

Abstract
The AAA+ family member KaiC is the central pacemaker for circadian rhythms in the cyanobacterium Synechococcus elongatus. Composed of two hexameric rings of adenosine triphosphatase (ATPase) domains with tightly coupled activities, KaiC undergoes a cycle of autophosphorylation and autodephosphorylation on its C-terminal (CII) domain that restricts binding of clock proteins on its N-terminal (CI) domain to the evening. Here, we use cryogenic-electron microscopy to investigate how daytime and nighttime states of CII regulate KaiB binding on CI. We find that the CII hexamer is destabilized during the day but takes on a rigidified C2-symmetric state at night, concomitant with ring-ring compression. Residues at the CI-CII interface are required for phospho-dependent KaiB association, coupling ATPase activity on CI to cooperative KaiB recruitment. Together, these studies clarify a key step in the regulation of cyanobacterial circadian rhythms by KaiC phosphorylation.
Cyanobacteria possess an internal circadian clock that temporally aligns gene expression with the solar day to maximize photosynthetic output and coordinate integrated metabolic processes1–3. The most basic manifestation of this timekeeping function is a cyclic pattern of autophosphorylation in CII domain of the hexameric clock protein KaiC4 that results in the following sequence of posttranslational modifications at residues S431 and T432: S/T ➝ S/pT ➝ pS/pT ➝ pS/T5,6. During the day, autophosphorylation is stimulated by another clock protein, KaiA, which binds to the C-terminal end of KaiC, known as the A-loop, to upregulate KaiC autophosphorylation7. At night, compression of the two KaiC rings partially opens the CI ring to expose binding sites8 that allow a third clock protein, KaiB, to be cooperatively recruited to KaiC9–12, where it undergoes a conformational change to bind and sequester KaiA13,14. Without KaiA bound to CII, the equilibrium of KaiC shifts toward CII autodephosphorylation7, which proceeds until affinity for KaiB on CI is lost. At this point, KaiA and KaiB dissociate from CI, as well as from each other, allowing KaiA to once again bind KaiC via its A-loops, thus completing a negative feedback loop that takes around 24 h per cycle.
Both in vitro and in vivo studies have linked the S/T and S/pT phosphostates of KaiC with the daytime when the CII domains are loosely bound to each other15 and the A-loops are exposed16, facilitating KaiA binding5,6. By contrast, the pS/pT and pS/T states are associated with the KaiB-bound nighttime state of KaiC, where KaiA is sequestered in its inactive form on the CI domain8,13,17. Formation of the repressive nighttime complex is linked to the adenosine triphosphatase (ATPase cycle of the CI domain, as mutations that block CI ATPase activity are deficient in KaiB binding18 and the KaiC CI domain is only found in the ADP-bound form when bound to KaiB13,17.
CI ATPase activity and CII autophosphorylation appear to be functionally linked as well because mutations in either domain can give rise to correlated changes in CI ATP hydrolysis and the overall period of biochemical oscillations19. In particular, substitutions of the CII residue Tyr402, located at the interface between CI and CII, gives rise to extreme changes in period and correlated ATPase activity depending on the amino acid substituted into this position20. This residue is poised on the CII-α8 helix near a patch of CI residues known as the arginine tetrad that are essential for circadian rhythms in vivo21, suggesting the existence of an allosteric conduit connecting the CI and CII active sites that runs through these regions. However, no direct structural evidence exists for this hypothesis, and it currently remains unclear how the information encoded by CII phosphorylation state is transduced roughly 70 Å to regulate KaiB binding on CI.
KaiC can be trapped in its daytime or nighttime-like states using phosphomimetic substitutions at the CII autophosphorylation sites6,22,23. Herein, we used KaiC-S431A-T432E as a daytime phosphomimetic (referred to as KaiC-AE, see Supplementary Table 1 for details on the constructs used) and KaiC-S431E-T432A to represent the nighttime variant (KaiC-EA) to study how phosphorylation influences conformational changes and coupling between CI and CII domains. Solution biophysical studies have demonstrated modest changes in the global conformation of KaiC throughout the phosphocycle24,25 as well as in the intra- (cis) and inter- (trans) subunit interactions of ATPase domains using these phosphomimetics mutations15,26.
Despite the stark differences in KaiC protein dynamics and clock protein association observed in solution studies, crystallographic experiments have not provided a suitable explanation for the distinct biochemical properties of KaiC phosphomimetics21,27, likely due to crystal packing forces. Given the intrinsically dynamic nature of KaiC25,28,29 and incident shallow conformational energy landscape, we sought to circumvent these issues by using cryogenic-electron microscopy (cryo-EM) to obtain structures of daytime and nighttime KaiC by single-particle reconstruction. This revealed previously unobserved conformations that demonstrate how information encoded by CII phosphorylation is transmitted through dynamic structural features to regulate biochemical activity on CI.
Results
Comparison of daytime and nighttime KaiC structures.
To establish a baseline for KaiB discrimination between the phosphomimetics, we first compared KaiB affinity in the daytime and nighttime-trapped forms of KaiC (Fig. 1a and Supplementary Fig. 1). We observed ≥30-fold tighter binding of KaiB to nighttime KaiC than the daytime variant, recapitulating the previously observed day/night distinction between KaiC-AE and KaiC-EA phosphomimetics6. Next, we subjected KaiC-EA and KaiC-AE to comparison by cryo-EM in the presence of saturating concentrations of ATP to identify potential structural differences between the phosphomimetic variants.
Fig. 1 |. Daytime and nighttime phosphomimetics confer distinct biochemical activities and global conformations to KaiC.
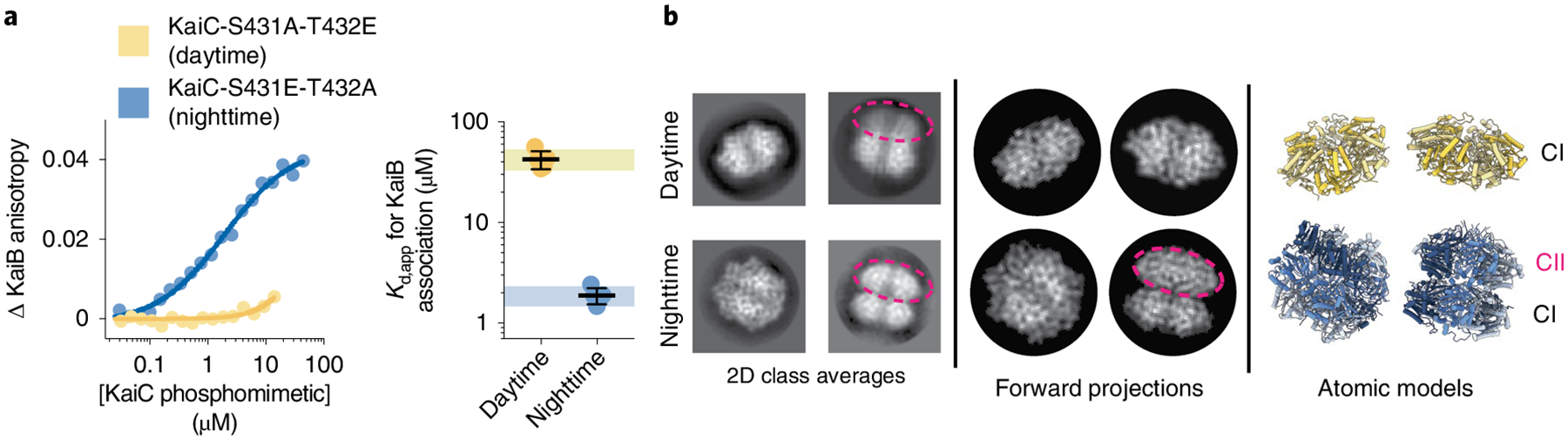
a, Titration curves and apparent binding constants (Kd,app) for KaiB association with daytime or nighttime KaiC phosphomimetics in units of protomer concentration, as measured using fluorescence anisotropy of labeled KaiB in KaiC titrations. Black lines and error bars represent mean ± standard deviation (s.d.) from n = 5 independent titrations. The hash (#) represents P < 0.0001 from a two-sided unpaired parametric t-test between the two phosphomimetics. 95% confidence interval for the measurements on the daytime and nighttime phosphomimetic variant are depicted as colored boxes (light blue for nighttime, light yellow for daytime). This depiction is used throughout the paper for comparison of other variants to the unsubstituted phosphomimetics. Mean Kd,app ± s.d. is 2 ± 0.4 μM for nighttime KaiC and ≥43 ± 8 μM for daytime KaiC. In cases where the upper bound is reported, Kd,app was too high to measure (Supplementary Fig. 1 and Methods). b, Reference-free two-dimensional class averages from electron micrographs of KaiC phosphomimetics. Dashed ovals indicate the CII rings, where visible, as inferred from forward projections obtained using the atomic models shown.
In daytime KaiC, we observed a decrease in well-defined cryo-EM density for the CII ring (Fig. 1b), consistent with previous solution studies where the S/pT state or the phosphomimetic AE mutant of KaiC exhibited an ‘open’ or destabilized CII hexamer15,30. Since flexible domains are often susceptible to damaging interactions with the hydrophobic air-water interface during sample preparation, we also characterized daytime KaiC in the presence of perfluorinated fos-choline, which limits air-water interface interactions31. KaiC phosphomimetics maintained their functional discrimination for KaiB binding in the presence of fos-choline (Supplementary Fig. 2), indicating that the structures observed under these conditions are functionally relevant. The inclusion of fos-choline stabilized the same ‘extended’ conformation in daytime KaiC that was originally observed by crystallography32 (Table 1, Fig. 2a and Extended Data Fig. 1), resolved to roughly 3.8 Å in our cryo-EM structure.
Table 1 |.
