

Abstract
Purpose:
The intestinal microbiome is critical for human health and preventing colonization by enteric pathogens. There are notable differences in the microbiota composition among individuals with and without enteric infections, though the impact that age and gender has on the composition and abundance of intestinal microbes is not known.
Methods:
A comparative 16S rRNA gene sequencing study was performed on stool DNA from 200 patients with enteric infections and 75 healthy family members representing both sexes and multiple age groups.
Results:
Microbial community profiles were affected by age and sex in patients with enteric infections and their healthy family members. Overall, we observed an increase in Bacteroides abundance and decrease in Escherichia abundance with age, though these differences were most apparent for patients with enteric infections. Genus Bacteroides was also higher in female communities while Escherichia predominated in males.
Conclusions:
Because Escherichia abundance was previously linked to symptom severity, children with enteric infections may be most susceptible to severe disease outcomes due to high and low abundance of Escherichia and Bacteroides, respectively. Future studies should focus on classifying specific differences in the microbiome using metagenomics and identifying novel methods aimed at shifting the intestinal microbiome to a healthy state.
Keywords: Diarrhea, Microbiota, 16S rRNA
Introduction
The composition of the intestinal microbiota is imperative for health as it plays a critical role in metabolism, digestion, and nutrient absorption as well as immune system modulation and prevention of pathogen colonization [1,2]. Age-related changes in gut physiology and alterations in the composition and function of the microbiota have been described previously [1,3]. Such changes were suggested to be partly due to dietary and physiological factors that can alter the metabolic capacity of the gut and lead to an increased susceptibility to gastrointestinal infections [4]. One study, for example, demonstrated that children between 16 months and 7 years had a significantly greater proportion of Enterobacteria when compared with adults between 21 and 34 years [4]. Moreover, the lowest abundance of Bifidobacteria, which has been shown to be protective and associated with intestinal health, was observed in the elderly population. Other studies have also reported sex-dependent effects in the intestinal microbiota composition as a result of gastrointestinal physiology [5] and diet, which affected males differently than females in vivo using mouse and stickleback models [6]. Similarly, another study demonstrated that commensal microbial communities altered sex hormone levels and could subsequently regulate immune cell responses [7]. These prior studies, which are summarized in Table 1, demonstrate that both age and sex are important factors to consider when conducting studies aimed at characterizing the microbiome.
Table 1.
Review of studies that have identified variation in microbiota profiles by sex or age group
| Date | Study population | Method | Effect of sex | Effect of age | Reference |
|---|---|---|---|---|---|
| 2000 | Children 16 mos–7 years (n = 10), adults 21–34 years (n = 7), elderly ≥ 67 years (n = 5) | 16S rRNA abundance | Not assessed | Greater abundance of Enterobacteria in children compared with adults | Hopkins et al. [4] |
| 2006 | Healthy adults between 20 and 50 years (n = 85), healthy adults, and elderly >60 years (n = 145) | Fluoresence in situ hybridization of 16S rRNA genes | Greater abundance of Bacteroides and Prevotella in males | Greater abundance of Enterobacteria in the elderly | Mueller et al. [8] |
| 2007 | Infants (n = 150), adults 25–35 years (n = 54), elderly 80–82 years (n = 45) | qPCR and fluorescence in situ hybridization of 16S rRNA genes specific for A. muciniphila | Not assessed | Decreased abundance of A. muciniphila, a type of mucin-degrading bacteria in the intestinal tract, in the elderly | Collado et al. [9] |
| 2009 | Adults 18–31 years (n = 17), institutionalized elderly 78–94 years (n = 17) | Denaturing gradient gel electrophoresis and qPCR | Not assessed | Greater abundance of Bacteroides and decreased abundance of Bifidobacteria in the elderly | Zwielehner et al. [10] |
| 2013 | Male (n = 123) and female (n = 132) nonobese diabetic germ-free and pathogen-free mice | 16S rRNA gene amplicon sequencing | Intestinal communities were distinct among male and female mice | Sex-specific differences in the intestinal community were most apparent in adult mice | Markle et al. [7] |
In our prior study of 200 patients with enteric infections caused by the four most common bacterial pathogens (Salmonella, Shiga toxin-producing Escherichia coli, Campylobacter, and Shigella) in Michigan and 75 healthy family members, we demonstrated that the intestinal microbial communities were variable across groups [11]. Specifically, patients with enteric infections had less diverse communities when compared with healthy family members as well as a significantly greater abundance of Escherichia that decreased after recovery. It is important to note that this increased abundance of Escherichia was observed among patients irrespective of the bacterial pathogen that caused each infection, suggesting that intestinal communities are altered in a similar way during the course of an enteric infection. After recovery from infection, patient Escherichia levels decreased in abundance and community diversity increased. As part of this prior study, however, we did not examine how the intestinal communities varied across age groups or other demographic characteristics including sex.
