5 μM) for 1 h at the end of the indicated time point following CD3/CD28 activation. Cy5-conjugated puromycin is readily incorporated in live cells without the need for methionine starvation that is required for Click-IT AHA (l-azidohomoalaine) labeling and therefore avoids additional effects of methionine starvation on global translation. Treatment groups included resting or activated OT-1 cells with or without 1 μM AMD (SRB00013, Sigma-Aldrich) for the indicated time points. Changes in mean fluorescence intensity of Cy5-conjugated puromycin as a measure of newly produced protein were analyzed by flow cytometric analysis.
Ribosomal profiling
The experimental procedure was adapted from a previous publication (40). Primed OT-1 cells were either activated with CD3/CD28 beads or left in a resting state for 2 h, followed by CHX treatment for 10 min. Total RNA and ribosome-protected fragments were isolated following a published protocol. Deep sequencing libraries were generated from these fragments and sequenced on the HiSeq 2000 platform.
Ribosomal profiling analysis
We mapped reads to transcripts using the XPRESS analytical pipeline (41) and removed reads mapping to ribosomal RNAs, noncoding RNAs, and duplicated reads. Read length across all samples is 31 bp and sequence quality is high. Before alignment, all ribosomal profiling samples had >2 million reads; however, after removal of duplicate reads and filtering, each sample was left with 180,000–280,000 reads. The number of reads limits the number of the gene, and we were still able to identify >1300 genes with at least 15 RP reads across all conditions. Read alignments are available on the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) under an accession number to be updated. The final alignment files used for quantifications are available on Gene Expression Omnibus (GEO) accession number GSE168476 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168476). RNA-seq and ribosomal footprinting reads were aligned using STAR v2.5 using the Ensembl mouse genome reference mm10 (GRCm38.Ensembl78). RNA-seq and ribosomal profiling reads were aligned using a singularity instance of XPRESSpipe, with the following parameters: 3′ adapter sequence: CTGTAGGCAC, minimum read length of 17, splice junction overhang (sjdbOverhang) of 49, removal of duplicates, removal of rRNA aligning sequences, and two-pass STAR alignment. The entire call for ribosomal read processing is as follows: xpresspipe riboseq: --input/data/in_dir\, --output/data/out_dir\, --reference/data/ref_dir\, --cdna_fasta/data/single_fasta/Mus_musculus.GRCm38.dna.chromosome.fa\, --gtf/data/ref_dir/transcripts_CT.gtf\, --experiment tcell_activation\, --a CTGTAGGCACCATCAAT\, --method RPM\, --sjdbOverhang 49\, --two-pass\, --remove_rrna\, --min_length 17\, --max_length 0\, --use_rmdup\, --feature_type CDS or TRANSCRIPT\, --deduplicate. The complete call for RNA-Seq read processing is as follows: xpresspipe peRNAseq: --input/data/in_dir\, --output/data/out_dir\, --reference/data/ref_dir\, --gtf/data/ref_dir/transcripts_CT.gtf\, --experiment tcell_activation\, --method RPM\, --adapter CTGTAGGCACCATCAAT CTGTAGGCACCATCAAT\, --sjdbOverhang 49\, --two-pass\, --remove_rrna\, --front_trim 0\, --min_length 0\, --max_length 1000\, --feature_type CDS or TRANSCRIPT\, --use_rmdup.
To rerun our analysis, the docker build environment is available on zenodo (10.5281/zenodo.4583883)
A forked version of RiboDiff v0.2.1, which can be found on GitHub (https://github.com/nrosed/RiboDiff), was used to estimate the change in translational efficiency between sample conditions. The forked version of this method uses the 85th quantile for library size normalization. Only protein-coding genes were considered in RiboDiff. The results of RiboDiff are available in GEO dataset accession number GSE168476 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168476). We used the following command line: ribodiff -p1 -s 15 -m BH.
Motif analysis
Motif analysis was performed using a single unique 5′ untranslated region (UTR) sequence for the top 250 translation efficiency (TE)-up genes. Five hundred genes were randomly selected from the RNA-seq analysis, and the 5′ UTR sequences were used as the background. For motif discovery, sensitive, thorough, rapid, enriched motif elicitation (STREME) and multiple expectation maximization for motif elicitation (MEME) analysis suites were used (42).
Luciferase assay
Luciferase assays were performed using the Dual-Luciferase reporter assay system (E1960; Promega) following the manufacturer’s instructions. Five tandem repeats of the identified motifs were cloned into the 5′ UTR of Renilla luciferase plasmid psiCHECK-2 vector. Motifs are as follows: GQ 5×, 5′-GGCGTCGGCG GCGGCGGCGGCAGCGGCTCCGGCCGAGGTGC-3′; GC-rich 5×, 5′-GCGTGTGCGGCAGCCGCAGCC-3′; pyrimidine-rich 5×, 5′-TTGTTGGTTTTCTTTTTCTTT-3′; A-rich 5×, 5′-AAAATTAAAAAATAAAAGAAAATAAAAGACTA-3′; T-rich 5×, 5′-TTTTAATTTTTATTTTCTTTTATTTTCTGAT-3′.
Plasmid and small interfering RNA transfections
Ten milliliters of OT-1 cells was nucleofected with 1 μg of each vector using a mouse T cell Nucleofector kit (VPA-1006, Lonza) and an Amaxa Nucleofector device (program X-01, unstimulated T cells). The nucleofected T cells were rested for 6 h in a mouse T cell Nucleofector medium (VZB-1001, Lonza) and stimulated with CD3/CD28 beads for 24 h before proceeding with a luciferase assay. For small interfering RNA (siRNA) silencing, 1 ml of OT-1 cells was resuspended in 750 μl of Accell siRNA delivery medium (Dharmacon, B-005000) containing 7.5 μl of each siRNA resuspended in 1× siRNA buffer (Dharmacon, B-002000-UB-100). After 48 h, the cells were activated and assayed for Western blot, activation, and proliferation. The following siRNA SMARTpool (Dharmacon) products were used: Notch1, E-041110; Il2rb, E-042082; Psat1, E-056759; Eef1a1, E-042142; Eef2, E-042517; importin 5 (Ipo5), E-042367; and nontargeting control, D-001810.
