

Abstract
Autophagy—the lysosomal degradation of cytoplasmic components via their sequestration into double-membraned autophagosomes—has not been detected non-invasively. Here we show that the flux of autophagosomes can be measured via magnetic resonance imaging or serial near-infrared fluorescence imaging of intravenously injected iron oxide nanoparticles decorated with cathepsin-cleavable arginine-rich peptides functionalized with the near-infrared fluorochrome Cy5.5 (the peptides facilitate the uptake of the nanoparticles by early autophagosomes, and are then cleaved by cathepsins in lysosomes). In the heart tissue of live mice, the nanoparticles enabled quantitative measurements of changes in autophagic flux, upregulated genetically, by ischaemia–reperfusion injury or via starvation, or inhibited via the administration of a chemotherapeutic or the antibiotic bafilomycin. In mice receiving doxorubicin, pre-starvation improved cardiac function and overall survival, suggesting that bursts of increased autophagic flux may have cardioprotective effects during chemotherapy. Autophagy-detecting nanoparticle probes may facilitate the further understanding of the roles of autophagy in disease.
Autophagy refers to the sequestration of a portion of the cytoplasm to form double-membraned autophagosomes, and the subsequent trafficking of these autophagosomes to lysosomes, where their contents are degraded1,2. Reduced levels of autophagy have been implicated in neurodegenerative disease, cardiovascular disease and cancer3–5. However, autophagy can also be a primary mechanism of cell death. A delicate balance exists between the protective and deleterious effects of autophagy6–8, and a compelling need, therefore, exists to detect and measure this process in vivo.
Fluorescent forms of LC3 (autophagy protein light chain 3) can be used to measure autophagy in cells and on the surface of exposed tissues9,10. However, this approach requires genetic transfection of cells or animals, does not always provide a measure of autophagic flux11 and does not support the detection of autophagy in vivo, particularly in deep tissues such as the heart. The aim of this study was, therefore, to develop an autophagy-detecting nanoparticle (ADN), capable of imaging autophagy and measuring its flux non-invasively in vivo.
In this Article, the validation of ADN was performed using classic stimulators and inhibitors of autophagy, a D-isomer of the probe, and cells with constitutive/induced deficiencies in critical autophagy proteins. The imaging of ADN in the intact heart was performed in mice after ischaemia–reperfusion (IR) and starvation and in a transgenic model with cardiomyocyte-specific upregulation of autophagy. The ability to detect the magnetic and fluorescent signatures of ADN in vivo was assessed, and the probe was used to measure autophagy flux in vivo under conditions that stimulate (starvation) or inhibit (bafilomycin (Baf) and doxorubicin (Dox)) the process. In addition, the imaging of ADN was used to guide the modulation of cardiomyocyte autophagy during cardiotoxic chemotherapy, and the impact of this strategy on cardiac function and overall survival was assessed.
Results
Rational design of the NPs.
The core of ADN consists of ferumoxytol (Feraheme, FH), an iron oxide nanoparticle (NP) that, we hypothesized, would be engulfed by autophagosomes when free in the cytoplasm12,13. To facilitate direct entry of the NP into the cell, the surface of FH was decorated with arginine-rich peptides14–16. This also enhances the uptake of the probe by early autophagosomes17,18, and promotes endosomal escape/release into the cytoplasm of NPs that enter the cell via endocytocis12,19–21. A near-infrared (NIR) fluorochrome (Cy5.5, Lumiprobe) was conjugated to the N-terminus of each peptide to provide an activatable fluorescent signature. The labelling of FH is covalent, with a copper-free click reaction, and resulted in an average of five peptide–fluorochrome moieties per NP (Fig. 1a). In parallel, a control NP (FH–Cy5.5) with similar physical properties (Supplementary Table 1 and Supplementary Fig. 1), but without the polyarginine linker, was also synthesized.
Fig. 1 |. Characterization of the ADN and its in vitro validation.
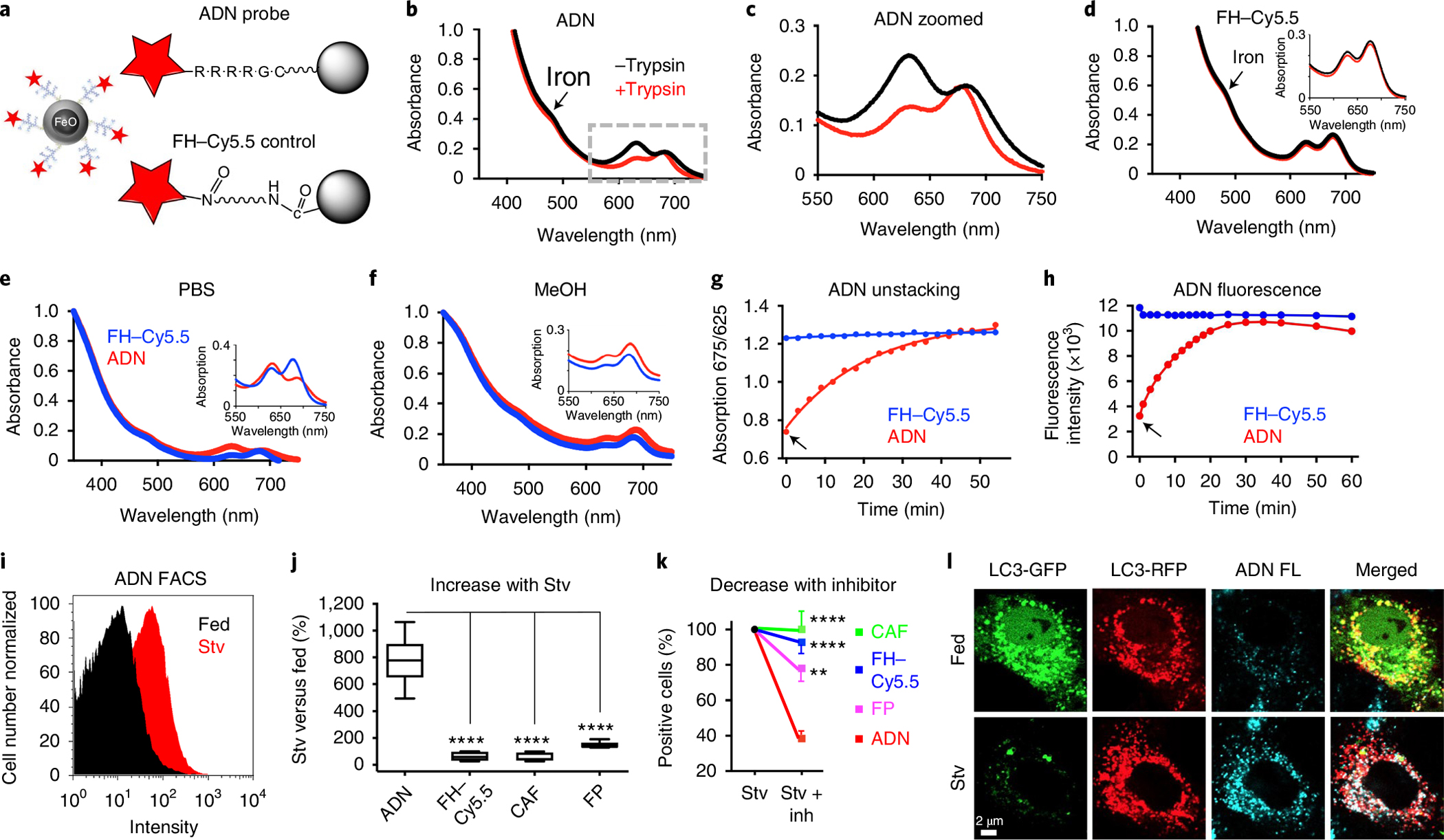
a, Schematic of ADN: the iron oxide NP ferumoxytol (FH) is decorated with cathepsin-cleavable polyarginine peptides, each conjugated to the NIR fluorochrome Cy5.5. For the control NP, Cy5.5 is conjugated directly to FH without an intervening peptide. b,c, Absorbance spectra of ADN showing a shift in the Cy5.5 peak (625 versus 675 nm) in its unactivated (black) and trypsin-activated (red) states, full spectrum (b) and zoomed in spectrum (c). d, No spectral shifts are seen with FH–Cy5.5. e,f, The absorbance spectra of unactivated ADN and FH–Cy5.5 differ in aqueous solution (e) but not in methanol (MeOH) (f). g,h, Time course of ADN activation following trypsin exposure (arrows) measured by the unstacking (spectral shift) of Cy5.5 and its signal intensity, respectively. i, FACS histogram showing ADN activation in starved (Stv) H9C2 cells. j, The increase in fluorescence in starved versus fed cells is significantly greater with ADN (n = 10) than other constructs: FH–Cy5.5 (n = 8), CAF (n = 6) and free fluorescent polyarginine peptide (FP; n = 7). In the plot, the box extends from the 25th to the 75th percentile, the line in the box indicates the median and the whiskers represent the minimum and maximum. k, Likewise, when the autophagy inhibitors E64d and pepstatin A are added to starved cells, the reduction in fluorescence (versus no inhibitors) is significantly greater with ADN (n = 7) compared with other constructs: FH–Cy5.5 (n = 7), CAF (n = 9) and FP (n = 7). Mean ± s.e.m. is plotted for Stv + Inh. For j and k, one-way ANOVA with Tukey’s post-test was performed. **P = 0.0017 and ****P < 0.0001. l, Confocal microscopy of H9C2 cells transfected with a dual GFP–RFP fluorescent LC3 plasmid (pTF-LC3). When autophagy flux is induced through starvation (Stv, bottom row), the LC3-GFP signal is lost owing to the low pH of the lysosome, while the LC3-RFP signal persists. ADN fluorescence (FL, blue) co-localizes with the red fluorescence from those autophagosomes that have undergone flux to the lysosome. The microscopy experiments were repeated in three biologically independent samples with consistent results. Scale bar, 2 μm.
The upregulation of cathepsin activity in the lysosomal compartment during autophagy22–24 is exploited by ADN through the attachment of an NIR fluorochrome (Cy5.5) to FH via the cathepsin-cleavable arginine-rich peptides. Absorption spectrophotometry revealed that the Cy5.5 fluorochrome on ADN had a prominent peak at 625 nm (Fig. 1b,c), which reverted to 675 nm upon activation of the probe with trypsin. This spectral shift, consistent with fluorochrome stacking and the quenching of Cy5.5 fluorescence in intact ADN, was not seen with the control FH–Cy5.5 (Fig. 1d), or when ADN was placed in methanol (Fig. 1e,f). Cleavage of the peptide by cathepsins/proteases resulted in the detachment of Cy5.5 from the FH–peptide construct and robust fluorescence. The time constant of ADN activation in vitro was similar when measured by the ratio of the 675/625 nm peaks (Fig. 1g) or the intensity of the fluorescence signal (Fig. 1h).
The NPs detect autophagy activation and flux in cardiomyocytes.
