

Abstract
Background and aim:
Disorders of sexual differentiation (DSD) with karyotype 46,XY include gonadal developmental differences such as complete gonadal dysgenesis, partial gonadal dysgenesis, testicular regression and ovotesticular sexual differentiation disorder, differences in androgen synthesis or action, such as androgen synthesis deficiency, androgen action deficits, LH receptor deficiency, AMH synthesis or action deficits, and other conditions such as severe hypospadias, cloaca estrophy, etc.
Methods:
A 17 years-old girl came to our attention for hirsutism, clitoral hypertrophy, primary amenorrhea, and bilateral mammary hypoplasia. According to clinical features and anamnesis, the diagnosis of 46, XY DSD was made. For diagnostic purposes, she underwent an extensive genetic analysis, hormone dosage and instrumental examinations. After a clitoridoplasty and hormone replacement treatment, the patient performs appropriate multidisciplinary follow-up and regular psychotherapy.
Results:
The clinical case reported falls, according to the recent classification developed by the Chicago Consensus, within the scope of DSD with karyotype 46, XY. About 160 cases of patients with 17β-HSD3 deficiency, diagnosed at a mean age of 12 years, are described in the literature, most of them coming from Western Asia and Europe and only three cases from Eastern Asia. Clinically, about 30% of patients showed virilization, 20% clitoromegaly, ambiguous genitalia, inguinal/labial mass, 16% primary amenorrhea, and 5% absence of mammary development, features that are partly traced in the case described here.
Conclusions:
This case underscores the complexity of managing individuals with DSD. Having acquired the concept that irreversible surgery should be avoided, except in cases where failure to do so would determine health risks, the primary objective of the medical decision lies in meeting conditions aimed at harmonious sexual identification, especially regarding sexual activity and fertility, involving a team of experienced professionals (psychologists, pediatricians, surgeons, endocrinologists, radiologists), capable of promptly identifying suggestive clinical signs. (www.actabiomedica.it).
Keywords: 46, XY DSD, 17β-HSD3 deficiency, chromosomal sex, phenotypic sex, multidisciplinary approach
Case presentation
We report the case of a 17-year-old girl who came to our attention, presenting clinical manifestations of hyperandrogenism such as significant hirsutism (Ferriman-Gallwey score > 15), labia majora, and clitoral hypertrophy (phallic-shape clitoris, 6.5 cm long) with urethral meatus and vaginal orifice opening very close to each other (Fig. 1). Her past medical history reported that she had never menstruated (primary amenorrhea) and that, one year before, she had undergone breast augmentation surgery for bilateral mammary hypoplasia. After an accurate physical examination, we decided to perform several investigations:
Figure 1.
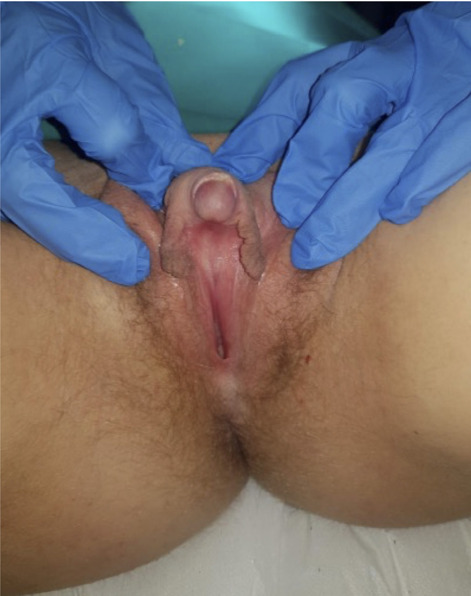
Genital anatomy
abdominal ultrasound (US) showed the absence of adrenal masses, the presence of two ovular formations suggestive of retained testicles, a 3.5 cm-long structure similar to an infantile uterus, and a 4 cm-long vaginal canal;
blood examinations with the evidence of normal hepatic and renal function, normal glucose metabolism, and vitamin D deficiency (blood 25(OH)D = 6 ng/ml);
tumoral markers (CEA, alpha , β-HCG, CA125, CA19.9, CA 15.3) were normal;
hormonal dosage revealed a low level of testosterone (total testosterone: 1.8 ng/ml, free testosterone: 47.8 pg/ml, dihydrotestosterone 729 pg/ml) and high level of D4-androstenedione (D4-A: 10 ng/ml), estradiol: 30.5 pg/ml, normal level of 17-OH-progesterone before and after ACTH stimulation test.