Real-time quantitative PCR assay
Total RNA was extracted using an RNeasy mini kit (74106, Qiagen). cDNA was made using a SuperScript III first-strand synthesis system (18080-051, Invitrogen, Thermo Fisher Scientific). Analysis was performed by ΔΔCt (Applied Biosystems). TaqMan gene expression assays were as follows: mouse Notch, Mm00627185_m1; human Notch, Hs01062014-m1; mouse Il2rb, Mm00434268_m1; human Il2rb, Hs01081697_m1; mouse Ifngr1, Mm00599890_m1; human Ifngr1, Hs00988304_m1; mouse Stat6, Mm01160477_m1; human Stat6, Hs00598625_m1; mouse Ipo5, Mm00659142_m1; human Ipo5, Hs00267008_m1; Shmt2, Mm00659512_g1; human Shmt2, Hs01059263_g1; mouse Psat1, Mm07293547_m1; human Psat1, Hs00795278_m1; mouse Eef1a1, Mm01973893_g1; human Eef1a1, Hs05015684_g1; mouse Eef2, Mm05700170_g1; human Eef2, Hs00157330_m1; mouse Gapdh, Mm99999915_g1; human actin, Hs01060665_g1.
Immunoblotting assay
Whole-cell lysates were prepared using TNN lysis buffer (50 mM Tris-Cl, 250 mM NaCl, 5 mM EDTA, and 0.5% Nonidet P-40 supplemented with protease inhibitor). Fifteen micrograms of protein was loaded onto SDS-PAGE gels and then transferred onto Immobilon-FL transfer membranes (IPFL00010, Millipore). The Abs used were Notch-1 (3608S), Stat6 (5397S), Shmt2 (33443), eEf2 (2332S), p-Jak family Ab sampler kit (97999T), and Jak3 (8863S) purchased from Cell Signaling Technology; Il2rb (PA5-86323) and Ifngr1 (PA5-96413) purchased from Thermo Fisher Scientific; Psat1 (ab96136), Ipo5 (ab137522), Eef1a1 (ab140632), and granzyme B (ab 255598) purchased from Abcam; and β-actin (A53160 purchased form Sigma-Aldrich). Immunoblots were quantified using ImageJ.
Statistical analysis
All tests for differential translational efficiency were corrected using the Benjamini–Hochberg procedure. For supplemental quality control figures, counts were library size normalized using the 85th quantile. In plotting the sample comparisons, counts were log10 transformed.
Data and software availability
All raw data can be accessed on NCBI SRA under the accession number to be updated. The quantifications for each sample are available on GEO with accession GSE168476. The GEO submission is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168476 and can be accessed by using the following token: yzmnwqyyfxwlzev. To reproduce our XPRESSpipe analysis, the docker image is available on zenodo (10.5281/zenodo.4583883). To rerun our RiboDiff analysis and recreate all plots, all code is provided on GitHub (https://github.com/ratschlab/tcell_activation_dockerized).
Results
First, we assessed the kinetics of signaling events upon CD8+ T cell activation and probed the contribution of transcription or translation using pharmacological inhibitors. We adapted the well-characterized MHC class I–restricted, OVA-specific CD8+ T cells (OT-1) system (43, 44) of ex vivo CD8+ T cell stimulation. Briefly, we expanded naive OT-1 TCR transgenic CD8+ T cells in the presence of OVA peptide–pulsed sublethally irradiated B cells for 7 d. We then cultured these Ag-experienced T cells (also known as central memory T cells based on CD44hi and CD62Lhi staining) in the absence of stimulus for 12 h, before either restimulating them with CD3/CD28 beads or leaving them untreated (Fig. 1A). Immunoblots using Abs against total and phosphorylated signaling proteins detect initial activation of kinase signals within 5–15 min of CD3/CD28 stimulation and activation indicated by Erk, S6, and 4EBP1 phosphorylation at 2 h (Fig. 1B). Flow cytometry confirms activation indicated by increased CD69 at the 2 h time point (p = 0.0001, n = 6) (Fig. 1C). Notably, CD69 detection is not affected by transcriptional block with AMD (1 µM) but is completely abrogated by translation inhibition with CHX (10 μg/ml) (Fig. 1C). The CD44 marker of activation followed the same pattern, and the activation-induced decrease in CD62L was equally unaffected by AMD but was blocked by the translation inhibitor CHX (data not shown). Hence, the early stages of T cell activation depend on translation and are unaffected by the transcriptional blockade.
FIGURE 1.
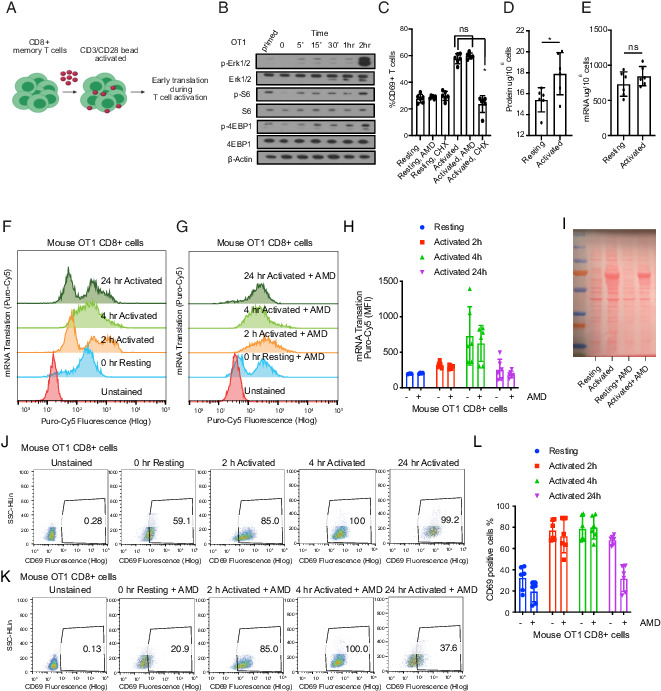
The early stages of T cell activation require translation and not transcription. (A) Schematic of OT-1 cell stimulation. CD8+ OT-1 T cells were primed ex vivo by an exposure to OVA-pulsed sublethally irradiated B cells, then placed to rest overnight and stimulated with CD3/CD28 beads. (B) Immunoblot of signal activation following bead stimulation to determine an early time point to measure translation activation. Similar results were obtained in three sets of experiments. (C) Flow cytometric quantification of T cell CD69 surface marker expression 2 h after activation with or without actinomycin D (AMD) (1 μM) or cycloheximide (CHX) (10 μg/ml) as indicated (n = 6). Individual data points and mean with SD are plotted. (D) Total protein and (E) total RNA content in 1 million OT-1 cells quantified by bicinchoninic acid (BCA) assay and a NanoDrop spectrophotometer, respectively. Data are representative of six experimental replicates. Individual data points and mean with SD are plotted. (F and G) Flow cytometry representative plot of two experiments showing incorporation of Cy5-conjugated puromycin (as mean fluorescence intensity [MFI]) in newly synthesized proteins in activated versus resting T cells, treated with AMD as indicated (data are representative of six biological replicates). Individual data points and mean with SD are plotted. (H) Quantification of flow cytometry data shown in (F) and (G). (I) Ponceau staining of the membrane containing protein lysates from 3 million OT-1 cells at indicated conditions. (J and K) Flow cytometry representative dot plot of two experiments showing CD69 positive staining in activated versus resting T cells, treated with actinomycin D as indicated. (L) Quantification of flow cytometry data shown in (J) and (K). CD69 staining was used (data are representative of six biological replicates). Individual data points and mean with SD are plotted. *p < 0.05.
