

Abstract
Zika virus (ZIKV) infection is one of the mosquito-borne flaviviruses of human importance with more than 2 million suspected cases and more than 1 million people infected in about 30 countries. There are reported inhibitors of the zika virus replication machinery, but no approved effective antiviral therapy including vaccines directed against the virus for treatment or prevention is currently available. The study investigated the chemoinformatic design and profiling of derivatives of dasabuvir, efavirenz, and tipranavir as potential inhibitors of the zika virus RNA-dependent RNA polymerase (RdRP) and/or methyltransferase (MTase). The three-dimensional (3D) coordinates of dasabuvir, efavirenz, and tipranavir were obtained from the PubChem database, and their respective derivatives were designed with DataWarrior-5.2.1 using an evolutionary algorithm. Derivatives that were not mutagenic, tumorigenic, or irritant were selected; docked into RdRP and MTase; and further subjected to absorption, distribution, metabolism, excretion, and toxicity (ADMET) evaluation with Swiss-ADME and pkCSM web tools. Some of the designed compounds are Lipinski’s rule-of-five compliant, with good synthetic accessibilities. Compounds 20d, 21d, 22d, and 1e are nontoxic with the only limitation of CYP1A2, CYP2C19, and/or CYP2C9 inhibition. Replacements of −CH3 and −NH– in the methanesulfonamide moiety of dasabuvir with −OH and −CH2– or −CH2CH2–, respectively, improved the safety/toxicity profile. Hepatotoxicity in 5d, 4d, and 18d is likely due to −NH– in their methanesulfonamide/sulfamic acid moieties. These compounds are potent inhibitors of N-7 and 2′-methylation activities of ZIKV methyltransferase and/or RNA synthesis through interactions with amino acid residues in the priming loop/“N-pocket” in the virus RdRP. Synthesis of these compounds and wet laboratory validation against ZIKV are recommended.
Introduction
Zika virus (ZIKV) is an arbovirus of the Flaviviridae family and flavivirus genus, primarily transmitted by the bite of infected Aedes spp. mosquitoes.1 ZIKV infection is one of the mosquito-borne flaviviruses of human importance2 first identified in 1947 in Uganda,3 and more than 2 million suspected cases have been reported4 with more than 1 million people infected in about 30 countries.5 The virus has resonated with global concern that the World Health Organization (WHO) on February 1, 2016 declared it “a public health emergency of international concern”.6 Also, a potential pandemic spread of nonvector-borne ZIKV transmission has been reported elsewhere.7 Again, the zika virus can cause several neurological complications, including microcephaly in newborns and Guillain–Barre syndrome in adults.8 There are conspiracy theories, controversies, and other pseudoscientific claims about ZIKV. A 2019 report showed belief in genetically modified mosquitoes as the cause of ZIKV,9,10 and the first genetically modified mosquitoes were released in 2021.11 The controversies, especially the controversial tissue research, could hamper essential research on ZIKV, like probing the link between the virus and birth defects12 including drug discovery. There are reported inhibitors of the zika virus replication/protein synthesis machinery including amodiaquine, hydroxychloroquine, dasabuvir, efavirenz, and tipranavir,2,13−17 but no approved effective antiviral therapy (drugs or vaccines) directed against the virus for treatment (or prevention) is currently available.2,18,19 Therefore, the discovery of novel antivirals against ZIKV is a research priority. To address this urgent medical need, we designed and profiled derivatives of dasabuvir, efavirenz, and tipranavir as potential inhibitors of the zika virus NS5 protein (RNA-dependent RNA polymerase (RdRP) and methyltransferase (MTase)). This is because the three approved drugs (dasabuvir, efavirenz, and tipranavir) have been reported to inhibit the replication of the zika virus.2 Again, they have shown some unfavorable therapeutic outcomes. For example, dasabuvir and tipranavir have been associated with mutations in hepatitis C NS5B polymerase and human immunodeficiency virus (HIV) protease, respectively, leading to resistance.20−22 Increased damage indexes and frequencies have been observed in the brain of mice treated with efavirenz,23 and WHO guidelines do not recommend efavirenz use during the first trimester of pregnancy due to concern for teratogenicity.24 Therefore, there is a need to discover/identify more potent and nontoxic/safer derivatives of the parent compounds.
The NS5 protein is the largest and most conserved flaviviral protein with an RNA-dependent RNA polymerase (RdRP) domain at its C-terminal end, which is vital for viral replication.25,26 Zika virus NS5 protein also has an N-terminal methyltransferase (MTase) domain that plays a critical role in viral RNA genome capping13,26 and interacts with the NS5-polymerase domain during RNA synthesis.5S-Adenosylmethionine (SAM) MTase substrate binding sites have been implicated in zika virus methyltransferase inhibition with sinefungin as its analogue.27 Specifically, the MTase domain of the the zika virus NS5 protein catalyzes methylation of the RNA 5′-cap structure to generate N7-methyl-guanosine and 2′-O-methyl adenosine (GpppA-RNA → m7GpppAm-RNA) using SAM as a methyl group donor. The NS5 protein has been validated as a promising antiviral drug target.5,26 Taken together, inhibition of both RNA-dependent RNA polymerase and methyltransferase activities of the zika virus will halt viral replication and appears a promising drug target option. Therefore, the study is aimed at chemoinformatic design and profiling of derivatives of dasabuvir, efavirenz, and tipranavir as potential inhibitors of the zika virus RNA-dependent RNA polymerase and/or methyltransferase. There are several computational techniques for drug discovery against diseases like the zika virus. They include, but are not limited to, virtual ligand screening, molecular docking, and molecular dynamics simulations including free energy and steered molecular dynamics simulations.28,29
Methods
Description of Data Sets
The data sets used for this study are dasabuvir (PubChem identifier: CID 56640146), efavirenz (PubChem identifier: CID 64139), and tipranavir (PubChem identifier: CID 54682461) and were obtained from the PubChem database.30 The drugs have been reported as inhibitors of zika virus replication at micromolar concentrations2 with selective toxicities in Vero, UKF-NB-4, and HBCA cells.2 Again, dasabuvir, efavirenz, and tipranavir have been reported to have some unfavorable therapeutic outcomes as mentioned earlier.
In Silico Design of the Hypothetical Compounds
The three-dimensional (3D) coordinates of dasabuvir (PubChem identifier: CID 56640146), efavirenz (PubChem identifier: CID 64139), and tipranavir (PubChem identifier: CID 54682461) were obtained from the PubChem database30 in the Structure Data File (SDF) format and used for the design of their respective derivatives. Briefly, approved druglike derivatives of dasabuvir, efavirenz, and tipranavir (Supporting Information 1) were designed with DataWarrior-5.2.131 using an evolutionary algorithm, SkelSphere descriptor, automatic number of generations, fitness criteria of structural (dis)similarity, 128 compounds per generation, and eight (8) compounds survival per generation. The number of heavy atoms, calculated logP (cLog P), and other pharmacokinetic/toxicity (e.g., mutagenic, tumorigenic, irritant) properties were calculated. The designed compounds that were not mutagenic, tumorigenic, and irritants (Supporting Information 1) were identified after extracting the calculated parameters, including their Simplified Molecular Input Line Entry System (SMILES) with a shell script (Supporting Information 2), and used for further studies.
Predocking Preparations of Designed Compounds and Target Proteins
The designed compounds were saved in the SDF format in the DataWarrior-5.2.1 platform. The files were separated into their compounds using Open Babel.32 Designed compounds that were not mutagenic, tumorigenic, and irritant (Supporting Information 1) were obtained and prepared for docking simulations using shell script (Code S2 in Supporting Information 2). The script utilized obminimize32 and General Amber Force Field (GAFF), which have parameters for most organic and pharmaceutical molecules.33 The compounds were subjected to 2500 steps of geometry optimization using the steepest descent algorithm before generating the PDBQT (Protein Data Bank, Partial Charge (Q), and Atom Type (T)) file format for docking simulation studies. Zika virus NS5-RNA-dependent RNA polymerase (PDB ID: 5U04) and NS5-methyltransferase (PDB ID: 5MRK) were used for the study. Their 3D atomic coordinates were obtained from Protein Data Bank34 and prepared for molecular docking simulations as previously reported, which involved the deletion of duplicate chains, heteromolecules, water molecules, and the addition of polar hydrogen.2
Validation of Docking Protocols
To substantiate our evidence, the experimental crystal complex of NS5-methyltransferase with sinefungin was reproduced in silico as previously reported2 using AutoDockVina.35 The sinefungin obtained in a complex with NS5-methyltransferase from the protein data bank was removed, optimized, and redocked with NS5-methyltransferase using the grid box sizes (13 × 18 × 15) and centers (18.216 × 7.699 × 4.793), xyz coordinates, at 1.0 Å grid space and 8 exhaustiveness, as reported in a previous study.2 Also, blind docking and redocking in the case of RNA-dependent RNA polymerase with sizes (12 × 16 × 16) and centers (25.036 × 68.817 × 103.577) at 1.0 Å grid space and 8 exhaustiveness were performed, as previously reported.2
Molecular Docking Simulations
The prepared designed compounds that were not mutagenic, tumorigenic, and irritant were docked into the zika virus NS5 protein (RNA-dependent RNA polymerase and methyltransferase) using AutoDockVina35 after validation of molecular docking protocols or blind docking and redocking simulations. Specifically, the compounds were docked into the priming loop/allosteric pocket (“N-pocket”) site16,36 in RNA-dependent RNA polymerase and substrate (S-adenosylmethionine) binding site in methyltransferase with docking parameters mentioned in the Validation of Docking Protocols section. Additionally, dasabuvir, efavirenz, and tipranavir, which were previously reported as inhibitors of flavivirus/zika virus replication in cell culture,2 were also docked into the zika virus NS5 protein as the control/standard. The simulations were performed locally on a Linux platform using a configuration file and script from a previous study2 containing information on the grid box centers and sizes of prepared receptors (RNA-dependent RNA polymerase and methyltransferase) and the designed compounds. The binding energies (BEs) (kcal/mol) (n = 4) from the molecular docking simulations were calculated and expressed as mean ± standard deviation. Designed compounds with better binding energies for RNA-dependent RNA polymerase and/or methyltransferase than their respective parent compounds (dasabuvir, efavirenz, and tipranavir) were identified, and their binding poses were visualized with Maestro, version 12.8, Schrödinger, LLC, New York, NY.
Calculation of Bioactivities
The AutoDockVina docking score corresponds to the predicted binding energy (BE).35 Inhibition constants (Ki) of the designed compounds with better BEs were obtained using eq 1 as reported elsewhere,37 where R is the gas constant (1.987 × 10–3 kcal/(K mol)) and T (298.15 K) is the absolute temperature of the protein–ligand complex. All ligand efficiency-related parameters38 were derived from the binding energy and thus calculated using eqs 2–5 as reported elsewhere.37
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
where LELP means ligand efficiency-dependent lipophilicity, FQ means fit quality, and HA means heavy atoms.
In Silico Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Predictions of the Designed Compounds
The designed compounds with better BEs than their respective parent compounds (dasabuvir, efavirenz, and tipranavir) were further subjected to absorption, distribution, metabolism, excretion, and toxicity (ADMET) and drug-likeness evaluation. The ADMET and drug-likeness predictions were performed with Swiss-ADME and pkCSM web tools.39,40
Results and Discussion
Designed Compounds
A total of 597, 582, and 609 approved druglike derivatives of dasabuvir, efavirenz, and tipranavir, respectively, were designed with DataWarrior-5.2.1. It is important to identify potentially mutagenic, tumorigenic, and irritant compounds and exclude them from the drug discovery compound library. Therefore, 77, 29, and 436 designed derivatives of dasabuvir, efavirenz, and tipranavir, representing 12.91, 4.98, and 71.59% of their respective designed derivatives, respectively, were identified as nonmutagenic, nontumorigenic, and nonirritant (Supporting Information 1). The summary of mutagenic, tumorigenic, and irritant assessments of all designed derivatives can be found in Figure S1 in Supporting Information 3. This study showed that dasabuvir is a low-mutagenic and highly tumorigenic agent but not an irritant (Supporting Information 1). This observation may be due to the amide group in the sulfonamide moiety of dasabuvir since amine/amide drugs/compounds and their nitrosation products are mutagenic.41,42 The previous reports have shown that treatment of hepatitis C with dasabuvir is associated with mutations including S556G and C316Y mutations in the viral NS5B polymerase, leading to resistance.20,21 Increased damage indexes and frequencies have been observed in the brain of mice treated with efavirenz,23 and findings from this study showed that efavirenz is a highly tumorigenic agent but not mutagenic nor irritant (Supporting Information 1). The tumorigenic effect of efavirenz may be responsible for adverse effects (e.g., headache, dizziness, insomnia, impaired concentration, agitation, amnesia, fatigue, and hallucinations) of efavirenz on the central nervous system.24 Also, the World Health Organization (WHO) guidelines does not recommend efavirenz use during the first trimester of pregnancy due to the concern for teratogenicity.24 This study showed that tipranavir was not mutagenic, tumorigenic, and irritant (Supporting Information 1). However, experimentally resolved structures of wild-type and L76V mutant of the HIV protease in a complex with tipranavir have been reported.22 All of the nonmutagenic, nontumorigenic, and nonirritant derivatives were selected as libraries28 and used for molecular docking simulations.