Cryo-EM data collection, refinement and validation statistics
| KaiC-EA compressed state (EMD-24850) (PDB 7S65) | KaiC-EA expanded state (EMD-24851) (PDB 7S66) | KaiC-AE expanded state (EMD-24852) (PDB 7S67) | |
|---|---|---|---|
| Data collection and processing | |||
| Microscope | Talos Arctica | Talos Arctica | Talos Arctica |
| Voltage (kV) | 200 | 200 | 200 |
| Detector | K2 Summit | K2 Summit | K2 Summit |
| Magnification (nominal/calibrated) | ×36,000/×43,478 | ×36,000/×43,478 | ×36,000/×43,478 |
| Exposure navigation | Image shift to 4 holes (tilted)/image shift to 16 holes (thin carbon) | Image shift to 4 holes (tilted)/image shift to 16 holes (thin carbon) | Image shift to 16 holes |
| Data acquisition software | Leginon | Leginon | Leginon |
| Electron exposure (e−/Å2) | 48 (tilted)/51 (thin carbon) | 48 (tilted)/51 (thin carbon) | 40.3 |
| Exposure rate (e−/pixel per s) | 7.94 (tilted)/7.5 (thin carbon) | 7.94 (tilted)/7.5 (thin carbon) | 8.6 |
| Frame length (ms) | 100 (tilted)/250 (thin carbon) | 100 (tilted)/250 (thin carbon) | 145 |
| Number of frames per micrograph | 80 (tilted)/36 (thin carbon) | 80 (tilted)/36 (thin carbon) | 62 |
| Pixel size (Å) | 1.15 | 1.15 | 1.15 |
| Defocus range (μm) | −0.8 to −1.7 (tilted)/−0.5 to −1.5 (thin carbon) | −0.8 to −1.7 (tilted)/−0.5 to −1.5 (thin carbon) | −0.5 to −2.0 |
| Micrographs collected (no.) | 1,137 (tilted)/8,405 (thin carbon) | 1,137 (tilted)/8,405 (thin carbon) | 1,541 |
| Reconstruction | |||
| Image-processing package | Relion | Relion | Relion/CryoSPARC |
| Total extracted particles (no.) | 2,016,826 (tilted)/6,896,832 (thin carbon) | 2,016,826 (tilted)/6,896,832 (thin carbon) | 427,620 |
| Refined particles (no.) | 1,341,217 (tilted)/2,856,972 (thin carbon) | 1,341,217 (tilted)/2,856,972 (thin carbon) | 367,247 |
| Final particles (no.) | 122,855 | 91,869 | 89,892 |
| Symmetry imposed | C2 | C6 | C6 |
| Global resolution (Å) | 3.2 | 2.8 | 3.8 |
| FSC 0.143 (unmasked/masked) | 3.3/3.2 | 2.9/2.9 | 3.9/3.8 |
| FSC 0.5 (unmasked/masked) | 3.5/3.4 | 3.2/3.1 | 4.1/4.0 |
| Map resolution range (local) (Å) | 3.0–4.0 | 2.7–4.0 | 3.6–4.7 |
| 3DFSC sphericity | 0.902 out of 1 | 0.979 out of 1 | 0.973 out of 1 |
| Model sharpening B factor (Å2) | −148.4 | −112.9 | −126.7 |
| Model composition | |||
| Protein residues | 2,765 | 2,910 | 2,892 |
| Ligands | 24 | 24 | 24 |
| Refinement | |||
| Refinement package | Phenix | Phenix | Phenix |
| CC (volume/mask) | 0.72/0.74 | 0.81/0.83 | 0.73/0.73 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.014 | 0.012 | 0.0013 |
| Bond angles (°) | 1.133 | 1.028 | 1.407 |
| Validation | |||
| Map-to-model FSC 0.5 | 3.3 | 3.1 | 3.9 |
| Ramachandran plot | |||
| Outliers | 0 | 0 | 0 |
| Allowed (%) | 1.93 | 1.45 | 1.46 |
| Favored (%) | 98.07 | 98.55 | 98.54 |
| MolProbity score | 1.4 | 0.99 | 1.28 |
| Poor rotamers (%) | 0.17 | 0 | 0 |
| Clashscore (all atoms) | 8.2 | 2.16 | 5.19 |
| C-beta deviations (%) | 0 | 0 | 0.23 |
| CaBLAM Outliers (%) | 0.8 | 1 | 0.8 |
| EMRinger score | 3.52 | 3.8 | 2.1 |
Fig. 2 |. Overview of three-dimensional models obtained for daytime and nighttime KaC variants.
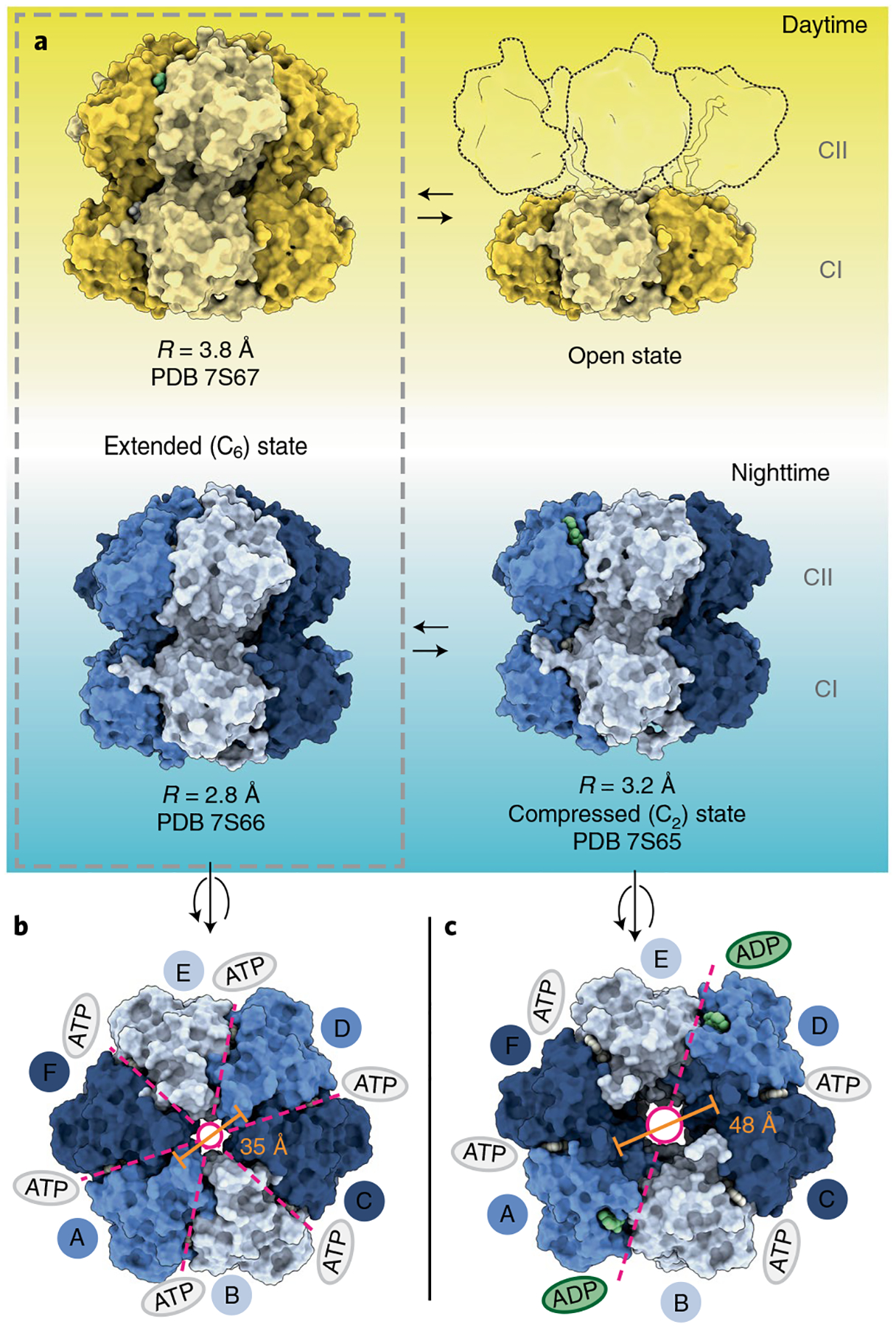
a, Space-filling depictions of daytime and nighttime KaiC conformations observed by cryo-EM. The ‘open’ daytime state is depicted with outlines to represent the destabilized CII protomers as rigid bodies flexibly tethered to the visible CI hexamer. b,c, Axial view of the extended (b) and compressed (c) nighttime KaiC hexamers, viewed from the CII side. Dashed lines indicate the axes of symmetry with colored ovals denoting nucleotide state in the CII ring. Pore diameters are reported as measured between Cα atoms of V433 on A and D protomers.
Although a saturating concentration of ATP was present in solution, we found ADP bound at CII-CII interfaces in the ‘extended’ conformation of daytime KaiC (Extended Data Fig. 2a). It should be noted that because both phosphorylation sites were mutated in our constructs, the ADP nucleotide accumulated at this site likely came from low concentrations of ADP present in solution, possibly arising from catalytic turnover at the CI domain. Because the original structures were obtained from crystals grown in the presence of nonhydrolyzable ATP analogs27,32, this suggests that the C6-symmetric ‘extended’ conformation can accommodate either ATP- or ADP-bound nucleotide state at CII in daytime KaiC, although ADP appears to be preferred. Notably, we also observed the C6-symmetric extended conformation favored by crystallography in nighttime KaiC, this time at a resolution of roughly 2.8 Å, but with ATP in the CII active sites (Extended Data Figs. 2b and 3). Because CII dephosphorylation proceeds through an ADP-bound intermediate33,34, this preference likely functions to delay dephosphorylation of the nighttime state until the ATP/ADP ratio has fallen sufficiently, aligning the phase of the oscillator with cellular metabolism3,35.
In the nighttime phosphomimetic, single-particle analysis also returned an additional distinct map with both the CI and CII rings resolved (Fig. 1b and Extended Data Fig. 3) in a new C2-symmetric structure that was resolved to roughly 3.2 Å. This new subpopulation of KaiC hexamers has a widened central pore (Fig. 2b,c), coupled with accumulation of ADP at two of the six CII-CII interfaces (Extended Data Fig. 2c,d) opposite each other in the hexameric ring. KaiC protomers adjacent to these sites adopt a ‘compressed’ conformation that brings the CI and CII domains together (Figs. 2a and 3a), whereas the other four protomers remain in the extended conformation. It should be noted that C2-symmetry is apparently rare in hexameric AAA+ proteins, with only a few other examples having been reported so far36–38. In contrast to these previously described ‘dimer of trimer’ states that each exhibit apo nucleotide pockets at their seam protomers (Extended Data Fig. 4), the C2-symmetric state of KaiC is unique in having its seam protomers occupied by ADP, suggesting that nucleotide binding on CII is an important regulator of CI-CII communication.
Fig. 3 |. Nucleotide interactions at CII-CII interfaces govern the transition between extended and compressed conformations.