Next, we measured changes in mRNA and protein contents during memory CD8+ T cell activation. At 2 h, we observed a 43% increase in mean total protein content (2.1 μg/106 cells without stimulus versus 3.7 μg/106 cells upon stimulation; p < 0.02, n = 6) (Fig. 1D). In contrast, we saw only a modest (∼10%) increase in total mean mRNA levels quantified by spectrophotometry (0.205 μg/106 for cells without stimulus versus 0.218 µg/106 for bead-stimulated cells, p < 0.24, n = 6) (Fig. 1E). We used metabolic labeling of newly synthesized proteins with Cy5-conjugated puromycin to separate changes in new protein production from changes in degradation in a time course with and without AMD (45). Quantification of Cy5-conjugated puromycin incorporation by flow cytometry shows a 1.5- to 2-fold increase in new protein synthesis at 2 and 4 h (2 h, p < 0.0010; 4 h, p < 0.06; 24 h, p < 0.5, versus 0 h, n = 6 for each time point), which was unaffected by transcription blockade with AMD at 2 and 4 h, indicating translation of pre-existing mRNAs during early activation (Fig. 1F, 1G, and quantification in (Fig. 1H). We confirmed this result using Ponceau staining of the membrane containing protein lysates from 3 × 106 CD8 T cells (Fig. 1I). Similarly, CD69 measurement by FACS showed that AMD does not affect CD8 T cell activation at the early time points of activation (2 h, p < 0.000022; 4 h, p < 0.000038; 24 h, p < 0.000028, versus 0 h, n = 6 for each time point) (Fig. 1J, 1K, and quantified in (Fig. 1L). In contrast, we saw no increase in total protein synthesis in the OT-1 T cells at these early time points despite evidence of activation such as a CD69 increase (Supplemental Fig. 1). The latter is most consistent with much slower and less efficient activation of total T cells compared with memory T cells, and consistently prior work on the translation in naive T cells had been performed several days after activation and not hours as in our study (19). Hence, memory CD8 T cells show the extensive and early translation of preformed mRNAs into protein within the initial hours following Ag encounter.
We performed transcriptome-scale ribosome footprinting to identify which mRNAs are most strongly recruited into translating ribosomes in the early phase of T cell activation. Briefly, the technique isolates changes in RNA translation by simultaneously measuring total RNA and ribosome-protected mRNA fragments (39, 40). In this manner, we can determine the TE for every mRNA by normalizing ribosome occupancy to the abundance and length of the transcript. We measured triplicates of stimulated (CD3/CD28 beads; 2 h) to unstimulated OT-1 memory T cells (Fig. 2A). Based on quality control data for the RP and RNA-seq and principal component analysis (Supplemental Fig. 2A–C) we excluded one replicate (t = 2 h; replicate no. 1) for the stimulated condition. We aligned ribosomal footprinting reads to the transcript and removed duplicates, noncoding sequences, and rRNA sequences as detailed in Materials and Methods. Read alignments are available on NCBI SRA number SRP309764. The final alignment files used for quantifications are available on GEO accession number GSE168476. The GEO submission is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168476 and can be accessed by using the following token: yzmnwqyyfxwlzev. As expected from the AHA metabolic labeling data, we detect a general shift of mRNAs toward higher translation efficiency indicating an overall increase in protein synthesis (Fig. 2B). For many transcripts (n = 1259) the increase in the ribosome occupancy corresponds to mRNA levels and does not constitute a change in their TE (TE unchanged) (q > 0.1; false discover rate [FDR] > 10%; the correlation between RNA and RF reads, r = 0.81) (Fig. 2B, Supplemental Fig. 2B; GEO accession number GSE168476, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168476). A complete list of differentially translated mRNA detected by RiboDiff analysis is shown in Supplemental Table I. Using strict statistical cutoffs (q < 0.1; FDR < 10%), we identifed 91 genes whose translation is most strongly increased (TE-up group). We also observed 35 genes that are relatively less translated (TE-down group) than expected based on their RNA abundance (Fig. 2B; GEO accession number GSE168476, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168476). The CHX treatment (Fig. 1C) indicates that the main and required effect for T cell activation is an increase in mRNA translation, and therefore we focused on translationally upregulated transcripts (TE-up).
FIGURE 2.
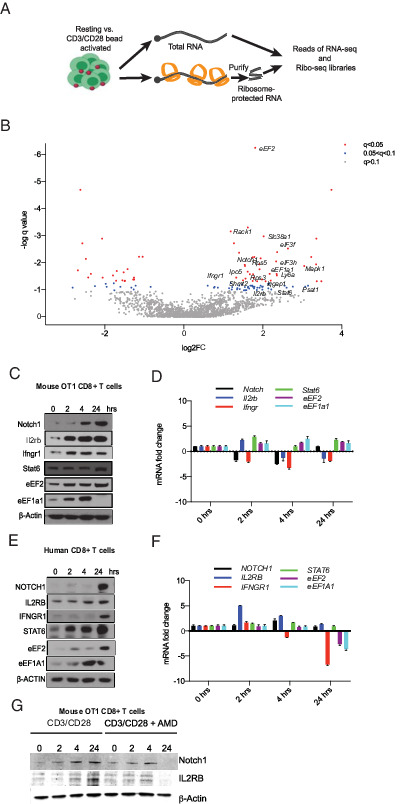
Rapid translational immune response program in CD8 T cells. (A) Schematic of ribosome footprinting study. We compared libraries of total mRNA (n = 3) against ribosome-protected (footprint) sequence reads (n = 3), which provided transcript-specific ribosome occupancy data as a surrogate of translation efficiency (TE). (B) Volcano plot of differential TE in resting and activated T cells, encompassing the q value and TE fold change. Blue dots indicate genes with 0.1 < q < 0.05, and red dots indicate genes with q < 0.05. (C) Western blot analysis of lysates from mouse OT-1 cells activated for the indicated time points. Similar results were obtained from three experiments. (D) mRNA expression levels of indicated genes in the same cells by real-time quantitative PCR (n = 3). (E) Western blot analysis of lysates from human CD8+ T cells activated for indicated time points. Representative blots from three experiments. (F) Corresponding mRNA expression levels (n = 3). (G) Western blot analysis of lysates from mouse OT-1 cells activated for indicated time points with and without actinomycin D (AMD). Similar results were obtained from three experiments.