Molecular Docking and Bioactivity Results of the Designed Compounds
The results of the molecular docking simulation binding energies/affinities (kcal/mol) of the parent compounds and their designed derivatives that were not mutagenic, tumorigenic, and irritant derivatives (Supporting Information 1) with the zika virus RNA-dependent RNA polymerase and methyltransferase are presented in Figure 1a,c and Tables S1–S4 in Supporting Information 3 and Supporting Information 1. The binding energy score is an indication of how strongly the compounds interact with drug targets. The lower the numeric value, the better the interaction. This study identified derivatives of dasabuvir 15d, efavirenz 3e, and tipranavir 21t, with binding energies for the zika virus RNA-dependent RNA polymerase only (41, 2, and 18, respectively), methyltransferase only (2, 2, and 50, respectively), and both RNA-dependent RNA polymerase and methyltransferase (14, 0, and 20, respectively) equivalent to or better than their respective parent compounds (dasabuvir, 15d; efavirenz, 3e; and tipranavir, 21t), as can be seen in Supporting Information 1. This is possibly due to the changes in the structures of the parent compounds. For example, some of the derivatives are due to the isosteric replacement of carbon with a nitrogen atom, and nitrogen-containing compounds have a reported exceptional broad-spectrum biological activity against viruses.43 The names and structures of some of the designed derivatives of dasabuvir 15d, efavirenz 3e, and tipranavir 21t, including their parent compounds (1d to 19d, 1t to 30t, 1e to 5e) were determined by their respective SMILES (Supporting Information 1) using ChemDraw-12.0 and are presented in Tables 1, 4–6, and Tables S7–S9 in Supporting Information 3. A previous study has identified and validated a derivative of dasabuvir, N-[4-[6-tert-butyl-5-methoxy-8-(6-methoxy-2-oxo-1H-pyridin-3-yl)-3 quinolyl]phenyl] methanesulfonamide (PubChem identifier: CID 49835560) against hepatitis C virus NS5B polymerase44 but not zika virus, derivatives of 1,2,4-oxadiazole as potential dengue virus NS5 inhibitors45 with better binding energies, and arylaldoxime/5-nitroimidazole small-molecule hybrids as potent microtubule affinity-regulating kinase 4 (MARK4) inhibitors and antiproliferative agents.46 This study designed derivatives of dasabuvir, efavirenz, and tipranavir as potential inhibitors of the zika virus RNA-dependent RNA polymerase and/or methyltransferase. Inhibition constants (Ki) of some of the derivatives including that of parent compounds (1d to 19d, 1t to 30t, 1e to 5e) are presented together with all ligand efficiency-related parameters derived from the binding energies in Figure 1b,d and Tables S5 and S6 in Supporting Information 3. It shows that 14 derivatives of dasabuvir (1d to 14d, see Tables 1, 5, and 6 for their names and Tables S7–S9 in Supporting Information 3 for their structures) are potential inhibitors of the zika virus RNA-dependent RNA polymerase and NS5-methyltransferase with Ki ranges of 2.461448198 ± 0.449621 to 8.642723911 ± 0.00000 μM and 0.076982246 ± 0.067006 to 0.180067848 ± 0.059893 μM, respectively, when compared with dasabuvir. It is important to mention that 18d and 19d showed Ki values of 0.177364835 ± 0.0000 and 0.488831033 ± 0.0000 μM, respectively, for the zika virus RNA-dependent RNA polymerase (Table S5 in Supporting Information 3 and Supporting Information 1), while dasabuvir showed a value of 9.795009846 ± 3.335809 μM. Again, some derivatives of tipranavir are better inhibitors of the zika virus RNA-dependent RNA polymerase and NS5-methyltransferase with Ki ranges of 1.005139848 ± 0.076595 to 3.503956799 ± 2.96929 μM and 0.056981919 ± 0.022164 to 0.076200557 ± 0.000000 μM, respectively, when compared with tipranavir (3.605182303 ± 2.139293 and 0.090227728 ± 0.000000 μM, respectively) (Table S6 in Supporting Information 3 and Supporting Information 1). It is important to mention that 22t to 28t (see Tables 4–6 for their names and Tables S7–S9 in Supporting Information 3 for their structures) showed a Ki range of 0.030192974 ± 0.002938 to 0.054349355 ± 0.000000 μM for the zika virus NS5-methyltransferase (Table S6 in Supporting Information 3 and Supporting Information 1). Also, there is no derivative of efavirenz that showed better binding energy and Ki for both zika virus RNA-dependent RNA polymerase and NS5-methyltransferase. However, 1e showed equivalent binding energy of −6.675 ± 0.05 kcal/mol and Ki of 12.67519392 ± 1.115312 μM for RNA-dependent RNA polymerase, while 4e showed equivalent binding energy of −7.6 ± 0.00 kcal/mol and Ki of 2.648363028 ± 0.000000 μM for NS5-methyltransferase when compared with efavirenz, 3e, (−6.60 ± 0.00 kcal/mol and 14.34816176 ± 0.000000 μM for RNA-dependent RNA polymerase; −7.43 ± 0.05 kcal/mol and 3.568824547 ± 0.288630 μM for NS5-methyltransferase) (Supporting Information 1).
Figure 1.
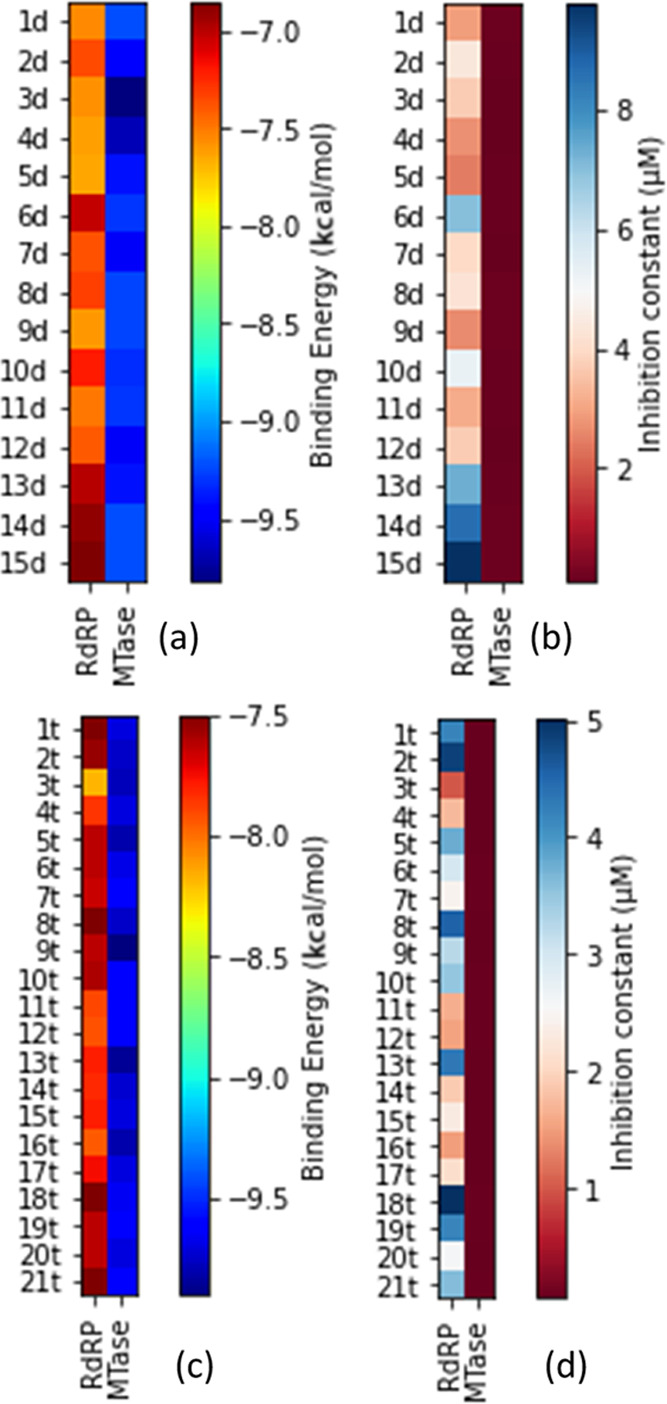
Heatmaps for (a) binding energies of dasabuvir and its derivatives, (b) inhibition constants of the designed derivatives of dasabuvir, 15d, (c) binding energies of tipranavir and its derivatives, and (d) inhibition constants of the designed derivatives of tipranavir, 21t, for both RNA-dependent RNA polymerase and methyltransferase.
Table 1. Drug-Likeness and Pharmacokinetic Parameters of Some of the Designed Derivatives of Dasabuvir Inhibitors of the Zika Virus RNA-Dependent RNA Polymerase (1d to 14d), Methyltransferase, and RNA-Dependent RNA Polymerase (16d to 17d) Evaluated Using Swiss-ADMEa.
| ID | MW (g/mol) | #rotatable bonds | #H-bond acceptors | #H-bond donors | tPSA (Å2) | consensus log P | GI absorption | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | #Lipinski violations | bioavailability score | #PAINS alerts | synthetic accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1d | 495.55 | 6 | 7 | 3 | 157.55 | 2.71 | low | no | no | no | no | no | no | 0 | 0.55 | 0 | 3.53 |
| 2d | 494.56 | 6 | 6 | 2 | 131.53 | 3.31 | low | no | no | yes | yes | no | yes | 0 | 0.55 | 0 | 3.58 |
| 3d | 493.58 | 6 | 6 | 3 | 138.31 | 3.36 | low | no | no | no | yes | no | yes | 0 | 0.55 | 0 | 3.68 |
| 4d | 493.58 | 6 | 6 | 3 | 138.31 | 3.38 | low | no | no | no | yes | no | yes | 0 | 0.55 | 0 | 3.73 |
| 5d | 496.54 | 6 | 7 | 3 | 151.76 | 2.85 | low | no | no | no | no | no | no | 0 | 0.11 | 0 | 3.55 |
| 6d | 429.51 | 5 | 4 | 2 | 90.11 | 3.94 | high | yes | no | no | yes | no | yes | 0 | 0.55 | 0 | 3.26 |
| 7d | 496.54 | 6 | 7 | 3 | 151.76 | 2.24 | low | yes | no | no | yes | no | yes | 0 | 0.55 | 0 | 3.56 |
| 8d | 493.58 | 6 | 5 | 3 | 134.33 | 3.21 | low | no | no | yes | yes | no | yes | 0 | 0.55 | 0 | 3.63 |
| 9d | 510.63 | 6 | 5 | 2 | 147.60 | 3.86 | low | no | no | yes | yes | no | yes | 1 | 0.55 | 0 | 3.69 |
| 10d | 509.58 | 6 | 6 | 3 | 157.55 | 2.78 | low | no | no | no | yes | no | yes | 1 | 0.55 | 0 | 3.68 |
| 11d | 509.58 | 6 | 6 | 3 | 157.55 | 2.77 | low | no | no | no | yes | no | yes | 1 | 0.55 | 0 | 3.70 |
| 12d | 512.55 | 6 | 7 | 2 | 131.53 | 3.62 | low | no | no | yes | yes | no | yes | 1 | 0.55 | 0 | 3.69 |
| 13d | 507.60 | 6 | 6 | 2 | 132.63 | 3.76 | low | no | no | yes | yes | no | yes | 1 | 0.55 | 0 | 3.69 |
| 14d | 494.56 | 6 | 7 | 2 | 145.52 | 2.58 | low | no | no | no | yes | no | no | 0 | 0.55 | 0 | 3.66 |
| 15d | 493.57 | 6 | 5 | 2 | 118.64 | 3.80 | low | no | no | yes | yes | no | yes | 0 | 0.55 | 0 | 3.46 |
| 16d | 508.59 | 7 | 6 | 2 | 131.53 | 3.49 | low | no | no | yes | yes | no | yes | 1 | 0.55 | 0 | 3.68 |
| 17d | 493.57 | 6 | 6 | 2 | 132.63 | 3.42 | low | no | no | yes | yes | no | yes | 0 | 0.55 | 0 | 3.54 |
N-{6-[3-tert-Butyl-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-2-methoxyphenyl]quinolin-2-yl}aminosulfonamide, 1d; N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)quinolin-2-yl)methanesulfonamide, 2d; N-(6-(3-(tert-butyl)-5-(2-imino-4-oxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)quinolin-2-yl)methanesulfonamide, 3d; N-(6-(3-(tert-butyl)-5-(4-imino-2-oxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)quinolin-2-yl)methanesulfonamide, 4d; (6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)quinolin-2-yl)sulfamic acid, 5d; 1-(3-(6-(aminomethyl)naphthalen-2-yl)-5-(tert-butyl)-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione, 6d; N-(6-(5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-(2-hydroxypropan-2-yl)-2-methoxyphenyl)quinolin-2-yl)methanesulfonamide, 7d; N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-(methylamino)phenyl)quinolin-2-yl)methanesulfonamide, 8d; N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-(methylthio)phenyl)quinolin-2-yl)methanesulfonamide, 9d; N-(3-amino-6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)quinolin-2-yl)methanesulfonamide, 10d; N-(8-amino-6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)quinolin-2-yl)methanesulfonamide, 11d; N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)-4-fluoroquinolin-2-yl)methanesulfonamide, 12d; (6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)-3-methylnaphthalen-2-yl)methanesulfonamide, 13d; (3-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)isoquinolin-7-yl)methanesulfonamide, 14d; N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (dasabuvir), 15d; N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxybenzyl)quinolin-2-yl)methanesulfonamide, 16d; (7-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide, 17d.