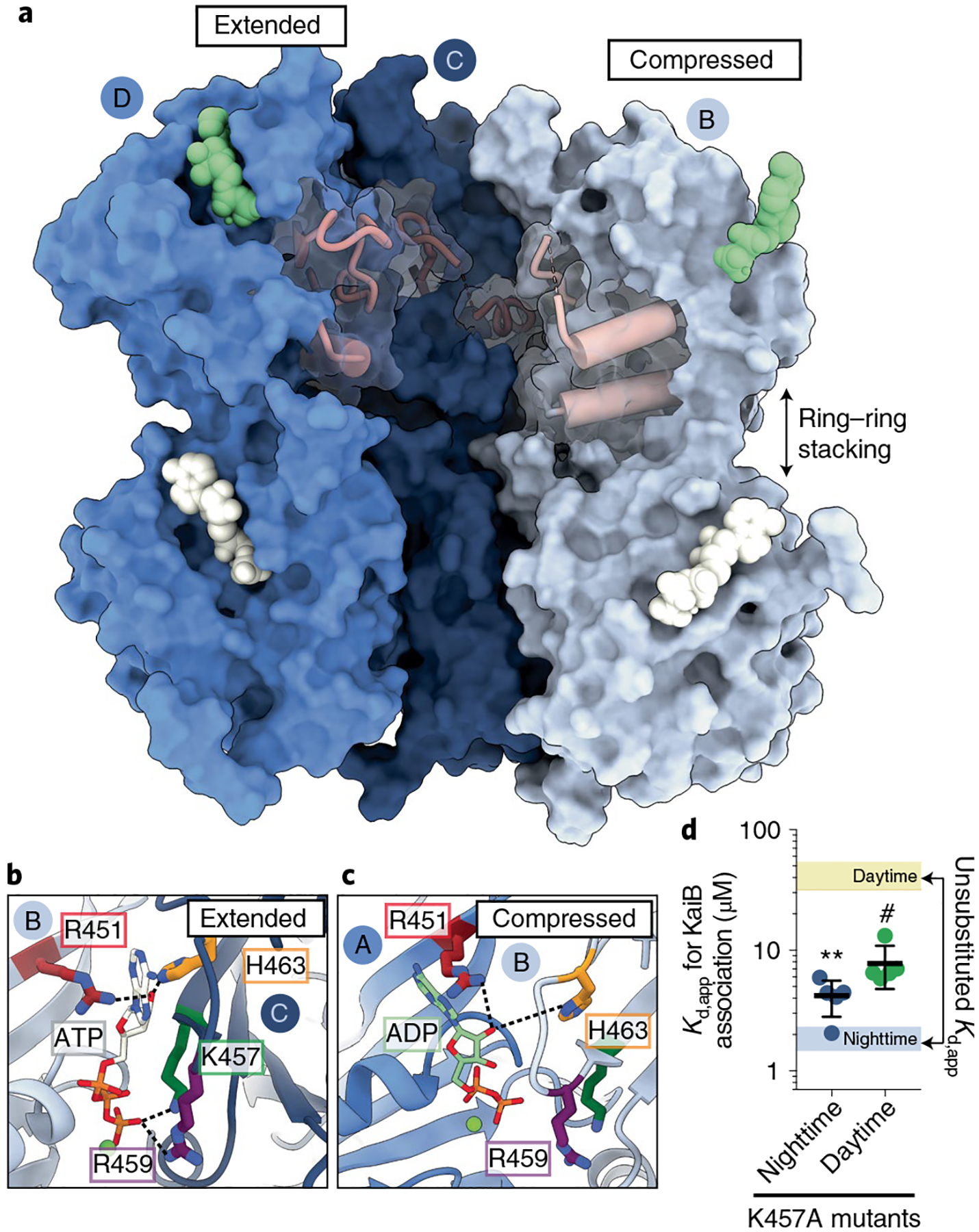
a, Surface representation of KaiC protomers B, C and D in the C2-symmetric hexamer with CII-α8 and CII-α9 depicted as pink cylinders. Three subunits (A, E and F) have been removed to view inside the hexamer. ATP nucleotides are depicted as white surfaces, and ADP in green. b,c, Close-ups of the CII nucleotide interface in the extended (b) and compressed (c) KaiC protomers. d, Kd,app for the interaction of KaiB with daytime (green) and nighttime (dark blue) KaiC variants the bearing K457A mutation. Black lines and error bars represent mean ± s.d. from n = 5 independent titrations. Colored bars representing the 95% confidence interval for analogous measurement on KaiC phosphomimetics without the mutation are provided for comparison. Symbols (#P < 0.0001; **P < 0.01) represent the results of two-sided unpaired parametric t-tests comparing each mutant to its respective unsubstituted phosphomimetic; exact P values reported in Supplementary Table 6. Mean Kd,app ± s.d. is 7.3 ± 1.9 μM for daytime KaiC-K457A and 4.6 ± 1.8 μM for nighttime KaiC-K457A.
Another feature of the C2-symmetric state we observed is the loss of A-loop interactions within the central pore of CII, driven by disruption of interactions across the compressed CII protomer interface (Extended Data Fig. 5a). To test whether these interactions influence catalytic activity on CII, we measured the phosphorylation state of several KaiC mutants in this region (Extended Data Fig. 5b,c). We found that the E444S mutant, which disrupts the hydrogen bond between the sidechain of E444 and the backbone amide of I490, significantly upregulated KaiC autophosphorylation in the absence of KaiA, similar to a previous study where an E444D mutation also resulted in constitutive hyperphosphorylation of KaiC7. Our observation of coupling between A-loop conformations across the hexamer in our C2-symmetric structure of nighttime KaiC suggests that coupling could also exist across KaiC protomers during the day, possibly explaining why only substoichiometric levels of KaiA are needed to maintain robust oscillations39.
Role of the compressed state in KaiB association.
These observations raise the question: how does ring compression influence KaiB association? In the extended KaiC protomers of both the C6- and C2-symmetric structures, the side chains of residues K457 and R459 interact directly with the γ-phosphate of ATP (Fig. 3b). However, contact with these side chains is lost at the ADP-occupied CII interfaces (Fig. 3c) and the CII domain on the compressed protomer breaks away from the adjacent CII interface. Computational studies have highlighted the importance of nucleotide interactions at CII interfaces on the global conformational dynamics of KaiC29. Given the apparent coupling between CII nucleotide interactions and the configuration of cis-interacting domains in the compressed protomers, we hypothesized that introducing an alanine residue in place of K457 would make this state accessible to the daytime variant to enhance KaiB association. Consistent with our prediction, the K457A mutation resulted in a roughly sixfold increase in KaiB affinity for daytime KaiC (Fig. 3d and Supplementary Fig. 1b), showing that KaiB association is bolstered by mutations that promote the compressed conformer in the daytime state. Furthermore, a modest loss of affinity was also observed in the nighttime K457A variant, suggesting that the extended conformations observed in the C2-symmetric structure, which are stabilized by K457 interactions, also play a role in efficient KaiB association.
These changes in CII nucleotide state and associated ‘breaking away’ of the CII-CII interface resulted changes to secondary structure in the compressed protomers of C2-symmetric KaiC structure. In particular, the CII-α9 helix, where the phosphomimetic-substituted autophosphorylation sites are located, comprises only a single turn in the extended protomers (Fig. 4a), but was lengthened in the ADP-bound, compressed protomers (Fig. 4b). When we inhibited this helical elongation by mutating I430 to glycine, we observed a ≥50-fold decrease in KaiB affinity (Fig. 4c and Supplementary Fig. 1c), demonstrating that KaiB association is dependent on helix extension near the CII autophosphorylation sites.
Fig. 4 |. Compression of the cis CI-CII interface primes nighttime KaiC for KaiB association.

a,b, EM density and atomic models for residues S424-T434 (pink) and CII-α8 helices in extended (a, contour, 0.0343) and compressed (b, contour, 0.0307) protomers of the C2-symmetric state of KaiC-EA. c, KaiB affinity for nighttime KaiC with variants with mutations\s of CII-α9 or arginine tetrad. Line and error represent mean ± s.d. for n = 5 (n = 4 for R216A) measurements. Symbols represent one-way analysis of variance (ANOVA) (#P < 0.0001) with Dunnett’s multiple comparisons for each mutant against nighttime KaiC. Mean Kd,app is ≥25 ± 12 μM (I430g), ≥25 ± 4 μM (R216A) and ≥13 ± 4 μM (R217A) for the nighttime KaiC mutants. d, Cylinder depictions of the CII-α8 helices from the three unique protomer conformations of the compressed hexamer, aligned about their CI domains. e, Proximity of CII-α8 to the arginine tetrad in the C2-symmetric state.
Lengthening of CII-α9 resulted in translocation of the nearby CII-α8 helix toward the CI-CII interface about 5 Å in our compressed KaiC structure (Fig. 4d). This is notable because a recent study identified the CII-α8 residue Tyrosine 402 as a key regulator of timing in KaiC20, with mutants at this position exhibiting extreme changes in CI ATPase activity and correlated in vivo circadian period (for example, 15 h to >6 days). We observed EM density for distinct rotamer conformations of Y402 only in the compressed protomers (Fig. 4e and Extended Data Fig. 6) situated near other period-determining residues on the linker connecting CI and CII19, indicating that period regulation by Tyrosine 402 likely occurs through dynamic interactions at the CI-CII interface.
Translocation of CII-α8 via C2-symmetric ring compression appears to regulate the biochemical activity of CI via four sequential, highly conserved arginine residues in the CI domain known as the ‘arginine tetrad’ (Fig. 4e and Extended Data Figs. 7 and 8a)21. These residues are essential for circadian rhythms and form a network of electrostatic interactions that join each CI protomer to both clockwise and counterclockwise neighbors (Extended Data Figs. 8b,c and Supplementary Table 2)21,40. This nexus of KaiC regulation is situated between the CI and CII domains at the tip of the CI-β9 hairpin that leads directly into the nucleotide binding pocket of CI (Extended Data Fig. 8b), where enzymatic activity is required for KaiB association18. We measured KaiB binding for several alanine mutants within the arginine tetrad and found that affinity was reduced by more than tenfold for R216A (Fig. 4c and Supplementary Fig. 1c) and by roughly sixfold for R217A, highlighting the importance of interdomain interactions in this region for KaiB association.
Transduction of phosphostate information throughout CI.