Examining intestinal microbial communities in a diverse population as in individuals with and without enteric infections, will enable the identification of microbial factors that are associated with disease susceptibility or more severe clinical symptoms. To better understand the impact of these demographics, we characterized the intestinal microbial communities of patients with enteric infections of varying age and sex by 16s rRNA sequencing, and made comparisons to intestinal communities from healthy family members. Analyzing the impact of epidemiologic factors on microbial communities of individuals with enteric infections may provide insight into disease susceptibilities in certain populations.
Material and methods
Sample collection and DNA isolation
Stool samples from patients with acute enteric infections caused by Salmonella, Shiga toxin-producing E. coli, Campylobacter, and Shigella (n = 200) and their otherwise healthy family members (n = 75) were collected via the Enteric Research Investigative Network at Michigan State University as described [11]. Family members represented individuals living in the same household who were related to the patients (e.g., parents, siblings, grandparents). Stools were homogenized, centrifuged, and stored in triplicate at 80°C immediately after receipt from the Michigan Department of Health and Human Services (MDHHS). Community DNA was isolated using the QIAamp kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol with a modification to the denaturation step (95°C) and inclusion of bead beating to enhance DNA isolation of gram-positive bacteria [11].
Pyrosequencing and microbial community analyses
PCR amplification of the 16S rRNA gene (V5eV3 regions) was performed using universal barcoded primers (357F and 926R) and the AccuPrime Taq DNA polymerase (Invitrogen, Waltham, MA, USA) in triplicate. The PCR protocol included an initial denaturation step (95°C for 2 mins) followed by 30 cycles of 95°C for 20 seconds, 50°C for 30 seconds, and 72°C for 5 minutes as described in our prior study [11]. Individual amplicon libraries were purified using Agencourt AMPure XP (Beckman Coulter, Inc., Indianapolis, IN, USA) beads, quantified using the Quant-iT PicoGreen dsDNA quantitation kit (Invitrogen), and normalized for pooling. Amplicon libraries were sequenced using the 454 GS Junior Titanium (Roche, Indianapolis, IN, USA).
Sequencing analysis and epidemiologic associations
As described previously [11], sequencing reads were filtered and processed using the Quantitative Insights Into Microbial Ecology (QIIME) program [12] followed by trimming and filtering by noise reduction using default parameters. Quality sequences were aligned followed by chimeric and singleton removal using USEARCH and classified using the Greengenes 16S rRNA gene reference database [12,13]. Taxonomical data was also analyzed using Paleontological Statistics Software Package For Education and Data Analysis (PAST) with the multivariate tests, Analysis of Similarities (ADONIS) and Permutational Multivariate Analysis Of Variance (PERMANOVA); neighbor-joining trees were constructed in FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Nonmetric Multidimensional Scaling (NMDS) plots were generated based on microbial abundance and composition using the BrayeCurtis and Jaccard similarity coefficients with 9999 permutations for a weighted and unweighted analysis, respectively [12].
The genera responsible for dissimilarities between groups were also identified by Similarity Percentages (SIMPER) based on the BrayeCurtis dissimilarity matrix [14], and linear discriminant analysis (LDA) effect size (LEfSe) was used to identify differentially abundant microbial features [15]. Data were analyzed in the following age groups: 0–10 years (n 91), 11–e18 years (n 32), 19–49 years (n = 84), 50–69 years (n = 45), and more than 70=years (n = 12; Table 2). With regard to sex, 141 intestinal communities originated from males and 126 originated from females. Age was missing from four healthy (uninfected) individuals, whereas sex was not available for eight individuals (three patients, five uninfected). Demographic characteristics were also examined after stratifying by the different microbial community clusters using the likelihood χ2 test with odds ratios and 95% confidence intervals.
