

Abstract
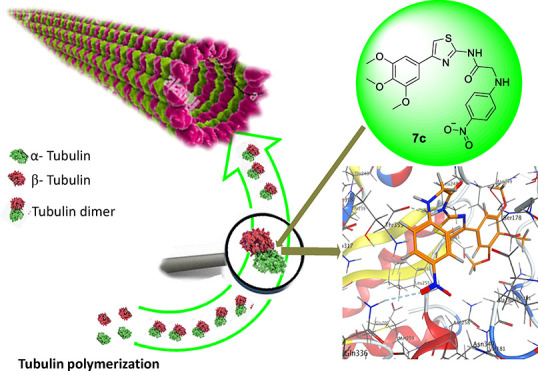
A new series of 2,4-disubstituted thiazole derivatives containing 4-(3,4,5-trimethoxyphenyl) moiety was synthesized and evaluated for their potential anticancer activity as tubulin polymerization inhibitors. All designed compounds were screened for cytotoxic activity against four human cancer cell lines, namely, HepG2, MCF-7, HCT116, and HeLa, using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay, with combretastatin A-4 as a reference drug. Compounds 5c, 6d, 7c, 8, and 9a,b showed superior activity against the tested cell lines, with IC50 values ranging from 3.35 ± 0.2 to 18.69 ± 0.9 μM. Further investigation for the most active cytotoxic agents as tubulin polymerization inhibitors was also performed in order to explore the mechanism of their antiproliferative activity. The obtained results suggested that compounds 5c, 7c, and 9a remarkably inhibit tubulin polymerization, with IC50 values of 2.95 ± 0.18, 2.00 ± 0.12, and 2.38 ± 0.14 μM, respectively, which exceeded that of the reference drug combretastatin A-4 (IC50 2.96 ± 0.18 μM). Molecular docking studies were also conducted to investigate the possible binding interactions between the targeted compounds and the tubulin active site. The interpretation of the results showed clearly that compounds 7c and 9a were identified as the most potent tubulin polymerization inhibitors with promising cytotoxic activity and excellent binding mode in the docking study.
1. Introduction
Cancer is a group of diseases characterized by uncoordinated and uncontrolled cell growth with the potential to spread into or invade nearby tissues in a process called metastasis, which is the major cause of death from cancer.1,2 Factors that are associated with the high risk of cancer are tobacco use, lack of physical activity, alcohol use, low vegetable and fruit intake, and obesity. These factors are thought to account for approximately one third of cancer mortalities.3 Cancer pathogenesis involves several gene mutations which eventually lead to abnormal cell proliferation.4 The increased cancer incidence worldwide led to an impressive progress in the discovery of new, safer, and curative chemotherapeutic agents.5 In the scope of designing novel antitumor agents, we are particularly interested in the present work with thiazoles as one of the bioactive heterocyclic compounds that exhibited countless biological activities such as antimicrobial,6,7 antiprotozoal,8,9 antitumor,10,11 antiviral,12,13 antioxidant,14 antidiabetic,15,16 anti-inflammatory,17,18 anticonvulsant,19 antipsychotic,20 and antihypertensive activities.21 Thiazole derivatives containing one or more thiazole rings represent a class of heterocyclic compounds of considerable importance as antitumor agents due to their remarkable affinity toward different biological targets involved in cancer pathogenesis such as tiazofurin22 and bleomycin.23 Additionally, considerable attention and extensive efforts have been made to enhance the antitumor activity of the 2-aminothiazole core in anticancer therapeutic areas as exemplified by dasatinib (A),24 thia-netropsin (B),25 and more recently alpelisib (C)26 that was approved for medical use in 2019. Moreover, some 2-amino-4-phenylthiazol derivatives have been reported to inhibit tubulin polymerization, disrupt microtubule formation, and suppress cell division.27,28 However, combretastatin A-4 (CA-4) (D) is a natural trimethoxyphenyl (TMP) containing stilbenoid29 that targets tubulin at the colchicine-binding site. CA-4 and other TMP-containing analogues such as combretoxazolone analogue (E) were reported as tubulin polymerization inhibitors.30,31 Inspired by the abovementioned facts, our objective was directed to gather two bioactive entities (3,4,5-trimethoxyphenyl and 2-aminothiazole) into one compact structure to be the main target skeleton for the development of new 2,4-disubstituted thiazole derivatives to suppress tubulin polymerization as an extremely attractive target for clinically effective anticancer drugs. Moreover, structure diversity was accomplished by introducing variable bioactive side arms at the 2-position of the selected backbone, particularly amide derivatives, as in the synthesized compounds 4, 5a–c, 6a–e, 7a–e, 8, and 9a,b, and urea derivatives as our target compounds 11, 12, and 13a,b. In addition, thiazolyl-2,4-diamine was also introduced at position 2 as in the new bis-thiazole compound 10. The cytotoxic activity of the tested compounds and their potential inhibitory effect on tubulin polymerization were evaluated. In addition, molecular docking methodology was conducted to identify the possible binding interactions between the target compounds and the tubulin active site. The structures of the reported lead compounds (A–E) and the newly designed targeted compounds are shown in Figure 1.
Figure 1.

Structures of the reported lead compounds (A–E) and the newly designed targeted compounds.
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis of Compounds 1–5 (Scheme 1)
Scheme 1. Synthesis of the Designed 2-Chloroacetamide Derivative 4 and 2-Substituted Amino Acetamides 5a–c.

Chloroacetylation of 2-aminothiazole 3 with chloroacetyl chloride was conducted in 1,4-dioxane in the presence of triethylamine to afford the chloroacetamide compound 4 as a key intermediate. It is noteworthy that the structure of compound 4 was verified by infrared (IR) spectroscopy that showed the appearance of a characteristic absorption band for the C=O group at 1690 cm–1, which was also confirmed by 13C NMR by a signal at δ 165.6 ppm.
The key intermediate 4 is considered as an α-halo amide that possesses key structural features, including an easily substituted chlorine atom, an active methylene group, and an amide moiety, which make it a versatile synthon in organic chemistry. Replacement of the α-halogen atom by different nitrogen nucleophiles is among the available methods for the synthesis of α-amino amides and creation of a new C–N bond. Hence, substitution reaction of compound 4 with primary aliphatic amines such as 2-aminoethanol, n-butylamine, or isopropylamine was proceeded successfully in dry dimethyl formamide (DMF) in the presence of anhydrous K2CO3 to afford compounds 5a–c.
2.1.2. Synthesis of Compounds 6–10 (Scheme 2)
Scheme 2. Synthesis of the Designed 2-Substituted Acetamides 6–9 and N4-Phenylthiazole-2,4-Diamine Derivative 10.

The target compounds 6a–e were obtained by the reaction of the chloroacetamide compound 4 with a variety of secondary heterocyclic amines in dry DMF, and 1H NMR spectra proved the inclusion of the secondary amines into their structures. For example, compounds 6a–d showed either two sets of multiplets, one multiplet, two sets of triplets, or two sets of multiplets integrated for eight protons of the piperazine ring, respectively.
Moreover, the key intermediate 4 was reacted with different aniline derivatives in ethanol in the presence of HCl to afford the corresponding anilinoacetamide derivatives 7a–e. The successful incorporation of the (un)substituted aniline moiety into the structure of compounds 7a–e was verified based on the spectral data. For example, the 1H NMR spectra of the later compounds showed the appearance of additional aromatic protons in the expected regions of the spectra. Moreover, their structures were further confirmed by mass spectroscopy that showed molecular ion peaks in accordance with the molecular formulae. In addition, compound 4 was treated with hydrazine hydrate in ethanol to furnish the corresponding hydrazinoacetamide 8, which was subsequently condensed with isatin derivatives in ethanol and in the presence of catalytic drops of acetic acid. The carbonyl group at position-3 of isatin is more reactive than the carbonyl group at position-2; hence, it is readily involved in the condensation reaction with hydrazine hydrate, affording isatinmonohydrazone derivatives 9a,b. The IR spectrum of compound 8 showed absorption bands at 3109 and 3188 cm–1, representing NH2 and NH of the hydrazine moiety, respectively. However, the protons of the hydrazine moiety resonated as two singlets at δ 3.40 and δ 4.20 ppm, representing NH2 and NH, respectively, which are D2O exchangeable. The successful condensation of isatin derivatives with compound 8 was proved on the basis of the spectral data of isatin hydrazone products 9a,b. Cyclization of the chloroacetamide compound 4 with thiourea was achieved by refluxing acetone in the presence of anhydrous K2CO3, yielding the corresponding N4-phenylthiazole-2,4-diamine derivative 10. The 1H NMR spectrum of the later compound confirmed the presence of thiazole diamine functionality, and the structure was further verified by the presence of a molecular ion peak at m/z 364 in mass spectroscopy.
2.1.3. Synthesis of Compounds 11–13 (Scheme 3)
Scheme 3. Synthesis of the Designed Urea Derivatives 11–13.

A successful synthesis of 1-(2-chloroethyl)-3-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)urea 11 was achieved through the addition of the aminothiazole derivative 3 to 2-chloroethylisocyanate in dry DMF in the presence of anhydrous K2CO3, and its structure was verified on the basis of the spectral data. The 2-chloroethyl urea derivative 11 was reacted with aniline, N-ethylpiperazine, or morpholine to afford the target compounds 12 and 13a,b, respectively.
2.2. Biological Screening
2.2.1. In Vitro Antitumor Evaluation
The in vitro cytotoxicity study of the newly synthesized compounds was performed on four different cell lines, namely, HepG2, HCT-116, MCF-7, and HeLa cell lines, by employing MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay32,33 using CA-4 as a reference drug. The tested compounds exhibited different degrees of cytotoxic activity against the tested cell lines, and the results are shown in Table 1.