The cooperative nature by which KaiB monomers are recruited to the KaiC hexamer is well established9–12. Given that the arginine tetrad is structurally poised to facilitate both cis and trans protomer regulation, we next wondered if the KaiC-R217A mutant would influence this cooperativity. To assess this, we updated our method for quantifying KaiB cooperativity11 to reflect the intrinsic homotropic cooperativity when unlabeled KaiB-I87A, often referred to as fold-switched or fsKaiB, is used to stimulate cooperative association of fluorescently labeled wild-type KaiB in KaiC binding assays (Extended Data Fig. 9a). Using this approach, we measured a KaiB cooperativity index of roughly 21 ± 2 for KaiC-EA (Fig. 5a and Extended Data Fig. 9b) using fsKaiB as a secondary titrant in KaiC affinity assays. Because wild-type KaiB takes on the structure of fsKaiB when associated with KaiC13,14,17, we can interpret this as a roughly 21-fold affinity enhancement for the subsequent association of KaiB after the first KaiB molecule binds to the hexamer. The R217A mutant showed a severe reduction of the cooperativity index (roughly 1.6 ± 0.5), suggesting that the arginine tetrad is essential not only for communication between CI and CII domains in cis, but that trans protomer communication must also run through this conduit. Another arginine tetrad residue, R218, interacts directly with the 2′ hydroxyl of the nucleotide at CI-CI interfaces, which is sandwiched between protomers by hydrogen bonds to R218 and H230 (Fig. 5b, Extended Data Fig. 10 and Supplementary Table 2). To test whether this nucleotide-mediated CI-CI interaction is important for KaiB association, we substituted H230 with alanine, and observed significantly diminished cooperativity (Fig. 5a), as well as about a fourfold reduction in overall affinity for KaiB (Fig. 5c and Supplementary Fig. 1d), demonstrating that the CI-CI interactions must traverse both the arginine tetrad and the nucleotide binding pocket for efficient KaiB association.
Fig. 5 |. Interactions in the CI nucleotide binding pocket link ATP hydrolysis to cooperative KaiB recruitment.

a, Cooperativity indices for nighttime KaiC variants determined with fsKaiB as secondary titrant. Lines and error bars represent mean ± s.d. for n = 3 independent titrations. Symbols represent one-way ANOVA (NS, not significant; **P < 0.01; #P < 0.0001) with Dunnett’s multiple comparisons to unsubstituted nighttime KaiC. b, Atomic models and electron density (Fc − F0, σ = 0.21) for nucleotide interactions from the prehydrolysis (PDB 4TL8, top) or posthydrolysis (PDB 4TLA chain C, bottom) CI domain crystal structures40. c, KaiB binding affinity for nighttime KaiC active site mutants. Lines and error bars represent mean and s.d. from n = 4 (n = 5 for K224A) independent titrations. Symbols represent statistical analysis identical to that described for a. Mean Kd,app values are 5.9 ± 1.5 μM (H230A), ≥38 ± 18 μM (R226A) and 4.6 ± 0.3 μM (K224A). d, Alignment of CI and CII protein sequences from S. elongatus. e, Turnover rate constants (kcat) for ATP hydrolysis by KaiC-EA variants. Lines and error bars represent mean values ± s.d. with statistical analysis as described for a; exact P values reported in Supplementary Table 6.
It should be noted that the overall architecture of the nucleotide binding pocket is well-conserved between the CI and CII domains of KaiC and is nearly invariant in KaiC across cyanobacteria (Fig. 5d and Extended Data Fig. 7). Residues R215, R216 and R217 of the arginine tetrad are unique to CI, indicative of their role in CI-CII and CI-CI interdomain regulation in KaiC. However, CI residues R218 and H230 have analogs in CII, R451 and H463 that play similar structural roles leading to trans CII-CII interdomain interactions between protomers that bridge the nucleotide (Fig. 3b). Furthermore, CI and CII domains also contain a conserved lysine/arginine pair (K224 and R226 in CI, K457 and R459 in CII) near the nucleotide binding site. Given the importance of nucleotide interactions we observed in the CII domain, we revisited the atomic resolution structures of CI hexamers previously reported by Abe et al.40. In these structures, K224 changes its orientation to coordinate the ultimate nucleotide phosphate at both pre- and posthydrolysis CI-CI interfaces (Fig. 5b, Extended Data Fig. 10 and Supplementary Table 3), whereas R226 coordinates nucleotide only in the ATP-bound state, playing a catalytic role in ATP hydrolysis40.
Given the importance of K457 in nucleotide-mediated CII-CII interactions and KaiB affinity, we next set out to explore the functional role of the analogous CI residue, K224, and its neighboring residues R226 and H230 in more detail. To determine how these mutations influence CI ATP hydrolysis, we measured the enzymatic turnover rate (kcat) of alanine substitution mutants at residues R226, K224 and H230 (Fig. 5e and Supplementary Fig. 3). As expected, the R226A mutation significantly decreased both ATPase activity and KaiB affinity, similar to other catalytically dead variants18. The H230A mutant exhibited increased ATPase activity that, combined with its diminished affinity for KaiB, shows that ATPase activity and KaiB binding are not simply correlated in a linear fashion. Notably, the K224A mutant had minor reductions in both KaiB affinity and ATPase activity (Fig. 5e) despite its role in the coordination of the nucleotide phosphates in CI. Both the H230A and K224A mutations essentially eliminated the cooperative recruitment of KaiB to the KaiC-EA hexamer (Fig. 5a), demonstrating that interactions around the CI nucleotide are critical for KaiB recruitment to KaiC through trans regulation of the CI domain.
The coupling of CI ATPase activity to cooperative KaiB recruitment could impart a switch-like nature to the assembly of this critical protein complex––a function that has been proposed for other RecA-like AAA+ proteins41,42. To assess the functional consequences of disrupting KaiB cooperativity, we set up in vitro oscillator reactions with the K224A or H230A KaiC mutants in the presence of KaiA and KaiB, and observed that both mutations resulted in a total loss of biochemical rhythms (Fig. 6a). This severe phenotype could potentially be explained by decreases in the affinity of KaiB for KaiC, although KaiC-EA K224A showed only a twofold decrease in affinity and weak rhythms would have been expected to persist11,39.
Fig. 6 |. Functional characterization of CI active site mutants.
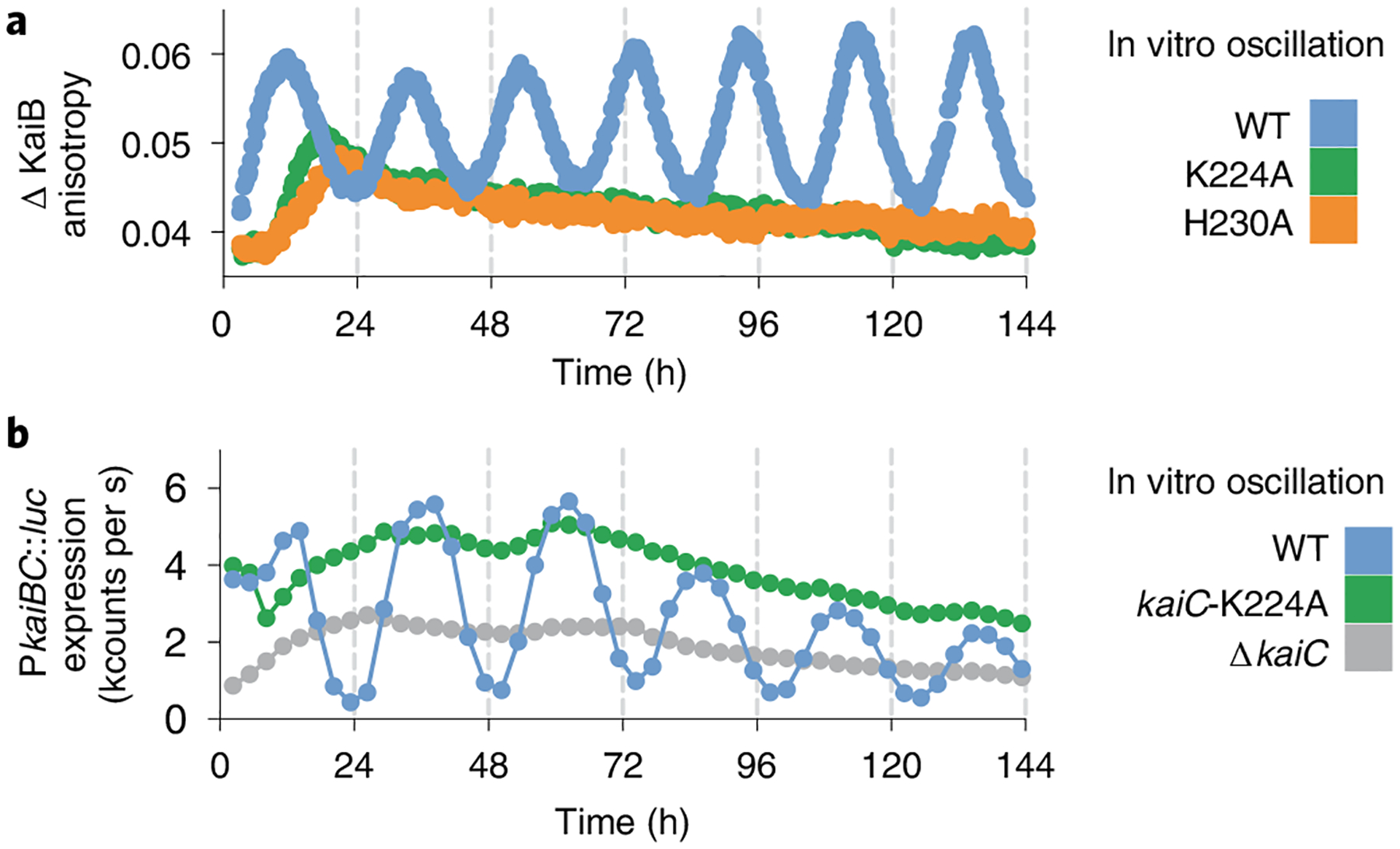
a, Representative in vitro oscillation reactions of KaiC in the presence of KaiA and KaiB measured by fluorescence polarization anisotropy of labeled KaiB (n = 3). b, Representative bioluminescence time courses from a luciferase reporter gene driven by PkaiBC in S. elongatus cultures entrained under 12-h light/dark cycles for 48-h and subsequently allowed to free-run in constant light (n = 6).
We recently reported that decreasing KaiB to subsaturating concentrations just below its Kd,app significantly reduced the amplitude of oscillations in vitro, but that rhythms could be rescued by the clock protein SasA, which uses heterotropic cooperativity to stimulate KaiB binding to KaiC11. The concentration of KaiC in vivo has been estimated at 6 ± 1 μM (ref.43) and KaiB is present at roughly twice that44; however, marked changes in the subcellular localization of clock proteins throughout the day45 could alter their local concentrations to influence clock protein assembly. Therefore, we introduced the K224A mutation into KaiC in S. elongatus using CRISPR–Cas12a and found that this KaiC variant could not support circadian rhythms in vivo (Fig. 6b). Therefore, no cellular factors can compensate for the modest reduction in KaiB affinity and loss of CI nucleotide sensing, demonstrating that trans regulation of the CI domain through K224 is essential for circadian rhythms.