Known regulators of T cell biology are among the genes that showed the strongest/most significant translational activation. These include Notch1 (a 3-fold increase in translation) and signaling molecules such as IFN-γR (Ifngr1; 2.3 fold), IL-2Rβ (Il2rb; 1.9 fold), and STAT6 (Stat6; 4-fold). We also saw increased production of key translation factors such as eukaryotic elongation factor α1 (eEF1a1; 4.9-fold) and eukaryotic elongation factor 2 (eEF2; 3.5-fold increase), several metabolic genes such as serine hydroxymethyltransferase 2 (Shmt2; 3.2 fold), phosphoserine aminotransferase 1 (Psat1; 2.8 fold), and multiple ribosomal proteins ribosomal protein S5 (Rps5; 2.9-fold) and S3 (Rps3; 3 fold) (Fig. 2B). We directly tested the effects on mRNA (n = 3) and protein levels for some key genes (Notch1, Ifngr, Il2r, Stat6, eEF1a1, eEF2) in the murine OT-1 cells (Fig. 2C, 2D). Next, we found differential effects on protein and RNA (n = 3) abundance in primary human CD8+ T cells isolated from peripheral blood of healthy volunteers (Fig. 2E, 2F). Further AMD treatment did not affect the translational induction of Notch1 and Il2rb at early time points (Fig. 2G).
Ribosome footprinting reveals a concise translational response program that comprises key regulators of T cell signaling, proliferation, and functional activation. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis further categorizes the 91 translationally increased genes into five groups: immune regulation, RNA processing/translation, metabolic, housekeeping, and cell division genes (Fig. 3A). For each transcript, we mapped the precise changes in ribosome occupancy across the entire transcript length. This readily illustrates the loss of ribosome occupancy across the coding sequences for key translation factors (eEF1a1, eEF2), enzymes of serine-glycine catabolism that yields active methyl groups for nucleotide synthesis (Psat1, Shmt2), as well as immune cell receptors (Ifngr1, Il2rb, and Notch1) (Fig. 3B–E). However, we observed no changes in ribosome occupancy in unaffected genes (p < 0.7) such as Tgfb1, Slc1a1, Hif1a, and Rpa1 (Supplemental Fig. 3A). We compared our data on translation efficiency with a recent proteomics (pulsed SILAC) study on activated T cells analyzed at 6 and 24 h (32) and found concordance, especially with respect to translation factors including eEF1A, eEF2, and ribosomal proteins (Supplemental Fig. 2D, 2E).
FIGURE 3.
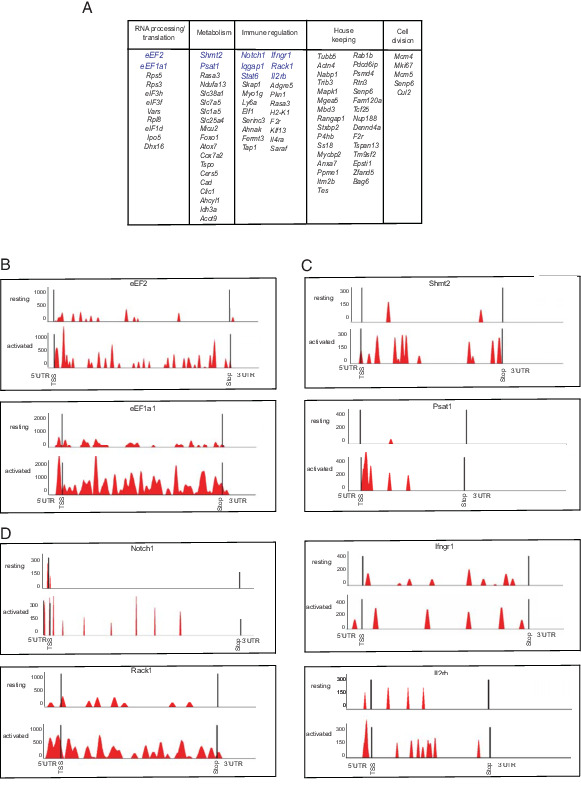
Key regulators of T cell biology are translational targets in the first hours following Ag exposure. (A) Gene Ontology classification of genes within the translation response signature adapted from KEGG pathway analysis (included genes up to q = 0.1; FDR 10%). (B–D) Ribosome coverage data for key transcripts in resting versus activated CD8 T cells based on ribosome footprinting data (n = 3) using gene coverage displays from XPRESSpipe and grouped by KEGG pathway categories.
The translation is primarily controlled at the initiation step through the sequence in the 5′ UTR and recruitment of relevant translation initiation factors (46). To better understand how translation is regulated in activated T cells, we performed an unbiased RNA motif search for differentially enriched or depleted 5′ UTR sequence elements among the TE-up group of genes. Briefly, we used STREME and MEME software algorithms (42) and analyzed an extended list of n = 250 TE-up genes under less stringent statistical criteria (q < 0.28). The 5′ and 3′ UTRs of affected genes showed no significant length prejudice (Supplemental Fig. 2F, 2G). In contrast, our unbiased sequence search against a background gene list (∼500 genes with comparable RNA levels) revealed that a large fraction of TE-up genes have known translation regulatory sequence elements, for example, 74.8% have 8-mer GC-rich motifs (47), 50.4% have 15-mer pyrimidine-rich motifs (48), 8% have mTOR responsive 5′ terminal oligopyrimidine (TOP) tracks sequences (49), and 11.2% have eIF4A RNA helicase–dependent G-quartet (GQ) elements (40) (Fig. 4A, GEO accession number GSE168476, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168476, and Supplemental Table II).
FIGURE 4.