Table 2.
Characteristics of the study population
| Characteristics | Patients with diarrhea (n = 200) | Otherwise healthy family members (n = 75) |
|---|---|---|
| No. (%) of individuals | No. (%) of individuals | |
| Age group | ||
| 0–10 years (n = 91) | 65 (32.5) | 26 (34.7) |
| 11–18 years (n = 32) | 21 (10.5) | 4 (5.3) |
| 19–49 years (n = 84) | 66 (33.0) | 32 (42.7) |
| 50–69 years (n = 45) | 37 (18.5) | 8 (10.7) |
| ≥ 70 years (n = 12) | 10 (5.0) | 2 (2.7) |
| Gender | ||
| Male (n = 141) | 104 (52.0) | 37 (49.3) |
| Female (n = 126) | 93 (46.5) | 33 (44.0) |
| Pathogen causing the infection* | ||
| Campylobacter (n = 89) | 71 (35.5) | 18 (24.0) |
| Salmonella (n = 98) | 66 (33.0) | 32 (42.7) |
| Shiga toxin-producing E. coli (n = 50) | 34 (17.0) | 16 (21.3) |
| Shigella (n = 38) | 29 (14.5) | 9 (12.0) |
Percentages represent the frequency of each characteristic out of the total number of individual per group. Denominators do not always add up to the total number of individuals in each column because of missing data.
Numbers in the otherwise healthy column refer to the pathogen that infected their family members.
Results
Association between the microbiota and demographics
Analysis of 275 intestinal communities from patients with enteric infections and healthy, uninfected individuals revealed significant differences in the abundance and composition of taxa (PERMANOVA P < .0001) when stratified by sex and specific age groups. Overall, sex had a significant impact on the abundance of taxa based on the BrayeCurtis dissimilarity index (PERMANOVA P = .008). The average dissimilarity for the intestinal communities originating from males and females was 63.0 as calculated by SIMPER. Genera Bacteroides and Escherichia contributed most to the dissimilarity between sexes. On average, females had a slightly higher abundance of Bacteroides (37.9%) than males (32.5%), though the males had higher Escherichia levels (18.8%) compared with females (12.3%); neither proportion was significantly different.
When stratified by age groups, no differences were identified in community composition or abundance based on the Jaccard similarity coefficient (P = .14) or Bray–Curtis dissimilarity index (P = .18), and clustering was not observed in the NMDS analysis. A single factorial analysis among different age groups, however, identified significant differences in taxa abundance among individuals between 11 and 18 years relative to 50–69 year olds by Analysis of Similarity (ANOSIM) (P = .04) Similarly, abundance profiles were significantly different by PERMANOVA (P = .04) between children aged 0–10 years and adults between 19 and 49 years. The average dissimilarity for all five age groups was 65.9. The same taxa, Bacteroides and Escherichia, identified to be most dissimilar between sexes, contributed most to the differences by age group. The mean abundance of Bacteroides was 33.0% and 31.2% in children aged 0–10 and 11–18 years, respectively, but was slightly higher in adults aged 19–49 (36.3%) and older than or equal to 50 years (38.6%); the difference in proportions of Bacteroides was not significant between any of these age groups. Comparatively, the proportion of Escherichia was slightly higher in children younger than or equal to 18 years (19.5%) relative to adults between 19 and 49 years (12.0%), 50–69 years (16.8%), and adults older than or equal to 70 years (6.0%), although the differences were not statistically significant. Similar trends were observed for Prevotella, which also contributed to the dissimilarity in the groups, with a mean abundance of 3.6% (0–10 years), 1.7% (11–18 years), 4.3% (19–49 years), 1.9% (50–69 years), and 0.03% (≥70 years).
