

Abstract
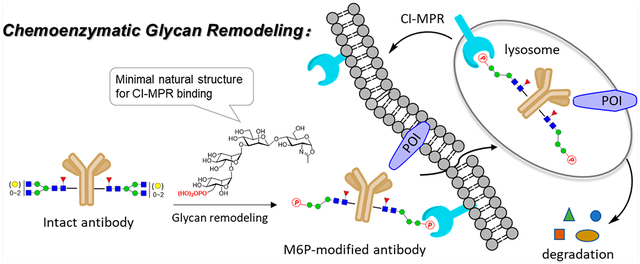
Lysosome-targeting chimeras (LYTACs) offer an opportunity for the degradation of extracellular and membrane-associated proteins of interest. Here, we report an efficient chemoenzymatic method that enables a single-step and site-specific conjugation of high-affinity mannose-6-phosphate (M6P) glycan ligands to antibodies without the need of protein engineering and conventional click reactions that would introduce “unnatural” moieties, yielding homogeneous antibody-M6P glycan conjugates for targeted degradation of membrane-associated proteins. Using trastuzumab and cetuximab as model antibodies, we showed that the wild-type endoglycosidase S (Endo-S) could efficiently perform the antibody deglycosylation and simultaneous transfer of an M6P–glycan from a synthetic M6P–glycan oxazoline to the deglycosylated antibody in a one-pot manner, giving structurally well-defined antibody–M6P glycan conjugates. A two-step procedure, using wild-type Endo-S2 for deglycosylation followed by transglycosylation with an Endo-S2 mutant (D184M), was also efficient to provide M6P glycan–antibody conjugates. The chemoenzymatic approach was highly specific for Fc glycan remodeling when both Fc and Fab domains were glycosylated, as exemplified by the selective Fc-glycan remodeling of cetuximab. SPR binding analysis indicated that the M6P conjugates possessed a nanomolar range of binding affinities for the cation-independent mannose-6-phosphate receptor (CI-MPR). Preliminary cell-based assays showed that the M6P–trastuzumab and M6P–cetuximab conjugates were able to selectively degrade the membrane-associated HER2 and EGFR, respectively. This modular glycan-remodeling strategy is expected to find wide applications for antibody-based lysosome-targeted degradation of extracellular and membrane proteins.
Graphical Abstract
INTRODUCTION
Proteolysis targeting chimeras (PROTACs)1–3 that degrade proteins of interest through the ubiquitin–proteasome system have been successfully employed in the degradation of different types of target proteins related to various diseases, including cancer,4–6 viral infection,7 immune disorders,8,9 and neuro-degenerative diseases.10 Different from the competitive- and occupancy-driven process of traditional inhibitors, PROTACs require only a transient interaction with the target protein and promote its degradation catalytically.11 However, this strategy is limited to targeting engagement within the cells for ubiquitination,12 leaving many membrane and extracellular proteins untargetable, thus the development of complementary strategies that include proteins without cytosolic binding domains is greatly needed.
Lysosomes are another major destination for protein degradation through autophagy and endocytosis.13 Unlike the proteasomal pathway, the lysosomal pathway for protein degradation is not limited to proteins that have intracellular domains. In 2020, Bertozzi et al. reported a novel strategy that explores lysosome-targeting chimeras (LYTACs) for degradation of extracellular and membrane-associated proteins through engagement of the cation-independent mannose-6-phosphate receptor (CI-MPR) expressed on the cell surface and lysosomes.14 The initial LYTACs consist of a small molecule or a specific antibody conjugated with a synthetic polypeptide carrying multiple C-linked mannose-6-phosphate (M6Pn) moieties for engaging the CI-MPR. The study provides proof of concept examples establishing a promising strategy for targeting extracellular and/or membrane-associated proteins for lysosomal degradation.14 Nevertheless, the first-generation CI-MPR-based LYTACs have relied on a random conjugation of the M6P-containing polypeptides to the antibodies, and the M6P-containing polypeptides synthesized are also heterogeneous, resulting in heterogeneous antibody–ligand conjugates that will complicate the downstream structure–activity relationship studies and pharmacokinetic analysis.14,15 More recently, several groups have extended the LYTACs strategy to engaging the asialoglycoprotein receptor (ASGPR), another lysosomal targeting receptor primarily expressed on hepatocytes, for delivering extracellular proteins of interest to liver cells for degradation.16–19 These LYTACS are bifunctional molecules composed of special tri-GalNAc ligands to engage the ASGPR and a small molecule binder or specific antibody to recognize the target protein. For the construction of the antibody-based LYTACs, the tri-GalNAc ligands have been linked to the antibody either by nonspecific lysine conjugation to give mixtures of heterogeneous antibody conjugates17, 18 or through a site-specific conjugation at the Fc domain involving genetic encoding of a reactive aldehyde handle on a specific site of the antibody followed by conjugation with the tri-GalNAc ligand via the oxime chemistry.18 It has been shown that the homogeneous cetuximab–tri–GalNAc conjugates obtained by the site-specific conjugation demonstrate improved pharmacokinetic properties in vivo over the heterogeneous antibody conjugates.18 This site-specific conjugation method should also be applicable for constructing antibody–M6P conjugates. Nevertheless, this site-specific antibody conjugation method requires genetic protein engineering of each antibody to encode the unnatural handle for subsequent ligand conjugation.
Various site-specific antibody conjugation methods have been explored in recent years, including antibody engineering for introducing a clickable handle and Fc glycan-mediated conjugation, for producing structurally well-defined homogeneous antibody–drug conjugates for improved stability, optimal pharmacokinetic properties, and overall efficacy.15, 20–24 Theoretically, most of these methods could be applied for making the LYTACs carrying the ligands for CI-MPR or ASGPR. In the present study, we sought to establish a general method for direct site-specific introduction of high-affinity M6P glycan ligands into an antibody to produce M6P ligand-based LYTACs that will have three features: (a) no need for protein engineering of the antibody for introducing a clickable handle, (b) no requirement of click chemistry that would introduce “unnatural moieties” in the construct, and (c) high-affinity “natural” M6P ligands instead of the unnatural, heterogeneous M6P-glycopeptide polymeric ligand (Figure 1). The Fairbank group and our lab have previously reported that some synthetic M6P oligosaccharide oxazolines corresponding to the N-glycan core could be transferred by endoglycosidase Endo-A and Endo-M for glycan remodeling of glycoproteins.25, 26 It has been shown that a single M6P moiety located at the low α−1,3-branch of the N-glycan context is sufficient for a high-affinity binding to receptor CI-MPR, while the presence of an M6P moiety at the α−1,6-branch is dispensable.26 More recently, we have synthesized an expanded panel of phosphorylated N-glycan oxazolines and have identified a minimal natural N-glycan structure carrying the essential Man6P-α1,2-Man moiety as a high-affinity ligand for CI-MPR. This minimal M6P glycan ligand has been successfully used for Endo-A and Endo-F3-catalyzed glycan remodeling of the recombinant human α-glucosidase (lumizyme).27 The M6P glycan-remodeled lumizyme has shown improved cellular uptake and significantly enhanced lysosomal degradation of glycogen in comparison with the lumizyme itself.27 On the other hand, we have previously reported that novel glycosynthase mutants derived from a class of bacterial endoglycosidases, including Endo-S, Endo-S2, and Endo-F3, can take native and azide-modified N-glycan oxazolines as donor substrates for transglycosylation of deglycosylated antibodies to give homogeneous antibody glycoforms.28–35 However, it is unclear if any of the above endoglycosidases could act on phosphorylated glycan oxazolines for antibody glycan remodeling, and if glycan remodeling is successful, the ability of the resulting antibody–M6P ligand conjugates, in which the M6P glycan is directly attached at the Fc glycosylation site, to recognize CI-MPR and to mediate targeted protein degradation has to be verified. We describe here the screening of a panel of endoglycosidases for their ability to transfer the M6P glycan ligands to therapeutic antibodies including trastuzumab (an anti-HER2 antibody) and cetuzumab (an anti-EGFR antibody), as well as the cell-based evaluation of the resulting homogeneous antibody–M6P glycan conjugates for targeted protein degradation. We found that only the Endo-S and Endo-S2 enzymes, but not other endoglycosidases tested, could efficiently use the synthetic phosphorylated glycan oxazolines as donor substrates for antibody glycan remodeling, enabling a highly efficient and site-selective bioconjugation of a high-affinity M6P glycan ligand to the antibody at the highly conserved Fc glycosylation site. The resulting homogeneous antibody–M6P ligand conjugates showed potent binding affinity for the M6P receptor, CI-MPR. A cell-based study demonstrated that the M6P glycan conjugates of cetuximab and trastuzumab could efficiently degrade cell-surface EGFR and HER2, respectively. The present study establishes a general chemoenzymatic method for constructing structurally well-defined antibody–M6P glycan conjugates from “off-the-shelf” IgG antibodies as LYTACs for CI-MPR mediated targeted protein degradation.
