

Abstract

Carbon monoxide (CO) is the second most abundant identified product of dissolved organic matter (DOM) photodegradation after CO2, but its formation mechanism remains unknown. Previous work showed that aqueous photodegradation of methoxy-substituted aromatics (ArOCH3) produces CO considerably more efficiently than aromatic carbonyls. Following on this precedent, we propose that the methoxy aromatic groups of lignin act as the C source for the photochemical formation of CO from terrestrial DOM via a two-step pathway: formal hydrolytic demethylation to methanol and methanol oxidation to CO. To test the reasonableness of this mechanism, we investigated the photochemistry of eight lignin model compounds. We first observed that initial CO production rates are positively correlated with initial substrate degradation rates only for models containing at least one ArOCH3 group, regardless of other structural features. We then confirmed that all ArOCH3-containing substrates undergo formal hydrolytic demethylation by detecting methanol and the corresponding phenolic transformation products. Finally, we showed that hydroxyl radicals, likely oxidants to initiate methanol oxidation to CO, form during irradiation of all models. This work proposes an explicit mechanism linking ubiquitous, abundant, and easily quantifiable DOM functionalities to CO photoproduction. Our results further hint that methanol may be an abundant (yet overlooked) DOM photoproduct and a likely precursor of formaldehyde, formic acid, and CO2 and that lignin photodegradation may represent a source of hydroxyl radicals.
Keywords: dissolved organic matter photodegradation, carbon monoxide, reaction mechanism, lignin, methanol, hydroxyl radicals, carbon cycling, methoxy-substituted aromatics
Short abstract
The chemical complexity of dissolved organic matter (DOM) hinders our understanding of its degradation processes. This study reports the first explicit mechanism for the formation of carbon monoxide from the photodegradation of terrestrial DOM.
Introduction
Carbon monoxide (CO) is a trace gas that plays an important role in modulating hydroxyl radical (OH•) concentrations in the troposphere, therefore influencing the residence time of greenhouse gases such as methane and halocarbons.1−4 Among other sources, CO can be produced from the photodegradation of dissolved organic matter (DOM). Albeit contributing only a minimal fraction of the global CO budget,1 photochemical production from DOM photolysis is relevant in remote ocean regions or in environments characterized by significant inputs of terrestrial DOM.1,5 CO is also involved in the production of carbonyl sulfide (OCS) from DOM photolysis.6 Photochemical processes are significant players in the global OCS budget, but their contributions are not yet well constrained.7−10 An improved understanding of OCS sources and sinks can indirectly contribute to better climate simulations, as this gas is used as a tracer of gross primary productivity.8,11 In addition, CO has been used as a proxy for the photoproduction of CO2, CH4, and biolabile organic carbon5,12−16 (even though this approach proved to be inaccurate for CO2)5,17,18 and for developing models of mixed layer processes.5,19,20
Despite the interest in CO biogeochemistry, little is currently known about its photochemical formation mechanism. Redden21 hypothesized that acetone, acetaldehyde, and other carbonyl-containing DOM photoproducts might undergo Norrish type I fragmentation and release CO. Others proposed that photosensitized degradation of α-ketoacids (e.g., pyruvic acid) may produce acetyl radicals, which then fragment to CO.6 Even if these processes are well-established in traditional photochemistry,22−27 low environmental concentrations and yields limit the role of low-molecular-weight carbonyls as CO precursors in natural environments.6,21 Furthermore, Stubbins et al.28 observed up to 300 times higher CO production from photolysis of ethoxy- and methoxy-substituted phenols compared to aromatic carbonyls. This finding reinforces the idea that carbonyls in general, not only low-molecular-weight ones, are secondary CO precursors and hints at an alternative production mechanism.
Following on the results reported by Stubbins et al.28, we propose that lignin is an environmentally relevant precursor of CO. Lignin is the second most abundant biopolymer on Earth after cellulose, consisting of three aromatic core units [p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S)] connected mostly via β-O-4 linkages (Figure 1).29−31G and S functionalities, which together account for 65 to 95% of the total core units across all plant types,29 contain one and two aromatic methoxy groups (ArOCH3), respectively. Lignin is a major component of terrestrial DOM32−34 and also comprises a small portion of marine DOM,33 making it a quantitatively relevant source of methoxy aromatics in aquatic ecosystems.
Figure 1.

Schematic of the proposed two-step mechanism. This example shows the reactivity of a G unit, but a similar process can be anticipated for the S residue. The molecule on the far left depicts a possible structure of native lignin, with highlighted an aromatic G core unit (gray highlight), β-O-4 bonds (bold bonds), and aromatic methoxy groups (orange highlights). In native lignin, the α-carbon is typically an alcohol; this group can be converted to a ketone during (photo)oxidation.31,35 Letters H, G, and S indicate p-hydroxyphenyl, guaiacyl, and syringyl units, respectively. RIs is an abbreviation for reactive intermediates.
We hypothesize that lignin’s aromatic methoxy groups act as the C source for the formation of CO via a two-step mechanism: conversion of the aromatic methoxy group to methanol via direct photolysis; and methanol oxidation to CO via indirect photolysis (Figure 1). Two sets of observations justify this hypothesis. Several authors reported evidence of ArO-CH3 and Ar-OCH3 cleavage of lignin model compounds,36−43 with consequent formation of methane, ethane, and chloroform (ArO-CH3 cleavage),38−40 or methanol (Ar-OCH3 cleavage).38,41 In particular, Dallin et al.38 showed that direct photolysis of syringic acid in D2O produced methanol (CH3OH) via singlet excited state protonation followed by nucleophilic attack by water (eq 1).
 |
1 |
In the presence of OH• or other reactive species, methanol can further oxidize to CO, as it has been observed in several systems.27,44−47 Our hypothesis that lignin is an important source of CO also agrees with the following observations: Apparent quantum yields for CO production are higher in freshwater compared to seawater (Mopper et al.5 and refs therein); both CO yields and lignin DOM content generally decrease along a salinity gradient;5,13,32,48,49 CO photoproduction rates28 and quantum yields49 are correlated with DOM aromaticity and the specific ultraviolet absorbance at 254 nm, respectively (even though this correlation is weak for large marine-based data sets);12,18 and, photodegradation of dead plant leaves can release CO.50,51 Finally, several authors reported loss of methoxy groups during photodegradation of lignin from different sources (Paulsson and Parkås52 and refs therein).
To evaluate the viability of this pathway, we investigated the photochemistry of eight lignin model compounds (Figure 2A) focusing on specific precursors and products. We first established structure-reactivity relationships between photochemical reactivity and CO production. We further sought evidence for the formation of methanol and aromatic 3-hydroxy groups via 1H NMR spectroscopy and mass spectrometry (MS), respectively. Finally, we quantified OH• to confirm that indirect photooxidation of methanol can occur under our experimental conditions. To the authors’ knowledge, this work is the first to propose an explicit mechanism for the formation of CO from the photodegradation of terrestrial DOM. We also unambiguously identified methanol as an abundant photoproduct of lignin model compounds and as a potential precursor of other ubiquitous C1 photoproducts, and we showed that photodegradation of lignin model compounds can be a source of OH•. Future work using natural lignin and more representative solution compositions will be needed to confirm the environmental relevance of these processes.
Figure 2.
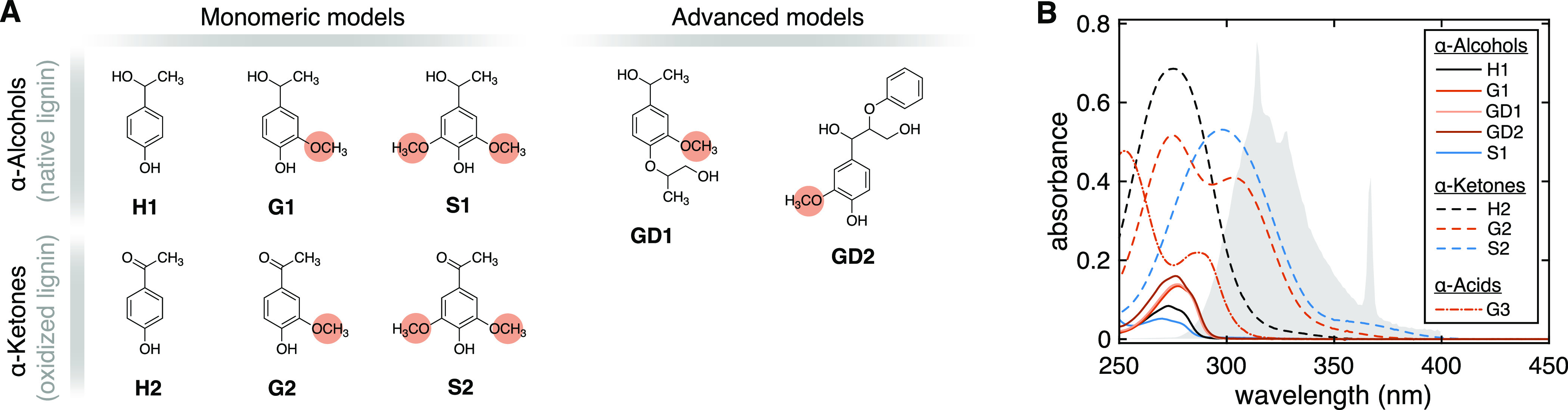
(A) Molecular structures and abbreviations of the lignin model compounds employed in this study. Orange highlights indicate the aromatic methoxy groups proposed as the CO precursors. (B) UV–vis absorption spectra of aqueous solutions used for irradiation experiments (≈50 μM, unbuffered). Each compound is identified by a line type (continuous line, α-alcohol; dashed line, α-ketone; dash-dot line, α-carboxylic acid) and a color (black, H = 0 × -OCH3; shades of red, G = 1 × -OCH3; blue, S = 2 × -OCH3). The gray area is the normalized UV lamp spectrum (max at 313 nm) in arbitrary units corrected for the absorption of the borosilicate test tubes. G3 is vanillic acid.