Subsequently, the lower abdomen magnetic resonance imaging (MRI) showed the presence of two undescended testicles, two hypoplastic corpora cavernosa, prostatic gland, and seminal vesicles in communication with the urethra wall (Fig. 2).
Figure 2.
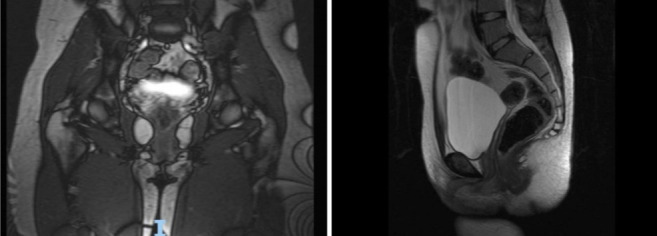
Abdominal MRI with the evident absence of structures morphologically referable to female internal genitalia.
According to the genetic evaluation, we performed the karyotype analysis that showed normal male karyotype (46 XY), Array-CGH that revealed only non-pathogenetic polymorphisms, and molecular investigation, using Next-Generation-Sequencing (NGS), with the evidence of compound heterozygous at the gene HSD17B3, associated with 17β-HSD3 deficiency. The molecular findings and the low testosterone/D4-androstenedione ratio, together with the clinical presentation, were sufficient for the diagnosis of 17β-HSD3 deficiency, without the need of HCG stimulation test.
According to multidisciplinary team composed of a Pediatric Endocrinologist, a Pediatric Surgeon with expertise in DSD, and a Mental Health Professional, we decided to maintain female sex, also considering patient gender perception after a preliminary psychotherapy.
Afterward, at the age of 18 years and 2-month, the patient underwent bilateral gonadectomy. Histological examination of the gonads revealed characteristics compatible with testicles. Therefore, we started a hormone replacement regime with estrogen and progesterone. We also recommended the need to keep on attending the psychological therapy. After three months, we performed Mollard clitoroplasty and prescribed vaginal enlargement utilizing progressively larger dilators (Fig. 3).
Figure 3.
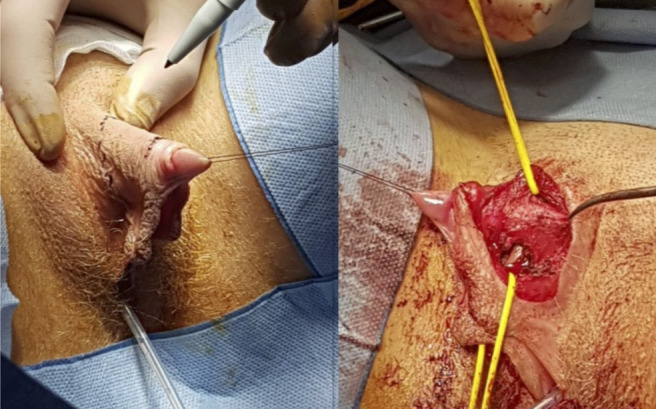
Mollard’s clitoroplasty.
Nowadays, she is in follow-up at our Unit of Pediatric Endocrinology, taken in charge by our multidisciplinary team dedicated to transitional care.
The parents provided written informed consent to publish the case with personal images.