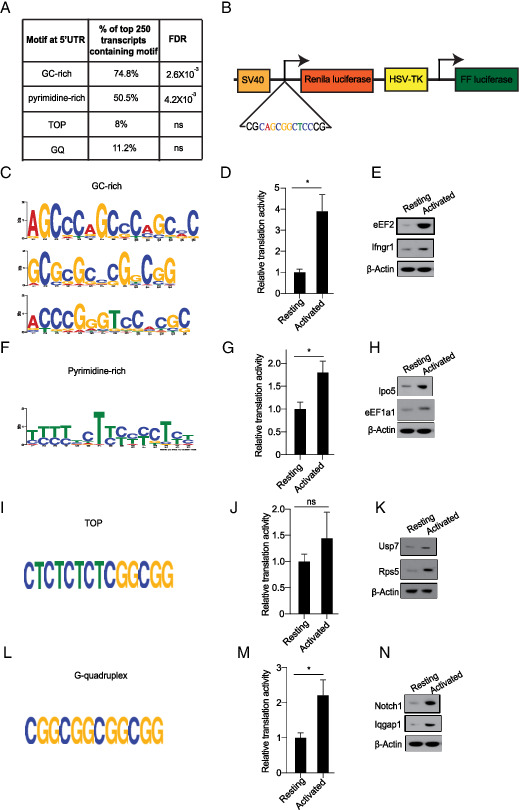
Identification and experimental evaluation of 5′ UTR sequence elements that are overrepresented in the response signature. (A) Percent distribution of indicated sequence motifs in the top 250 genes with increased TE. (B) Graphic representation of a dual-luciferase reporter to test translational function of indicated 5′ UTR elements during OT-1 cell stimulation. (C) GC-rich motifs were identified through STREME analysis. (D) Reporter assay for GC-rich sequences in resting versus activated OT-1 cells. (E) Western blot analysis of indicated genes containing GC-rich motif. (F) Pyrimidine-rich motifs were identified through MEME analysis. (G) Relative translational activity of pyrimidine-rich motif in resting versus activated T cells, quantified as in (D). (H) Western blot analysis of indicated genes containing pyrimidine-rich motif. (I) Schematic representation of TOP motif. (J) The relative translational activity of TOP motif in resting versus activated T cells. (K) Western blot analysis of indicated genes containing TOP motif. (L) Schematic representation of G-quadruplex (GQ) motif. (M) Relative translational activity of GQ motif in resting versus activated T cells. (N) Western blot analysis of indicated genes containing GQ motif. All of the Western blot data are representative of three independent experiments. For all of the luciferase reporter assays, data are representative of three independent experiments. Mean with SD is plotted. *p < 0.05.
Next, we tested the role of specific 5′ UTR sequences that are enriched among translationally upregulated genes in T cells. We built dual (Renilla/firefly) luciferase translation reporters where an experimental UTR use is normalized to the intraplasmid expression of firefly luciferase under the HSV thymidine kinase promoter (Fig. 4B). First, we found a 4-fold induction of translation from a 5′ UTR reporter encoding a GC-rich sequence compared with the control 5′ UTR (n = 3), and this effect is reflected in increased levels of the Ifngr1 and eEF2 proteins upon OT-1 stimulation (Fig. 4C–E). Similarly, we observed significant enrichment of pyrimidine-rich elements, and the translation reporter showed 1.8-fold activation (n = 3) and a corresponding increase in eEF1a1 and Ipo5 proteins that harbor these elements (Fig. 4F–H). The mTOR-responsive TOP element has been implicated in T cell activation (19, 32, 49), and although its enrichment does not reach significance in our profiling data, we can confirm a modest (1.5-fold) activation of a TOP-driven translation reporter (n = 3) and a corresponding increase in TOP-responsive proteins such as Rps5 and the ubiquitin-specific peptidase 7 (Usp7) (Fig. 4I–K). Similarly, eIF4A-dependent translation of GQ-containing UTRs was present in 11% of activated mRNAs but fell short of statistical significance. Testing in the GQ reporter showed a 2.2-fold induction (n = 3) during OT-1 stimulation and increased production of GQ-responsive proteins such as Notch1 and the GTPase-activating protein 1 Iqgap1 (Fig. 4I–N). Other translation reporters encoding A-rich stretches, T-rich stretches, or an internal ribosome entry site were not enriched or activated during OT-1 cell stimulation (Supplemental Fig. 4A–C). Consistently, internal ribosome entry site–controlled proteins such as eukaryotic translation initiation factor 4γ 1 and 2 (eIF4g1 and eIF4g2) showed no increase in protein expression as observed by immunoblot analysis (Supplemental Fig. 4D). Hence, T cell activation triggers translation from several 5′ UTR elements, leading to increased production of the Ifngr1, Notch, eEF2, and eEF1a1 proteins.
We speculate that the early increase in the translation of cytokine receptor genes augments subsequent signaling effects during CD8 T cell activation. To test this translational feed-forward mechanism, we tested to what extent translation and transcription contribute to the expression and signaling activities of the Ifngr1 and Il2 receptors (Fig. 5A). FACS measurements readily confirm increased levels of surface Ifngr1 and Il2rb within 2 h of OT-1 cell activation (n = 3), and this effect was sensitive to CHX and was not affected by AMD, indicating a translational mechanism of receptor production (Fig. 5B–D). Consistently, immunoblots for downstream signaling molecules p-Jak1–p-Jak3 showed the expected increase in phosphorylation upon activation, which is unaltered by AMD and largely blocked by CHX treatment (Fig. 5E and quantified in (Fig. 5F. To further dissect this global effect on mRNA translation, we used siRNAs to target individual proteins whose translation is activated upon T cell stimulation (Fig. 5G). Measurements of Cell Trace Violet dilution showed that only loss of the translation initiation and elongation factors eEF1A1 and eEF2 reduced OT-1 cell proliferation (Fig. 5H). However, knockdown of Notch1, Il2rb, Ipo5, Psat1, and the translation factors eEF1A1 and eEF1 each caused a partial defect in T cell activation with reduced CD69, Il2, and IFN-γ production (p < 0.05, n = 4) (Fig. 5I–K). A more pronounced defect in granzyme B production was observed at 24 h poststimulation, consistent with decimated CD8 T cell function (Fig. 5L).
FIGURE 5.