To determine the ability of ADN to detect cardiomyocyte autophagy, we exposed H9C2 cells to 24 h of nutrient deprivation in serum-free medium, a potent stimulator of autophagy25,26. Flow cytometry showed that ADN fluorescence was shifted to the right in starved H9C2 cells, compared with fed cells (Fig. 1I), consistent with the uptake and activation of the probe. The number of ADN-positive cells increased 7.7 ± 1.8-fold with starvation (Stv), which was significantly greater (P < 0.0001) than the change seen with the control constructs including FH–Cy5.5, a cathepsin-activatable fluorochrome (CAF; ProSense, PerkinElmer) or the free polyarginine–Cy5.5 peptide (Fig. 1j). The increase in ADN fluorescence in starved cells was attenuated by the autophagy/lysosomal protease inhibitors E64d and pepstatin A, demonstrating the molecular specificity of ADN activation (Fig. 1k). To further confirm the specificity of ADN for autophagy, we transfected H9C2s with the dual green/red fluorescent LC3 plasmid10. In fed cells, both green and red fluorescent LC3 was evident, and little ADN fluorescence was seen (Fig. 1l). Upon starvation, LC3 is translocated to the acidic lysosomal environment, where the pH-sensitive green fluorescence disappears, while the pH-insensitive red fluorescent LC3 persists. A high degree of co-localization was seen between ADN and red fluorescent LC3, confirming that ADN is a marker of cells with enhanced autophagic flux.
Specificity of the NPs for autophagy.
To further demonstrate the specificity of ADN for autophagy, we synthesized a D-isomer of the probe (FH-D-iso), in which the peptides in the amino acid were changed from an L-configuration (cathepsin-cleavable) to a D-configuration (non-cleavable). The structure of the active and D-isomer probes was otherwise identical (Supplementary Fig. 2). Unlike ADN, the exposure of FH-D-iso to protease/trypsin did not alter its absorbance spectra or result in the emission of fluorescence (Fig. 2a,b). In H9C2 cells, the signal from ADN but not from FH-D-iso increased with starvation (Fig. 2c,d and Extended Data Figs. 1 and 2).
Fig. 2 |. Specificity of ADN activation and its uptake in early autophagosomes.
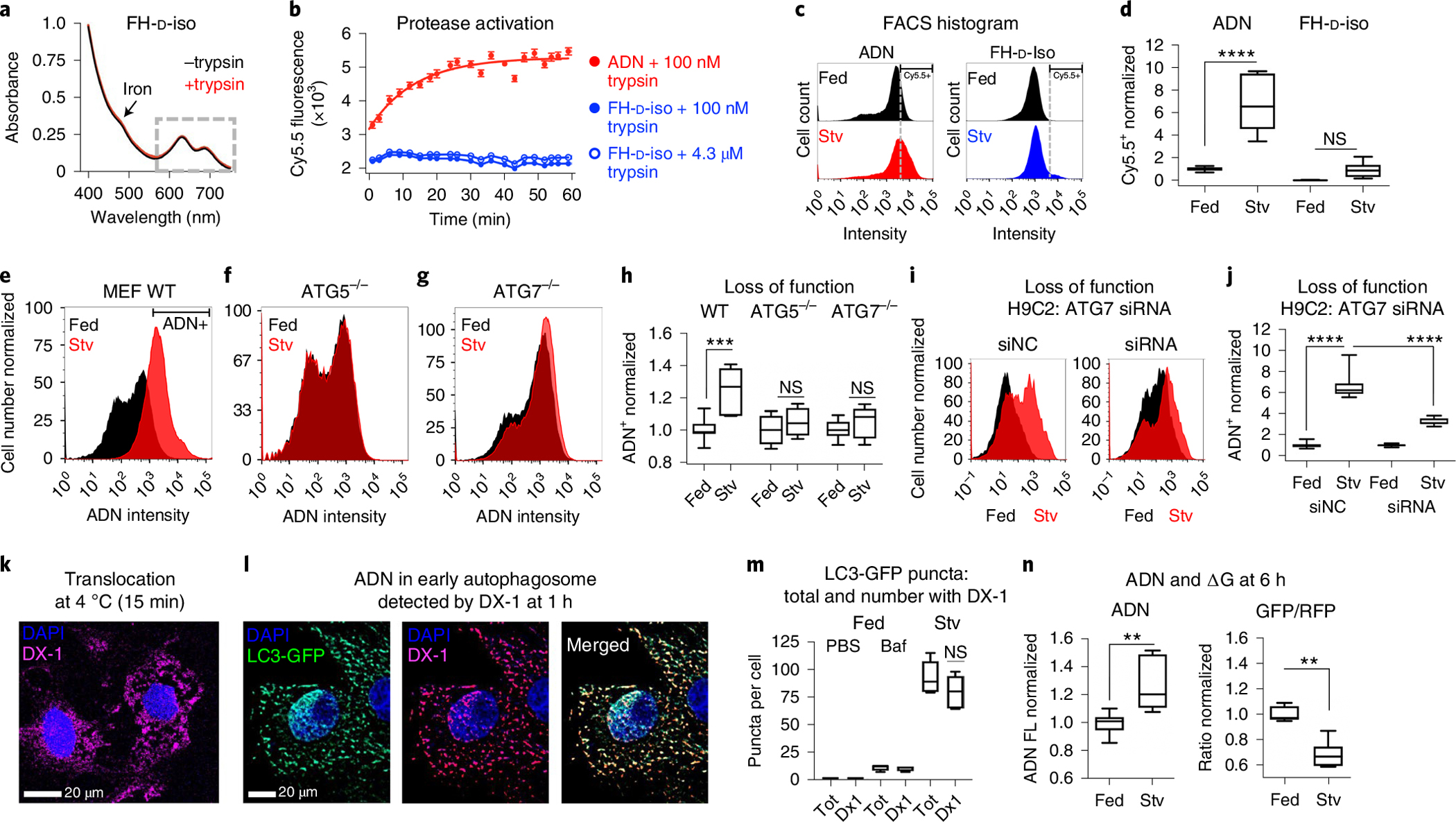
a–d, Comparison of ADN and its D-isomer (FH-D-iso): no shift in the absorbance spectra of FH-D-Iso is seen after trypsin treatment (a); while trypsin activates ADN fluorescence, no increase in FH-D-Iso fluorescence is seen, even at high concentrations of trypsin (n = 3 for each condition; data are shown as mean ± s.e.m.) (b); in H9C2 cells, robust activation of ADN (n = 7), but not FH-D-Iso (n = 8), is seen with starvation (Stv, dashed line indicates Cy5.5 threshold; one-way ANOVA with Tukey post-test was performed, ****P < 0.0001; box plots show minimum, maximum, median and 25–75% percentiles) (c,d). NS, not significant. e–j, ADN is not activated in MEFs (e–h) or H9C2 cells (i,j) deficient in ATG5 or ATG7: ADN is strongly activated by wild-type (WT, n = 7) MEFs upon starvation (e) but not by ATG5−/−, n = 4 (f), or ATG7−/−, n = 8 MEFs (g). h, Quantification of ADN activation in WT MEFs versus ATG5−/− or ATG7−/− MEFs. Comparison with one-way ANOVA with Tukey post-test, ***P = 0.0004. Box plots show minimum, maximum, median and 25–75% percentiles. In i–j, H9C2 cells were treated with non-coding siRNA (siNC, n = 11) and siRNA to ATG7 (n = 11). ADN activation with starvation is robust with exposure to siNC but highly attenuated by ATG7-targeted siRNA. Comparison with one-way ANOVA with Tukey post-test, ****P < 0.0001. Box plots show minimum, maximum, median and 25–75%. k–m, Uptake of ADN by early autophagosomes detected via the DX1 antibody to its surface dextran. ADN can enter the cytoplasm via endosomal escape or by direct translocation: within 15 min, the probe is diffusely distributed in the cytoplasm of starved H9C2 cells that were pre-cooled to 4 °C for 2 h to eliminate endocytosis (k); at 1 h, the ADN in starved H9C2s that express LC3-GFP, a marker of early autophagosome formation, is punctate and co-localizes strongly with LC3-GFP, where the number of LC3-GFP puncta with co-localized DX-1(ADN) signal increased from a median [and interquartile range] of 1 [1–2] in fed H9C2 cells to 11 [8–12] in fed H9C2 cells exposed to Baf, and 80 [65–94] in starved H9C2 cells, and a median of 92% [90–100%] of the LC3-GFP puncta in the baf-exposed cells, and 85% [81–90%] in the starved cells, stained positively for DX-1 (ADN) (comparison with one-way ANOVA with Tukey post-test; box plots show minimum, maximum, median and 25–75% percentiles) (l,m). Tot, Total. n = 5 biologically independent samples with consistent results. n, Activation of ADN (Cy5.5 fluorescence) after 6 h of starvation in H9C2 cells expressing the ATG4B-sensitive dual-fluorescent-LC3 ΔG construct. The activation of ADN (n = 7 fed versus n = 7 Stv, **P = 0.0012, two-tailed Mann–Whitney test) correlates well with the reduction in the GFP/RFP ratio of the ΔG reporter (n = 6 fed versus n = 7 Stv, **P = 0.0012, two-tailed Mann–Whitney test) consistent with autophagic flux. Box plots show minimum, maximum, median and 25–75% percentiles.
We next tested the response of ADN to genetic deficiency of the autophagy proteins ATG5 and ATG7. In wild-type murine embryonic fibroblasts (MEFs), ADN was strongly activated by starvation (Fig. 2e and Extended Data Fig. 3), while in MEFs with genetic deletion of ATG5 or ATG7 (ref. 27), no significant change in the signal was seen (Fig. 2f–h and Extended Data Figs. 4 and 5). A lack of ADN activation was also seen in H9C2 cells with ATG7 knockdown by siRNA (Fig. 2i,j, Extended Data Fig. 6 and Supplementary Fig. 3). To better characterize the kinetics of ADN before lysosomal activation, an antibody (DX-1) (ref. 28) to its surface dextran coating was used. Large amounts of ADN were seen in the cytoplasm of starved H9C2 cells within 15 min of exposure, even in cells pre-cooled to 4 °C to eliminate endocytocis (Fig. 2k). By 1 h, ADN was concentrated into early autophagosomes and co-localized with >80% of LC3-GFP punctates in the cytoplasm (Fig. 2l,m). Electron microscopy confirmed the uptake of ADN by early autophagosomes (Supplementary Fig. 4).
In an additional validation step, we compared the signal from ADN activation in lysosomes with changes in H9C2s expressing the ATG4B protease-specific fluorescent ΔG reporter (Fig. 2n)29. The increase in ADN signal closely mirrored the reduction in the LC3 GFP/RFP ratio in the ΔG probe due to autophagic flux. Collectively, these data show that ADN can enter cells via direct translocation and is internalized by early autophagosomes, trafficked to the lysosome and activated there by lysosomal cathepsins.
Validation of the NPs in murine models of cardiomyocyte autophagy.