Effect of sex on the intestinal microbiota from infected and uninfected individuals
To determine whether the microbial communities from individuals of varying age and sex were impacted by intestinal health, the communities from uninfected and infected individuals were analyzed separately. Among the 70 healthy individuals of known sex, no difference was observed in abundance or composition. The average dissimilarity was 50.6 using SIMPER with genera Bacteroides (22.3%), Prevotella (11.3%), other Rikenellaceae (5.5%), Parabacteroides (5.0%), Faecalibacterium, and other Ruminococcaceae (4.4%) predominantly contributing to this difference. LEfSe identified genus Paraprevotella to be differentially abundant in healthy intestinal communities from males (LDA > 3), whereas Dialister, Lactobacillus, unclassified Lactobacillaceae, unclassified Comamonadaceae, and Comamonas were differentially abundant in females (LDA > 3). In addition, hierarchical clustering based on the BrayeCurtis dissimilarity matrix demonstrated that the healthy intestinal communities clustered differently when analyzed separately without infected communities as was done previously [11]. Five clusters were defined among the uninfected communities, with the previously defined Cluster IV separating in to four groups (Clusters IVaeIVd; Fig. 1A). No significant differences were observed between clusters by sex.
Fig. 1.
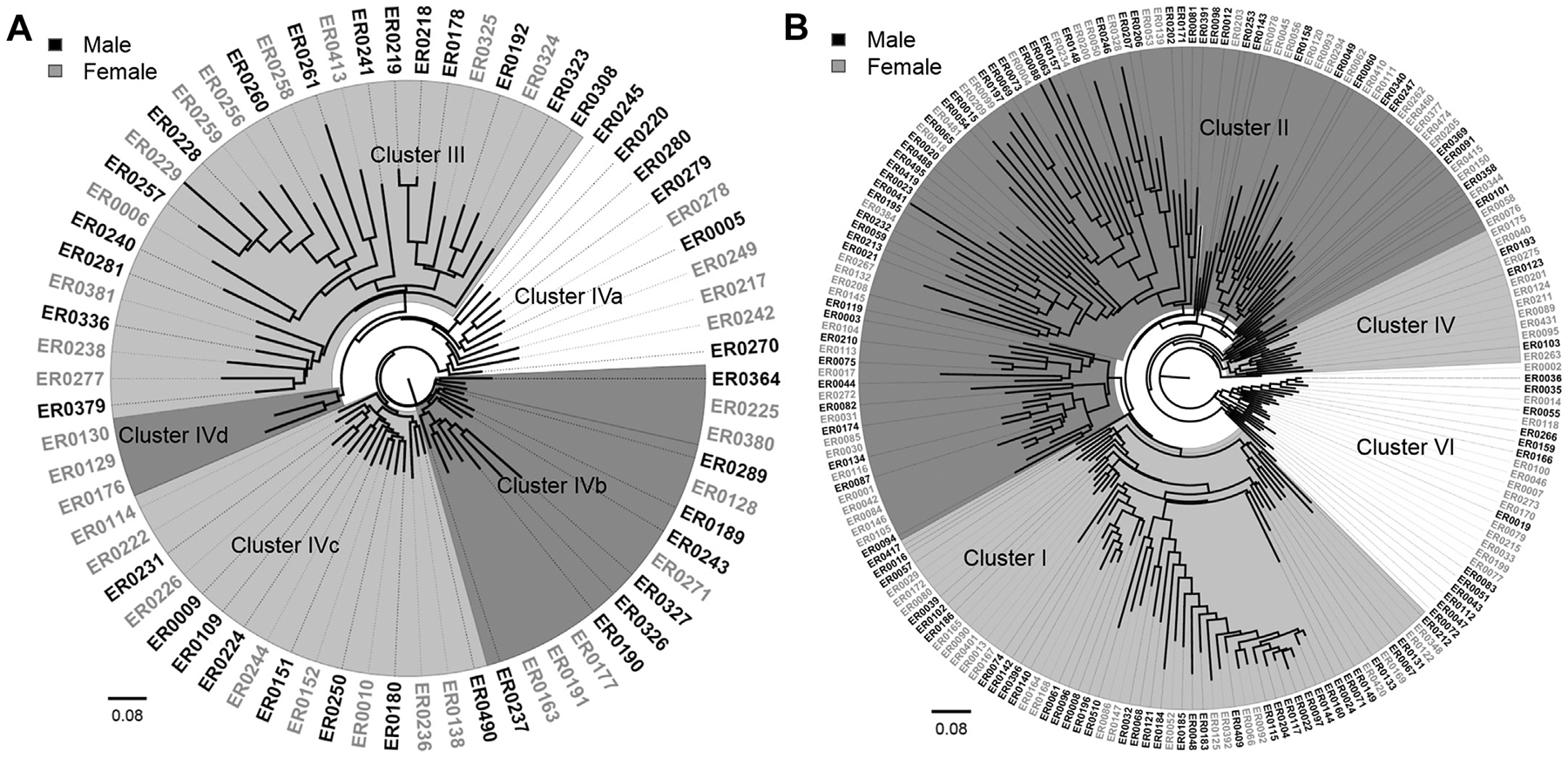
Phylogeny of intestinal microbiota profiles by sex. Hierarchical clustering identified three distinct clusters in (A) healthy family members of patients with enteric infections, and five clusters in (B) the patients. The Neighbor-Joining tree was constructed based on the BrayeCurtis dissimilarity index with 1000 bootstrap replications. Communities from males are labeled in black outside the phylogeny, whereas female communities are labeled in gray.