Materials and Methods
Chemicals
Chemicals were purchased from commercial vendors (Text S1) or synthesized via modified literature procedures (Text S2, Figure S1). Experimental solutions were obtained upon dilution of concentrated stocks in nanopure water (Text S1). At the experimental pH (5.8–6.5), all phenols were present in their protonated form (≥97%, Text S3).
Photolysis Experiments
We conducted all photochemistry experiments inside a photochemical reactor (Southern New England Ultraviolet Company, USA) equipped with a fan, a motorized turntable, and UV bulbs (Southern New England Ultraviolet Co., RPR 3000A; Figure 2B). Most experiments were conducted with 6 lamps; in selected cases (specified in the text), we used 12 bulbs to accelerate photochemical rates. When using 6 lamps, the solution temperature was ≈29–31 °C.
To quantify CO, we irradiated aqueous solutions of each model compound (50 μM) in headspace-free borosilicate test tubes (Pyrex, 15 × 85 mm; disposable) capped with rubber septa (Text S4.1). At each time point, one test tube was removed from the carousel and replaced with a water-filled tube. At the end of the irradiation, ≈6 mL of each solution was transferred into a nitrogen-flushed 20-mL serum vial for headspace gas chromatography (GC) measurements (Text S4.2); the leftover volume (150 μL) was analyzed via ultra-high-performance liquid chromatography (UPLC).
In 1H NMR experiments, we irradiated solutions containing 0.8–1.1 mM of a substrate and 0.48 mM of methanesulfonate (internal standard) in D2O in an NMR tube. Every 30 min, each tube was shaken vigorously to assure mixing (Text S5, Figure S2A). At selected time points, we removed the tube from the photoreactor, collected a 1H NMR spectrum, and then continued the irradiation. At the beginning and end of each experiment, we collected an aliquot (in triplicate), diluted it 20- or 40-fold, and quantified the substrate concentration via UPLC.
For MS measurements, we employed borosilicate test tubes containing aqueous solutions of each model compound (50 μM; 12 lamps). At each time point, we collected aliquots for UPLC (150 μL) and MS (800 μL) analyses. To quantify [OH•]ss, we used the same setup (with 6 lamps), but experimental solutions contained benzoate (BA; 10 μM or 1 mM) in addition to the model compound.53,54 Unlike experiments for CO quantification, the borosilicate test tubes were left uncapped.
All experiments were performed at least in duplicate, except for 1H NMR and MS, which we carried out as single measurements. Controls confirmed that chemical changes were solely due to photochemical processes (Text S6, Figures S3 and S4).
Sample and Data Analysis
Lignin Model Compounds
Lignin model compounds were analyzed by UPLC (Waters ACQUITY) with a C18 column (Acquity, BEH130 C18, 1.7 μm; 2.1 × 150 mm) and a photodiode array detector. Analyses were performed in isocratic mode using different combinations of aqueous and organic eluents, 5 μL injection volume, and flow rates of 0.15–0.20 mL min–1. Details for each compound are given in Table S1.
In CO production experiments, data from at least duplicate experiments were pooled together and fitted to eq 2 (Matlab R2018b), where kobs (h–1) is the pseudo-first-order photodegradation rate constant of substrate A.
| 2 |
The initial parent compound degradation rate was calculated as Rparent0 = kobs[A]0, where [A]0 was measured via UPLC.
Carbon Monoxide
Headspace CO concentrations in the serum vials (pCO,hs) were quantified via GC analyses as described in Borduas-Dedekind et al.55 (Text S7.1) and were then converted to aqueous-phase CO concentrations ([CO]t, in μM) in the headspace-free test tubes used for irradiation via eq 3.
| 3 |
where pCO,hs′ is the headspace serum vial concentration converted in atm, R is the gas constant, T is the air temperature, Vhs is the headspace volume in the serum vial, Vaq is the volume of liquid in the same vial, and KCO is the temperature-corrected Henry’s Law constant for CO56 (details are given in Text S7.2). For each compound, we calculated the CO conversion efficiency after 6 h of irradiation (YCO) via eq 4.
| 4 |
where [CO]6h,corr = [CO]6h – [CO]0 – [CO]6hblank is the amount of photoproduced CO corrected for time zero ([CO]0 = 0–0.20 μM) and blank contributions ([CO]6h = 1.25–2.62 μM, depending on the number of lamps), while [A]0 and [A]6h are initial and final substrate concentrations quantified via UPLC, respectively. Blank contributions are caused by photodegradation of organic carbon leached from the rubber stoppers (Text S7.3, Figure S5). In addition to YCO, we fitted the CO production kinetics to obtain the initial CO production rate as RCO0 = ab[A]0, where a and b are the fitting parameters of eq 5, [A]0 is the initial substrate concentration obtained via UPLC, and [CO]t is the total CO produced during irradiation.
| 5 |
For a few compounds, we fitted the CO kinetics with a linear model because the exponential fit did not converge; in this case, we obtained RCO0 from the slope of the linear regression line.
Methanol
We performed 1H NMR analyses on a Bruker AVANCE III-400 spectrometer using D2O as the solvent and methanesulfonate (2.81 ppm) as the internal standard. Each spectrum was analyzed with MestReNova 14.2.0 (Mestrelab Research S.L., Spain) and referenced to the chemical shift of the residual D2O signal at 4.79 ppm. Using a calibration line (Text S5, Figure S2B), we converted relative areas into concentrations. The value at end of the irradiation ([CH3OH]end) was used to compute the methanol conversion efficiency (YCH3OH) via eq 6, where [A]0 and [A]end are the initial and final substrate concentrations quantified by UPLC.
| 6 |
Liquid Chromatography Coupled to High-Resolution MS
Samples were injected (50 μL) at a flow rate of 0.3 mL min–1 into an LC system (UltiMate 3000 UHPLC system, Dionex) equipped with a C18 column (3 μm particle size, 3 × 150 mm, Atlantis) at 30 °C. Analyses were performed using the following gradient (A = 0.1% formic acid in ultrapure water; B = 0.1% formic acid in methanol): 0–1 min, A:B = 95:5; 1–17 min, from 95:5 to 5:95; 17–25 min, 5:95; 25–27 min, from 5:95 to 95:5; 27–30 min, 95:5. The LC was coupled to a quadrupole-Orbitrap mass spectrometer (Thermo Scientific, Bremen) operated with an electrospray ionization source in positive (4 kV spray voltage, 250 °C capillary temperature, 40 sheath gas flow rate, 10 auxiliary gas flow rate) or negative mode (GD2 only; 3 kV spray voltage, 325 °C capillary temperature, 40 sheath gas flow rate, 10 auxiliary gas flow rate). Data were acquired in full scan mode with m/z = 100–1000 at 140,000 resolution (at 200 m/z). MS2 data were obtained with a top-5 experiment with normalized collision energy of 30 at 17,500 resolution (at 200 m/z).
We used Compound Discoverer 3.2 (CD 3.2, Thermo Scientific, Germany) to identify features generated during irradiation and to assign molecular formulas. A list of expected masses and chemical structures of transformation products (TPs) was included in the data analysis workflow in CD3.2. For each molecular formula, we confirmed that its peak area was absent before irradiation, was at least 5 times higher in irradiated samples compared to the blank, and showed growing or growth-and-decay patterns in peak area as a function of time. Using Freestyle (Thermo Scientific, Germany), we extracted peak areas, total ion chromatograms, MS, and MS2 spectra of the main TPs. We then compared MS2 fragmentation patterns to in silico predictions obtained with MetFrag,57,58 as TPs library spectra were not available. This information was used to assess the level of confidence according to Schymanski et al.59
Hydroxyl Radicals (OH•)
BA was quantified via UPLC (Table S1). Its pseudo-first-order degradation rate constant (kobs,BA) obtained from the slope of a ln([BA]/[BA]0) vs time plot was employed to calculate [OH•]ss via eq 7.
| 7 |
where krxn,OH•BA = 5.9 × 109 M–1 s–1 is the second-order rate constant for the reaction of OH• with BA.60 For compounds not inducing BA decay, [OH•]ss was estimated from the ratio of their p-hydroxybenzoic acid production rate (RpHBA) to that of GD1, that is, [OH•]ss ≈ RpHBA/RpHBA · [OH•]ssGD1.
Other Instrumental Analyses
UV–vis measurements were performed with a Varian 100 Bio spectrometer and a 1-cm pathlength quartz cuvette. The instrument was run in double beam mode using nanopure water as the blank.
Results
Description of the Lignin Model Compounds
The selected model compounds reproduce several aspects of lignin chemistry, namely, the substitution pattern of the aromatic core unit (H, G, or S), the oxidation state of the α-carbon (α-alcohol or α-ketone), and the presence of ancillary groups (Figure 2A). Based on the proposed mechanism, we expected the presence or absence of methoxy groups to be the most relevant variable controlling photochemical CO production. Specifically, guaiacyl (G1, G2, GD1, and GD2) and syringyl (S1 and S2) models are expected to produce CO, while the p-hydroxyphenyl derivatives (H1 and H2) are not. The effect of the α-carbon oxidation state was less clear prior to the study. We anticipated methoxy-substituted ketones to be more photoreactive than the corresponding alcohols in the 290–400 nm range due to the n → π* carbonyl absorption band,40 but we could not anticipate how this fact would impact CO production.
Correlation between Chemical Structure and CO Production
To assess the plausibility of the proposed mechanism, we first established qualitative and quantitative correlations between molecular structure and CO photoproduction. To this aim, we irradiated an aqueous solution of each model compound using a headspace-free setup designed to limit CO partitioning to the gas phase. We then quantified substrate loss and CO production via liquid chromatography with UV detection and headspace GC, respectively.