We next tested the ability of ADN to image cardiomyocyte autophagy in the murine heart ex vivo using three model systems with increasing specificity for autophagy: (1) IR injury, (2) starvation/fasting and (3) transgenic mice with cardiomyocyte-specific upregulation of autophagy. The mice with IR were co-injected with ADN and a CAF at the onset of reperfusion and imaged within 4 h (Fig. 3a)23. This is well before the onset of an inflammatory cell infiltrate in the myocardium, which would reduce the specificity of ADN activation23,30. At 4 h after reperfusion, ADN but not CAF fluorescence was seen in the ischaemic area at risk, delineated by the lack of fluorescent microspheres (Fig. 3b,c). The target-to-background ratio (TBR) between the area at risk and the un-injured septal wall was significantly elevated with ADN but not CAF (4.6 ± 1.3 versus 1.4 ± 0.6; P < 0.01).
Fig. 3 |. Imaging of cardiomyocyte autophagy ex vivo with ADN is both sensitive and specific.
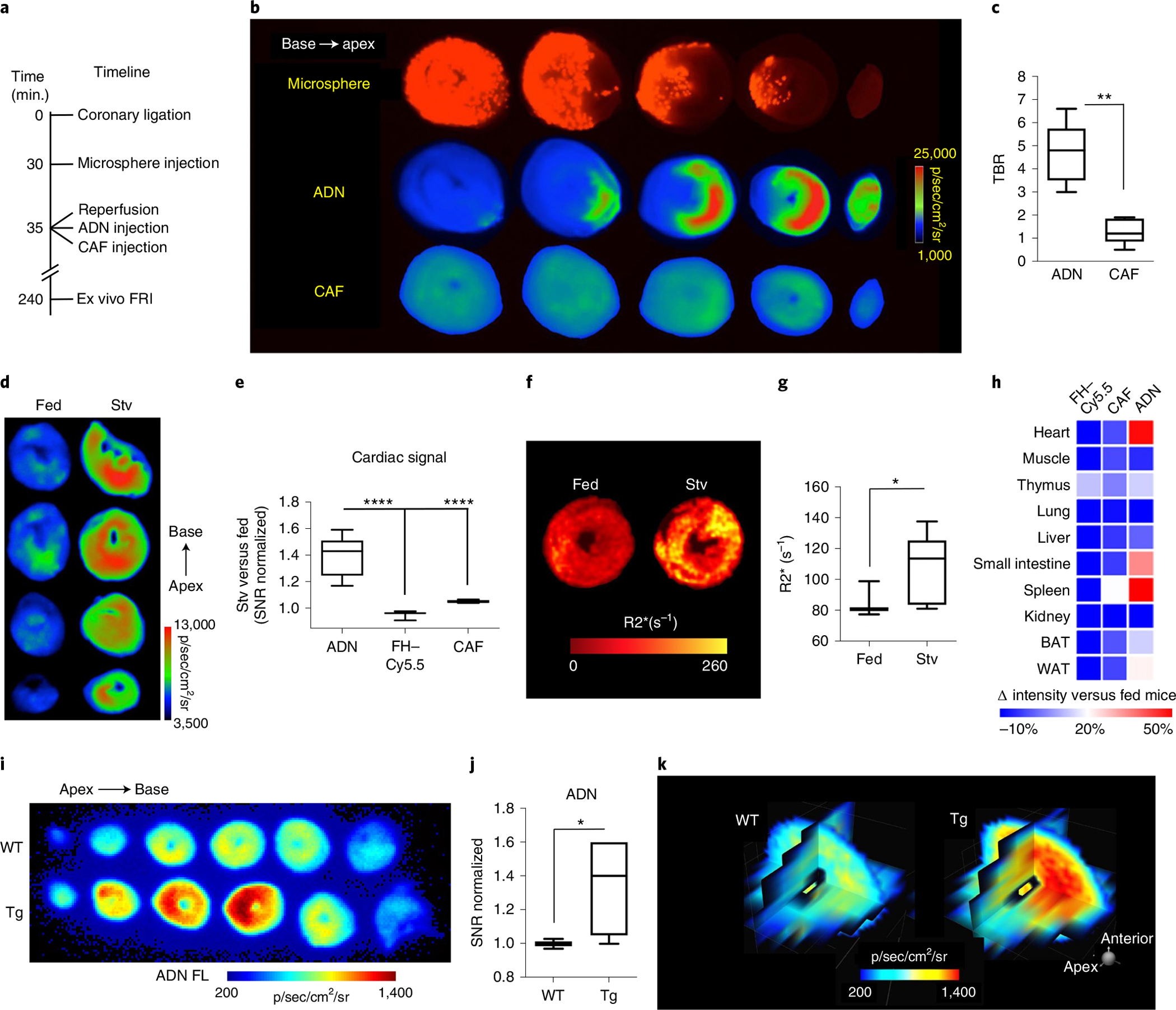
a–c, IR: experimental scheme (a); short-axis slices of a mouse heart showing robust ADN activation during IR, where the area at risk is delineated by the absence of microspheres and correlates strongly with the region of ADN activation and, in contrast, very low fluorescence is seen with CAF in the same mouse heart. Fluorescence intensity is expressed as p/sec/cm2/sr (radiance unit of photons per second per square centimeter per steradian) (b); the sensitivity of ADN to IR-induced autophagy is significantly higher than CAF (c). N = 5 mice for ADN and CAF, **P = 0.0079, two-tailed Mann–Whitney test. Box plots show minimum, maximum, median and 25–75% percentiles. d–h, ADN activation in mice with starvation-induced autophagy: an increase in cardiac fluorescence is seen only in mice injected with ADN after starvation (n = 15 mice), and not in fed mice (n = 10) or starved mice injected with CAF (n = 6 mice) or FH-Cy5.5 (n = 3 mice) (****P < 0.0001, one-way ANOVA with Tukey post-test; box plots show minimum, maximum, median and 25–75% percentiles (d,e); detection of ADN with MRI via its effect on transverse magnetic relaxation, with short-axis R2* maps at the mid-left ventricle (LV) showing a significant increase in R2* in starved versus fed mice (N = 7 mice in each of the fed and the Stv groups, *P = 0.0262, two-tailed Mann–Whitney test; box plots show minimum, maximum, median and 25–75% percentiles) (f,g); systemic profiling of ADN, CAF and FH-Cy5.5 in starved mice versus fed controls, where the activation of ADN in the heart, small intestine and spleen reflects the upregulation of autophagy in these organs during starvation (h). i–k, Transgenic mouse model characterized by cardiomyocyte-specific upregulation of autophagy: ex vivo short-axis slices of a littermate negative (WT, top) and DDiT4L+ mouse (Tg, bottom) (i); robust activation of ADN is seen in the DDiT4L-overexpressing mice, confirming the molecular specificity of the probe (N = 4 Tg versus N = 5 WT mice, *P = 0.033, two-tailed unpaired t-test. Box plots show minimum, maximum, median and 25–75% percentiles) (j); multiplanar reformats of a wild-type (WT) and transgenic heart (Tg) (k).
In mice exposed to 24 h of starvation (Stv), ADN activation was seen throughout the myocardium and fluorescence intensity was 1.4 ± 0.1-fold (P < 0.0001) higher in starved mice than in fed mice (Fig. 3d,e). No increase in fluorescence was seen in starved mice with the control FH–Cy5.5 or CAF constructs. As ADN contains a superparamagnetic NP, we sought to determine whether the increase in autophagy during starvation would result in probe aggregation (Supplementary Fig. 5) that could be detected with magnetic resonance (Fig. 3f,g). Maps of the transverse magnetic relaxation rate (R2*) in the heart showed significant differences between the starved and fed mice (107.1 ± 22.7 s−1 versus 83.0 ± 7.1 s−1, P < 0.05). ADN fluorescence during starvation was most increased in the heart, small intestine and spleen, suggesting that autophagy is particularly upregulated in these tissues during starvation (Fig. 3h). Starvation did not produce an inflammatory response in the heart (Supplementary Fig. 6).
We have recently reported the development of a transgenic mouse with cardiomyocyte-specific overexpression of the transcription factor DNA-damage-inducible transcript 4-like (DDiT4L) and autophagy31. In DDiT4L transgenic mice, ADN fluorescence was significantly increased in the myocardium (1.26 ± 0.21 versus 1.00 ± 0.03, P < 0.05) normalized to littermate controls (Fig. 3i,j). Three-dimensional multiplanar representation of the heart showed robust ADN fluorescence in transgenic mice but not in control mice (Fig. 3k). This result, in conjunction with the results obtained in IR, starvation and in H9C2 cells, demonstrates that ADN is a sensitive and specific marker of autophagic flux.
The NPs detect reduced autophagy flux in myocytes exposed to Dox.
While some reports have suggested that Dox toxicity may be caused by an increase in cardiomyocyte autophagy32, others have suggested that it reduces flux33,34. We aimed here to further interrogate this using ADN. H9C2 cells were exposed to Dox for 24 h, and autophagy flux was measured by western blot and fluorescence-activated cell sorting (FACS). As shown in Fig. 4a–d, Dox exposure reduced the autophagy protein LC3 I (19.2 ± 0.1% versus vehicle control; P < 0.05), had no effect on LC3 II (P = 0.32) and significantly increased p62 levels (8.3 ± 0.03% versus vehicle control; P < 0.01). Exposure of H9C2 cells to Dox significantly reduced the number of ADN-positive cells by FACS and significantly increased the annexin-V-positive population (Fig. 4e,f and Extended Data Figs. 7 and 8). These data are consistent with Dox causing a late block in autophagy flux11 and an increase in apoptotic cell death.
Fig. 4 |. ADN detects a reduction in autophagy when H9C2 cardiomyocytes are exposed to Dox and a cytoprotective effect when autophagy is restored.