Discussion
We presented an example of 46 XY DSD condition management. DSD is a various group of diseases that result in discordance between chromosomal sex, gonadic sex, and anatomic sex (1); among these conditions, there are 46 XY DSD, defined as DSD with 46 XY chromosome complement (2,3). Clinical presentation varies among the spectrum of 46 XY DSD: it can be characterized by ambiguous or female appearance of external genitalia with variable degrees of development of the Wolffian and Müllerian structures (4-7). Testes are present in many patients regardless their degree of undervirilization (6-8). The virilization stages influnce diagnostic timing: partially virilized patients with anomalies in external genitalia are often noted at birth (9-11), while patients with complete lack of virilization and female-typical external genitalia come to the attention of the clinicians at puberty, due to primary amenorrhea and breast hypoplasia (9). The underlying etiologies of the conditions labeled as 46XY DSD are various: anomalies during gonadal development (Complete Gonadal Dysgenesis/ Swyer Syndrome, Partial Gonadal Dysgenesis, Testicular Regression Syndrome and Ovotesticular DSD), defects in androgen production ( deficit of 17beta-HSD3, deficit of 5alfa-reductase, StAR mutations), impaired androgen action (Complete /Partial Androgen Insensitivity Syndrome), a deficit of LH receptor, a deficit in production or action of the Anti Müllerian Hormone (AMH) or other conditions such as Cloacal Exstrophy or severe Hypospadias (12). The diagnostic flow-chart always starts with accurate documentation of the family history, taking into consideration consanguinity, history of previous neonatal deaths, history of siblings and other family members diagnosed with DSD, presenting DSD features or infertility, followed by a careful general physical examination with particular attention in evaluation of the external genitalia and secondary sexual characters (11). The karyotype is often the first test performed to classify DSD. Array comparative genomic hybridization (aCGH) and single-nucleotide polymorphism (SNP) array are useful too, especially in patients with malformations or syndromic characteristics, to detect pathogenic copy number variations (CNVs). Introduction of Next Generation Sequencing (NGS) has given a fundamental contribution in diagnosing 46XY DSD, giving the possibility to analyze panels of DSD candidate genes and allowing better prognostic predictions, genetic counseling, and individualized management (12). Although no definitive diagnosis is based only on endocrine tests, they often provide a suggestive diagnosis (11); these tests include serum FSH, LH, and testosterone and measure the rise in serum testosterone delta-4-androstenedione and dihydrotestosterone after HCG stimulation. For example, flat response in testosterone after stimulation suggests the presence of testosterone biosynthetic defect or Leydig-cell hypoplasia, reduced testosterone/delta-4-androstenedione ratio suggest a deficit of 17 beta HSD type 3, altered testosterone/dihydrotestosterone ratio indicates the possibility of 5-alfa-reductase deficiency and, in the end, an elevated basal and stimulated testosterone levels are suggestive for Partial Androgen Insensitivity syndrome. Imaging studies are the other cornerstone for the DSD diagnostic process for prenatal and postnatal diagnosis. The prenatal US for fetal sex phenotype identification and sex genotype identification, utilizing noninvasive prenatal testing (NIPT), shows genotype/phenotype discordance in some cases. As far as postnatal diagnosis is concerned, the goal for imaging includes visualization of uterus, vagina/urogenital sinus, and localization of gonads, if present. Imaging may also be necessary to visualize kidneys, adrenals, and the terminal spine.
Ultrasound and magnetic MRI are considered equally sensitive to evaluate pelvic structures, but US depends more on the evaluator’s expertise and the device used for the analysis. Gonads are studied better with MRI; however, streak gonads are challenging to detect with MRI, and laparoscopy is the gold standard. Alternatively, genitography, cystoscopy/genitoscopy can be used to visualize the internal anatomy; nevertheless, the latter two techniques are usually performed during surgical procedures, owing to their invasive nature.
Once the diagnosis is established, other pivotal step in 46XY DSD management is the assignment of sex. This decision should consider that gender identity often disagrees with genetic, gonadal, or anatomic sex and that sexual attraction is challenging to predict according to sex phenotype. Since older children recognize and reveal their gender identity, the greater complexity lies in the attribution of sex of rearing for newborns (12). Firstly, it is crucial to gather a multidisciplinary team, including a mental-health specialist, to avoid distress about the implications of their child’s diagnosis, often reported by parents. Thus, a straightforward and clear explanation about their child’s future, development, fertility, sexual function, and need for medical/surgical intervention must be given to the parents before deciding the sex of rearing for their child. An important issue, written by senior endocrinologists based on more than 300 years of experience in treating DSD, offers indications on the decision of sex of rearing among various cultures and countries (12). For instance, existing data support female sex of rearing for newborns with CAIS or complete gonadal dysgenesis, while data report high male reassignment for female-reared patients affected by 17b HSD type3 deficiency or by 5a-RD2 deficiency. Determining factors influencing psychosexual development have been founded during the last years, such as early androgen exposure, especially during the prenatal period and sex of rearing that is the best predictor for gender development. Before deciding on the sex of rearing, it is also fundamental to consider condition-specific outcomes related to psychosexual development, anatomy, fertility potential, and the need for medical and/or surgical treatment. Finally, parents’ cultural and religious beliefs must be respected when deciding gender assignment. At this point, another critical component for 46 XY DSD management is hormone replacement. The goals for this treatment are the induction and maintenance of secondary sex characteristics, growth, bone mineralization, and ameliorating psychosocial and psychosexual development and general well-being. Induction and maintenance of puberty are essential despite the gender identity, and the pediatric endocrinologist must consider specific indications depending on the underlying etiologies.