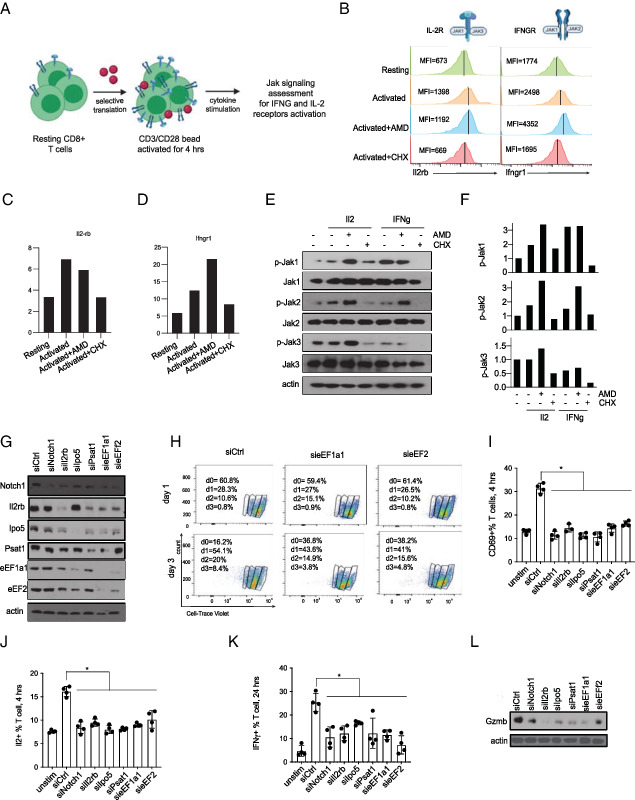
The early translation of cytokine receptors augments subsequent signals and functional CD8 T cell activation. (A) Schematic representation of cytokine stimulation assay to determine the impact of early Il2rb and Ifngr1 translation on subsequent signaling activities. (B) Flow cytometry analysis of Il2rb and Ifngr1 expression (measured as mean fluorescence intensity [MFI]) on OT-1 cells activated for 4 h with or without actinomycin D (AMD) (1 μM) or cycloheximide (CHX) (10 μg/ml) as indicated. Representative histogram from three independent experiments. (C and D) Quantification of the MFI ratio of the Il2rb (C) and Ifngr1(D) receptor expression to unstained control (n = 3). (E) Western blot analysis of lysates from mouse T cell activated for 4 h with or without AMD (1 μM) or CHX (10 μg/ml) as indicated. Mouse recombinant Il2 or IFN-γ was added in the last 30 min before cell collection. (F) Quantification of immunoblot in (E). (G) siRNA-mediated knockdown of indicated proteins; representative blot from three independent experiments. (H) Proliferation of OT-1 cells treated with the indicated siRNAs measured by dilution of Cell Trace Violet stain. The proliferation was assessed by staining OT1 T cells with the fluorescent dye Cell Trace Violet (CTV) and determining the CTV content after 1 or 3 d of stimulation with CD3/CD28 beads. siRNA knockdown for eEF1A1 and eEF2 in T cells leads to reduced ability of T cells to proliferate as indicated with more cells in day 0 at 3 d postactivation. Data are representative of three experiments. (I) FACS analysis for CD69 as a marker of early T cell activation on OT-1 control cells compared with indicated siRNAs (n = 4); individual data points and mean with SD are plotted. (J) FACS analysis for intracellular Il2 as a marker of early T cell functionality in OT-1 control cells compared with indicated siRNAs (n = 4). Individual data points and mean with SD are plotted. (K) FACS analysis for intracellular IFN-γ as a marker of T cell functionality on OT-1 control cells compared with indicated siRNAs (n = 4). Individual data points and mean with SD are plotted. (L) Representatives immunoblot for granzyme B as a late indicator of T cell effector function in control and siRNA-treated cells. *p < 0.05.
Discussion
Our findings provide insight into the early events during CD8 memory T cell activation following Ag encounter. Prior work has described the transcriptional and translational changes 24 h and several days following naive T cell activation (19, 20, 29, 50). We reasoned that much earlier events are likely important in the acute activation of memory CD8 T cells, and new methodologies such as ribosome footprinting and simultaneous RNA-seq allow us to measure gene expression and mRNA translation at these earlier time points. Indeed, the first signaling changes are detectable within minutes of Ag encountered (7, 8), and synthesis of new proteins increases significantly within 2 h and in the absence of transcription, which is a much slower process. This is consistent with the view of translationally poised T cells that contain large amounts of idling ribosomes and a reservoir of mRNAs ready for immediate translation (31–33). Previous studies have relied on the fractionation of heavy and light polysome fractions to identify which RNAs are translated (19, 20). However, the method is cumbersome with low resolution and high experimental variability. The newer method of ribosome footprinting (sometimes called ribosome profiling) has not been applied to acute T cell activation. The method allows precise mapping of ribosomes across every transcript, and the number of ribosomes unit of transcript length yields a measure of the transcript’s translation efficiency (39, 40, 51). Using this approach, we identified a relatively small, high-confidence group of ∼100 mRNAs that are recruited into ribosomes within the first 2 h of Ag exposure. This early translational immune response program includes regulators of T cell functions of known importance such as the RNAs encoding the IFNγR, Il2RB, and Notch1 receptors, or signaling molecules such as Stat6 and Rack1, and metabolic enzymes and transporters (Shmt2, Psat, Slc38a1, Slc7a5, Slc25a4). We also saw an increased production of serval translation factors (eEF2, eEF1a1, eIF3H, eIF3F, eIF1D) and ribosomal proteins (Rps5, Rps3, and Rpl8). Components of this signature indicate activation of an mTOR-related program to increase overall translation capacity (19, 20, 49), and of the serine-glycine catabolism pathway that produces activated methyl groups for biosynthetic and epigenetic reactions (52). Our findings further indicate that the swift production of key cytokine receptors amplifies subsequent signaling events and contributes to the functional activation of memory CD8 T cells. This is consistent with mathematical modeling studies that indicate the particular importance of cellular feed-forward signals in amplifying rapid cellular responses such as memory T cell activation (53–55).
Supplementary Material
Acknowledgments
We thank Morgan Huse (Memorial Sloan Kettering Cancer Center) for advice and reagents. Thanks to all the members of Memorial Sloan Kettering Flow Cytometry Core for support in the processing biological samples. Illustrations were made using BioRender.
This work was supported by the Lymphoma Research Foundation (to H.-G.W.), the Cycle for Survival (to H.-G.W.), the Steven A. Greenberg Foundation (to H.-G.W.), Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, National Institutes of Health Grants RO1CA248168-01, 1RO1CA207217-05, and 1R35CA252982-01 (to H.-G.W.), Starr Cancer Consortium GC260079_(HGW) Core Grant P30 CA008748 (to H.-G.W.), National Institutes of Health Spore P50 CA192937-01A1, Leukemia and Lymphoma Society Score GC227729 (to H.-G.W.), and by the Geoffrey Beene Cancer Research Center (Grant GC261180). D.S. was supported by the Translational Research in Oncology Training Fellowship (Memorial Sloan Kettering Cancer Center). H.-G.W. is a Scholar of the Leukemia and Lymphoma Society. G.R. and N.R.D. received core funding from the Memorial Sloan Kettering Cancer Center and ETH Zürich. V.R.S. is a recipient of the Rally Foundation’s Young Investigator Award and the Department of Defense’s Concept and Career Development Awards.
The online version of this article contains supplemental material.
