

Abstract
Phosphoenolpyruvate synthetase (PpsA) was purified from the hyperthermophilic archaeon Pyrococcus furiosus. This enzyme catalyzes the conversion of pyruvate and ATP to phosphoenolpyruvate (PEP), AMP, and phosphate and is thought to function in gluconeogenesis. PpsA has a subunit molecular mass of 92 kDa and contains one calcium and one phosphorus atom per subunit. The active form has a molecular mass of 690 ± 20 kDa and is assumed to be octomeric, while approximately 30% of the protein is purified as a large (∼1.6 MDa) complex that is not active. The apparent Km values and catalytic efficiencies for the substrates pyruvate and ATP (at 80°C, pH 8.4) were 0.11 mM and 1.43 × 104 mM−1 · s−1 and 0.39 mM and 3.40 × 103 mM−1 · s−1, respectively. Maximal activity was measured at pH 9.0 (at 80°C) and at 90°C (at pH 8.4). The enzyme also catalyzed the reverse reaction, but the catalytic efficiency with PEP was very low [kcat/Km = 32 (mM · s)−1]. In contrast to several other nucleotide-dependent enzymes from P. furiosus, PpsA has an absolute specificity for ATP as the phosphate-donating substrate. This is the first PpsA from a nonmethanogenic archaeon to be biochemically characterized. Its kinetic properties are consistent with a role in gluconeogenesis, although its relatively high cellular concentration (∼5% of the cytoplasmic protein) suggests an additional function possibly related to energy spilling. It is not known whether interconversion between the smaller, active and larger, inactive forms of the enzyme has any functional role.
A number of unique microorganisms that thrive at temperatures of 90°C or higher have been isolated in the last 2 decades. One of the best studied of these so-called hyperthermophiles is the anaerobic archaeon Pyrococcus furiosus, which grows optimally at 100°C by the fermentation of carbohydrates and peptides (15). The organism uses a modified Embden-Meyerhof glycolytic pathway that contains several novel enzymes (13, 23). For example, both hexokinase and phosphofructokinase are ADP- rather than ATP-dependent enzymes (24, 48). In addition, the expected glyceraldehyde-3-phosphate dehydrogenase and phosphoglycerate kinase enzymes, which would convert glyceraldehyde-3-phosphate to 3-phosphoglycerate with the concomitant phosphorylation of ADP to ATP and reduction of NAD+ to NADH, appear to be replaced by a single enzyme, glyceraldehyde-3-phosphate:ferredoxin oxidoreductase (GAPOR). GAPOR oxidizes glyceraldehyde-3-phosphate directly to 3-phosphoglycerate and uses ferredoxin rather than NAD+ as the electron acceptor. Purified GAPOR is a unique tungsten-containing enzyme that has no known analog in mesophilic archaea or bacteria (31). Another unusual step in glucose catabolism includes the conversion of acetyl coenzyme A (acetyl-CoA) to acetate. In anaerobic bacteria, this is a two-step process via acetyl phosphate catalyzed by phosphotransacetylase and acetate kinase. In P. furiosus, however, these two enzymes are replaced by acetyl-CoA synthetase, which converts acetyl-CoA directly to acetate and phosphorylates ADP to ATP (29). Other enzymes involved in glucose oxidation that have been purified from Pyrococcus species include pyruvate:ferredoxin oxidoreductase (4), enolase (36), and triosephosphate isomerase (25). In contrast to the enzymes described previously, these are quite similar (except in thermostability) to their mesophilic counterparts.
P. furiosus also synthesizes glucose during growth on peptide-derived amino acids (15), but less work has been done to explore the gluconeogenic pathway. This is thought to occur by a reversal of the conventional Embden-Meyerhof pathway, and the activities of the enzymes proposed to be involved have been measured in cell extracts (44). Of the gluconeogenic-specific enzymes, only phosphoglycerate kinase (19) has been purified from Pyrococcus species. Herein we focus on the enzyme that carries out the first step in the conversion of pyruvate to glucose, phosphoenolpyruvate (PEP) synthetase, which catalyzes the phosphorylation of pyruvate according to equation 1:
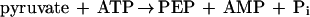 |
1 |
Transcriptional analyses indicated that the cellular concentration of PEP synthetase increased when a glycolytic substrate such as maltose was added to the growth medium of P. furiosus (38, 39). To explain this result, it was suggested that this enzyme functions in glycolysis (reverse of equation 1) as well as in gluconeogenesis (39). However, there are no kinetic data to support this contention. It was, therefore, of some relevance to determine the catalytic properties of PEP synthetase from this organism.
Surprisingly, little is known about PEP synthetases from prokaryotes, in spite of their importance in controlling carbon flow during glucose metabolism (20, 28, 37, 45, 47). The only one to be studied extensively is that from Escherichia coli (10, 11, 32, 33, 34). It is a cofactorless homodimer with a molecular mass of 150 kDa and catalyzes the phosphorylation of pyruvate by using ATP to generate PEP, AMP, and phosphate. Only two other PEP synthetases have been purified and examined to any extent, and both are from archaea, the moderately thermophilic methanogen Methanobacterium thermoautotrophicum (14) and the heterotrophic hyperthermophile Staphylothermus marinus (8, 9, 16, 17). The quaternary structure of the former enzyme was not reported, but S. marinus PEP synthetase exists as a large homomultimeric complex containing 24 subunits (17). Conversely, while the kinetic properties of the latter enzyme are unknown, those of the M. thermoautotrophicum enzyme are similar to those of E. coli PEP synthetase (14). It was therefore of some interest to investigate the physical and catalytic properties of the enzyme from P. furiosus, especially as the enzyme has been reported to be sensitive to inactivation by oxygen (44). For example, did the enzyme exist as a large complex, did it function in both gluconeogenesis and glycolysis, did it utilize nucleotides other than ATP as the phosphate donor for PEP synthesis, and what may be the cause of its oxygen sensitivity?
MATERIALS AND METHODS
Enzyme assays.