CI ATP hydrolysis is required for KaiB association18 and ADP-bound CI interfaces have been observed in all KaiB-bound KaiC structures solved so far13,17. However, we did not observe ADP-bound CI domains in our cryo-EM structures of isolated KaiC, suggesting that the phospho-dependent regulation of KaiB association by CII does not occur by simply trapping the ADP-bound state in CI before KaiB association. Rather, we propose that ADP is replaced with ATP after hydrolysis unless KaiB association locks the CI domain into the posthydrolysis state by inhibiting ADP release.
Discussion
This work provides an important mechanistic link between the daily phosphorylation cycle of KaiC and quaternary structural changes that mediate negative feedback in the cyanobacterial circadian pacemaker. Our cryo-EM studies on the daytime state corroborate previous solution studies that observed a destabilized CII ring in the S/pT phosphostate8,15,26. These results indicate that the destabilized CII state lacks the ability to engage the CI domain in a way that is permissive for initial KaiB association, that is, through the C2-symmetric compressed state (Fig. 7a). Because the compressed state is important for KaiB association, we propose that both the dusk-like pSpT and nighttime pST phosphostates are able to take on this conformation, as our previous studies on a KaiC-EE (pS/pT) phosphomimetic showed a similar affinity and cooperativity of KaiB binding compared to the nighttime state11. Our results therefore support a model where phosphorylation of S431 regulates the CI domain through elongation of the CII-α9 helix, leading to cis domain compression and translocation of CII-α8 toward the arginine tetrad on CI (Fig. 7b). From there, dynamic interactions radiate down into the CI active site, as well as to adjacent protomers, where ATP hydrolysis and CI nucleotide state are coupled with KaiB occupancy on both cis and trans CI domains. The fact that this occurs in the context of a C2-symmetric intermediate further reinforces the cooperative aspect of this phenomenon, because the direct priming of KaiC for KaiB association occurs in concert for two protomers across the hexamer.
Fig. 7 |. Model for regulation of KaiB association by KaiC phosphostate.

a, Cartoon depiction of the KaiB association pathway. Daytime (yellow) and nighttime (blue) states of KaiC both adopt an extended C6-symmetric ground state. During the day, the extended state is in equilibrium with an open state where the CII hexamer is destabilized relative to the CI domain. At night, excursions from the extended state result in a C2-symmetric hexamer where two subunits of the CII domain are compressed to prime KaiC for KaiB association. An initial KaiB binding event takes place through this compressed intermediate to pave the way for cooperative saturation of the CI hexamer with KaiB molecules. b, Schematic depiction of the regulatory conduit through which the CI and CII domains communicate to regulate KaiB association according to clock phase.
The presumptive role of KaiC-K224 in coupling of ATP hydrolysis to switch-like biochemical activity of KaiB binding bears a striking similarity to a recently reported ‘arginine coupler’ in the bacterial helicase loader DnaC42. In this system, alanine substation of a positively charged amino acid just upstream of the arginine finger was found to increase ATPase activity, while being essential for the switch-like loading activity of DnaC. Notably, this motif is conserved in mammalian initiator ATPases42. Our observation of a similar mechanism in KaiC in regulation of the cyanobacterial clock suggests that this may be an even more widespread design feature within the AAA+ family.
Although CI ATPase activity is correlated with circadian period in vivo and in vitro for many KaiC mutants19,20,40,46, no precise chemo-mechanical mechanism has yet been described to explain this. Our results, combined with crystal structures from the literature, demonstrate that the enzymatic cycle on CI is coupled to the CII active site through a dynamic allosteric conduit involving the arginine tetrad and CII-α8. This could explain why the most extreme period-altering mutations in KaiC are located on CII-α8 or nearby in the linker that connects the CI and CII domains19,20. Mutations such as the Tyr402 substitutions reported by Ito-Miwa et al. likely alter interactions between the arginine tetrad and CII-α8 that couple CI ATPase activity to the CII domain to give rise to such dramatic period effects. Together with the associated biochemistry, our structures identify the main waypoints in the allosteric pathway connecting the CI and CII domains, thus paving the way for a comprehensive understanding of the posttranslational oscillator from both modeling and reverse-engineering perspectives.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41594-022-00803-w.
Methods
Site-directed mutagenesis and protein expression/purification.
All protein constructs were expressed from a kanamycin-resistant pET-28b plasmid with an N-terminal His-singly occupied molecular orbital tag for affinity purification. Mutations were introduced by amplification of the whole vector, installing point mutations via primers using the method described by Liu et al.47. KaiA and KaiB were purified as described previously8,15. KaiC constructs were expressed in BL21(DE3) cells (NEB). Cultures were grown in M9 medium under antibiotic selection to an optical density (wavelength λ = 600 nm) between 0.6 and 0.8 by shaking at 37 °C, and were subsequently cooled to 18 °C before induction with 200 μM isopropyl-β-d-thiogalactoside and shaking overnight at 18 °C.
Next, the cultures were spun down and resuspended in 50 mM NaH2PO4 pH 8.0, 500 mM NaCl, 20 mM imidazole and 5 mM β-mercaptoethanol (Ni2+ Buffer A). For KaiC, all buffers also contained fresh stocks of 1 mM ATP and 1 mM MgCl2. Cell suspensions were either frozen for later purification or lysed immediately by passing through an Emulsiflex high pressure homogenizer (Avestin) ≥10 times at ≥10,000 psi. The soluble portion of the lysate was recovered by centrifugation for 45 min at 45,000g at 4 °C. Clarified lysate was loaded onto Ni-NTA agarose (Qiagen, 5 ml per 2 l of E. coli culture) that had been equilibrated with 10 column volumes of Ni2+ Buffer A.
The Ni-NTA resin loaded with lysate was washed with ≥10 column volumes of Ni2+ Buffer A and bound proteins were eluted with 30 ml (per 5 ml Ni-NTA) of Ni2+ Buffer containing 250 mM imidazole. The eluted protein was then treated with roughly 10 nmol Ulp1 enzyme for ≥30 min at room temperature to cleave the singly occupied molecular orbital tag. The cleaved KaiC was then concentrated to ≤2 ml using a 30 kDa centrifugal filter (Millipore) and injected on a Superdex 200 gel filtration column (GE Healthcare) equilibrated with 20 mM Tris pH 7.4, 150 mM NaCl, 1 mM ATP, 1 mM MgCl2 and 1 mM TCEP (KaiC buffer). Fractions containing KaiC hexamer were quantified using absorbance readings (λ = 280 nm) using the calculated extinction coefficient, frozen in liquid nitrogen and stored at −70 °C.
KaiB binding assays.
KaiB binding assays were performed generally as described in Chavan et al.11. Briefly, 30 μl of KaiC buffer containing 0.1% Tween and 50 nM fluorescently labeled KaiB was pipetted into 21 adjacent wells of a 384-well flat-bottom black polystyrene assay plate (Corning). KaiC aliquots were then thawed and supplemented with 0.1% Tween and 50 nM fluorescently labeled KaiB before 90 μl portions were added to the plate in line with the 21 buffer wells. Serial dilution was executed by transferring 60 μl of KaiC mixture to the first buffer well, mixing by pipetting in and out ≥6 times and then transferring 60 μl from that well to the subsequent well and repeating this process for all 21 wells. The plates were sealed and incubated overnight (roughly 15 h) at room temperature. Fluorescence anisotropy was measured on uncovered plates using Gen5 software on a SYNERGY2 microplate reader (BioTek). Between 10 and 20 measurements were collected and averaged for each well, and data were analyzed using Prism v.9 (GraphPad). Data were fit to the Langmuir isotherm:
where a is anisotropy, Δatotal is the change in anisotropy between bound and unbound KaiB, [KaiC] is the concentration of KaiC variant, Kd,app is the apparent dissociation equilibrium constant and BG is the background anisotropy. In cases where saturation could not be reached, as evidenced by the lack of stable anisotropy value as a function of KaiC concentration, a lower limit for Kd,app was estimated by holding Δatotal constant at 0.043 anisotropy units. The raw anisotropy values were subsequently converted to change in anisotropy by subtracting the BG term from the anisotropy values in each titration curve (Supplementary Data and Source Data for Fig. 1). The reported Kd,app values were then determined by refitting the background-subtracted change in anisotropy values to the Langmuir function above with the BG term omitted.
Error arising from differences in protein stability between mutants, as well as the inherent error in our curve-fitting method, was estimated by performing replicate titrations using protein stocks that were thawed on different days. Replicate Kd,app values from derived from curve-fitting on separate days are logarithmically transformed and pooled before statistical comparison.
Electron microscopy.
Sample preparation.
KaiC-EA or KaiC-AE was briefly incubated on ice in 20 mM Tris pH 7.4, 150 mM NaCl and 1 mM each of ATP, MgCl2 and TCEP. Sample (2.5 μl at 1.5 mg ml−1) was then applied to an UltraAuFoil R1.2/1.3 300-mesh grid (Electron Microscopy Services), which was previously plasma-cleaned using a Gatan Solarus (75% argon/2% oxygen atmosphere, 15 W for 7 s). To overcome orientation bias in ice, an additional dataset was obtained by applying KaiC-EA (2.5 μl at 0.2 mg ml−1) to holey C-flat grids, which previously had thin carbon floated on them. These grids were pretreated with 5 μl of 0.1% (w/v) poly-l -lysine hydrobromine (Polysciences), blotted to dryness and then washed three times with 10 μl of water. Grids with applied sample were then manually blotted with filter paper (Whatman No. 1) for roughly 3 s in a 4 °C cold room before plunge freezing in liquid ethane cooled by liquid nitrogen.
For the detergent dataset, KaiC-AE was incubated on ice with 4 mM fluorinated fos-choline-8 (Anatrace) added to the sample buffer. 2.5 μl of 6 mg ml−1 sample was added to UltraAuFoil R1.2/1.3 300-mesh grids (Electron Microscopy Services)that were pretreated with the addition of a graphene monolayer, using a modified protocol48 for the deposition of graphene and made hydrophilic via ultraviolet/ozone cleaner (UVOCS T10×10 system) as previously described49, and then incubated for 4 min in a cold room to concentrate particles on the surface of the grid before manual blotting for roughly 3 s followed by plunge freezing into liquid ethane.
Data acquisition.
All cryo-EM data were acquired using the Leginon automated acquisition software50. For KaiC-EA, all real-time image preprocessing, consisting of frame alignment, local contrast transfer function (CTF) estimation and particle picking were performed using the Appion image-processing pipeline during data collection51. For KaiC-AE, all preprocessing was performed using Relion v.3.0 (ref.52).
