

Abstract
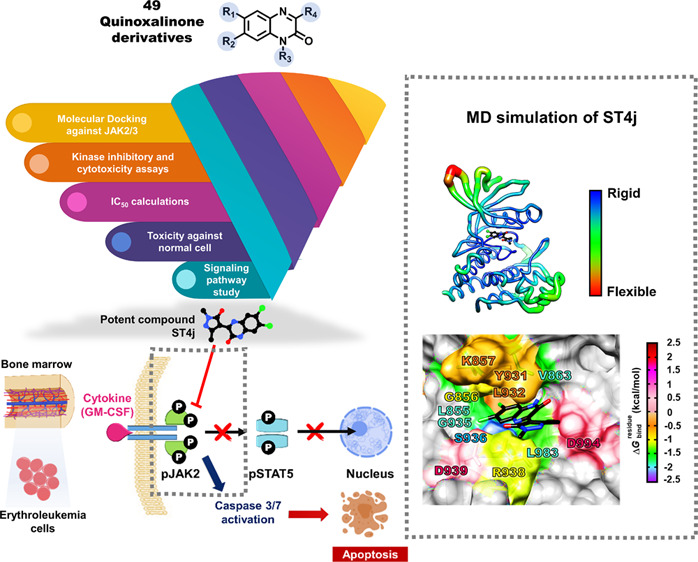
Janus kinases (JAKs) are involved in a wide variety of cell signaling associated with T-cell and B-cell mediated diseases. The pathogenesis of common lymphoid-derived diseases and leukemia cancer has been implicated in JAK2 and JAK3. Therefore, to decrease the risk of these diseases, targeting this pathway using JAK2/3 inhibitors could serve as a valuable research tool. Herein, we used a combination of the computational and biological approaches to identify the quinoxalinone-based dual inhibitors of JAK2/3. First, an in-house library of 49 quinoxalinones was screened by molecular docking. Then, the inhibitory activities of 17 screened compounds against both JAKs as well as against two human erythroleukemia cell lines, TF1 and HEL were examined. The obtained results revealed that several quinoxalinones could potentially inhibit JAK2/3, and among them, ST4j showed strong inhibition against JAKs with the IC50 values of 13.00 ± 1.31 nM for JAK2 and 14.86 ± 1.29 nM for JAK3, which are better than ruxolitinib and tofacitinib. In addition, ST4j potentially inhibited TF1 cells (IC50 of 15.53 ± 0.82 μM) and HEL cells (IC50 of 17.90 ± 1.36 μM), similar to both tofacitinib ruxolitinib. Mechanistically, ST4j inhibited JAK2 autophosphorylation and induced cell apoptosis in dose- and time-dependent manners. From molecular dynamics simulations, ST4j was mainly stabilized by van der Waals interactions, and its hydroxyl group could form hydrogen bonds in the hinge region at residues S936 and R938 of JAK2. This research highlights the potential of ST4j to be a novel therapeutic agent for the treatment of lymphoid-derived diseases and leukemia cancer.
1. Introduction
Janus kinases (JAKs, Figure 1) are intracellular tyrosine kinases that bind to cell surface receptors through cytokine receptors which are involved in cell growth, survival, development, and cell differentiation.1 In addition, signal transducer and activator of transcription (STAT) proteins are eventually phosphorylated and transported to the nucleus to trigger target gene transcription. In several immune-related diseases and cancers, aberrant JAK-STAT signaling is observed.2 Inhibitions of JAKs involved in cytokine-mediating signaling as secondary messengers are important for the prevention or regulation of the aforementioned diseases. The JAKs family consists of four types including JAK1, JAK2, JAK3, and TYK2.1 Among JAKs, JAK2 is involved in multiple cytokine pathways, for example, IL-6-mediated signaling as well as a granulocyte-macrophage colony-stimulating factor (GM-CSF), which are critical for different physiological processes, such as bone metabolism, hematopoiesis, and differentiation of B-cells.3,4 The mutation of JAK2 (V617F) has been shown to cause hematological malignancies, particularly in chronic myeloproliferative neoplasms.5 JAK3 also plays a crucial role in the production of lymphoid through the IL-2 pathway, which controls the function of various populations of lymphoid cells, including T and B lymphocytes and natural killer (NK) cells. The dysfunction of JAK3 (R925S and Q988P) leads to acute lymphoblastic leukemia (T-ALL) and lymphoid-derived disease pathogenesis.6,7 The development of dual JAK2/3 inhibitors may therefore be an important remedy for reducing the number of lymphoid-derived diseases that are dependent on the signaling cascade of JAK2 and JAK3.8
Figure 1.
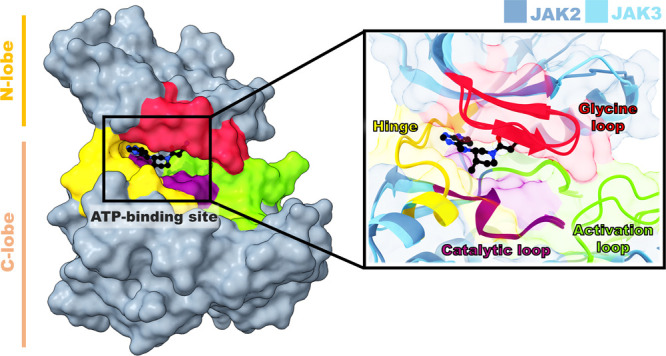
Superimposition structures of JAK2 and JAK3 complexed with tofacitinib.
A dual JAK2/3 inhibitor, AG-490, in clinical trials effectively inhibits unregulated development of B-cells in patients with acute lymphoblastic leukemia by inhibiting the irregular constitutive activation of JAK2.9 In addition, this inhibitor also prevents phytohemagglutinin (PHA) or antigen-specific-activated human T cells from IL-2-mediated cell growth by inactivating the signaling cascade for JAK3 and STAT5.10 The FDA approved drugs tofacitinib and ruxolitinib have been used for rheumatoid arthritis and myeloproliferative treatments, respectively.11 Tofacitinib has also been reported as a potent inhibitor against JAK1/2/3 (IC50 = 1.7–3.7 nM for JAK1, 1.8–4.1 nM for JAK2 and 0.75–1.6 nM for JAK3).12−15 In addition, ruxolitinib is an effective inhibitor for JAK1 (IC50 = 0.09 nM) and JAK2 (IC50 = 0.036 nM).16 Apart from drugs that can inhibit JAKs, other molecules such as quinazoline, quinoxalinones, indoles, and pyrazolones have been reported as inhibitors of JAKs.8,17−19 Quinoxalinones possess a wide range of biological activities such as anticancer, antibacterial, and anti-inflammatory activities.20,21 JAKs inhibitors derived from quinoxalinone derivatives have been reported. Two new designs of JAK2 inhibitor derived from scaffold morphing including amino benzoxazole scaffold (IC50 = 12 nM) and 6,6-fused heterocyclic 2,8-diaryl-quinoxaline (IC50 = 12 nM) have been investigated by enzymatic assay.8,22 In addition, they reported that the 2-amino substituent and ring as well as the backbone carbonyl group of amino benzoxazole scaffold and the nitrogen atom in the quinoxaline ring form a hydrogen bond with L932 in the hinge region of JAK2.8,22 Moreover, inhibitors derived from dimethoxy and amino-phenol quinazoline derivatives (WHI-P131) showed potent JAK3 (IC50 = 9 μM) kinase inhibition.17 Other interesting JAKs inhibitors are indoles and pyrazolone. The pyrazolone derivative of pyrimidine analogues can inhibit the activity of JAK2 (IC50 = 45.3 nM) as well as HEL cell growth with JAK2-overexpressing human erythroleukemia (IC50 = 12.2 μM).23 The indol-4-carboxamide core showed JAK2 inhibition (IC50 = 18 nM).19 Sulfoximines have emerged as new and valuable isosteres of the sulfur-containing functional groups, such as proline-rich tyrosine kinase 2 (PYK2) and Brc-Abl tyrosine kinase, which are anticancer.24,25
In this work, quinoxalinone derivatives including NH-sulfoximine, indole, and pyrazolone groups (Figure 2),26−28 which have never been reported on JAKs inhibition were selected. First, the quinoxalinone compounds (49) were screened for JAK2/3 inhibition by molecular docking. Subsequently, the screened compounds were tested for kinase inhibition and cytotoxicity in the human erythroleukemia cell lines expressing JAK2, TF1 (WT), and HEL (V617F). The potent compound was investigated for the inhibition of JAK2 phosphorylation in the TF1 cell using Western blot analysis. Dose- and time-dependent apoptosis was determined using flow cytometric analysis. Finally, the molecular interactions of the potent compound with JAK2 were studied by molecular dynamics simulations and free energy calculation.