The methods used to characterize P. furiosus PEP synthetase required discontinuous assays where reaction substrates or products were measured at 23°C after catalytic conversion at 80°C. One unit of PEP synthetase activity is equivalent to 1 μmol min−1 of product (PEP or phosphate) formed in the forward reaction (equation 1) or of pyruvate formed in the reverse reaction. To measure phosphate formation, the enzyme sample and 4 mM pyruvate were incubated at 80°C in 1 ml of 10 mM MgCl2, 200 mM KCl, 50 mM N-2-hydroxyethylpiperazine-N′-3-propanesulfonic acid (EPPS) buffer (pH 8.4). ATP (4 mM) was added to start the reaction, and 0.2 ml of 5 M H2SO4 was added after 2 min to quench it. The amount of phosphate produced was measured as described previously (18).
The method used to determine the amount of PEP formed was modified from that reported previously (14). The reaction was carried out as described above, and the residual pyruvate was first converted to lactate by lactate dehydrogenase (LDH) (see equation 2 below). Aliquots were added to a reaction mixture containing 2 ml of 20 mM MgCl2 and 40 μM NADH and 5 U of LDH in 100 mM EPPS buffer (pH 8.0) maintained at 23°C. The PEP concentration was then determined by adding 1 mM ADP and 5 U of pyruvate kinase (PK; Sigma) to this reaction mixture. The amount of NADH oxidized by LDH was determined by the decrease in absorption at 340 nm as pyruvate formed from PEP (see equation 3 below) is converted to lactate (equation 2). A molar absorbance of 6,220 M−1 · cm−1 was used for NADH.
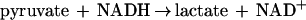 |
2 |
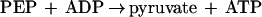 |
3 |
Pyruvate formation by PEP synthetase was measured using a method based on the previously published assay (14). The 1-ml assay mixture contained the enzyme sample, 4 mM AMP, 10 mM phosphate, 10 mM MgCl2, and 100 mM KCl in 50 mM EPPS buffer (pH 8.4) at 80°C. The reaction was initiated by the addition of 4 mM PEP (reverse reaction of equation 1). After 2 min, the reaction mixture was placed on ice to quench the reaction. Samples of the reaction mixture were transferred to cuvettes containing 40 μM NADH, 10 mM MgCl2, and 100 mM KCl in 50 mM EPPS buffer (pH 7.5) in a final volume of 2 ml. The pyruvate formed was measured by adding LDH (5 U) and determining the amount of NADH oxidized (equation 2).
PEP synthetase was also examined for its ability to catalyze pyruvate phosphate dikinase (PPDK) (equation 4) and PK (equation 5) reactions.
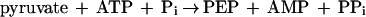 |
4 |
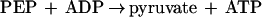 |
5 |
The PPDK activity was measured by adding 10 mM phosphate as substrate in the PEP formation assay. The PK activity was measured with a modified pyruvate formation assay; AMP and phosphate were replaced with ADP (4 mM) as the substrate. The effect of pH on enzyme activity was tested using the following buffers at a concentration of 50 mM (at the indicated pH values): sodium citrate (pH 4.0 to 5.5), 2-(N-morpholino)ethanesulfonic acid (MES) (pH 5.5 to 7.0), morpholinepropanesulfonic acid (MOPS) (pH 7.0 to 8.0), EPPS (pH 8.0 to 9.0), and 2-(N-cyclohexylamino)ethanesulfonic acid (CHES) (pH 9.0 to 10.0).
Purification of PEP synthetase.
P. furiosus (DSM 3638) was grown with maltose as the carbon and energy source in a 600-liter fermentor, as described previously (6). All of the purification steps were carried out at 23°C. The cell extract and the first chromatography step were anoxic, since other enzymes of interest that are oxygen sensitive were purified from the same cell batch. The buffers for these steps were repeatedly degassed and flushed with Ar, contained sodium dithionite (2 mM) and dithiothreitol (2 mM), and were kept under positive Ar pressure. Thereafter, the purification was performed aerobically. P. furiosus cells (200 g [wet weight]) were suspended in 600 ml of 50 mM Tris buffer (pH 8.0) with DNase I (0.5 μg/ml) and were stirred for 2 h at 37°C to suspend and lyse the cells. The cytosolic portion of the cell extract was separated from the membranes by centrifugation at 50,000 × g for 2 h. This was applied to a column (10 by 14 cm) of DEAE Sepharose fast-flow (Pharmacia) equilibrated with 50 mM Tris buffer (pH 8.0) by using a Fast Protein Liquid Chromatography system (Pharmacia). The extract was diluted threefold with equilibration buffer as it was loaded onto the column at a rate of 15 ml/min. PEP synthetase did not bind to the column and was collected in the pass-through. This was filtered through a 0.2-μm-pore-size filter (Maxiculture Capsule; Fisher) and was applied to an ion exchange column (5 by 35 cm) of quaternary ammonium (Q)-Sepharose fast-flow (Pharmacia) equilibrated with 50 mM glycine buffer (pH 9.5). The filtered protein was diluted fourfold with equilibration buffer and loaded onto the column. The bound protein was eluted with a 0 to 0.5 M NaCl linear gradient (3,500 ml) in 50 mM glycine (pH 9.5) using a flow rate of 5 ml/min. PEP synthetase activity was measured in fractions collected from the column during the 0.38 to 0.43 M NaCl portion of the gradient. The fractions with specific activity above 7 U/mg were pooled and concentrated by ultrafiltration (PM 30; Amicon). The concentrated sample was applied to a column (3.5 by 60 cm) of Superdex 200 (Pharmacia) equilibrated with 50 mM Tris buffer (pH 8.2) containing 0.2 M NaCl at 0.7 ml/min. PEP synthetase eluted in the void volume. Those fractions judged to be pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were concentrated and stored in liquid N2.
Other methods.