All compounds with at least one aromatic methoxy group produced CO after 6 h of UV irradiation, with CO conversion efficiencies (YCO) ranging from 10.2 to 17.6% (Table 1; YCO is defined as moles of CO produced per mole of substrate degraded, eq 4). Comparison between YCO values of G (11.5–17.6%) and S models (10.2–11.2%) suggests that the presence of at least one ArOCH3 group, not the number of such functionalities, is the most relevant CO predictor. All guaiacyl α-alcohols have comparable YCO values (11.5–14.4%), hinting that ancillary groups do not considerably influence CO photoproduction in this class of compounds.
Table 1. Overview of the Main Photodegradation Products for the Lignin Model Compounds of Figure 2Aa.
| GC analyses |
1H NMR analyses |
RIs analyses | ||||
|---|---|---|---|---|---|---|
| compound | α-carbon | # -OCH3 | YCO (%) | YCH3OH (%) | other products | [OH•]ss (10–16 M) |
| H1 | alcohol | 0 | n.s. | n.d. | ≈1.5¤ | |
| G1 | alcohol | 1 | 11.5 ± 1.4, 12.6 ± 2.6§ | 28.8 ± 3.2 | 4.5 ± 0.3 | |
| GD1 | alcohol | 1 | 11.7 ± 0.5§ | 36.3 ± 1.1 | 1,2-propanediol | 3.7 ± 0.5 |
| GD2 | alcohol | 1 | 14.4 ± 0.2§ | 18.7 ± 1.3 | ≈1.8¤ | |
| S1 | alcohol | 2 | 10.2 ± 1.4 | 73.7 ± 2.8 | 2.6 ± 0.6 | |
| H2 | ketone | 0 | 2.2 ± 0.7 | n.d. | acetate | 14.3 ± 1.0 |
| G2 | ketone | 1 | 17.6 ± 2.1 | 23.1 ± 1.5† | acetate | 23.4 ± 1.5 |
| S2 | ketone | 2 | 11.2 ± 0.2 | 73.4 ± 1.4†† | acetate | 31.0 ± 2.5 |
CO conversion efficiencies (YCO) were calculated via eq 4 with the blank-corrected CO concentration after 6 h of UV irradiation (no mark, 6 lamps; §, 12 lamps; n.s., not significant). CH3OH conversion efficiencies (YCH3OH) were obtained via eq 6 using the methanol concentration at the end of the experiment (no mark, 6 h irradiation; †, 4 h irradiation; ††, 2 h irradiation; n.d., not detected). [OH•]ss were obtained from BA degradation via eq 7 (no mark) or were estimated from the p-hydroxybenzoate production rate of GD1 (¤). All errors are obtained from error propagation.
The α-ketone H2 also produced CO (2.2%), albeit with lower efficiency compared to methoxy-substituted compounds (≥10.2%). This result indicates that a secondary CO production pathway, perhaps a Norrish type I reaction,61,62 may be operative during UV irradiation of this molecule. Along the same lines, the α-ketone G2 had slightly higher YCO (17.6%) compared to the corresponding α-alcohol G1 (≈12%); however, this difference was negligible for S compounds. We highlight that the comparable YCO values of alcohols and ketones with the same substitution pattern is a strong indication that the α-carbonyl is not involved in the primary CO production pathway. This conclusion also agrees with Stubbins et al.,28 who showed that aromatic ketones lacking ArOCH3 groups are not efficient CO precursors. Trace amounts of CO were also detected during UV irradiation of H1 but controls indicated that ≥85% of the gas originated from the photodegradation of the organic carbon leached from the stoppers into the experimental solution, not from the substrate (Figure S5).
To gain further insight into the mechanism, we fitted the kinetics to obtain initial substrate degradation rates (Rparent0, in μM h–1) and initial CO production rates (RCO, in μM h–1; Figures S6 and S7, Table S2). We found a strong positive correlation (R2 = 0.97, N = 5) between Rparent0 and RCO for models with at least one ArOCH3 group, regardless of their α-carbon functionality (Figure 3). In this analysis, we also included vanillic acid, a guaiacyl monomer with an absorption onset intermediate between G1 and G2 (Figure 2B). From the linear regression line, we obtained a slope of (5.7 ± 0.5)%, which represents the initial, mean YCO of the process. The fact that 6 h conversion efficiencies (Table 1) are higher than this value indicates that CO is not a first-generation product (Text S8, Figure S8).
Figure 3.

Correlation between initial CO production rate (RCO0) and initial substrate degradation rate (Rparent; numeric data in Table S2). Experiments were performed with 6 (filled symbols) or 12 (empty symbols) UV lamps. Data for S and G monomers measured with six lamps were fitted to a linear regression model (starred in legend), yielding y = (0.057 ± 0.005) · x + (0.19 ± 0.36) (R2 = 0.97, p < 0.0017, N = 5; gray dashed line).
The three guaiacyl α-alcohols had comparable Rparent0 (10.5–19.0 μM h–1) and RCO values (1.65–2.00 μM h–1; Table S2), which mirrors similarities in YCO values (Table 1) and absorption spectra (Figure 2B). These compounds were irradiated with 12 (instead of 6) UV lamps to speed up their photodegradation; as we expect blank CO production (i.e., the intercept of the regression line) to vary with light intensity, these data were not included in the regression analysis. In p-hydroxyphenyl models, the increase in Rparent0 did not lead to an enhanced ability to produce CO (Figure 3, black circles). Control measurements indicated that RCO for H1 and the blank were within their experimental error (0.218 (±0.009) vs 0.197 (±0.011) μM h–1), while the value for H2 was slightly higher than the blank (0.41 (±0.02) μM h–1), in agreement with 6 h YCO data.
Detection of Methanol Via 1H NMR
Motivated by the correlation between the presence of ArOCH3 functionalities and CO photoproduction, we collected evidence supporting the occurrence of formal hydrolytic demethylation, the substitution of a methoxy by a hydroxy group (Figure 1). We posited that the direct photolysis of guaiacyl and syringyl models releases methanol, while photodegradation of H1 and H2, which lack ArOCH3 groups, do not. To evaluate this hypothesis, we irradiated a solution of each model compound in D2O, and we followed the reaction via 1H NMR as in Dallin et al.38
A qualitative analysis of 1H NMR spectra during UV irradiation confirmed our hypothesis. Irradiated solutions of G and S models showed a singlet at 3.36 ppm, corresponding to CH3OH (further confirmed with a methanol standard spike), with increasing intensity as a function of irradiation time (Figures 4A, S9B–F). This peak was not detected during irradiation of H2 and H1 (Figures 4B, S9A). In addition, all α-ketones produced acetate, while GD1 generated 1,2-propanediol (Table 1, Figure S9C). In almost all experiments, we detected acetone, but controls indicated its production to be an experimental artifact—as acetone was used to clean NMR tubes and caps prior to irradiation. In agreement with previous literature,38 the presence of acetone during irradiation had negligible influence on the results (Text S6.2).
Figure 4.

1H NMR spectra of G2 (A) and H2 (B) in D2O during 4 h of UV irradiation (dark gray traces). At the end of the experiment, we added methanol (+0.18 mM) to confirm its occurrence as a reaction product (orange traces). Gray areas represent CH3OH (3.36 ppm), blue areas denote the internal standard (CH3SO3–, 2.81 ppm), and orange areas represent the resonance range of ArOCH3 protons. Additional relevant signals are indicated with a filled circle (acetate, 2.03–2.08 ppm) or an empty circle (acetone, 2.23 ppm). Note that acetone is not a reaction product. 1H NMR spectra of other substrates are in Figure S9.
Using the data at the end of irradiation, we calculated the CH3OH conversion efficiency (YCH3OH), defined as moles of methanol produced per moles of substrate degraded (eq 6). This calculation assumes that CH3OH degradation is minimal during the experimental time span, which we confirmed experimentally (Text S6.2). This fact is not in contrast with the proposed mechanism, as we observed no OH• production at the high substrate concentrations used in these experiments (≈1 mM), likely due to light screening limitations (Text S6.2). We obtained YCH3OH values of 19–36% and 73–74% for G and S models, respectively (Table 1), indicating that the number of ArOCH3 groups defines the conversion efficiency. Like CO photoproduction, α-carbon functionality and presence of ancillary groups have a small influence on YCH3OH. Further analyses revealed that concentrations of methanol and methoxy aromatic protons are always linearly correlated (R2 ≥ 0.94; Figure S10), supporting the idea that CH3OH is a first-generation product (Text S9, Figure S11). Furthermore, the fact that YCH3OH > YCO (19–74% > 10.2–17.6%) agrees with methanol being a precursor of CO and suggests that this gas is not its only oxidation product, in agreement with previous findings.44
Detection of Hydroxylated Photoproducts Via LC-HRMS Measurements
Based on our proposed mechanism, formal hydrolytic demethylation leads to production of an aromatic hydroxy group (Figure 1). To confirm the occurrence of this process, we screened for 3-hydroxylated TPs via LC-HRMS (analyses performed only for G and S models). To increase the confidence of our results, we also collected MS2 data and compared it to in silico predictions obtained with MetFrag.57,59
MS signals corresponding to the expected TPs were detected for all tested compounds (Table S3, Figures S12–S17). Upon irradiation of G2, we observed a signal at m/z = 153.0550 corresponding to the [M + H]+ adduct of G2 TP1, the TP of formal hydrolytic demethylation (Figure 5). Its signal was detected at the same retention time (r.t.) of a commercial standard analyzed under the same conditions, and its MS2 spectrum was in good agreement with the standard and in silico predictions (Figure S12F). We also detected 3-hydroxylated TPs during irradiation of G1 (m/z = 137.0601, detected as [M – H2O + H]+; Figure S13) and GD2 (m/z = 257.0828, detected as [M – H]−; Figure S14). For the two syringyl compounds, we observed both the mono- and bis-demethylated species (Figures S15 and S16), which mirrors their higher YCH3OH values as compared to G models (Table 1). For GD1, the only compound without a free phenol, we observed loss of both alkoxy substituents (Figure S17), in agreement with detection of both methanol and 1,2-propanediol via 1H NMR (Table 1). For monomeric compounds, the formation of 3-hydroxylated TPs was assessed with relatively high confidence (level 1–2 according to Schymanski et al.59), while for GD1 and GD2, the confidence was lower (level 3–4; Table S3).