Figure 1.
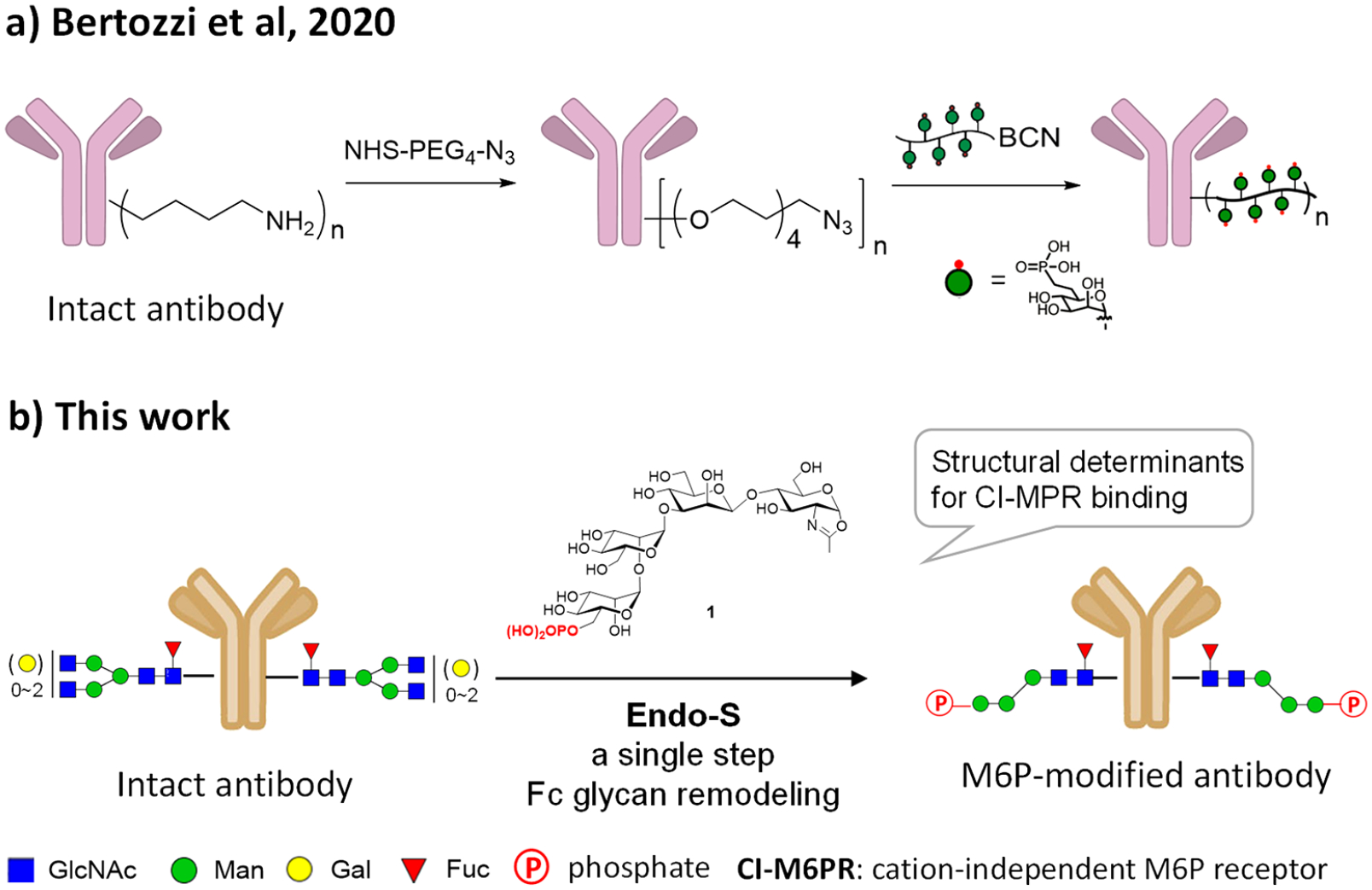
Lysosome-targeting chimera (LYTAC) strategy employing CI-MPR-mediated trafficking of extracellular proteins to lysosomes. (a) The conjugation method reported by Bertozzi et al.14 (b) The site-specific chemoenzymatic method described in the present study.
RESULTS AND DISCUSSION
Screening of Endoglycosidases for Site-Specific Enzymatic Transfer of Phosphorylated Glycans to Antibodies.
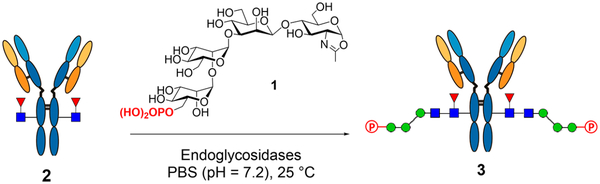
We have recently identified the phosphorylated tetrasaccharide oxazoline (1) corresponding to the α−1,3-branch of the N-glycans as a good substrate of wild-type Endo-A and Endo-F3 for transglycosylation to yield high-affinity ligands for CI-MPR.27 To test if this M6P-glycan oxazoline can be efficiently transferred to an intact antibody by an enzyme, we sought to screen a panel of endoglycosidases using the deglycosylated trastuzumab (Herceptin) as the acceptor and the phosphorylated tetrasaccharide oxazoline (1) as the donor substrate. The results are summarized in Table 1. We started from wild-type Endo S that has been previously found to be active and specific for Fc deglycosylation of intact IgG antibodies and also for enzymatic transfer of truncated N-glycans.28, 32, 36 With a catalytic amount of enzyme (0.2% w/w), we found that Endo S-WT was quite efficient for transglycosylation, and the reaction went to completion within 2 h when a total of 30 mol equiv of the M6P tetrasaccharide oxazoline (1) was added in two portions. Only marginal (less than 1%) hydrolysis of the attached glycans was observed under this condition, which allowed for the accumulation of product. Considering the potent transglycosylation activity, together with its efficient hydrolysis of the complex-type N-glycans from the commercial antibody, wild-type Endo-S provides a practical one-pot strategy for remodeling of heterogeneous glycoforms of the antibody to produce a homogeneous M6P–antibody conjugate, as the deglycosylation and transglycosylation could be performed in a one-pot manner without the need of isolating the wild-type enzyme and the deglycosylated antibody intermediate. We then tested its mutant, Endo-S D233Q, which could efficiently transfer large biantennary glycan oxazolines to antibodies.28 We found that the enzymatic reaction with the D233Q mutant was much slower than that of the wild type Endo-S, and it required a larger amount of enzyme and sugar oxazoline to drive the reaction to completion. Endo-S2, another endoglycosidase from the Streptococcus pyogenes of serotype M49,37 also exhibited rapid transglycosylation of the phosphorylated glycan (1) to the antibody when a catalytic amount of enzyme (0.1%, w/w) was used. But it was found that the wild-type Endo-S2 also hydrolyzed the glycan oxazoline (1) rapidly, although its hydrolysis of the resulting “ground-state” transglycosylation product was relatively slow. Thus, the wild type Endo-S2 was less efficient in this case than the Endo-S enzyme. To reduce the hydrolysis of glycan oxazoline substrate, we tested the D184 M mutant of Endo-S2, a glycosynthase with broad substrate specificity and diminished hydrolytic activity.29 We found that the D184 M mutant exhibited excellent transglycosylation activity. With a catalytic amount of the enzyme (0.1–0.2% w/w) and even reduced equivalents of the oxazoline (1) (10 equiv), the reaction went on smoothly to afford the transglycosylation product within 1 h. As expected, no additional oxazoline was needed, and the transglycosylation product was resistant to hydrolysis by the mutant. Thus, the Endo-S2 D184 M mutant provided another practical method for the production of M6P-containing antibodies. In addition to Endo-S and Endo-S2 and their mutants, we also tested several other endoglycosidases. Endo-F3, which prefers to hydrolyze core-fucosylated complex-type N-glycans30 and has been successfully used for the M6P-glycan remodeling of a multiply glycosylated protein,27 showed only a slow transglycosylation, even when a large amount of enzyme (10%, w/w) was added. The Endo-F3 D165A mutant, a glycosynthase mutant that has been used for remodeling of both Fab and Fc glycans of a therapeutic antibody,33 did not show apparent activity toward this phosphorylated tetrasaccharide, as only a trace amount of the transglycosylation product was detected (Table 1). As for Endo-A, Endo-D, and Endo-CC that prefer nonfucosylated substrates, the GNF-trastuzumab (2) was treated with a fucosidase (BfFucH) to produce an afucosylated Fc glycoform. However, all three of these enzymes could not yield any product even when a large amount of the enzyme (10%, w/w) was used (Table 1). Taken together, our results suggest that the wild-type Endo-S and the glycosynthase mutant (D184M) of Endo-S2 are the best enzymes for antibody bioconjugation using the synthetic phosphorylated N-glycan oxazoline (1) as the donor substrate, and the wild-type Endo-S could perform simultaneous deglycosylation and glycosylation, allowing a one-pot bioconjugation with an M6P glycan ligand to produce a homogeneous antibody–M6P glycan conjugate.