The molecular weight of the holoenzyme was determined using an analytical Superose 6 (Pharmacia) gel filtration column (1 by 28 cm) equilibrated with 50 mM Tris buffer (pH 8.4) containing 0.2 M NaCl. The standards used to calibrate the column were alcohol dehydrogenase (150 kDa), β-amylase (200 kDa), catalase (223 kDa), apoferritin (440 kDa), thyroglobin (663 kDa), and human immunoglobulin M (900 kDa). The subunit molecular weight was determined by SDS-PAGE with an 8% acrylamide–Tris-glycine-buffered gel, as described previously (26). The thermal stability of PEP synthetase was determined by incubating samples at 90°C for various time intervals in 50 mM EPPS buffer (pH 8.0) containing 0, 0.2, or 0.5 M KCl. The samples were placed on ice and were assayed immediately by pyruvate formation at 80°C. Metal content was determined by inductively coupled plasma emission spectroscopy (ICP) on a Jarrel Ash Plasma Comp 750 instrument at the Chemical Analysis Laboratory of the University of Georgia. The N-terminal amino acid sequence of the protein was determined (using an Applied Biosystems model 477 sequencer) by the Molecular Genetics Instrumentation Facility at the University of Georgia. The PEP synthetase sample was prepared for sequencing by electrophoresis on an SDS–15% acrylamide Tris-glycine gel followed by electroblotting (Bio-Rad blotting system) onto a polyvinylidene difluoride sequencing membrane. The electroblotting was carried out at 200 mA for 7 h in 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) buffer (pH 11.0) containing 10% (vol/vol) methanol. Protein concentrations were estimated by the method described by Bradford (5).
Substrate requirements and kinetic constants of PEP synthetase were determined using the phosphate formation assay for the forward reaction and the pyruvate formation assay for the reverse reaction. The effects of the reducing agents dithiothreitol (5 mM) and β-mercaptoethanol (40 mM) were tested by incubating them with the enzyme followed by anaerobic PEP formation assay; vials containing reaction mixture were degassed and placed under Ar with either dithiothreitol or β-mercaptoethanol. The specificity of the PEP synthetase for α-keto acids, phosphate donors, and phosphate acceptors was determined by using both the phosphate formation and pyruvate formation assays. The PEP formation assay was used to study the effect of divalent and monovalent cations on enzyme activity. The calcium requirement of PEP synthetase was determined by first removing calcium by EGTA treatment. The enzyme (1 mg ml−1 in 50 mM Tris [pH 8.0]) was incubated with 1 mM EGTA for 30 min at 25°C or for 5 min at 80°C and was then passed through a Sephadex G-25 column (5 ml) equilibrated with 50 mM Tris, pH 8.0. The metal content was measured by ICP analysis, and the catalytic activity of the EGTA-treated enzyme was determined by the phosphate formation assay.
RESULTS
Purification of PEP synthetase.
Cell extracts of P. furiosus cells grown with maltose as the carbon source contained high PEP synthetase activity. Values of 1.2 ± 0.5 U mg−1 were obtained using the phosphate formation assay. The results of a typical purification procedure are summarized in Table 1. The yield of the pure enzyme from 200 g (wet weight) of cells was approximately 50 mg with a 20-fold purification factor, suggesting that PEP synthetase represents a significant portion (∼5%) of the cytoplasmic protein (Table 1). The final specific activity of the protein that was ≥90% homogeneous as judged by SDS-PAGE (see below) was 18 ± 4 U mg−1 using the phosphate formation assay. During the last steps of the purification, the concentration of the PEP synthetase had to be maintained below 10 mg ml−1 (pH 7.5 to 8.5); otherwise the solution became viscous and the protein precipitated. In catalyzing PEP formation, the purified enzyme was dependent on both ATP and pyruvate for activity and there was no increase in activity when phosphate was added as an additional substrate. This result indicates that the enzyme is a PEP synthetase (equation 1) rather than a PPDK (equation 4). The enzyme also catalyzed the reverse reaction (the formation of pyruvate from PEP, AMP, and phosphate), and AMP and phosphate could not be replaced by ADP, showing that the enzyme is not a PK (equation 5).
TABLE 1.
Purification of P. furiosus PEP synthetasea
| Step | Activity (U) | Protein (mg) | Sp act (U mg−1) | Recovery (%) | Purification (fold) |
|---|---|---|---|---|---|
| Cytoplasmic extract | 9,450 | 12,300 | 0.77 | 100 | 1 |
| DEAE-Sepharose | 5,710 | 1,741 | 3.28 | 60 | 4.3 |
| Q-Sepharose | 2,060 | 286 | 7.21 | 22 | 9.4 |
| Superdex 200 | 730 | 49 | 14.9 | 7.7 | 19.4 |
From 200 g (wet weight) of cells.
Physical properties.
During the final purification step, PEP synthetase eluted in the void volume of a Superdex 200 gel filtration column, indicating that it had a molecular mass of at least 600 kDa. Subsequent chromatography of the purified protein on a column of Superose 6 (with an exclusion limit of 40 MDa) gave rise to two protein fractions. One eluted with an apparent molecular mass of 690 ± 20 kDa, while the other had a molecular mass of 1.64 ± 0.01 MDa. The two forms were indistinguishable by SDS-PAGE analysis, and their N-terminal amino acid sequences were identical (see below). The larger form accounted for about 30% of the total protein applied to the column. However, only the smaller of the two protein forms had PEP synthetase activity; no activity (<0.01 U mg−1 in the phosphate release assay) was detected with the larger form. Removal of the inactive 1.6-MDa form resulted in an increase in the specific activity of the “pure” protein by 1.7-fold.