Table 4. Predicted Toxicity of Some of the Designed Derivatives of Tipranavir Inhibitors of the Zika Virus RNA-Dependent RNA Polymerase and Methyltransferases (1t to 20t and 22t to 26t) Evaluated Using pkCSMa.
| ID | formula | AMES toxicity | max. tolerated dose (human) (log mg/kg/day) | hERG I inhibitor | hERG II inhibitor | oral rat acute toxicity (LD50) (mol/kg) | oral rat chronic toxicity (LOAEL) (log mg/kg_bw/day) | hepatotoxicity | skin sensitization |
|---|---|---|---|---|---|---|---|---|---|
| 1t | C31H33F3N2O5S | no | 0.051 | no | yes | 2.736 | 1.75 | yes | no |
| 2t | C33H37F3N2O5S | no | –0.207 | no | yes | 1.911 | 2.264 | yes | no |
| 3t | C30H31F3N2O5S | no | –0.379 | no | yes | 2.698 | 2.244 | yes | no |
| 4t | C31H34F3N3O5S | no | –0.116 | no | yes | 2.593 | 2.476 | yes | no |
| 5t | C32H36F3N3O5S | no | –0.115 | no | yes | 2.634 | 2.447 | yes | no |
| 6t | C33H37F3N2O5S | no | 0.061 | no | yes | 2.721 | 1.74 | yes | no |
| 7t | C30H30F4N2O5S | no | 0.206 | no | yes | 2.732 | 1.837 | yes | no |
| 8t | C32H36F3N3O5S | no | –0.276 | no | yes | 2.559 | 1.854 | yes | no |
| 9t | C31H33F3N2O5S2 | no | 0.063 | no | yes | 2.664 | 2.509 | yes | no |
| 10t | C32H33F3N2O5S | no | –0.034 | no | yes | 1.987 | 2.111 | yes | no |
| 11t | C32H31F3N2O5S | no | –0.11 | no | yes | 1.893 | 2.154 | yes | no |
| 12t | C32H36F2N2O5S | no | 0.053 | no | yes | 2.719 | 1.759 | yes | no |
| 13t | C30H32F3N3O5S | no | –0.035 | no | yes | 2.786 | 1.719 | yes | no |
| 14t | C32H31F3N2O5S | no | –0.121 | no | yes | 1.904 | 2.161 | yes | no |
| 15t | C31H34F3N3O5S | no | –0.004 | no | yes | 2.628 | 1.983 | yes | no |
| 16t | C30H32F3N3O5S | no | 0.027 | no | yes | 2.654 | 1.975 | yes | no |
| 17t | C32H33F3N2O5S | no | –0.095 | no | yes | 1.9 | 2.118 | yes | no |
| 18t | C33H35F3N2O5S | no | –0.114 | no | yes | 1.996 | 2.099 | yes | no |
| 19t | C29H29F4N3O5S | no | 0.384 | no | yes | 3.246 | 1.88 | yes | no |
| 20t | C32H35F3N2O6S | no | –0.042 | no | yes | 2.715 | 1.866 | yes | no |
| 21t | C31H33F3N2O5S | no | –0.354 | no | yes | 2.367 | 2.326 | yes | no |
| 22t | C30H32F3N3O5S | no | –0.172 | no | yes | 2.866 | 2.027 | yes | no |
| 23t | C29H29F4N3O5S | no | 0.413 | no | yes | 3.038 | 1.716 | yes | no |
| 24t | C32H33F3N2O7S | no | –0.004 | no | no | 3.097 | 2.138 | yes | no |
| 25t | C31H29F3N2O5S | no | –0.061 | no | yes | 1.958 | 2.188 | yes | no |
| 26t | C32H35F3N2O5S | no | –0.143 | no | yes | 1.843 | 2.197 | yes | no |
N-(3-((R)-1-((R)-4-Hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 1t; N-(3-((R)-1-((R)-4-hydroxy-6-isopentyl-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 2t; N-(3-((R)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)ethyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 3t; N-(3-((R)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)-2-(methylamino)ethyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 4t; N-(3-((R)-2-(dimethylamino)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)ethyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 5t; N-(3-((R)-2-fluoro-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)ethyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 6t; N-(3-((R)-4-amino-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)butyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 7t; N-(3-((R)-1-((S)-4-hydroxy-6-isobutyl-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)-2-mercaptoethyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 8t; N-(3-((R)-1-((S)-4-hydroxy-6-(2-methylallyl)-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 9t; N-(3-((R)-1-((R)-6-(but-3-yn-1-yl)-4-hydroxy-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 10t; 5-(1,1-difluoroethyl)-N-(3-((R)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)pyridine-2-sulfonamide, 11t; N-(3-((R)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyrazine-2-sulfonamide, 12t; N-(3-((R)-1-((S)-6-(but-2-yn-1-yl)-4-hydroxy-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 13t; N-(3-((S)-1-((S)-6-butyl-4-hydroxy-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-6-(trifluoromethyl)pyridazine-3-sulfonamide, 14t; N-(3-((R)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-6-(trifluoromethyl)pyridazine-3-sulfonamide, 15t; N-(3-((R)-1-((S)-6-((E)-but-2-en-1-yl)-4-hydroxy-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 16t; N-(3-((R)-1-((R)-4-hydroxy-6-(3-methylbut-3-en-1-yl)-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 17t; N-(3-((R)-1-((S)-4-hydroxy-6-((R)-2-methylbutyl)-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 18t; N-(5-((S)-2-fluoro-1-((S)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)ethyl)pyridin-3-yl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 19t; N-(3-((R)-4-hydroxy-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)butyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 20t; N-(3-((R)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide (tipranavir), 21t; N-(6-((S)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)propyl)pyridin-2-yl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 22t; N-(6-((S)-2-fluoro-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)ethyl)pyridin-2-yl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 23t, (R)-4-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)-4-(3-(5-(trifluoromethyl)pyridine-2-sulfonamido)phenyl)butanoic acid, 24t; N-(3-((R)-1-((S)-4-hydroxy-2-oxo-6-phenethyl-6-(prop-2-yn-1-yl)-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 25t; N-(3-((R)-1-((S)-4-hydroxy-6-isobutyl-2-oxo-6-phenethyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide, 26t.
Table 6. Predicted Toxicity of Designed Derivatives of Dasabuvir, Efavirenz, and Tipranavir That Are Not Inhibitors of the Human Ether-a-go-go-Related Gene (hERG) Nor Induce Hepatotoxicitya.
| ID | formula | AMES toxicity | max. tolerated dose (human) (log mg/kg/day) | hERG I inhibitor | hERG II inhibitor | oral rat acute toxicity (LD50) (mol/kg) | oral rat chronic toxicity (LOAEL) (log mg/kg_bw/day) | hepatotoxicity | skin sensitization |
|---|---|---|---|---|---|---|---|---|---|
| 19d | C28H30N2O6S | yes | 0.231 | no | no | 2.588 | 2.096 | no | no |
| 20d | C26H26N2O6S | no | 0.447 | no | no | 2.623 | 2.026 | no | no |
| 21d | C27H28N2O6S | no | 0.373 | no | no | 2.679 | 1.653 | no | no |
| 22d | C27H28N2O6S | no | 0.325 | no | no | 2.606 | 2.018 | no | no |
| 1e | C15H11NOClF3S | no | –0.150 | no | no | 3.002 | 0.915 | no | no |
3-(6-(3-(tert-Butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)propane-1-sulfonic acid, 19d, (6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonic acid, 20d, (6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-ethoxyphenyl)naphthalen-2-yl)methanesulfonic acid, 21d, 2-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)ethanesulfonic acid, 22d, (S)-6-chloro-4-(3-cyclopropylprop-2-yn-1-yl)-4-(trifluoromethyl)-1H-benzo[d][1,3]oxazine-2(4H)-thione, 1e.
Table 5. Predicted Toxicity of Designed Derivatives of Dasabuvir, Efavirenz, and Tipranavir That Are Noninhibitors of the Human Ether-a-go-go-Related Gene (hERG)a.
| ID | formula | AMES toxicity | max. tolerated dose (human) (log mg/kg/day) | hERG I inhibitor | hERG II inhibitor | oral rat acute toxicity (LD50) (mol/kg) | oral rat chronic toxicity (LOAEL) (log mg/kg_bw/day) | hepatotoxicity | skin sensitization |
|---|---|---|---|---|---|---|---|---|---|
| 5d | C24H24N4O6S | no | 0.652 | no | no | 2.658 | 1.956 | yes | no |
| 4d | C25H27N5O4S | no | 0.756 | no | no | 2.511 | 2.2 | yes | no |
| 18d | C24H24N4O6S | no | 0.554 | no | no | 2.507 | 2.273 | yes | no |
| 24t | C32H33F3N2O7S | no | –0.004 | no | no | 3.097 | 2.138 | yes | no |
| 5e | C12H7N3OClF3S | no | –0.427 | no | no | 2.581 | 1.199 | yes | no |
| 4e | C12H7N3OF4S | no | 0.031 | no | no | 2.858 | 1.322 | yes | no |
| 2e | C14H10N2O2ClF3 | no | –0.417 | no | no | 2.839 | 0.512 | yes | no |
| 3e | C14H9NO2ClF3 | no | 0.111 | no | no | 2.768 | 1.243 | no | no |
| 15d | C26H27N3O5S | no | 0.149 | no | yes | 3.081 | 3.139 | yes | no |
| 21t | C31H33F3N2O5S | no | –0.354 | no | yes | 2.367 | 2.326 | yes | no |
N-(6-(3-(tert-Butyl)-5-(4-imino-2-oxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)quinolin-2-yl)methanesulfonamide, 4d, (6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)quinolin-2-yl)sulfamic acid, 5d; N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (dasabuvir), 15d; (7-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)isoquinolin-3-yl)sulfamic acid, 18d, N-(3-((R)-1-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)propyl)phenyl)-5-(trifluoromethyl)pyridine-2-sulfonamide (tipranavir), 21t; (R)-4-((R)-4-hydroxy-2-oxo-6-phenethyl-6-propyl-5,6-dihydro-2H-pyran-3-yl)-4-(3-(5-(trifluoromethyl)pyridine-2-sulfonamido)phenyl)butanoic acid, 24t; (S)-6-chloro-4-(3-cyclopropylprop-2-yn-1-yl)-4-(trifluoromethyl)-1H-pyrido[3,4-d][1,3]oxazin-2(4H)-one, 2e; (S)-6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-1H-benzo[d][1,3]oxazin-2(4H)-one (efavirenz), 3e; (S)-5-(cyclopropylethynyl)-3-fluoro-5-(trifluoromethyl)-5H-pyridazino[3,4-d][1,3]oxazine-7(8H)-thione, 4e; (S)-3-chloro-5-(cyclopropylethynyl)-5-(trifluoromethyl)-5H-pyridazino[3,4-d][1,3]oxazine-7(8H)-thione, 5e.
Previous studies have reported inhibitors of the zika virus replication/NS5-RNA-dependent RNA polymerase and/or NS5-methyltransferase2,13,15−19,36,47,48 using various strategies. This study identified some derivatives of the said parent compounds as potential inhibitors of the virus NS5 protein, and a previous report had validated N-[4-[6-tert-butyl-5-methoxy-8-(6-methoxy-2-oxo-1H-pyridin-3-yl)-3 quinolyl] phenyl] methanesulfonamide (PubChem identifier: CID 49835560), a derivative of dasabuvir, against hepatitis C virus NS5B polymerase44 but not zika virus. Again, novel guanosine derivatives against the zika virus polymerase have been reported.49 The inhibition constant value in the micromolar range, fit quality (FQ) of ≥0.8, and the range of LELP between −10 and 10 have been reported for acceptable lead/druglike compounds.37,50 The majority of the designed derivatives (1d to 14d, 16d to 22d, 1t to 20t, 22t to 26t, 1e, 2e, 3e, and 4e) have better Ki, FQ, and LELP values when compared with their respective parent compounds (Tables S6 and S5 in Supporting Information 3). This suggests that they may have better antiviral activities against the virus than their respective parent compounds. Also, they have good pharmacokinetic parameters, drug-likeness, and toxicity profiles (Tables 1–10), including synthetic accessibilities.
Table 10. Drug-Likeness and Pharmacokinetic Parameters of Derivatives of Dasabuvir, Efavirenz, and Tipranavir That Are Not Inhibitors of the Human Ether-a-go-go-Related Gene (hERG) Nor Induce Hepatotoxicity.
| ID | MW (g/mol) | #rotatable bonds | #H-bond acceptors | #H-bond donors | TPSA (Å2) | consensus log P | GI absorption | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | #Lipinski violations | bioavailability score | #PAINS alerts | synthetic accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 19d | 522.61 | 8 | 6 | 2 | 126.84 | 4.32 | low | no | no | yes | yes | no | no | 1 | 0.56 | 0 | 3.78 |
| 22d | 508.59 | 7 | 6 | 2 | 126.84 | 4.02 | low | no | no | yes | yes | no | no | 1 | 0.56 | 0 | 3.63 |
| 20d | 494.56 | 6 | 6 | 2 | 126.84 | 3.48 | low | no | no | yes | no | no | no | 0 | 0.56 | 0 | 3.57 |
| 21d | 508.59 | 7 | 6 | 2 | 126.84 | 4.06 | low | no | no | yes | yes | no | no | 1 | 0.56 | 0 | 3.71 |
| 1e | 345.77 | 2 | 4 | 1 | 53.35 | 4.59 | high | yes | yes | yes | yes | no | no | 0 | 0.55 | 0 | 3.66 |
Molecular Interactions
Molecular interactions of two representatives of derivatives of dasabuvir (3d and 5d) with better binding energies and Ki for both zika virus methyltransferase and RNA-dependent RNA polymerase (Figure 1a,b) than dasabuvir, 15d, are presented in Figure 2. It is important to mention that some derivatives of dasabuvir (16d and 17d) showed better binding energies and Ki (Tables S1 and S5 in Supporting Information 3) than 3d and 5d for RNA-dependent RNA polymerase but not for methyltransferase. Figure 3 shows the molecular interactions of 16d and 17d with RNA-dependent RNA polymerase. Also, molecular interactions of two representatives of derivatives of tipranavir (3t and 10t) with better binding energies and Ki for both zika virus methyltransferase and RNA-dependent RNA polymerase (Figure 1c,d) than tipranavir, 21t, are presented in Figure 5. Again, some derivatives of tipranavir (e.g., 22t to 26t) showed better binding energies (−10.00 ± 0.00 to −10.25 ± 0.006 kcal/mol) and Ki (0.030192974 ± 0.002938 to 0.045899983 ± 0.000000 μM) (Tables S2 and S6 in Supporting Information 3) than 3t and 10t for methyltransferase but not for RNA-dependent RNA polymerase. Figures 4 and S2 in Supporting Information 3 show the molecular interactions of 22t to 26t with zika virus NS5-methyltransferase.
Figure 2.