Table 1.
Screening of the Transglycosylation Conditions
|
|||||
|---|---|---|---|---|---|
| transglycosylation | |||||
| ENGases | oxazoline (eq)a | enzyme (w/w) | time | conversion yield | product hydrolysis |
| Endo-S | 20 + 10 | 0.2% | 2 h | 98% | <1%d |
| Endo-S D233Q | 20 + 20 | 2% | 2 h | 75% | no |
| Endo-S2 | 20 + 10 | 0.1% | 2 h | 70%b | 15%d |
| Endo-S2 D184M | 10 | 0.1%~0.2% | 1 h | 98% | <1%d |
| Endo-F3 | 20 + 20 | 10% | 2 h | 55% | no |
| Endo-F3 D165A | 20 | 10% | 3 h | 10% | no |
| Endo-A | 20 | 10% | 3 h | <5%c | −e |
| Endo-D | 20 | 10% | 3 h | <5%c | −e |
| Endo-CC | 20 | 10% | 3 h | <5%c | −e |
Based on the reaction sites.
Due to simultaneous hydrolysis of the transglycosylation product by the wild-type enzyme.
Nonfucosylated trastuzumab was used.
After 3 h at RT.
Not determined.
Evaluation of Additional M6P–Glycan Oxazolines As Donor Substrates for Antibody Glycan Remodeling.
After identifying the D184 M mutant of Endo-S2 as an efficient mutant for glycosylation of trastuzumab with synthetic M6P-tetrasaccharide oxazoline (1), we examined and compared the glycosylation of the deglycosylated trastuzumab with two additional phosphorylated N-glycan oxazolines (4 and 5) that we have previously synthesized.26 As expected, the D184 M mutant of Endo-S2 could efficiently act on glycan oxazoline 1 to produce the glycoform (3) carrying the corresponding M6P-containing N-glycan at the Fc domain (Scheme 1). The D184 M mutant could also use the larger bis-phosphorylated glycans (4 and 5) as donor substrates for transfer to give the two M6P antibody glycoforms (6 and 7), respectively, but a relatively large amount of enzyme and higher concentration of the antibody were required to drive the reaction to completion. These results suggest that the glycan oxazolines carrying two M6P moieties are not as good as the M6P tetrasaccharide oxazoline (1) as the donor substrate. Nevertheless, the data indicate that the Endo-S2 mutant can tolerate the modifications on the glycans to afford antibody glycoforms with different patterns of phosphorylation. The antibody products were purified on a protein A column, and their identities were confirmed by LC-ESI-MS analysis (Figures S1–S3). Finally, excess glycan oxazolines were recovered in the form of free oligosaccharides during purification, which were readily converted into the glycan oxazolines in a single step with DMC/Et3N,38 thus permitting the recycling of glycan oxazolines for transglycosylation.
Scheme 1.
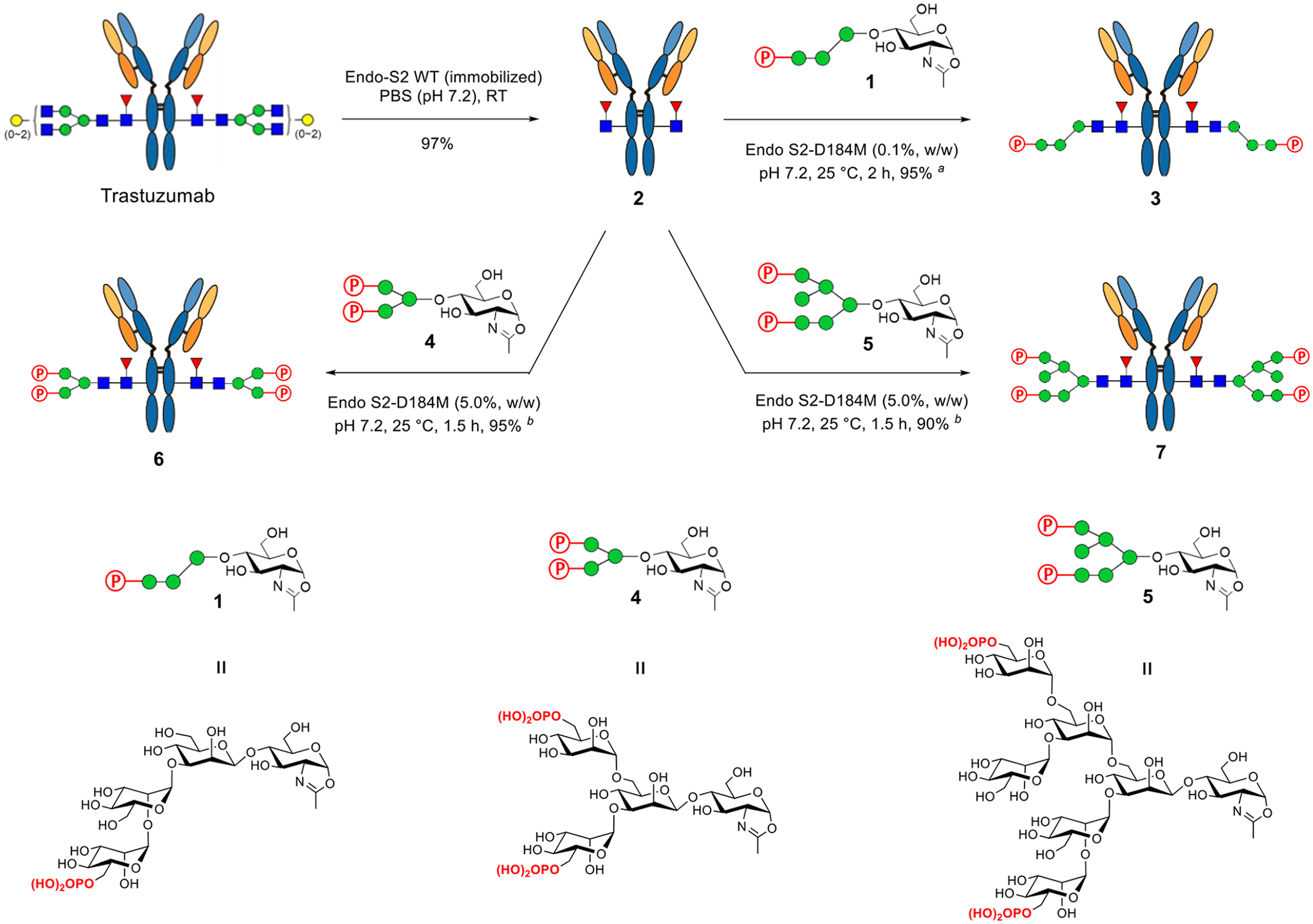
Chemoenzymatic Glycan Remodeling of Trastuzumab with Phosphorylated Glycan Oxazolinesa
aThe reactions were conducted in PBS buffer, and the yields were based on protein A purification. (a) 10 equiv of oxazoline with an antibody concentration of 10 mg/mL; (b) 30 equiv of oxazoline with an antibody concentration of 20 mg/mL.