The purified protein gave rise after SDS-PAGE analysis to one band that corresponded to a molecular mass near 92 kDa (Fig. 1). However, on some occasions, a second band of comparable size was observed with some enzyme samples. This result is consistent with the behavior of the S. marinus enzyme (8). The specific activities of the various preparations (with one or two bands) were comparable, and the two forms had the same N-terminal amino acid sequence (AYRFIKGFEELSKNDVPLVG), showing that they are different forms of the same enzyme. These are thought to be phosphorylated and nonphosphorylated forms, as discussed below. Their sequences closely matched that previously deduced from the gene sequence of P. furiosus PEP synthetase (19 of 20 residues [21, 39]), with the exception of the encoded N-terminal methionine, which was not present (21). The calculated molecular mass of the subunit (without the N-terminal methionine) from the gene sequence is 90,346 Da, which is approximately the size of the subunit as determined by gel electrophoresis. The active form of the enzyme (690 kDa) therefore appears to be a homooctomer. ICP spectroscopic analysis of the pure protein giving rise to a single major band on the SDS gel revealed 1.1 ± 0.40 g-atoms of Ca and 1.1 ± 0.43 g-atoms of phosphorus per 92,000 Da of protein. Metals such as Zn, Co, and Cu were not present in significant amounts (<0.1 g-atoms subunit−1). All preparations of PEP synthetase examined contained iron in amounts of 0.1 to 0.4 g-atoms subunit−1. However, the enzyme specific activity did not correlate with the iron concentration, so it is assumed that this metal is not a functional part of the enzyme. The presence of phosphorus is consistent with previous data on the PEP synthetase from E. coli. This enzyme can be isolated in a phosphorylated form, which is an intermediate in the enzyme's phosphate transfer mechanism (3), although the E. coli protein has not been reported to contain calcium. To determine if calcium was adventitiously bound to P. furiosus PEP synthetase, the enzyme was incubated with EGTA at 23°C or at 80°C for 30 min and then separated from the chelator by ion-exchange chromatography (Pharmacia). The enzyme did not lose activity during either treatment, and ICP analysis showed that the calcium content remained the same (∼1 g-atom subunit−1). This element therefore appears to be an integral part of the P. furiosus PEP synthetase.
FIG. 1.

SDS-PAGE of P. furiosus PEP synthetase. The right lane contained purified PEP synthetase, and the left lane contained marker proteins with molecular masses (in kDa) indicated at the left.
Previous researchers concluded that PEP synthetase might be redox active, since the enzyme from P. furiosus was oxygen sensitive (44) and the enzyme from methanogens required reducing agents for maximum activity (14). However, the pure P. furiosus enzyme lost no activity when stored overnight under air (or Ar), and incubation for 30 min with either 5 mM dithiothreitol or 40 mM β-mercaptoethanol, either at 23 or 80°C, had no affect on its activity. The thermal stability of the enzyme (1 mg ml−1 in 50 mM EPPS, pH 8.0) was measured at 90°C, and the time required to lose 50% of its activity (t1/2) was about 9 h. The presence of KCl, either at 0.2 or 0.5 M, had no effect on the t1/2 value. To investigate whether thermal inactivation was due to a change in the quaternary structure of the enzyme, a sample (1 mg ml−1 in 50 mM Tris, pH 8.0) that had been incubated at 90°C for 10 h was cooled on ice and then applied to a Superose 6 gel filtration column. The proportion of the larger, inactive form of the enzyme remained about the same (increasing from 29 to 34% of the total), indicating that aggregation of the active 690-kDa form was not the main cause of loss of enzyme activity.
Catalytic properties.
PEP synthetase required divalent metal cations in the assay mixture for catalysis as measured by the PEP formation assay; no activity was detected if they were omitted. The optimal Mg2+ concentration was 10 mM (range tested, 0 to 50 mM). At Mg2+ concentrations above 10 mM, there was slight inhibition. The activity of the enzyme was also measured in the presence of Ca2+, Sr2+, Mn2+, Co2+, and Ni2+ (2 mM, as the chloride salts) to see if they could substitute for Mg2+. This was the case with Mn2+ (at 95% of Mg2+ activity) and Co2+ (93%) ions, while Ni2+ (15%) was not as effective and both Sr2+ (4%) and Ca2+ (3%) showed only minor activation. The effects of the monovalent cations Na+, K+, and NH4+ (chloride salts) on PEP synthetase were measured in the presence of 10 mM Mg2+ ions. Enzyme activity was stimulated by the addition of NH4+ (48% increase with 100 mM) or K+ (50% with 100 mM) ions, but Na+ (100 mM) had no effect. The potassium ion concentration for optimal enzyme activity was between 30 and 250 mM, within the intracellular range (30). The enzyme showed an optimal pH near 9.0 in the PEP formation assay, while the optimal concentration of the pyruvate formation reaction was 7.5 (Fig. 2). The optimal temperature for PEP formation over a 2-min period was 90°C. The enzyme exhibited measurable activity at 30°C, but it was only 0.2% of that measured at 90°C (data not shown).
FIG. 2.

pH dependence of the pyruvate (empty symbols) and PEP (solid symbols) formation reactions of P. furiosus PEP synthetase. The enzyme activities were determined under the conditions described in Materials and Methods but at the indicated pH.
Kinetic constants were determined for pyruvate and ATP in the forward reaction and for PEP, AMP, and phosphate in the reverse reaction. The substrate concentrations of the invariant substrates were 4 mM except in the case of phosphate, which was used at 10 mM. The variable substrates were used at 0.004 to 4 mM in all cases except for phosphate, where the substrate concentration varied between 0.1 and 100 mM. As shown in Table 2, the enzyme catalyzed PEP formation with a catalytic efficiency that is at least 100-fold greater than that measured for the reverse reaction, consistent with the proposed physiological role of the enzyme in gluconeogenesis. The specificity of PEP synthetase was examined with a number of 2-ketoacids in addition to pyruvate, and these included 2-keto-3-methylvalerate, 2-ketoisocaproate, 2-ketoisovalerate, 2-ketocaproate, 2-ketobutyrate, 2-ketoglutarate, 2-ketomalonate, phenylpyruvate, hydroxyphenylpyruvate, imidazolpyruvate, and indole-3-pyruvate. The enzyme has a strong preference for pyruvate and only a slight amount of phosphorylation of 2-ketobutyrate (2 mM, showing 8% of the activity measured with pyruvate) and 2-ketoglutarate (2 mM, 5.3%). The substrate specificity is reasonable since it is not clear what the cell would do with phosphorylated forms of butyrate and glutarate.
TABLE 2.