Figure 5.
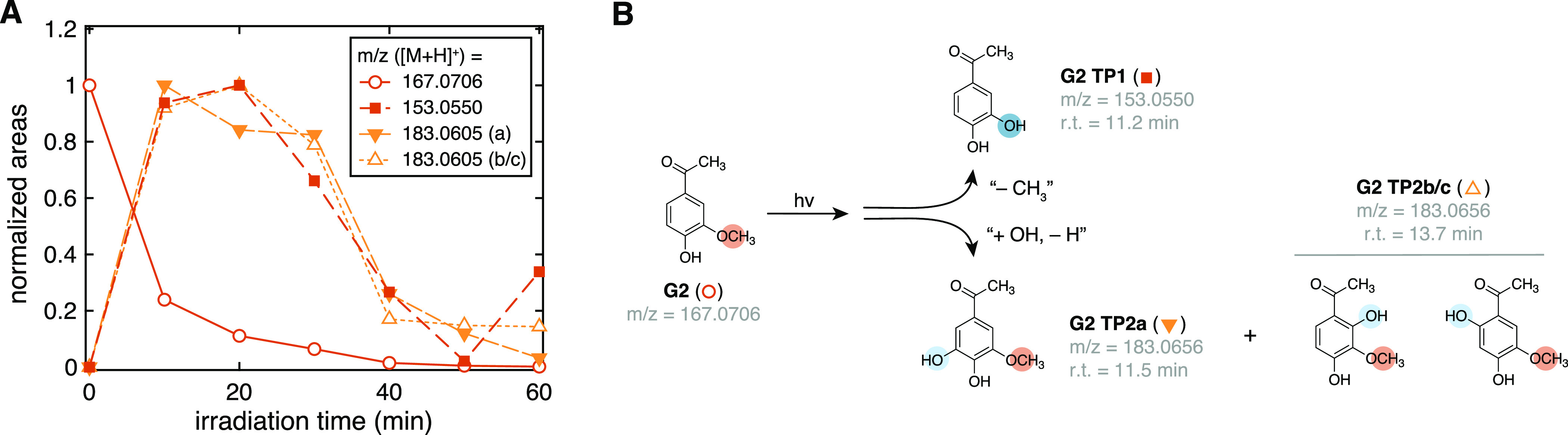
(A) Kinetic traces of G2 and relevant degradation products detected via LC-HRMS as [M + H]+ adducts (12 UV lamps). All traces are reported in normalized areas to aid comparison; absolute areas and MS2 data are in Figure S12. (B) Schematic of the photodegradation process of G2 based on the data in panel (A). Note that three different isomers can have m/z = 183.0656; when possible, structures are assigned based on differences in retention time. Data for other substrates are given in Figures S13–S17.
During irradiation of some G models, we also observed hydroxylated TPs that retained the ArOCH3 group. When irradiating G2, we also detected m/z = 183.0656 at two different retention times. We assigned the signal at 11.5 min to the 5-hydroxylated species (G2 TP2a; Figure 5B) based on similarities in r.t. and MS2 spectrum with the mono-demethylated product of S2, while the second can be the 2- or 6-hydroxylated species (G2 TP2b/c; Figure 5B). For G1, we only observed the 2- and 6-hydroxylated products (G1 TD2a/b; Figure S13), while for GD2, we observed mono- (GD2 TP2a/b) and bis-hydroxylation (GD2 TP3; Table S3). These additional products were not detected during irradiation of GD1 nor for syringyl models.
A qualitative analysis of the signals’ kinetics indicates that all detected TPs are photolabile; thus, they are transient intermediates. All species showed growth-and-decay kinetics, and their signal intensity was typically 2–3 orders of magnitude smaller than the corresponding substrate (Figures S12–S17). For G2 TP1, the only compound for which a commercial standard was available, we calculated a maximum concentration of 0.36 μM after 20 min of irradiation, corresponding to [G2 TP1]max/[G2]0 = 7 × 10–3. The photochemical instability of G2 TP1 was further confirmed experimentally by irradiating a solution of this compound in deionized water, obtaining direct photolysis rate constants of (10.7 ± 0.1) × 10–2 min–1 and (12.1 ± 0.3) × 10–2 min–1 for G2 TP1 and G2, respectively.
Detection of Hydroxyl Radicals
The second step of the proposed mechanism involves methanol oxidation to CO (Figure 1). As methanol does not absorb UV light, we hypothesized that reactive intermediates formed during photodegradation of lignin models trigger this reaction. We focused on the hydroxyl radical (OH•) due to its well-acknowledged reactivity with methanol, both in laboratory and environmental settings.44,45,60,63 Literature precedents also indicate that OH• can be produced during irradiation of hydroxybenzoic acids.53,64,65 To investigate whether the substrates of this study can be sources of OH•, we used BA as a hydroxyl radical probe.63,66
Our measurements confirmed that UV irradiation of lignin model compounds produces OH•. Most substrates induced a measurable BA depletion, which could be fitted to a monoexponential decay function (R2 = 0.90–0.98; Figure S18) to yield [OH•]ss = 2.6–31.0 × 10–16 M (eq 7; Table 1). H1 and GD2 did not induce significant BA loss but produced p-, m-, and o-hydroxybenzoic acids, the expected hydroxylation products,63 in experiments employing higher BA concentration (Figure S19). Compared to the other substrates, hydroxylated products were generated in lower concentrations, indicating that lack of BA decay was caused by OH• production below the method’s detection limit. For H1 and GD2, we used the p-hydroxybenzoic acid production kinetics to estimate [OH•]ss = 1.5–1.8 × 10–16 M (Table 1).
Control experiments further corroborated the formation of OH•. We first confirmed the photochemical stability of BA under UV light (Figure S18I) and that hydroxylated products cannot be produced when the probe is irradiated in the absence of lignin models (Figure S19I). We also irradiated each substrate in the presence of BA and isopropanol (1% v/v), a common OH• quencher, observing suppression of the probe’s decay (Figure S18). The latter further hints that BA is degraded by free OH•, not by other hydroxylating species or reactive intermediates,53,67,68 similar to what was reported for other hydroxybenzoic acids.64
Discussion
Step 1: Formal Hydrolytic Demethylation of Lignin Model Compounds Via Direct Photolysis
1H NMR and LC-HRMS measurements provided unequivocal evidence for the occurrence of formal hydrolytic demethylation via direct photolysis. All G and S models produced CH3OH (Figures 4, S9) and the corresponding 3-hydroxylated TP (Figures S12–S17) regardless of their α-carbon oxidation state or presence of ancillary substituents, while CH3OH was not detected during irradiation of compounds lacking the ArOCH3 group. In addition, YCH3OH values of S compounds (73–74%) were considerably higher than G models (19–36%; Table 1); correspondingly, MS data revealed that both mono- and bis-demethoxylated products formed during irradiation of S1 and S2 (Figure S15 and S16). Thus, all available information is consistent with CH3OH originating from cleavage of ArOCH3 groups. Notably, CH3OH is also a major microbial degradation product of lignin,69 which is in agreement with the recent work by Nalven et al.70 showing that sunlight and microbial enzymes induce similar chemical changes in DOM.
Based on the available data, we speculate that CH3OH is released via the same pathway proposed by Dallin et al.38 for syringic acid: excited state protonation of the aromatic ring followed by nucleophilic attack by water and subsequent loss of CH3OH via Ar-OCH3 cleavage (eq 1). While confirmation of this hypothesis requires a dedicated study, our observations disfavor alternative options, namely, ArO-CH3 cleavage followed by CH4 release (reported in vacuum in the solid state)40 and OH•-induced cleavage (reported during fungal demethylation69 and chemical oxidation43). Specifically, 1H NMR measurements showed that CH3OH is a major product (YCH3OH = 19–73%) and that it is produced via an apparent one-step reaction (Figures S10 and S11) under conditions that would favor CH4 accumulation, that is, when OH• is not formed (Figure S4B). Furthermore, the occurrence of hydroxylation at multiple ring sites (e.g., G2 TP2a/b/c; Figure 5) and the detection of products originating from cleavage of other aromatic functionalities (i.e., 1,2-propanediol and acetic acid; Table 1) hint that all sterically accessible ring locations, and not only the ones adjacent to the ArOCH3 group, may undergo this process. Finally, control experiments showed minimal changes in substrates’ photodegradation kinetics in the presence and absence of isopropanol (Figure S20), ruling out OH• as a major trigger of hydrolytic demethylation. Note that formation of hydroxylated products without the intermediacy of OH• is not uncommon during direct photolysis of aromatic compounds.38,53,71−73
Step 2: Methanol Oxidation to CO Via Indirect Photolysis
Even though CO and OH• production data support the occurrence of the second step (i.e., CH3OH + OH• → CO), more work is needed for its confirmation and for uncovering its mechanistic details. Literature precedents show that methanol oxidation in water is a complex process, as its kinetics and product yields depend on CH3OH concentration, pH, [OH•]ss, and presence of other radicals.44,74 Furthermore, gas-phase studies suggest that CO is generated via oxidation of formaldehyde,27,46 a methanol oxidation product in both aqueous and gas phase,44−46,74 not directly from CH3OH. Thus, detection of OH• radicals during UV irradiation of lignin model compounds only indicates that this reaction is plausible but falls short of definitive proof. In addition to OH•, other reactive intermediates formed during photodegradation of lignin model compounds may trigger the conversion of CH3OH to CO. While we discarded singlet oxygen and triplet excited states due to their low reactivity toward methanol,75,76 peroxy radicals may play a more important role.77−79 Peroxy radicals form when a carbon-centered radical reacts with oxygen80 and have been detected during photooxidation of lignin and its model compounds (reviewed by Heitner81), OH• oxidation of methanol,44,74 and DOM photolysis.82 Ongoing work in our lab is elucidating identity and yields of CH3OH photooxidation products under conditions relevant to sunlit surface waters.