In addition to trastuzumab, we also performed M6P-glycan remodeling of cetuximab (Scheme 2), a therapeutic antibody that targets the epidermal growth factor receptor (EGFR) for the treatment of colorectal cancer and squamous-cell carcinoma.39 Cetuximab is glycosylated in both Fab and Fc domains with tremendous heterogeneity in the N-glycan structures.40 Previous studies have shown that wild-type Endo-S2 is highly specific for hydrolyzing the Fc glycans.33 Thus, the commercial cetuximab was first treated with wild-type Endo-S2 to produce the deglycosylated Fc glycoform. Then, the resulting GNF–cetuximab (8) was used as an acceptor for Endo-S2 D184M-catalyzed transglycosylation with glycan oxazoline 1 and 5, affording the M6P–glycan remodeled cetuximab 9 and 10, respectively. We found that the M6P–glycan remodeling of cetuximab was equally as efficient as that of trastuzumab, and the products (9 and 10) were isolated in over 90% yield after protein A purification. To further verify that the M6P glycans were specifically conjugated to the Fc domain, the antibody products (9 and 10) were digested with the protease IdeS followed by LC-ESI-MS analysis of the Fab and Fc domains.41 LC-ESI-MS analysis showed that the change of molecular weight of the Fc domain monomer was consistent with the attachment of the corresponding M6P glycan in antibodies 9 and 10, respectively, but the Fab domains appeared as a mixture of glycoforms that did not change before and after the enzymatic glycoengineering procedures (Figures S4 and S5). These results confirm that the chemoenzymatic glycan remodeling approach is highly selective at the Fc domain without modification of the Fab domains. In addition, MALDI-TOF MS analysis of the N-glycans released from the Fc and Fab domains of antibody 9 further confirmed that the Fc domain of 9 carried a single M6P glycan, while the Fab glycans were intact before and after the glycan remodeling (Figure 2).
Scheme 2.
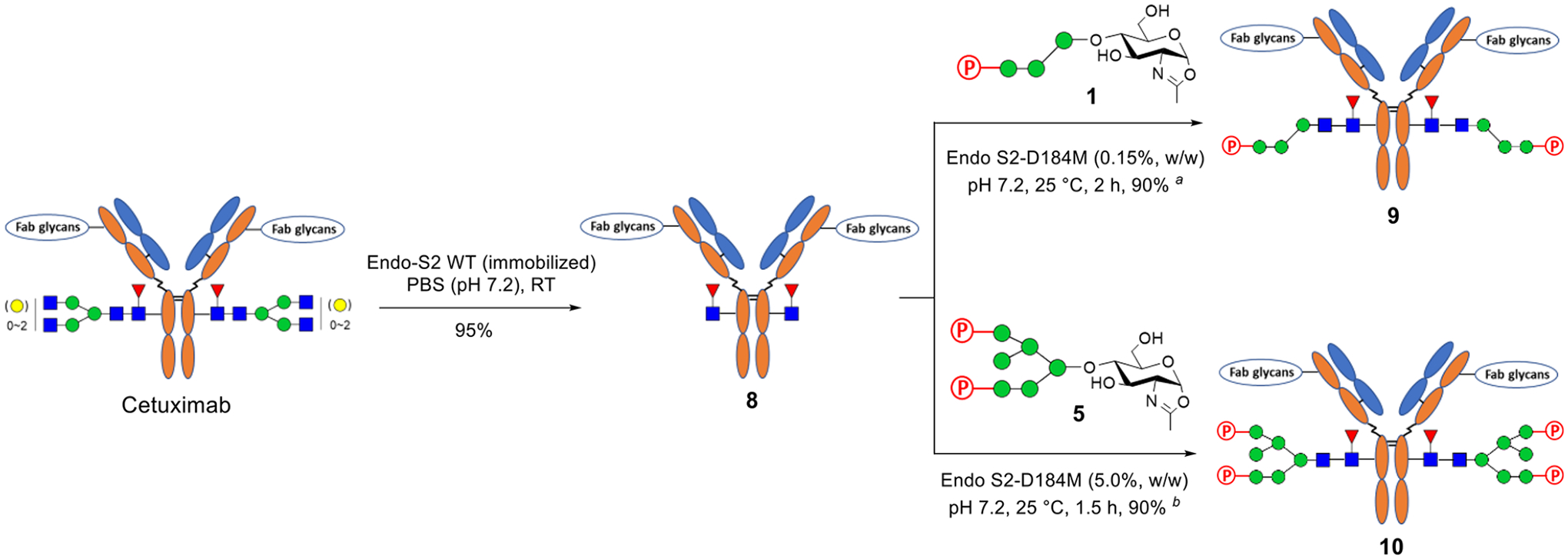
Chemoenzymatic Glycan Remodeling of Cetuximaba
aThe reactions were conducted in PBS buffer, and the yields were based on protein A purification. (a) 10 eqiuv of the glycan oxazoline (1) and an antibody concentration of 10 mg/mL were used. (b) 30 equiv of the glycan oxazoline (5) and an antibody concentration of 20 mg/mL were used.
Figure 2.
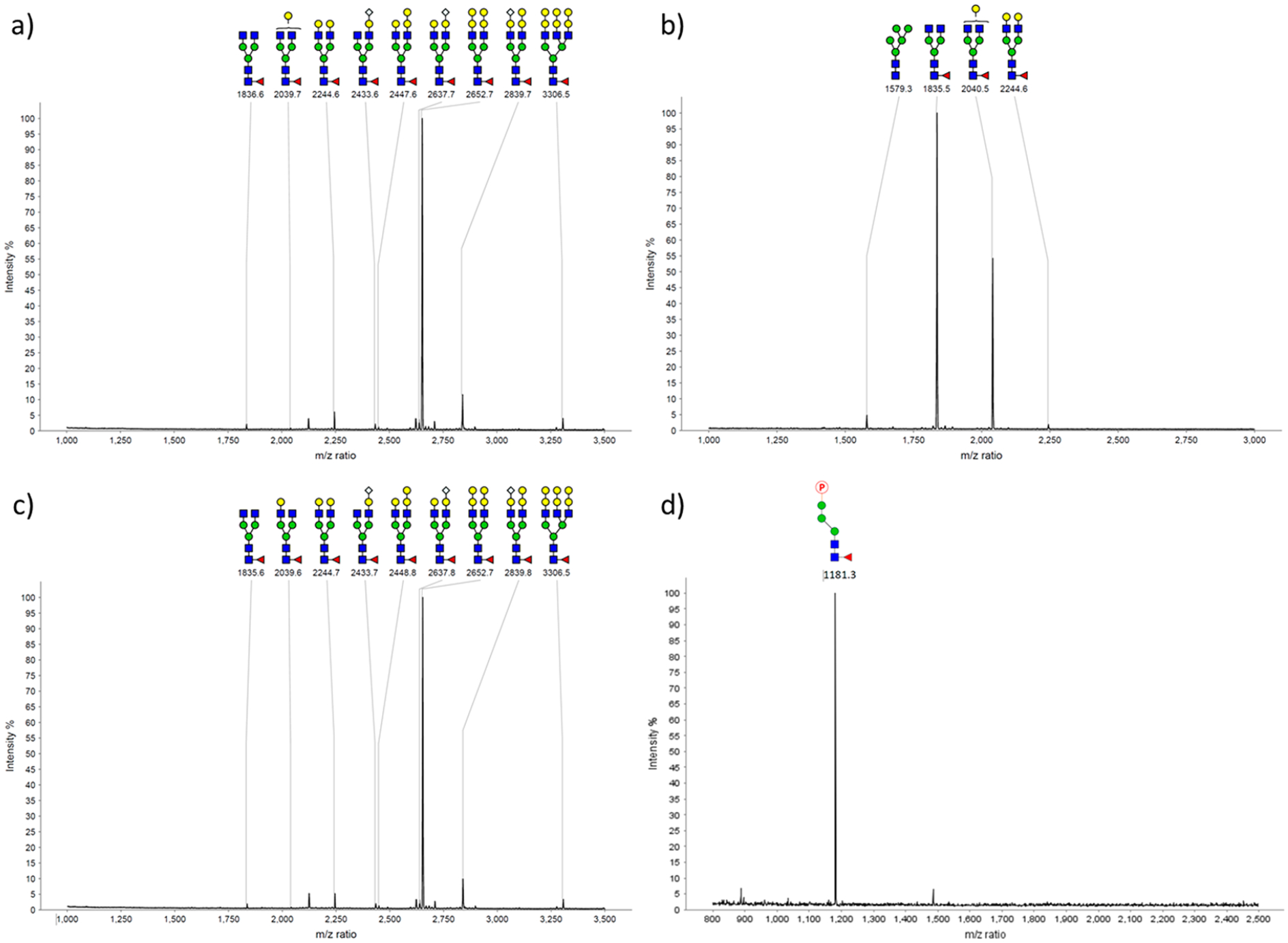
MALDI-TOF-MS analysis (positive-ion mode) of the Fc and Fab N-glycans of the commercial cetuximab and the glycoengineered cetuximab (9). The Fab and Fc domains were disconnected by IdeS treatment followed by protein L separation. The N-glycans were then released by PNGase F treatment, followed by permethylation before MALDI-TOF MS analysis, except for the M6P-containing glycan, which resulted in a loss of the phosphate group. (a) Fab glycans from commercial cetuximab; (b) Fc glycans from commercial cetuximab; (c) Fab glycans from glycoengineered cetuximab 9; (d) Fc glycans from glycoengineered cetuximab 9 (without permethylation).