Kinetic constants of P. furiosus PEP synthetase
| Reaction | Apparent Km (mM) | Apparent kcat (s−1) | Apparent kcat/Km (mM−1 · s−1) |
|---|---|---|---|
| PEP formationa | |||
| Pyruvate | 0.11 | 1,573 | 1.43 × 104 |
| ATP | 0.39 | 1,326 | 3.40 × 103 |
| Pyruvate formationb | |||
| PEP | 0.40 | 12.6 | 31.5 |
| AMP | 1.00 | 8.7 | 8.7 |
| Phosphate | 38.4 | 11.9 | 0.315 |
Determined using the phosphate formation assay.
Determined using the pyruvate formation assay.
PEP synthetase was tested for the ability to use alternate phosphate donors and acceptors, since there is a precedent for unusual nucleotide usage of enzymes in P. furiosus (24, 29, 48). Of the phosphate donors tried, ATP was the only substrate that the enzyme would use to catalyze PEP formation. The activity with ADP, GDP, or GTP (each at 2 mM) was less than 1% of the activity of the enzyme with ATP. In the reverse direction, ADP, AMP, GDP, and GMP (2 mM), with or without phosphate, were tested as phosphate acceptors for PEP synthetase. The enzyme had a strong preference for AMP with phosphate as a substrate, although there was significant activity (20% of that with AMP) using ADP with phosphate.
DISCUSSION
Unlike several other nucleotide-utilizing enzymes recently purified from P. furiosus, such as hexokinase (glucokinase), phosphofructokinase, and acetyl-CoA synthetase (24, 29, 48), PEP synthetase is typical in that it uses the nucleotides expected from studies of gluconeogenesis in mesophilic organisms. Pyruvate and ATP are used to generate PEP, AMP, and Pi, and catalytic efficiencies suggest that the reaction occurs in the gluconeogenic direction. The enzyme does not catalyze to any extent PPDK (equation 4) or PK (equation 5) reactions. However, the enzyme was unique relative to its mesophilic counterparts in that it was a homooctomer and comprised a large proportion (∼5%) of the total cytosolic protein.
Little is known about PEP synthetases in general, so there is only limited information with which to compare the properties of the P. furiosus enzyme (Table 3). The most obvious difference between those purified thus far is size. The homomultimeric enzymes from the hyperthermophilic archaea S. marinus and P. furiosus are much larger than the homodimeric E. coli protein. Elegant and extensive structural studies on S. marinus PEP synthetase by electron microscopy have shown that it contains 24 subunits. In contrast, the P. furiosus enzyme is purified in what appear to be 8- and 18-subunit forms, with only the homooctomer possessing enzymatic activity. Unfortunately, whether S. marinus PEP synthetase is active has not been reported, as it was purified according to its size (8), and it is not known whether this organism contains a smaller (and active) version of the enzyme. It has been suggested that the increase in the number of subunits of hyperthermophilic enzymes compared to that for their mesophilic analogs might contribute to their increased thermostability (2, 12, 27, 50). Whether the larger and seemingly inactive version of P. furiosus PEP synthetase is an artifact of purification or whether it has a physiological significance remains to be determined.
TABLE 3.
Properties of purified PEP synthetasesa
| Property | Properties of PEP synthetases for:
|
|||
|---|---|---|---|---|
| E. coli | M. thermoautotrophicum | S. marinus | P. furiosus | |
| Molecular mass (kDa) | 150 | NRb | 2,250 | 690 |
| No. of subunits | 2 | NR | 24 | 8 (18c) |
| Subunit size (kDa)c | 87 | 75.6 | 93 | 90.4 |
| Sequence similarity (%)c | 52 | 60 | 61 | 100 |
| Km for pyruvate (mM) | 0.28 | 0.04 | NR | 0.11 |
| Km for ATP (mM) | 0.19 | 0.7 | NR | 0.13 |
| Metal content (per subunit) | NR | NR | NR | 1 Ca |
| M2+ requirement | Mg or Mn | Mg | NR | Mg, Mn, or Co |
| M+ requirement | None | NH4 or K | NR | NH4 or K |
| Reductant required | None | DTT or MEd | NR | None |
| Optimal pH (forward) | 8.4 | 7.8 | NR | 9.0 |
| Optimal pH (reverse) | 6.8 | 6.6 | NR | 7.5 |
Data for the enzymes from E. coli (11, 32, 34), M. thermoautotrophicum (14, 46), and S. marinus (8, 17) were taken from the indicated references.
NR, not reported.
Number of subunits for larger, inactive subunit is in parenthesis.
Calculated from the gene sequences.
DTT, dithiothreitol; ME, β-mercaptoethanol.
Studies on the response of growing P. furiosus on maltose showed that maltose addition caused an increase in the concentration of the mRNA encoding PEP synthetase (38, 39). However, PEP synthetase activity varied less than twofold in cultures grown on maltose versus on peptides (1). To explain the first result, it was suggested that this enzyme not only played a role in gluconeogenesis but also catalyzed the conversion of PEP to pyruvate in the glycolytic pathway of P. furiosus, a conversion normally catalyzed by PK (38, 39). However, the kinetic properties of the P. furiosus PEP synthetase (Table 2) indicate that the gluconeogenic reaction is greatly favored and that the enzyme has extremely low PK activity in comparison.