The pediatric surgeon should be included in the multidisciplinary team to provide information on the surgical possibilities and the subsequent consequences from childhood to adulthood. In this context, surgery aims to change the appearance of the external genitalia to suit the sex of rearing, correct the anatomy to avoid urinary and genital complications and promote fertility and sexual function. The right time for surgery has been a topic of discussion since the first guidelines in the 1950s. If the American Academy of Pediatrics and the original Consensus recommend early surgery, on the other hand, human rights organizations, ethicists, and patient advocates oppose such suggestion. It is reported that early surgery is preferred by some surgeons and parents because younger children are thought to heal better from the intervention and to prevent the stigma associated with living with atypical anatomy and because it relieves parent’s and child’s distress. However, there is no systematic evidence supporting this belief (1). Conversely, some retrospective studies describe poor cosmetic and functional outcomes and the irreversibility of these procedures for patients too young to give their informed consent. At present, this decision remains debatable, and clinicians and parents are left to decide on a case by case.
Conflict of Interest:
Each author declares that they do not have commercial associations that might pose a conflict of interest in connection with the submitted article.
References
- Hughes IA, Houk C, Ahmed SF, et al. ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91:554–63. doi: 10.1136/adc.2006.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdoğan S, Kara C, Uçaktürk A, et al. Etiological classification and clinical assessment of children and adolescents with disorders of sex development. J Clin Res Pediatr Endocrinol. 2011;3(2):77–83. doi: 10.4274/jcrpe.v3i2.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutney K, Konczal L, Kaminski B, et al. Challenges in the diagnosis and management of disorders of sex development. Birth Defects Res C Embryo Today. 2016;108(4):293–308. doi: 10.1002/bdrc.21147. [DOI] [PubMed] [Google Scholar]
- Looijenga LH, Hersmus R, Oosterhuis JW, et al. Tumor risk in disorders of sex development (DSD) Best Pract Res Clin Endocrinol Metab. 2007;21(3):480–95. doi: 10.1016/j.beem.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Imperato-McGinley J, Zhu YS. Androgens and male physiology the syndrome of 5a-reductase-2 deficiency. Mol Cell Endocrinol. 2002;198(1–2):51–9. doi: 10.1016/s0303-7207(02)00368-4. [DOI] [PubMed] [Google Scholar]
- Mendonca BB, Domenice S, Arnhold IJ, et al. 46, XY disorders of sex development (DSD) Clin Endocrinol (Oxf) 2009;70(2):173–87. doi: 10.1111/j.1365-2265.2008.03392.x. [DOI] [PubMed] [Google Scholar]
- Domenice S, Arnhold IJP, Costa EMF, et al. 46, XY Disorders of Sexual Development. 2017 May 3. Endotext [Internet] South Dartmouth (MA): MDText.com, Inc; 2000. [Google Scholar]
- Wilson JD. Sexual differentiation of the gonads and the reproductive tract. Biol Neonate. 1989;55(6):322–30. doi: 10.1159/000242936. [DOI] [PubMed] [Google Scholar]
- Domenice S, Machado AZ, Ferreira FM, et al. Wide spectrum of NR5A1-related phenotypes in 46, XY and 46, XX individuals. Birth Defects Res C Embryo Today. 2016;108(4):309–20. doi: 10.1002/bdrc.21145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markosyan R, Ahmed SF. Sex assignment in conditions affecting sex development. J Clin Res Pediatr Endocrinol. 2017;9(Suppl 2):106–12. doi: 10.4274/jcrpe.2017.S009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashamboo A, Brauner R, Bignon-Topalovic J, et al. Mutations in the FOG2/ZFPM2 gene are associated with anomalies of human testis determination. Hum Mol Genet. 2014;23(14):3657–65. doi: 10.1093/hmg/ddu074. [DOI] [PubMed] [Google Scholar]
- Raza J, Zaidi SZ, Warne GL. Management of disorders of sex development - With a focus on development of the child and adolescent through the pubertal years. Best Pract Res Clin Endocrinol Metab. 2019;33(3):101297. doi: 10.1016/j.beem.2019.101297. [DOI] [PubMed] [Google Scholar]