Image collection was conducted using a Thermo Fischer Talos Arctica operating at 200 keV and equipped with a Gatan K2 Summit DED. We used a nominal magnification of ×36,000, corresponding to a pixel size of 1.15 Å at the detector. For KaiC-EA, 1,137 videos were collected at a 40° tilt to overcome preferred orientation53 with an exposure time of 8 s and an exposure rate of 7.94 e−/pixels per s and a total exposure of 48 e−/Å2 (0.6 e− per frame) with a nominal defocus range of 0.8–1.7 μM. For the thin carbon KaiC-EA dataset, we collected 8,405 videos with no tilt and with an exposure time of 9 s. We used an exposure rate of 7.5 e−/pixels per s for a total exposure of 51 e−/Å2 (1.4 e− per frame) with a nominal defocus range of 0.5–1.5 μM. In the tilted KaiC-AE dataset, 1,132 videos were collected at 40° tilt with an exposure time of 11 s and exposure rate of 5.71 e−/pixels per s for a total exposure of 48 e−/Å2 (0.9 e− per frame) and with a nominal defocus range of 0.8–1.7 μM. For the KaiC-AE detergent dataset, 1,541 videos were collected with no tilt, using an exposure time of 6.2 s and exposure rate of 8.61 e−/pixels per s for a total exposure of 40.32 e−/Å2 (0.7 e− per frame) with a nominal defocus range of 0.5–2.0 μM.
Image processing.
Videos of all KaiC datasets were aligned and dose-weighted in the Appion pipeline using Motioncorr2. For the tilted KaiC-EA dataset, 239 images were used for automated particle picking using the difference of Gaussians picker to yield an initial 151,640 particles (Fig. 5a)54. Gctf was used for local CTF estimation, using an increased raster spacing of 500 to accommodate the defocus gradient resulting from the tilted data collection55. Fourier-binned 4 × 4 particles were then subjected to reference-free 2D classification with multivariate statistical analysis and multi-reference alignment in the Appion pipeline51. The two best classes representing tilted and side views were selected for template-based particle picking using FindEM56, resulting in 2,016,826 particle picks from the entire dataset.
For initial particle curation, the roughly 2 million particle picks were subjected to two rounds of reference-free 2D classification in Relion52, leaving 1,341,217 particles bearing structural details. A previous crystal structure of KaiC (Protein Data Bank (PDB) 3K0C) was used to generate an initial model using the molmap feature in UCSF Chimera57. This model was low-pass filtered to 20 Å resolution for 3D autorefinement in Relion, which resulted in a roughly 9.6 Å map that did not have discernable density for most of the CII ring. 3D classification into six classes without alignment resulted in two classes with mostly intact CII and CI rings, totaling 579,706 particles. These particles were recentered and reextracted at 2.3 Å per pixel.
For the thin carbon dataset, FindEM was used along with the same tilted and side view templates from the tilted dataset, resulting in 2,856,972 particle picks. CTFFIND4 was used for CTF estimation before extracting particles. Particles were Fourier-binned 4 × 4 and then subjected to one round of 2D classification in Relion, allowing for the removal of false particle picks and for the identification and selection of side views. Then 2,856,972 particles that belonged to exemplary side views were recentered and reextracted at 2.3 Å per pixel and then combined with the 579,706 particles from the tilted dataset. 3D classification in Relion with alignment, asking for four classes, gave a single well-resolved class comprising 1,002,777 particles. 3D autorefinement of the well-resolved class resulted in a map with a global resolution of roughly 4.7 Å. These particles were recentered and reextracted at 1.15 Å per pixel before undergoing a second 3D autorefinement, giving a new map with a global resolution of roughly 4.1 Å. Additional 3D classification, without alignment and using a Tau value of 10 and asking for four classes, generating three high resolution classes: two seemingly C2-symmetric and one seemingly C6-symmetric.
The 658,212 particles belonging to the two seemingly C2-symmetric classes were joined for one round of 3D classification into three classes with no alignment and a Tau value of 20. One class containing the highest resolution features was selected for further processing. These 122,855 particles underwent 3D autorefinement with applied C2 symmetry, giving a map with a global resolution of roughly 3.7 Å. An in-house python script was used to group particles by image shift, which then underwent iterative rounds of CTF refinement in Relion v.3.1 refining both beam tilt and per-particle astigmatism correction. Subsequent 3D autorefinement with applied C2 symmetry and a soft mask around the CI ring resulted in a map at roughly 3.5 Å. Postprocessing in Relion yielded a final C2-state map at roughly 3.2 Å global resolution.
Particles from the seemingly C6-symmetric class from the earlier 3D classification without alignment underwent 3D autorefinement, resulting in a roughly 4.1 Å map with 91,869 particles. 3D autorefinement with applied C6 symmetry returned a roughly 3.7 Å map. Image shift grouping and iterative CTF refinement in Relion v.3.1, followed by 3D autorefinement with applied C6 symmetry and a soft mask around the CI ring resulted in a roughly 3.2 Å map. Postprocessing resulted in a final map for the C6-state at roughly 2.8 Å resolution.
Particle picking for the detergent dataset for KaiC-AE was done using FindEM template picking, using a single axial and side view from the earlier datasets as templates, resulting in 427,620 particle picks. Particles were extracted at 2.3 Å per pixel and then underwent 2D alignment with cryoSPARC. Particles belonging to classes that showed secondary structure were selected as input particles for ab initio reconstruction, asking for three classes. One class, containing 111,877 particles, resembled the stacked ring shape of KaiC and was selected for homogeneous refinement resulting in a map at roughly 6.3 Å resolution. These particles then underwent a second round of ab initio reconstruction with three classes, with one class containing 57,067 particles that resolved to roughly 6.7 Å after homogenous refinement. These particles were subjected to one final round of ab initio reconstruction with two classes, resulting in one class showing detailed structural features, consisting of 42,838 particles. Homogeneous refinement using C1-symmetry resulted in a roughly 6.5 Å reconstruction, followed by homogenous refinement with C6-symmetry, yielding a map at roughly 4.8 Å resolution. The full dataset (427,620 particle picks) was extracted, unbinned at 1.15 Å per pixel and underwent 2D classification in Relion. The selected 367,247 particles were then subjected to 3D classification in Relion with a limited resolution E-step of 7 Å, using the roughly 4.8 Å map obtained from cryoSPARC as an initial model. Of the four resulting classes, one was selected for further processing with 89,892 particles. These particles underwent 3D autorefinement, resulting in a roughly 5.6 Å map, followed by CTF refinement and 3D autorefinement once again, resulting in a roughly 4.8 Å map. Another round of CTF refinement was conducted, followed by 3D autorefinement with C6-symmetry resulting in a roughly 4.2 Å map that led to a final sharpened map at roughly 3.8 Å after postprocessing. All reported resolution are according to the Fourier shell correlation (FSC) at a cutoff of 0.143.
Atomic model building and refinement.
For both the extended and compressed state models of KaiC-EA, a crystal structure of the phosphomimetic mutant S431A/T432E was used as a starting point for model building (PDB 3K0C)27 with the C-terminal A-loops deleted. For the C2-state, which showed large-scale repositioning of the C-terminal domains, proSMART local restraints58 were generated in Coot59 and then used to refit the domains into the cryo-EM density. After one round of real-space refinement in Phenix using default parameters and five macrocycles60, Coot and ISOLDE61 were used to improve main chains and side chains. The Molprobity server62 (http://molprobity.biochem.duke.edu/) and PDB validation service server (https://validate-rcsb-1.wwpdb.org/) were used to identify problem regions for subsequent correction in Coot.
KaiC autophosphorylation assays.
Samples of each KaiC variant were freshly prepared in KaiC buffer and mixed with KaiA to generate a final concentration of 1.5 μM KaiA and 3.4 μM KaiC. Samples were incubated at 30 °C for 24 h, at which point 50 μl was removed and quenched with 10 μl of 6× SDS before incubating at 95 °C for 5 min.
Phos-Tag acrylamide gels were prepared with a resolving gel consisting of 10% (w/v) acrylamide (29:1 acrylamide:bis-acrylamide) containing Tris-HCl pH 8.8 supplemented with 50 μM Phos-tag reagent and 100 μM Mn2+. Gels were run at constant 25 mA with a 165 V limit for 35 min before loading any samples into the gel. Samples were loaded and run at room temperature with the same mA and voltage parameters as stated above for 170–190 min. Gels were stained using SYPRO Orange following the Bio-Rad protocol and density was analyzed using ImageJ software63. Error was estimated as the standard deviation from n = 3 replicate measurements analyzed separately.
Multiple sequence alignments.
KaiC protein sequences were obtained from the UniProt database64 and aligned using Clustal Omega65.
KaiC ATPase activity assays.
After the initial protein purification, frozen aliquots of each KaiC variant were thawed and run over a Sephadex 200 size-exclusion column (GE Healthcare) to exchange them into fresh buffer containing 50 mM MOPS pH 7.4, 150 mM NaCl2, 1 mM TCEP, 1 mM MgCl2, 400 μM ATP (ATPase buffer). Fractions containing KaiC hexamer were diluted to working assay concentrations with ATPase buffer for a final volume of 50 μl. Samples were then incubated at 30 °C and 5 μl aliquots were removed at 0, 4 and 8 h timepoints and quenched using the ADP-Glo assay kit (Promega) according to the manufacturer’s instructions. Luminescence measurements (in relative light units, RLU) were taken at room temperature with a SYNERGY2 microplate reader in 384-well microplates. Data analysis was performed using Prism (GraphPad).
Standard curves were generated each day in ATPase buffer containing 20, 40, 60, 80 and 100% ADP while maintaining 400 μM total nucleotide concentration with ATP. Aliquots (5 μl) of these samples were mixed with ADP-Glo reagent as described above. Plotting RLU versus μM ADP and solving for the x intercept gives an equation that converts RLU values to μM ADP.
Because the concentration of substrate (ATP) was in vast excess of the KM of 2 μM (ref.66). the Michaelis–Menten equation simplifies to the following:
and the slope of the v0 is the initial velocity, kcat is the catalytic turnover rate and [enzyme] is, in this case, the concentration of KaiC variant in the assay. Thus, we plotted v0 for each concentration of a given mutant against [KaiC] and, using the equation above, extracted kcat as the slope of the resulting line.
Thermodynamic modeling of cooperativity indices.