When the 197 infected male and female communities were examined, stratifying by sex significantly affected the composition (P = .04) and abundance (P = .002) by PERMANOVA; only the abundance was affected (P = .02) by ANOSIM. The average dissimilarity between the 104 males and 93 females was 63.0 by SIMPER. In males, Bacteroides (30.9%) was most abundant followed by Escherichia (25.1%), unclassified Enterobacteriaceae (7.0%), and Faecalibacterium (2.8%). The same genera predominated in female communities and the abundance of Bacteroides (38.0%), unclassified Enterobacteriaceae (7.3%), and Faecalibacterium (3.5%) was similar when compared with males. The Escherichia (16.1%) abundance, however, was lower in infected females, although the difference in proportions was not statistically significant (P = .12). In addition, LEfSe detected 54 differentially abundant microbial features (LDA > 2.4) in the male and female patient communities, which is more than 10 times was detected in the healthy communities. Enterobacteriaceae predominated among the 11 differentially abundant features detected in males, whereas Bacteroidaceae predominated among the 43 differentially abundant features in females.
Unlike the hierarchical clustering, results for the healthy, uninfected communities, those communities from infected males and females more closely matched one of the three previously defined Clusters [11]. The few communities that previously classified as Cluster III were an exception as they were scattered throughout the neighbor-joining phylogeny after excluding the healthy communities (Fig. 1B). Infected males were significantly more likely to have intestinal communities representing Cluster I (odds ratio: 1.9; 95% confidence interval: 1.01–3.56) relative to females. On the other hand, infected females were more likely to have communities belonging to Cluster IV (Fisher’s exact P = .04), though the sample size was low. An equal number of male and female communities comprised Clusters II and IV.
Effect of age on the intestinal microbiota of infected and uninfected individuals
Among the 72 communities from uninfected individuals with age data available, composition differences were identified between certain age groups, though some age groups had very low sample sizes. Intestinal communities of children between 0 and 10 years (n = 26), for example, were significantly different from 50 to 69eyear olds (n = 8) by PERMANOVA (P = .03), whereas children between 11 and 18 years (n = 4) were significantly different from 50 to 69eyear olds (n = 8) by PERMANOVA (P = .03). Among all uninfected communities, only genus Lachnospira was differentially abundant by LEfSe in individuals between 50 and 69 years (LDA = 4). Hierarchical clustering did not uncover any age-specific clusters in the neighbor-joining phylogeny (data not shown).