Molecular interactions of two representatives of derivatives of dasabuvir and dasabuvir, 15d, with zika virus methyltransferase and RNA-dependent RNA polymerase. The first panel (a–c) shows the interactions of 3d, 5d, and dasabuvir with methyltransferase, while the second panel (d–f) shows the interactions with RNA-dependent RNA polymerase, respectively.
Figure 3.

Molecular interactions of the designed derivatives of dasabuvir (16d and 17d) with the zika virus RNA-dependent RNA polymerase: Interactions of 16d (a) and 17d (b) with RNA-dependent RNA polymerase.
Figure 5.

Molecular interactions of two representatives of derivatives of tipranavir (3t and 10t) and tipranavir, 21t, with the zika virus methyltransferase and RNA-dependent RNA polymerase. The first panel (a–c) shows the interactions of 3t, 10t, and tipranavir with methyltransferase, while the second panel (d–f) shows the interactions with the RNA-dependent RNA polymerase, respectively.
Figure 4.
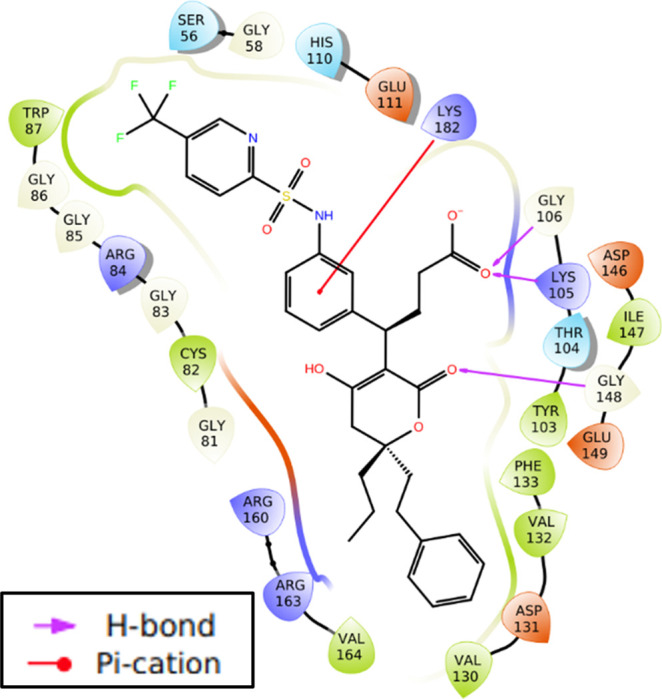
Molecular interactions of 24t with zika virus methyltransferase.
Docking results showed that derivatives of dasabuvir (3d, 5d, 16d, and 17d) and tipranavir (3t, 10t, 22t to 26t) complexes with methyltransferase and RNA-dependent RNA polymerase are stabilized by a significant number of noncovalent interactions (Figures 2–5). Specifically, there are four intermolecular hydrogen bonds between four amino acid residues (Ser56, Gly85, Gly86, and Asp131) in the methyltransferase and two derivatives of dasabuvir (3d, 5d) compared with three (Ser56, Gly85, and Gly86) between methyltransferase and dasabuvir (Figure 2a–c). Also, the study observed a hydrogen bond between Gly148 in the methyltransferase with tipranavir derivatives (3t, 10t, 22t to 26t) (Figures 4 and 5a–c). Additionally, other amino acid residues in the methyltransferase with direct contacts (hydrogen bond, π–π stacking, and/or π–cation) with 3t, 10t, 22t to 26t are Lys105, Gly106, Glu111, Phe133, Asp146, and/or Lys182 (Figures 4, 5a–c, and S2 in Supporting Information 3). Similar observations have been made for a reference inhibitor (2-(4-((4-phenylpiperazin-1-yl) sulfonyl)phenethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione) of zika virus MTase.47 Again, a previous study has reported intermolecular interactions between S-adenosylmethionine (SAM) with Lys105, Ile147, Val132, Asp131, Gly106, Glu111, Thr104, Ser56, Asp146, Gly86, and Trp87 in zika virus methyltransferase.51 Also, interactions between Gly148, Cys82, Arg84, Lys105, Val132, and Gly77 in zika virus MTase with in vitro validated inhibitors of the virus MTase have been reported.47 This suggests that the designed derivatives of dasabuvir and tipranavir bind to the SAM binding site and are possible inhibitors of zika virus methyltransferase. The possible inhibition of N-7 and 2′ methylation activities of the virus MTase by the designed compounds due to the mentioned hydrogen bonds implies virus death through host immune response. This is because methylation at the N-7 position is essential for viral replication and methylation at the 2′-O position helps RNA evade host immune response,52 and mutating Gly148 to Ala148 abolishes the N-7, and 2′-O methylation capacity of dengue virus MTase activity.47 This may be the case for the zika virus MTase. Again, Arg160, and Arg163 are important for N-7 methylation, while Phe133 is important for 2′-O methylation.47 This study observed intermolecular interactions between Phe133, Arg160, and Arg163 in the methyltransferase with the designed compounds (Figures 2 and 4). Other compounds like theaflavin, a natural compound, have been reported to directly bind to Asp146 of zika virus NS5 MTase to inhibit replication.48 Asp146 in zika virus is part of the Lys61–Asp146–Lys182–Glu218 tetrad motif that is important for both N-7 and 2′O methylation,51 and the designed compounds (3d, 5d, 16d, 17d, 3t, 10t, 22t to 26t) showed interaction with Asp146 (Figures 2a–c, 4, 5a–c, and S2 in Supporting Information 3). This implies that the designed derivatives of dasabuvir and tipranavir are possible inhibitors of methylation at the N-7 position, which is essential for viral replication and methylation at the 2′-O position and helps the virus evade the host immune response.52 However, there is a need for in vitro validations of the designed compounds against zika virus methyltransferase activity.
In the case of interactions of derivatives of dasabuvir and tipranavir with the virus RNA-dependent RNA polymerase, this study observed hydrogen-bonding or π–cation interaction between Arg731 and dasabuvir and its derivatives (3d, 5d, 16d, 17d) (Figures 2d–f and 3). Other amino acid residues in the RNA-dependent RNA polymerase that have direct contacts (hydrogen bonding, π–π stacking, and/or π–cation) with dasabuvir and its derivatives (3d, 5d, 16d, 17d) including tipranavir and its derivatives (3t and 10t) are Tyr609, Arg612, Asp665, Cys711, Ser712, Arg739, Arg794, Trp797, and/or Ser798 (Figures 2d–f, 3, and 5d–f). Intermolecular hydrogen bonds have been reported between the sulfonamide moiety of nonnucleoside inhibitors and Arg739, Trp797, Arg731, and Thr796 residues in the allosteric pocket (“N-pocket”) of the zika virus RNA-dependent RNA polymerase.16 Also, similar interactions have been reported for novel guanosine derivatives and nonnucleoside inhibitors like 5-(5-fluorothiophen-2-yl)-2-hydroxy-4-methoxy-N-((3-(trifluoromethyl)phenyl)sulfonyl) benzamide against the zika virus polymerase.16,49 This largely validates the designed compounds as potential inhibitors of the zika virus RNA-dependent RNA polymerase. It has been reported elsewhere that fidaxomicin inhibits zika virus replication and prevents the virus-infected mice from death.36 Also, strong binding between fidaxomicin and zika virus RNA-dependent RNA polymerase has implicated Thr795 and Ser798 to be located in the priming loop (residues 787–809), an important stacking platform for initiation of nucleotide triphosphate entry into de novo RNA synthesis.36 This suggests that the designed compounds have the potential for in vitro and in vivo activities against the zika virus since they showed intermolecular hydrogen bond(s) with amino acid residues from the priming loop region. However, there is a need for laboratory confirmations of the activities of the designed compounds against the zika virus RNA-dependent RNA polymerase. A previous report showed that tipranavir contains stereocentres at both C-3α and C-6 and a robust resistance profile to HIV. Tipranavir binding with few hydrogen bonds creates increased flexibility to adjust to amino acid changes in the HIV protease active site, which is largely responsible for its resistance profile.53 This may explain why this study observed fewer hydrogen bonds in the interactions of the designed derivatives of tipranavir with RNA-dependent RNA polymerase and methyltransferase (Figures 4 and 5) when compared with those of dasabuvir (Figures 2 and 3). Generally, some of the designed derivatives of dasabuvir and tipranavir showed better/improved binding energies/inhibition constants than their respective parent compounds (dasabuvir and tipranavir). However, derivatives of efavirenz (1e, 2e, 4e, and 4e) did not show appreciable improvement in binding energies and inhibition constants (Table 7). Interestingly, atoms/groups or substituents in these designed derivatives with better/improved binding energies and inhibition constants that make them different from their parent compounds were not directly involved in intermolecular interactions except in the case of a few of the designed derivatives such as 16d and 24t, interacting with RNA-dependent RNA polymerase and methyltransferase, respectively (Figures 3a and 4). Quinoline nitrogen (N) and oxygen (O) atoms in carboxylic functional groups of 16d and 24t are involved in interactions with RNA-dependent RNA polymerase and methyltransferase, respectively, when compared with dasabuvir and tipranavir (Figures 2c and 5c). It is possible that the atoms/groups or substituents in these designed derivatives indirectly influence the binding environments to achieve better binding energies/inhibition constants since they either showed additional/different types of interaction or more interacting amino acid residues than the parent compounds. Two of the designed derivatives (PubChem identifiers: CID 25234840, CID 25234911) of dasabuvir with better binding energies have been previously reported, while the derivatives of tipranavir previously reported are four (PubChem identifiers: CID 58746420, CID 59131653, CID 162581045 and CID 71211717).
Table 7. Binding Energies and Bioactivities of Derivatives of Dasabuvir, Efavirenz, and Tipranavir That Are Noninhibitors of the Human Ether-a-go-go-Related Gene (hERG).
| ID | binding energy for RdRP (kcal/mol) | Ki for RdRP (μM) | LE (kcal/mol/heavy atom) | LEscale | LELP | FQ |
|---|---|---|---|---|---|---|
| 5d | –7.65 ± 0.10 | 2.461448198 ± 0.449621 | 0.218571 | 0.287404 | 9.16817 | 0.760503 |
| 4d | –7.63 ± 0.25 | 2.719231219 ± 1.199306 | 0.217857 | 0.287404 | 14.66603 | 0.758018 |
| 18d | –7.30 ± 0.00 | 4.396662616 ± 0.000000 | 0.208571 | 0.287404 | 9.020411 | 0.725709 |
| 2e | –6.65 ± 0.06 | 13.23284987 ± 1.287851 | 0.302273 | 0.428717 | 10.62716 | 0.705063 |
| 3e | –6.60 ± 0.00 | 14.34816176 ± 0.000000 | 0.314286 | 0.441696 | 11.64991 | 0.711543 |
| 15d | –6.85 ± 0.19 | 9.795009846 ± 3.335809 | 0.195714 | 0.287404 | 21.05672 | 0.680974 |
| 21t | –7.50 ± 0.34 | 3.605182303 ± 2.139293 | 0.178571 | 0.228931 | 41.0228 | 0.780024 |
| ID | binding energy for MTase (kcal/mol) | Ki for MTase (μM) | LE | LEscale | LELP | FQ |
|---|---|---|---|---|---|---|
| 5d | –9.40 ± 0.20 | 0.132631428 ± 0.051589 | 0.268571 | 0.287404 | 7.46133 | 0.934475 |
| 4d | –9.68 ± 0.05 | 0.07970735 ± 0.0070140 | 0.276429 | 0.287404 | 11.5585 | 0.961813 |
| 24t | –10.08 ± 0.05 | 0.040548134 ± 0.003568 | 0.223889 | 0.20695 | 23.04893 | 1.081848 |
| 5e | –7.50 ± 0.00 | 3.135879681 ± 0.000000 | 0.357143 | 0.441696 | 7.39452 | 0.808572 |
| 4e | –7.60 ± 0.00 | 2.648363028 ± 0.000000 | 0.361905 | 0.441696 | 6.603118 | 0.819353 |
| 3e | –7.43 ± 0.05 | 3.568824547 ± 0.288630 | 0.353571 | 0.441696 | 10.35547 | 0.800486 |
| 15d | –9.23 ± 0.25 | 0.180067848 ± 0.059893 | 0.263571 | 0.287404 | 15.63561 | 0.917078 |
| 21t | –9.60 ± 0.00 | 0.090227728 ± 0.000000 | 0.228571 | 0.228931 | 32.04906 | 0.998431 |
Results of In Silico ADMET Predictions of the Designed Compounds
The predicted drug-likeness, pharmacokinetics, and ADMET parameters of the designed compounds with better binding energies than dasabuvir or tipranavir are presented in Tables 1–6, 9, and 10 and Supporting Information 1. Tables 1, 3, 9, and 10 illustrate the parameters suggested by Lipinski drug-likeness and oral bioavailability, gastrointestinal adsorption (GIA), pan-assay interference compounds (PAINS) alert, synthetic accessibility, and bioavailability score. Tables 2 and 4–6 present some selected toxicity profiles of the designed compounds with better binding energies than dasabuvir or tipranavir. Determination of the drug-likeness of any potential drug candidate is paramount in the drug discovery/development process. The drug-likeness properties for potential drug candidates proposed by Lipinski40,54 and implemented in the Swiss-ADME web tool were utilized. It could be observed from Tables 1, 3, 9, and 10 that the designed compounds with better binding energies/inhibition constants than dasabuvir or tipranavir did not violate more than one of the rules proposed by Lipinski. This suggests good drug-likeness and oral bioavailability54 of the compounds. Other information in Tables 1, 3, 9, and 10 includes glycoprotein (P-gp) substrate, gastrointestinal adsorption (GIA), pan-assay interference compounds (PAINS) alert, bioavailability score, and synthetic accessibility. It is clear from Tables 1, 9, and 10 that the derivatives of dasabuvir are all not substrates for permeability glycoprotein (P-gp), the most important member of ATP-binding cassette (ABC) transporters for increasing active efflux through biological membranes,40 except 6d and 7d. Then, all of the derivatives of tipranavir (Tables 3 and 9) are substrates of P-gp. Again, derivatives of efavirenz (2e, 4e, and 5e) are P-gp substrates just like efavirenz (Tables 9 and 10). Previous reports have shown that P-gp inhibitors can overcome multidrug resistance and poor bioavailability problems of therapeutic P-gp substrates.55,56 The designed derivatives of dasabuvir and tipranavir showed low gastrointestinal absorption (GIA) like their parent compounds, except 23d, 24d, and 6d, while all derivatives of efavirenz showed high GIA like their parent compound (efavirenz) (Tables 9 and 10). The high GIA observed in the derivatives of dasabuvir was due to methylene (−CH2−) (Figure S3 in Supporting Information 3) with increased cLog P compared with that of dasabuvir (Tables S10 and S11). This is possible because the methyl group has been associated with increased lipophilicity, log P, of compounds,57 which can lead to their solubility in biomembranes. This has been implicated in the passage of compounds from the gastrointestinal tract to target tissues and transport through the bloodstream.57 However, 23d, 24d, and 6d were positive for Ames toxicity, hERG II inhibition, and hepatotoxicity (Figure S3 in Supporting Information 3). The designed compounds were predicted to possess no PAINS alert (Tables 1, 3, 9, and 10). This suggests that the designed compounds are not frequent hitters or promiscuous compounds and do not contain substructures that will show potent responses in assays irrespective of the protein target.40
Table 9. Drug-Likeness and Pharmacokinetic Parameters of Derivatives of Dasabuvir, Efavirenz, and Tipranavir That Are Noninhibitors of the Human Ether-a-go-go-Related Gene (hERG).