SPR Binding Studies.
Next, we evaluated the binding affinities of the M6P-modified antibodies to the lysosomal-targeting receptor (CI-MPR) using the SPR technology. We found that the antibodies carrying the M6P tetrasaccharide moiety corresponding to the α−1,3-branch (3 and 9) and the bis-phosphorylated heptasaccharide moiety (7 and 10) showed high-affinity for the M6P receptor, CI-MPR (Figure 3). In the case of trastuzumab, the KD values of binding were determined to be 28.6 nM and 5.4 nM for glycoforms 3 and 7, respectively. The data suggest that the affinity of the glycoform (7) carrying the bis-phosphorylated heptasaccharide moiety was about 4-fold higher than that of the glycoform (3) carrying the M6P tetrasaccharide moiety. On the other hand, the KD values for the two cetuximab glycoforms (9 and 10) were 70.3 nM and 29.6 nM, respectively. Thus, the affinity of the glycoform (10) carrying the large bis-phosphorylated N-glycan was about 2-fold that of the one carrying the smaller M6P tetrasaccahride moiety (9). These data suggest that the nature of the antibody context also has a moderate effect on the affinity of the attached M6P glycan for the M6P receptor (CI-MPR). Interestingly, the antibody glycoform carrying the M6P-Man3GlcNAc2 glycan (6) showed only weak affinity for CI-MPR under the SPR measurement conditions. While the global fitting using the Langmuir binding model did not give reliable kinetic data for glycoform 6, a steady-state fitting gave an estimated KD value of ca. 0.70 μM. This result is consistent with our previous observation on the affinity of different M6P glycan ligands for CI-MPR.27 Our previous structure–affinity relationship studies of the M6P–glycopeptides have shown that a Man6P-α1, 2-Man disaccharide moiety constitutes an essential structural motif that retains strong binding affinity for the CI-MPR receptor. The glycopeptide carrying the M6P-Man3GlcNAc glycan lacks this motif and shows only weak binding to CI-MPR, with a KD of ca. 0.84 μM, the affinity of which is much lower than that of the glycopeptides carrying the glycan with a Man6P-α1, 2-Man disaccharide moiety.27 Thus, the minimal M6P-tetrasaccharide oxazoline (1) that carries the essential high-affinity Man6P-α1, 2-Man motif and is much easier to synthesize than the larger bis-M6P glycan oxazoline (5) was identified as a suitable donor substrate for direct Fc glycan remodeling to construct antibody–M6P–glycan conjugates with high affinity for CI-MPR.
Figure 3.
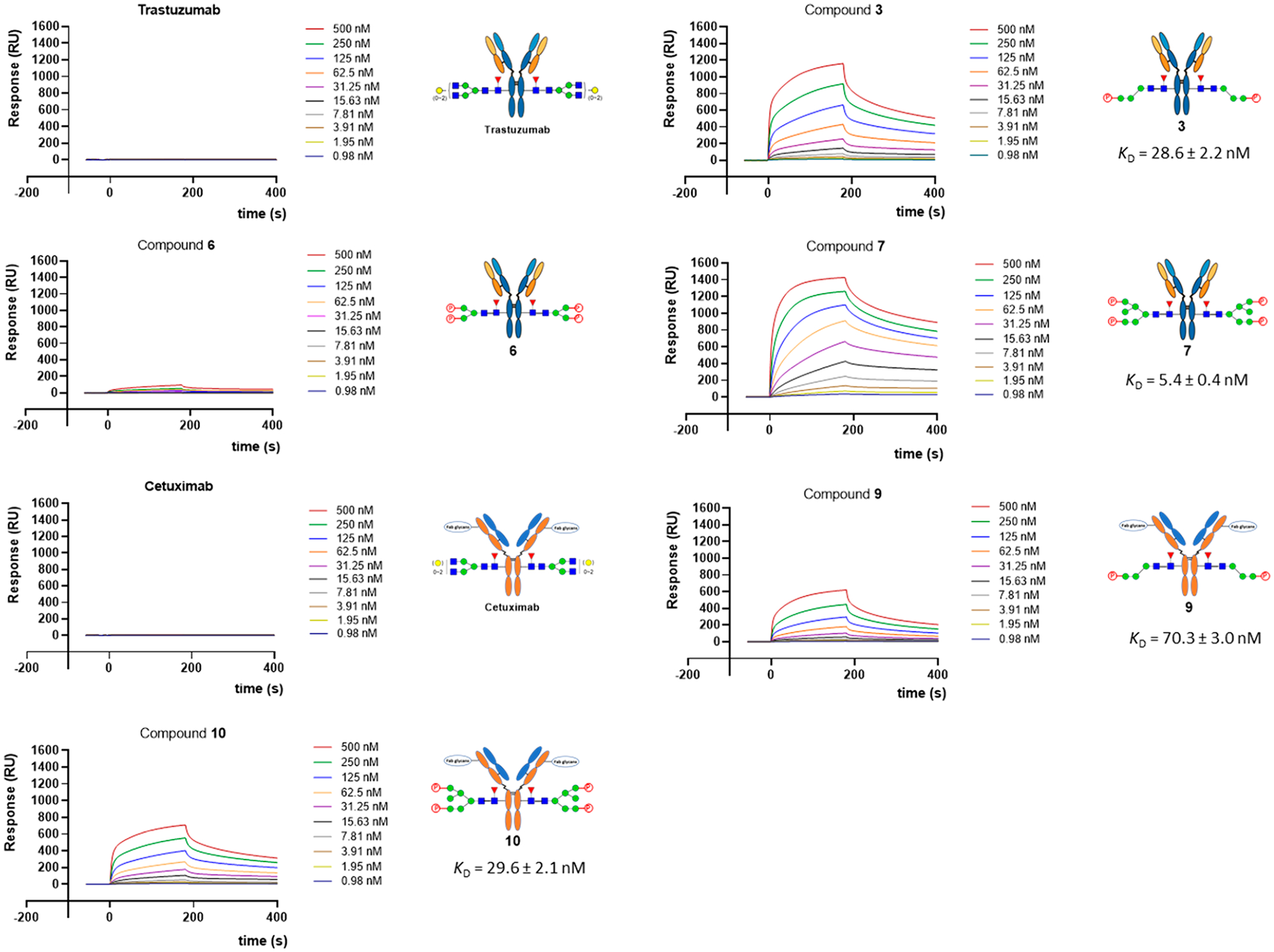
SPR binding data of the CI-MPR with M6P-containing antibodies. The analytes were flowed over an immobilized chip with 2-fold serial dilution of the highest concentration of 500 nM. The sensorgrams are representative of two independent experiments.
Degradation of Membrane-Associated Antigens with the M6P Glycan-Remodeled Antibodies.
We examined two antibodies, the M6P-modified trastuzumab (3) that recognizes HER2 and the M6P-modified cetuximab (9) that targets EGFR, for their potential in targeted degradation of the respective antigens in a cell-based assay. Thus, BT474 (with endogenous HER2 expression) and HepG2 (with endogenous EGFR expression) cell lines were treated with 3 and 9 for 48 h, respectively, and the total antigen levels were measured by Western blot. As indicated by independent experiments (Figure S7), we found that M6P-modified trastuzumab (3) could degrade about 55% of HER2 with a concentration as low as 10 nM, while trastuzumab alone degraded HER2 at ca. 18% (Figure 4a and b). Similar degradation of EGFR was observed by the treatment with M6P-modified cetuximab (9), which resulted in ca. 57% reduction of the total EGFR after treatment, while cetuximab itself gave about 12% degradation of the total EGFR (Figure 4c and d). The targeted degradation of EGFR by the cetuximab-M6P glycan conjugate (9) produced in the present method was comparable to the cetuximab conjugate carrying the polymeric M6Pn-glycopep-tides, where ca. 70% EGFR degradation was observed.14
Figure 4.