D.S. and K.S. designed, performed, analyzed experiments, and cowrote the manuscript. N.R.D. and D.K. performed the computational analysis. L.C. performed the footprinting experiment. M.T.L. isolated and provided the OT1 cells for all the experiments. V.R.S. cloned the reporter construct. G.R. supervised the computational analysis. M.J. provided technical assistance. A.V. performed the HiSeq 2000. H.-G.W. designed the study and cowrote the paper and acts as the lead contact.
- AMD
- actinomycin D
- CHX
- cycloheximide
- eEF1a1
- eukaryotic elongation factor α1
- eEF2
- eukaryotic elongation factor 2
- FDR
- false discovery rate
- GEO
- Gene Expression Omnibus
- GQ
- G-quartet
- Ipo5
- importin 5
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- MEME
- multiple expectation maximization for motif elicitation
- NCBI SRA
- National Center for Biotechnology Information Sequence Read Archive
- Psat1
- phosphoserine aminotransferase 1
- RNA-seq
- RNA sequencing
- Rps5
- ribosomal protein S5
- siRNA
- small interfering RNA
- Shmt2
- serine hydroxymethyltransferase 2
- STREME
- sensitive thorough rapid enriched motif elicitation
- TE
- translation efficiency
- TOP
- 5′ terminal oligopyrimidine
- UTR
- untranslated region
Disclosures
The authors have no financial conflicts of interest.
References
- 1. Kay J. E. 1968. Phytohaemagglutinin: an early effect on lymphocyte lipid metabolism. Nature 219: 172–173. [DOI] [PubMed] [Google Scholar]
- 2. Michie C. A., McLean A., Alcock C., Beverley P. C.. 1992. Lifespan of human lymphocyte subsets defined by CD45 isoforms. Nature 360: 264–265. [DOI] [PubMed] [Google Scholar]
- 3. Miyamoto S., Qin J., Safer B.. 2001. Detection of early gene expression changes during activation of human primary lymphocytes by in vitro synthesis of proteins from polysome-associated mRNAs. Protein Sci. 10: 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Radvanyi L. G., Shi Y., Vaziri H., Sharma A., Dhala R., Mills G. B., Miller R. G.. 1996. CD28 costimulation inhibits TCR-induced apoptosis during a primary T cell response. J. Immunol. 156: 1788–1798. [PubMed] [Google Scholar]
- 5. Noel P. J., Boise L. H., Green J. M., Thompson C. B.. 1996. CD28 costimulation prevents cell death during primary T cell activation. J. Immunol. 157: 636–642. [PubMed] [Google Scholar]
- 6. Imboden J. B., Weiss A., Stobo J. D.. 1985. The antigen receptor on a human T cell line initiates activation by increasing cytoplasmic free calcium. J. Immunol. 134: 663–665. [PubMed] [Google Scholar]
- 7. Mayya V., Lundgren D. H., Hwang S. I., Rezaul K., Wu L., Eng J. K., Rodionov V., Han D. K.. 2009. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal. 2: ra46. [DOI] [PubMed] [Google Scholar]
- 8. Navarro M. N., Goebel J., Hukelmann J. L., Cantrell D. A.. 2014. Quantitative phosphoproteomics of cytotoxic T cells to reveal protein kinase D 2 regulated networks. Mol. Cell. Proteomics 13: 3544–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Preston G. C., Sinclair L. V., Kaskar A., Hukelmann J. L., Navarro M. N., Ferrero I., MacDonald H. R., Cowling V. H., Cantrell D. A.. 2015. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. 34: 2008–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lord J. D., McIntosh B. C., Greenberg P. D., Nelson B. H.. 2000. The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans-activation domain of Stat5. J. Immunol. 164: 2533–2541. [DOI] [PubMed] [Google Scholar]
- 11. Wang R., Green D. R.. 2012. Metabolic checkpoints in activated T cells. Nat. Immunol. 13: 907–915. [DOI] [PubMed] [Google Scholar]
- 12. Chapman N. M., Boothby M. R., Chi H.. 2020. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 20: 55–70. [DOI] [PubMed] [Google Scholar]
- 13. Yoon H., Kim T. S., Braciale T. J.. 2010. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One 5: e15423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kay J. E., Ahern T., Atkins M.. 1971. Control of protein synthesis during the activation of lymphocytes by phytohaemagglutinin. Biochim. Biophys. Acta 247: 322–334. [DOI] [PubMed] [Google Scholar]
- 15. Condotta S. A., Richer M. J.. 2017. The immune battlefield: the impact of inflammatory cytokines on CD8+ T-cell immunity. PLoS Pathog. 13: e1006618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pennock N. D., White J. T., Cross E. W., Cheney E. E., Tamburini B. A., Kedl R. M.. 2013. T cell responses: naive to memory and everything in between. Adv. Physiol. Educ. 37: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson M. O., Siska P. J., Contreras D. C., Rathmell J. C.. 2016. Nutrients and the microenvironment to feed a T cell army. Semin. Immunol. 28: 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rathmell J. C. 2012. Metabolism and autophagy in the immune system: immunometabolism comes of age. Immunol. Rev. 249: 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Araki K., Morita M., Bederman A. G., Konieczny B. T., Kissick H. T., Sonenberg N., Ahmed R.. 2017. Translation is actively regulated during the differentiation of CD8+ effector T cells. Nat. Immunol. 18: 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan T. C. J., Knight J., Sbarrato T., Dudek K., Willis A. E., Zamoyska R.. 2017. Suboptimal T-cell receptor signaling compromises protein translation, ribosome biogenesis, and proliferation of mouse CD8 T cells. Proc. Natl. Acad. Sci. USA 114: E6117–E6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kouzine F., Wojtowicz D., Yamane A., Resch W., Kieffer-Kwon K. R., Bandle R., Nelson S., Nakahashi H., Awasthi P., Feigenbaum L., et al. 2013. Global regulation of promoter melting in naive lymphocytes. Cell 153: 988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scharer C. D., Barwick B. G., Youngblood B. A., Ahmed R., Boss J. M.. 2013. Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 191: 3419–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Russ B. E., Olshanksy M., Smallwood H. S., Li J., Denton A. E., Prier J. E., Stock A. T., Croom H. A., Cullen J. G., Nguyen M. L., et al. 2014. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8+ T cell differentiation. [Published erratum appears in 2014 Immunity 41: 1064.] Immunity 41: 853–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goodbourn S. 1994. T-cell activation: transcriptional regulation in activated T cells. Curr. Biol. 4: 930–932. [DOI] [PubMed] [Google Scholar]
- 25. Cheadle C., Fan J., Cho-Chung Y. S., Werner T., Ray J., Do L., Gorospe M., Becker K. G.. 2005. Control of gene expression during T cell activation: alternate regulation of mRNA transcription and mRNA stability. BMC Genomics 6: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crabtree G. R. 1989. Contingent genetic regulatory events in T lymphocyte activation. Science 243: 355–361. [DOI] [PubMed] [Google Scholar]