| ID | MW (g/mol) | #rotatable bonds | #H-bond acceptors | #H-bond donors | TPSA (Å2) | consensus log P | GI absorption | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | #Lipinski violations | bioavailability score | #PAINS alerts | synthetic accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5d | 496.54 | 6 | 7 | 3 | 151.76 | 2.85 | low | no | no | no | no | no | no | 0 | 0.11 | 0 | 3.55 |
| 4d | 493.58 | 6 | 6 | 3 | 138.31 | 3.38 | low | no | no | no | yes | no | yes | 0 | 0.55 | 0 | 3.73 |
| 18d | 496.54 | 6 | 7 | 3 | 151.76 | 2.72 | low | no | no | no | no | no | no | 0 | 0.11 | 0 | 3.55 |
| 24t | 646.67 | 14 | 11 | 3 | 151.27 | 5.27 | low | yes | no | no | yes | yes | yes | 1 | 0.11 | 0 | 5.44 |
| 5e | 333.72 | 1 | 6 | 1 | 79.13 | 3.3 | high | yes | yes | yes | yes | no | no | 0 | 0.55 | 0 | 3.76 |
| 4e | 317.26 | 1 | 7 | 1 | 79.13 | 3 | high | yes | no | yes | yes | no | no | 0 | 0.55 | 0 | 3.83 |
| 2e | 330.69 | 2 | 6 | 1 | 51.22 | 3.39 | high | yes | yes | yes | yes | no | yes | 0 | 0.55 | 0 | 3.61 |
| 15d | 493.57 | 6 | 5 | 2 | 118.64 | 3.8 | low | no | no | yes | yes | no | yes | 0 | 0.55 | 0 | 3.46 |
| 3e | 315.67 | 1 | 5 | 1 | 38.33 | 3.8 | high | yes | yes | yes | yes | no | no | 0 | 0.55 | 0 | 3.56 |
| 21t | 602.66 | 12 | 9 | 2 | 113.97 | 6.06 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.29 |
Table 3. Drug-Likeness and Pharmacokinetic Parameters of Some of the Designed Derivatives of Tipranavir Inhibitors of the Zika Virus RNA-Dependent RNA Polymerase and Methyltransferases (1t to 20t and 22t to 26t) Evaluated Using Swiss-ADME.
| ID | MW (g/mol) | #rotatable bonds | #H-bond acceptors | #H-bond donors | TPSA (Å2) | consensus log P | GI absorption | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | #Lipinski violations | Bioavailability score | #PAINS alerts | synthetic accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1t | 602.66 | 12 | 9 | 2 | 113.97 | 6.02 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.29 |
| 2t | 630.72 | 13 | 9 | 2 | 113.97 | 6.66 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.54 |
| 3t | 588.64 | 11 | 9 | 2 | 113.97 | 5.75 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.17 |
| 4t | 617.68 | 13 | 10 | 3 | 126 | 5.27 | low | yes | no | yes | no | yes | yes | 1 | 0.55 | 0 | 5.41 |
| 5t | 631.71 | 13 | 10 | 2 | 117.21 | 5.4 | low | yes | no | yes | no | yes | yes | 1 | 0.55 | 0 | 5.53 |
| 6t | 630.72 | 13 | 9 | 2 | 113.97 | 6.61 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.72 |
| 7t | 606.63 | 12 | 10 | 2 | 113.97 | 5.88 | low | yes | yes | yes | no | yes | yes | 1 | 0.56 | 0 | 5.28 |
| 8t | 631.71 | 14 | 10 | 3 | 139.99 | 5.45 | low | yes | no | yes | no | yes | yes | 1 | 0.55 | 0 | 5.47 |
| 9t | 634.73 | 12 | 9 | 2 | 152.77 | 5.87 | low | yes | no | yes | no | yes | yes | 1 | 0.11 | 0 | 5.42 |
| 10t | 614.68 | 12 | 9 | 2 | 113.97 | 6.29 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.36 |
| 11t | 612.66 | 12 | 9 | 2 | 113.97 | 6.03 | low | yes | no | yes | yes | yes | yes | 1 | 0.56 | 0 | 5.35 |
| 12t | 598.7 | 12 | 8 | 2 | 113.97 | 6.04 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.42 |
| 13t | 603.65 | 12 | 10 | 2 | 126.86 | 5.43 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.32 |
| 14t | 612.66 | 11 | 9 | 2 | 113.97 | 6 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.39 |
| 15t | 617.68 | 13 | 10 | 2 | 126.86 | 5.79 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.38 |
| 16t | 603.65 | 12 | 10 | 2 | 126.86 | 5.66 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.26 |
| 17t | 614.68 | 12 | 9 | 2 | 113.97 | 6.12 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.36 |
| 18t | 628.7 | 13 | 9 | 2 | 113.97 | 6.6 | low | yes | yes | yes | no | yes | yes | 1 | 0.56 | 0 | 5.46 |
| 19t | 607.62 | 12 | 11 | 2 | 126.86 | 5.14 | low | yes | no | yes | yes | yes | yes | 1 | 0.56 | 0 | 5.31 |
| 20t | 632.69 | 14 | 10 | 3 | 134.2 | 5.54 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.47 |
| 21t | 602.66 | 12 | 9 | 2 | 113.97 | 6.06 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.29 |
| 22t | 603.65 | 12 | 10 | 2 | 126.86 | 5.41 | low | yes | no | yes | yes | yes | yes | 1 | 0.56 | 0 | 5.41 |
| 23t | 607.62 | 12 | 11 | 2 | 126.86 | 5.16 | low | yes | no | yes | yes | yes | yes | 1 | 0.56 | 0 | 5.37 |
| 24t | 646.67 | 14 | 11 | 3 | 151.27 | 5.27 | low | yes | no | no | yes | yes | yes | 1 | 0.11 | 0 | 5.44 |
| 25t | 598.63 | 11 | 9 | 2 | 113.97 | 5.7 | low | yes | no | yes | yes | yes | yes | 1 | 0.56 | 0 | 5.23 |
| 26t | 616.69 | 12 | 9 | 2 | 113.97 | 6.21 | low | yes | no | yes | no | yes | yes | 1 | 0.56 | 0 | 5.41 |
Table 2. Predicted Toxicity of Some of the Designed Derivatives of Dasabuvir Inhibitors of the Zika Virus RNA-Dependent RNA Polymerase, Methyltransferase (1d to 14d), and RNA-Dependent RNA Polymerase (16d to 17d) Evaluated Using pkCSM.
| ID | formula | AMES toxicity | max. tolerated dose (human) (log mg/kg/day) | hERG I inhibitor | hERG II inhibitor | oral rat acute toxicity (LD50) (mol/kg) | oral rat chronic toxicity (LOAEL) (log mg/kg_bw/day) | hepatotoxicity | skin sensitization |
|---|---|---|---|---|---|---|---|---|---|
| 1d | C24H25N5O5S | no | 0.56 | no | yes | 3.084 | 1.771 | yes | no |
| 2d | C25H26N4O5S | no | 0.623 | no | yes | 2.947 | 1.138 | yes | no |
| 3d | C25H27N5O4S | no | 0.539 | no | yes | 2.971 | 0.679 | yes | no |
| 4d | C25H27N5O4S | no | 0.756 | no | no | 2.511 | 2.200 | yes | no |
| 5d | C24H24N4O6S | no | 0.652 | no | no | 2.658 | 1.956 | yes | no |
| 6d | C26H27N3O3 | yes | 0.375 | no | yes | 2.566 | 1.658 | yes | no |
| 7d | C24H24N4O6S | no | 0.716 | no | yes | 3.14 | 1.613 | yes | no |
| 8d | C25H27N5O4S | no | 0.64 | no | yes | 2.614 | 1.053 | yes | no |
| 9d | C25H26N4O4S2 | no | 0.273 | no | yes | 3.054 | 0.813 | yes | no |
| 10d | C25H27N5O5S | no | 0.519 | no | yes | 2.504 | 1.593 | yes | no |
| 11d | C25H27N5O5S | no | 0.658 | no | yes | 2.392 | 2.356 | yes | no |
| 12d | C25H25FN4O5S | no | 0.27 | no | yes | 2.862 | 1.564 | yes | no |
| 13d | C27H29N3O5S | yes | 0.166 | no | yes | 2.934 | 1.711 | yes | no |
| 14d | C25H26N4O5S | no | 0.324 | no | yes | 3.149 | 1.773 | yes | no |
| 15d | C26H27N3O5S | no | 0.149 | no | yes | 3.081 | 3.139 | yes | no |
| 16d | C26H28N4O5S | no | –0.044 | no | yes | 2.905 | 2.138 | yes | no |
| 17d | C26H27N3O5S | yes | 0.223 | no | yes | 2.943 | 1.928 | yes | no |
Prediction of synthetic accessibility (ease of synthesis) of bioactive compounds possessing drug-likeness is paramount for the selection of the most promising molecules for syntheses, biological assays, or other experiments.40,58 A good number of the designed compounds, especially 6d (3.26) and 3t (5.17), demonstrated ease of synthesis due to their lower synthetic accessibility score (Tables 1, 3, 9, and 10) compared with dasabuvir (3.46) and tipranavir (5.29), respectively. This is because the smaller the value, the easier the synthesis of a compound.58 Synthetic accessibility of designed derivatives of 1,2,4-oxadiazole has been reported elsewhere.45 The bioavailability scores of the designed compounds are similar to those of their parent compounds and within the probability of 55% or more, except 5d, 9t, 18d, and 24t, which showed 11%58 each (Tables 1, 3, 9, and 10). Similar observations have been made elsewhere.45
Prediction of interactions of molecules/compounds with cytochromes P450 (CYP) during drug discovery/development is very important. This is because cytochrome P450 and its isoenzymes play a key role in drug elimination through metabolic biotransformation.40 The propensity with which the designed compounds caused drug interactions through inhibition of CYPs, and the isoforms affected,40 was evaluated, and it shows that they are inhibitors of at least one CYP450 isoform, except 1d, 5d, and 18d, which are not inhibitors of any of the isoforms (Tables 1, 3, 9, and 10). However, the bioavailability score of 5d and 18d is 0.11 each, while that of 1d is 0.55 compared with 0.55 of dasabuvir. Previous reports revealed that many therapeutic compounds are a substrate of one of the five major cytochrome P450 isoforms (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4).59,60 Therefore, inhibition of one or more of the isoforms could lead to pharmacokinetics-related drug–drug interactions,61−63 which may result in toxic or other unwanted adverse effects.64 This suggests that 1d, 5d, and 18d, which are not inhibitors of any of the isoforms, will not lead to pharmacokinetics-related drug–drug interactions and toxic or other unwanted adverse effects. However, the isoforms not inhibited by the designed derivatives may selectively process the compounds for elimination through metabolic biotransformation such that inhibition by a specific inhibitor will lead to an increase in the exposure to the substrate.61 Also, CYP and P-gp can process small molecules synergistically to improve the protection of tissues and organisms.65 Again, 5d and 18d showed lower bioavailability scores (Tables 1 and 9).
The toxicity of the designed compounds with binding energies equal to or better than their respective parent compounds (dasabuvir, efavirenz, and tipranavir) was further assessed using the pkCSM web tool, and the results are presented in Tables 2 and 4–6 and Supporting Information 1. The results showed that six of the designed derivatives of dasabuvir (6d, 13d, 17d, 19d, 23d, and 24d) showed Ames toxicity, whereas derivatives of efavirenz and tipranavir did not (Tables 2 and 4–6). The Ames test is a short-term bacterial reverse mutation assay detecting a large number of compounds that can induce genetic damage and frameshift mutations.66 Toxicity is a leading cause of attrition at all stages of drug development,66 and the identification of compounds capable of inducing mutations has become an important procedure in safety assessment67 during drug discovery/development. This suggests that the designed derivatives without Ames toxicity are relatively safe than those that show Ames toxicity. No skin sensitization was observed in the designed derivatives of dasabuvir, efavirenz, and tipranavir (Tables 2 and4–6). This suggests that the compounds will not cause allergic reactions such as rash, blisters, or itch when in contact with the skin.68 Few of the designed compounds (12) with better binding energies are noninhibitors of human ether-a-go-go-related genes I and II (hERG I and hERG II) (Tables 5 and 6). The rest of the designed compounds are inhibitors of hERG II but not hERG I (Tables 2 and 4–6). Activation of hERG channels mediates cardiac action potential repolarization.69 Loss-of-function mutations in hERG and its inhibition can lead to inherited and acquired long QT syndrome (LQTS), respectively,69−71 which can in turn lead to fatal cardiac arrhythmia70 and sudden death.69 However, small molecules that increase/activate hERG have found application in the treatment of LQTS.69,71 This suggests that the identified noninhibitors of hERG I and hERG II are less cardiovascular toxic, making them safer than noninhibitors of only hERG I. Similar observations have been made for derivatives of 1,2,4-oxadiazole.45 Also, the predicted toxicities of designed derivatives of dasabuvir, efavirenz, and tipranavir that are noninhibitors of the human ether-a-go-go-related gene (hERG) and/or noninducers of hepatotoxicity, binding energies, bioactivities, drug-likeness, and pharmacokinetic parameters are presented in Tables 5–10. They showed no Ames toxicity, except 19d, and no skin sensitization (Table 7).