Limitations of this Work
The use of simple substrates is an intrinsic limitation of all model compound studies. In this work, we mitigated this drawback by mimicking as accurately as possible the key structural elements of natural lignin (Figures 1 and 2A). Furthermore, our results strongly suggested that the proposed mechanism is controlled by the chemical structure of the aromatic core unit, which is the same in our models and in the natural polymer. As a matter of fact, the three guaiacyl α-alcohols (G1, GD1, and GD2) always showed the same reactivity pattern. Lignin is also known to depolymerize during photodegradation,48,81 thereby releasing monomers with the same aromatic core units of our models. Indeed, irradiation of GD1 released 1,2-propanediol (Table 1) and formed GD1 TP2 (Figure S17), the same TP detected during irradiation of G1 (i.e., G1 TP1; Figure S13). Gao and Zepp83 also showed that that low-molecular-weight fractions (<1 kDa) have 2.5–3 times higher apparent CO quantum yields than unfractionated DOM, which fits with the yet untested hypothesis that the proposed mechanism is mostly effective after (partial or total) depolymerization. Thus, despite the promising results, follow-up investigations need to confirm our findings using natural lignin.
Additional limitations include substrates’ concentrations and the use of deionized water as solvent. The choice of 50 μM (i.e., 6.9–14.5 mg L–1 or 4.8–9.6 mgC L–1) as our initial concentration was dictated by the constraints of the CO measurement setup, that is, the ≈1 μM CO background and YCO values of 2–18%. Depending on the selected lignin proxy, these concentrations are either comparable or at least one order of magnitude higher than natural lignin (Section S10). Whereas we expect variations in concentration to have minimal impact on the direct photolysis step, rates and yields of methanol oxidation to CO may change.44 This step is also susceptible to matrix variations, including nature and concentrations of salts, metals, and additional DOM components (see the previous section). Solution temperature is an additional variable that can influence methanol oxidation to CO, as we propose this to be a bimolecular reaction. Apparent CO quantum yields of DOM have a temperature dependence;49 confirming this dependence in our model system can help to establish its environmental relevance.
Environmental Significance
This work presents data supporting the role of ArOCH3 groups as the precursors of CO via a two-step mechanism involving a combination of direct and indirect photolysis. This result builds on previous work by Stubbins et al.28 and, to the authors’ knowledge, is the first explicit mechanism justifying CO production from DOM photodegradation. Albeit further investigations need to confirm its natural occurrence and elucidate specific mechanistic details, this work sets the basis for the development of predictive models directly applicable to DOM. A key finding is that initial CO photoproduction rates can be predicted from the initial substrate photodegradation rate and the substitution pattern of the aromatic core unit (Figure 3). If this correlation proves valid also for natural lignin and DOM, it will allow estimating CO photoproduction rates from the number of G + S core units and the direct photolysis degradation rate, two parameters that can realistically be obtained using established techniques (e.g., Yan and Kaiser84). This information will allow one to predict variations in apparent CO production quantum yields as a function of DOM type and thus improve current estimates of photochemical CO production in coastal regions and freshwaters. These environments are potential production hotspots not accurately described in global CO photoproduction models, which rely on apparent CO quantum yield parametrizations based on marine data.1,85,86
An unanticipated finding of our work is the potential of lignin to act as the precursor of CH3OH and other C1-containing compounds, and as a source of OH•. So far, CH3OH has not been reported as a DOM photodegradation product, even though its formation was hypothesized.87−89 The few studies investigating this process focused on the ocean,88,89 where DOM and lignin concentrations are low and thus where their contribution is easy to overlook. To the authors’ knowledge, photoproduction of CH3OH from terrestrial DOM has never been reported nor investigated, perhaps due to analytical difficulties in detecting this alcohol88 and/or the presence of removal mechanisms (e.g., OH• oxidation) that rapidly consume it.44,45 Given lignin’s abundance in terrestrial DOM and the high methanol yields that we measured, a reassessment of photochemical CH3OH sources is warranted.
The proposed mechanism may also represent a possible production pathway of formaldehyde, formic acid, and CO2, abundant and ubiquitous DOM photoproducts.5,90 Monod et al.44 showed that OH• oxidation of CH3OH under cloud-water conditions produces formaldehyde and formic acid in 49% yield, while the remaining 51% was hypothesized to be CO and CO2. This fact agrees with previous work indicating that OH• may be involved in the production of at least a fraction of the formaldehyde,91 formic acid,92−94 and CO292,93 generated during DOM photooxidation. Other oxidants present in sunlit surface waters and capable of oxidizing CH3OH (e.g., Cl•)95 may lead to the same compounds.
Lastly, lignin photodegradation may represent an environmentally relevant source of OH•, the reactive intermediate whose formation mechanism is less known.53,64,68 Previous authors showed that irradiation of quinones,53 mono- and diphenols,64 hydroxybenzoic acids,53,64,65 and hydroxybenzoic aldehydes64 can produce OH• and/or other hydroxylating species. For quinones, a mechanism involving an exciplex between triplet excited states and water has been proposed.53,64,96 An alternative hypothesis that has not yet been thoroughly considered (albeit once proposed64) is that hydroxyl radicals are generated during photolysis of hydroxylated aromatics. This hypothesis fits with the general understanding that (at least part of) OH• in sunlit surface waters form during the photodegradation of aromatic components of DOM92,96−98 and that model compounds that generate OH• are typically degraded during irradiation.53,65 Dallin et al.38 also proposed that, in addition to releasing methanol, the intermediate formed after excited state protonation of syringic acid releases OH• and forms another of their observed products. Furthermore, C-centered radicals formed during lignin photodegradation (e.g., upon H-abstraction from α-alcohols)37 can react with O2 yielding a peroxy radicals, which further fragment to form α-carbonyls and superoxide radicals.37 The latter can dismutate to yield H2O2, a precursor of OH•, and H2O.97,99
These unforeseen insights on long-standing questions related to DOM photochemistry call for a thorough reassessment of the light-triggered reactivity of lignin and its degradation products in aquatic systems.
Acknowledgments
We thank Martin H. Schroth for his support on GC-FID analyses and Bernadette Vogler for guidance on LC-HRMS data analysis. We also acknowledge the anonymous reviewers for their insightful comments that helped improve the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.2c03762.
Sources and synthetic procedures of used chemicals; compound speciation at the experimental pH; supplementary details, graphs, and tables on experiments detecting CO, methanol, hydroxylated transformation products, and OH•; description of control experiments; details of UPLC methods; supplementary calculations on kinetic parameters; and estimation of lignin concentrations in natural waters. (PDF)
Author Present Address
⊥ Centre for Microbiology and Environmental Systems Science, University of Vienna, Vienna 1090, Austria (R.G.)
Author Present Address
∥ Department of Chemistry, Colorado State University, Fort Collins 80523, Colorado, United States (R.O.)
Author Contributions
R.G. and J.H. contributed equally to the work. R.O. conceived and designed the project with contributions from R.G., J.H., and K.M. R.G. performed the chemical syntheses under the supervision of A.M. R.O. and R.G. performed all photochemistry experiments and associated UPLC analyses. M.V. performed preliminary experiments for OH detection. R.G. performed GC-FID analyses, R.O. performed 1H NMR measurements, and J.H. carried out LC-HRMS analyses. R.O., R.G., and J.H. analyzed the data. R.O. wrote the manuscript with contribution from all authors.
The authors gratefully acknowledge the Swiss National Science Foundation (Grant no. 200020_188565) for funding.
The authors declare no competing financial interest.
Supplementary Material
References
- Conte L.; Szopa S.; Séférian R.; Bopp L. The Oceanic Cycle of Carbon Monoxide and Its Emissions to the Atmosphere. Biogeosciences 2019, 16, 881–902. 10.5194/bg-16-881-2019. [DOI] [Google Scholar]
- Khalil M. A. K.; Rasmussen R. A. The Global Cycle of Carbon Monoxide: Trends and Mass Balance. Chemosphere 1990, 20, 227–242. 10.1016/0045-6535(90)90098-E. [DOI] [Google Scholar]
- Gaubert B.; Worden H. M.; Arellano A. F. J.; Emmons L. K.; Tilmes S.; Barré J.; Alonso S. M.; Vitt F.; Anderson J. L.; Alkemade F.; Houweling S.; Edwards D. P. Chemical Feedback From Decreasing Carbon Monoxide Emissions. Geophys. Res. Lett. 2017, 44, 9985–9995. 10.1002/2017GL074987. [DOI] [Google Scholar]
- Hartmann D. L.; Klein Tank A. M. G.; Rusticucci M.; Alexander L. V.; Brönnimann S.; Charabi Y.; Dentener F. J.; Dlugokencky E. J.; Easterling D. R.; Kaplan A.; Soden B. J.; Thorne P. W.; Wild M.; Zhai P. M.. Observations: Atmosphere and Surface. In IPCC, 2013: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, 2013.