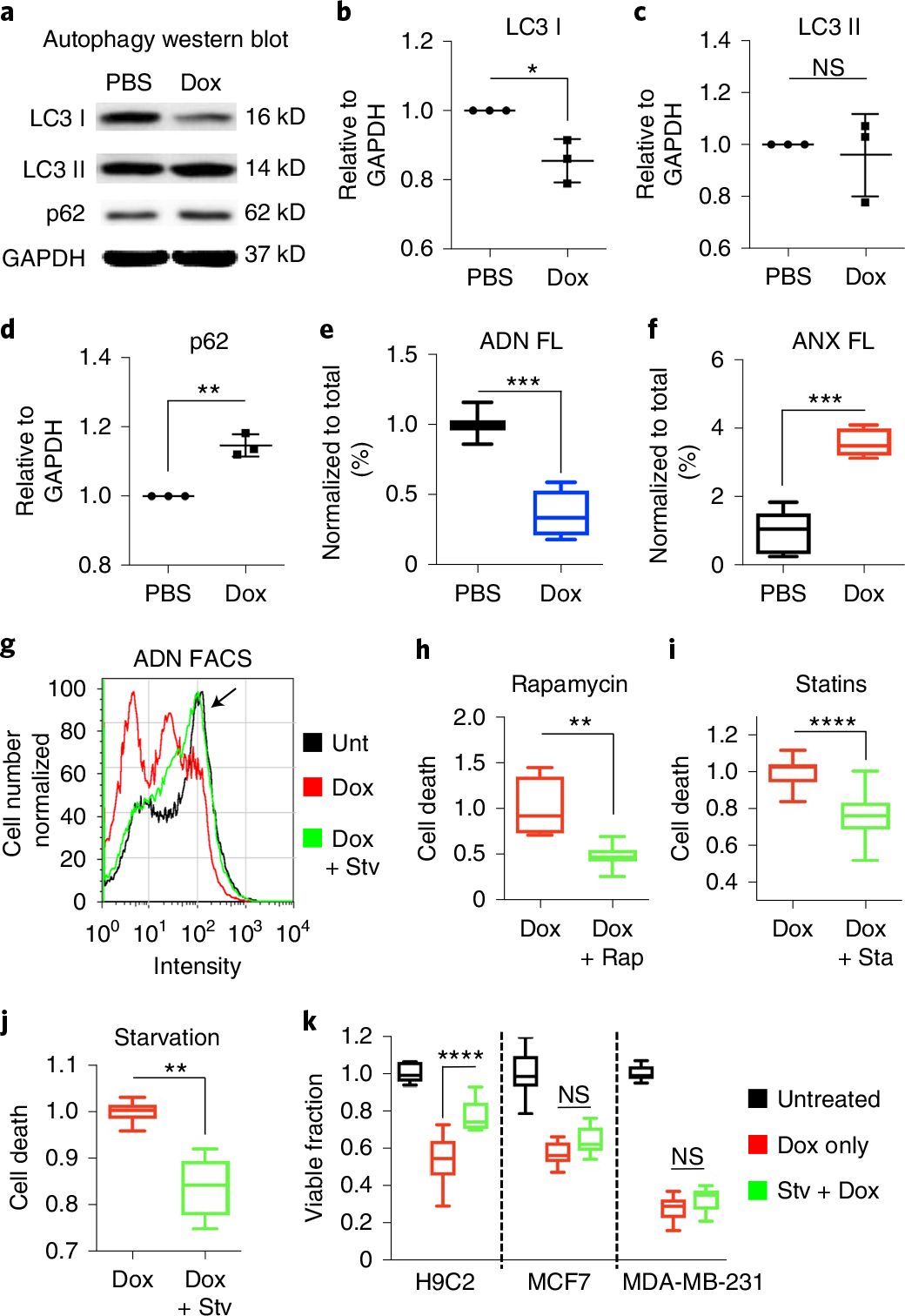
a–e, Dox exposure inhibits autophagy in H9C2 cells: LC3 I is reduced (n = 3 in each PBS/Dox group, *P = 0.0162, two-tailed unpaired t-test), LC3 II is unchanged, p62 is increased (n = 3 in each PBS/Dox group, **P = 0.0014, two-tailed unpaired t-test) and ADN fluorescence is more than twofold decreased (n = 7 in PBS and n = 9 Dox groups, ***P = 0.0002, two-tailed Mann–Whitney test), consistent with a late block of autophagy flux. Uncropped gels are shown in Source Data Fig. 1. In b–d, mean ± s.d. is plotted; in e and f, box plots show minimum, maximum, median and 25–75% percentiles. f, A corresponding increase in cell death (ANX fluorescence) is seen (n = 6 in PBS and n = 9 in Dox groups, ***P = 0.0004, two-tailed Mann–Whitney test). g, Histogram of ADN fluorescence in H9C2 cells exposed to PBS, Dox alone and Dox plus nutrient deprivation/starvation. The peak in the histogram of the Dox + Stv cells is shifted back towards the ADN peak of untreated cells (arrow), consistent with a restoration of autophagy. h–j, The stimulation of autophagy by rapamycin (Dox n = 3 and Dox + Rap n = 9, **P = 0.0091, two-tailed Mann–Whitney test) (h), statins (Dox n = 12 and Dox + Sta n = 12, ****P = < 0.0001, two-tailed Mann–Whitney test) (i) and starvation (Dox n = 6, Dox + Stv n = 6, **P = 0.0022, two-tailed Mann–Whitney test) (j) all reduce cell death (annexin-positive fraction) in H9C2 cells exposed to Dox. Box plots show minimum, maximum, median and 25–75% percentiles. k, In H9C2 cells, starvation also increases cell survival/viability by methylthiazol tetrazolium (MTT) after Dox exposure. However, in common human cancer cell lines (MCF7 and MDA-MB-231), no attenuation in the cytotoxicity (loss of viability by MTT) of Dox is seen with the induction of autophagy through starvation. H9C2 n = 6, MCF7 n = 9 and MDA-MB-231 n = 12 for all conditions. One-way ANOVA with Tukey post-test was performed. ****P < 0.0001. Box plots show minimum, maximum, median and 25–75% percentiles.
We next exposed Dox-treated H9C2 cells to autophagy-promoting stimuli including starvation, rapamycin and statins35. The activation of autophagy by these stimuli was highly detectable by ADN. Autophagy activation by starvation restored the ADN-positive population on FACS to levels comparable to cells not exposed to Dox (Fig. 4g). The induction of autophagy with starvation, rapamycin and statins all significantly reduced the population of annexin-positive cells (Fig. 4h–j and Extended Data Fig. 9). Starvation, for instance, produced a 16% reduction in cell death (P < 0.001) in Dox-treated H9C2 cells (Fig. 4j). Starvation had no impact on the cytotoxic effects of Dox on two common human cancer cell lines, MCF-7 and MDA-MB-231 (Fig. 4k).
Imaging of autophagy in mice dosed with Dox correlates with animal survival.
The impact of Dox on cardiomyocyte autophagy in vivo was tested in mice under two dosing conditions: a single high dose followed by imaging 24 h later and a chronic dosing regimen over 5 weeks. In mice challenged with a single dose of Dox (15 mg kg−1), ADN and fluorescent annexin V (ANX) were injected intravenously at 24 h and the heart was imaged ex vivo 4 h later by reflectance imaging (Fig. 5a)36. The inherent fluorescence of Dox was exploited to create distribution maps of its uptake in the heart37, which was highly homogeneous (Fig. 5a). Dox significantly increased ANX fluorescence and decreased ADN fluorescence (Fig. 5b,c). Starvation for 24 h to activate autophagy before the Dox was given restored the ADN signal to basal levels, comparable to that in control mice injected with PBS. The rescue of autophagy with pre-starvation was also associated with a reduction in cardiomyocyte apoptosis (Fig. 5a–c). The beneficial result of starvation was not due to reduced uptake or retention of Dox in the myocardium, since Dox fluorescence was similar in the fed and starved mice (Fig. 5d). In both the fed and the starved mice, the degree of Dox fluorescence correlated inversely with the signal from ADN (Fig. 5e).
Fig. 5 |. Starvation before Dox challenge in mice restores basal levels of autophagy, attenuates apoptosis, enhances cardiac function and improves overall survival.
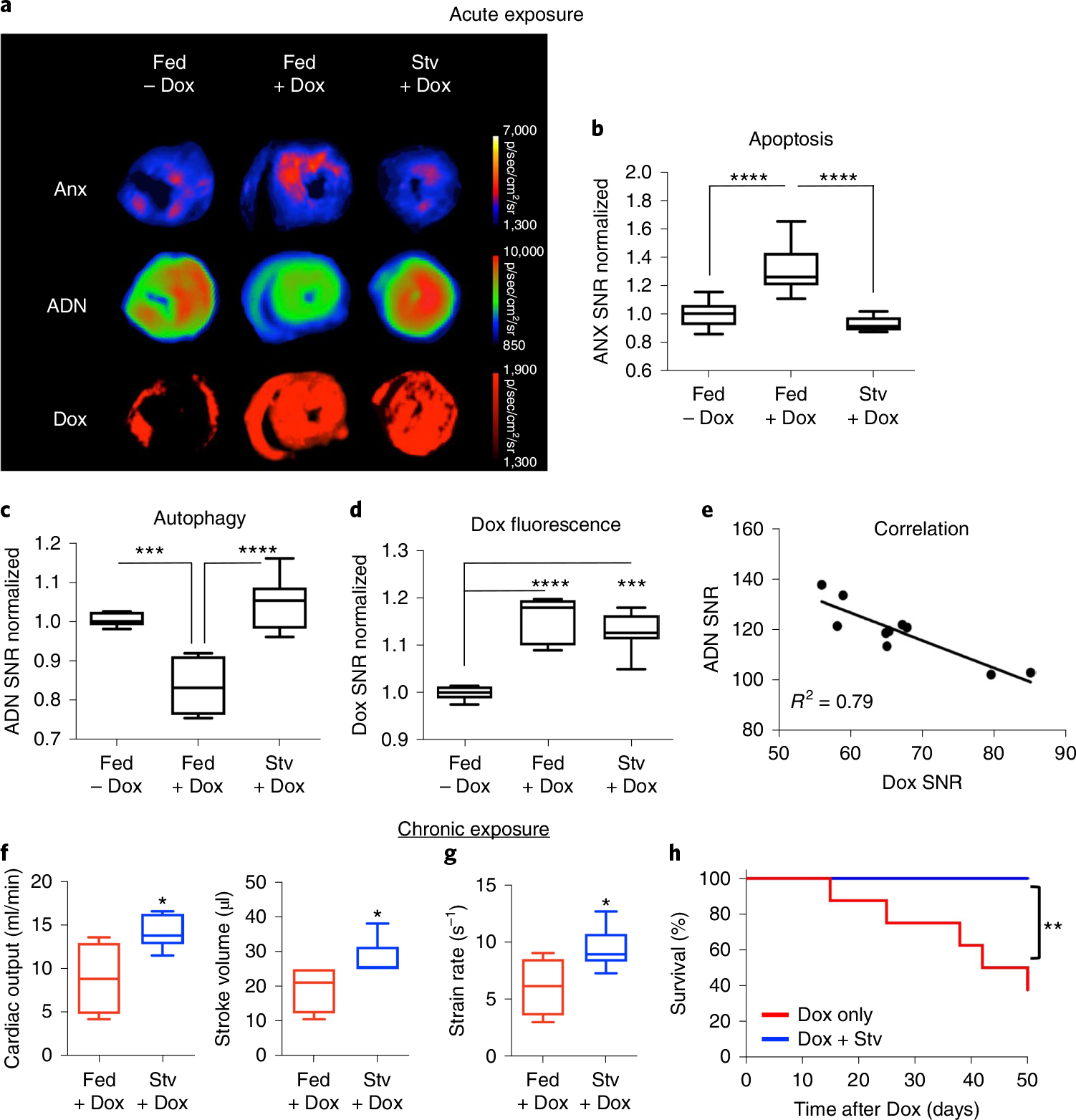
a–c, Dox administration is associated with increased apoptosis and reduced autophagy. This pattern is reversed by starvation (Stv) before Dox administration, which restores basal levels of autophagy and attenuates apoptosis. Fed n = 9, Fed + Dox n = 9 and Stv + Dox n = 7. One-way ANOVA with Tukey post-test, ***P = 0.0005 and ****P < 0.0001. Box plots show minimum, maximum, median and 25–75% percentiles. In a, fluorescence maps show ADN activation, ANX and Dox distribution in the heart. d, The protective effect of pre-starvation is not due to reduced uptake or retention of Dox in the heart, which is similar in fed (n = 9) and starved (n = 7) mice. One-way ANOVA with Tukey post-test, ***P = 0.0002 and ****P < 0.0001. Box plots show minimum, maximum, median and 25–75% percentiles. e, An inverse correlation is seen between ADN activation and Dox fluorescence in the heart. f,g, Indices of cardiac function including cardiac output, stroke volume (f) and strain rate (g) were significantly improved in mice exposed to pre-starvation (n = 7) before weekly doses of Dox versus fed mice exposed to Dox (n = 4). Two-tailed Mann–Whitney tests were performed, *P = 0.042 (cardiac output), *P = 0.027 (stroke volume) and *P = 0.030 (strain rate). Box plots show minimum, maximum, median and 25–75% percentiles. h, Pre-starvation in this chronic Dox administration model (n = 8 in both groups) also improved the overall survival of the mice. Comparison of survival curves was performed using a log-rank Mantel–Cox test. **P = 0.0085.