Within the 200 communities from patients, different age groups were also associated with specific community profiles. Pairwise comparison of the intestinal community composition of infected children between 0 and 10 years (n = 65) detected significant differences relative to patients between 19 and 49 years (n = 66) by ANOSIM (P = .02) and PERMANOVA (P = .01). Community composition of 19–49 year olds also differed significantly from individuals older than 70 years (n = 10) by PERMANOVA (P = .04). The abundance profiles varied across age groups as well, with communities from patients between 11 and 18 years (n = 21) differing signifi cantly from communities of 50e69 year olds (n = 37) by ANOSIM (P = .01). Communities from infected children 0–10 years of age also differed relative to 70–90 year olds (n = 10) by PERMANOVA (P = .01). Additional comparisons using SIMPER resulted in an overall average dissimilarity of 62.6. Bacteroides, Escherichia, unclassified Enterobacteriaceae, Faecalibacterium, and Prevotella predominated and were responsible for the differences between the communities, although Escherichia and Bacteroides contributed most to the dissimilarity. Bacteroides increased slightly by age group and was found in 31.7% (0–10 years), 28.0% (11–18 years), 35.9% (19–49 years), 38.7% (50–69 years), and 38.4% (≥70 years) of infected communities (Fig. 2). In contrast, Escherichia abundance gradually decreased with age and was found in 27.6% (0–10 years), 20.8% (11–18 years), 17.1% (19–49 years), 20.0% (50–69 years), and 7.3% (70–90 years) of the infected communities. Nonetheless, no significant differences in the proportion of either Bacteroides or Escherichia were detected across groups, and similar to the uninfected communities, the intestinal communities from patients did not cluster together by age group in the neighbor-joining phylogeny.
Fig. 2.
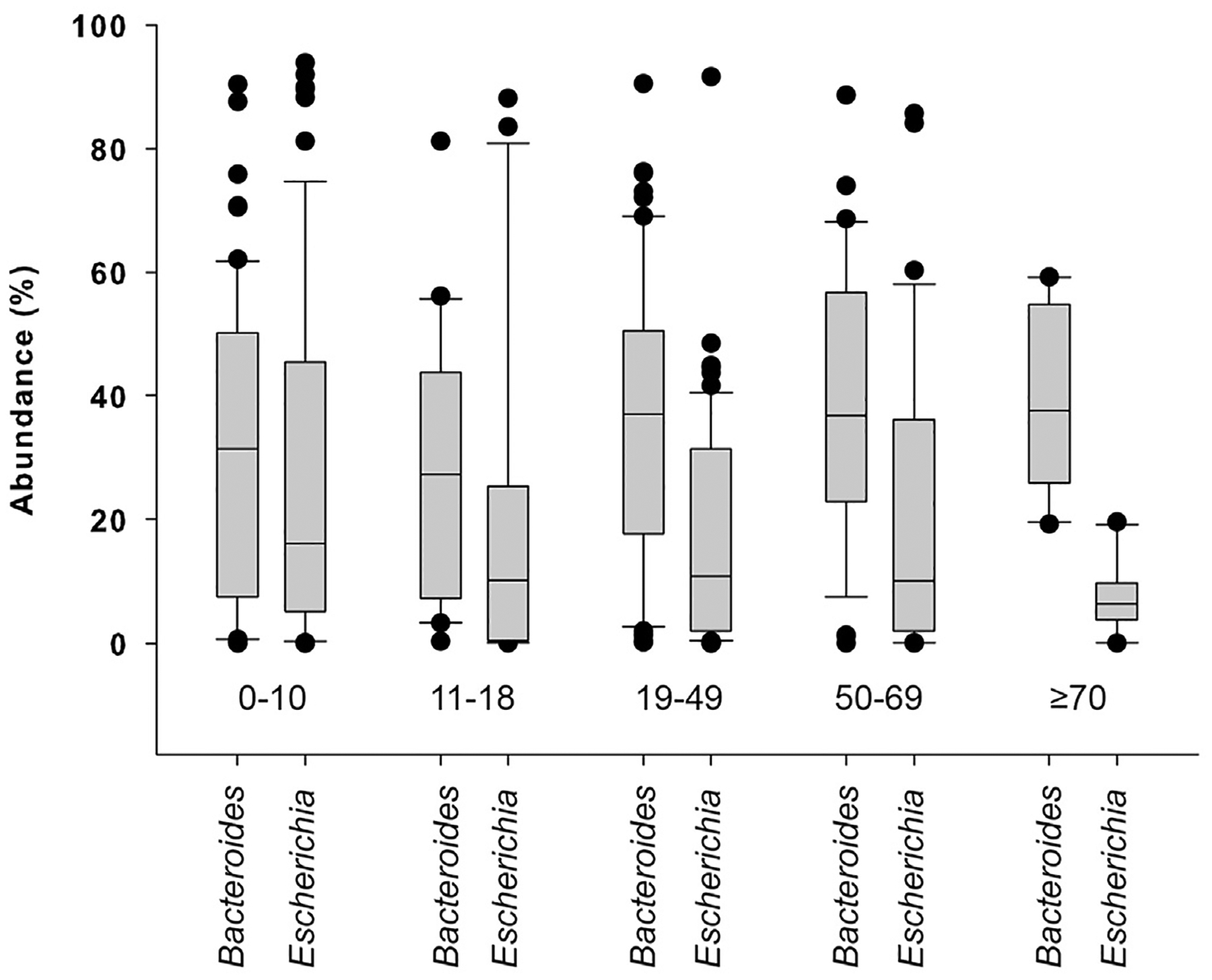
Abundance of Escherichia and Bacteroides in the intestinal communities of patients with enteric infections. The box plot represents the relative abundances of genera Escherichia and Bacteroides in infected patient communities across different age groups.