- Mopper K.; Kieber D. J.; Stubbins A.Chapter 8 - Marine Photochemistry of Organic Matter: Processes and Impacts A2 - Hansell, Dennis A. In Biogeochemistry of Marine Dissolved Organic Matter (Second Edition); Carlson C. A., Ed.; Academic Press: Boston, 2015; 389–450. [Google Scholar]
- Pos W. H.; Riemer D. D.; Zika R. G. Carbonyl Sulfide (OCS) and Carbon Monoxide (CO) in Natural Waters: Evidence of a Coupled Production Pathway. Mar. Chem. 1998, 62, 89–101. 10.1016/S0304-4203(98)00025-5. [DOI] [Google Scholar]
- Lennartz S. T.; Marandino C. A.; von Hobe M.; Cortes P.; Quack B.; Simo R.; Booge D.; Pozzer A.; Steinhoff T.; Arevalo-Martinez D. L.; Kloss C.; Bracher A.; Röttgers R.; Atlas E.; Krüger K. Direct Oceanic Emissions Unlikely to Account for the Missing Source of Atmospheric Carbonyl Sulfide. Atmos. Chem. Phys. 2017, 17, 385–402. 10.5194/acp-17-385-2017. [DOI] [Google Scholar]
- Whelan M. E.; Lennartz S. T.; Gimeno T. E.; Wehr R.; Wohlfahrt G.; Wang Y.; Kooijmans L. M. J.; Hilton T. W.; Belviso S.; Peylin P.; Commane R.; Sun W.; Chen H.; Kuai L.; Mammarella I.; Maseyk K.; Berkelhammer M.; Li K.-F.; Yakir D.; Zumkehr A.; Katayama Y.; Ogée J.; Spielmann F. M.; Kitz F.; Rastogi B.; Kesselmeier J.; Marshall J.; Erkkilä K.-M.; Wingate L.; Meredith L. K.; He W.; Bunk R.; Launois T.; Vesala T.; Schmidt J. A.; Fichot C. G.; Seibt U.; Saleska S.; Saltzman E. S.; Montzka S. A.; Berry J. A.; Campbell J. E. Reviews and Syntheses: Carbonyl Sulfide as a Multi-Scale Tracer for Carbon and Water Cycles. Biogeosciences 2018, 15, 3625–3657. 10.5194/bg-15-3625-2018. [DOI] [Google Scholar]
- Launois T.; Belviso S.; Bopp L.; Fichot C. G.; Peylin P. A New Model for the Global Biogeochemical Cycle of Carbonyl Sulfide - Part 1: Assessment of Direct Marine Emissions with an Oceanic General Circulation and Biogeochemistry Model. Atmos. Chem. Phys. 2015, 15, 2295–2312. 10.5194/acp-15-2295-2015. [DOI] [Google Scholar]
- Lennartz S. T.; Gauss M.; von Hobe M.; Marandino C. A. Monthly Resolved Modelled Oceanic Emissions of Carbonyl Sulfide and Carbon Disulfide for the Period 2000-2019. Earth Syst. Sci. Data Discuss. 2020, 13, 2095–2110. 10.5194/essd-13-2095-2021. [DOI] [Google Scholar]
- Asaf D.; Rotenberg E.; Tatarinov F.; Dicken U.; Montzka S. A.; Yakir D. Ecosystem Photosynthesis Inferred from Measurements of Carbonyl Sulphide Flux. Nat. Geosci. 2013, 6, 186–190. 10.1038/ngeo1730. [DOI] [Google Scholar]
- Reader H. E.; Miller W. L. Variability of Carbon Monoxide and Carbon Dioxide Apparent Quantum Yield Spectra in Three Coastal Estuaries of the South Atlantic Bight. Biogeosciences 2012, 9, 4279–4294. 10.5194/bg-9-4279-2012. [DOI] [Google Scholar]
- White E. M.; Kieber D. J.; Sherrard J.; Miller W. L.; Mopper K. Carbon Dioxide and Carbon Monoxide Photoproduction Quantum Yields in the Delaware Estuary. Mar. Chem. 2010, 118, 11–21. 10.1016/j.marchem.2009.10.001. [DOI] [Google Scholar]
- Miller W. L.; Zepp R. G. Photochemical Production of Dissolved Inorganic Carbon from Terrestrial Organic Matter: Significance to the Oceanic Organic Carbon Cycle. Geophys. Res. Lett. 1995, 22, 417–420. 10.1029/94GL03344. [DOI] [Google Scholar]
- Miller W. L.; Moran M.; Sheldon W. M.; Zepp R. G.; Opsahl S. Determination of Apparent Quantum Yield Spectra for the Formation of Biologically Labile Photoproducts. Limnol. Oceanogr. 2002, 47, 343–352. 10.4319/lo.2002.47.2.0343. [DOI] [Google Scholar]
- Li Y.; Fichot C. G.; Geng L.; Scarratt M. G.; Xie H. The Contribution of Methane Photoproduction to the Oceanic Methane Paradox. Geophys. Res. Lett. 2020, 47, e2020GL088362 10.1029/2020GL088362. [DOI] [Google Scholar]
- Powers L. C.; Miller W. L. Hydrogen Peroxide and Superoxide Photoproduction in Diverse Marine Waters: A Simple Proxy for Estimating Direct CO2 Photochemical Fluxes. Geophys. Res. Lett. 2015, 42, 7696–7704. 10.1002/2015GL065669. [DOI] [Google Scholar]
- Powers L. C.; Miller W. L. Photochemical Production of CO and CO2 in the Northern Gulf of Mexico: Estimates and Challenges for Quantifying the Impact of Photochemistry on Carbon Cycles. Mar. Chem. 2015, 171, 21–35. 10.1016/j.marchem.2015.02.004. [DOI] [Google Scholar]
- Kettle A. J. Diurnal Cycling of Carbon Monoxide (CO) in the Upper Ocean near Bermuda. Ocean Model. 2005, 8, 337–367. 10.1016/j.ocemod.2004.01.003. [DOI] [Google Scholar]
- Doney S. C.; Najjar R. G.; Stewart S. Photochemistry, Mixing and Diurnal Cycles in the Upper Ocean. J. Mar. Res. 1995, 53, 341–369. 10.1357/0022240953213133. [DOI] [Google Scholar]
- Redden G. D.Characteristics of Photochemical Production of Carbon Monoxide in Seawater; Master of Science in Oceanography, Oregon State University, 1982. [Google Scholar]
- Kagan J.Photochemistry of the Carbonyl Group. In Organic Photochemistry; Kagan J., Ed.; Academic Press: San Diego, 1993; 55–64. [Google Scholar]
- Eugene A. J.; Xia S.-S.; Guzman M. I. Aqueous Photochemistry of Glyoxylic Acid. J. Phys. Chem. A 2016, 120, 3817–3826. 10.1021/acs.jpca.6b00225. [DOI] [PubMed] [Google Scholar]
- Chen J. C.; Volman D. H. The Photochemical Decomposition of Acetaldehyde in Aqueous Solutions of Allyl Alcohol at 2537 Å.1. J. Am. Chem. Soc. 1961, 83, 1047–1049. 10.1021/ja01466a008. [DOI] [Google Scholar]
- Volman D. H.; Swanson L. W. The Photochemical Decomposition of Acetone in Aqueous Solutions of Allyl Alcohol at 2537 Å.1. J. Am. Chem. Soc. 1960, 82, 4141–4144. 10.1021/ja01501a003. [DOI] [Google Scholar]
- Gierczak T.; Burkholder J. B.; Bauerle S.; Ravishankara A. R. Photochemistry of Acetone under Tropospheric Conditions. Chem. Phys. 1998, 231, 229–244. 10.1016/S0301-0104(98)00006-8. [DOI] [Google Scholar]
- Saunders S. M.; Jenkin M. E.; Derwent R. G.; Pilling M. J. Protocol for the Development of the Master Chemical Mechanism, MCM v3 (Part A): Tropospheric Degradation of Non-Aromatic Volatile Organic Compounds. Atmos. Chem. Phys. 2003, 3, 161–180. 10.5194/acp-3-161-2003. [DOI] [Google Scholar]
- Stubbins A.; Hubbard V.; Uher G.; Law C. S.; Upstill-Goddard R. C.; Aiken G. R.; Mopper K. Relating Carbon Monoxide Photoproduction to Dissolved Organic Matter Functionality. Environ. Sci. Technol. 2008, 42, 3271–3276. 10.1021/es703014q. [DOI] [PubMed] [Google Scholar]
- Calvo-Flores F. G.; Dobado J. A.; Isac-García J.; Martín-Martínez F. J.. Structure and Physicochemical Properties. In Lignin and Lignans as Renewable Raw Materials; John Wiley & Sons, Ltd, 2015; 9–48. [Google Scholar]
- Lancefield C. S.; Westwood N. J. The Synthesis and Analysis of Advanced Lignin Model Polymers. Green Chem. 2015, 17, 4980–4990. 10.1039/C5GC01334H. [DOI] [Google Scholar]
- Brunow G.; Lundquist K.. Functional Groups and Bonding Patterns in Lignin (Including the Lignin-Carbohydrate Complexes). In Lignin and Lignans; CRC Press, 2010; 267–299. [Google Scholar]
- Lu C.-J.; Benner R.; Fichot C. G.; Fukuda H.; Yamashita Y.; Ogawa H.. Sources and Transformations of Dissolved Lignin Phenols and Chromophoric Dissolved Organic Matter in Otsuchi Bay, Japan.Front. Mar. Sci. 2016, 3, 85 10.3389/fmars.2016.00085. [DOI] [Google Scholar]
- Opsahl S.; Benner R. Distribution and Cycling of Terrigenous Dissolved Organic Matter in the Ocean. Nature 1997, 386, 480–482. 10.1038/386480a0. [DOI] [Google Scholar]
- Hernes P. J.; Benner R. Photochemical and Microbial Degradation of Dissolved Lignin Phenols: Implications for the Fate of Terrigenous Dissolved Organic Matter in Marine Environments. J. Geophys. Res. Oceans 2003, 108, 3291. 10.1029/2002JC001421. [DOI] [Google Scholar]