Table 1. In Vitro Cytotoxic Activity of the Designed Compounds.
| IC50 (μM)a |
||||
|---|---|---|---|---|
| comp. no | HePG2b | HCT-116c | MCF-7d | HeLae |
| CA-4f | 4.50 ± 0.2 | 5.23 ± 0.3 | 4.17 ± 0.2 | 5.57 ± 0.3 |
| 4 | 62.57 ± 3.1 | 69.03 ± 3.5 | 62.91 ± 3.1 | 76.92 ± 3.8 |
| 5a | 22.03 ± 1.1 | 29.36 ± 1.5 | 30.00 ± 1.5 | 50.86 ± 2.5 |
| 5b | 10.92 ± 0.5 | 24.38 ± 1.2 | 22.80 ± 1.1 | 28.30 ± 1.4 |
| 5c | 11.87 ± 0.6 | 12.88 ± 0.6 | 15.49 ± 0.8 | 16.02 ± 0.8 |
| 6a | 8.17 ± 0.4 | 28.94 ± 1.4 | 40.87 ± 2.0 | 47.74 ± 2.4 |
| 6b | 74.61 ± 3.7 | 83.68 ± 4.2 | 65.27 ± 3.3 | 87.32 ± 4.4 |
| 6c | 33.86 ± 1.7 | 37.18 ± 1.9 | 41.36 ± 2.1 | 48.19 ± 2.4 |
| 6d | 4.03 ± 0.2 | 15.74 ± 0.8 | 9.64 ± 0.5 | 18.69 ± 0.9 |
| 6e | 19.76 ± 1.0 | 18.03 ± 0.9 | 39.08 ± 2.0 | 58.84 ± 2.9 |
| 7a | 94.13 ± 4.7 | 52.48 ± 2.6 | >100 | 54.09 ± 2.7 |
| 7b | 34.17 ± 1.7 | 9.35 ± 0.5 | 44.84 ± 2.2 | 13.12 ± 0.7 |
| 7c | 6.26 ± 0.3 | 7.30 ± 0.4 | 10.01 ± 0.5 | 9.85 ± 0.5 |
| 7d | 28.10 ± 1.4 | 33.10 ± 1.7 | 41.42 ± 2.1 | 29.78 ± 1.5 |
| 7e | 25.65 ± 1.3 | 31.15 ± 1.6 | 34.98 ± 1.7 | 41.45 ± 2.1 |
| 8 | 3.35 ± 0.2 | 5.32 ± 0.3 | 8.66 ± 0.4 | 6.15 ± 0.3 |
| 9a | 7.32 ± 0.4 | 10.13 ± 0.5 | 9.80 ± 0.5 | 10.59 ± 0.5 |
| 9b | 8.04 ± 0.4 | 9.93 ± 0.5 | 11.55 ± 0.6 | 13.42 ± 0.7 |
| 10 | 49.62 ± 2.5 | 55.30 ± 2.8 | 60.62 ± 3.0 | 48.03 ± 2.4 |
| 11 | 92.97 ± 4.6 | 86.69 ± 4.3 | 95.77 ± 4.8 | 89.86 ± 4.5 |
| 12 | >100 | 88.46 ± 4.4 | >100 | 91.82 ± 4.6 |
| 13a | 14.17 ± 0.7 | 20.08 ± 1.0 | 16.95 ± 0.8 | 32.39 ± 1.6 |
| 13b | 25.67 ± 1.3 | 27.42 ± 1.4 | 32.03 ± 1.6 | 43.93 ± 2.2 |
IC50 (μM): expressed as mean ± SD 1–10 (very strong); 11–20 (strong); 21–50 (moderate); 51–100 (weak); and above 100 (noncytotoxic).
Human hepatocellular carcinoma cell line (HepG2).
Human breast adenocarcinoma cell line (MCF-7).
Human colorectal carcinoma cell line (HCT-116).
Epitheloid cervix carcinoma cell line (HeLa).
CA-4: combretastatin A-4.
2.2.1.1. Cytotoxic Activity of the Amide-Based Targeted Compounds (4–9)
According to the results of biological screening, the key intermediate 2-chloroacetamide derivative 4 showed a weak cytotoxic effect (Table 1, Scheme 1). However, compounds 5a–c generally showed strong-to-moderate activity against the four tested cell lines. Among these compounds, compound 5a, with a hydroxyethyl amino acetamido side chain, showed moderate cytotoxic activity against the tested cell lines, while compound 5b with a butylamino substituent showed a strong lethal effect on the HepG2 cell line with IC50 value of 10.92 ± 0.5 μM and moderate effect on the other tested cell lines. Substitution with an isopropylamino acetamido side chain as in compound 5c exploited a strong cytotoxic effect against HepG2, HCT-116, MCF-7, and HeLa cell lines with IC50 values of 11.87 ± 0.6, 12.88 ± 0.6, 15.49 ± 0.8, and 16.02 ± 0.8 μM, respectively (Table 1, Scheme 1). Compound 6a with an unsubstituted piperazine moiety showed selective cytotoxicity against the HepG2 cell line with IC50 value of 8.17 ± 0.4 μM and moderate activity against the rest of the tested cell lines. It was noticed that substituting the piperazine moiety with a methyl or ethyl group as in compounds 6b and 6c, respectively, decreased their cytotoxic activity. However, connecting 4-fluorophenylpiperazine moiety to the acetamido side chain as in compound 6d greatly enhanced its lethal effect over HepG2 with IC50 values of 4.03 ± 0.2 μM, which is better than that of the reference drug CA-4 (IC50 4.50 ± 0.2 μM). Moreover, compound 6d showed high cytotoxic activity over HCT-116, MCF-7, and HeLa cell lines with IC50 values of 15.74 ± 0.8 and 9.64 ± 0.5, and 18.69 ± 0.9 μM, respectively. Replacing the piperazine moiety by morpholine afforded compound 6e with strong cytotoxic activity against HepG2 and HCT-116. In compound 6e, the piperazine ring was replaced by morpholine (Table 1, Scheme 2). Reaction of 2-chloro-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide 4 with different (un)substituted aniline derivatives afforded the corresponding anilinoacetamides 7a–e representing the aromatic substitution. The results from biological screening revealed that the unsubstituted aniline derivative 7a showed weak cytotoxic activities against the HepG2, HCT-116, and MCF-7 cell lines and that it was noncytotoxic on the HeLa cell line. However, 4-substitution of the aniline moiety with bromo, nitro, or morpholino groups exerted a positive impact on increasing the activity as in compounds 7b, 7c, and 7e, respectively. For example, compound 7b, with a bromo substituent, showed a selective cytotoxic activity against HCT-116 and HeLa cell lines with IC50 values of 9.35 ± 0.5 and 13.12 ± 0.7 μM, respectively. Moreover, compound 7c, with 4-nitroanilino acetamide side arm, displayed a notable cytotoxic and broad-spectrum activity among this series, with IC50 values ranging from 6.26 ± 0.3 to 10.01 ± 0.5 μM against the four tested cell lines. However, disubstitution of the aniline moiety as in compound 7d decreases the cytotoxic activity level comparable to the monosubstituted derivatives, while it still has more lethal effect than the unsubstituted compound 7a (Table 1, Scheme 2). Remarkable cytotoxic activity was noticed for compound 8 obtained by hydrazinolysis of the chloroacetamide derivative 4 and was identified as the most potent and broad-spectrum cytotoxic agent among all the tested compounds. Compound 8 showed the most lethal effect over the HepG2 cell line with IC50 value of 3.35 ± 0.2 μM which exceeded that of CA-4 (IC50 value of 4.50 ± 0.2 μM). Moreover, this compound showed an equipotent cytotoxic activity against HCT-116 with IC50 value of 5.32 ± 0.3 μM, which is comparable to that of the reference drug (IC50 value of 5.23 ± 0.3 μM). In addition, marked cytotoxic activity level was observed for compound 8 over both MCF-7 and HeLa cell lines with IC50 values of 8.66 ± 0.4 and 6.15 ± 0.3 μM, respectively (Table 1, Scheme 2). Regarding the isatinhydrazone derivatives, both methyl and chloro compounds 9a and 9b showed potent cytotoxicity against the four tested cancer cell lines with IC50 values ranging from 7.32 ± 0.4 to 13.42 ± 0.7 μM (Table 1, Scheme 2).
2.2.1.2. Cytotoxic Activity of the Bis-Thiazole Compound 10
Compound 10 with two thiazole moieties in its core structure showed moderate-to-weak cytotoxicity against the tested cell lines, as revealed from the IC50 values (Table 1, Scheme 2).
2.2.1.3. Cytotoxic Activity of the Urea-Based Target Compounds (11–13)
According to the results of biological screening, 1-(chloromethyl)-3-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)urea 11 showed a weak cytotoxic effect. Compound 12, in which an aniline moiety was connected to the methylurea side arm, showed the least activity against HCT-116 and HeLa cell lines among the tested compounds and complete loss of cytotoxicity over HepG2 and MCF-7 cell lines. However, the corresponding piperazinylmethyl urea derivative 13a showed strong cytotoxicity over HepG2, HCT-116, and MCF-7 cell lines with IC50 values of 14.17 ± 0.7, 20.08 ± 1.0, and 16.95 ± 0.8 μM, respectively. This compound also exerted a moderate effect against the HeLa cell line with IC50 values of 32.39 ± 1.6 μM, and the activity slightly decreased when the piperazine ring was replaced by morpholine as in compound 13b (Table 1, Scheme 3).
It is conspicuously observed that most of our newly synthesized compounds (16 out of 22) showed their best antiproliferative activity against the HePG2 cell line and the rest of the compounds; 6e, 7a, 7b, 11, and 12 displayed their best antiproliferative activity against the HCT-116 cell line, whereas compound 6b was seen to be the only compound that exhibited its best antiproliferative activity against the MCF-7 cell line. However, among these aforementioned 18 compounds selective for the HePG2 cell line, there are five compounds 6d, 7c, and 8–9b that showed very strong activity (below 10 μM) in the HePG2 cell line, with consistently good IC50 values across the investigated series of cell lines. Notably, all of these comprise two hydrogen-bond donors, for example, 7c and 8, or three hydrogen-bond donors, for example, 9a–b, with the exception of 6d that has only one.
2.2.2. In Vitro Tubulin Polymerization Inhibition Assay
To investigate whether the antitumor activity of the proposed compounds was related to the inhibition of the tubulin system, an in vitro tubulin polymerization inhibition assay was performed. Ten selected cytotoxic compounds including 5c, 6a,d,e, 7c,d, 8 and 9a,b and 13a were evaluated for their tubulin polymerization inhibition activity, with CA-4 as the reference drug. IC50 values of the tested compounds were calculated and listed in Table 2 and graphically represented in Figure 2. Compounds 5c, 7c, and 9a exhibited the highest inhibition of tubulin assembly among the tested compounds with IC50 values 2.95 ± 0.18, 2.00 ± 0.12, and 2.38 ± 0.14 μM, respectively, which are better than that of the standard drug (IC50 = 2.96 ± 0.18 μM). The rest of the tested compounds showed a certain level of inhibition, with IC50 values ranging from 3.42 ± 0.21 to 5.11 ± 0.31 μM.
Table 2. Results of the in Vitro Tubulin Polymerization Inhibition Assay and Docking Interaction Energy.
| comp. no | IC50 (μM)a tubulin polymerization inhibition | docking interaction energy(kcal/mol) |
|---|---|---|
| CA-4b | 2.96 ± 0.18 | –13.42 |
| 5c | 2.95 ± 0.18 | –13.88 |
| 6a | 4.50 ± 0.27 | –13.41 |
| 6d | 5.00 ± 0.30 | –13.09 |
| 6e | 3.72 ± 0.23 | –13.99 |
| 7c | 2.00 ± 0.12 | –14.15 |
| 7d | 3.42 ± 0.21 | –13.91 |
| 8 | 3.91 ± 0.24 | –13.20 |
| 9a | 2.38 ± 0.14 | –14.50 |
| 9b | 4.21 ± 0.26 | –14.20 |
| 13a | 5.11 ± 0.31 | –13.25 |
IC50 (μM): expressed as mean ± SD.
CA-4: combretastatin A-4.
Figure 2.

Tubulin polymerization inhibition chart of the tested compounds vs CA-4 (reference drug) expressed as IC50 (μM).