Discussion
In this analysis, we have demonstrated that intestinal microbial communities vary across individuals and that intestinal health is the most important factor contributing to community composition differences by sex and age group. This result is consistent with prior studies that have detected variation in community composition across individuals [6,16] as well as our prior study, which observed greater diversity and richness in healthy versus infected intestinal communities [11].
Among the healthy individuals examined herein, Lactobacillus and Comamonas were differentially abundant in the female relative to male communities, which may be linked to diet and nutrition. Comamonas has been identified as a member of the microbiome comprising murine intestinal crypts, which are important for epithelial regeneration and health [17], whereas members of Lactobacillus have been linked to probiotic use. Such foods are likely to be differentially consumed by males and females. Because no difference in the abundance of either Lactobacillus or Comamonas was observed by sex in patients with enteric infections, the benefits are unclear. Indeed, the probiotic effects of lactobacilli were suggested to be strain specific and not broadly applicable to one genus [18]; hence, additional longitudinal studies using metagenomics are required to assess whether certain bacterial species are linked to disease susceptibility and may be differentially present in males and females. Similar findings were observed among healthy individuals representing different age groups, although only Lachnospira was differentially abundant in individuals between 50 and 69 years of age. The biological implications of these findings are not understood, though the small number of healthy communities examined in this study may have prevented the identification of meaningful differences across healthy individuals of varying ages.
In contrast to the healthy, uninfected communities, more differences were identified in patients after stratifying by both sex and age. Interestingly, Escherichia, which we previously reported to be higher in patients than healthy individuals [11], was more abundant in the male communities than female communities. Communities from males were also overrepresented in Cluster I of the neighbor-joining phylogeny; Cluster I communities were associated with more severe symptoms in our prior analysis [11]. It is therefore possible that males are more susceptible to increases in Escherichia levels, which is correlated with decreases in Bacteroides and negatively impacts intestinal health. Although these data are consistent with prior studies demonstrating that sex-dependent differences are linked to hormones [7], physiology [5], and diet [6], additional studies are required to test the effect of sex on the abundance of specific microbial populations during an enteric infection and throughout the recovery period.
Similar findings were observed after stratifying by age group as children younger than 18 years had the highest abundance of Escherichia and lowest abundance of Bacteroides. Indeed, as age increased, the abundance of Escherichia decreased and the abundance of Bacteroides increased. These data suggest that children with enteric infections, particularly those younger than or equal to 10 years, may be more susceptible to increases in the Escherichia population and may partly explain why children are more prone to severe clinical outcomes. Prior studies have suggested that dietary factors combined with age-associated changes in gut physiology as well as the composition and function of the microbiota can impact susceptibility to enteric infections [3,4]. In one study, healthy children between 16 months and 7 years had a greater proportion of Enterobacteria when compared with young adults, whereas the lowest abundance of Bifidobacteria was detected in elderly subjects [4]. If children have more abundant populations of Enterobacteria (e.g., Escherichia) before an infection, then it is possible that increases in the resident Escherichia population will be more pronounced in infected children relative to infected adults with lower baseline levels. Given that community members belonging to Bacteroidetes (e.g., Bacteroides), which were less abundant in infected children, cannot use nitrate, then it is possible that over-growth of Escherichia in certain individuals is linked to nitrate respiration [19]. It is interesting to note that the abundance profiles observed in the elderly patients were opposite the profiles observed in children and suggests that other factors besides the microbiota composition may work in concert to enhance susceptibility to disease in the elderly.