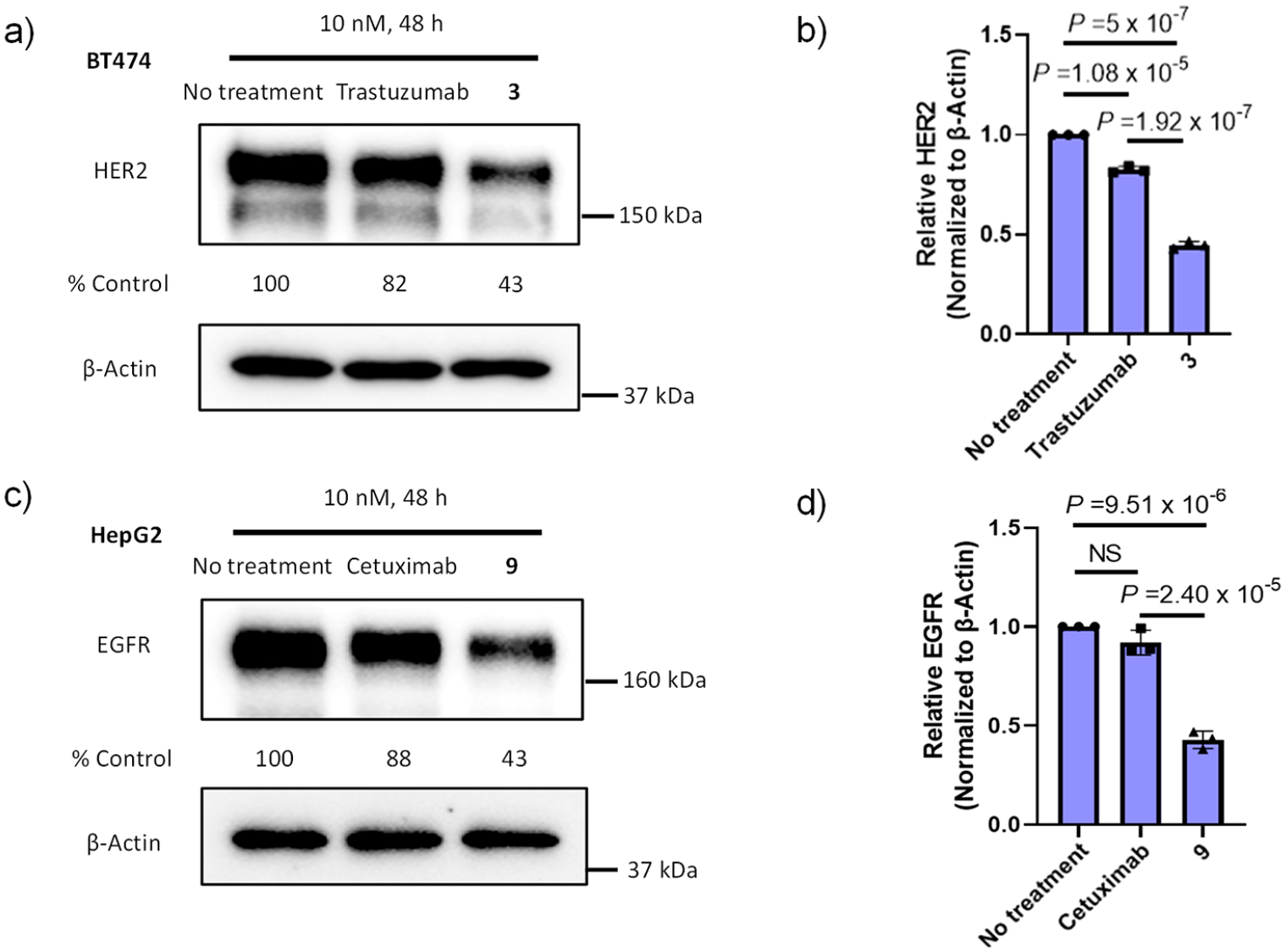
Western blot analysis of total HER2 and EGFR levels. Cells were treated with native or M6P-modified antibodies at a final concentration of 10 nM, then lysed in RIPA buffer after 48 h. Cells without antibody treatment were set as a control. (a) Representative degradation of HER2 by treatment with trastuzumab or 3. (b) HER2 levels after treatment with trastuzumab or 3. (c) Representative degradation of EGFR by treatment with cetuximab or 9. (d) EGFR levels after treatment with cetuximab or 9. All assays were performed in triplicate. P values were determined by one-way ANOVA multiple comparison tests. NS, not significant; a P value of >0.05 was considered not significant.
We also performed a preliminary analysis of cell surface HER2 and EGFR by flow cytometry after the cells were treated with native or M6P-modified antibodies (Figure S8). The results showed that both unmodified and M6P-modified trastuzumab could significantly reduce surface HER2 and the M6P-trastuzumab demonstrated further reduction of cell surface HER2 in comparison with the unmodified tratuzumab. This result was consistent with previous report that anti-HER2 antibodies such as trastuzumab could efficiently internalize surface HER2 into cells, leading to up to a 50% reduction after treatment with trastuzumab for 2 h.42, 43 However, the significant reduced total HER2 by M6P-modified trastuzumab (3) over the unmodified trastuzumab, as indicated by the Western blot (Figure 4a and b), suggests that only the M6P-modified trastuzumab led to eventual degradation of the internalized HER2. In the case of M6P-modified cetuximab, a significant reduction of cell surface EGFR by both unmodified and M6P-modified cetuximab was also observed (Figure S8). However, this result was unexpected, as a previous report has shown that cetuximab alone triggers very little endocytosis of cell surface EGFR.44, 45 To address this apparent discrepancy, we performed a competitive binding analysis of cetuximab with the detecting PE antihuman EGFR antibody (BioLegend, Cat.# 352903). We found that cetuximab showed apparent competitive binding with the detecting antibody (Figure S9), resulting in significant underestimated quantitation of cell surface EGFR. Thus, the result shown in Figure S8c and d did not reflect the actual level of cell surface EGFR after cetuximab treatment. An anti-EGFR detecting antibody that would not compete with cetuximab should be used for the flow cytometry. Nevertheless, both the M6P-modified trastuzumab (3) and M6P-modified cetuximab (9) showed much better effects than the unmodified antibodies on reducing the total HER2 and EGFR, respectively, as probed by Western blot analysis (Figure 4). The data suggest that the M6P-modified antibodies could efficiently drive the antigens to lysosomes for degradation. These results confirm the feasibility of the structurally well-defined M6P glycan remodeled antibodies, prepared by the present single-step glycan remodeling method, for degradation of membrane-associated proteins. Future work will be directed to identifying more potent M6P glycan ligands for the chemoenzymatic glycan remodeling of antibodies and to testing an expanded panel of antibodies for examining the LYTAC approach.
CONCLUSION
A highly efficient chemoenzymatic method for site-specific bioconjugation of high-affinity M6P glycan ligands to antibodies is established. The method provides structurally well-defined homogeneous M6P glycan–antibody conjugates that show high affinity for the CI-MPR. The preliminary cell-based assays indicate that the M6P–trastuzumab and M6P–cetuximab conjugates can selectively degrade the membrane-associated HER2 and EGFR, respectively. This study provides the first example of endoglycosidase-catalyzed transfer of synthetic phosphorylated N-glycans to antibodies. This modular glycan-remodeling strategy enables the construction of homogeneous antibody–M6P–glycan conjugates in a single-step and site-specific manner without the need of protein engineering and conventional click reactions that would introduce “unnatural” moieties. It is expected that this method could be equally applicable to other antibodies to generate M6P–antibody conjugates for targeted degradation of extracellular and membrane proteins of interest.
METHODS
Enzymatic Glycosylation of the Deglycosylated Antibodies with Phosphorylated Glycan Oxazolines As the Donor Substrates.
General.
LC-ESI-MS analysis was performed on an Ultimate 3000 HPLC system coupled to an Exactive Plus Orbitrap mass spectrometer (Thermo Fischer Scientific) with a C4 (whole antibody, gradient, 5–95% aq MeCN containing 0.1% FA for 6 min, 0.4 mL/min) or C8 (IdeS digestion, gradient, 25–35% aq MeCN containing 0.1% FA for 6 min, 0.4 mL/min) column. Deconvolution data were transformed with MagTran software.
Synthesis of Trastuzumab M6P–Glycoform 3.
A solution of the deglycosylated trastuzumab (2; 2.1 mg) and oxazoline 1 (220 μg, 10 eq per reaction site) was incubated with Endo S2-D184 M (2.1 μg) at 25 °C in 210 μL of 150 mM PBS buffer (pH = 7.2), and the reaction was monitored by LC-ESI-MS of the aliquots. Within 2 h, LC-ESI-MS analysis indicated the completion of the transglycosylation; the product was purified using protein A chromatography to give 3 (2.0 mg, 95%). ESI-MS, calculated for whole antibody: M = 147 403 Da. Found (m/z): 147 406 (deconvolution data). Calculated for the Fc monomer after IdeS digestion: M = 24903 Da. Found (m/z): 24 904 (deconvolution data).