- Guo H.; Miles-Barrett D. M.; Neal A. R.; Zhang T.; Li C.; Westwood N. J. Unravelling the Enigma of LigninOX: Can the Oxidation of Lignin Be Controlled?. Chem. Sci. 2018, 9, 702–711. 10.1039/C7SC03520A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang H.; Zhang Q.; Wang H.; Cai D.; Ma Y.; Li L.; Li K.; Lu X.; Chen H.; Yang X.; Chen J. Photochemical Aging of Guaiacol by Fe(III)–Oxalate Complexes in Atmospheric Aqueous Phase. Environ. Sci. Technol. 2019, 53, 127–136. 10.1021/acs.est.8b04507. [DOI] [PubMed] [Google Scholar]
- Lanzalunga O.; Bietti M. Photo- and Radiation Chemical Induced Degradation of Lignin Model Compounds. J. Photochem. Photobiol. B 2000, 56, 85–108. 10.1016/S1011-1344(00)00054-3. [DOI] [PubMed] [Google Scholar]
- Dallin E.; Wan P.; Krogh E.; Gill C.; Moore R. M. New PH-Dependent Photosubstitution Pathways of Syringic Acid in Aqueous Solution: Relevance in Environmental Photochemistry. J. Photochem. Photobiol., A 2009, 207, 297–305. 10.1016/j.jphotochem.2009.07.023. [DOI] [Google Scholar]
- Moore R. M. A Photochemical Source of Methyl Chloride in Saline Waters. Environ. Sci. Technol. 2008, 42, 1933–1937. 10.1021/es071920l. [DOI] [PubMed] [Google Scholar]
- Weir N. A.; Arct J.; Ceccarelli A. Photodecomposition of Lignin Model Compounds: Part I. Polym. Degrad. Stab. 1994, 43, 409–414. 10.1016/0141-3910(94)90013-2. [DOI] [Google Scholar]
- Noutary C.; de Violet P. F.; Vercauteren J.; Castellan A. Photochemical Studies on a Phenolic Phenylcoumarone Lignin Model Molecule in Relation to the Photodegradation of Lignocellulosic Materials. Part 2. Photophysical and Photochemical Studies. Res. Chem. Intermed. 1995, 21, 247. 10.1007/BF03052256. [DOI] [Google Scholar]
- Argyropoulos D. S.; Sun Y. Photochemically Induced Solid-State Degradation, Condensation, and Rearrangement Reactions in Lignin Model Compounds and Milled Wood Lignin. Photochem. Photobiol. 1996, 64, 510–517. 10.1111/j.1751-1097.1996.tb03098.x. [DOI] [Google Scholar]
- Gierer J. Formation and Involvement of Superoxide (O2-/HO2•) and Hydroxyl (OH•) Radicals in TCF Bleaching Processes: A Review. Holzforschung 1997, 51, 34–46. 10.1515/hfsg.1997.51.1.34. [DOI] [Google Scholar]
- Monod A.; Chebbi A.; Durand-Jolibois R.; Carlier P. Oxidation of Methanol by Hydroxyl Radicals in Aqueous Solution under Simulated Cloud Droplet Conditions. Atmos. Environ. 2000, 34, 5283–5294. 10.1016/S1352-2310(00)00191-6. [DOI] [Google Scholar]
- Bates K. H.; Jacob D. J.; Wang S.; Hornbrook R. S.; Apel E. C.; Kim M. J.; Millet D. B.; Wells K. C.; Chen X.; Brewer J. F.; Ray E. A.; Commane R.; Diskin G. S.; Wofsy S. C. The Global Budget of Atmospheric Methanol: New Constraints on Secondary, Oceanic, and Terrestrial Sources. J. Geophys. Res. Atmos. 2021, 126, e2020JD033439 10.1029/2020JD033439. [DOI] [Google Scholar]
- Held T. J.; Dryer F. L. A Comprehensive Mechanism for Methanol Oxidation. Int. J. Chem. Kinet. 1998, 30, 805–830. . [DOI] [Google Scholar]
- Yuda A.; Ashok A.; Kumar A. A Comprehensive and Critical Review on Recent Progress in Anode Catalyst for Methanol Oxidation Reaction. Catal. Rev. 2022, 64, 126–228. 10.1080/01614940.2020.1802811. [DOI] [Google Scholar]
- Opsahl S.; Benner R. Photochemical Reactivity of Dissolved Lignin in River and Ocean Waters. Limnol. Oceanogr. 1998, 43, 1297–1304. 10.4319/lo.1998.43.6.1297. [DOI] [Google Scholar]
- Zhang Y.; Xie H.; Chen G. Factors Affecting the Efficiency of Carbon Monoxide Photoproduction in the St. Lawrence Estuarine System (Canada). Environ. Sci. Technol. 2006, 40, 7771–7777. 10.1021/es0615268. [DOI] [PubMed] [Google Scholar]
- Tarr M. A.; Miller W. L.; Zepp R. G. Direct Carbon Monoxide Photoproduction from Plant Matter. J. Geophys. Res. Atmos. 1995, 100, 11403–11413. 10.1029/94JD03324. [DOI] [Google Scholar]
- Sulzberger B.; Austin A. T.; Cory R. M.; Zepp R. G.; Paul N. D. Solar UV Radiation in a Changing World: Roles of Cryosphere–Land–Water–Atmosphere Interfaces in Global Biogeochemical Cycles. Photochem. Photobiol. Sci. 2019, 18, 747–774. 10.1039/C8PP90063A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsson M.; Parkås J. Review: Light-Induced Yellowing Of Lignocellulosic Pulps – Mechanisms And Preventive Methods. BioResources 2012, 7, 5995–6040. 10.15376/biores.7.4.5995-6040. [DOI] [Google Scholar]
- Couch K.; Leresche F.; Farmer C.; McKay G.; Rosario-Ortiz F. L. Assessing the Source of the Photochemical Formation of Hydroxylating Species from Dissolved Organic Matter Using Model Sensitizers. Environ. Sci. Process. Impacts 2022, 24, 102–115. 10.1039/D1EM00345C. [DOI] [PubMed] [Google Scholar]
- Cai Y.; Apell J. N.; Pflug N. C.; McNeill K.; Bollmann U. E. Photochemical Fate of Medetomidine in Coastal and Marine Environments. Water Res. 2021, 191, 116791 10.1016/j.watres.2020.116791. [DOI] [PubMed] [Google Scholar]
- Borduas-Dedekind N.; Ossola R.; David R. O.; Boynton L. S.; Weichlinger V.; Kanji Z. A.; McNeill K. Photomineralization Mechanism Changes the Ability of Dissolved Organic Matter to Activate Cloud Droplets and to Nucleate Ice Crystals. Atmos. Chem. Phys. 2019, 19, 12397–12412. 10.5194/acp-19-12397-2019. [DOI] [Google Scholar]
- Fry V. A.; Istok J. D.; Semprini L.; O’Reilly K. T.; Buscheck T. E. Retardation of Dissolved Oxygen Due to a Trapped Gas Phase in Porous Media. Ground Water 1995, 33, 391–398. 10.1111/j.1745-6584.1995.tb00295.x. [DOI] [Google Scholar]
- Ruttkies C.; Schymanski E. L.; Wolf S.; Hollender J.; Neumann S. MetFrag Relaunched: Incorporating Strategies beyond in Silico Fragmentation. J. Cheminformatics 2016, 8, 3. 10.1186/s13321-016-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S.; Schmidt S.; Müller-Hannemann M.; Neumann S. In Silico Fragmentation for Computer Assisted Identification of Metabolite Mass Spectra. BMC Bioinformatics 2010, 11, 148. 10.1186/1471-2105-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymanski E. L.; Jeon J.; Gulde R.; Fenner K.; Ruff M.; Singer H. P.; Hollender J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. 10.1021/es5002105. [DOI] [PubMed] [Google Scholar]
- Buxton G. V.; Greenstock C. L.; Helman W. P.; Ross A. B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (•OH/•O– in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. 10.1063/1.555805. [DOI] [Google Scholar]
- Norrish Type I Reaction. In Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Ltd, 2010; 2062–2066. [Google Scholar]
- Wagner P. J. Chemistry of Excited Triplet Organic Carbonyl Compounds. Top. Curr. Chem. 1976, 66, 1–52. 10.1007/BFb0047763. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Mopper K. Determination of Photochemically Produced Hydroxyl Radicals in Seawater and Freshwater. Mar. Chem. 1990, 30, 71–88. 10.1016/0304-4203(90)90062-H. [DOI] [Google Scholar]
- Sun L.; Qian J.; Blough N. V.; Mopper K. Insights into the Photoproduction Sites of Hydroxyl Radicals by Dissolved Organic Matter in Natural Waters. Environ. Sci. Technol. Lett. 2015, 2, 352–356. 10.1021/acs.estlett.5b00294. [DOI] [Google Scholar]
- Tafer R.; Sleiman M.; Boulkamh A.; Richard C. Photomineralization of Aqueous Salicylic Acids. Photoproducts Characterization and Formation of Light Induced Secondary OH Precursors (LIS-OH). Water Res. 2016, 106, 496–506. 10.1016/j.watres.2016.10.038. [DOI] [PubMed] [Google Scholar]
- Rosario-Ortiz F. L.; Canonica S. Probe Compounds to Assess the Photochemical Activity of Dissolved Organic Matter. Environ. Sci. Technol. 2016, 50, 12532–12547. 10.1021/acs.est.6b02776. [DOI] [PubMed] [Google Scholar]
- Vione D.; Ponzo M.; Bagnus D.; Maurino V.; Minero C.; Carlotti M. E. Comparison of Different Probe Molecules for the Quantification of Hydroxyl Radicals in Aqueous Solution. Environ. Chem. Lett. 2010, 8, 95–100. 10.1007/s10311-008-0197-3. [DOI] [Google Scholar]