- 27. Diehn M., Alizadeh A. A., Rando O. J., Liu C. L., Stankunas K., Botstein D., Crabtree G. R., Brown P. O.. 2002. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc. Natl. Acad. Sci. USA 99: 11796–11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Best J. A., Blair D. A., Knell J., Yang E., Mayya V., Doedens A., Dustin M. L., Goldrath A. W., Immunological Genome Project Consortium . 2013. Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat. Immunol. 14: 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaech S. M., Hemby S., Kersh E., Ahmed R.. 2002. Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111: 837–851. [DOI] [PubMed] [Google Scholar]
- 30. Wang R., Dillon C. P., Shi L. Z., Milasta S., Carter R., Finkelstein D., McCormick L. L., Fitzgerald P., Chi H., Munger J., Green D. R.. 2011. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Welsh G. I., Miyamoto S., Price N. T., Safer B., Proud C. G.. 1996. T-cell activation leads to rapid stimulation of translation initiation factor eIF2B and inactivation of glycogen synthase kinase-3. J. Biol. Chem. 271: 11410–11413. [DOI] [PubMed] [Google Scholar]
- 32. Wolf T., Jin W., Zoppi G., Vogel I. A., Akhmedov M., Bleck C. K. E., Beltraminelli T., Rieckmann J. C., Ramirez N. J., Benevento M., et al. 2020. Dynamics in protein translation sustaining T cell preparedness. Nat. Immunol. 21: 927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ricciardi S., Manfrini N., Alfieri R., Calamita P., Crosti M. C., Gallo S., Müller R., Pagani M., Abrignani S., Biffo S.. 2018. The translational machinery of human CD4+ T cells is poised for activation and controls the switch from quiescence to metabolic remodeling. [Published erratum appears in 2018 Cell Metab. 28: 961.] Cell Metab. 28: 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piccirillo C. A., Bjur E., Topisirovic I., Sonenberg N., Larsson O.. 2014. Translational control of immune responses: from transcripts to translatomes. Nat. Immunol. 15: 503–511. [DOI] [PubMed] [Google Scholar]
- 35. Araki K., Turner A. P., Shaffer V. O., Gangappa S., Keller S. A., Bachmann M. F., Larsen C. P., Ahmed R.. 2009. mTOR regulates memory CD8 T-cell differentiation. Nature 460: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hukelmann J. L., Anderson K. E., Sinclair L. V., Grzes K. M., Murillo A. B., Hawkins P. T., Stephens L. R., Lamond A. I., Cantrell D. A.. 2016. The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat. Immunol. 17: 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delgoffe G. M., Kole T. P., Zheng Y., Zarek P. E., Matthews K. L., Xiao B., Worley P. F., Kozma S. C., Powell J. D.. 2009. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30: 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Powell J. D., Delgoffe G. M.. 2010. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 33: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ingolia N. T., Ghaemmaghami S., Newman J. R., Weissman J. S.. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wolfe A. L., Singh K., Zhong Y., Drewe P., Rajasekhar V. K., Sanghvi V. R., Mavrakis K. J., Jiang M., Roderick J. E., Van der Meulen J., et al. 2014. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 513: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berg J. A., Belyeu J. R., Morgan J. T., Ouyang Y., Bott A. J., Quinlan A. R., Gertz J., Rutter J.. 2020. XPRESSyourself: enhancing, standardizing, and automating ribosome profiling computational analyses yields improved insight into data. PLOS Comput. Biol. 16: e1007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bailey T. L., Boden M., Buske F. A., Frith M., Grant C. E., Clementi L., Ren J., Li W. W., Noble W. S.. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37(Web Server): W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hogquist K. A., Jameson S. C., Heath W. R., Howard J. L., Bevan M. J., Carbone F. R.. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76: 17–27. [DOI] [PubMed] [Google Scholar]
- 44. Clarke S. R., Barnden M., Kurts C., Carbone F. R., Miller J. F., Heath W. R.. 2000. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol. Cell Biol. 78: 110–117. [DOI] [PubMed] [Google Scholar]
- 45. Liu J., Xu Y., Stoleru D., Salic A.. 2012. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc. Natl. Acad. Sci. USA 109: 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hinnebusch A. G., Ivanov I. P., Sonenberg N.. 2016. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352: 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Courel M., Clément Y., Bossevain C., Foretek D., Vidal Cruchez O., Yi Z., Bénard M., Benassy M. N., Kress M., Vindry C., et al. 2019. GC content shapes mRNA storage and decay in human cells. eLife 8: e49708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hsieh A. C., Liu Y., Edlind M. P., Ingolia N. T., Janes M. R., Sher A., Shi E. Y., Stumpf C. R., Christensen C., Bonham M. J., et al. 2012. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thoreen C. C., Chantranupong L., Keys H. R., Wang T., Gray N. S., Sabatini D. M.. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485: 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaech S. M., Cui W.. 2012. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Singh K., Lin J., Zhong Y., Burčul A., Mohan P., Jiang M., Sun L., Yong-Gonzalez V., Viale A., Cross J. R., et al. 2019. c-MYC regulates mRNA translation efficiency and start-site selection in lymphoma. J. Exp. Med. 216: 1509–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Parsa S., Ortega-Molina A., Ying H. Y., Jiang M., Teater M., Wang J., Zhao C., Reznik E., Pasion J. P., Kuo D., et al. 2020. The serine hydroxymethyltransferase-2 (SHMT2) initiates lymphoma development through epigenetic tumor suppressor silencing. Nat. Can. 1: 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alon U. 2007. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8: 450–461. [DOI] [PubMed] [Google Scholar]
- 54. Altan-Bonnet G., Mukherjee R.. 2019. Cytokine-mediated communication: a quantitative appraisal of immune complexity. Nat. Rev. Immunol. 19: 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Coward J., Germain R. N., Altan-Bonnet G.. 2010. Perspectives for computer modeling in the study of T cell activation. Cold Spring Harb. Perspect. Biol. 2: a005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data can be accessed on NCBI SRA under the accession number to be updated. The quantifications for each sample are available on GEO with accession GSE168476. The GEO submission is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168476 and can be accessed by using the following token: yzmnwqyyfxwlzev. To reproduce our XPRESSpipe analysis, the docker image is available on zenodo (10.5281/zenodo.4583883). To rerun our RiboDiff analysis and recreate all plots, all code is provided on GitHub (https://github.com/ratschlab/tcell_activation_dockerized).