Synthesis of Trastuzumab M6P–Glycoform 6.
A solution of the deglycosylated trastuzumab (2; 300 μg) and oxazoline 4 (70 μg, 20 equiv per reaction site) was incubated with Endo S2-D184 M (15 μg) at 25 °C in 15 μL of 150 mM PBS buffer (pH = 7.2), and the reaction was monitored with LC-ESI-MS of the aliquots. After 1 h, another portion of oxazoline 4 (35 μg, 10 eq per reaction site) was added, and the reaction was carried out for another 30 min when LC-ESI-MS indicated the completion of transglycosylation. The product was purified on a protein A column to give 6 (285 μg, 95%). ESI-MS calculated for whole antibody: M = 147563 Da. Found (m/z): 147 564 (deconvolution data). Calculated for the Fc monomer after IdeS digestion: M = 24983 Da. Found (m/z): 24 984 (deconvolution data).
Synthesis of Trastuzumab M6P–Glycoform 7.
A solution of the deglycosylated trastuzumab (2; 300 μg) and oxazoline 5 (110 μg, 20 equiv per reaction site) was incubated with Endo S2-D184 M (15 μg) at 25 °C in 15 μL of 150 mM PBS buffer (pH = 7.2), and the reaction was monitored by LC-ESI-MS of the aliquots. After 1 h, another portion of oxazoline 5 (55 μg, 10 eq per reaction site) was added, and the reaction was run for another 30 min when LC-ESI-MS indicated the completion of the transglycosylation reaction. The product was purified using protein A chromatography to give 7 (270 μg, 90%). ESI-MS calculated for whole antibody: M = 148535 Da. Found (m/z): 148 537 (deconvolution data). Calculated for the Fc monomer after IdeS digestion: M = 25 470 Da. Found (m/z): 25 470 (deconvolution data).
Synthesis of Cetuximab M6P–Glycoform 9.
A solution of the deglycosylated cutuximab (8; 2.1 mg) and the M6P tetrasaccharide oxazoline (1; 220 μg, 20 equiv per reaction site) was incubated with Endo S2-D184 M (3.0 μg) at 25 °C in 210 μL of 150 mM PBS buffer (pH = 7.2), and the reaction was monitored by LC-ESI-MS of the aliquots. The reaction was complete after 2 h, and the product was purified using protein A chromatography to give 9 (1.9 mg, 90%). ESI-MS calculated for the Fc monomer after IdeS digestion: M = 24903 Da. Found (m/z): 24 904 (deconvolution data).
Synthesis of Cetuximab M6P–Glycoform 10.
A solution of the deglycosylated cutuximab (8; 300 μg) and the glycan oxazoline (5; 110 μg, 20 equiv per reaction site) was incubated with Endo S2-D184 M (15 μg) at 25 °C in 15 μL of 150 mM PBS buffer (pH = 7.2), and the reaction was monitored using LC-ESI-MS of the aliquots. After 1 h, another portion of oxazoline 5 (55 μg, 10 eq per reaction site) was added, and the mixture was incubated for another 30 min when LC-ESI-MS indicated the completion of the reaction. The product was purified using protein A chromatography to give 10 (270 μg, 90%). ESI-MS calculated for the Fc monomer after IdeS digestion: M = 25470 Da. Found (m/z): 25 470 (deconvolution data).
Surface Plasmon Resonance (SPR) Measurements.
SPR experiments were performed on a Biacore T200 instrument (GE Healthcare). Recombinant human IGF-II R (CI-MPR) was purchased from R&D Systems. Approximately 6000 resonance units (RU) of CI-MPR were immobilized on a CM5 sensor chip in a sodium acetate buffer (25 μg/mL, pH 4.0) at 25 °C, using the amine coupling kit provided by the manufacturer. M6P-modified antibodies were determined at 25 °C under a flow rate of 10 μL/min. HBS-P+ buffer (10 mM HEPES, 150 mM NaCl, 0.05% surfactant P20, pH 7.4) was used as the sample buffer and running buffer. Association was measured for 3 min and dissociation for 10 min at the same flow rate (10 μL/min). The surface regeneration was performed using 2 M MgCl2 at a flow rate of 10 μL/min for 60 s. Antibody analytes were flowed over an immobilized chip with 2-fold serial dilution of the highest concentration of 500 nM. Kinetic analyses were performed by global fitting of the binding data to a 1:1 Langmuir binding model using BIAcore T200 evaluation software.
Western Blot.
BT474 (ATCC HTB-20) or HepG2 (ATCC HB-8065) cells were treated with native or M6P-modified antibodies at a final concentration of 10 nM; whole-cell lysate in Laemmli sample buffer was subjected to SDS-PAGE and Western blotting. The primary antibodies used in this study were against EGFR (Cell Signaling Technology), HER2 (Cell Signaling Technology), and beta-actin (Life Technology). Horseradish peroxidase-conjugated anti-rabbit IgG was used in this study as the secondary antibody. The specific reactions were detected with chemiluminescence substrate, and the signal was recorded digitally using the ChemiDoc MP Imaging System (Bio-Rad). Relative band intensity was calculated using ImageJ software (NIH). All statistical analysis was performed with one-way ANOVA multiple comparison tests. A P value of >0.05 was considered “not significant”.
Flow Cytometry.
HER2 or EGFR expression on the target cell surface was examined by flow cytometry. BT474 or HepG2 cells were trypsinized, centrifuged for 5 min at 2000 rpm, then washed with PBS. Cells were stained with PE conjugated antihuman CD340 (erbB2/HER2) antibody (BioLegend, Cat.# 324405), PE conjugated antihuman EGFR (BioLegend, Cat.# 352903), and the PE conjugated isotype control antibody in PBS at 4 °C for 30 min. After staining, cells were washed with PBS, then fixed with 2.5% formalin. Flow cytometry was performed using a FACSCanto II cell sorter (BD); data were analyzed using FlowJo software (BD).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH grants R01AI155716 and R01GM096973 to L.X.W.)
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00751.
Figures S1–S9; procedures for the preparation of deglycosylated antibodies; PNGase F-catalyzed release and enrichment of N-glycans from the Fab and Fc domains; additional Western blot and flow cytometry data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.1c00751
The authors declare the following competing financial interest(s): L.X.W. is the founder and a major shareholder of GlycoT Therapeutics LLC. The other authors declare no conflict of interest.
Contributor Information
Xiao Zhang, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Huiying Liu, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Jia He, Division of Virology, Pathogenesis, and Cancer, Institute of Human Virology, Departments of Pharmacology, Microbiology, and Immunology, University of Maryland School of Medicine, Baltimore, Maryland 21201, United States.
Chong Ou, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Thomas C. Donahue, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States
Musleh M. Muthana, Division of Virology, Pathogenesis, and Cancer, Institute of Human Virology, Departments of Pharmacology, Microbiology, and Immunology, University of Maryland School of Medicine, Baltimore, Maryland 21201, United States
Lishan Su, Division of Virology, Pathogenesis, and Cancer, Institute of Human Virology, Departments of Pharmacology, Microbiology, and Immunology, University of Maryland School of Medicine, Baltimore, Maryland 21201, United States.