There is some confusion about which enzyme catalyzes the conversion of PEP to pyruvate in P. furiosus. It was recently suggested that the organism contains a novel AMP-dependent PK that forms pyruvate and ATP from PEP, AMP, and Pi, the reverse reaction of PEP synthetase (42). This is highly significant, as such an enzyme would have the effect of regenerating ATP from the AMP formed by the ADP-dependent kinases in the earlier steps of the glycolytic pathway. However, the genome sequence of P. furiosus (http://comb5-156.umbi.umd.edu/genemate) contains an open reading frame that is homologous to conventional PKs (e.g., it shows 48% similarity to the amino acid sequence of PK from Bacillus stearothermophilus), and cell extracts of P. furiosus contain significant ADP-dependent PK activity (1.4 U mg−1 at 100°C [43]). Interestingly, the novel AMP-dependent PK exhibits Km values for PEP, AMP, and Pi not too dissimilar from those reported here for PEP synthetase (although they were measured at pH 6.5 and 50°C [42]). Moreover, although the subunit size of this novel PK (78 kDa) is much smaller than that of PEP synthetase (92 kDa by SDS-PAGE; 90.3 kDa from the sequence), its N-terminal amino acid sequence reportedly (T. Ohshima, H. Sakuraba, H. J. Schreier, E. Utsumi, and N. Nunoura-Kominato, Abstr. 3rd Int. Congr. Extremophiles, abstr. L30, 2000) matches that of the PEP synthetase described herein, an enzyme whose complete amino acid sequence is homologous to those for other known PEP synthetases (21). Thus, while the true nature of the novel PK (42) is unclear, the results presented herein are fully consistent with PEP conversion to pyruvate and pyruvate conversion to PEP occurring in P. furiosus through conventional PK and PEP synthetase reactions, respectively. In addition, extracts of P. furiosus also contain significant adenylate kinase activity (0.22 to 0.37 U mg−1 [1]), suggesting that the interconversion of AMP, ADP, and ATP also occurs by a conventional mechanism.
What reasons are there, then, for such a relatively large amount of PEP synthetase in P. furiosus (∼5% of the cellular protein)? Under such conditions it seems unreasonable that the PEP produced by the PEP synthetase reaction would be required for biosynthesis, since sufficient PEP should be formed in the glycolytic pathway. One possible explanation is that a futile cycle between PEP and pyruvate exists in this organism when grown under our conditions. It has been suggested that such futile cycles can be used by some bacteria when they are grown in the presence of high carbohydrate concentrations to remove excess energy, as this can be harmful to the cell (40). Such organisms experience an imbalance in metabolism, as the energy produced from carbohydrate oxidation via glycolysis is much greater than the energy required for cell maintenance and growth (41, 51). The disposal of the catabolic energy generated in excess of the cells' anabolic needs has been termed “energy spilling” (41). The P. furiosus cells used in the current study were grown in a medium with a high maltose concentration (6), so the cells could well experience the metabolic imbalance that makes energy spilling advantageous. However, futile cycling between PEP and pyruvate via PEP synthetase has not yet been demonstrated in any organism. In E. coli, for example, such cycling between glycolytic and gluconeogenic enzymes is prevented by the tight regulation of these pathways (7, 35). However, regulation in P. furiosus seems to occur only at the level of glyceraldehyde-3-phosphate, via glyceraldehyde-3-phosphate dehydrogenase and the novel enzyme GAPOR (48, 49). Therefore, it is possible that PEP synthetase could be functioning in P. furiosus as an energy-spilling mechanism. Growth studies similar to those reported previously (22) but using an extended range of carbohydrate concentrations are required to determine whether this is the case and to see whether differences in growth yield accompany changes in PEP synthetase activity. Such studies are underway (38, 39). Whether regulation also involves the interconversion between the smaller, active and larger, inactive forms remains to be established.
ACKNOWLEDGMENTS
We thank Marc Verhagen for helpful discussions and suggestions.
This research was supported by grants from the National Science Foundation (MCB 9809060 and BES-0004257).
REFERENCES
- 1.Adams, M. W. W., J. F. Holden, A. Lal Menon, G. J. Schut, A. M. Grunden, C. Hou, A. M. Hutchins, F. E. Jenney, Jr., C. Kim, K. Ma, G. Pan, R. Roy, R. Sapra, S. V. Story, and M. F. J. M. Verhagen. Key role for sulfur in peptide metabolism and in the regulation of three hydrogenases in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 183:716–724. [DOI] [PMC free article] [PubMed]
- 2.Backmann J, Schäfer G, Wyns L, Bönisch H. Thermodynamics and kinetics of unfolding of the thermostable trimeric adenylate kinase from the archaeon Sulfolobus acidocaldarius. J Mol Biol. 1998;284:817–833. doi: 10.1006/jmbi.1998.2216. [DOI] [PubMed] [Google Scholar]
- 3.Berman K M, Cohn M. Phosphoenolpyruvate synthetase: partial reactions studied with adenosine triphosphate analogues and the inorganic phosphate-H218O exchange reaction. J Biol Chem. 1970;245:5319–5325. [PubMed] [Google Scholar]
- 4.Blamey J M, Adams M W W. Purification and characterization of pyruvate ferredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus. Biochim Biophys Acta. 1993;1161:19–27. doi: 10.1016/0167-4838(93)90190-3. [DOI] [PubMed] [Google Scholar]
- 5.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 6.Bryant F O, Adams M W W. Characterization of hydrogenase from the hyperthermophilic archaebacterium, Pyrococcus furiosus. J Biol Chem. 1989;264:5070–5079. [PubMed] [Google Scholar]