The predicted toxicity of designed derivatives of dasabuvir, efavirenz, and tipranavir that are noninhibitors of the human ether-a-go-go-related gene (hERG) and noninducers of hepatotoxicity is presented in Table 6. The rest of the designed compounds showed hepatoxicity just like their parent compounds, except efavirenz 3e. Reports have shown that drug-induced hepatotoxicity is the main cause of drug nonapproval and/or withdrawal.72,73 Therefore, the identification of non-hepatotoxic compounds early in drug discovery will offer drugs devoid of hepatotoxicity. This is important because drug-induced liver injury may lead to acute hepatitis and/or acute liver failures.72,74,75 Interestingly, only five compounds (19d, 20d, 21d, 22d, 1e) showed no hepatotoxicity (Table 6). They also showed no Ames toxicity, except 19d, no skin sensitization, hERG inhibition, and better Ki for RdRP (Table 8) but not MTase. Above all, they did not violate more than one Lipinski rule and showed no PAINS alerts (Table 10). Based on the toxicity profile of the designed compounds, it may be reasonable to classify 20d, 21d, 22d, and 1e as nontoxic and those in Tables 5, 7, and 9 (5d, 4d, 18d, 24t, 5e, 4e, and 2e) as moderately toxic (i.e., only hepatotoxic) than the rest of the compounds.
Table 8. Binding Energies and Bioactivities of Derivatives of Dasabuvir, Efavirenz, and Tipranavir That Are Not Inhibitors of the Human Ether-a-go-go-Related Gene (hERG) Nor Induce Hepatotoxicity.
| ID | binding energy for RdRP (kcal/mol) | Ki for RdRP (μM) | LE (kcal/mol/heavy atom) | LEscale | LELP | FQ |
|---|---|---|---|---|---|---|
| 19d | –7.55 ± 0.10 | 2.911069187 ± 0.449621 | 0.204054 | 0.269598 | 19.53404 | 0.756884 |
| 22d | –7.30 ± 0.00 | 4.396662616 ± 0.00000 | 0.202778 | 0.278385 | 17.41611 | 0.728408 |
| 20d | –7.10 ± 0.00 | 6.164344338 ± 0.00000 | 0.202857 | 0.287404 | 15.16782 | 0.705827 |
| 21d | –6.90 ± 0.00 | 8.642723911 ± 0.00000 | 0.191667 | 0.278385 | 18.17322 | 0.688495 |
| 1e | –6.68 ± 0.05 | 12.67519392 ± 1.115312 | 0.303409 | 0.428717 | 14.75335 | 0.707714 |
Structural Alerts for hERG II Inhibition and Hepatotoxicity
Molecular structures of the designed compounds that showed no hERG inhibition and/or hepatotoxicity (Table 10) including high GIA were visualized to identify structural alerts/features associated with the observed hERG II inhibition, high GIA, and/or hepatotoxicity (Figures 6–8 and S3 in Supporting Information 3). Such structural alerts are indicative of probable hERG II inhibition, high GIA, or hepatotoxicity. However, it is also possible that certain substitutions can block the resulting toxicity, absorption, or inhibition.76 It can be observed from Figure 6 that compounds 4d, 5d, and 18d contain quinoline or isoquinoline moieties. Liver tumors have been observed in rats and mice exposed to quinoline.77,78 This may be the case for compounds 4d, 5d, and 18d. However, this is unlikely since dasabuvir, which also showed hepatotoxicity, does not have a quinoline or isoquinoline moiety (Figure 6). Structural alerts for hepatotoxicity due to para-aminophenylsulphonamides such as sulfadimethoxine, sulfaphenazole, and sulfamethoxazole have been reported elsewhere.76 Since −NH– in the methanesulfonamide/sulfamic acid moieties (red color in Figure 6) and attached to the quinoline/isoquinoline moieties (green color in Figure 6) is common in the para-aminophenylsulphonamides and compounds 4d, 5d, 15d, and 18d, it may be responsible for the observed hepatotoxicity. Moreover, its absence in 19d to 22d (Figure 7) supports this view. Interestingly, sulfonamides/nitromethanesulfonamides have been linked to cases of hepatic injury and acute liver failure.76,79−81 It is unlikely that −NH– in the pyrimidine moiety is responsible for hepatotoxicity because it is also present in compounds 19d to 22d, which did not show hepatotoxicity. Ingestion/overdose of other drugs like acetaminophen (a drug containing −NH−) has been associated with hepatotoxicity82 through their conversion to a toxic metabolite, N-acetyl-p-benzoquinone imine.83 Again, high acute toxicity has been reported for some piperine amide analogues containing either −NH or −N–.84 Therefore, the observed hepatotoxicity of 4d, 5d, 15d, and 18d is likely due to the presence of −NH– in the methanesulfonamide/sulfamic acid moieties. Introduction of hydrophilic and/or polar hydroxyl groups (−NH, −N–, and/or −OH) in dasabuvir results in compounds 4d, 5d, and 18d with no hERG inhibition and containing quinoline or isoquinoline instead of the naphthalene moiety (Figure 6). Similar findings have been reported elsewhere.85 These modifications made 4d, 5d, and 18d have cLog P values of 3.38, 2.85, and 2.73, respectively, compared with 3.8 for dasabuvir (Table 6). It is widely admitted that compounds that have higher log P and pKa tend to be at higher risk of hERG inhibition,85 and this may be the case for dasabuvir. Quinoline-based kinase inhibitors with reduced hERG activity have been reported elsewhere.86 Again, drugs with a quinoline structure, particularly with high molecular polarity, can exert a significant potential hERG inhibitory activity.87 This may be the case for 4d, 5d, 18d, 19d, 20d, 21d, and 22d since they are more polar than dasabuvir. It is important to note that some hERG inhibition cannot be explained by the decrease in lipophilicity or basicity but by subtle changes in the compound that can lead to the flexibility of groups like methylene.85 The introduction of two fluorine (F) atoms and replacement of piperidine with piperazine leading to methylene (−CH2−) flexibility have been used to overcome hERG inhibition.85 Indeed, 19d, 20d, 21d, and 22d were a result of replacements of −NH– with −CH2–, −CH2CH2– or −CH2CH2CH2–, and −CH3 with −OH including replacement of the hydrogen atom (H) with −CH3 in the case of 21d. A previous study showed that modification of aryl-1H-imidazole compounds with the gem-dimethyl moiety between hydroxyl and amide groups has resulted in compounds with reduced hERG channel interactions.85 A closer look at Figures 4 and 5c shows that 24t was derived from tipranavir, 21t, by replacement of a hydrogen atom (H) with −CH2COOH. This caused a decrease in cLog P from 6.06 to 5.27 (Table 9), making 24t more polar with the added benefit of being a noninhibitor of hERG II (Table 5). However, this modification caused a decrease in the 24t bioavailability score, which is 0.11 compared with 0.56 of tipranavir. Also, the bioavailability score of 5d and 18d is 0.11 each compared with 0.55 of dasabuvir. The modifications in 5d, 18d, and 1d did not lead to inhibition of any of the CYP450 isoforms. This suggests that they will not lead to pharmacokinetics-related drug–drug interactions,61−63 which may result in toxic or other unwanted adverse effects.64 However, they showed a smaller bioavailability score than dasabuvir (Figure 8). Molecular interactions of 19d, 20d, 21d, and 22d with RNA-dependent RNA polymerase (Figures 9 and S4 in Supporting Information 3) involve hydrogen bonding with Arg731 and Arg739, just like designed compounds with better binding energies than 19d, 20d, 21d, and 22d. Introduction of sulfur (S), nitrogen (N), and fluorine (F) atoms in efavirenz led to 1e, 2e, and 4e (Figure 8), with cLog P values of 4.59, 2.39, and 3.0, respectively (Tables 9 and 10) when compared with 3.8 of efavirenz. These changes were due to the replacement of the oxygen (O) atom with S, Cl with F, and H with N. Compound 1e had higher cLog P because the S atom is less electronegative than the O atom. This implies hERG inhibition. Fluorine incorporation in 1,4-substituted piperidines during optimization showed improved hERG channel selectivity,85 while that of nitrogen atom on 7,9-dihydro-8H-purin-8-ones led to its triazolopyridine analogue, which keeps its activity toward DNA-dependent protein kinase with reduced hERG potency.85 This may be the case for the derivatives during interactions with zika virus RNA-dependent RNA polymerase and methyltransferase in vivo.
Figure 6.
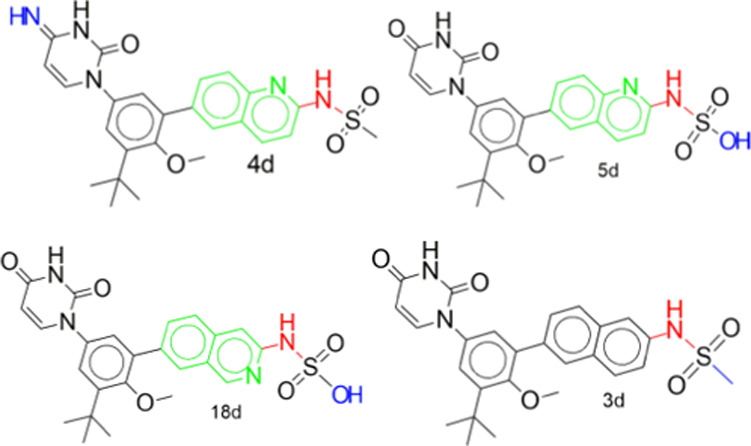
Hepatotoxic structural alerts for derivatives of dasabuvir. Compounds 4d, 5d, and 18d are noninhibitors of the human ether-a-go-go-related gene (hERG) but showed hepatotoxicity. Also, dasabuvir (15d) is the parent compound that showed both hERG II inhibition and hepatotoxicity.
Figure 8.
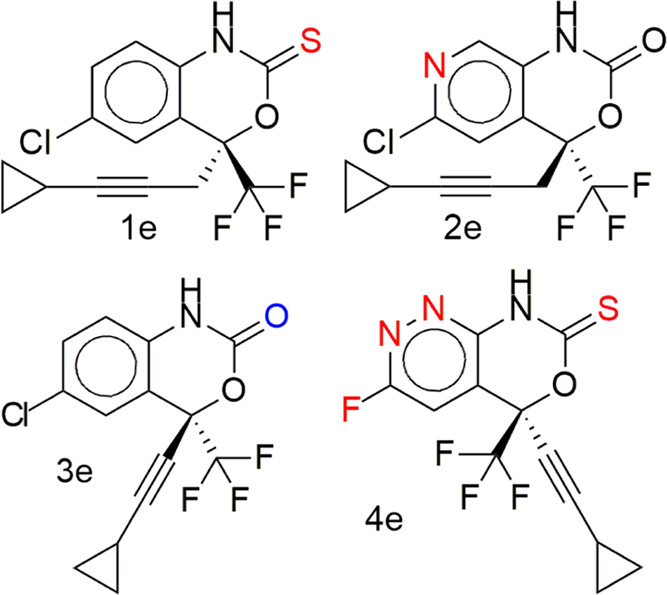
Structural alerts for derivatives of efavirenz. Compounds 1e and efavirenz (3e) are noninhibitors of the human ether-a-go-go-related gene (hERG) and do not show hepatotoxicity, while compounds 2e and 4e are noninhibitors of hERG but showed hepatotoxicity. It is also possible that pkCSM did not classify as a hepatotoxic agent due to certain substructural elements, but this is unlikely because pkCSM relies on learning patterns between chemical composition, similarity, pharmacokinetics, and safety properties for predictive models capable of identifying implicit patterns consistent and valid for unseen data.39 Again, the generation of toxic/reactive metabolites is a possibility for liver failure due to efavirenz.
Figure 7.
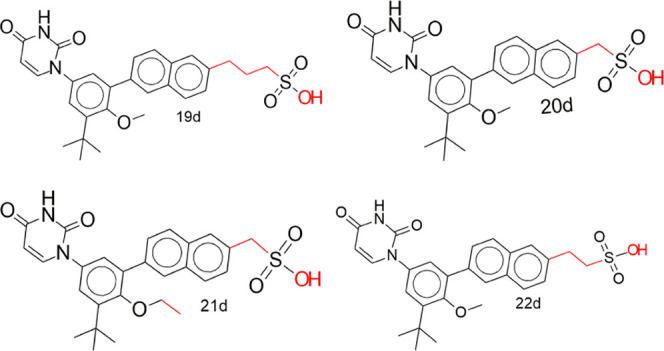
Structural determinants of noninhibition of hERG and no hepatotoxicity in derivatives of dasabuvir (19d, 20d, 21, and 22d) compared with dasabuvir (15d) in Figure 6, which showed both hERG II inhibition and hepatotoxicity.
Figure 9.

Molecular interactions of the designed derivatives of dasabuvir (19d and 22d) with the zika virus RNA-dependent RNA polymerase. Interactions of 19d (a) and 22d (b) with RNA-dependent RNA polymerase.