Collectively, these data highlight the significant perturbations that occur in the intestinal microbial communities of patients with enteric infections, and further demonstrate that both age and sex can impact the abundance of specific taxa. Overgrowth of potentially harmful bacterial populations such as Escherichia and decreases in beneficial populations like Bacteroides may be important for both disease susceptibility and time to recovery. Although we previously found that decreases in Escherichia and increases in Bacteroides occurred in patients postrecovery [11], additional studies are needed to better understand the factors that contribute to major shifts in these populations during infections, particularly among specific individuals at the risk of more severe disease.
Acknowledgments
This work was supported by the NIH Enterics Research Investigational Network Cooperative Research Center at Michigan State University (U19AI090872) and in part by the USDA NIFA (2011-67005-30004). We would like to thank Dr. Sandip Shah, Dr. James Rudrik, Ben Hutton, Jason Wholehan, and Sheri Robeson at the Michigan Department of Health and Human Services for specimen processing as well as the participating clinical laboratories. We also thank Rebekah Mosci, Katherine Jernigan, and Angela Zell for assistance with sample preparation, sequencing, and data management.
References
- [1].Cummings JH, Macfarlane GT. Role of intestinal bacteria in nutrient metabolism. JPEN J Parenter Enteral Nutr 1997;21(6):357e65. [DOI] [PubMed] [Google Scholar]
- [2].Sonnenburg JL, Angenent LT, Gordon JI. Getting a grip on things: how do communities of bacterial symbionts become established in our intestine? Nat Immunol 2004;5(6):569e73. [DOI] [PubMed] [Google Scholar]
- [3].Lovat LB. Age related changes in gut physiology and nutritional status. Gut 1996;38(3):306e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hopkins MJ, Sharp R, Macfarlane GT. Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundance, and community cellular fatty acid profiles. Gut 2001;48(2):198e205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Freire AC, Basit AW, Choudhary R, Piong CW, Merchant HA. Does sex matter? The influence of gender on gastrointestinal physiology and drug delivery. Int J Pharm 2011;415(1–2):15e28. [DOI] [PubMed] [Google Scholar]
- [6].Bolnick DI, Snowberg LK, Hirsch PE, Lauber CL, Org E, Parks B, et al. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat Commun 2014;5:4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013;339(6123):1084e8. [DOI] [PubMed] [Google Scholar]
- [8].Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T, et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol 2006;72(2):1027e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl Environ Microbiol 2007;73(23):7767e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zwielehner J, Liszt K, Handschur M, Lassl C, Lapin A, Haslberger AG. Combined PCR-DGGE fingerprinting and quantitative-PCR indicates shifts in fecal population sizes and diversity of Bacteroides, Bifidobacteria and Clostridium cluster IV in institutionalized elderly. Exp Gerontol 2009;44(6–7):440e6. [DOI] [PubMed] [Google Scholar]
- [11].Singh P, Teal TK, Marsh TL, Tiedje JM, Mosci R, Jernigan K, et al. Intestinal microbial communities associated with acute enteric infections and disease recovery. Microbiome 2015;3(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7(5):335e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26(19):2460e1. [DOI] [PubMed] [Google Scholar]
- [14].Hammer O, Harper DAT, Ryan PD. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol Electron 2001;4(1):9. [Google Scholar]
- [15].Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12(6):R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Backhed F, Fraser CM, Ringel Y, Sanders ME, Sartor RB, Sherman PM, et al. Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 2012;12(5):611e22. [DOI] [PubMed] [Google Scholar]
- [17].Pedron T, Mulet C, Dauga C, Frangeul L, Chervaux C, Grompone G, et al. A crypt-specific core microbiota resides in the mouse colon. mBio 2012;3(3). e00116–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Herbel SR, Vahjen W, Wieler LH, Guenther S. Timely approaches to identify probiotic species of the genus Lactobacillus. Gut Pathog 2013;5(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 2013;339(6120):708e11. [DOI] [PMC free article] [PubMed] [Google Scholar]