2.3. Docking Studies
Molecular docking was carried out for all the newly synthesized compounds into the colchicine binding site of our molecular target tubulin to confirm their capability for binding and explain their binding modes in comparison with CA-4 (Supporting Information data and Table 2). The values of free binding energies of the docked compounds 5c, 6e, 7b–d, 9a,b, and 12 ranged from −13.88 to −14.50 kcal/mol, which were higher than that of CA-4 (−13.42 kcal/mol). Compounds 5b, 6a,b,d, 7a, 8, and 13a scored free binding energy in the range of −13.02 to −13.41 kcal/mol, while the rest of docked compounds showed less free binding energies (−12.99 to −12.09 kcal/mol) than that of CA-4. The high level of tubulin polymerization inhibition by the promising compounds 5c, 7c, and 9a prompted us to use these compounds along with the least active 13a as representative examples to obtain a consistent and more precise picture of the biological active molecules at the atomic level and provide new insights that can be used to design novel therapeutic agents (Table 2 and Figures 3 and 4).
Figure 3.

2D and 3D interactions of CA-4 (upper panel), compounds 5c (middle panel), and 7c (lower panel) with the receptor pocket of tubulin (PDB ID: 4O2B).
Figure 4.

2D and 3D interactions of compounds 9a (upper panel) and 13a (lower panel) with the receptor pocket of tubulin (PDB ID: 4O2B).
In the binding profile of the reference drug CA-4 (Figure 3, upper panel), the terminal 2-methoxylated phenol moiety constructed a conspicuous H-bond via its hydroxyl group that acted as a H-donor with the H-acceptor-conserved amino acid SerA178 along with strong hydrophobic/hydrophilic interactions in between, easily detected from the deep blue and cyan shadows around OH and SerA178, respectively. Having an olefinic bridge, CA-4 is trapped in cis conformer that gave rise to the confrontation of 3,4,5-trimethoxy groups to the conserved amino acids LeuB248, LysB254 LeuB255, AlaB316, LysB352, and ThrB353, establishing intense hydrophobic/hydrophilic interactions, paving the way for the stability of the ligand/complex to score free binding energy of −13.42 kcal/mol. The molecular docking results of compound 5c (Figure 3, middle panel) demonstrated a thiazole ring strongly involved in noncovalent bindings: sulfur bond with AsnB249 and arene-H bond with AsnA101. The amidic nitrogen formed a H-bond with the H-acceptor-conserved amino acid SerA178 in the pocket core. Additionally, the noticeable hydrophobic/hydrophilic interactions appeared in both terminals of the ligand (trimethoxy groups and isopropyl fragment) as a blue shadow, improved the overall ligand recognition, and augmented the ligand/receptor complex stability with free binding energy of −13.88 kcal/mol. However, compound 7c (Figure 3, lower panel) showed a higher stability of ligand/receptor complex with free binding energy of −14.15 kcal/mol. ThrB353 built a bifurcated bond with sulfur atom and amidic NH, GlnB247 acted as a H-bond donor to amidic carbonyl, and GlnB336 acted as a H-bond donor to the double-bound oxygen atom of the terminal nitro group.
Furthermore, 5-chloro-3-oxoindolin-2-ylidene in compound 9a (Figure 4, upper panel) formed an arene-H bond through its pyrrole ring that acted as a nonclassical Lewis base with the H-donor-conserved amino acid AsnB249. Moreover, the hydrogen atom of the amino group of the hydrazino moiety established a H-bond with ThrA179. Besides, the excellent binding mode of thiazole moiety formed two different types of interactions; its sulfur established a noncovalent bond with AsnA101, and its aromaticity with its electron cloud as a nonclassical Lewis base formed an arene-H bond with LeuB248, improving significantly the stability of the ligand/receptor complex to score the best free binding energy of −14.50 kcal/mol among the docked compounds. Docking of urea derivative 13a (Figure 4, lower panel), which showed the least tubulin inhibition activity among the tested compounds, demonstrated that ThrB353 acted as the wedge to fix two sites of the ligand; it bound with thiazole sulfur and the hydrogen atom of the interior amino group of urea moiety by the sulfur-bond and H-bond, respectively. In spite of the cyan shadow around LeuB28, LysB352, LeuB255, and AsnA10 that reflected the hydrophobic/hydrophilic interactions between the ligand and the tubulin hot spot, the ligand scored free binding energy of −13.25 kcal/mol, comparatively lower than those of 5c, 7c, and 9a. As per the molecular docking results, it can be stated that the thiazole ring with its variable bioactive side arms has played a great impact in destabilising tubulin through a variety of binding interactions. Sulfur has σ-hole features that give rise to the formation of σ-hole bonding as a recently recognized noncovalent bonding type similar to halogen bond and thus has attracted the attention in medicinal chemistry34 and became a fundamental tool in rational drug design.35−39 Furthermore, TMP with its steric effect has steered efficiently the ligands into the appropriate binding mode by its miscellaneous interactions, so it is a successful pharmacophore to inhibit the tubulin assembly, in agreement with Tron et al.40 CA-4 as an olefinic structure with cis-restricted rotation has two obvious hindrances; first, it can convert rapidly to a trans-isomer which is 100-fold less active, by the effect of light, heat, or even protic solvent,41 that is, having poor metabolic stability ascribed to its olefinic bridge, and second, it is weakly soluble in water.42 Therefore, there is an eminent need for alternatives; so, we recommend, herein, that compounds 7c and 9a are excellent candidates for further clinical studies as tubulin assembly inhibitors.
3. Conclusions
In this study, newly synthesized 2-substituted-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide, 2-substituted-4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)urea, and (4-(3,4,5-trimethoxyphenyl) thiazol-2-yl)thiazole-2,4-diamine were subjected to antitumor evaluation as potential tubulin polymerization inhibitors. The obtained data revealed that compounds 5c, 6d, 7c, 8, and 9a,b showed strong activity against the tested cell lines (HepG2, MCF-7, HCT116, and HeLa in comparison with CA-4. In addition, the high level of inhibition of tubulin polymerization by compounds 5c, 7c, and 9a with IC50 better than that of the CA-4 reference drug indicated that their antitumor activity may be mediated through this mechanism, and this was further supported by the docking study. There was good agreement between the results of the docking study and the tubulin inhibition assay. The promising cytotoxic compounds 7c and 9a inhibit tubulin polymerization with IC50 2.00 ± 0.12 and 2.38 ± 0.14 μM, respectively, and they also score free binding energy of −14.15 and −14.50 kcal/mol, respectively. Hence, these new compounds might be used as the leading units to develop novel tubulin polymerization inhibitors as potential anticancer agents.
4. Experimental Section
4.1. Chemistry
Melting points (°C) were recorded using a Stuart melting point apparatus and are uncorrected. IR spectra were recorded on a Mattson 5000 FT-IR spectrophotometer (ν in cm–1) using a potassium bromide disk at the Faculty of Pharmacy, Mansoura University.1H NMR and 13C NMR spectra were determined by using a NMR spectrometer, Bruker 400, UK, at the Faculty of Pharmacy, Mansoura University. A proper amount of each compound was dissolved in either DMSO-d6 or CDCl3 before measurement. Tetramethylsilane was used as the internal standard. MS was performed on a Hewlett Packard 5988 spectrometer, at Al-Azhar University, Cairo, Egypt. Elemental microanalyses of the synthesized compounds were performed by the method proposed by Sullivan et al., at Al-Azhar University, Cairo, Egypt. The completion of reactions was monitored using thin-layer chromatography (TLC) plates, precoated with silica gel 60 F254 (E. Merck), and the spots were visualized under UV light (254, 365 nm). Pet. ether/EtOAc (1:1) or (3:1) were adopted as elution solvents. All chemicals and reagents were purchased from Sigma-Aldrich, and the solvents were obtained from El-Gomhouria Company for Pharmaceuticals and Chemicals. 2-Bromo-1-(3,4,5-trimethoxyphenyl)ethan-1-one (2)43 and 4-(3,4,5-trimethoxyphenyl)thiazol-2-amine (3)44 were prepared according to the previous report.
4.1.1. Procedure for the Synthesis of 2-Chloro-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (4)
To a mixture of compound 3 (2.6 g, 0.01 mol) in 1,4-dioxane (25 mL) and triethylamine (1 mL), chloroacetyl chloride (0.8 mL, 0.01 mol) was added dropwise at (5–10) °C. The reaction mixture was stirred at 70 °C for 6 h. After the completion of the reaction, the solvent was reduced and then the reaction mixture was poured into crushed ice, and the obtained solid was recrystallized from ethanol.
Brown crystals, mp 173–176 °C, yield (70%), IR (KBr, ν, cm–1): 3450 (N–H amide), 1690 (C=O amide), 1658 (C=N), 1592 (C=C), 1236 (C–O), 761 (C–Cl), 1H NMR (400 MHz, DMSO-d6): δ 3.67 (s, 3H, p-OCH3), 3.82 (s, 6H, m-OCH3), 4.39 (s, 2H, COCH2Cl), 7.18 (s, 2H, H-2 and H-6 Ar), 7.71 (s, 1H, thiazole-H), 12.50 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 42.7 (CH2Cl), 56.4 (m-OCH3), 60.6 (p-OCH3), 103.6 (2,6-Ar-C), 108.7 (thiazole-C-5), 130.3 (1-Ar-C), 137.9 (4-Ar-C), 149.4 (thiazole-C-4), 153.6 (3,5-Ar-C), 157.7 (thiazole-C-2), 165.6 (C=O), MS (m/z, Rel. Abundance): 342.07 (M+, 55.31), 343.16 (M + 1, 15.91), 344.25 (M + 2, 15.75), 109.08 (100), Anal. Calcd for C14H15ClN2O4S calculated %: C, 49.05; H, 4.41; N, 8.17; S, 9.35. Found %: C, 49.08; H, 4.45; N, 8.14; S, 9.33.
4.1.2. General Procedure for the Synthesis of 2-Substituted N-(4-(3,4,5-Trimethoxyphenyl)thiazol-2-yl)acetamide Derivatives (5a–c)
A mixture of compound 4 (0.34 g, 0.001 mol), aliphatic amines (0.001 mol), and anhydrous K2CO3 (0.14 g, 0.001 mol) in dry DMF (10 mL) was heated under reflux for 8–9 h. After the reaction completion, the reaction mixture was poured onto crushed ice, and the precipitated product was filtered, dried, and recrystallized from acetone.