The chronic low-dose regimen involved a weekly injection of Dox (4 mg kg−1) for 5 weeks. Half the mice (n = 8) were starved for 24 h before each dose of Dox, while the other half (n = 8) were fed normally. In the mice pre-starved before each Dox injection, echocardiography 8 weeks after the last Dox dose revealed a significant improvement in cardiac output, stroke volume and strain rate (Fig. 5f,g). In addition, a significant decrease in mortality (0/8 versus 5/8, P < 0.01) was seen in the pre-starved mice during the 8 weeks of follow-up (Fig. 5h).
Imaging of cardiomyocyte autophagy and flux in mice.
We next used ADN to image autophagy in the heart in vivo. C57BL/6 mice were starved for 24 h and then injected with ADN (n = 4) or ADN plus Baf (n = 3). The mice were imaged with planar reflectance imaging for 6 h, which allowed images of ADN to be acquired at multiple timepoints during this window. Three-dimensional fluorescence imaging, co-registered with x-ray computed tomography (CT), was performed at 4–6 h and confirmed that the elevated thoracic signal in the starved mice emanated from the heart (Fig. 6a). A strong fluorescence signal in the sternum was seen in the planar images of the C57BL/6 mice, allowing the left/right (L/R) signal ratio in the thorax to be calculated (Fig. 6b). The L/R ratio did not increase over time in the mice co-injected with ADN and Baf, but increased steadily in the mice injected only with ADN (Fig. 6c).
Fig. 6 |. In vivo imaging of autophagy with ADN.
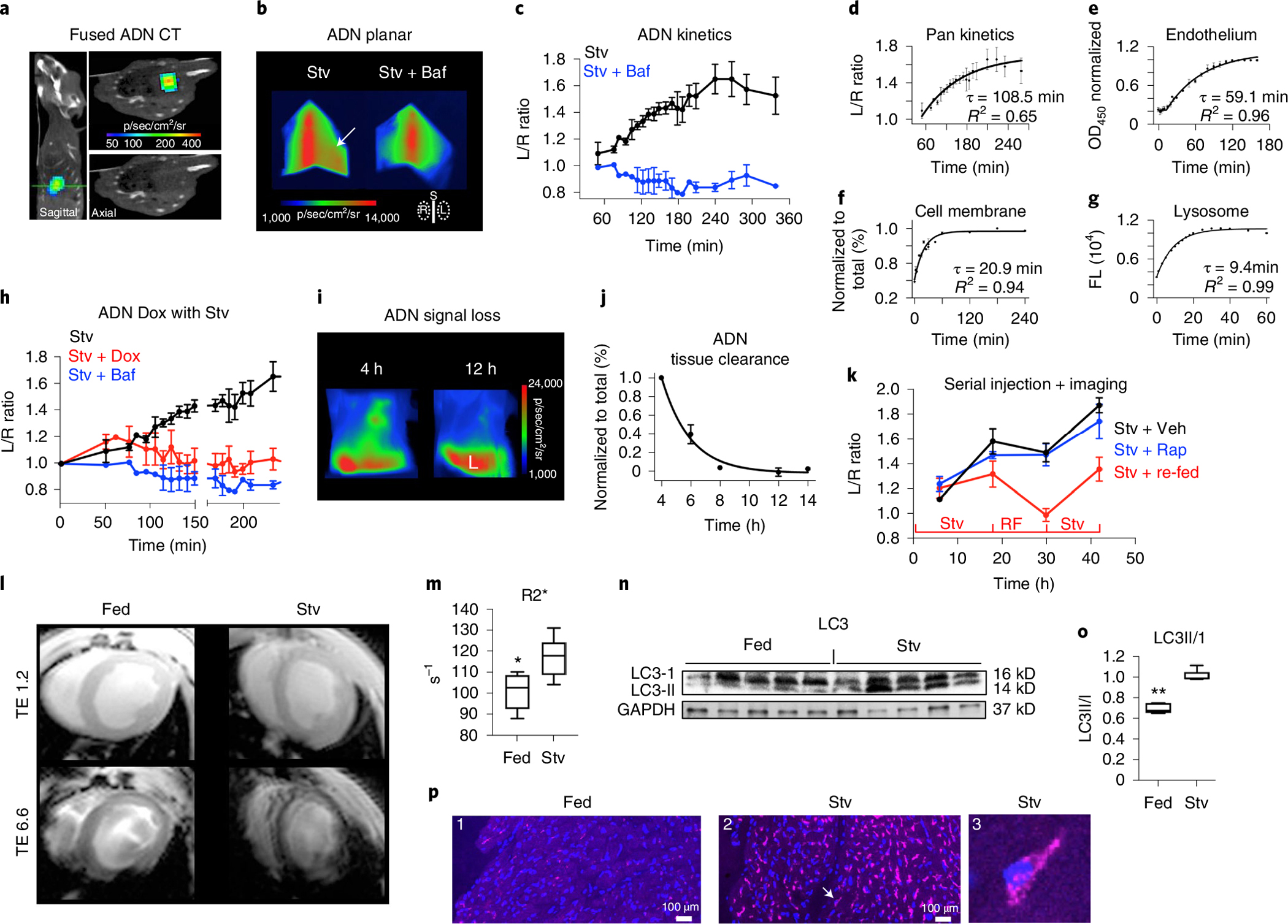
a, Three-dimensional fluorescence and computed tomography in a starved C57BL/6 mouse. Sagittal and axial (green line) images, shown with and without fused fluorescence, confirm that the signal from ADN localizes strongly with the heart. b,c, Kinetics of ADN activation in the heart in vivo, determined with serial high-throughput planar reflectance imaging. Intense sternal (S) fluorescence in the planar images facilitates image segmentation and allows the cardiac signal to be estimated from L/R thoracic fluorescence. This ratio is elevated in starved mice (Stv, n = 4 mice) injected with ADN (arrow) but not in those co-injected with Baf (n = 3 mice). Mean ± s.e.m. values are plotted. d–g, Compartmental analysis of ADN kinetics with associated time constants (t): fit of kinetic curve in c, reflecting all compartments (n = 4 mice) (d); passage of ADN across the endothelial (n = 4 biologically independent experiments) and cell membranes (n = 3 biologically independent experiments) (e,f); mean ± s.e.m. values are plotted (d–f); activation of ADN by lysosomal cathepsins/proteases (average values are plotted of n = 4 biologically independent experiments) (g). The in vivo time constant of autophagic flux (tFlux) is calculated from d–g and equals 20 min. h, The in vivo kinetics of ADN are initially unperturbed by Dox (given 2 h before), but within 4 h show an incomplete but severe block. n = 4 mice (Stv), n = 4 mice (Stv + Dox) and n = 3 mice (Stv + Baf). Mean ± s.e.m. values are plotted. i–k, In vivo imaging in starved nude mice over an extended time course (>40 h): the cardiac signal following a single dose of ADN is cleared/undetectable within 12 h (n = 4 mice) (mean ± s.e.m. values are plotted; L, liver) (i,j); ADN activation is strongly modulated by cycles of starvation (Stv) and re-feeding (RF) but not by the addition of rapamycin to starved mice (n = 3 mice in each group; mean ± s.e.m. values are plotted; Veh, vehicle) (k). l,m, In vivo MRI of ADN in fed (n = 6) and starved (n = 6) mice. The signal in the myocardium at the short TE (ms) is similar, but at the longer TE the accumulation of ADN in the myocardium of the starved mouse causes its MRI signal to dephase rapidly. In m, in vivo R2* in the myocardium was significantly higher in starved mice (*P = 0.0108, two-tailed Mann–Whitney test). Box plots show minimum, maximum, median and 25–75% percentiles. n,o, Western blots of the excised hearts confirm that autophagy (LC3II/I) is increased by starvation. n = 5 mice per group, **P = 0.0079, two-tailed Mann–Whitney test. Box plots show minimum, maximum, median and 25–75% percentiles. Uncropped gels are shown in Source Data Fig. 2. p, Confocal microscopy shows little activation of ADN in a fed mouse (P1) but profound activation in a starved mouse (P2–3). The arrow in P2 points to the cardiomyocyte shown in P3, where activated ADN is visible in perinuclear punctates. Microscopy was performed in fed (n = 3) and Stv (n = 4) mice with consistent results.
The time constant of ADN signal evolution (tADN) in vivo was 109 min (Fig. 6d). This was used to calculate the time constant of autophagic flux from the following relationship: tFlux = tADN − (tEndoth + tCell_Memb + tLyso). More detail on the derivation of these time constants (Fig. 6e–g) is provided in Supplementary Fig. 7 and Supplementary Equations 1–11. In brief, the passage of ADN across the endothelium (tEndoth) was measured in a two-compartment well separated by a barrier of endothelial cells, while the movement of ADN across the cell membrane (tCell_Memb) was measured by flow cytometry of H9C2 cells. The time constant of ADN activation by lysosomal proteases (tLyso) was derived from Fig. 1h. The values of these time constants (tEndoth, tCell_Memb and tLyso) averaged 59, 21 and 9 min, respectively. This yielded an in vivo time constant of autophagic flux (tFlux) of approximately 20 min. Further kinetic modelling (Supplementary Fig. 8) revealed that starvation increased autophagic flux to >40 autophagosomes per cell per hour and resulted in the accumulation of over 130 autophagosomes in each cardiomyocyte. While Baf completely blocked autophagic flux, the administration of Dox to starved mice severely attenuated, but did not completely eliminate, the upregulation of autophagy (Fig. 6h).
We next sought to measure dynamic changes in autophagy in vivo over a more extended period (48 h). This required the use of nude mice (Fig. 6i–k), since removing surface fur in C57BL/6 mice results in a local inflammatory response within 12–48 h. Serial imaging after a single ADN injection revealed that the probe was cleared from the heart within 8–12 h. Serial injections were therefore performed at 12-h intervals in (1) starved mice, (2) starved mice injected with rapamycin and (3) mice exposed to dynamic cycles of starvation and feeding. The modulation of autophagy in the heart by both starvation and re-feeding was rapid and robust (Fig. 6k). In contrast, the addition of rapamycin to the starved mice had little if any impact.