Figure 2.

2D structure of quinoxalinone derivatives (49).
2. Results and Discussion
2.1. Compound Screening by Molecular Docking
A total of 49 quinoxalinone derivatives consisting of three groups including sulfoximines, indoles, and pyrazolones (Figure 2) were used to screen the potent compounds toward JAK2/3 by molecular docking (Figures 3 and S1). Our results showed that the binding energies of quinoxalinone derivatives between JAK2 and JAK3 were similar (Figure 3A). Based on the criteria of binding energies lower than the known drugs (tofacitinib and ruxolitinib), 29 compounds showed good binding efficiency against JAK2, while 17 compounds exhibited good binding efficiency against JAK3 (Figure 3B). In addition, we found that 17 compounds could likely inhibit both JAK2 and JAK3. Therefore, these compounds were selected for further analyses.
Figure 3.

(A) Docking results of quinoxalinone derivatives toward JAK2/3 derived from FlexX docking. (B) Summary of screened compounds toward JAK2/3.
2.2. Janus Kinase 2/3 Inhibitory Activity
The 17 screened compounds from in silico screening were tested in vitro for kinase inhibition ability toward JAK2/3 (Figure 4). All 17 compounds with derivatives of indoles (MN series) and pyrazolones (ST series) can inhibit both JAKs, in which several compounds could inhibit both JAKs similar to the known drugs, tofacitinib, and ruxolitinib. The four compounds (MN390, MN341P, ST4j, and ST3i) were selected by the top 25% of the ability to inhibit JAK2/3 and were used for the measurement of IC50 values (Figures 5A and S2). In JAK2 inhibition, the MN341P compound (20.19 ± 1.71 nM) gave an IC50 value close to MN390 (19.77 ± 1.24 nM), while ST3i (28.50 ± 0.70 nM) gave the highest IC50 value. Moreover, it was found that these three compounds displayed a similar inhibition toward JAK3 (∼20 to 28 nM). Interestingly, the ST4j compound showed the lowest IC50 value for both JAKs (13.00 ± 1.31 nM for JAK2 and 14.86 ± 1.29 nM for JAK3), similar to the previous report demonstrating that the 2,8-diaryl-quinoxalines consisting of 4-phenylacetic morpholine amide can inhibit JAK2 with an IC50 value of 13 nM.29 Compared with the known drug, ST4j inhibits JAK2 at a similar level to ruxolitinib (14.50 ± 0.90 nM) but significantly lower than tofacitinib by 2-fold (29.09 ± 0.30 nM). In addition, the IC50 for JAK3 inhibition by ST4j (14.86 ± 1.29 nM) is significantly lower than tofacitinib (20.86 ± 0.81 nM). Altogether, these results suggested that ST4j could be a potent compound targeting JAK2 and JAK3.
Figure 4.

Janus kinase inhibitory activity screening of quinoxalinone derivatives at 1 μM concentrations. * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001 vs tofacitinib, #p ≤ 0.05, ##p ≤ 0.01 and ###p ≤ 0.001 vs ruxolitinib.
Figure 5.

In vitro study of focused compounds toward JAK2/3 by kinase assay and cell-based assay. (A) In vitro IC50 values of the potent quinoxalinone derivatives (MN341P, MN390, ST3i, and ST4j) and drugs (tofacitinib and ruxolitinib) toward JAK2/3 (ND = not detected), the IC50 value of ST4j compound and drugs toward (B) TF1 and HEL, (C) Vero and HepG2 cells, and (D) Western blot analysis in TF1 cells after treatment with ST4j and drugs at various concentrations. * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001 vs tofacitinib, #p ≤ 0.05 and ###p ≤ 0.001 vs ruxolitinib.
2.3. Cytotoxicity
The 17 screened compounds were also tested for cytotoxicity at 10 μM toward the wild-type (TF1) and mutant (HEL) forms of JAK2-overexpressing human erythroleukemia cells (Figure S3). Among them, MN341P, MN390, ST3i, and ST4j were susceptible to mutant HEL cells, which is consistent with the JAK2 inhibitory activity results (Figure 4). In addition, almost all compounds from computational screening (except ST4j) were more susceptible to mutant cells than to wild-type cells. This is because the V617F mutation near the hinge region maintains its open conformation of the activation loop (A loop), resulting in a higher occupancy of the ligand.30 The ST4j (IC50 = 15.53 ± 0.82 μM) was found to be a potent compound against the TF1 cells significantly better than tofacitinib (IC50 = 25.13 ± 0.68 μM) by ∼1-fold but lower than ruxolitinib (IC50 = 10.66 ± 0.88 μM). In the case of HEL cells, ST4j (17.90 ± 1.36 μM) showed cell growth inhibition at a similar level to both drugs, tofacitinib (23.28 ± 1.55 μM), and ruxolitinib (19.28 ± 0.64 μM) (Figure S4). ST4j, a pyrazolone derivative, can inhibit the HEL cell growth with an IC50 value of 17.90 ± 1.36 μM, similar to the reported pyrazolone derivative of pyrimidine analogues (IC50 = 12.2 μM).23
Several quinoxaline analogues have been reported to inhibit leukemia cells growth such as pyrrolo[1,2-a]quinoxaline carboxylate that can inhibit many types of myeloid and lymphoid leukemia cells growths (K652, chronic myelogenous leukemia IC50 = 12 μM; HL60 acute promyelocytic leukemia IC50 = 24 μM and Jurkat acute T cell leukemia IC50 = 5 μM).31 In addition, isoindolo[2,1-a]quinoxaline inhibited Jurkat cell growth with an IC50 of 12 μM.32 Although we used cell lines different from the previous reports mentioned above, quinoxaline analogues have been proved to inhibit the growth of leukemia cells with JAK2 expression. Therefore, the ST4j compound could be a good compound for inhibitions of TF1 as well as HEL cells.
Furthermore, the ST4j compound was selected to investigate the toxicity toward Vero (monkey kidney epithelial cells) and HepG2 (liver hepatocellular cells) cells (Figures 5C and S4). The quinoxaline-1,4-di-N-oxide derivative in the previous report showed low Vero cell growth inhibition (IC50 > 100 μM).33 Consistent with this finding, we found that ST4j was low toxic to normal Vero cells (IC50 of >100 μM in Vero cells and of >50 μM in HepG2 cells, similar to both known drugs (Figures 5C and S4). The results suggested that ST4j specifically inhibited human erythroleukemia cells with low toxicity to the normal cells.