4.1.2.1. 2-((2-Hydroxyethyl)amino)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (5a)
Brown crystals, mp 85–88 °C, yield (71%), IR (KBr, ν, cm–1): 3410 (OH), 3347 (N–H amide), 3114 (2° amine NH), 1662 (C=O amide), 1589 (C=C), 1234 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 3.09 (s, 2H, COCH2), 3.40 (t, J = 12.80 Hz, 2H, NHCH2), 3.57 (t, J = 17.60 Hz, 2H, CH2OH), 3.67 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 4.80 (s, 1H, OH, D2O exchangeable), 5.20 (s, 1H, CH2NH, D2O exchangeable), 7.10 (s, 2H, H-2 and H-6 Ar), 7.20 (s, 1H, thiazole-H), 12.50 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 52.0 (NHCH2), 56.3 (m-OCH3), 60.5 (p-OCH3), 60.5 (CH2OH), 61.6 (COCH2–NH), 101.6 (thiazole-C-5), 103.3 (2,6-Ar-C), 131.2 (1-Ar-C), 137.3 (4-Ar-C), 150.2 (thiazole-C-4), 153.3 (3,5-Ar-C), 168.5 (thiazole-C-2), 171.0 (C=O), MS (m/z, Rel. Abundance): 367.1 (M+, 33.67), 265.9 (100), Anal. Calcd for C16H21N3O5S calculated %: C, 52.30; H, 5.76; N, 11.44; S, 8.73. Found %: C, 52.34; H, 5.72; N, 11.41; S, 8.72.
4.1.2.2. 2-(Butylamino)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (5b)
Brown crystals, mp 87–92 °C, yield (64%), IR (KBr, ν, cm–1): 3490 (N–H amide), 3331 (2° amine NH), 1655 (C=O amide), 1590 (C=C), 1235 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 0.88 (t, J = 16.00 Hz, 3H, CH2CH3), 1.32 (m, 4H, CH2CH2CH3), 2.70 (t, J = 16.00 Hz, 2H, NHCH2), 3.40 (s, 2H, COCH2), 3.52 (s, 1H, NH, D2O exchangeable), 3.70 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 7.10 (s, 2H, H-2 and H-6 Ar), 7.64 (s, 1H, thiazole-H), 12.50 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 13.5 (butyl amine-C-4), 20.3 (butyl amine-C-3), 32.4 (butyl amine-C-2), 49.1 (butyl amine-C-1), 52.8 (COCH2), 56.9 (m-OCH3), 60.7 (p-OCH3), 101.5 (2,6-Ar-C), 107.2 (thiazole-C-5), 128.1 (1-Ar-C), 141.1 (4-Ar-C), 151.1 (thiazole-C-4), 154.1 (3,5-Ar-C), 165.2 (thiazole-C-2), 169.6 (C=O), MS (m/z, Rel. Abundance): 381.62 (M+, 19.52), 379.29 (100), Anal. Calcd for C18H25N3O4S calculated %: C, 56.97; H, 6.64; N, 11.07; S, 8.45. Found %: C, 57.01; H, 6.67; N, 11.05; S, 8.46.
4.1.2.3. 2-(Isopropylamino)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (5c)
Black crystals, mp 178–181 °C, yield (68%), IR (KBr, ν, cm–1): 3424 (N–H amide), 3331 (2° amine NH), 1655 (C=O amide), 1588 (C=C), 1235 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 1.01 (d, J = 6.40 Hz, 6H, CH3), 1.05 (m, 1H, CH3 CHCH3), 3.20 (s, 2H, COCH2), 3.40 (s, 1H, CH2NHD2O exchangeable), 3.70 (s, 3H, p-OCH3), 3.83 (s, 6H, m-OCH3), 7.15 (s, 2H, H-2 and H-6 Ar), 7.26 (s, 1H, thiazole-H), 12.50 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 23.2 (isopropyl-CH3), 50.5 (COCH2–NH), 51.3 (isopropyl-CH), 57.34 (m-OCH3), 61.9 (p-OCH3), 100.5 (2,6-Ar-C), 105.2 (thiazole-C-4), 127.5 (1-Ar-C), 139.1 (4-Ar-C), 150.3 (thiazole-C-5), 153.3 (3,5-Ar-C), 164.5 (thiazole-C-2), 168.7 (C=O), MS (m/z, Rel. Abundance): 364.45 (M+, 37.24), 109.38 (100), Anal. Calcd for C17H23N3O4S calculated %: C, 55.87; H, 6.34; N, 11.50; S, 8.77. Found %: C, 55.84; H, 6.35; N, 11.48; S, 8.81.
4.1.3. General Procedure for the Synthesis of Substituted (4-(3,4,5-Trimethoxyphenyl)thiazol-2-yl)acetamide Derivatives (6a–e)
A mixture of compound 4 (1 g, 0.003 mol) and the appropriate secondary amines (0.003 mol) in dry DMF (10 mL) containing few drops of triethylamine (3–4 drops) was heated under reflux for 8 h. The reaction mixture was left to cool, then poured onto ice-cold water, and the formed precipitate was filtered, dried, and recrystallized from ethanol.
4.1.3.1. 2-(Piperazin-1-yl)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (6a)
Brown crystals, mp 258–261 °C, yield (59%), IR (KBr, ν, cm–1): 3449 (N–H amide), 3114 (2° amine NH), 1656 (C=O amide), 1555 (C=N), 1267 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 1.78 (s, 1H, piperazine-NH, D2O exchangeable), 2.47 (m, 4H, piperazine-CH2), 2.75 (m, 4H, piperazine-CH2), 3.28 (s, 2H, COCH2), 3.69 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 7.21 (s, 2H, H-2 and H-6 Ar), 7.64 (s, 1H, thiazole-H), 12.50 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 44.1 (3,5-piperazine-CH2), 55.7 (m-OCH3), 56.1 (2,6-piperazine-CH2), 60.9 (p-OCH3), 63.0 (COCH2), 101.3 (2,6-Ar-C), 106.1 (thiazole-C-5), 128.5 (1-Ar-C), 139.7 (4-Ar-C), 149.9 (thiazole-C-4), 153.6 (3,5-Ar-C), 163.7 (thiazole-C-2), 169.1 (C=O), MS (m/z, Rel. Abundance): 392.97 (M+, 42.88), 392 (100), Anal. Calcd for C18H24N4O4S calculated %: C, 55.09; H, 6.16; N, 14.28; S, 8.17. Found %: C, 55.07; H, 6.12; N, 14.31; S, 8.20.
4.1.3.2. 2-(4-Methylpiperazin-1-yl)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (6b)
Brown crystals, mp 135–138 °C, yield (66%), IR (KBr, ν, cm–1): 3451 (N–H amide), 1682 (C=O amide), 1658 (C=N), 1588 (C=C), 1455 (N–CH3) 1236 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 2.15 (s, 3H, NCH3), 2.33 (m, 8H, piperazine-CH2), 3.29 (s, 2H, COCH2), 3.69 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 7.21 (s, 2H, H-2 and H-6 Ar), 7.64 (s, 1H, thiazole-H), 12.50 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 46.2 (N–CH3), 52.9 (2,6-piperazine-CH2), 55.1 (m-OCH3), 56.4 (3,5-piperazine-CH2), 60.5 (p-OCH3), 60.7 (CH2–N), 103.5 (2,6-Ar-C), 108.1 (thiazole-C-5), 130.5 (1-Ar-C), 137.7 (4-Ar-C), 149.2 (thiazole-C-4), 153.3 (3,5-Ar-C), 158.1 (thiazole-C-2), 169.3 (C=O), MS (m/z, Rel. Abundance): 406.02 (M+, 43), 407.25 (M+ + 1, 28.64) 336.1 (100), Anal. Calcd for C19H26N4O4S calculated %: C, 56.14; H, 6.45; N, 13.78; S, 7.89. Found %: C, 56.11; H, 6.48; N, 13.75; S, 7.85.
4.1.3.3. 2-(4-Ethylpiperazin-1-yl)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (6c)
Black crystals, mp 133–136 °C, yield (62%), IR (KBr, ν, cm–1): 3446 (N–H amide), 1653 (C=O amide), 1591 (C=C), 1239 (C–O), 1H NMR (400 MHz, CDCl3): δ 1.25 (t, J = 14.50 Hz, 3H, CH2CH3), 2.43 (q, 2H, CH2CH3), 2.54 (t, J = 9.50 Hz, 4H, piperazine-CH2), 2.69 (t, J = 8.00 Hz, 4H, piperazine-CH2), 3.32 (s, 2H, COCH2), 3.80 (s, 3H, p-OCH3), 3.95 (s, 6H, m-OCH3), 7.04 (s, 2H, H-2 and H-6 Ar), 8.00 (s, 1H, thiazole-H), 12.50 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 15.4 (N–CH2–CH3), 49.1 (N–CH2CH3), 54.3 (2,6-piperazine-CH2), 55.7 (m-OCH3), 56.9 (3,5-piperazine-CH2), 60.3 (p-OCH3), 62.5 (COCH2), 101.3 (2,6-Ar-C), 106.1 (thiazole-C-5), 128.5 (1-Ar-C), 138.7 (4-Ar-C), 149.6 (thiazole-C-4), 152.4 (3,5-Ar-C), 160.5 (thiazole-C-2), 168.1 (C=O), MS (m/z, Rel. Abundance): 420.07 (M+, 100), Anal. Calcd for C20H28N4O4S calculated %: C, 57.12; H, 6.71; N, 13.32; S, 7.62. Found %: C, 57.15; H, 6.75; N, 13.31; S, 7.64.
4.1.3.4. 2-(4-(4-Fluorophenyl)piperazin-1-yl)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (6d)
Dark yellow crystals, mp 97–100 °C, yield: (60%), IR (KBr, ν, cm–1): 3386 (N–H amide), 1679 (C=O amide), 1590 (C=C), 1230 (C–O), 1126 (C–F), 1H NMR (400 MHz, DMSO-d6): δ 2.37 (m, 4H, piperazine-CH2), 2.54 (m, 4H, piperazine-CH2), 3.30 (s, 2H, COCH2), 3.70 (s, 3H, p-OCH3), 3.85 (s, 6H, m-OCH3), 7.09 (d, 2H, J = 16.00 Hz, fluorophenyl H-2, H-6), 7.22 (s, 2H, H-2 and H-6 Ar), 7.28 (d, 2H, J = 12.00 Hz, fluorophenyl H-3, H-5), 7.65 (s, 1H, thiazole-H), 12.06 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 52.9 (3,5-piperazine-CH2), 53.0 (m-OCH3), 56.4 (2,6-piperazine-CH2), 60.5 (p-OCH3), 61.6 (COCH2–N), 103.6 (2,6-Ar-C), 108.1 (thiazole-C-5), 115.2 (2,6-fluoro-phenyl c), 115.4 (3,5-fluoro-phenyl c), 130.5 (1-Ar-C), 134.8 (thiazole-C-4), 137.8 (4-Ar-C), 149.3 (1-fluoro-phenyl c), 153.6 (3,5-Ar-C), 157.7 (4-fluoro-phenyl c), 160.5 (thiazole-C-2), 162.9 (C=O), MS (m/z, Rel. Abundance): 486.42 (M+, 34.40), 487.06 (M + 1, 6.53), 109.16 (100), Anal. Calcd for C24H27 FN4O4S calculated %: C, 59.25; H, 5.59; N, 11.52; S, 6.59. Found %: C, 59.28; H, 5.56; N, 11.50; S, 6.61.