In vivo detection of the probe in starved mice was also performed with magnetic resonance imaging (MRI) using a multi-echo gradient echo sequence (Fig. 6l,m). R2* in the myocardium of the starved mice (n = 6) was significantly higher (117.2 ± 9.4 versus 100.8 ± 8.4; P < 0.05) than that of the fed mice (n = 6). Western blots and confocal microscopy of excised hearts (Fig. 6n–p) showed that the ratio of LC3II/I, indicative of autophagy, was increased in starved mice and that activation of the probe was robust in starved myocardium. Collectively, the in vivo data show that ADN can be detected by magnetic resonance or fluorescence, that the response of the heart to starvation is both dynamic and profound, that autophagic flux is a rapid and efficient process and that the stimulation of autophagy by starvation can partially mitigate its inhibition by cardiotoxic drugs. A similar protective effect during chemotherapy was seen with statins (Supplementary Fig. 9).
Discussion
We have shown that a rationally designed ADN can image autophagy in vivo with a high degree of sensitivity and specificity. The ADN allowed us to show that autophagy may be inhibited during cardiotoxic chemotherapy and that its restoration in these circumstances is protective. The ADN produces fluorescent and magnetic resonance readouts of cardiomyocyte autophagy that can be imaged in deep tissues. ADNs could be further developed for the cellular and translational investigation of autophagy in cardiovascular disease.
ADN can enter the cytoplasm via direct translocation across the cell membrane14–16, or via endocytosis and subsequent endosomal escape19–21. Both processes are facilitated by decorating NPs with cell-penetrating peptides such as polyarginine. Once in the cytoplasm, we have shown that ADN is rapidly taken up by LC3-GFP-positive early autophagosomes, which are avid for free NPs12,13, and polyarginine17,18. The polyarginine peptides on ADN thus serve multiple functions including cell penetration, endosomal escape, stimulation of autophagosome uptake and cleavage by lysosomal cathepsins resulting in the activation of fluorescence.
The importance of measuring the flux of autophagosomes is being increasingly realized11,38. The uptake of ADN by autophagosomes or lysosomes results in focal aggregation of the NPs and a dramatic increase in their magnetic relaxivity, facilitating their detection with MRI39,40. The fluorescence signal of ADN provides an even more robust readout of flux since only those ADN-containing autophagosomes that are trafficked to the lysosome undergo cathepsin-mediated cleavage of their surface peptides and emit fluorescence23. Our data suggest that cardiomyocytes are able to traffic >40 autophagosomes per hour and digest potentially harmful cytoplasmic contents within 20 min. This is probably rapid enough to effectively neutralize most pro-apoptotic stimuli, while avoiding excessive energy expenditure by the cell.
The use of CAFs designed to detect inflammation41 does not support the detection of autophagy. Our prior experience with these agents showed that the simultaneous and artificial superposition of multiple autophagy-inducing stimuli was required to detect any signal23. This finding was replicated here, where autophagy induced by ischaemia, starvation or rapamycin could be easily detected by ADN but not by CAFs. ADN is thus able to non-invasively detect autophagy under physiologically meaningful and relevant conditions. Fluorescence microscopy and electron microscopy of ADN were performed separately in this study. Future work with correlative light-electron microscopy could provide valuable insights.
Starvation is a classic stimulator of autophagy25,26, and resulted in marked activation of ADN both in vitro and in vivo. Starvation did not increase endothelial permeability or the number of CD68 positive cells in the myocardium (Supplementary Fig. 6), confirming that the activation of ADN in starvation was not due to an inflammatory response42. The activation of ADN in the heart in vivo could be completely blocked by Baf, and dynamically modulated by cycles of starvation and re-feeding, consistent with specificity for autophagy.
The activation of ADN in DDiT4L-overexpressing mice further demonstrates its specificity for autophagy. Tetracycline withdrawal in these mice drives the α-myosin heavy chain promoter and results in cardiomyocyte-specific overexpression of DDiT4L (ref. 31). The DDiT4L protein complexes with and inhibits mTORC1 on the early endosome, resulting in an increase in autophagic flux, which was robustly detected with ADN. The crossing of DDiT4L and beclin+/− knockout mice completely eliminates the molecular signatures of increased autophagy and normalizes the cardiac phenotype31, underscoring the suitability of the model to validate ADN.
Dox inhibits lysosomal acidification and results in the accumulation of autophagosomes due to a block in their breakdown33,34. This reduction in flux was well detected with ADN. In vivo imaging with ADN also revealed that the stimulation of flux by pre-starvation remained intact for 4 h following Dox administration, and thereafter was severely attenuated but not eliminated. This response was sufficient to significantly improve cardiac function and overall survival in the pre-starved mice, suggesting that even a brief upstream burst of autophagy may allow cardiomyocytes to assume a sustained protective posture.
The pleiotropic effect of statins is frequently ascribed to their anti-inflammatory effects. Our results, however, suggest that their stimulation of autophagy may be a key factor as well. Importantly, the stimulation of autophagy in human cancer cell lines did not affect the cytotoxic effects of Dox in this study. However, the role of autophagy in human cancers is complex, and its induction for cardioprotection will need to be carefully studied.
Autophagy has hitherto not been measurable in vivo. We have developed a rationally designed ADN that can be detected with either fluorescent or magnetic resonance imaging. We show with ADN that cardiomyocytes are able to rapidly traffic autophagosomes to the lysosome, but that this process is suppressed by several common cardiotoxic chemotherapies. We further show that even short upstream bursts of autophagy during chemotherapy can be highly cardioprotective. ADN provides an accurate readout of autophagy flux and could facilitate an improved understanding of the role of autophagy in a broad range of disease states.
Methods
A list of cells and reagents used and descriptions of the methods used for the cell viability assay, protein immunoblotting assay, electron microscopy of ADN, endothelial passage of ADN and murine echocardiography are provided in Supplementary Information.
ADN synthesis.
The US Food and Drug Administration-approved NP ferumoxytol (Feraheme, FH; Advanced Magnetics) was surface-functionalized with azides as previously described43. Fluorescent poly-L-arginine peptides with the DBCO moiety were synthesized, and 0.3 μmol was prepared in 15 μl anhydrous DMSO, and added in small fractions (3 μl each time) to 5 mg of azide–FH in 1.1 ml of 1× PBS buffer, pH 7, well stirring. The exact peptide sequence was R-R-R-R-G-C, in an l-isomer configuration (Supplementary Figs. 1 and 2). The reaction was light-protected and incubated at room temperature with vigorous stirring for 1 h, concentrated with Amicon centrifugal filters with 50 kDa cut-off (EMD Millipore), loaded on PD-10 columns (GE Healthcare), and eluted with 1× PBS, pH 7. The non-activatable FH-d-iso control NP was synthesized identically but using the d-isomer of the R-R-R-R-G-C peptide. FH–Cy5.5 was similarly synthesized by reacting Cy5.5–DBCO (Lumiprobe) with azide–FH under the same conditions above but without any polyarginine peptides.
ADN characterization and fluorescence activation assay.
UV spectra of ADN and the control FH–Cy5.5 NP were measured by absorption spectrophotometry, with a wavelength range of 350–750 nm, on an Evolution 300 UV-Vis spectrometer (Thermo Fisher Scientific). An absorbance peak at 450 nm was used to measure iron concentration as previously described43. The absorbance peak at 675 nm was used to calculate Cy5.5 concentration on the basis of an extinction coefficient of 250,000 M−1cm−1. To assay for fluorescence activation of ADN through protease exposure, 0.0125% of trypsin–EDTA solution (Sigma-Aldrich) was added to 0.04 mg iron ml−1 solution prepared in 1× PBS, mixed and incubated at room temperature. ADN fluorescence intensity was measured on a Cary Eclipse Fluorescence Spectrophotometer (Agilent) with filter settings of 645 nm excitation wavelength, and 665–850 nm emission wavelength with peak integration. FH–Cy5.5 solution was similarly treated with trypsin, and fluorescence intensity was measured as above.
FACS analysis.
Analytical FACS was performed using standard protocols. Briefly, cells were seeded on 24-well plates at 1 × 105 cells per well. Autophagy was induced by incubating the cells in minimal DMEM without serum (starvation) for 20 h. In select experiments, the autophagy inhibitors E64d (10 μg ml−1) and pepstatin A (10 μg ml−1) were added to the medium. In all cases, ADN (10 μg Fe ml−1) was added to the medium for an additional 4 h. To examine the effects of chemotherapy agents on autophagy and cell fate, cells were treated for 24 h with either Dox (5 μM) or dasatinib (10 μM), with or without starvation (6-h pre-treatment), rapamycin (1 μM), atorvastatin (1 μM) or simvastatin (1 μM). ADN 10 μg ml−1, FH–Cy5.5 (10 μg Fe ml−1) or CAF (4 μM) were added to the cells 4 h before FACS. For cell death assays, cells were trypsinized and fluorescent ANX (5 μg ml−1) was added in annexin binding buffer (Thermo Fisher Scientific) to the cells for 15 min at 37 °C. Cells were immediately analyzed on a four-laser LSRII system (BD Biosciences). A compensation matrix was applied to correct for spectral overlap and signal spill-over. However, the degree of spectral overlap between Dox and propidium iodide precluded the use of propidium iodide to measure cell death. Therefore, in cells exposed to Dox, the readout of cell death was provided by annexin. The cytometry data were normalized to cells exposed to baseline conditions. The data were analyzed and histograms were generated using Flowjo (Tree Star).
The duration of starvation before FACS in all the cell-based experiments in the study was 24 h. The one exception to this was the comparison of ADN with the ΔG reporter, which was performed after only 6 h of starvation in order to replicate the conditions from prior studies with the ΔG probe29. To compare ADN activation with the fluorescent ΔG reporter29, H9C2 cells were transfected with the pMRX-IP-GFP-LC3-RFP-LC3ΔG plasmid (Addgene) with lipofectamine per the manufacturer’s instructions. After transfection of the plasmid, cells were pre-starved for 2 h and then incubated with ADN (10 μg Fe ml−1) for an additional 4 h for a total of 6 h of starvation. Cells were then collected for FACS analysis of the GFP, RFP and Cy5.5 signals and analyzed using Flowjo.
Autophagy loss-of-function cell systems.
MEFs either wild type or ATG5−/− (originated from the Noboru Mizushima laboratory) or ATG7−/− (originated from the Masaaki Komatsu laboratory), were provided courtesy of the Patrice Codogno laboratory27. The cells were cultured in DMEM supplemented with 10% foetal bovine serum. Starvation was induced with MEM medium with 1% glucose and no serum for 24 h. FACS was performed as above.
For ATG7 knockdown, the siRNA construct as well as the scrambled non-coding control (siNC) were purchased from Integrated DNA Technologies. H9C2 cells were transfected with the siRNA/siNC (50 nM) using Lipofectamine RNAiMax Transfection Reagent (Thermo Fisher, cat. no. 13778150) per the manufacturer’s instructions. Forty-eight hours after transfection, H9C2 cells were starved as before for 20 h, followed by ADN incubation for 4 h. ATG7 knockdown efficiency was confirmed by qRT–PCR with the following primer pairs: rq-ATG7-F: AGA AGG ATT CAC CTT CCT GGC, rq-ATG7-R: CAC TCA TGT CCC AGA TCT CAG C, rq-betaActin-F: TTC AAC ACC CCA GCC ATG TA, rq-betaActin-R: GTG GTG GTG AAG CTG TAG CC, purchased from Integrated DNA Technologies, using the SsoAdvanced Universal SYBR Green Supermix (Bio-rad, cat. no. 1725275) and run on the QuantStudio 6 Flex Real-Time PCR Systems (Invitrogen). The reduction in autophagy was confirmed by western blot of LC3 (Cell Signaling, cat. no. 12741S) normalized to β-actin (Sigma, cat. no. A5316).