2.4. Modulation of JAK2 Signaling Pathways by ST4j
To investigate whether the JAK2 autophosphorylation could be inhibited by ST4j, the phosphorylation status of JAK2 and its downstream proteins were determined in TF1 cells using Western blotting. As shown in Figure 5D, we found that ruxolitinib at the IC50 value completely abolished phosphorylation of JAK2 (Y1007/1008) as well as STAT5 (Y694). Previous studies have shown that quinoxalinone-containing compounds, including quinoxaline pyrazole morpholine dihydrochloride derivative (NVP-BSK805)34 and imidazoquinoline derivative,35 can inhibit JAK2 and STAT5 pathways in many kinds of cancers. Corresponding with these reports, we found that ST4j at IC25, IC50, and IC75 inhibits phosphorylation of JAK2 as well as STAT5. Therefore, this finding indicated that ST4j inhibits human erythroleukemia TF1 cell growth via the JAK2/STAT5 signaling pathway.
2.5. Apoptosis Induction by ST4j
To explore the apoptosis mechanism of TF1 cells induced by ST4j, we performed flow cytometric analysis at 24, 48, and 72 h (Figures 6A,B and S5–S6). The flip out of phosphatidylserine on the cell surface and the activation of caspase contribute to the production of signaling cascades responsible for apoptotic processes.36,37 In the annexin V assay, after a 24-h incubation (Figures 6A and S5), the total apoptotic cells were increased by ∼30% for ST4j and ∼35% for both drugs, tofacitinib, and ruxolitinib treatment. At 72-h incubation, the apoptotic cell populations of ST4j treated cells were quite stable at ∼40%, whereas those of drug-treated cells were increased by ∼50%. Our results showed that apoptotic cell populations from annexin V of ST4j and drug treatments in TF1 cells were increased in a time-dependent manner, which corresponds with results from the caspase 3/7 activation (Figures 6B and S5).
Figure 6.

Flow cytometric analysis of time-dependent induced-apoptotic TF1 cells treated with IC50 values of ST4j compound and drugs (tofacitinib and ruxolitinib) for 24, 48, and 72 h, respectively. Representative figures showing the population of living, apoptosis, and dead cells of (A) annexin V, (B) caspase3/7, and (C) proposed mechanisms of ST4j treatment in TF1 cells, in which ST4j promotes cell death via apoptosis by the inhibition of pJAK2.
Caspase 3/7 is an executioner caspase in apoptosis due to its role in coordinating the destruction of cellular structures.38 The total number of apoptotic cells of ST4j treatment was ∼1% at 24-h incubation. After 48- and 72-h incubations, the apoptotic cells were increased by ∼40% and ∼70%, respectively. In addition, the apoptotic cells of drugs, tofacitinib, and ruxolitinib at 24-h incubations were ∼20%. Afterward, the total number of apoptotic cells at 48- and 72-h incubations increased ∼2-fold (∼40%) and ∼2-fold (∼50%), respectively, from 24-h incubations. These findings indicate that ST4j induced cytotoxicity via apoptosis in a time-dependent manner.
We further examined the dose-dependent effect at IC25, IC50, and IC75 of ST4j in comparison with drugs at 24-h incubation on TF1 cells by annexin V (Figure S6). In positive control conditions, tofacitinib and ruxolitinib treatment, the percentage of cell apoptosis was increased in a dose-dependent manner. Supportively, several previous works showed that ruxolitinib induced apoptotic cell death in dose-dependent effect.39−41 For ST4j treatment, the total apoptosis was increased by ∼15%, 20%, and 30% after treatment with IC25, IC50, and IC75, respectively. Our research implies that ST4j induced cytotoxicity via apoptosis in a dose-dependent manner.
Altogether, Figure 6C shows the proposed mechanisms of ST4j toward the TF1 cell line. It is represented that ST4j treatment inhibited phosphorylation of JAK2, resulting in decreased phosphorylation of STAT5. This leads to caspase 3/7 activation and apoptosis induction in dose- and time-dependent manners. Similarly, previous studies demonstrated that quinoxaline as pyrazole morpholine dihydrochloride derivative (NVP-BSK805) induced apoptosis in SET-2 cells, a human megakaryoblastic cell line.34
2.6. Key Binding Affinity
Because ST4j showed potent inhibition toward JAK2 from kinase inhibition and a cell-based assay, the key binding affinity of ST4j against JAK2 was assessed. The binding affinity between ST4j and JAK2 was investigated in comparison with JAK2/ruxolitinib complex by three independent MD simulations for 500 ns. The ΔGbind,residue calculations based on the MM/GBSA method were performed on the 100 snapshots obtained from the last 100 ns of all simulations. Note that each simulation system including number of hydrogen bonds and atom contacts showed quite similar phenomena (Figures S7–S8). The complexation of three JAK2/ST4j systems (∼1–6) demonstrated the higher number of hydrogen bonds than JAK2/ruxolitinib (∼1–4). Therefore, only one simulation (run1 from JAK2/ruxolitinib and run2 from JAK2/ST4j systems) of energy per residue and compound binding mode inside the binding pocket is illustrated in Figure 7.
Figure 7.

(A) Per-residue decomposition free energy (ΔGbindresidue), the van der Waals (vdW) and electrostatic energy contributions of the domain of JAK2 for the binding of tofacitinib and ST4j. (B) the binding orientation of ruxolitinib and ST4j within the binding pocket drawn from the last MD snapshot. The lowest and highest energies are colored from purple to dark red, respectively.
The hydrophobic residues L855, V863, Y931, L932, and L983 of JAK2 could interact with ruxolitinib, while L855, G856, G935, S936, V863, and L983 of JAK2 could interact with ST4j, which exhibited an energy contribution of less than −1.0 kcal/mol (Figure 7A,B). Interestingly, the complexes between JAK2/ruxolitinib and JAK2/ST4j were strongly stabilized by two regions as follows: (i) G loop, L855 and V863 residues; and (ii) catalytic loop, L983 residue. The docking orientation of the quinoxaline derivative of benzoxazole in the previous report formed a hydrophobic interaction with V863 in the G loop, which corresponds with this result.42 The Cl derivative in the quinoxalinone ring of ST4j interacted with residue G935 at the hinge region (green, −1.63 kcal/mol). In drug design, the incorporation of halogen atoms in lead candidates is often found, since they are highly electronegative and reactive atoms that could interact well within the binding pocket of the enzyme.43 For ruxolitinib binding with JAK2, the cyclopentyl showed destabilization with K882. However, the pyrrolopyrimidine and pyrazole of ruxolitinib were braced between the hydrophobic residues, A880, M929, G856, V911, and S862, while the cyanide group points to G993 in the C lobe. Besides ruxolitinib, tofacitinib which consists of pyrrolopyrimidine core was also stabilized within the JAK2 pocket by hydrophobic interactions with Y931 and L932 residues, similar to JAK2/ruxolitinib in this work.44 However, tofacitinib inhibited JAK3 higher than JAK2 since the methyl side chain of the piperidine ring protrudes to the C909 residue, effectively enhancing the hydrophobicity of the JAK3 binding site compared with the other isotypes where the serine residue occupies this position (JAK1; S963, JAK2; S936 and TYK2; S985).16,44
Apart from hydrophobic interactions, electrostatic interactions are also important for ST4j and ruxolitinib binding. The S936 and R938 residues at the tail hinge region of JAK2 also played an important role for stabilizing JAK2/ST4j. The strong electrostatic contributions for ruxolitinib binding with JAK2 were observed at E930 at the deep hinge region and D994 residue at the A loop. Our results showed that ruxolitinib and ST4j were stabilized within JAK2 in the hinge region. However, the drug binds to the hinge region deeper than ST4j. For energy contributions (Figure 7A), the vdW interactions of ruxolitinib and ST4j binding at the conserved regions were predominant rather than electrostatic interactions.