4.1.3.5. 2-Morpholino-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (6e)
Black crystals, mp 102–104 °C, yield: (65%), IR (KBr, ν, cm–1): 3450 (N–H amide), 1637 (C=O amide), 1530 (C=C), 1235 (C–O), 1H NMR (400 MHz, CDCl3): δ 3.20 (s, 2H, COCH2), 3.40 (t, 4H, J = 10.00 Hz, morpholine-CH2), 3.65 (t, 4H, J = 10.50 Hz, morpholine-CH2), 3.72 (s, 3H, p-OCH3), 3.86 (s, 6H, m-OCH3), 7.05 (s, 2H, H-2 and H-6 Ar), 7.08 (s, 1H, thiazole-H), 12.40 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 55.6 (3,5-morpholine-CH2), 56.4 (m-OCH3), 61.3 (p-OCH3), 62.6 (COCH2), 64.2 (2,6-morpholine-CH2), 103.5 (2,6-Ar-C), 107.1 (thiazole-C-5), 130.5 (1-Ar-C), 137.7 (4-Ar-C), 149.2 (thiazole-C-4), 153.6 (3,5-Ar-C), 158.1 (thiazole-C-2), 169.3 (C=O), MS (m/z, Rel. Abundance): 394.17 (M+, 41.68), 393.33 (100), Anal. Calcd for C18H23 N3O5S calculated %: C, 54.95; H, 5.89; N, 10.68; S, 8.15. Found %: C, 54.97; H, 5.87; N, 10.64; S, 8.11.
4.1.4. General Procedure for the Synthesis of 2-Substituted N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide Derivatives (7a–e)
A mixture of compound 4 (1 g, 0.003 mol) and substituted anilines (0.003 mol) in absolute ethanol (10 mL) and few drops of HCl (5–6 drops) was refluxed overnight. The reaction progress was monitored by TLC after the completion of the reaction. The reaction mixture was poured into ice-cold water, and the resultant precipitate was filtered, dried, and recrystallized from methanol.
4.1.4.1. 2-(Phenylamino)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (7a)
Black crystals, mp 165–168 °C, yield (65%), IR (KBr, ν, cm–1): 3340 (N–H amide), 3091 (2° amine NH), 1629 (C=O amide), 1583 (C=C), 1251 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 3.70 (s, 3H, p-OCH3), 3.77 (s, 2H, COCH2), 3.86 (s, 6H, m-OCH3), 6.70 (s, 1H, aniline-NH, D2O exchangeable), 7.18 (s, 2H, H-2 and H-6 Ar), 7.35 (s, 1H, thiazole-H),7.41–7.50 (m, 5H, aniline-H), 10.70 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 54.1 (CH2–N), 56.6 (m-OCH3), 60.2 (p-OCH3), 100.3 (2,6-Ar-C), 105.8 (thiazole-C-5), 114.6 (aniline-C-2,6), 119.5 (aniline-C-4), 127.8 (1-Ar-C), 130.0 (aniline-C-3,5), 140.2 (4-Ar-C), 147.1 (aniline-C-1), 150.5 (thiazole-C-4), 153.6 (3,5-Ar-C), 164.8 (thiazole-C-2), 168.7 (C=O), MS (m/z, Rel. Abundance): 399.14 (M+, 5.83), 109.02 (100), Anal. Calcd for C20H21 N3O4S calculated %: C, 60.14; H, 5.30; N, 10.52; S, 8.03. Found %: C, 60.18; H, 5.32; N, 10.56; S, 8.01.
4.1.4.2. 2-((4-Bromophenyl)amino)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (7b)
Brown crystals, mp 147–150 °C, yield (64%), IR (KBr, ν, cm–1): 3340 (N–H amide), 3192 (2° amine NH), 1630 (C=O amide), 1588 (C=N), 1560 (C=C), 1251 (C–O), 657 (C–Br), 1H NMR (400 MHz, DMSO-d6): δ 3.70 (s, 3H, p-OCH3), 3.78 (s, 2H, COCH2), 3.86 (s, 6H, m-OCH3), 6.70 (s, 1H, aniline-NH, D2O exchangeable), 7.17 (s, 2H, H-2, H-6 Ar), 7.32 (s, 1H, thiazole-H), 7.34 (d, J = 4.00 Hz, 2H, aniline-H), 7.65 (d, J = 8.00 Hz, 2H, aniline-H), 9.07 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 55.6 (CH2–N), 56.4 (m-OCH3), 60.0 (p-OCH3), 100.5 (2,6-Ar-C), 105.0 (thiazole-C-5), 114.1 (aniline-C-2,6), 115.9 (aniline-C-4), 127.5 (1-Ar-C), 133.1 (aniline-C-3,5), 139.7 (4-Ar-C), 146.9 (aniline-C-1), 150.4 (thiazole-C-4), 153.8 (3,5-Ar-C), 164.5 (thiazole-C-2), 168.4 (C=O), MS (m/z, Rel. Abundance): 478.86 (M+, 32.51), 479.55 (M + 1, 18.52), 480.75 (M + 2, 30.58), 267.55 (100), Anal. Calcd for C20H20BrN3O4S calculated %: C, 50.22; H, 4.21; N, 8.78; S, 6.70. Found %: C, 50.25; H, 4.24; N, 8.74; S, 6.72.
4.1.4.3. 2-((4-Nitrophenyl)amino)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (7c)
Black crystals, mp 100–103 °C, yield (60%), IR (KBr, ν, cm–1): 3479 (N–H amide), 3362 (2° amine NH), 1631 (C=O amide), 1595 (C=C), 1501 (N–O), 1H NMR (400 MHz, DMSO-d6): δ 3.71 (s, 3H, p-OCH3), 3.78 (s, 2H, COCH2), 3.85 (s, 6H, m-OCH3), 6.59 (t, J = 13.60 Hz, 2H, aniline-H), 6.90 (s, 1H, aniline-NH, D2O exchangeable), 7.10 (s, 2H, H-2, H-6 Ar), 7.40 (s, 1H, thiazole-H), 7.93 (t, J = 14.00 Hz, 2H, aniline-H), 12.50 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 55.4 (CH2–N), 56.3 (m-OCH3), 60.7 (p-OCH3), 100.5 (2,6-Ar-C), 105.3 (thiazole-C-4), 115.6 (aniline-C-2,6), 127.3 (1-Ar-C), 128.2 (aniline-C-3,5), 136.4 (aniline-C-4), 140.3 (4-Ar-C), 151.2 (thiazole-C-5), 153.0 (3,5-Ar-C), 153.8 (aniline-C-1), 164.8 (thiazole-C-2), 168.7 (C=O), MS (m/z, Rel. Abundance): 444.02 (M+, 7.47), 52.08 (100), Anal. Calcd for C20H20N4O6S calculated %: C, 54.05; H, 4.54; N, 12.61; S, 7.21. Found %: C, 54.02; H, 4.58; N, 12.63; S, 7.23.
4.1.4.4. 2-((4-Bromo-3-(trifluoromethyl)phenyl)amino)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (7d)
Black crystals, mp 103–105 °C, yield (57%), IR (KBr, ν, cm–1): 3336 (N–H amide), 3084 (2° amine NH), 1628 (C=O amide), 1560 (C=C), 1257 (C–O), 1126 (C–F), 662 (C–Br), 1H NMR (400 MHz, DMSO-d6): δ 3.70 (s, 2H, COCH2), 3.78 (s, 3H, p-OCH3), 3.86 (s, 6H, m-OCH3), 6.74 (s, 1H, aniline NH, D2O exchangeable), 6.92 (d, J = 6.00 Hz, 2H, aniline-H), 7.21 (s, 2H, H-2, H-6 Ar), 7.50 (s, 1H, thiazole-H), 7.54 (d, J = 7.60 Hz, 1H, aniline-H), 12.50 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 56.3 (CH2–N), 57.3 (m-OCH3), 61.2 (p-OCH3), 100.8 (2,6-Ar-C), 105.3 (thiazole-C-4), 109.4 (aniline-C-4), 112.1 (aniline-C-2), 118.1 (aniline-C-6), 123.5 (CF3), 127.8 (1-Ar-C), 132.0 (aniline-C-3), 133.1 (aniline-C-5), 139.6 (4-Ar-C), 147.1 (aniline-C-1), 150.7 (thiazole-C-5), 154.0 (3,5-Ar-C), 164.3 (thiazole-C-2), 168.7 (C=O), MS (m/z, Rel. Abundance): 546.33 (M+, 41.60), 548.01 (M + 2, 37.77), 91.88 (100), Anal. Calcd for C21H19BrF3N3O4S calculated %: C, 46.17; H, 3.51; N, 7.69; S, 5.87. Found %: C, 46.20; H, 3.53; N, 7.68; S, 5.90.
4.1.4.5. 2-((4-Morpholinophenyl)amino)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (7e)
Gray crystals, mp 239–242 °C, yield (60%), IR (KBr, ν, cm–1): 3453 (N–H), 3362 (2° amine NH), 1672 (C=O amide), 1655 (C=N), 1585 (C=C), 1234 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 3.2 (m, 4H, morpholine H), 3.70 (s, 3H, p-OCH3), 3.74 (m, 4H, morpholine-H), 3.75 (s, 2H, COCH2), 3.86 (s, 6H, m-OCH3), 5.70 (s, 1H, aniline NH, D2O exchangeable), 6.50 (m, 2H, aniline-H), 6.80 (m, 2H, aniline-H), 7.15 (s, 2H, H-2, H-6 Ar), 7.28 (s, 1H, thiazole-H), 12.50 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 53.7 (morpholine-C-3,5), 55.5 (CH2–N), 56.7 (m-OCH3), 60.4 (p-OCH3), 67.0 (morpholine-C-2,6), 100.5 (2,6-Ar-C), 105.1 (thiazole-C-5), 117.5 (aniline-C-2,6), 127.5 (1-Ar-C), 129.1 (aniline-C-3,5), 137.6 (aniline-C-1), 138.8 (aniline-C-4), 139.5 (4-Ar-C), 150.3 (thiazole-C-4), 153.5 (3,5-Ar-C), 164.6 (thiazole-C-2), 168.4 (C=O), MS (m/z, Rel. Abundance): 484.41 (M+, 53.45), 90.97 (100).
4.1.5. Procedure for the Synthesis of 2-Hydrazinyl-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (8)
Hydrazine hydrate 64–65% (1.5 mL, 0.03 mol) was added to a stirred solution of compound 4 (1 g, 0.003 mol) in absolute ethanol (10 mL). The reaction mixture was refluxed for 6 h. The reaction mixture was left to cool, and the precipitate was collected by filtration and recrystallized from methanol.