Hepatotoxicity of compounds 2e and 4e may be similar to that of adenosine-based reverse transcriptase inhibitors such as tenofovir, vidarabine, and clofarabine76 due to the substituted nitrogen atom(s). Efavirenz can be considered a safer drug for the liver.88 However, there are reported cases of acute liver failure among patients on efavirenz-based antiretroviral therapy due to efavirenz,89 but our findings did not mark efavirenz as a hepatotoxic agent.
Conclusions
Some of the designed compounds follow Lipinski’s rule-of-five, with no mutagenic, tumorigenic, or irritant properties. Again, the compounds did not show skin sensitization and PAINS alert while possessing good synthetic accessibilities. Compounds 20b, 21b, and 22b are noninhibitors of hERG while possessing no hepatotoxicity and Ames toxicity. Based on the toxicity profile of the designed compounds, 20d, 21d, 22d, and 1e are classified as nontoxic with the only limitation of CYP1A2, CYP2C19, and/or CYP2C9 inhibition, which can be managed with careful administration to avoid pharmacokinetics-related drug–drug interactions and toxicity or other unwanted adverse effects when developed and brought to the clinic. Replacement of −CH3 and −NH– in the methanesulfonamide moiety of dasabuvir with −OH and −CH2– or −CH2CH2– respectively improved the safety/toxicity profiles of 20d, 21d, and 22d. Hepatotoxicity observed in some of the derivatives of dasabuvir (5d, 4d, 18d) is likely due to the presence of −NH– in their methanesulfonamide or sulfamic acid moieties. The designed compounds are potent inhibitors of N-7 and 2′ methylation activities of zika virus MTase, which can lead to virus death and/or the ability to not evade host immune response. Also, they are potent inhibitors of the virus RNA synthesis through interactions with amino acid residues in the priming loop and/or “N-pocket” in the virus RNA-dependent RNA polymerase. Synthesis and wet laboratory validations of the designed compounds against the zika virus are recommended.
Acknowledgments
F.C.E. is grateful for the 2020 African-German Network of Excellence in Science (AGNES) Grant for Junior Researchers from the African-German Network of Excellence in Science supported by Alexander von Humboldt Foundation (AvH) and the German Federal Ministry of Education and Research (BMBF).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c03945.
Designed derivatives of dasabuvir, efavirenz, and tipranavir; their binding energies and bioactivities; predicted ADME properties evaluated with Swiss-ADME; and toxicity and other parameters evaluated with pkCSM (Supporting Information 1) (XLSX)
Shell scripts used for extraction of calculated parameters and preparation of designed derivatives for docking simulations (Supporting Information 2) (TXT)
Various tables and figures supporting the study (Supporting Information 3) (PDF)
The authors declare no competing financial interest.
Notes
The primary data set (dasabuvir [PubChem identifier: CID 56640146], efavirenz [PubChem identifier: CID 64139] and tipranavir [PubChem identifier: CID 54682461]) used is available in the public domain at https://pubchem.ncbi.nlm.nih.gov/.
Notes
The data that support the findings of this study are available as supporting information/data and may be found in the online version of the article at the publisher’s website.
Supplementary Material
References
- Roiz D.; Wilson A. L.; Scott T. W.; Fonseca D. M.; Jourdain F.; Müller P.; Velayudhan R.; Corbel V. Integrated Aedes management for the control of Aedes-borne diseases. PLoS Neglected Trop. Dis. 2018, 12, e0006845 10.1371/journal.pntd.0006845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanik M.; Valdes J. J.; Ezebuo F. C.; Haviernik J.; Uzochukwu I. C.; Fojtikova M.; Salat J.; Eyer L.; Ruzek D. FDA-approved drugs efavirenz, tipranavir, and dasabuvir inhibit replication of multiple flaviviruses in vero cells. Microorganisms 2020, 8, 599 10.3390/microorganisms8040599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubry F.; Darmuzey M.; Lequime S.; Delang L.; Romero-vivas C. M.; Jarman R. G.; Diagne C. T.; Faye O.; Faye O.; Sall A. A.; Neyts J.; Nguyen L.; Kaptein S. J. F.; Lambrechts L.; et al. Recent African strains of Zika virus display higher transmissibility and fetal pathogenicity than Asian strains. Nat. Commun. 2021, 12, 916 10.1038/s41467-021-21199-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard B.; Barral K.; Lichière J.; Selisko B.; Martin B.; Aouadi W.; Lombardia M. O.; Debart F.; Vasseur J.-J.; Guillemot J. C.; Canard B.; Decroly E. Zika Virus Methyltransferase: Structure and Functions for Drug Design Perspectives. J. Virol. 2017, 91, e02202-16 10.1128/jvi.02202-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.; Yi G.; Du F.; Chuang Y.; Vaughan R. C.; Sankaran B.; Kao C. C.; Li P. Structure and function of the Zika virus full-length NS5 protein. Nat. Commun. 2017, 8, 14762 10.1038/ncomms14762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shwe D.; Shehu N.; Pam V.; Keneth O.; Gomerep S.; Isa S.; Yilgwan C.; Okolo O.; Abok I.; Obishakin E.; Egah D.; Oguche S. Epidemiologic review of zika virus disease. Niger. J. Med. 2018, 27, 180. 10.4103/1115-2613.278777. [DOI] [Google Scholar]
- Lucey D.; Gostin L. O. A yellow fever epidemic. a new global health emergency?. JAMA, J. Am. Med. Assoc. 2016, 315, 2661–2662. 10.1001/jama.2016.6606. [DOI] [PubMed] [Google Scholar]
- Herrera B. B.; Chang C. A.; Hamel D. J.; Mboup S.; Ndiaye D.; Imade G.; Okpokwu J.; Agbaji O.; Bei A. K.; Kanki P. J. Continued Transmission of Zika Virus in Humans in West Africa, 1992-2016. J. Infect. Dis. 2017, 215, 1546–1550. 10.1093/infdis/jix182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klofstad C. A.; Uscinski J. E.; Connolly J. M.; West J. P. What drives people to believe in Zika conspiracy theories?. Palgrave Commun. 2019, 5, 1–8. 10.1057/s41599-019-0243-8. [DOI] [Google Scholar]
- Phillips S. Teaching Public, Scientific Controversy: Genetically Modified Mosquitoes in the Technical Writing Classroom. J. Tech. Writing Commun. 2019, 49, 51–69. 10.1177/0047281617744507. [DOI] [Google Scholar]
- Waltz E. First genetically modified mosquitoes released in the United States. Nature 2021, 593, 175–176. 10.1038/d41586-021-01186-6. [DOI] [PubMed] [Google Scholar]
- Hayden E. C. Zika highlights role of fetal-tissue research. Nature 2016, 532, 16. 10.1038/nature.2016.19655. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Feng T.; Cheng J.; Li Y.; Yin X.; Zeng W.; Jin X.; Li Y.; Guo F.; Jin T. Structure of the NS5 methyltransferase from Zika virus and implications in inhibitor design. Biochem. Biophys. Res. Commun. 2017, 492, 624–630. 10.1016/j.bbrc.2016.11.098. [DOI] [PubMed] [Google Scholar]
- Ramharack P.; Soliman M. E. S. Zika Virus NS5 Protein Potential Inhibitors: An Enhanced In silico Approach in Drug Discovery. J. Biomol. Struct. Dyn. 2018, 36, 1118–1133. 10.1080/07391102.2017.1313175. [DOI] [PubMed] [Google Scholar]
- Noreen; Ali R.; Lal Badshah S.; Faheem M.; Wajid Abbasi S.; Ullah R.; Bari A.; Babar Jamal S.; Majid Mahmood H.; Haider A.; Haider S. Identification of potential inhibitors of Zika virus NS5 RNA-dependent RNA polymerase through virtual screening and molecular dynamic simulations. Saudi Pharm. J. 2020, 28, 1580–1591. 10.1016/j.jsps.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharbi-Ayachi A.; Santhanakrishnan S.; Wong Y. H.; Chan K. W. K.; Tan S. T.; Bates R. W.; Vasudevan S. G.; El Sahili A.; Lescar J. Non-nucleoside Inhibitors of Zika Virus RNA-Dependent RNA Polymerase. J. Virol. 2020, 94, e00794-20 10.1128/jvi.00794-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spizzichino S.; Mattedi G.; Lauder K.; Valle C.; Aouadi W.; Canard B.; Decroly E.; Kaptein S. J. F.; Neyts J.; Graham C.; Sule Z.; Barlow D. J.; Silvestri R.; Castagnolo D. Design, Synthesis and Discovery of N,N’-Carbazoyl-aryl-urea Inhibitors of Zika NS5 Methyltransferase and Virus Replication. ChemMedChem 2020, 15, 385–390. 10.1002/cmdc.201900533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatchez J. A.; Tran L. T.; Li J.; Luan Y.; Siqueira-Neto J. L.; Li R. Drugs for the Treatment of Zika Virus Infection. J. Med. Chem. 2020, 63, 470–489. 10.1021/acs.jmedchem.9b00775. [DOI] [PubMed] [Google Scholar]
- Song W.; Zhang H.; Zhang Y.; Li R.; Han Y.; Lin Y.; Jiang J. Repurposing clinical drugs is a promising strategy to discover drugs against Zika virus infection. Front. Med. 2021, 15, 404–415. 10.1007/s11684-021-0834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivella J. P.; Gutierrez J.; Martin P. Dasabuvir: A new direct antiviral agent for the treatment of hepatitis C. Expert Opin. Pharmacother. 2015, 16, 617–624. 10.1517/14656566.2015.1012493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Awady M. K.; Dawood R. M.. Resistance to Direct-Acting Antiviral Agents in Treatment of Hepatitis C Virus Infections. Update on Hepatitis C; IntechOpen, 2017; pp 93–108. [Google Scholar]
- Bastys T.; Gapsys V.; Walter H.; Heger E.; Doncheva N. T.; Kaiser R.; De Groot B. L.; Kalinina O. V. Non-active site mutants of HIV-1 protease influence resistance and sensitisation towards protease inhibitors. Retrovirology 2020, 17, 13 10.1186/s12977-020-00520-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moraes Filho A. V.; Carvalho C. d. J. S.; Carneiro C. C.; do Vale C. R.; da Silva Lima D. C.; Carvalho W. F.; Vieira T. B.; De Silva D. M. E.; Cunha K. S.; Chen-Chen L. Genotoxic and cytotoxic effects of antiretroviral combinations in mice bone marrow. PLoS One 2016, 11, e0165706 10.1371/journal.pone.0165706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usach I.; Melis V.; Peris J. E. Non-nucleoside reverse transcriptase inhibitors: A review on pharmacokinetics, pharmacodynamics, safety and tolerability. J. Int. AIDS Soc. 2013, 16, 18567 10.7448/IAS.16.1.18567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Thurmond S.; Hai R.; Song J. Structure and function of Zika virus NS5 protein: perspectives for drug design. Cell. Mol. Life Sci. 2018, 75, 1723–1736. 10.1007/s00018-018-2751-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshahawi H.; Hassan S. S.; Balasubramaniam V. Importance of Zika virus NS5 protein for viral replication. Pathogens 2019, 8, 169 10.3390/pathogens8040169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hercik K.; Brynda J.; Nencka R.; Boura E. Structural basis of Zika virus methyltransferase inhibition by sinefungin. Arch Virol. 2017, 162, 2091–2096. 10.1007/s00705-017-3345-x. [DOI] [PubMed] [Google Scholar]
- Ezebuo F. C.; Kushwaha P. P.; Singh A. K.. In-Silico Methods of Drug Design: Molecular Simulations and Free Energy Calculations. In Phytochemistry: An In-Silico and In-Vitro Update; Kumar S.; Egbuna C., Eds.; Springer Nature: Singapore, 2019; pp 521–533. [Google Scholar]
- Kumar S.; Bhardwaj V. K.; Singh R.; Das P.; Purohit R. Identification of acridinedione scaffolds as potential inhibitor of DENV-2 C protein: An in silico strategy to combat dengue. J. Cell. Biochem. 2022, 123, 935–946. 10.1002/jcb.30237. [DOI] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; Zaslavsky L.; Zhang J.; Bolton E. E. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander T.; Freyss J.; Von Korff M.; Rufener C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An Open chemical toolbox. J. Cheminf. 2011, 3, 33 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Battistuz T.; Bhat T. N.; Bluhm W. F.; Bourne P. E.; Burkhardt K.; Feng Z.; Gilliland G. L.; Iype L.; Jain S.; Fagan P.; Marvin J.; Padilla D.; Ravichandran V.; Schneider B.; Thanki N.; Weissig H.; Westbrook J. D.; Zardecki C. The protein data bank. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 899–907. 10.1107/S0907444902003451. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J.; Yu J.; Huang Y.; He Z.; Luo J.; Wu Y.; Zheng Y.; Wu J.; Zhu X.; Wang H.; Li M. Antibiotic fidaxomicin is an RdRp inhibitor as a potential new therapeutic agent against Zika virus. BMC Med. 2020, 18, 204 10.1186/s12916-020-01663-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulaiman K. O.; Kolapo T. U.; Onawole A. T.; Islam M. A.; Adegoke R. O.; Badmus S. O. Molecular dynamics and combined docking studies for the identification of Zaire ebola virus inhibitors. J. Biomol. Struct. Dyn. 2019, 37, 3029–3040. 10.1080/07391102.2018.1506362. [DOI] [PubMed] [Google Scholar]