Confocal microscopy of ADN.
Detection before probe activation by DX1 staining.
Experiments were performed 15 min and 1 h after ADN exposure: To determine whether ADN entry into cells can be independent of endocytosis, H9C2 cells were starved for 22 h and then incubated in a 4 °C incubator for 2 h to inhibit endocytosis without inducing apoptosis. The cells were then incubated with ADN (10 μg Fe ml−1) in 4 °C medium for 15 min. To characterize the uptake of ADN by early autophagosomes, H9C2 cells were transfected with the pTF-LC3 plasmid and cultured under fed or starved conditions for 24 h. In each condition (fed or starved), half the cells were exposed to Baf A1 (0.1 μM) during the last 2 h of culture. ADN (10 μg Fe ml−1) was added to all cells during the final hour of culture. To visualize ADN before activation in the lysosome, the cells were washed, fixed, permeabilized and stained with the DX-1 antibody followed by a secondary fluorescent antibody. After nuclear staining with DAPI, the cells were imaged on a Leica SP8 inverted microscopy system equipped with an automated stage, spectral scanner system ranging from 400 to 850 nm, and Navigator software, at 1,024 by 1,024 resolution using preset DAPI, GFP, RFP and Alexa 790 (for DX-1 signal) scan settings. The total number of LC3-GFP puncta per cell and the number of LC3-GFP puncta co-localizing with DX-1 were visualized in ImageJ and manually counted by two independent researchers. The analysis was performed in five independent cultures or replicates.
Detection after probe activation by Cy5.5 fluorescence.
Imaging was performed after 4 h of ADN exposure. Two sets of experiments were performed. To correlate the signal from activated ADN with a dual fluorescent LC3 plasmid, H9C2 cells were transfected with a dual fluorescent LC3 plasmid (pTF-LC3) from Addgene, with lipofectamine per the manufacturer’s instructions. After transfection, cells were cultured on glass chamber slides for imaging. ADN (10 μg Fe ml−1) was added to the transfected cells for 4 h before imaging. Confocal fluorescent images were acquired on a Nikon A1R system attached to an inverted microscope, automated stage, and a stage-top incubator optimized for live cell imaging. ADN was imaged with the 640 nm laser for excitation, GFP was imaged with the 488 nm laser and 525/50 nm emission filter cube, and RFP was imaged with the 561 nm filter and 595/50 nm emission filter cube. The acquired images were processed in ImageJ. The second set of experiments were performed to correlate the signal from activated ADN with its detection using the DX1 antibody. Imaging was performed on a Leica SP8 microscopy system, as described above, using DAPI, Alexa 680 (for Cy5.5 signal) and Alexa 790 (for DX-1 signal) scan settings.
Ex vivo microscopy of ADN activation.
The distribution of ADN in the myocardium was assessed with confocal microscopy in C57BL/6 mice injected with ADN (5 mg kg−1, i.v.). The mice were either fed (n = 3) or starved (n = 4) for 24 h, with injection of the probe performed at the 20-h mark to allow 4 h of circulation. The mice were then killed, and the excised hearts were flushed with PBS, bisected and embedded in optimal cutting temperature compound (Fisher Scientific). The embedded hearts were cut in 8-μm sections and stained with DAPI (Thermo Fisher) before imaging on a Nikon Eclipse Ti confocal inverted microscope. ADN was excited using a 640 nm laser and Cy5 HYQ emission filter (663–738 nm), while DAPI was imaged with 405 nm laser excitation and 460/60 emission filter. Images were acquired with a 40× objective lens, at 0.63 μm per pixel resolution.
Animal models.
C57BL/6 mice between 8 and 10 weeks of age were purchased from Jackson Laboratories, and DDiT4L mice bred in-house were housed in ventilated cages on standard corncob bedding and cotton nestlets. NU mice from Charles River Laboratories were housed in ventilated cages on Alpha-Dri bedding with Enviro-dri enrichment for dust control. All mice were maintained on 12-h alternating light and dark cycles, with free access to water and food (Teklad 2916 irradiated diet, Envigo) unless otherwise noted for the starvation experiments. All experiments and procedures were performed in accordance with the National Institutes of Health’s ‘Guide for the Care and Use of Laboratory Animals’ and were approved by the Institutional Animal Care and Use Committee of the Massachusetts General Hospital and Tufts University (B2020-03).
A total of 160 mice (138 C57BL/6, 9 DDiT4L/FVB and 13 NU mice) were included in the study. A mix of male and female mice was used. Unless otherwise stated, the mice below are C57BL/6:
IR injury: thoracotomy and occlusion of the left coronary artery for 35 min, before reperfusion, was performed in mice (n = 5, female, 10–12 weeks of age), as previously described42. At 5 min before reperfusion, fluorescent microspheres (Thermo Fisher) were injected directly in the left ventricle42. At the time of reperfusion, 5 mg Fe kg−1 of ADN and 2 nmol of CAF were co-injected intravenously via the tail vein, and allowed to circulate for 4 h. After 4 h, the hearts were collected and processed for imaging. In addition, mice (n = 3, female, 10–12 weeks of age) 24 h after transient coronary ligation were used as positive controls to assess CD68 staining in the myocardium (Supplementary Fig. 6).
-
Starvation-induced autophagy: mice (n = 30, equal ratio of female/male mice at 14 ± 4 weeks of age) were fasted (without food, but with unlimited access to water) for 20 h, while fed control mice (n = 20) were fed ad libitum. ADN was then injected (5 mg kg−1, i.v.) and allowed to circulate for 4 h. Some of these mice (n = 15 starved, n = 10 fed) were used for fluorescence reflectance imaging (FRI), some (n = 7 starved, n = 7 fed) for MRI and the remainder (n = 8 starved, n = 3 fed) for fluorescence microscopy.
Additional mice (22 total of equal female/male mice at 14 ± 4 weeks of age) were intravenously injected with 2 nmol of CAF (Prosense, PerkinElmer; n = 6 starved, n = 7 fed) or with 5 mg kg−1 of FH–Cy5.5 (n = 3 starved, n = 6 fed), and imaged ex vivo by FRI.
In the in vivo fluorescence imaging arm, a total of 11 C57BL/6 mice, of equal female/male ratio at 14 ± 4 weeks of age, underwent pre-starvation for 20 h, were then injected intravenously with 10 mg kg−1 of ADN and randomized to either starvation alone (n = 4), starvation plus 1.5 mg kg−1 i.p. of Baf (n = 3) or starvation plus 5 mg kg−1 i.p. of Dox (n = 4, males, 8–10 weeks of age). Baf and Dox were injected 2 h before ADN injection. Imaging was performed for 6 h after ADN injection.
In the in vivo MR imaging arm, an additional six C57BL/6 mice, of equal female/male ratio at 14 ± 4 weeks of age, underwent pre-starvation for 20 h and six were fed controls (matched for age and gender to the pre-starvation mice). The mice were injected intravenously with 10 mg kg−1 of ADN and in vivo MRI was performed 6 h after ADN injection.
Acute Dox model: mice (n = 13, male, 8–10 weeks of age) were injected with 15 mg kg−1 of Dox i.p36. Of these 13 mice, a subset (n = 7) were pre-starved before the Dox injection for 24 h and the remainder (n = 6) fed normally before Dox. An additional six mice were fed controls injected with PBS. All 19 mice were co-injected with ADN (5 mg kg−1, i.v.) 4 h before FRI and with fluorescent ANX (PerkinElmer; 4 μl g−1, i.v.) 2 h before FRI.
Chronic Dox model: mice (n = 16, male, 8–10 weeks of age) received weekly injections of Dox (4 mg kg−1, i.p.) over 5 weeks (20 mg kg−1 total)33. Half of the cohort were pre-starved for 24 h before each Dox challenge. All mice were imaged by echo at 8 weeks after the final dose of Dox.
A total of nine DDiT4L mice (four transgenic mice and five littermate controls carrying the empty cassette without the transgene) including both females (n = 5) and males (n = 4) bred on the FVB/NJ background were imaged. Autophagy was induced by activation of the DDiT4L transgene, produced by doxycycline exposure until 4 weeks of age, followed by 8 weeks without exposure to induce DDiT4L overexpression31. At the end of the 8-week doxycycline withdrawal, ADN (5 mg kg−1, i.v.) was injected and allowed to circulate for 4 h before sacrifice and tissue preparation for FRI.
To investigate the clearance of ADN from the myocardium, and the response of the heart to sequential cycles of starvation, a total of 13 NU mice (both females and males at 12 ± 4 weeks of age) were starved, and randomized into the following four groups: (1) tissue clearance group (n = 4), (2) starvation plus vehicle (DMSO, n = 3), (3) starvation plus rapamycin (2 mg kg−1 in DMSO, i.p., n = 3) and (4) starvation plus re-feeding (n = 3).
In the tissue clearance group, starved mice were injected with ADN and imaged with serial in vivo FRI 4, 6, 8, 12 and 14 h after injection. The total thoracic signal (L + R) in each mouse was normalized to the signal at 4 h and followed a mono-exponential decay. The clearance experiments showed that ADN was completely cleared from the heart within 10–12 h. The serial imaging experiments that followed were therefore performed by injecting ADN into the same mouse at 12-h increments, with the imaging performed 4 h after each injection at 6, 18, 30 and 42 h. At each timepoint, the L/R thoracic ratio was calculated, using sternal autofluorescence and/or the white light images to guide segmentation. The timelines for the three groups of serial experiments were as follows:
Stv + Rap: rapamycin (2 mg kg−1, mixed with 5% DMSO) was injected 2 h before the initiation of starvation, which was continued uninterrupted for 42 h. The mice had unrestricted access to water during starvation.
Stv: the mice were injected with DMSO (to match conditions in the Stv + Rap group) 2 h before the initiation of starvation, which was continued uninterrupted for 42 h. The mice had unrestricted access to water during starvation.
Stv + re-feeding: The mice were starved for 18 h, re-fed for the next 12 h and then starved again for the last 12 h. The mice had unrestricted access to water during the periods of starvation. DMSO was not injected in this cohort of mice.
FRI ex vivo.
Hearts collected from the mice were sectioned into 1-mm axial slices and imaged ex vivo in reflectance mode on the IVIS spectrum system (PerkinElmer), as well as on a high-resolution fluorescence lifetime imaging system, as previously described42. The time domain fluorescence system utilizes a Ti:Sapphire laser (SpectraPhysics, MaiTai) and a gated intensified CCD camera (Picostar HRI, LaVision Gmbh). ADN fluorescence was acquired with 675 nm excitation and 720 nm emission filter settings. Annexin fluorescence was acquired with 745 nm excitation and 800 nm emission filters. Signal-to-noise ratio (SNR) and TBR were calculated using standard definitions, as previously described37,42.