Lai-Xi Wang, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
REFERENCES
- (1).Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol 2015, 22 (6), 755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (15), 8554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348 (6241), 1376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov 2019, 18 (12), 949–963. [DOI] [PubMed] [Google Scholar]
- (5).Zou Y; Ma D; Wang Y The PROTAC technology in drug development. Cell Biochem. Funct 2019, 37 (1), 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cotton AD; Nguyen DP; Gramespacher JA; Seiple IB; Wells JA Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc 2021, 143 (2), 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Montrose K; Krissansen GW Design of a PROTAC that antagonizes and destroys the cancer-forming X-protein of the hepatitis B virus. Biochem. Biophys. Res. Commun 2014, 453 (4), 735–740. [DOI] [PubMed] [Google Scholar]
- (8).Kolb R; De U; Khan S; Luo Y; Kim M-C; Yu H; Wu C; Mo J; Zhang X; Zhang P; Zhang X; Borcherding N; Koppel D; Fu Y-X; Zheng SG; Avram D; Zheng G; Zhou D; Zhang W Proteolysis-targeting chimera against BCL-XL destroys tumor-infiltrating regulatory T cells. Nat. Commun 2021, 12 (1), 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).McCoull W; Cheung T; Anderson E; Barton P; Burgess J; Byth K; Cao Q; Castaldi MP; Chen H; Chiarparin E; et al. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol 2018, 13 (11), 3131–3141. [DOI] [PubMed] [Google Scholar]
- (10).Tomoshige S; Ishikawa M PROTACs and Other Chemical Protein Degradation Technologies for the Treatment of Neuro-degenerative Disorders. Angew. Chem. Int. Ed 2021, 60 (7), 3346–3354. [DOI] [PubMed] [Google Scholar]
- (11).Bondeson DP; Mares A; Smith IE; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 2015, 11 (8), 611–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cromm PM; Crews CM Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem. Biol 2017, 24 (9), 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lamb CA; Dooley HC; Tooze SA Endocytosis and autophagy: Shared machinery for degradation. Bioessays 2013, 35 (1), 34–45. [DOI] [PubMed] [Google Scholar]
- (14).Banik SM; Pedram K; Wisnovsky S; Ahn G; Riley NM; Bertozzi CR Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584 (7820), 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Walsh SJ; Bargh JD; Dannheim FM; Hanby AR; Seki H; Counsell AJ; Ou X; Fowler E; Ashman N; Takada Y; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev 2021, 50 (2), 1305–1353. [DOI] [PubMed] [Google Scholar]
- (16).Caianiello DF; Zhang M; Ray JD; Howell RA; Swartzel JC; Branham EMJ; Chirkin E; Sabbasani VR; Gong AZ; McDonald DM; et al. Bifunctional small molecules that mediate the degradation of extracellular proteins. Nat. Chem. Biol 2021, 17 (9), 947–953. [DOI] [PubMed] [Google Scholar]
- (17).Zhou Y; Teng P; Montgomery NT; Li X; Tang W Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent. Sci 2021, 7 (3), 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ahn G; Banik SM; Miller CL; Riley NM; Cochran JR; Bertozzi CR LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol 2021, 17, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ahn G; Banik SM; Bertozzi CR Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem. Biol 2021, 28 (7), 1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sadiki A; Vaidya SR; Abdollahi M; Bhardwaj G; Dolan ME; Turna H; Arora V; Sanjeev A; Robinson TD; Koid A; et al. Site-specific conjugation of native antibody. Antib. Ther 2020, 3 (4), 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang LX; Tong X; Li C; Giddens JP; Li T Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem 2019, 88, 433–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chari RV; Miller ML; Widdison WC Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chem. Int. Ed 2014, 53 (15), 3796–827. [DOI] [PubMed] [Google Scholar]
- (23).Beck A; Goetsch L; Dumontet C; Corvaïa N Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov 2017, 16 (5), 315–337. [DOI] [PubMed] [Google Scholar]
- (24).do Pazo C; Nawaz K; Webster RM The oncology market for antibody-drug conjugates. Nat. Rev. Drug Discov 2021, 20 (8), 583–584. [DOI] [PubMed] [Google Scholar]
- (25).Priyanka P; Parsons TB; Miller A; Platt FM; Fairbanks AJ Chemoenzymatic Synthesis of a Phosphorylated Glycoprotein. Angew. Chem., Int. Ed 2016, 55 (16), 5058–61. [DOI] [PubMed] [Google Scholar]
- (26).Yamaguchi T; Amin MN; Toonstra C; Wang LX Chemoenzymatic Synthesis and Receptor Binding of Mannose-6-Phosphate (M6P)-Containing Glycoprotein Ligands Reveal Unusual Structural Requirements for M6P Receptor Recognition. J. Am. Chem. Soc 2016, 138 (38), 12472–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang X; Liu H; Meena N; Li C; Zong G; Raben N; Puertollano R; Wang LX Chemoenzymatic glycan-selective remodeling of a therapeutic lysosomal enzyme with high-affinity M6P-glycan ligands. Enzyme substrate specificity is the name of the game. Chem. Sci 2021, 12 (37), 12451–12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huang W; Giddens J; Fan SQ; Toonstra C; Wang LX Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J. Am. Chem. Soc 2012, 134 (29), 12308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Li T; Tong X; Yang Q; Giddens JP; Wang LX Glycosynthase Mutants of Endoglycosidase S2 Show Potent Transglycosylation Activity and Remarkably Relaxed Substrate Specificity for Antibody Glycosylation Remodeling. J. Biol. Chem 2016, 291 (32), 16508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Giddens JP; Lomino JV; Amin MN; Wang L-X Endo-F3 Glycosynthase Mutants Enable Chemoenzymatic Synthesis of Core-fucosylated Triantennary Complex Type Glycopeptides and Glycoproteins. J. Biol. Chem 2016, 291 (17), 9356–9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li T; DiLillo DJ; Bournazos S; Giddens JP; Ravetch JV; Wang LX Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (13), 3485–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tong X; Li T; Orwenyo J; Toonstra C; Wang LX One-pot enzymatic glycan remodeling of a therapeutic monoclonal antibody by endoglycosidase S (Endo-S) from Streptococcus pyogenes. Bioorg. Med. Chem 2018, 26, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Giddens JP; Lomino JV; DiLillo DJ; Ravetch JV; Wang LX Site-selective chemoenzymatic glycoengineering of Fab and Fc glycans of a therapeutic antibody. Proc. Natl. Acad. Sci. U.S.A 2018, 115 (47), 12023–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ou C; Li C; Zhang R; Yang Q; Zong G; Dai Y; Francis RL; Bournazos S; Ravetch JV; Wang LX One-Pot Conversion of Free Sialoglycans to Functionalized Glycan Oxazolines and Efficient Synthesis of Homogeneous Antibody-Drug Conjugates through Site-Specific Chemoenzymatic Glycan Remodeling. Bioconjug. Chem 2021, 32 (8), 1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zhang X; Ou C; Liu H; Prabhu SK; Li C; Yang Q; Wang LX General and Robust Chemoenzymatic Method for Glycan-Mediated Site-Specific Labeling and Conjugation of Antibodies: Facile Synthesis of Homogeneous Antibody-Drug Conjugates. ACS Chem. Biol 2021, 16 (11), 2502–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Collin M; Olsen A EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J 2001, 20 (12), 3046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sjogren J; Struwe WB; Cosgrave EF; Rudd PM; Stervander M; Allhorn M; Hollands A; Nizet V; Collin M EndoS2 is a unique and conserved enzyme of serotype M49 group A Streptococcus that hydrolyses N-linked glycans on IgG and alpha1-acid glycoprotein. Biochem. J 2013, 455 (1), 107–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Noguchi M; Tanaka T; Gyakushi H; Kobayashi A; Shoda S -i. Efficient Synthesis of Sugar Oxazolines from Unprotected N-Acetyl-2-amino Sugars by Using Chloroformamidinium Reagent in Water. J. Org. Chem 2009, 74 (5), 2210–2212. [DOI] [PubMed] [Google Scholar]
- (39).Chung CH; Mirakhur B; Chan E; Le Q-T; Berlin J; Morse M; Murphy BA; Satinover SM; Hosen J; Mauro D; et al. Cetuximab-Induced Anaphylaxis and IgE Specific for Galactose-α−1,3-Galactose. N. Engl. J. Med 2008, 358 (11), 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Qian J; Liu T; Yang L; Daus A; Crowley R; Zhou Q Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal. Biochem 2007, 364 (1), 8–18. [DOI] [PubMed] [Google Scholar]
- (41).Chevreux G; Tilly N; Bihoreau N Fast analysis of recombinant monoclonal antibodies using IdeS proteolytic digestion and electrospray mass spectrometry. Anal. Biochem 2011, 415 (2), 212–4. [DOI] [PubMed] [Google Scholar]
- (42).Klapper LN; Waterman H; Sela M; Yarden Y Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res 2000, 60 (13), 3384–8. [PubMed] [Google Scholar]
- (43).Baselga J; Albanell J Mechanism of action of anti-HER2 monoclonal antibodies. Ann. Oncol 2001, 12, S35–S41. [DOI] [PubMed] [Google Scholar]
- (44).Spangler JB; Neil JR; Abramovitch S; Yarden Y; White FM; Lauffenburger DA; Wittrup KD Combination antibody treatment down-regulates epidermal growth factor receptor by inhibiting endosomal recycling. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (30), 13252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jones S; King PJ; Antonescu CN; Sugiyama MG; Bhamra A; Surinova S; Angelopoulos N; Kragh M; Pedersen MW; Hartley JA; Futter CE; Hochhauser D Targeting of EGFR by a combination of antibodies mediates unconventional EGFR trafficking and degradation. Sci. Rep 2020, 10 (1), 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.