- 7.Chao Y-P, Liao J C. Metabolic responses to substrate futile cycling in Escherichia coli. J Biol Chem. 1994;269:5122–5126. [PubMed] [Google Scholar]
- 8.Cicicopol C, Peters J, Kellermann J, Baumeister W. Primary structure of multimeric protein, homologous to the PEP-utilizing enzyme family and isolated from a hyperthermophilic archaebacterium. FEBS Lett. 1994;356:345–350. doi: 10.1016/0014-5793(94)01304-7. [DOI] [PubMed] [Google Scholar]
- 9.Cicicopol C, Peters J, Lupas A, Cejka Z, Müller S A, Golbik R, Pfeifer G, Lilie H, Engel A, Baumeister W. Novel molecular architecture of the multimeric archaeal PEP-synthase homologue (MAPS) from Staphylothermus marinus. J Mol Biol. 1999;290:347–361. doi: 10.1006/jmbi.1999.2878. [DOI] [PubMed] [Google Scholar]
- 10.Cooper R A, Kornberg H L. Net formation of phosphoenolpyruvate from pyruvate by Escherichia coli. Biochim Biophys Acta. 1965;104:618–620. doi: 10.1016/0304-4165(65)90374-0. [DOI] [PubMed] [Google Scholar]
- 11.Cooper R A, Kornberg H L. Phosphoenolpyruvate synthetase. Methods Enzymol. 1969;13:309–314. [Google Scholar]
- 12.Delboni L F, Mande S C, Rentier-Delrue F, Mainfroid V, Turley S, Vellieux F M D, Martial J A, Hol W G J. Crystal structure of recombinant triosephosphate isomerase from Bacillus stearothermophilus. An analysis of potential thermostability factors in six isomerases with known three-dimensional structures points to the importance of hydrophobic interactions. Protein Sci. 1995;4:2594–2604. doi: 10.1002/pro.5560041217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vos W M, Kengen S W M, Voorhorst W G B, van der Oost J. Sugar utilization and its control in hyperthermophiles. Extremophiles. 1998;2:201–205. doi: 10.1007/s007920050061. [DOI] [PubMed] [Google Scholar]
- 14.Ezyaguirre J, Jansen K, Fuchs G. Phosphoenolpyruvate synthetase in Methanobacterium thermoautotrophicum. Arch Microbiol. 1982;132:67–74. [Google Scholar]
- 15.Fiala G, Stetter K O. Pyrococcus furiosus sp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100°C. Arch Microbiol. 1986;145:56–61. [Google Scholar]
- 16.Harauz G. Symmetry in the 2.25 MDa homomultimeric phosphoenolpyruvate synthase from Staphylothermus marinus: analyses of negatively stained preparations. Micron. 1998;29:161–173. [Google Scholar]
- 17.Harauz G, Cicicopol C, Hegerl R, Cejka Z, Goldie K, Santarius U, Engel A, Baumeister W. Structural studies on the 2.25 MDa homomultimeric phosphoenolpyruvate synthase from Staphylothermus marinus. J Struct Biol. 1996;116:290–301. doi: 10.1006/jsbi.1996.0044. [DOI] [PubMed] [Google Scholar]
- 18.Hasegawa H, Parniak M, Kaufman S. Determination of the phosphate content of purified proteins. Anal Biochem. 1982;120:360–364. doi: 10.1016/0003-2697(82)90358-x. [DOI] [PubMed] [Google Scholar]
- 19.Hess D, Krüger K, Knappik A, Palm P, Hensel R. Dimeric 3-phosphoglycerate kinases from hyperthermophilic archaea. Eur J Biochem. 1995;233:227–237. doi: 10.1111/j.1432-1033.1995.227_1.x. [DOI] [PubMed] [Google Scholar]
- 20.Jetten M S M, Pitoc G A, Follettie M T, Sinskey A J. Regulation of phospho(enol)-pyruvate- and oxaloacetate-converting enzymes in Corynebacterium glutamicum. Appl Microbiol Biotechnol. 1994;41:47–52. [Google Scholar]
- 21.Jones C E, Fleming T M, Piper P W, Littlechild J A, Cowan D A. Cloning and sequencing of a gene from the archaeon Pyrococcus furiosus with high homology to a gene encoding phosphoenolpyruvate synthetase from Escherichia coli. Gene. 1995;160:101–103. doi: 10.1016/0378-1119(95)00128-s. [DOI] [PubMed] [Google Scholar]
- 22.Kengen S W M, Stams A J M. Growth and energy-conservation in batch cultures of Pyrococcus furiosus. FEMS Microbiol Lett. 1994;117:305–309. [Google Scholar]
- 23.Kengen S W M, Stams A J M, de Vos W M. Sugar metabolism of hyperthermophiles. FEMS Microbiol Rev. 1996;18:119–137. [Google Scholar]
- 24.Kengen S W M, Tuininga J E, de Bok F A M, Stams A J M, de Vos W M. Purification and characterization of a novel ADP-dependent glucokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem. 1995;270:30453–30457. doi: 10.1074/jbc.270.51.30453. [DOI] [PubMed] [Google Scholar]
- 25.Kohlhoff M, Dahm A, Hensel R. Tetrameric triosephosphate isomerase from hyperthermophilic archaea. FEBS Lett. 1996;383:245–250. doi: 10.1016/0014-5793(96)00249-9. [DOI] [PubMed] [Google Scholar]
- 26.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 27.Lebbink J H G, Kengen S W M, van der Oost J, de Vos W M. Glutamate dehydrogenase from hyperthermophilic bacteria and archaea: determinants of thermostability and catalysis at extremely high temperatures. J Mol Catal B Enzym. 1999;7:133–145. [Google Scholar]
- 28.Lebloas P, Lindley N D, Loubiere P. Regulation of carbon and energy metabolism during the linear growth phase in batch fermentations of the acetogeneic methylotroph Eubacterium limosum on methanol/CO2. Enzyme Microb Technol. 1996;19:187–195. [Google Scholar]
- 29.Mai X, Adams M W W. Purification and characterization of two reversible and ADP-dependent acetyl coenzyme A synthetases from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol. 1996;178:5897–5903. doi: 10.1128/jb.178.20.5897-5903.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martins L O, Huber R, Huber H, Stetter K O, da Costa M S, Santos H. Organic solutes in hyperthermophilic archaea. Appl Environ Microbiol. 1997;63:896–902. doi: 10.1128/aem.63.3.896-902.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukund S, Adams M W W. Glyceraldehyde-3-phosphate ferredoxin oxidoreductase, a novel tungsten-containing enzyme with a potential glycolytic role in the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem. 1995;270:8389–8392. doi: 10.1074/jbc.270.15.8389. [DOI] [PubMed] [Google Scholar]