Brown crystals, mp 135–138 °C, yield (75%), IR (KBr, ν, cm–1): 3403 (N–H amide), 3188 (2° amine NH), 3109 (1° amine NH), 1635 (C=O amide), 1590 (C=C), 1234 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 3.40 (s, 2H, NH2, D2O exchangeable), 3.67 (s, 2H, COCH2), 3.69 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 4.20 (s, 1H, NH, D2O exchangeable), 7.19 (s, 2H, H-2 and H-6 Ar), 7.62 (s, 1H, thiazole-H), 12.30 (s, 1H, NHCO D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 56.3 (m-OCH3), 60.5 (p-OCH3), 60.6 (COCH2–NH), 101.7 (thiazole-C-5), 103.5 (2,6-Ar-C), 130.5 (1-Ar-C), 137.4 (4-Ar-C), 149.1 (thiazole-C-4), 153.4 (3,5-Ar-C), 168.5 (thiazole-C-2), 169.2 (C=O), MS (m/z, Rel. Abundance): 338.35 (M+, 50.13), 308.27, (100), Anal. Calcd for C14H18N4O4 scalculated %: C, 49.69; H, 5.36; N, 16.56; S, 9.47. Found %: C, 49.66; H, 5.39; N, 16.59; S, 9.44.
4.1.6. Procedure for the Synthesis of 2-(2-Substituted hydrazinyl)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide Derivatives (9a–b)
A mixture of hydrazide 8 (0.44 g, 0.002 mol) and isatin derivative (0.002 mol) was refluxed in absolute ethanol (10 mL) in the presence of few drops of glacial acetic acid (5–6 drops) for 24 h. Progress of the reaction was monitored through TLC. After the completion of the reaction, the solvent was evaporated to afford the target product. The product was filtered and recrystallized from methanol.
4.1.6.1. (Z)-2-(2-(5-Chloro-2-oxoindolin-3-ylidene)hydrazinyl)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (9a)
Orange crystals, mp 170–173 °C, yield (60%), IR (KBr, ν, cm–1): 3400 (N–H amide), 3280 (N–H isatin), 3239 (N–H. hydrazine), 1686 (C=O amide), 1587 (C=C), 1237 (C–O), 812 (C–Cl), 1H NMR (400 MHz, DMSO-d6): δ 3.50 (s, 2H, COCH2), 3.70 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 7.19 (s, 2H, H-2 and H-6 Ar), 7.30 (m, 1H, NH–N, D2O exchangeable), 7.42 (s, 1H, isatin-H), 7.68 (m, 1H, isatin-H) 7.80 (s, 1H, thiazole-H), 7.90 (m, 1H, isatin-H), 10.80 (s, 1H, isatin NH, D2O exchangeable), 12.50 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 53.5 (COCH2–NH), 56.4 (m-OCH3), 60.6 (p-OCH3), 103.5 (2,6-Ar-C), 111.9 (thiazole-C-5), 117.4 (3a-isatin C) 123.8 (7-isatin C), 126.8 (4-isatin C), 127.2 (1-Ar-C), 127.5 (3-isatin C), 129.2 (5-isatin C), 132.1 (6-isatin C), 137.6 (4-Ar-C), 137.9 (2-isatin C), 138.2 (thiazole-C-4), 153.3 (3,5-Ar-C), 153.6 (7a-isatin C), 163.1 (thiazole-C-2), 168.6 (C=O), MS (m/z, Rel. Abundance): 501.53 (M+, 14.29), 503.59 (M + 1, 6.05) 72.08 (100), Anal. Calcd for C22H20ClN5O5S calculated %: C, 52.64; H, 4.02; N, 13.95; S, 6.39. Found %: C, 52.68; H, 4.06; N, 13.93; S, 6.35.
4.1.6.2. (Z)-2-(2-(5-Methyl-2-oxoindolin-3-ylidene)hydrazinyl)-N-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)acetamide (9b)
Orange crystals, mp139–142 °C, yield (57%), IR (KBr, ν, cm–1): 3392 (N–H amide), 3337 (N–H hydrazine), 3235 (N–H isatin), 1687 (C=O amide), 1626 (C=N), 1591 (C=C), 1450 (CH3), 1241 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 2.27 (s, 3H, CH3), 3.50 (s, 2H, COCH2), 3.69 (s, 3H, p-OCH3), 3.85 (s, 6H, m-OCH3), 6.50 (d, 1H, isatin H-7), 7.18 (s, 2H, H-2 and H-6 Ar), 7.20 (s, 1H, NH=N, D2O exchangeable), 7.30 (m, 1H, isatin H-6), 7.40 (s, 1H, isatin H-4), 7.80 (s, 1H, thiazole-H), 10.60 (s, 1H, isatin NH, D2O exchangeable), 12.50 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 22.0 (CH3 isatin), 53.6 (COCH2–NH), 56.3 (m-OCH3), 60.4 (p-OCH3), 102.9 (2,6-Ar-C), 108.9 (thiazole-C-5), 117.5 (3a-isatin C), 122.6 (7-isatin C), 127.1 (1-Ar-C), 129.2 (4-isatin C), 130.4 (6-isatin C), 133.1 (3-isatin C), 135.5 (5-isatin C), 136.8 (7a-isatin C), 137.9 (2-isatin C), 138.5 (4-Ar-C), 145.2 (thiazole-C-4), 153.3 (3,5-Ar-C), 163.7 (thiazole-C-2), 168.6 (C=O), MS (m/z, Rel. Abundance): 481.74 (M+, 11.24), 485.6 (M + 1, 6.28), 118.23 (100), Anal. Calcd for C23H23N5O5S calculated %: C, 57.37; H, 4.81; N, 14.54; S, 6.66. Found %: C, 57.33; H, 4.84; N, 14.57; S, 6.64.
4.1.7. Procedure for the Synthesis of N4-(4-(3,4,5-Trimethoxyphenyl)thiazol-2-yl)thiazole-2,4-diamine (10)
A mixture of compound 4 (0.34 g, 0.001 mol) and thiourea (0.08 g, 0.001 mol) and anhydrous K2CO3 (0.14 g, 0.001 mol) in acetone (10 mL) was refluxed overnight. After the completion of the reaction, the solvent was evaporated to afford the target product. The product was filtered and recrystallized from methanol.
Brown crystals, mp 180–183 °C, yield (66%), IR (KBr, ν, cm–1): 3421 (2° amine N–H), 3188 (1° amine NH2), 2160 (N=C=S), 1589 (C=C), 1125 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 3.69 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 5.30 (s, 1H, thiazole′-H), 7.05 (s, 2H, H-2 and H-6 Ar), 7.20 (s, 2H, NH2, D2O exchangeable), 7.60 (s, 1H, thiazole-H), 9.00 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 56.3 (m-OCH3), 60.5 (p-OCH3), 103.5 (2,6-Ar-C), 108.4 (thiazole-C-4), 110.5 (thiazole′-C-4), 130.4 (1-Ar-C), 137.8 (4-Ar-C), 142.1 (thiazole′-C-5), 149.3 (thiazole-C-5), 153.6 (3,5-Ar-C), 158.0 (thiazole-C-2), 168.4 (thiazole′-C-2), MS (m/z, Rel. Abundance): 364 (M+, 33.12), 149 (100), Anal. Calcd for C15H16N4O3S2 calculated %: C, 49.44; H, 4.43; N, 15.37; S, 17.59. Found %: C, 49.47; H, 4.40; N, 15.39; S, 17.62.
4.1.8. Procedure for the Synthesis of 1-(2-Chloroethyl)-3-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)urea (11)
A mixture of compound 3 (0.5 g, 0.002 mol), 2-chloroethyl isocyanate (0.3 mL, 0.002 mol), and anhydrous K2CO3 (0.3 g, 0.002 mol) was heated under reflux in dry DMF (10 mL) for 12 h. The reaction mixture was left to cool, concentrated under reduced pressure, and poured onto crushed ice. The separated precipitate was filtered and dried.
Brown crystals, mp 205–208 °C, yield (52%), IR (KBr, ν, cm–1): 3487, 3328 (NH amide), 1657 (C=O amide), 1589 (C=C), 1238 (C–O), 738 (C–Cl), 1H NMR (400 MHz, (CD3)2CO): δ 3.65 (t, J = 10.00 Hz, 2H, NHCH2), 3.70 (t, J = 10.00 Hz, 2H, CH2Cl), 3.73 (s, 3H, p-OCH3), 3.87 (s, 6H, m-OCH3), 7.28 (s, 2H, H-2 and H-6 Ar), 7.52 (s, 1H, thiazole-H), 8.20 (t, J = 15.00 Hz, 1H, NHCH2, D2O exchangeable), 9.60 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 42.3 (NHCH2CH2), 42.9 (CH2Cl), 56.5 (m-OCH3), 60.9 (p-OCH3), 100.5 (2,6-Ar-C), 105.3 (thiazole-C-4), 127.9 (1-Ar-C), 139.1 (4-Ar-C), 149.9 (thiazole-C-5), 153.6 (3,5-Ar-C), 154.5 (C=O), 164.4 (thiazole-C-2), MS (m/z, Rel. Abundance): 371.96 (M+, 18.84), 373.73 (M + 1, 11.96), 169.22 (100), Anal. Calcd for C15H18ClN3O4S calculated %: C, 48.45; H, 4.88; N, 11.30; S, 8.62. Found %: C, 48.47; H, 4.86; N, 11.34; S, 8.59.
4.1.9. Procedure for the Synthesis of 1-(2-(Phenylamino)ethyl)-3-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)urea (12)
A mixture of compound 11 (0.5 g, 0.001 mol) and aniline (0.1 mL, 0.001 mol) in absolute ethanol (10 mL) and few drops of HCl (5–6 drops) was heated under reflux overnight. The reaction progress was monitored by TLC; after the completion of the reaction, the reaction mixture was poured into ice-cold water, and the precipitate was filtered, dried, and recrystallized from methanol.
Brown crystals, mp 278–281 °C, yield: (70%), IR (KBr, ν, cm–1): 3451, 3380 (N–H amide), 3250 (2° amine N–H), 1682 (C=O amide), 1658 (C=N), 1588 (C=C), 1236 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 3.50 (t, 2H, J = 16.80 Hz, CH2N), 3.69 (s, 3H, p-OCH3), 3.70 (t, 2H, J = 17.60 Hz, CONHCH2), 3.84 (s, 6H, m-OCH3), 4.07 (t, 1H, J = 15.60 Hz, CONHCH2, D2O exchangeable), 4.14 (t, 1H, J = 16.00 Hz, CH2NH, D2O exchangeable), 7.15 (m, 3H, aniline H-2, H-4, H-6), 7.21 (s, 2H, H-2 and H-6 Ar), 7.55 (s, 2H, aniline H-3, H-5), 7.60 (s, 1H, thiazole-H), 9.80 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 41.8 (ethyl-C-1), 43.5 (ethyl-C-2), 56.7 (m-OCH3), 61.2 (p-OCH3), 101.2 (2,6-Ar-C), 105.2 (thiazole-C-4), 114.3 (aniline-C-2,6), 121.3 (aniline-C-4), 127.8 (1-Ar-C), 130.0 (aniline-C-3,5), 139.7 (4-Ar-C), 148.0 (aniline-C-1), 150.7 (thiazole-C-5), 153.8 (3,5-Ar-C), 154.7 (C=O), 164.8 (thiazole-C-2), MS (m/z, Rel. Abundance): 428.39 (M+, 13.94), 343.66 (100), Anal. Calcd for C21H24N4O4S calculated %: C, 58.86; H, 5.65; N, 13.08; S, 7.48. Found %: C, 58.83; H, 5.67; N, 13.06; S, 7.45.