2.7. Ligand-Protein Hydrogen Bonding and Mobility of Protein
One of the intermolecular attractions for determining the ligand’s binding strength within the protein is the hydrogen bond. In this study, the hydrogen bond occupations of JAK2/ruxolitinib (run 1) and JAK2/ST4j (run 2) complexes from the last 100 ns simulation were performed (Figure 8A). Our results showed that hydrogen bond formation of ruxolitinib with the E930 (99.99%) and L932 (90.45%), located in the hinge region, provide strong stabilization. This result corresponds with the hydrogen bonding of the pyrrolopyrimidine ring in ruxolitinib from Glide docking16 as well as tofacitinib from MD simulations and with both residues in JAK2.44 ST4j formed hydrogen bonds with residues S936 (51.34%) and R938 (72.74%) at the hinge region. In addition, this compound could form a hydrogen bond with K857 (20.45%) at the G loop. In the previous report, the amide group of 2,4-dihydroxyphenyl-isonicotinohydrazide quinazoline (3b) formed a hydrogen bond with K857 of JAK2.45 The ruxolitinib forms hydrogen bonds with JAK2 at the deep hinge region, while hydrogen bond formation of JAK2/ST4j was displayed at the edge of the hinge region opposite in the G loop. Although no hydrogen bonds are shown in the same amino acid position between ruxolitinib and ST4j, low mobility of ST4j within the JAK2′s ATP-binding pocket was observed (Figure 8B), suggesting that ST4j was stabilized well within the JAK2, similar to ruxolitinib. Therefore, the quinoxalinone analogue ST4j could be used as a JAK2 inhibitor.
Figure 8.
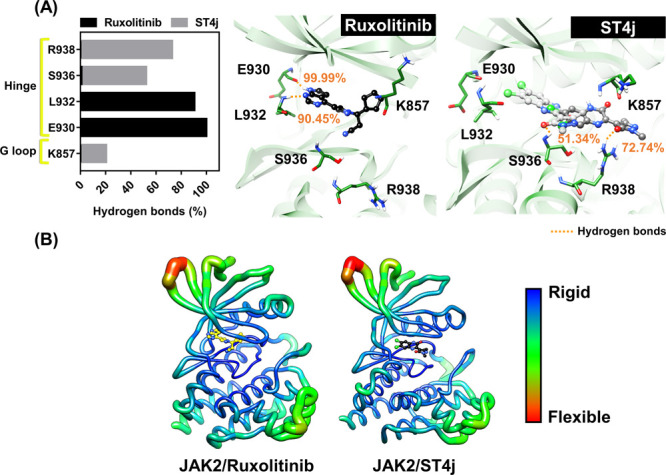
(A) Percentage of hydrogen bond occupation of ruxolitinib and ST4j within the JAK2 binding pocket. Note that the hydrogen bond with value >50% was selected to represent in 3D and (B) B factor of JAK2, the flexible and rigid regions are ranged from blue to green and red, respectively. The data were derived from the last 100 ns of the one simulation of JAK2 with ruxolitinib (run 1) and ST4j (run 2).
3. Conclusions
The molecular docking approach was used to screen for the quinoxalinone compounds that could inhibit JAK2/3. We obtained the quinoxalinone with derivatives of pyrazolone and chlorine atom (ST4j) as a novel inhibitor of JAK2/3. This compound showed strong inhibition against JAK2/3 in the kinase assay. The ST4j showed inhibition of TF1 (IC50 of 15.53 ± 0.82 μM) and HEL (IC50 of 17.90 ± 1.36 μM) cell growths, similar to the commercial drugs tofacitinib and ruxolitinib. Furthermore, ST4j treatment in TF1 cells, which overexpress JAK2, strongly inhibited JAK2 phosphorylation and the subsequent STAT5 phosphorylation, and induced cell death via apoptosis in dose and time-dependent manners. From MD simulations, the core quinoxalinone of ST4j occupied hydrophobic regions such as L855, V863, and L932 and were stabilized by hydrogen bonds with S936 and R938 residues. Hence, the ST4j compounds could serve as a promising lead for the development of anticancer drug targeting JAK2/3.
4. Material and Methods
Human erythroleukemia TF1 (ATCC CRL-2003) and HEL 92.1.7 (ATCC TIB-180) cell lines, human liver cancer HepG2 cell line (ATCC HB-8065) and monkey (Cercopithecus aethiops) kidney Vero cell line (ATCC CCL-81) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Primary antibodies against JAK2 (ab108596), p-JAK2 (ab32101; Y1007/1008) were purchased from Abcam. Primary antibody of p-STAT5 (#9351; Y694), β-actin (#4970) and secondary antibody, antirabbit IgG, HRP-linked (#7074) were purchased from Cell Signaling Technology. Poly(glu-tyr) Peptide(P61–58) was purchased from SignalChem Biotech (Canada). Muse annexin V and dead cell kit (MCH100105) and caspase-3/7 kit (MCH100108) were purchased from Merk (Germany). Quinoxalinones were synthesized and obtained from Associate Professor Dr. Sirilata Yotphan, Department of Chemistry, Faculty of Science, Mahidol University.26−28
4.1. Experimental Methods
4.1.1. Kinase Inhibitory Activity
The inhibitions of kinase toward JAK2/3 (Sigma-Aldrich: SRP0171, SRP0173) of screened compounds were measured by using the ADP-Glo Kinase Assay Kit (Promega).46,47 The reaction consists of 2.5 ng/μL of JAK2/3, screened compounds at 1 μM, 5 μM ATP and 2 ng/μLpoly(glu·tyr) in a buffer (40 mM Tris–HCI pH 7.5, 20 mM MgCl2, and 0.1 mg/mL BSA). The mixture was incubated for 1 h at room temperature. The ADP-Glo reagent was then added to 5 μL and incubated for 40 min. After that, 10 μL of kinase detection reagent was added and incubated at room temperature for 30 min. Finally, the ATP product’s luminescence analysis was carried using a microplate spectrophotometer (Synergy HTX Multi-Mode Reader, BioTek). The compound inhibition (%) was measured in comparison to the control of the reaction without an inhibitor. The IC50 inhibitions of compounds were assayed by 2-fold dilution.
4.1.2. Cell Cultures
The TF1 cell was grown in RPMI (Roswell Park Memorial Institute medium) supplemented with 2 ng/mL GM-CSF (Granulocyte-macrophage colony-stimulating factor), 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin, and HEL cell was grown in RPMI supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. HepG2 and Vero cells were grown in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. All cells were maintained at 37 °C in a humidified 5% CO2 atmosphere.