- Cavalluzzi M. M.; Mangiatordi G. F.; Nicolotti O.; Lentini G. Ligand efficiency metrics in drug discovery: the pros and cons from a practical perspective. Expert Opin. Drug Discovery 2017, 12, 1087–1104. 10.1080/17460441.2017.1365056. [DOI] [PubMed] [Google Scholar]
- Pires D. E. V.; Blundell T. L.; Ascher D. B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews A. W.; Lijinsky W.; Snyder S. W. Mutagenicity of amine drugs and their products of nitrosation. Mutat. Res., Genet. Toxicol. 1984, 135, 105–108. 10.1016/0165-1218(84)90162-9. [DOI] [PubMed] [Google Scholar]
- Nohmi T.; Watanabe M. Mutagenicity of carcinogenic heterocyclic amines in Salmonella typhimurium YG strains and transgenic rodents including gpt delta. Genes Environ. 2021, 43, 38 10.1186/s41021-021-00207-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T. N.; Henary M. Synthesis and Applications of Nitrogen-Containing Heterocycles as Antiviral Agents. Molecules 2022, 27, 2700 10.3390/molecules27092700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talamas F. X.; Abbot S. C.; Anand S.; Brameld K. A.; Carter D. S.; Chen J.; Davis D.; de Vicente J.; Fung A. D.; Gong L.; Harris S. F.; Inbar P.; Labadie S. S.; Lee E. K.; Lemoine R.; Le Pogam S.; Leveque V.; Li J.; McIntosh J.; Nájera I.; Park J.; Railkar A.; Rajyaguru S.; Sangi M.; Schoenfeld R. C.; Staben L. R.; Tan Y.; Taygerly J. P.; Villaseñor A. G.; Weller P. E. Discovery of N-[4-[6-tert-Butyl-5-methoxy-8-(6-methoxy-2-oxo-1H-pyridin-3-yl)-3-quinolyl]phenyl] methanesulfonamide (RG7109), a Potent Inhibitor of the Hepatitis C Virus NS5B Polymerase. J. Med. Chem. 2014, 57, 1914–1931. 10.1021/jm401329s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adawara S. N.; Shallangwa G. A.; Mamza P. A.; Abdulkadir I. Chemoinformatic design and profiling of some derivatives of 1, 2, 4-oxadiazole as potential dengue virus NS-5 inhibitors. Bull. Natl. Res. Cent. 2022, 46, 65 10.1186/s42269-022-00755-7. [DOI] [Google Scholar]
- Peerzada M. N.; Khan P.; Khan N. S.; Avecilla F.; Siddiqui S. M.; Hassan M. I.; Azam A. Design and development of small-molecule arylaldoxime/5-nitroimidazole hybrids as potent inhibitors of MARK4: A promising approach for target-based cancer therapy. ACS Omega 2020, 5, 22759–22771. 10.1021/acsomega.0c01703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen P.; Baz M.; Boivin G.; Lin S. X. Structural Insight into NS5 of Zika Virus Leading to the Discovery of MTase Inhibitors. J. Am. Chem. Soc. 2016, 138, 16212–16215. 10.1021/jacs.6b10399. [DOI] [PubMed] [Google Scholar]
- Song W.; Zhang H.; Zhang Y.; Chen Y.; Lin Y.; Han Y.; Jiang J. Identification and Characterization of Zika Virus NS5 Methyltransferase Inhibitors. Front. Cell. Infect. Microbiol. 2021, 11, 665379 10.3389/fcimb.2021.665379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. Novel guanosine derivatives against Zika virus polymerase in silico. J. Med. Virol. 2020, 92, 11–16. 10.1002/jmv.25573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egieyeh S. A.; Syce J.; Malan S. F.; Christoffels A. Prioritization of anti-malarial hits from nature: Chemo-informatic profiling of natural products with in vitro antiplasmodial activities and currently registered anti-malarial drugs. Malar. J. 2016, 15, 50 10.1186/s12936-016-1087-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coloma J.; Jain R.; Rajashankar K. R.; García-Sastre A.; Aggarwal A. K. Structures of NS5 Methyltransferase from Zika Virus. Cell Rep. 2016, 16, 3097–3102. 10.1016/j.celrep.2016.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams R. P. M.; Solis J.; Nath A. Therapeutic Approaches for Zika Virus Infection of the Nervous System. Neurotherapeutics 2017, 14, 1027–1048. 10.1007/s13311-017-0575-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schake D. How flexible is tipranavir in complex with the HIV-1 protease active site?. AIDS 2004, 18, 579–580. 10.1097/00002030-200402200-00033. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Delivery Rev. 2016, 101, 34–41. 10.1016/j.addr.2016.04.029. [DOI] [PubMed] [Google Scholar]
- Srivalli K. M. R.; Lakshmi P. K. Overview of P-glycoprotein inhibitors: a rational outlook. Braz. J. Pharm. Sci. 2012, 48, 353–367. 10.1590/S1984-82502012000300002. [DOI] [Google Scholar]
- Nanayakkara A. K.; Follit C. A.; Gang C.; Williams N. S.; et al. Targeted inhibitors of P-glycoprotein increase mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967 10.1038/s41598-018-19325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro E. J.; Kümmerle A. E.; Fraga C. A. M. The methylation effect in medicinal chemistry. Chem. Rev. 2011, 111, 5215–5246. 10.1021/cr200060g. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminf. 2009, 1, 8 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C. R.; Smith G.; Smith R. L. Pharmacogenetics. Br. Med. J. 2000, 320, 987–990. 10.1136/bmj.320.7240.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L. The role of drug metabolizing enzymes in clearance. Expert Opin. Drug Metab. Toxicol. 2014, 10, 379–393. 10.1517/17425255.2014.876006. [DOI] [PubMed] [Google Scholar]
- Hakkola J.; Hukkanen J.; Turpeinen M.; Pelkonen O. Inhibition and induction of CYP enzymes in humans: an update. Arch. Toxicol. 2020, 94, 3671. 10.1007/s00204-020-02936-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg P. F. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab. Rev. 2002, 34, 17–35. 10.1081/DMR-120001387. [DOI] [PubMed] [Google Scholar]
- Huang S. M.; Strong J. M.; Zhang L.; Reynolds K. S.; Nallani S.; Temple R.; Abraham S.; Al Habet S.; Baweja R. K.; Burckart G. J.; Chung S.; Colangelo P.; Frucht D.; Green M. D.; Hepp P.; Karnaukhova E.; Ko H. S.; Lee J. I.; Marroum P. J.; Norden J. M.; Qiu W.; Rahman A.; Sobel S.; Stifano T.; Thummel K.; Wei X.; Yasuda S.; Zheng J. H.; Zhao H.; Lesko L. J. Drug interactions/review: New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 2008, 48, 662–670. 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- Kirchmair J.; Göller A. H.; Lang D.; Kunze J.; Testa B.; Wilson I. D.; Glen R. C.; Schneider G. Predicting drug metabolism: Experiment and/or computation?. Nat. Rev. Drug Discovery 2015, 14, 387–404. 10.1038/nrd4581. [DOI] [PubMed] [Google Scholar]
- van Waterschoot R. A. B.; Schinkel A. H. A Critical Analysis of the Interplay between Cytochrome P450 3A and P-Glycoprotein: Recent insights from knockout and transgenic mice. Pharmacol. Rev. 2011, 63, 390–410. 10.1124/pr.110.002584. [DOI] [PubMed] [Google Scholar]
- Xu C.; Cheng F.; Chen L.; Du Z.; Li W.; Liu G.; Lee P. W.; Tang Y. In silico Prediction of Chemical Ames Mutagenicity. J. Chem. Inf. Model. 2012, 52, 2840–2847. 10.1021/ci300400a. [DOI] [PubMed] [Google Scholar]
- Mortelmans K.; Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2000, 455, 29–60. 10.1016/S0027-5107(00)00064-6. [DOI] [PubMed] [Google Scholar]
- Ta G. H.; Weng C.; Leong M. K. In silico Prediction of Skin Sensitization: Quo vadis?. Front. Pharmacol. 2021, 12, 655771 10.3389/fphar.2021.655771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M.; Sachse F. B.; Sanguinetti M. C. Structural basis of action for a human ether-a-go-go-related gene 1 potassium channel activator. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 13827–13832. 10.1073/pnas.0703934104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Balakin K. V.; Savchuk N.; Ivanenkov Y. Insights for Human Ether-a-Go-Go-Related Gene Potassium Channel Inhibition Using Recursive Partitioning and Kohonen and Sammon Mapping Techniques. J. Med. Chem. 2006, 49, 5059–5071. 10.1021/jm060076r. [DOI] [PubMed] [Google Scholar]
- Zhou P.-z.; Babcock J.; Liu L.; Li M.; Gao Z. Activation of human ether-a-go-go related gene (hERG) potassium channels by small molecules. Acta Pharmacol. Sin. 2011, 32, 781–788. 10.1038/aps.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D.; Cho W. C.; Upadhyay G. Drug-Induced Liver Toxicity and Prevention by Herbal Antioxidants: An Overview. Front. Physiol. 2016, 6, 363 10.3389/fphys.2015.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier L.; Larrey D. Drug-induced liver injury: Biomarkers, requirements, candidates, and validation. Front. Pharmacol. 2019, 10, 1482 10.3389/fphar.2019.01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray K. F.; Hadzic N.; Wirth S.; Bassett M.; Kelly D. Drug-related hepatotoxicity and acute liver failure. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 395–405. 10.1097/mpg.0b013e3181709464. [DOI] [PubMed] [Google Scholar]
- Pandit A.; Sachdeva T.; Bafna P. Drug-induced hepatotoxicity: A review. J. Appl. Pharm. Sci. 2012, 2, 233–243. 10.7324/JAPS.2012.2541. [DOI] [Google Scholar]
- Hewitt M.; Enoch S. J.; Madden J. C.; Przybylak K. R.; Cronin M. T. D. Hepatotoxicity: A scheme for generating chemical categories for read-across, structural alerts and insights into mechanism(s) of action. Crit. Rev. Toxicol. 2013, 43, 537–558. 10.3109/10408444.2013.811215. [DOI] [PubMed] [Google Scholar]
- Reign G.; McMahon H.; Ishizaki M.; Ohara T.; Shimane K.; Esumi Y.; Green C.; Tyson C.; Ninomiya S. I. Cytochrome P450 species involved in the metabolism of quinoline. Carcinogenesis 1996, 17, 1989–1996. 10.1093/carcin/17.9.1989. [DOI] [PubMed] [Google Scholar]
- EPA . Toxicological Review of Quinoline, Assessment (Issue 91), EPA/635/R-01/005; U.S. Environmental Protection Agency, 2001.
- Dujovne C.; Chan C. H.; Zimmerman H. J. Sulfonamide hepatic injury. review of the literature and report of a case due to sulfamethoxazole. N. Engl. J. Med. 1967, 277, 785–788. 10.1056/NEJM196710122771503. [DOI] [PubMed] [Google Scholar]
- Qazi A. F.; Shaikh D. I. N. M.; Shaikh H. Z. I. A. The Homeostasis Boosting Effects of Curcuma Longa in Nitro-Methane Sulfonamide Induced Hepatic Stress in Male Albino Wistar Rats. Isra Med. J. 2014, 6, 146–150. [Google Scholar]
- Qazi A. F.; Shaikh D. M. Curcuma Longa Against Nitromethanesulfonamide Induced Hepatic Injury In A Rat Model. IOSR J. Dent. Med. Sci. 2017, 16, 71–74. 10.9790/0853-1601067174. [DOI] [Google Scholar]
- Yoon E.; Babar A.; Choudhary M.; Kutner M.; Pyrsopoulos N. Acetaminophen-induced hepatotoxicity: A comprehensive update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. 10.14218/JCTH.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender R. P.; Lindsey R. H.; Burden D. A.; Osheroff N. N-Acetyl-p-benzoquinone Imine, the Toxic Metabolite of Acetaminophen, Is a Topoisomerase II Poison. Biochemistry 2004, 43, 3731–3739. 10.1021/bi036107r. [DOI] [PubMed] [Google Scholar]
- Pereira E.; Farias E.; Ribeiro A.; Alvarenga E.; Aguiar A.; Ferreira J.; Picanço M. Toxicity of piperine amide analogs toward the tomato pinworm tuta absoluta (lepidoptera: Gelechiidae) and risk assessment for two predators. Horticulturae 2019, 5, 70 10.3390/horticulturae5040070. [DOI] [Google Scholar]
- Garrido A.; Lepailleur A.; Mignani S. M.; Dallemagne P.; Rochais C. hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem. 2020, 195, 112290 10.1016/j.ejmech.2020.112290. [DOI] [PubMed] [Google Scholar]
- Haile P. A.; Casillas L. N.; Bury M. J.; Mehlmann J. F.; Singhaus R.; Charnley A. K.; Hughes T. V.; Demartino M. P.; Wang G. Z.; Romano J. J.; Dong X.; Plotnikov N. V.; Lakdawala A. S.; Duraiswami C.; Convery M. A.; Votta B. J.; Lipshutz D. B.; Desai B. M.; Swift B.; Capriotti C. A.; Berger S. B.; Mahajan M. K.; Reilly M. A.; Rivera E. J.; Sun H. H.; Nagilla R.; LePage C.; Ouellette M. T.; Totoritis R. D.; Donovan B. T.; Brown B. S.; Chaudhary K. W.; Gough P. J.; Bertin J.; Marquis R. W. Erratum: Identification of Quinoline-Based RIP2 Kinase Inhibitors with an Improved Therapeutic Index to the hERG Ion Channel (ACS Med. Chem. Lett. (2018) 9:10 (1039-1044) DOI: 10.1021/acsmedchemlett.8b00344). ACS Med. Chem. Lett. 2020, 11, 1353. 10.1021/acsmedchemlett.0c00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R.; Li P. Computational and experimental studies on the inhibitory mechanism of hydroxychloroquine on hERG. Toxicology 2021, 458, 152822 10.1016/j.tox.2021.152822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero A.; Mira J. A.; Pineda J. A. Liver toxicity induced by non-nucleoside reverse transcriptase inhibitors. J. Antimicrob. Chemother. 2007, 59, 342–346. 10.1093/jac/dkl524. [DOI] [PubMed] [Google Scholar]
- Segamwenge I. L.; Bernard M. K. Acute Liver Failure among Patients on Efavirenz-Based Antiretroviral Therapy. Case Rep. Hepatol. 2018, 2018, 1270716 10.1155/2018/1270716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.