4.1.10. General Procedure for the Synthesis of 1-Substituted-3-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)urea (13a–b)
A mixture of compound 11 (0.5 g, 0.001 mol) and appropriate secondary amines (0.001 mol), N-ethyl piperazine (0.001 mol), and morpholine (0.001 mol) in DMF (10 mL) containing few drops of triethylamine was refluxed for 8 h. After the completion of the reaction, the reaction mixture was cooled and poured onto ice-cold water, and the formed precipitate was filtered, dried, and recrystallized from ethanol.
4.1.10.1. 1-(2-(4-Ethylpiperazin-1-yl)ethyl)-3-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)urea (13a)
Black crystals, mp 217–220 °C, yield (58%), IR (KBr, ν, cm–1): 3421, 3255 (N–H amide), 1657 (C=O amide), 1586 (C=C), 1408 (CH3), 1235 (C–O), 1H NMR (400 MHz, DMSO-d6): δ 1.01 (t, 3H, J = 14.00 Hz, CH2CH3), 2.28 (m, 8H, piperazine-Hs), 3.20 (q, 2H, CH2CH3), 3.50 (t, 2H, J = 16.00 Hz, CH2N), 3.68 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 4.13 (t, 2H, J = 16.00 Hz, NHCH2), 7.21 (s, 2H, H-2 and H-6 Ar), 7.56 (s, 1H, thiazole-H), 8.30 (s, 1H, NHCH2, D2O exchangeable), 9.70 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 12.4 (CH2CH3), 37.8 (NHCH2CH2), 45.0 (m-OCH3), 52.1 (NHCH2CH2), 53.4 (CH2CH3), 56.4 (piperazine-C), 60.6 (p-OCH3), 103.7 (2,6-Ar-C), 107.3 (thiazole-C-5), 130.6 (1-Ar-C), 137.7 (4-Ar-C), 149.0 (thiazole-C-4), 153.5 (3,5-Ar-C), 157.6 (C=O), 158.7 (thiazole-C-2), MS (m/z, Rel. Abundance): 449.86 (M+, 13.79), 333.93 (100), Anal. Calcd for C21H31N5O4S calculated %: C, 56.10; H, 6.95; N, 15.58; S, 7.13. Found %: C, 56.13; H, 6.92; N, 15.61; S, 7.09.
4.1.10.2. 1-(2-Morpholinoethyl)-3-(4-(3,4,5-trimethoxyphenyl)thiazol-2-yl)urea (13b)
Brown crystals, mp 236–239 °C, yield (70%), IR (KBr, ν, cm–1): 3420, 3258 (N–H amide), 1655 (C=O amide), 1589 (C=C), 1235 (C–O), 1000 (C–O morpholine), 1H NMR (400 MHz, DMSO-d6): 3.20 (m, 4H, morpholine-H), 3.30 (m, 2H, CH2N), 3.50 (m, 4H, morpholine-H), 3.60 (t, 2H, J = 16.00 Hz, NHCH2), 3.69 (s, 3H, p-OCH3), 3.84 (s, 6H, m-OCH3), 7.21 (s, 2H, H-2 and H-6 Ar), 7.56 (s, 1H, thiazole-H), 8.50 (s, 1H, NHCH2, D2O exchangeable), 9.70 (s, 1H, NHCO, D2O exchangeable), 13C NMR (100 MHz, DMSO-d6): δ 53.4 (NHCH2CH2), 56.4 (NHCH2CH2), 58.6 (m-OCH3), 60.5 (p-OCH3), 60.9 (3,5-morpholine-C), 66.0 (2,6-morpholine-C), 103.7 (2,6-Ar-C), 107.3 (thiazole-C-5), 130.6 (1-Ar-C), 137.7 (4-Ar-C), 149 (thiazole-C-4), 153.5 (3,5-Ar-C), 158.3 (C=O), 158.7 (thiazole-C-2), MS (m/z, Rel. Abundance): 422.53 (M+, 31.40), 125.73 (100), Anal. Calcd for C19H26N4O5S calculated %: C, 54.01; H, 6.20; N, 13.26; O, 18.93; S, 7.59. Found %: C, 54.05; H, 6.23; N, 13.22; S, 7.61.
4.2. Biological Screening
4.2.1. Materials
The cell lines and the reference drug CA-4 were obtained from ATCC via the Holding Company for Biological Products and Vaccines (VACSERA), Cairo, Egypt. Fetal bovine serum was obtained from GIBCO, UK, and the other reagents such as RPMI-1640 medium, MTT, and dimethyl sulfoxide (DMSO) were obtained from Sigma Co., St. Louis, USA.
4.2.2. In Vitro Antitumor Evaluation (MTT Assay)
All the synthesized compounds (4–13) were evaluated for their inhibition of the growth of a panel of four different human cancer cell lines, HEPG-2, HCT-116, MCF-7, and HeLa in comparison with CA-4 as positive controls using the MTT assay.32,33 This colorimetric assay is based on the conversion of the yellow tetrazolium bromide (MTT) to a purple formazan derivative by mitochondrial succinate dehydrogenase in viable cells. Cell lines were cultured in RPMI-1640 medium with 10% fetal bovine serum. Antibiotics added were 100 units/mL penicillin and 100 μg/mL streptomycin at 37 °C in a 5% CO2 incubator. The cell lines were seeded in a 96-well plate at a density of 1.0 × 104 cells/well at 37 °C for 48 h under 5% CO2. After incubation, the cells were treated with different concentrations of compounds and incubated for 24 h. After 24 h of drug treatment, 20 μL of MTT solution at 5 mg/mL was added and incubated for 4 h. DMSO in a volume of 100 μL is added into each well to dissolve the purple formazan formed. Colorimetric assay is performed and recorded at an absorbance of 570 nm using a plate reader (EXL 800, USA). The cytotoxic activity was expressed as the concentration of the compound that caused 50% growth inhibition (IC50, mean ± SD).
4.2.3. In Vitro Tubulin Polymerization Inhibition Assay
To further characterize the interaction of the proposed compounds with the microtubule system, the selected 10 compounds including 5c, 6a,d,e, 7b,c, 8, 9a,b, and 13a were evaluated for their in vitro inhibition of tubulin polymerization, with CA-4 as the reference drug.
Standard polymerization reactions (minus tubulin ligands) were carried out as described in the polymerization protocol. Briefly, the standard polymerization reaction contains 100 μL volume of 4 mg/mL tubulin in 80 mM PIPES, pH 6.9, 0.5 mM EGTA, 2 mM MgCl2, and 1 mM GTP. Polymerization was started by incubation at 37 °C, followed by absorption readings at 340 nm. Under these conditions, polymerization will reach a maximal OD340 between 0.15 and 0.25 within 30 min. The three phases of polymerization are shown: I (nucleation), II (growth), and III (steady state). In this experimental setup (100 μL volume in a spectrophotometer with a 0.5 cm pathlength), OD340 of 0.1 is approximately equal to 1 mg/mL of polymer mass. Thus, under the conditions described, approximately 40% of tubulin is polymerized, leaving the flexibility for detecting the enhancers and inhibitors of polymerization. The tubulin polymerization inhibition activity for each compound was expressed as IC50 values, and the data were represented as mean ± SD from three independent experiments.
4.3. Docking Methodology
The coordinates of the target co-crystallized with colchicine were retrieved from the Protein Data Bank (PDB ID: 4O2B).45 All hydrogen atoms were added the crystal 3D structure of the protein with their standard geometry, followed by their energy minimization. The tested compounds and CA-4 were drawn into Marvin Sketch of Marvin suite (http://www.chemaxon.com) to generate the lowest energy conformer. Dock module of MOE (Molecular Operating Environment) version MOE 2019.0102,246 on a computer having Pentium 1.6 GHz workstation, 512 MB memory using windows operating system, was utilized in docking studies. Our tested compounds were docked into the rigid binding pocket, the active site of colchicine, of the protein using flexible ligand mode. From the ligand conformations, the placement phase generates poses. The free energy of binding of the ligand from a given pose is estimated using GBVI/WSA ΔG as a force field-based scoring function.47
Acknowledgments
Our honest appreciation to the Holding Company for Biological Products and Vaccines (VACSERA), Cairo, Egypt, for performing the biological screening.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c05077.
The authors declare no competing financial interest.