- 32.Narindrasorasak S, Bridger W A. Phosphoenolpyruvate synthetase of Escherichia coli: molecular weight, subunit composition, and identification of phosphohistidine in phosphoenzyme intermediate. J Biol Chem. 1977;252:3121–3127. [PubMed] [Google Scholar]
- 33.Nègre D, Oudot C, Prost J-F, Murakami K, Ishihama A, Cozzone A J, Cortay J-C. FruR-mediated transcriptional activation at the ppsA promoter of Escherichia coli. J Mol Biol. 1998;276:355–365. doi: 10.1006/jmbi.1997.1548. [DOI] [PubMed] [Google Scholar]
- 34.Niersbach M, Kreuzaler F, Geerse R H, Postma P W, Hirsch H J. Cloning and nucleotide sequence of the Escherichia coli K-12 ppsA gene, encoding PEP synthase. Mol Gen Genet. 1992;231:332–336. doi: 10.1007/BF00279808. [DOI] [PubMed] [Google Scholar]
- 35.Patnaik R, Roof W D, Young R F, Liao J C. Stimulation of glucose catabolism in Escherichia coli by a potential futile cycle. J Bacteriol. 1992;174:7527–7532. doi: 10.1128/jb.174.23.7527-7532.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peak M J, Peak J G, Stevens F J, Blamey J, Mai X, Zhou Z H, Adams M W W. The hyperthermophilic glycolytic enzyme enolase in the archaeon, Pyrococcus furiosus: comparison with mesophilic enolases. Arch Biochem Biophys. 1994;313:280–286. doi: 10.1006/abbi.1994.1389. [DOI] [PubMed] [Google Scholar]
- 37.Ramseier T M, Nègre D, Cortay J-C, Scarabel M, Cozzone A J, Saier M H., Jr In vitro binding of the pleiotropic transcriptional regulatory protein, FruR, to the fru, pps, ace, pts and icd operons of Escherichia coli and Salmonella typhimurium. J Mol Biol. 1993;234:28–44. doi: 10.1006/jmbi.1993.1561. [DOI] [PubMed] [Google Scholar]
- 38.Robinson K A, Robb F T, Schreier H J. Isolation of maltose-regulated genes from the hyperthermophilic archaeum, Pyrococcus furiosus, by subtractive hybridization. Gene. 1994;148:137–141. doi: 10.1016/0378-1119(94)90247-x. [DOI] [PubMed] [Google Scholar]
- 39.Robinson K A, Schreier H J. Isolation, sequence and characterization of the maltose-regulated mlrA gene from the hyperthermophilic archaeum Pyrococcus furiosus. Gene. 1994;151:173–176. doi: 10.1016/0378-1119(94)90651-3. [DOI] [PubMed] [Google Scholar]
- 40.Russell J B. Strategies that ruminal bacteria use to handle excess carbohydrate. J Anim Sci. 1998;76:1955–1963. doi: 10.2527/1998.7671955x. [DOI] [PubMed] [Google Scholar]
- 41.Russell J B, Cook G M. Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol Rev. 1995;59:48–62. doi: 10.1128/mr.59.1.48-62.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakuraba H, Utsumi E, Kujo C, Ohshima T. An AMP-dependent (ATP-forming) kinase in the hyperthermophilic archaeon Pyrococcus furiosus: characterization and novel physiological role. Arch Biochem Biophys. 1999;364:125–128. doi: 10.1006/abbi.1999.1121. [DOI] [PubMed] [Google Scholar]
- 43.Schäfer T, Schönheit P. Maltose fermentation to acetate, CO2 and H2 in the anaerobic hyperthermophilic archaeon Pyrococcus furiosus: evidence for the operation of a novel sugar fermentation pathway. Arch Microbiol. 1992;158:188–202. [Google Scholar]
- 44.Schäfer T, Schönheit P. Gluconeogenesis from pyruvate in the hyperthermophilic archaeon Pyrococcus furiosus: involvement of reactions of the Embden-Meyerhof pathway. Arch Microbiol. 1993;159:354–363. [Google Scholar]
- 45.Schobert P, Bowien B. Unusual C3 and C4 metabolism in the chemoautotroph Alcaligenes eutrophus. J Bacteriol. 1984;159:167–172. doi: 10.1128/jb.159.1.167-172.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith D R, Doucette-Stamm L A, Deloughery C, Lee H, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbet K, Harrison D, Hoang L, Keagle P, Lumm W, Pothier B, Qiu D, Spadafora R, Vicaire R, Wang Y, Wierzbowski J, Gibson R, Jiwani N, Caruso A, Bush D, Safer H, Patwell D, Prabhakar S, McDougall S, Shimer G, Goyal A, Pietrokovski S, Church G M, Daniels C J, Mao J-I, Rice P, Nölling J, Reeve J N. Complete genome sequence of Methanobacterium thermoautotrophicum ΔH: functional analysis and comparative genomics. J Bacteriol. 1997;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smyer J R, Jeter R M. Characterization of phosphoenolpyruvate synthase mutants in Salmonella typhimurium. Arch Microbiol. 1989;153:26–32. doi: 10.1007/BF00277536. [DOI] [PubMed] [Google Scholar]
- 48.Tuininga J E, Verhees C H, van der Oost J, Kengen S W M, Stams A J M, de Vos W M. Molecular and biochemical characterization of the ADP-dependent phosphofructokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem. 1999;274:21023–21028. doi: 10.1074/jbc.274.30.21023. [DOI] [PubMed] [Google Scholar]
- 49.van der Oost J, Schut G, Kengen S W M, Hagen W R, Thomm M, de Vos W M. The ferredoxin-dependent conversion of glyceraldehyde-3-phosphate in the hyperthermophilic archaeon Pyrococcus furiosus represents a novel site of glycolytic regulation. J Biol Chem. 1998;273:28149–28154. doi: 10.1074/jbc.273.43.28149. [DOI] [PubMed] [Google Scholar]
- 50.Villeret V, Clantin B, Tricot C, Legrain C, Roovers M, Stalon V, Glansdorff N, Van Beeumen J. The crystal structure of Pyrococcus furiosus ornithine carbamoyltransferase reveals a key role for oligomerization in enzyme stability at extremely high temperatures. Proc Natl Acad Sci USA. 1998;95:2801–2806. doi: 10.1073/pnas.95.6.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng A-P, Deckwer W-D. A kinetic model for substrate and energy consumption of microbial growth under substrate-sufficient conditions. Biotechnol Prog. 1995;11:71–79. doi: 10.1021/bp00031a010. [DOI] [PubMed] [Google Scholar]