4.1.3. Cytotoxicity
Cells were seeded into 96-well plates and were incubated overnight. The density of cells was used including 50 000 cells/well for TF1; 25 000 cells/well for HEL; 7000 cells/well for HepG2 and 2000 cells/well for Vero. After that, cells were treated with screened compounds at serial 2-fold dilutions for 72 h. The TF1 and HEL cells which are suspension cells were assessed using the PrestoBlue assay; 10 μL of resazurin solution was added and incubated for 1 h. Both HepG2 and Vero cells, which are adherent cells, were assessed using the MTT assay; 100 μL of MTT solution (5 mg/mL) was added and then incubated for 3 h. The medium was removed, and 50 μL of DMSO was added subsequently. Finally, the resorufin of the PrestoBlue assay and formazan of the MTT assay product absorbance measurement was performed at 570 nm using a microplate spectrophotometer.48,49 The vehicle control was treated without inhibitor and the reaction contains 0.2% DMSO of all wells.
4.1.4. Apoptosis Analysis
The TF1 cell treatments with inhibitors in (i) various times (24, 48, and 72 h) at the IC50 value and (ii) various concentrations (IC25, IC50 and IC75) at 24 h were gently trypsinized for 5 min. Then, the cells were centrifuged at 1000 rpm for 5 min. The pelleted cells were collected. Then, the pelleted were resuspended and incubated for 25 min at room temperature in the dark with annexin V and dead cell reagent.
In addition, the TF1 cell treatments with an IC50 value of inhibitor at various times including 24, 48, and 72 h at the IC50 value was performed to assess apoptotic cells by caspase3/7. After trypsinization and centrifugation in the method above, the pellet was removed. Pellets were resuspended and incubated with 5 μL of caspase 3/7 reagent. Reactions were then incubated at 37 °C for 30 min and then mixed with 7-aminoactinomycin D (7-ADD). Finally, the number of apoptosis cells (%) of annexin V and caspase3/7 methods were analyzed by flow cytometry using cell analyzer MUSE (Merck-MiIIipore, MA).
4.1.5. Immunoblotting
The TF1 cells in RPMI containing 0.1% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at a density of 300 000 cells/well were seeded into a 24-well plate and were incubated overnight. Then, cells were treated with the designated compounds for 2 h (IC25, IC50, and IC75 values for ST4j compound and IC50 value for ruxolitinib). Subsequently, the treated cells were stimulated for 20 min with 2 ng/mL of GM-CSF, and the cells were collected by centrifugation at 1000 rpm for 5 min. The pelleted cells were rinsed twice with cold PBS with 1 mM sodium orthovanadate by centrifugation at 1000 rpm for 5 min. Homogenized pelleted cells by adding the RIPA buffer containing a protease inhibitor and incubated on ice for 1 h. Cells were sonicated for 10 min and centrifuged for 20 min. The supernatants were collected, and the protein concentration was determined by the Bradford assay.50 Total protein (20 μg) were separated on 12% SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked with 3% BSA in TBST (10 mm Tris–HCl, pH 8, 150 mM NaCl, and 0.1% Tween 20) for 1 h. Subsequently, primary antibodies in 5% TBST including β-actin, JAK2, pJAK2, and pSTAT5 were incubated at 4 °C overnight. After incubation, the membrane was washed twice for 5 min with the TBST buffer and incubated for 1 h at room temperature with the secondary antirabbit IgG antibody. The immunoreactive bands were detected using the ECL kit (Amersham, Little Chalfont, UK).
4.1.6. Statistical Analysis
Data from three independent experiments which were evaluated using GraphPad Prism 7.0 software were expressed by mean ± standard error of mean (SEM). For statistical analysis, a one-way and two-way analysis of variance (ANOVA) in which Tukey’s multiple comparisons test. p ≤ 0.05 was chosen as the significance level for all analyses performed.
4.2. Computational Methods
4.2.1. Molecular Docking
Tofacitinib complexes with JAK2 (code 3FUP) and JAK3 (code 3LXK) were downloaded from Protein Data Bank.12,51 For protein preparations, all missing residues 920–923 of JAK2 were built using the SWISS-MODEL server.52 The protonation states were predicted by ionizable amino acids at physiological pH 7.4 using PROPKA3.1.53 For ligand preparations, the 3D structures of known drugs, ruxolitinib and tofacitinib, were downloaded from the ZINC database54 and in-house database quinoxalinones were generated from the Gaussian09 program.55 The protonation state of ligands was characterized by ChemAxon.56
Tofacitinib was defined in both JAKs as the center of the active site in the FlexX docking ATP-binding pocket.57 The docking parameter was used as 12 Å of sphere radius, 100 independent runs, Gasteiger–Marsili formalism atomic charges.58 The results of docking were visualized for binding patterns by the UCSF Chimera package59 and Accelrys Discovery Studio 2019 (Accelrys Inc., San Diego, CA, USA).60
4.2.2. Molecular Dynamics Simulations
All-atom MD simulations with three different initial velocities including ruxolitinib/JAK2 and focused compound/JAK2 were simulated using periodic boundary condition with the isothermal–isobaric ensemble (NPT) at a constant pressure of 1 atm equilibrated at 310 K in AMBER16 through pmemd CUDA.61 The force fields of JAK2 and inhibitors were FF14SB62 and GAFF2,63 respectively. The optimization of inhibitors in the HF/6-31g(d) level was adopted as previous studies44,64−68 by the Gaussian09 program.55 Consequently, the electrostatic potential (ESP) charges and restrained ESP (RESP) were computed in AMBER16. All systems were solvated in explicit water using the TIP3P model. For nonbonded interactions, the cutoff was 12 Å, while long-range electrostatic interactions were applied by Ewald’s method.69 All covalent bonds involving hydrogen atoms were constrained by the SHAKE algorithm.70 The temperature was controlled by a Langevin thermostat with a collision frequency of 2.0 ps. Finally, the unrestrained NPT simulation was simulated for 500 ns, which were recorded the MD trajectories every 500 steps. The system analysis was performed by intermolecular HB occupation, atom contacts, and B factor using the CPPTRAJ module.71 Besides, decomposition free energy (ΔGbindresidue) was calculated by the MM/PBSA.py module.
Acknowledgments
This research is funded by Thailand Science Research and Innovation Fund Chulalongkorn University (CU) (CUFRB65_hea(69)_132_23_62) (Fundamental Fund 2565, CU for T.R.; National Research council of Thailand (NRCT5-RSA63002-07) and Kasetsart University Research and Development Institutes (KURDI (FF(KU)6.64)) for K.C. K.S. thanks the Science Achievement Scholarship of Thailand. The authors express their gratitude to the Research Unit for Natural Product Biotechnology, Faculty of Pharmaceutical Sciences, Chulalongkorn University and Prof. Wanchai De-Eknamkul for providing FlexX software and technical assistance with LeadIT.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04769.