KaiC titrations were performed as described above, but with various concentrations of KaiB-I87A (fsKaiB) in both the KaiC and diluent stocks. Background anisotropy was subtracted from these data before analysis. Least-squares fitting was performed using DynaFit (BioKin) as described in Chavan et al. with slight modifications (see Supplementary Software for DynaFit scripts). Specifically, the assumption that K5 = K1 was replaced by the assumption that K5 = K3 because that study used the KaiB-related protein SasA as a secondary titrant, whereas here we used fsKaiB. Because wild-type KaiB assumes the same fold-switched conformation when in complex with KaiC13, the assumption that K5 = K3 is therefore appropriate when fsKaiB is used as the secondary titrant.
Anisotropy values were allowed to float initially, and then restricted for extraction of the final model. The two highest fsKaiB concentrations were also allowed to float when fitting data for the mutant KaiC variants. Coefficients of variation for all parameters were found to be near or below 50% in almost all cases (source data for Extended Data Fig. 9), indicating that the standard error from the regression is small compared to the best-fit value, and that the least-squares fitting gave reliable convergence to the thermodynamic model. Best-fit estimates for the equilibrium constants K1, K2 and K4 were obtained, with K3 being defined in this case by the thermodynamic balance equation,
referring to the reaction scheme in Extended Data Fig. 9a (see ref.67 for derivation and additional details). Cooperativity indices, defined as K1/K3, were then calculated by dividing K2 by K4, which is equal to the cooperativity index by rearranging the thermodynamic balance equation to
The error from least-squares fitting was estimated by propagation of the standard errors via the equation:
where Kx refers to the best-fit value for a given output and σx refers to the standard error for that particular parameter. In the 2D titrations with KaiC-EA the absolute values of K2 and K4 were not well-defined by the data, with CV% greater than 100 observed. However, cooperativity indices of these estimates were consistent between replicate titrations, and Monte Carlo simulation of n = 1,000 fits good correlation between the values of K2 and K4. Error was therefore estimated by calculating the cooperativity index for each individual Monte Carlo simulation, sorting them by magnitude and taking n = 25 and 975 as the upper and lower bound of the 95% confidence interval, respectively, and taking n = 500 as the average.
In vitro oscillation assays.
KaiABC oscillator reactions were performed as described previously11,68. Briefly, purified protein stocks were mixed in buffer containing 20 mM Tris pH 8.0, 150 mM NaCl, 5 mM MgCl2 and 1 mM ATP to a final concentration of 3.5 μM KaiC, 3.5 μM KaiB, 1.2 μM KaiA and 50 nM KaiB-K25C-fluorescein. Fluorescence anisotropy of was monitored at 30 °C using a CLARIOstar (BMG Labtech) microplate reader. All data collection was performed using the fluorescein channel (λex, 490 ± 5 nm; λem, 520 ± 5 nm), with a measurement taken every 15 min.
Strains and culture conditions for in vivo experiments.
Markerless incorporation of the K224A point mutation into kaiC of S. elongatus PCC 7942 was performed by CRISPR–Cas12a engineering as described previously69. The plasmids and primers used for generating the kaiC-K224A strain are listed in Supplementary Table 4.
Characterization of circadian rhythms in the kaiC-K224A strain were performed as described in Chavan et al.11. Briefly, bioluminescence was monitored from the PkaiBC∷luc fusion reporter, inserted into a neutral site of the S. elongatus chromosome as previously described70. Strains to be monitored were diluted to an optical density (OD750) of 0.2. Aliquots (20 μl) of the suspension were added to 280 μl pads of BG-11 agar with 3.5 mM firefly luciferin in 96-well plates. Cells were entrained under 12-h light-dark cycles (80 μmol m−2 s−1) to synchronize clock phases. After 48 h of entrainment, cells were released into continuous light (30 μmol m−2 s−1). Plates were transferred to a lighted stacker (40 μE light) attached to a Tecan Infinite M200 Pro and bioluminescence monitored every 2–3 h. All strains used in this study are listed in Supplementary Table 5.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Image processing pipeline and validation of C6 symmetric KaiC-AE structure.

a, Image processing pipeline for KaiC-EA datasets with fluorinated fos-choline-8. b, Local resolution estimation of cryo-EM reconstruction calculated by RELION52. c, Euler distribution plots depicting particle orientations present in final reconstruction. More populated views are colored in red while less populated are depicted in blue. d, 3D Fourier Shell Correlation (3DFSC)53 of final daytime state reconstruction, with a global resolution of 3.8 Å at FSC = 0.143.
Extended Data Fig. 2 |. CII nucleotide state in extended and compressed KaiC structures.

a, Electron volume and atomic model for CII nucleotide in the KaiC-AE structure determined in the presence of 4 mM fos-choline, b the C6-symmetric KaiC-EA structure and c extended or d compressed subunits of the C2-symmetric nighttime KaiC structure.
Extended Data Fig. 3 |. Image processing pipeline and validation of KaiC-EA.

a, Image processing pipeline for two combined KaiC-EA datasets: a 40° tilted dataset, and one collected on thin carbon. b, Local resolution estimation of cryo-EM reconstructions calculated by RELION52. c, Euler distribution plots depicting particle orientations present in final reconstructions. More populated views are colored in red. d, 3D Fourier Shell Correlation (3DFSC)53 of final nighttime state reconstructions, with a global resolution of 2.8 Å for the C6-state, and 3.2 Å for the C2-state at FSC = 0.143.
Extended Data Fig. 4 |. Structural comparison of C2-symmetric ATPase structures.

Seam protomers are depicted in blue while ATP and ADP nucleotides are shown in white and green, respectively. Apo nucleotide pockets are indicated with dashed circles. While other C2-symmetric ATPases are unliganded at their seam protomers, KaiC is ADP-bound at these subunits.
Extended Data Fig. 5 |. Allostery about the CII ring regulates KaiC autophosphorylation.

a, In the expanded state, the A-loop of an adjacent protomer (yellow) is positioned near the phosphosite-adjacent 422-loop through a hydrophobic interaction between I490 and M420. E444’s sidechain forms a hydrogen bond with the mainchain nitrogen of I490 in trans. In the compressed state of KaiC-EA, E444 in the ADP-bound protomers is positioned away from the pore and no longer stabilizes the A-loops, allowing them to become disordered and causing the 422-loops to collapse towards phosphosite–containing α9. Binding of KaiA to the C-terminus of KaiC may disrupt this tripartite interaction and stimulate hyperphosphorylation. b, Sypro orange fluorescence of KaiC mutants that were run on a 10% denaturing polyacrylamide gel containing 50 μM Phos-tag™ reagent and 100 μM Mn2+. c, Densitometric analysis of bands corresponding to phosphorylated and unphosphorylated KaiC. Gray bars represent mean ± standard deviation for n = 3 repeats.
Extended Data Fig. 6 |. KaiC residue Y402 exhibits rotameric heterogeneity in the compressed conformation.

a, Close-up view of the two Y402 rotamer conformations from the compressed protomer of the C2-symmetric KaiC-EA structure. Cryo-EM density is depicted in white and semi-transparent for clarity. Analysis from the modeling software Coot59 indicates that both rotamers are allowed with similar probabilities. Model vs. Data Cross Correlation in Phenix60 indicates that Rotamer 1 has a higher correlation compared to Rotamer 2, but that both are favorable. b, Superposition of compressed protomer B with expanded protomer A, aligned by the CII domain. The rotameric heterogeneity of Y402 in the compressed protomer is associated with rotameric deviations in nearby residues in comparison to an expanded protomer.
Extended Data Fig. 7 |. Multiple sequence alignment of key KaiC regions from various species of cyanobacteria.

a, Protein sequences of the CI and b CII regions of KaiC from eight distinct strains of cyanobacteria. Sequences are presented with each domain grouped to together and aligned amongst the species, and also aligned about the CI and CII domains within a given protein sequence. Key residues are highlighted with colors illustrating their conservation between the CI and CII domains.
Extended Data Fig. 8 |. The arginine tetrad creates a network of CI-CI trans interactions.

a, The 4TL8 structure40 is shown as light blue ribbons with the P-loops displayed in darker blue. Arginine tetrad side chains at clockwise (b) or counterclockwise (c) subunits as well as their electrostatic interaction partners are shown in brown. Average interatomic distance from the six interfaces are shown, as tabulated in Supplementary Table 3.
Extended Data Fig. 9 |. Cooperativity analysis of KaiC mutants.

a, Thermodynamic model, based on the one originally derived for heterotropic cooperativity in Chavan et al.34. In this case, the heterotropic cooperativity factor, S, is replaced with the homotropic factor represented by unlabeled KaiB-I87A (fsKaiB), which binds much tighter to KaiC than wild-type KaiB10. Wild-type KaiB is monitored, as indicated by an orange asterisk. b, 2-dimensional titrations are overlaid with titration curves derived from least-squares fitting of the data from each individual dataset. For unsubstituted (WT) KaiC-EA, the 95% confidence intervals from n = 1,000 Monte Carlo simulation is reported. These represent the n = 25 and n = 975 values from the simulation, which gave median (n = 500) values of 22, 16 and 25 (top panel to bottom panel). For each 2D-titration of the mutant nighttime KaiC variants, best fit cooperativity indices from least-squares analysis are reported as best fit ± standard error. See methods for more information on how these values and standard errors were determined.
Extended Data Fig. 10 |. Analysis of CI-CI nucleotide contacts from PDB 4TLA.

a, Top-down view of the CI domain from the mixed nucleotide state CI structure reported in Abe et al.40. with the nucleotides observed at each interface labeled. Schematic depictions of the specific atomic interactions with the 2′ hydroxyls as well as residues K224 and R226 observed at each interface exhibiting either two (b), one (c) or no (d) electrostatic interactions with the nucleotide phosphates.
Supplementary Material
Acknowledgements
We thank J.C. Ducom at Scripps Research High Performance Computing for computational support, as well as B. Anderson at the Scripps Research Electron Microscopy Facility for microscopy support. Funding for this work was provided through National Institutes of Health grant nos. R01GM121507 and R35GM141849 to C.L.P., R01NS095892 and R21GM142196 to G.C.L., R35GM118290 to S.S.G., R01GM107521 to A.L. and F32GM130070 to D.E., as well as the National Science Foundation grant no. NSF-CREST: Center for Cellular and Biomolecular Machines at the University of California, Merced (grant no. NSF-HRD-1547848) to A.L.
Footnotes
Competing interests
The authors declare no competing interests.
Extended data are available for this paper at https://doi.org/10.1038/s41594-022-00803-w.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41594-022-00803-w.