Supplementary Material
References
- Su Z.; Yang Z.; Xu Y.; Chen Y.; Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. 10.1186/s12943-015-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipiro J. T.; Talbert R. L.; Yee G. C.; Matzke G. R.; Wells B. G.; Posey L. M.. Pharmacotherapy: A Pathophysiologic Approach; Appleton and Lange: Connecticut, 2014; Vol. 4, pp 141–142. [Google Scholar]
- Bukowski K.; Kciuk M.; Kontek R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. 10.3390/ijms21093233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanpour S. H.; Dehghani M. Review of cancer from perspective of molecular. J. Cancer Res. Pract. 2017, 4, 127–129. 10.1016/j.jcrpr.2017.07.001. [DOI] [Google Scholar]
- Al-Suwaidan I. A.; Alanazi A. M.; Abdel-Aziz A.-M.; Mohamed M. A.; El-Azab A. S. Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: molecular docking study. Bioorg. Med. Chem. Lett. 2013, 23, 3935–3941. 10.1016/j.bmcl.2013.04.056. [DOI] [PubMed] [Google Scholar]
- Gümüş M.; Yakan M.; Koca İ. Recent advances of thiazole hybrids in biological applications. Future Med. Chem. 2019, 11, 1979–1998. 10.4155/fmc-2018-0196. [DOI] [PubMed] [Google Scholar]
- Edrees M. M.; Melha S. A.; Saad A. M.; Kheder N. A.; Gomha S. M.; Muhammad Z. A. Eco-friendly synthesis, characterization and biological evaluation of some novel pyrazolines containing thiazole moiety as potential anticancer and antimicrobial agents. Molecules 2018, 23, 2970. 10.3390/molecules23112970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu S.; Ghosh S. K.; Gahtori P.; Pratap Singh U. P.; Bhattacharyya D. R.; Bhat H. R. In silico ADMET study, docking, synthesis and antimalarial evaluation of thiazole-1,3,5-triazine derivatives as Pf-DHFR inhibitor. Pharmacol. Rep. 2019, 71, 762–767. 10.1016/j.pharep.2019.04.006. [DOI] [PubMed] [Google Scholar]
- de Oliveira Filho G. B.; Cardoso M. V.; Espíndola J. W. P.; Oliveira e Silva D. A. O.; Ferreira R. S.; Coelho P. L.; Anjos P. S.; Santos E.; Meira C. S.; Moreira D. R. M.; Soares M. B. P.; Leite A. C. L. Structural design, synthesis and pharmacological evaluation of thiazoles against Trypanosoma cruzi. Eur. J. Med. Chem. 2017, 141, 346–361. 10.1016/j.ejmech.2017.09.047. [DOI] [PubMed] [Google Scholar]
- Gagic Z.; Ruzic D.; Djokovic N.; Djikic T.; Nikolic K. In silico methods for design of kinase inhibitors as anticancer drugs. Front. Chem. 2020, 7, 873. 10.3389/fchem.2019.00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Siqueira L. R. P.; de Moraes Gomes P. A. T.; de Lima Ferreira L. P.; de Melo Rêgo M. J. B.; Leite A. C. L. Multi-target compounds acting in cancer progression: Focus on thiosemicarbazone, thiazole and thiazolidinone analogues. Eur. J. Med. Chem. 2019, 170, 237–260. 10.1016/j.ejmech.2019.03.024. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S.; May B. C.; Renslo A. R.; Prusiner S. B. Discovery of 2-aminothiazoles as potent antiprion compounds. J. Virol. 2010, 84, 3408–3412. 10.1128/JVI.02145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova A. S.; Yarovaya O. I.; Bormotov N. I.; Shishkina L. N.; Salakhutdinov N. F. Synthesis and antiviral activity of camphor-based 1,3-thiazolidin-4-one and thiazole derivatives as Orthopoxvirus-reproduction inhibitors. Med. Chem. Commun. 2018, 9, 1746–1753. 10.1039/C8MD00347E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic M.; Fesatidou M.; Xenikakis I.; Geronikaki A.; Angelova V. T.; Savic V.; Pasic M.; Krilovic B.; Djukic D.; Gobeljic B. In vitro antioxidant activity of thiazolidinone derivatives of 1,3-thiazole and 1,3,4-thiadiazole. Chem.-Biol. Interact. 2018, 286, 119–131. 10.1016/j.cbi.2018.03.013. [DOI] [PubMed] [Google Scholar]
- Iino T.; Tsukahara D.; Kamata K.; Sasaki K.; Ohyama S.; Hosaka H.; Hasegawa T.; Chiba M.; Nagata Y.; Eiki J.-i.; Nishimura T. Discovery of potent and orally active 3-alkoxy-5-phenoxy-N-thiazolyl benzamides as novel allosteric glucokinase activators. Bioorg. Med. Chem. 2009, 17, 2733–2743. 10.1016/j.bmc.2009.02.038. [DOI] [PubMed] [Google Scholar]
- Khatik G. L.; Datusalia A. K.; Ahsan W.; Kaur P.; Vyas M.; Mittal A.; Nayak S. K. A retrospect study on thiazole derivatives as the potential antidiabetic agents in drug discovery and developments. Curr. Drug Discovery Technol. 2018, 15, 163–177. 10.2174/1570163814666170915134018. [DOI] [PubMed] [Google Scholar]
- Giri R. S.; Thaker H. M.; Giordano T.; Williams J.; Rogers D.; Sudersanam V.; Vasu K. K. Design, synthesis and characterization of novel 2-(2,4-disubstituted-thiazole-5-yl)-3-aryl-3H-quinazoline-4-one derivatives as inhibitors of NF-κB and AP-1 mediated transcription activation and as potential anti-inflammatory agents. Eur. J. Med. Chem. 2009, 44, 2184–2189. 10.1016/j.ejmech.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Kumar G.; Singh N. Synthesis, anti-inflammatory and analgesic evaluation of thiazole/oxazole substituted benzothiazole derivatives. Bioorg. Chem. 2021, 107, 104608. 10.1016/j.bioorg.2020.104608. [DOI] [PubMed] [Google Scholar]
- Siddiqui N.; Ahsan W. Synthesis, anticonvulsant and toxicity screening of thiazolyl-thiadiazole derivatives. Med. Chem. Res. 2011, 20, 261–268. 10.1007/s00044-010-9313-6. [DOI] [Google Scholar]
- Chandra Sekhar K. V. G.; Rao V. S.; Deuther-Conrad W.; Sridhar D.; Nagesh H. N.; Kumar V. S.; Brust P.; Kumar M. M. K. Design, synthesis, and preliminary in vitro and in vivo pharmacological evaluation of 4-{4-[2-(4-(2-substitutedquinoxalin-3-yl)piperazin-1-yl)ethyl]phenyl}thiazoles as atypical antipsychotic agents. Med. Chem. Res. 2013, 22, 1660–1673. 10.1007/s00044-012-0164-1. [DOI] [Google Scholar]
- Omarx A. M. M.; Eshba N. H. Synthesis and Biological Evaluation of New 2,3-Dihydrothiazole Derivatives for Antimicrobial, Antihypertensive, and Anticonvulsant Activities. J. Pharm. Sci. 1984, 73, 1166–1168. 10.1002/jps.2600730837. [DOI] [PubMed] [Google Scholar]
- Franchetti P.; Cappellacci L.; Grifantini M.; Barzi A.; Nocentini G.; Yang H.; O’Connor A.; Jayaram H. N.; Carrell C.; Goldstein B. M. Furanfurin and thiophenfurin: two novel tiazofurin analogs. Synthesis, structure, antitumor activity, and interactions with inosine monophosphate dehydrogenase. J. Med. Chem. 1995, 38, 3829–3837. 10.1021/jm00019a013. [DOI] [PubMed] [Google Scholar]
- Abraham A. T.; Lin J.-J.; Newton D. L.; Rybak S.; Hecht S. M. RNA cleavage and inhibition of protein synthesis by bleomycin. Chem. Biol. 2003, 10, 45–52. 10.1016/S1074-5521(02)00306-X. [DOI] [PubMed] [Google Scholar]
- Saha S. K.; Gordan J. D.; Kleinstiver B. P.; Vu P.; Najem M. S.; Yeo J.-C.; Shi L.; Kato Y.; Levin R. S.; Webber J. T. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discovery 2016, 6, 727–739. 10.1158/2159-8290.CD-15-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouvier B.; Houssin R.; Bailly C.; Hénichart J. P. Synthesis and DNA-binding study of A thiazole-containing analog of netropsin. J. Heterocycl. Chem. 1989, 26, 1643–1647. 10.1002/jhet.5570260625. [DOI] [Google Scholar]
- Juric D.; Janku F.; Rodón J.; Burris H. A.; Mayer I. A.; Schuler M.; Seggewiss-Bernhardt R.; Gil-Martin M.; Middleton M. R.; Baselga J.; Bootle D.; Demanse D.; Blumenstein L.; Schumacher K.; Huang A.; Quadt C.; Rugo H. S. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer. JAMA Oncol. 2019, 5, e184475 10.1001/jamaoncol.2018.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Kim S. J.; Choi H.; Kim Y. H.; Lim I. T.; Yang H.-m.; Lee C. S.; Kang H. R.; Ahn S. K.; Moon S. K.; Kim D.-H.; Lee S.; Choi N. S.; Lee K. J. Identification of CKD-516: A Potent Tubulin Polymerization Inhibitor with Marked Antitumor Activity against Murine and Human Solid Tumors. J. Med. Chem. 2010, 53, 6337–6354. 10.1021/jm1002414. [DOI] [PubMed] [Google Scholar]
- Sun M.; Xu Q.; Xu J.; Wu Y.; Wang Y.; Zuo D.; Guan Q.; Bao K.; Wang J.; Wu Y.; Zhang W. Synthesis and bioevaluation of N,4-diaryl-1,3-thiazole-2-amines as tubulin inhibitors with potent antiproliferative activity. PLoS One 2017, 12, e0174006 10.1371/journal.pone.0174006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirla A.; Mann J. Combretastatins: from natural products to drug discovery. Nat. Prod. Rep. 2003, 20, 558–564. 10.1039/B306797C. [DOI] [PubMed] [Google Scholar]
- Marrelli M.; Conforti F.; Statti G. A.; Cachet X.; Michel S.; Tillequin F.; Menichini F. Biological potential and structure-activity relationships of most recently developed vascular disrupting agents: an overview of new derivatives of natural combretastatin A-4. Curr. Med. Chem. 2011, 18, 3035–3081. 10.2174/092986711796391642. [DOI] [PubMed] [Google Scholar]
- Nam N.-H.; Kim Y.; You Y.-J.; Hong D.-H.; Kim H.-M.; Ahn B.-Z. Combretoxazolones: synthesis, cytotoxicity and antitumor activity. Bioorg. Med. Chem. Lett. 2001, 11, 3073–3076. 10.1016/S0960-894X(01)00622-9. [DOI] [PubMed] [Google Scholar]
- Denizot F.; Lang R. Rapid colorimetric assay for cell growth and survival. J. Immunol. Methods 1986, 89, 271–277. 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Feng M.; Tang B.; Liang S. H.; Jiang X. Sulfur containing scaffolds in drugs: synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. 10.2174/1568026615666150915111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirimulla S.; Bailey J. B.; Vegesna R.; Narayan M. Halogen Interactions in Protein-Ligand Complexes: Implications of Halogen Bonding for Rational Drug Design. J. Chem. Inf. Model. 2013, 53, 2781–2791. 10.1021/ci400257k. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Schyman P. Treatment of halogen bonding in the OPLS-AA force field: application to potent anti-HIV agents. J. Chem. Theory Comput. 2012, 8, 3895–3901. 10.1021/ct300180w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Auffinger P.; Hays F. A.; Westhof E.; Ho P. S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 16789–16794. 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Shi T.; Wang Y.; Yang H.; Yan X.; Luo X.; Jiang H.; Zhu W. Halogen Bonding-A Novel Interaction for Rational Drug Design?. J. Med. Chem. 2009, 52, 2854–2862. 10.1021/jm9000133. [DOI] [PubMed] [Google Scholar]
- Tron G. C.; Pirali T.; Sorba G.; Pagliai F.; Busacca S.; Genazzani A. A. Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 2006, 49, 3033–3044. 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- Nam N. H. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr. Med. Chem. 2003, 10, 1697–1722. 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- Aprile S.; Del Grosso E.; Tron G. C.; Grosa G. In vitro metabolism study of combretastatin A-4 in rat and human liver microsomes. Drug Metab. Dispos. 2007, 35, 2252–2261. 10.1124/dmd.107.016998. [DOI] [PubMed] [Google Scholar]
- Lawrence N. J.; Patterson R. P.; Ooi L.-L.; Cook D.; Ducki S. Effects of α-substitutions on structure and biological activity of anticancer chalcones. Bioorg. Med. Chem. Lett. 2006, 16, 5844–5848. 10.1016/j.bmcl.2006.08.065. [DOI] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Brancale A.; Ricci A.; Hamel E.; Bortolozzi R.; Basso G.; Viola G. Convergent Synthesis and Biological Evaluation of 2-Amino-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl Thiazoles as Microtubule Targeting Agents. J. Med. Chem. 2011, 54, 5144–5153. 10.1021/jm200392p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prota A. E.; Danel F.; Bachmann F.; Bargsten K.; Buey R. M.; Pohlmann J.; Reinelt S.; Lane H.; Steinmetz M. O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. 10.1016/j.jmb.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE) version 2019.0102; Chemical Computing Group Inc.: 1010 Sherbooke St. West, Suite# 910, Montreal, 2019. [Google Scholar]
- Labute P. The generalized Born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. 10.1002/jcc.20933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.