Supplemental figures: S1. Superimposition of tofacitinib toward JAK2/3 between X-ray structure and FlexX docking generation; S2. The IC50 curve of potent compounds (MN341P, MN390, ST3i, and ST4j) and drugs (tofacitinib and ruxolitinib) toward JAK2/3; S3. Cellular cytotoxicity assay of TF1 and HEL cells treated with quinoxalinone derivatives at 10 μM concentrations; S4. Cell viability after treatment for 72 h of cell lines; S5. Flow cytometry analysis of time-dependent induced-apoptotic TF1 cells treated with IC50 values of ST4j compound and drugs (tofacitinib and ruxolitinib) for 24, 48, and 72 h, respectively; S6. Flow cytometry analysis from annexin V of dose-dependent induced-apoptotic TF1 cells treated with various concentrations (IC25, IC50 and IC75 values) of the ST4j compound and drugs (tofacitinib and ruxolitinib) for 24 h; S7. The number of hydrogen bonds formed b,etween JAK2 and ruxolitinib/ST4j along 500 ns of simulation; S8. The number of atom contacts between JAK2 and ruxolitinib/ST4j along 500 ns of simulation (PDF)
Author Contributions
T.R. and K.C. conceived and designed the experiments. K.S. conducted theoretical and experimental studies. T.K. and S.Y. synthesized all compounds. T.R., K.C., K.S., P.M., L.T., S.S., T.K., and S. Y. analyzed the data. K.S. and P.M. wrote the original manuscript. All authors reviewed and edited the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ghoreschi K.; Laurence A.; O’Shea J. J. Janus kinases in immune cell signaling. Immunol Rev. 2009, 228, 273–287. 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea J. J.; Schwartz D. M.; Villarino A. V.; Gadina M.; McInnes I. B.; Laurence A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. 10.1146/annurev-med-051113-024537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich P. C.; Behrmann I.; Muller-Newen G.; Schaper F.; Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y. F.; Lew A. M.; Chopin M. The Pleiotropic Effects of the GM-CSF Rheostat on Myeloid Cell Differentiation and Function: More Than a Numbers Game. Front Immunol 2019, 10, 2679. 10.3389/fimmu.2019.02679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A.; Kantarjian H.; Cortes J.; Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat. Rev. Drug Discov 2011, 10 (2), 127–140. 10.1038/nrd3264. [DOI] [PubMed] [Google Scholar]
- Smith K. A. Interleukin-2: inception, impact, and implications. Science 1988, 240 (4856), 1169–76. 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- Hammaren H. M.; Virtanen A. T.; Raivola J.; Silvennoinen O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. 10.1016/j.cyto.2018.03.041. [DOI] [PubMed] [Google Scholar]
- Menet C. J.; Van Rompaey L.; Geney R. Advances in the Discovery of Selective JAK Inhibitors. Prog. Med. Chem 2013, 52, 153–223. 10.1016/B978-0-444-62652-3.00004-1. [DOI] [PubMed] [Google Scholar]
- Vainchenker W.; Leroy E.; Gilles L.; Marty C.; Plo I.; Constantinescu S. N. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res. 2018, 7, 82. 10.12688/f1000research.13167.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirken R. A.; Erwin R. A.; Taub D.; Murphy W. J.; Behbod F.; Wang L. H.; Pericle F.; Farrar W. L. Tyrphostin AG-490 inhibits cytokine-mediated JAK3/STAT5a/b signal transduction and cellular proliferation of antigen-activated human T cells. J. Leukocyte Biol. 1999, 65 (6), 891–899. 10.1002/jlb.65.6.891. [DOI] [PubMed] [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36 (7), 422–439. 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Chrencik J. E.; Patny A.; Leung I. K.; Korniski B.; Emmons T. L.; Hall T.; Weinberg R. A.; Gormley J. A.; Williams J. M.; Day J. E.; Hirsch J. L.; Kiefer J. R.; Leone J. W.; Fischer H. D.; Sommers C. D.; Huang H. C.; Jacobsen E. J.; Tenbrink R. E.; Tomasselli A. G.; Benson T. E. Structural and Thermodynamic Characterization of the TYK2 and JAK3 Kinase Domains in Complex with CP-690550 and CMP-6. J. Mol. Biol. 2010, 400 (3), 413–433. 10.1016/j.jmb.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Boluda J. C. H.; Gomez M.; Perez A. JAK2 inhibitors. Med. Clin. 2016, 147 (2), 70–75. 10.1016/j.medcli.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Changelian P. S.; Flanagan M. E.; Ball D. J.; Kent C. R.; Magnuson K. S.; Martin W. H.; Rizzuti B. J.; Sawyer P. S.; Perry B. D.; Brissette W. H.; McCurdy S. P.; Kudlacz E. M.; Conklyn M. J.; Elliott E. A.; Koslov E. R.; Fisher M. B.; Strelevitz T. J.; Yoon K.; Whipple D. A.; Sun J. M.; Munchhof M. J.; Doty J. L.; Casavant J. M.; Blumenkopf T. A.; Hines M.; Brown M. F.; Lillie B. M.; Subramanyam C.; Shang-Poa C.; Milici A. J.; Beckius G. E.; Moyer J. D.; Su C. Y.; Woodworth T. G.; Gaweco A. S.; Beals C. R.; Littman B. H.; Fisher D. A.; Smith J. F.; Zagouras P.; Magna H. A.; Saltarelli M. J.; Johnson K. S.; Nelms L. F.; Des Etages S. G.; Hayes L. S.; Kawabata T. T.; Finco-Kent D.; Baker D. L.; Larson M.; Si M. S.; Paniagua R.; Higgins J.; Holm B.; Reitz B.; Zhou Y. J.; Morris R. E.; O’Shea J. J.; Borie D. C. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003, 302 (5646), 875–878. 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- Meyer D. M.; Jesson M. I.; Li X. O.; Elrick M. M.; Funckes-Shippy C. L.; Warner J. D.; Gross C. J.; Dowty M. E.; Ramaiah S. K.; Hirsch J. L.; Saabye M. J.; Barks J. L.; Kishore N.; Morris D. L. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J. Inflamm 2010, 7, 41. 10.1186/1476-9255-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. 10.1016/j.phrs.2016.07.038. [DOI] [PubMed] [Google Scholar]
- Wilson L. J. Recent patents in the discovery of small molecule inhibitors of JAK3. Expert Opin Ther Pat 2010, 20 (5), 609–623. 10.1517/13543771003709767. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Chen C. J.; Yu R. N.; Shu L.; Zhang T. T.; Zhang D. Y. Discovery of novel selective Janus kinase 2 (JAK2) inhibitors bearing a 1H-pyrazolo[3,4-d]pyrimidin-4-amino scaffold. Bioorg. Med. Chem. 2019, 27 (8), 1562–1576. 10.1016/j.bmc.2019.02.054. [DOI] [PubMed] [Google Scholar]
- Lim J.; Taoka B.; Otte R. D.; Spencer K.; Dinsmore C. J.; Altman M. D.; Chan G.; Rosenstein C.; Sharma S.; Su H. P.; Szewczak A. A.; Xu L.; Yin H.; Zugay-Murphy J.; Marshall C. G.; Young J. R. Discovery of 1-amino-5H-pyrido[4,3-b]indol-4-carboxamide inhibitors of Janus kinase 2 (JAK2) for the treatment of myeloproliferative disorders. J. Med. Chem. 2011, 54 (20), 7334–49. 10.1021/jm200909u. [DOI] [PubMed] [Google Scholar]
- El Newahie A. M. S.; Ismail N. S. M.; Abou El Ella D. A.; Abouzid K. A. M. Quinoxaline-Based Scaffolds Targeting Tyrosine Kinases and Their Potential Anticancer Activity. Arch Pharm. 2016, 349 (5), 309–326. 10.1002/ardp.201500468. [DOI] [PubMed] [Google Scholar]