- Page S. E.; Arnold W. A.; McNeill K. Assessing the Contribution of Free Hydroxyl Radical in Organic Matter-Sensitized Photohydroxylation Reactions. Environ. Sci. Technol. 2011, 45, 2818–2825. 10.1021/es2000694. [DOI] [PubMed] [Google Scholar]
- Venkatesagowda B.; Dekker R. F. H. Microbial Demethylation of Lignin: Evidence of Enzymes Participating in the Removal of Methyl/Methoxyl Groups. Enzyme Microb. Technol. 2021, 147, 109780 10.1016/j.enzmictec.2021.109780. [DOI] [PubMed] [Google Scholar]
- Nalven S. G.; Ward C. P.; Payet J. P.; Cory R. M.; Kling G. W.; Sharpton T. J.; Sullivan C. M.; Crump B. C. Experimental Metatranscriptomics Reveals the Costs and Benefits of Dissolved Organic Matter Photo-Alteration for Freshwater Microbes. Environ. Microbiol. 2020, 22, 3505–3521. 10.1111/1462-2920.15121. [DOI] [PubMed] [Google Scholar]
- von Sonntag J.; Mvula E.; Hildenbrand K.; von Sonntag C. Photohydroxylation of 1,4-Benzoquinone in Aqueous Solution Revisited. Chem. – Eur. J. 2004, 10, 440–451. 10.1002/chem.200305136. [DOI] [PubMed] [Google Scholar]
- Matsuura T.; Omura K. Photochemical Hydroxylation of Aromatic Compounds. Synthesis 1974, 1974, 173–184. 10.1055/s-1974-23269. [DOI] [Google Scholar]
- Durai Manickam M. C.; Pitchumani K.; Srinivasan C. Selectivity in Photohydroxylation of 4-Nitroveratrole and Nitroanisoles Catalysed by Cyclodextrins. J. Photochem. Photobiol., A 2002, 149, 131–137. 10.1016/S1010-6030(02)00016-3. [DOI] [Google Scholar]
- Ulanski P.; von Sonntag C. The OH Radical-Induced Chain Reactions of Methanol with Hydrogen Peroxide and with Peroxodisulfate. J. Chem. Soc. Perkin Trans. 2 1999, 2, 165–168. 10.1039/a808543i. [DOI] [Google Scholar]
- Wilkinson F.; Helman W. P.; Ross A. B. Rate Constants for the Decay and Reactions of the Lowest Electronically Excited Singlet State of Molecular Oxygen in Solution. An Expanded and Revised Compilation. J. Phys. Chem. Ref. Data 1995, 24, 663–677. 10.1063/1.555965. [DOI] [Google Scholar]
- Peon J.; Tan X.; Hoerner J. D.; Xia C.; Luk Y. F.; Kohler B. Excited State Dynamics of Methyl Viologen. Ultrafast Photoreduction in Methanol and Fluorescence in Acetonitrile. J. Phys. Chem. A 2001, 105, 5768–5777. 10.1021/jp010084d. [DOI] [Google Scholar]
- Watkins M. A.; Pitzenberger S.; Harmon P. A. Direct Evidence of 2-Cyano-2-Propoxy Radical Activity During AIBN-Based Oxidative Stress Testing in Acetonitrile–Water Solvent Systems. J. Pharm. Sci. 2013, 102, 1554–1568. 10.1002/jps.23500. [DOI] [PubMed] [Google Scholar]
- Paul H.; Small R. D. Jr.; Scaiano J. C. Hydrogen Abstraction by Tert-Butoxy Radicals. A Laser Photolysis and Electron Spin Resonance Study. J. Am. Chem. Soc. 1978, 100, 4520–4527. 10.1021/ja00482a033. [DOI] [Google Scholar]
- Banks J. T.; Scaiano J. C. The Laser-Drop Method: A New Approach to Induce Multiple Photon Chemistry with Pulsed Lasers. Examples Involving Reactions of Diphenylmethyl and Cumyloxyl Radicals. J. Am. Chem. Soc. 1993, 115, 6409–6413. 10.1021/ja00067a067. [DOI] [Google Scholar]
- Orlando J. J.; Tyndall G. S. Laboratory Studies of Organic Peroxy Radical Chemistry: An Overview with Emphasis on Recent Issues of Atmospheric Significance. Chem. Soc. Rev. 2012, 41, 6294–6317. 10.1039/C2CS35166H. [DOI] [PubMed] [Google Scholar]
- Heitner C.The Photochemistry of Lignin. In Lignin and Lignans; CRC Press, 2010; 555–584. [Google Scholar]
- Mill T.; Hendry D. G.; Richardson H. Free-Radical Oxidants in Natural Waters. Science 1980, 207, 886–887. 10.1126/science.207.4433.886. [DOI] [PubMed] [Google Scholar]
- Gao H.; Zepp R. G. Factors Influencing Photoreactions of Dissolved Organic Matter in a Coastal River of the Southeastern United States. Environ. Sci. Technol. 1998, 32, 2940–2946. 10.1021/es9803660. [DOI] [Google Scholar]
- Yan G.; Kaiser K. A Rapid and Sensitive Method for the Analysis of Lignin Phenols in Environmental Samples Using Ultra-High Performance Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry with Multiple Reaction Monitoring. Anal. Chim. Acta 2018, 1023, 74–80. 10.1016/j.aca.2018.03.054. [DOI] [PubMed] [Google Scholar]
- Valentine R. L.; Zepp R. G. Formation of Carbon Monoxide from the Photodegradation of Terrestrial Dissolved Organic Carbon in Natural Waters. Environ. Sci. Technol. 1993, 27, 409–412. 10.1021/es00039a023. [DOI] [Google Scholar]
- Fichot C. G.; Miller W. L. An Approach to Quantify Depth-Resolved Marine Photochemical Fluxes Using Remote Sensing: Application to Carbon Monoxide (CO) Photoproduction. Remote Sens. Environ. 2010, 114, 1363–1377. 10.1016/j.rse.2010.01.019. [DOI] [Google Scholar]
- Blough N. V.Photochemistry in the Sea-Surface Microlayer. In The Sea Surface and Global Change; Liss P. S., Duce R. A., Eds.; Cambridge University Press, 1997; 383–424. [Google Scholar]
- Carpenter L. J.; Nightingale P. D. Chemistry and Release of Gases from the Surface Ocean. Chem. Rev. 2015, 115, 4015–4034. 10.1021/cr5007123. [DOI] [PubMed] [Google Scholar]
- Dixon J. L.; Beale R.; Nightingale P. D. Production of Methanol, Acetaldehyde, and Acetone in the Atlantic Ocean. Geophys. Res. Lett. 2013, 40, 4700–4705. 10.1002/grl.50922. [DOI] [Google Scholar]
- Moran M. A.; Zepp R. G. Role of Photoreactions in the Formation of Biologically Labile Compounds from Dissolved Organic Matter. Limnol. Oceanogr. 1997, 42, 1307–1316. 10.4319/lo.1997.42.6.1307. [DOI] [Google Scholar]
- de Bruyn W. J.; Clark C. D.; Pagel L.; Takehara C. Photochemical Production of Formaldehyde, Acetaldehyde and Acetone from Chromophoric Dissolved Organic Matter in Coastal Waters. J. Photochem. Photobiol., A 2011, 226, 16–22. 10.1016/j.jphotochem.2011.10.002. [DOI] [Google Scholar]
- Goldstone J. V.; Pullin M. J.; Bertilsson S.; Voelker B. M. Reactions of Hydroxyl Radical with Humic Substances: Bleaching, Mineralization, and Production of Bioavailable Carbon Substrates. Environ. Sci. Technol. 2002, 36, 364–372. 10.1021/es0109646. [DOI] [PubMed] [Google Scholar]
- Pullin M. J.; Bertilsson S.; Goldstone J. V.; Voelker B. M. Effects of Sunlight and Hydroxyl Radical on Dissolved Organic Matter: Bacterial Growth Efficiency and Production of Carboxylic Acids and Other Substrates. Limnol. Oceanogr. 2004, 49, 2011–2022. 10.4319/lo.2004.49.6.2011. [DOI] [Google Scholar]
- Brinkmann T.; Hörsch P.; Sartorius D.; Frimmel F. H. Photoformation of Low-Molecular-Weight Organic Acids from Brown Water Dissolved Organic Matter. Environ. Sci. Technol. 2003, 37, 4190–4198. 10.1021/es0263339. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Parker K. M. Halogen Radical Oxidants in Natural and Engineered Aquatic Systems. Environ. Sci. Technol. 2018, 52, 9579–9594. 10.1021/acs.est.8b02219. [DOI] [PubMed] [Google Scholar]
- Vaughan P. P.; Blough N. V. Photochemical Formation of Hydroxyl Radical by Constituents of Natural Waters. Environ. Sci. Technol. 1998, 32, 2947–2953. 10.1021/es9710417. [DOI] [Google Scholar]
- Gligorovski S.; Strekowski R.; Barbati S.; Vione D. Environmental Implications of Hydroxyl Radicals (•OH). Chem. Rev. 2015, 115, 13051–13092. 10.1021/cr500310b. [DOI] [PubMed] [Google Scholar]
- Page S. E.; Logan J. R.; Cory R. M.; McNeill K. Evidence for Dissolved Organic Matter as the Primary Source and Sink of Photochemically Produced Hydroxyl Radical in Arctic Surface Waters. Environ. Sci. Process. Impacts 2014, 16, 807–822. 10.1039/C3EM00596H. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Del Vecchio R.; Blough N. V. Investigating the Mechanism of Hydrogen Peroxide Photoproduction by Humic Substances. Environ. Sci. Technol. 2012, 46, 11836–11843. 10.1021/es3029582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.