Fluorescence planar reflectance imaging in vivo.
Mice were injected with 10 mg kg−1 i.v. of ADN and, in the C57BL/6 mice, fur over the thorax was removed with nair. The mice were image supine on an IVIS spectrum system (PerkinElmer), anaesthetized with isoflurane and maintained at 37 °C in a heated chamber. Fluorescence reflectance images were acquired with 675 nm excitation/720 nm emission filter settings, with 68 μm resolution and a 30 s exposure. White light images were taken for each fluorescence imaging dataset. ADN fluorescence signal was quantified in ImageJ (National Institute of Health) in regions of interest over the left and right side of the thoracic cavity, as shown schematically in Fig. 6b. Segmentation was guided using sternal fluorescence and/or the white light images of the mouse.
Three-dimensional fluorescence imaging in vivo.
Three-dimensional fluorescence tomography of ADN, integrated with x-ray CT, was performed on the IVIS Spectrum (PerkinElmer) system according to the manufacturer’s instructions. Briefly, the mice were anaesthetized with isoflurane and positioned in the prone position on the heated stage maintained at 37 °C. Fluorescence images were acquired with 675 nm excitation, 720 nm emission filter settings in trans-illumination mode. Trans-illumination light sources were selected to cover the entire thorax of the mouse and normalized transmission fluorescence efficiency images, in which the fluorescent emission image is normalized by the transmission image, were acquired for 3D reconstruction. Reconstruction was performed using the Living Image software (version 4.7.4, Caliper Life Sciences), with the image sequences set to minimum threshold (0.1% of maximum intensity), and with the non-negative least squares algorithm optimized for mouse tissue. The fluorescence tomography reconstruction was then co-registered to the surface tomography image of the mouse, and the quality of the 3D reconstruction was validated by photon density profiling.
MRI ex vivo.
ADN (5 mg kg−1) was injected intravenously into C57BL/6 mice and allowed to circulate. After 4 h, mice were killed and the hearts were perfused with PBS, bisected along the short axis, embedded in optimal cutting temperature compound (Tissue-Tek, Sakura) and stored at −80 °C. At the time of imaging, the heart tissue was thawed, placed in fomblin (Solvay), and MRI was performed on a 9.4 Tesla horizontal bore magnet (Biospec, Bruker). Transverse relaxivity (R2*) maps were generated from multi-echo gradient echo images, acquired in the short axis of the left ventricle, with the following settings: repetition time (TR) 500 ms, 14 echo times (TE) beginning at 1.784 ms with an increment of 1.25 ms, four averages, field of view 28.8 mm × 28.8 mm, matrix dimension 192 × 192, spatial resolution 0.15 mm × 0.15 mm, slice thickness 0.5 mm, flip angle 10°.
MRI in vivo.
In vivo MRI was performed on a 9.4 Tesla horizontal bore magnet (Biospec, Bruker) 6 h after the injection of 10 mg Fe kg−1 of ADN. An electrocardiogram-gated multi-echo gradient echo sequence was acquired in the short axis of the left ventricle in mid-systole. Parameters included: field of view 23 × 23 mm, matrix 128 × 128, spatial resolution 0.179 × 0.179 mm2, slice thickness 1 mm, flip angle 30°, earliest TE 1.17 ms, echo spacing 0.78 ms, number of echoes 8, averages 4. R2* was measured, as per clinical guidelines44, in those segments not corrupted by susceptibility artefacts from air tissue interfaces. In addition, the large amount of ADN uptake in the liver created susceptibility artefacts in the inferoseptum. R2* was, therefore, measured in the antero-septum and anterior wall in two to three slices at the level of the papillary muscles using MATLAB and averaged into a single R2* value for each mouse.
Statistical analysis.
Statistical analysis of the data, and the generation of graphs, was performed using Prism (GraphPad). All data were tested for normality using the D’Agostino-Pearson omnibus normality test, the Shapiro–Wilk test and the Kolmogorov–Smirnov test. Comparison between two groups was performed with a two-tailed unpaired t-test if normal and a Mann–Whitney test if non-Gaussian. Paired comparisons between the total number of GFP puncta per cell and those co-localizing with DX-1 were performed with the Wilcoxon signed rank test. Analysis of variance (ANOVA) was used for comparisons of three or more groups, followed by a Tukey post-test. The data in the text are reported as mean ± standard deviation (s.d.) if normal, and as median plus interquartile range if non-Gaussian.
The data in the figures are graphed using box and whisker plots, which depict the median, interquartile range, minimum and maximum values for both normal and non-Gaussian data or as mean ± standard error of the mean (s.e.m.). The Pearson coefficient was used to correlate datasets that follow a normal distribution. Comparison of survival curves was performed using a log-rank Mantel–Cox test. For all cell-based assays, experiments were performed in at least three biologically independent samples. If multiple measurements of the same biological sample were obtained (Source Data Fig. 4a), these were combined into a single average value for that sample. The differences between groups were considered statistically significant at P < 0.05. No animals or data points were excluded from the analyses. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those reported in previous publications23,37,42. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This study was supported in part by the following grants from the National Institutes of Health: K99/R00HL121152 (to H.H.C.), R01HL112831 and R01HL141563 (to D.E.S.), R01HL122547 and R01HL102368-06A1 (to S.D.), R01HL131635 (to C.M.) and R01HL131831 (to R.M.B.). Electron microscopy was performed in the Microscopy Core of the Center for Systems Biology/Program in Membrane Biology, which is partially supported by an Inflammatory Bowel Disease Grant (DK043351) and a Boston Area Diabetes and Endocrinology Research Center (BADERC) award (DK057521). The funders had no role in the design of the study, in data collection and analysis, in the decision to publish the manuscript or in its preparation.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-022-00904-3.
Peer review information Nature Biomedical Engineering thanks Richard Kitsis, Ben Loos and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analyzed datasets generated during the study are too large to be publicly shared but are available for research purposes from the corresponding authors on reasonable request. Source data are provided with this paper.
References
- 1.Levine B & Klionsky DJ Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Levine B Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120, 159–162 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Choi AM, Ryter SW & Levine B Autophagy in human health and disease. N. Engl. J. Med 368, 651–662 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Nishino I et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Mizushima N & Levine B Autophagy in human diseases. N. Engl. J. Med 383, 1564–1576 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Nah J, Fernandez AF, Kitsis RN, Levine B & Sadoshima J Does autophagy mediate cardiac myocyte death during stress? Circ. Res 119, 893–895 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doherty J & Baehrecke EH Life, death and autophagy. Nat. Cell Biol 20, 1110–1117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savini M & Wang MC Does autophagy promote longevity? It depends. Cell 177, 221–222 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Kabeya Y et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19, 5720–5728 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura S, Noda T & Yoshimori T Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Gottlieb RA, Andres AM, Sin J & Taylor DP Untangling autophagy measurements: all fluxed up. Circ. Res 116, 504–514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi S et al. Artificial induction of autophagy around polystyrene beads in nonphagocytic cells. Autophagy 6, 36–45 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Remaut K, Oorschot V, Braeckmans K, Klumperman J & De Smedt SC Lysosomal capturing of cytoplasmic injected nanoparticles by autophagy: an additional barrier to non viral gene delivery. J. Control. Release 195, 29–36 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Lewin M et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol 18, 410–414 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Schmidt N, Mishra A, Lai GH & Wong GC Arginine-rich cell-penetrating peptides. FEBS Lett 584, 1806–1813 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Nakase I et al. Arginine-rich cell-penetrating peptide-modified extracellular vesicles for active macropinocytosis induction and efficient intracellular delivery. Sci. Rep 7, 1991 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dowaidar M et al. Role of autophagy in cell-penetrating peptide transfection model. Sci. Rep 7, 12635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robison AD et al. Polyarginine interacts more strongly and cooperatively than polylysine with phospholipid bilayers. J. Phys. Chem. B 120, 9287–9296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Najjar K et al. Unlocking endosomal entrapment with supercharged arginine-rich peptides. Bioconjug. Chem 28, 2932–2941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Appelbaum JS et al. Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chem. Biol 19, 819–830 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian Z et al. Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry 53, 4034–4046 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uchiyama Y Autophagic cell death and its execution by lysosomal cathepsins. Arch. Histol. Cytol 64, 233–246 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Chen HH et al. Fluorescence tomography of rapamycin-induced autophagy and cardioprotection in vivo. Circ. Cardiovasc. Imaging 6, 441–447 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu P et al. Myocardial upregulation of cathepsin D by ischemic heart disease promotes autophagic flux and protects against cardiac remodeling and heart failure. Circ. Heart Fail 10, e004044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuma A et al. The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Mizushima N, Yamamoto A, Matsui M, Yoshimori T & Ohsumi Y In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J et al. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome–lysosome fusion. Cell Res 23, 508–523 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McConnell HL et al. Ferumoxytol nanoparticle uptake in brain during acute neuroinflammation is cell-specific. Nanomedicine 12, 1535–1542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morishita H, Kaizuka T, Hama Y & Mizushima N A new probe to measure autophagic flux in vitro and in vivo. Autophagy 13, 757–758 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nahrendorf M et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med 204, 3037–3047 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simonson B et al. DDiT4L promotes autophagy and inhibits pathological cardiac hypertrophy in response to stress. Sci. Signal 10, eaaf5967 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dhingra R et al. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc. Natl Acad. Sci. USA 111, E5537–E5544 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li DL et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133, 1668–1687 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdullah CS et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep 9, 2002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitehead NP, Kim MJ, Bible KL, Adams ME & Froehner SC A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc. Natl Acad. Sci. USA 112, 12864–12869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y et al. Visnagin protects against doxorubicin-induced cardiomyopathy through modulation of mitochondrial malate dehydrogenase. Sci. Transl. Med 6, 266ra170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asnani A et al. Highly potent visnagin derivatives inhibit Cyp1 and prevent doxorubicin cardiotoxicity. JCI Insight 3, e96753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klionsky DJ et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17, 1–382 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez JM, Josephson L, O’Loughlin T, Hogemann D & Weissleder R Magnetic relaxation switches capable of sensing molecular interactions. Nat. Biotechnol 20, 816–820 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Taktak S, Sosnovik D, Cima MJ, Weissleder R & Josephson L Multiparameter magnetic relaxation switch assays. Anal. Chem 79, 8863–8869 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Nahrendorf M et al. Dual channel optical tomographic imaging of leukocyte recruitment and protease activity in the healing myocardial infarct. Circ. Res 100, 1218–1225 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Chen HH et al. Theranostic nucleic acid binding nanoprobe exerts anti-inflammatory and cytoprotective effects in ischemic injury. Theranostics 7, 814–825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan H et al. Heat-induced-radiolabeling and click chemistry: a powerful combination for generating multifunctional nanomaterials. PLoS ONE 12, e0172722 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Messroghli DR et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: a consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J. Cardiovasc. Magn. Reson 19, 75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analyzed datasets generated during the study are too large to be publicly shared but are available for research purposes from the corresponding authors on reasonable request. Source data are provided with this paper.