- Tariq S.; Somakala K.; Amir M. Quinoxaline: An insight into the recent pharmacological advances. Eur. J. Med. Chem. 2018, 143, 542–557. 10.1016/j.ejmech.2017.11.064. [DOI] [PubMed] [Google Scholar]
- Furet P.; Gerspacher M.; Pissot-Soldermann C. Design of two new chemotypes for inhibiting the Janus kinase 2 by scaffold morphing. Bioorg. Med. Chem. Lett. 2010, 20 (6), 1858–1860. 10.1016/j.bmcl.2010.01.151. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Chen C. J.; Yu R. N.; Shu L.; Zhang T. T.; Zhang D. Y. Discovery of novel selective Janus kinase 2 (JAK2) inhibitors bearing a 1H-pyrazolo[3,4-d]pyrimidin-4-amino scaffold. Bioorgan Med. Chem. 2019, 27 (8), 1562–1576. 10.1016/j.bmc.2019.02.054. [DOI] [PubMed] [Google Scholar]
- Mader P.; Kattner L. Sulfoximines as Rising Stars in Modern Drug Discovery? Current Status and Perspective on an Emerging Functional Group in Medicinal Chemistry. J. Med. Chem. 2020, 63 (23), 14243–14275. 10.1021/acs.jmedchem.0c00960. [DOI] [PubMed] [Google Scholar]
- Sirvent J. A.; Lucking U. Novel Pieces for the Emerging Picture of Sulfoximines in Drug Discovery: Synthesis and Evaluation of Sulfoximine Analogues of Marketed Drugs and Advanced Clinical Candidates. Chemmedchem 2017, 12 (7), 487–501. 10.1002/cmdc.201700044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonchue S.; Sumunnee L.; Phomphrai K.; Yotphan S. Metal-free direct oxidative C-C bond coupling of pyrazolones and quinoxalinones. Org. Chem. Front 2018, 5 (12), 1928–1932. 10.1039/C8QO00328A. [DOI] [Google Scholar]
- Sumunnee L.; Pimpasri C.; Noikham M.; Yotphan S. Persulfate-promoted oxidative C-N bond coupling of quinoxalinones and NH-sulfoximines. Org. Biomol Chem. 2018, 16 (15), 2697–2704. 10.1039/C8OB00375K. [DOI] [PubMed] [Google Scholar]
- Noikham M.; Kittikool T.; Yotphan S. Iodine-Catalyzed Oxidative Cross-Dehydrogenative Coupling of Quinoxalinones and Indoles: Synthesis of 3-(Indol-2-yl)quinoxalin-2-one under Mild and Ambient Conditions. Synthesis-Stuttgart 2018, 50 (12), 2337–2346. 10.1055/s-0037-1609445. [DOI] [Google Scholar]
- Pissot-Soldermann C.; Gerspacher M.; Furet P.; Gaul C.; Holzer P.; McCarthy C.; Radimerski T.; Regnier C. H.; Baffert F.; Drueckes P.; Tavares G. A.; Vangrevelinghe E.; Blasco F.; Ottaviani G.; Ossola F.; Scesa J.; Reetz J. Discovery and SAR of potent, orally available 2,8-diaryl-quinoxalines as a new class of JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20 (8), 2609–2613. 10.1016/j.bmcl.2010.02.056. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O.; Hubbard S. R. Molecular insights into regulation of JAK2 in myeloproliferative neoplasms. Blood 2015, 125 (22), 3388–3392. 10.1182/blood-2015-01-621110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplat V.; Vincenzi M.; Lucas R.; Moreau S.; Savrimoutou S.; Pinaud N.; Lesbordes J.; Peyrilles E.; Marchivie M.; Routier S.; Sonnet P.; Rossi F.; Ronga L.; Guillon J. Synthesis and evaluation of the cytotoxic activity of novel ethyl 4-[4-(4-substitutedpiperidin-1-yl)]benzyl-phenylpyrrolo[1,2-a]quinoxaline-carboxy late derivatives in myeloid and lymphoid leukemia cell lines. Eur. J. Med. Chem. 2016, 113, 214–27. 10.1016/j.ejmech.2016.02.047. [DOI] [PubMed] [Google Scholar]
- Desplat V.; Moreau S.; Belisle-Fabre S.; Thiolat D.; Uranga J.; Lucas R.; de Moor L.; Massip S.; Jarry C.; Mossalayi D. M.; Sonnet P.; Deleris G.; Guillon J. Synthesis and evaluation of the antiproliferative activity of novel isoindolo[2,1-a]quinoxaline and indolo[1,2-a]quinoxaline derivatives. J. Enzyme Inhib Med. Chem. 2011, 26 (5), 657–667. 10.3109/14756366.2010.548326. [DOI] [PubMed] [Google Scholar]
- Pan Y. H.; Li P. P.; Xie S. Y.; Tao Y. F.; Chen D. M.; Dai M. H.; Hao H. H.; Huang L. L.; Wang Y. L.; Wang L. Y.; Liu Z. L.; Yuan Z. H. Synthesis, 3D-QSAR analysis and biological evaluation of quinoxaline 1,4-di-N-oxide derivatives as antituberculosis agents. Bioorg. Med. Chem. Lett. 2016, 26 (16), 4146–4153. 10.1016/j.bmcl.2016.01.066. [DOI] [PubMed] [Google Scholar]
- Baffert F.; Regnier C. H.; De Pover A.; Pissot-Soldermann C.; Tavares G. A.; Blasco F.; Brueggen J.; Chene P.; Drueckes P.; Erdmann D.; Furet P.; Gerspacher M.; Lang M.; Ledieu D.; Nolan L.; Ruetz S.; Trappe J.; Vangrevelinghe E.; Wartmann M.; Wyder L.; Hofmann F.; Radimerski T. Potent and Selective Inhibition of Polycythemia by the Quinoxaline JAK2 Inhibitor NVP-BSK805. Mol. Cancer Ther 2010, 9 (7), 1945–1955. 10.1158/1535-7163.MCT-10-0053. [DOI] [PubMed] [Google Scholar]
- Britschgi A.; Andraos R.; Brinkhaus H.; Klebba I.; Romanet V.; Muller U.; Murakami M.; Radimerski T.; Bentires-Alj M. JAK2/STAT5 Inhibition Circumvents Resistance to PI3K/mTOR Blockade: A Rationale for Cotargeting These Pathways in Metastatic Breast Cancer. Cancer Cell 2012, 22 (6), 796–811. 10.1016/j.ccr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- McIlwain D. R.; Berger T.; Mak T. W. Caspase Functions in Cell Death and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5 (4), a008656. 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu G.; Chen X. Y. Apoptosis Imaging: Beyond Annexin V. J. Nucl. Med. 2010, 51 (11), 1659–1662. 10.2967/jnumed.110.078584. [DOI] [PubMed] [Google Scholar]
- Walsh J. G.; Cullen S. P.; Sheridan C.; Luthi A. U.; Gerner C.; Martin S. J. Executioner caspase-3 and caspase-7 are functionally distinct proteases. P Natl. Acad. Sci. USA 2008, 105 (35), 12815–12819. 10.1073/pnas.0707715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Neto J. A.; de Melo Campos P.; Favaro P.; Lazarini M.; da Silva Santos Duarte A.; Lorand-Metze I.; Costa F. F.; Olalla Saad S. T.; Traina F. Stathmin 1 inhibition amplifies ruxolitinib-induced apoptosis in JAK2V617F cells. Oncotarget 2015, 6 (30), 29573–29584. 10.18632/oncotarget.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo Campos P.; Machado-Neto J. A.; Eide C. A.; Savage S. L.; Scopim-Ribeiro R.; da Silva Souza Duarte A.; Favaro P.; Lorand-Metze I.; Costa F. F.; Tognon C. E.; Druker B. J.; Saad S. T. O.; Traina F. IRS2 silencing increases apoptosis and potentiates the effects of ruxolitinib in JAK2(V617F)-positive myeloproliferative neoplasms. Oncotarget 2016, 7 (6), 6948–6959. 10.18632/oncotarget.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima K.; Carlos J. A. E. G.; Alves-Paiva R. D.; Vicari H. P.; Santos F. P. D.; Hamerschlak N.; Costa-Lotufo L. V.; Traina F.; Machado-Neto J. A. Reversine exhibits antineoplastic activity in JAK2(V)(617)(F)-positive myeloproliferative neoplasms. Sci. Rep-Uk 2019, 9, 9895. 