Data availability
EMDB and PDB depositions: Compressed conformation of nighttime KaiC (PDB 7S65, EMDB-24850), extended conformation of nighttime KaiC (PDB 7S66, EMDB-24851) and extended conformation of daytime state KaiC (PDB 7S67, EMDB-24852). All data are available in the main text or the supplementary information. Source data are provided with this paper.
References
- 1.Ishiura M et al. Expression of a gene cluster kaiABC as a circadian feedback process in cyanobacteria. Science 281, 1519–1523 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Diamond S, Jun D, Rubin BE & Golden SS The circadian oscillator in Synechococcus elongatus controls metabolite partitioning during diurnal growth. Proc. Natl Acad. Sci. USA 112, E1916–E1925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pattanayak GK, Phong C & Rust MJ Rhythms in energy storage control the ability of the cyanobacterial circadian clock to reset. Curr. Biol 24, 1934–1938 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayashi F et al. Roles of two ATPase-motif-containing domains in cyanobacterial circadian clock protein KaiC. J. Biol. Chem 279, 52331–52337 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Rust MJ, Markson JS, Lane WS, Fisher DS & O’Shea EK Ordered phosphorylation governs oscillation of a three-protein circadian clock. Science 318, 809–812 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishiwaki T et al. A sequential program of dual phosphorylation of KaiC as a basis for circadian rhythm in cyanobacteria. EMBO J. 26, 4029–4037 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim YI, Dong G, Carruthers CW Jr., Golden SS & LiWang A The day/night switch in KaiC, a central oscillator component of the circadian clock of cyanobacteria. Proc. Natl Acad. Sci. USA 105, 12825–12830 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang YG, Tseng R, Kuo NW & LiWang A Rhythmic ring-ring stacking drives the circadian oscillator clockwise. Proc. Natl Acad. Sci. USA 109, 16847–16851 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snijder J et al. Insight into cyanobacterial circadian timing from structural details of the KaiB-KaiC interaction. Proc. Natl Acad. Sci. USA 111, 1379–1384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murakami R et al. Cooperative binding of KaiB to the KaiC Hexamer ensures accurate circadian clock oscillation in cyanobacteria. Int. J. Mol. Sci 10.3390/ijms20184550 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chavan AG et al. Reconstitution of an intact clock reveals mechanisms of circadian timekeeping. Science 374, eabd4453 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow GK et al. A night-time edge site intermediate in the cyanobacterial circadian clock identified by EPR spectroscopy. J. Am. Chem. Soc 144, 184–194 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Tseng R et al. Structural basis of the day-night transition in a bacterial circadian clock. Science 355, 1174–1180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang YG et al. Circadian rhythms. A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science 349, 324–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang YG, Kuo NW, Tseng R & LiWang A Flexibility of the C-terminal, or CII, ring of KaiC governs the rhythm of the circadian clock of cyanobacteria. Proc. Natl Acad. Sci. USA 108, 14431–14436 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tseng R et al. Cooperative KaiA-KaiB-KaiC interactions affect KaiB/SasA competition in the circadian clock of cyanobacteria. J. Mol. Biol 426, 389–402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snijder J et al. Structures of the cyanobacterial circadian oscillator frozen in a fully assembled state. Science 355, 1181–1184 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Phong C, Markson JS, Wilhoite CM & Rust MJ Robust and tunable circadian rhythms from differentially sensitive catalytic domains. Proc. Natl Acad. Sci. USA 110, 1124–1129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terauchi K et al. ATPase activity of KaiC determines the basic timing for circadian clock of cyanobacteria. Proc. Natl Acad. Sci. USA 104, 16377–16381 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito-Miwa K, Furuike Y, Akiyama S & Kondo T Tuning the circadian period of cyanobacteria up to 6.6 days by the single amino acid substitutions in KaiC. Proc. Natl Acad. Sci. USA 117, 20926–20931 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pattanayek R, Xu Y, Lamichhane A, Johnson CH & Egli M An arginine tetrad as mediator of input-dependent and input-independent ATPases in the clock protein KaiC. Acta Crystallogr. D. Biol. Crystallogr 70, 1375–1390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong G et al. Elevated ATPase activity of KaiC applies a circadian checkpoint on cell division in Synechococcus elongatus. Cell 140, 529–539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murakami R et al. The roles of the dimeric and tetrameric structures of the clock protein KaiB in the generation of circadian oscillations in cyanobacteria. J. Biol. Chem 287, 29506–29515 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murayama Y et al. Tracking and visualizing the circadian ticking of the cyanobacterial clock protein KaiC in solution. EMBO J. 30, 68–78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukaiyama A et al. Conformational rearrangements of the C1 ring in KaiC measure the timing of assembly with KaiB. Sci. Rep 8, 8803 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oyama K, Azai C, Matsuyama J & Terauchi K Phosphorylation at Thr432 induces structural destabilization of the CII ring in the circadian oscillator KaiC. FEBS Lett. 592, 36–45 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Pattanayek R et al. Structures of KaiC circadian clock mutant proteins: a new phosphorylation site at T426 and mechanisms of kinase, ATPase and phosphatase. PLoS ONE 4, e7529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito H et al. Autonomous synchronization of the circadian KaiC phosphorylation rhythm. Nat. Struct. Mol. Biol 14, 1084–1088 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Hong L, Vani BP, Thiede EH, Rust MJ & Dinner AR Molecular dynamics simulations of nucleotide release from the circadian clock protein KaiC reveal atomic-resolution functional insights. Proc. Natl Acad. Sci. USA 115, E11475–E11484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oyama K, Azai C, Nakamura K, Tanaka S & Terauchi K Conversion between two conformational states of KaiC is induced by ATP hydrolysis as a trigger for cyanobacterial circadian oscillation. Sci. Rep 6, 32443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Efremov RG, Leitner A, Aebersold R & Raunser S Architecture and conformational switch mechanism of the ryanodine receptor. Nature 517, 39–43 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Pattanayek R et al. Visualizing a circadian clock protein: crystal structure of KaiC and functional insights. Mol. Cell 15, 375–388 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Nishiwaki T & Kondo T Circadian autodephosphorylation of cyanobacterial clock protein KaiC occurs via formation of ATP as intermediate. J. Biol. Chem 287, 18030–18035 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishiwaki-Ohkawa T, Kitayama Y, Ochiai E & Kondo T Exchange of ADP with ATP in the CII ATPase domain promotes autophosphorylation of cyanobacterial clock protein KaiC. Proc. Natl Acad. Sci. USA 111, 4455–4460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leypunskiy E et al. The cyanobacterial circadian clock follows midday in vivo and in vitro. eLife 10.7554/eLife.23539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jessop M et al. Structural insights into ATP hydrolysis by the MoxR ATPase RavA and the LdcI-RavA cage-like complex. Commun. Biol 3, 46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye QZ et al. TRIP13 is a protein-remodeling AAA plus ATPase that catalyzes MAD2 conformation switching. eLife 4, e0736710.7554/eLife.07367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glynn SE, Nager AR, Baker TA & Sauer RT Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat. Struct. Mol. Biol 19, 616–622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakajima M, Ito H & Kondo T In vitro regulation of circadian phosphorylation rhythm of cyanobacterial clock protein KaiC by KaiA and KaiB. FEBS Lett. 584, 898–902 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Abe J et al. Circadian rhythms. Atomic-scale origins of slowness in the cyanobacterial circadian clock. Science 349, 312–316 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Erzberger JP & Berger JM Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct 35, 93–114 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Puri N et al. The molecular coupling between substrate recognition and ATP turnover in a AAA+ hexameric helicase loader. eLife 10.7554/eLife.64232 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutu A & O’Shea EK Two antagonistic clock-regulated histidine kinases time the activation of circadian gene expression. Mol. Cell 50, 288–294 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitayama Y, Iwasaki H, Nishiwaki T & Kondo T KaiB functions as an attenuator of KaiC phosphorylation in the cyanobacterial circadian clock system. EMBO J. 22, 2127–2134 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen SE et al. Dynamic localization of the cyanobacterial circadian clock proteins. Curr. Biol 24, 1836–1844 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouyang D et al. Development and optimization of expression, purification, and ATPase assay of KaiC for medium-throughput screening of circadian clock mutants in cyanobacteria. Int. J. Mol. Sci 10.3390/ijms20112789 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu H & Naismith JH An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naydenova K, Peet MJ & Russo CJ Multifunctional graphene supports for electron cryomicroscopy. Proc. Natl Acad. Sci. USA 116, 11718–11724 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han Y et al. High-yield monolayer graphene grids for near-atomic resolution cryoelectron microscopy. Proc. Natl Acad. Sci. USA 117, 1009–1014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carragher B et al. Leginon: an automated system for acquisition of images from vitreous ice specimens. J. Struct. Biol 132, 33–45 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Lander GC et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan YZ et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voss NR, Yoshioka CK, Radermacher M, Potter CS & Carragher B DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol 166, 205–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang K Gctf: real-time CTF determination and correction. J. Struct. Biol 10.1016/j.jsb.2015.11.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roseman AM FindEM–a fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol 145, 91–99 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Pettersen EF et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Nicholls RA, Fischer M, McNicholas S & Murshudov GN Conformation-independent structural comparison of macromolecules with ProSMART. Acta Crystallogr. D. Biol. Crystallogr 70, 2487–2499 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Croll TI ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D. Struct. Biol 74, 519–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.UniProt C UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sievers F & Higgins DG The clustal omega multiple alignment package. Methods Mol. Biol 2231, 3–16 (2021). [DOI] [PubMed] [Google Scholar]
- 66.Hayashi F et al. ATP-induced hexameric ring structure of the cyanobacterial circadian clock protein KaiC. Genes Cells 8, 287–296 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Bagshaw CR Biomolecular Kinetics: A Step-By-Step Guide (CRC Press, Taylor & Francis Group, 2017). [Google Scholar]
- 68.Heisler J, Chavan A, Chang YG & LiWang A Real-time in vitro fluorescence anisotropy of the cyanobacterial circadian clock. Methods Protoc. 10.3390/mps2020042 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ungerer J & Pakrasi HB Cpf1 is a versatile tool for CRISPR genome editing across diverse species of cyanobacteria. Sci. Rep 6, 39681 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mackey SR & Golden SS Winding up the cyanobacterial circadian clock. Trends Microbiol. 15, 381–388 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
EMDB and PDB depositions: Compressed conformation of nighttime KaiC (PDB 7S65, EMDB-24850), extended conformation of nighttime KaiC (PDB 7S66, EMDB-24851) and extended conformation of daytime state KaiC (PDB 7S67, EMDB-24852). All data are available in the main text or the supplementary information. Source data are provided with this paper.