10.1038/s41598-019-46163-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin R.; Majumder A.; Sayeski P. P. The Recent Medicinal Chemistry Development of Jak2 Tyrosine Kinase Small Molecule Inhibitors. Curr. Med. Chem. 2010, 17 (36), 4551–4558. 10.2174/092986710794182953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandes M. Z.; Cavalcanti S. M. T.; Moreira D. R. M.; de Azevedo W. F.; Leite A. C. L. Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11 (3), 303–314. 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- Sanachai K.; Mahalapbutr P.; Choowongkomon K.; Poo-Arporn R. P.; Wolschann P.; Rungrotmongkol T. Insights into the Binding Recognition and Susceptibility of Tofacitinib toward Janus Kinases. ACS Omega 2020, 5 (1), 369–377. 10.1021/acsomega.9b02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggana S. J.; Paturi M. C.; Perka H.; Reddy G. D.; Prasad V. V. S. R. Novel 2,4-disubstituted quinazolines as cytotoxic agents and JAK2 inhibitors: Synthesis, in vitro evaluation and molecular dynamics studies. Comput. Biol. Chem. 2019, 79, 110–118. 10.1016/j.compbiolchem.2019.01.008. [DOI] [PubMed] [Google Scholar]
- Seetaha S.; Ratanabanyong S.; Choowongkomon K. Expression, purification, and characterization of the native intracellular domain of human epidermal growth factor receptors 1 and 2 in Escherichia coli. Appl. Microbiol. Biotechnol. 2019, 103 (20), 8427–8438. 10.1007/s00253-019-10116-6. [DOI] [PubMed] [Google Scholar]
- Seetaha S.; Boonyarit B.; Tongsima S.; Songtawee N.; Choowongkomon K. Potential tripeptides against the tyrosine kinase domain of human epidermal growth factor receptor (HER) 2 through computational and kinase assay approaches. J. Mol. Graph Model 2020, 97, 107564. 10.1016/j.jmgm.2020.107564. [DOI] [PubMed] [Google Scholar]
- Sangpheak K.; Tabtimmai L.; Seetaha S.; Rungnim C.; Chavasiri W.; Wolschann P.; Choowongkomon K.; Rungrotmongkol T. Biological Evaluation and Molecular Dynamics Simulation of Chalcone Derivatives as Epidermal Growth Factor-Tyrosine Kinase Inhibitors. Molecules 2019, 24 (6), 1092. 10.3390/molecules24061092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obounchoey P.; Tabtimmai L.; Suphakun P.; Thongkhao K.; Eurtivong C.; Gleeson M. P.; Choowongkomon K. In silico identification and in vitro validation of nogalamycin N-oxide (NSC116555) as a potent anticancer compound against non-small-cell lung cancer cells. J. Cell Biochem 2019, 120 (3), 3353–3361. 10.1002/jcb.27605. [DOI] [PubMed] [Google Scholar]
- Cheng Y. F.; Wei H. M.; Sun R.; Tian Z. G.; Zheng X. D. Rapid method for protein quantitation by Bradford assay after elimination of the interference of polysorbate 80. Anal. Biochem. 2016, 494, 37–39. 10.1016/j.ab.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Williams N. K.; Bamert R. S.; Patel O.; Wang C.; Walden P. M.; Wilks A. F.; Fantino E.; Rossjohn J.; Lucet I. S. Dissecting Specificity in the Janus Kinases: The Structures of JAK-Specific Inhibitors Complexed to the JAK1 and JAK2 Protein Tyrosine Kinase Domains. J. Mol. Biol. 2009, 387 (1), 219–232. 10.1016/j.jmb.2009.01.041. [DOI] [PubMed] [Google Scholar]
- Waterhouse A.; Bertoni M.; Bienert S.; Studer G.; Tauriello G.; Gumienny R.; Heer F. T.; de Beer T. A. P.; Rempfer C.; Bordoli L.; Lepore R.; Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46 (W1), W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostkowski M.; Olsson M. H. M.; Sondergaard C. R.; Jensen J. H. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 2011, 11, 6. 10.1186/1472-6807-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC - A free database of commercially available compounds for virtual screening. J. Chem. Inf Model 2005, 45 (1), 177–182. 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA. 2009.
- Marvin 17.21.0; Marvin was used for drawing, displaying ,and characterizing chemical structures, substructures, and reactions; ChemAxon: Budapest, Hungary, 2017. (https://www.chemaxon.com).
- Rarey M.; Kramer B.; Lengauer T.; Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261 (3), 470–489. 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- Shulga D. A.; Oliferenko A. A.; Pisarev S. A.; Palyulin V. A.; Zefirov N. S. Fast tools for calculation of atomic charges well suited for drug design. Sar Qsar Environ. Res. 2008, 19 (1–2), 153–165. 10.1080/10629360701844142. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- BIOVIA; Dassault Systèmes, discovery studio visualizer software, version 2019 client; Dassault Systèmes: San Diego, 2019. (http://www.accelrys.com).
- Gotz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput 2012, 8 (5), 1542–1555. 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. M.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25 (9), 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Sanachai K.; Mahalapbutr P.; Sanghiran Lee V.; Rungrotmongkol T.; Hannongbua S. In Silico Elucidation of Potent Inhibitors and Rational Drug Design against SARS-CoV-2 Papain-like Protease. J. Phys. Chem. B 2021, 125 (50), 13644–13656. 10.1021/acs.jpcb.1c07060. [DOI] [PubMed] [Google Scholar]
- Mahalapbutr P.; Lee V. S.; Rungrotmongkol T. Binding Hotspot and Activation Mechanism of Maltitol and Lactitol toward the Human Sweet Taste Receptor. J. Agric. Food Chem. 2020, 68 (30), 7974–7983. 10.1021/acs.jafc.0c02580. [DOI] [PubMed] [Google Scholar]
- Mahalapbutr P.; Sangkhawasi M.; Kammarabutr J.; Chamni S.; Rungrotmongkol T. Rosmarinic Acid as a Potent Influenza Neuraminidase Inhibitor: In Vitro and In Silico Study. Curr. Top Med. Chem. 2020, 20 (23), 2046–2055. 10.2174/1568026619666191118110155. [DOI] [PubMed] [Google Scholar]
- Somboon T.; Mahalapbutr P.; Sanachai K.; Maitarad P.; Lee V. S.; Hannongbua S.; Rungrotmongkol T. Computational study on peptidomimetic inhibitors against SARS-CoV-2 main protease. J. Mol. Liq. 2021, 322, 114999. 10.1016/j.molliq.2020.114999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalapbutr P.; Kongtaworn N.; Rungrotmongkol T. Structural insight into the recognition of S-adenosyl-L-homocysteine and sinefungin in SARS-CoV-2 Nsp16/Nsp10 RNA cap 2′-O-Methyltransferase. Comput. Struct Biotechnol J. 2020, 18, 2757–2765. 10.1016/j.csbj.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York D. M.; Darden T. A.; Pedersen L. G. The Effect of Long-Range Electrostatic Interactions in Simulations of Macromolecular Crystals - a Comparison of the Ewald and Truncated List Methods. J. Chem. Phys. 1993, 99 (10), 8345–8348. 10.1063/1.465608. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18 (12), 1463–1472. . [DOI] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9 (7), 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.