

Patient presentation: A 24-year-old man, who smoked 1 pack a day of cigarettes, presented to the emergency department with sudden onset of 7/10 chest pain radiating to the back. His medical history was significant for anxiety and depression and a bicuspid aortic valve (BAV) diagnosed at age 5 years because of an auscultatory click. Outside transthoracic echocardiogram 3 years previously had shown normal valve function and aortic sinuses (root) of 42 mm. His family history was notable for a mother and uncle with BAV and aortic dilatation, but there was no early sudden cardiac death or aortic dissection in the family. The patient denied illicit drug use. On examination, he was in mild distress from chest pain. His heart rate was 75 beats/minute, his blood pressure was 124/72 mm Hg and equal in both arms, and room air oxygen saturation was 96%. Physical examination was unremarkable with normal cardiac auscultation. The ECG showed an early repolarization pattern and troponins were undetectable. A triple-rule-out computed tomography angiogram (CTA) showed no evidence of pulmonary embolus, coronary disease, or aortic dissection. The aortic root measured 48 mm with asymmetric noncoronary sinus dilatation (Figure 1) and no aortic coarctation. The remainder of the thoracic aorta was normal. After resolution of chest pain had occurred with analgesics administration, the patient was discharged and scheduled for outpatient cardiology follow-up in 2 weeks. The cause of his chest pain remained unknown.
Figure 1. Chest computed tomography angiography and brain magnetic resonance angiography.
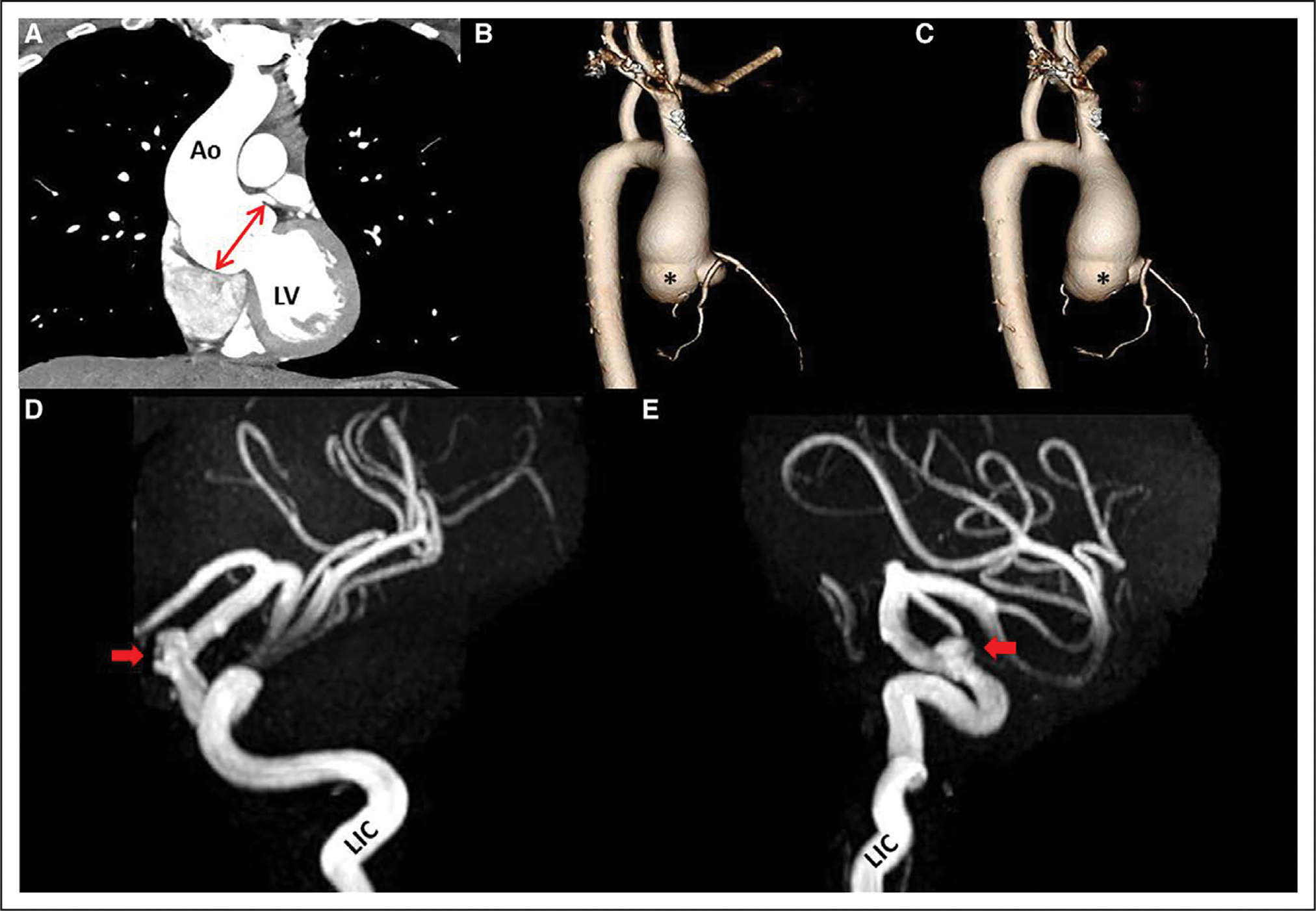
A, Chest computed tomography angiogram demonstrates left ventricular (LV) outflow with dilated root (double-headed arrow) and proximal ascending aorta (Ao); 3-dimensional reconstruction shows root phenotype with enlarged sinuses in lateral view (B) and posterior view (C) with proximal ascending involvement. Note disproportional dilatation of noncoronary sinus (asterisk). The remainder of the distal thoracic aorta was normal. Magnetic resonance angiogram of the brain shows left supraclinoid intracranial aneurysm (arrows), originating from the left internal carotid artery (LIC), in lateral (D) and left anterior oblique (E) views.
Dr Michelena: The congenital BAV condition affects 1% to 2% of the population and is fundamentally a valvulo-aortopathy characterized by substantial heterogeneity of its valvular and aortic phenotypic expressions, associated disorders, complications, and prognosis.1 From a nosology perspective, the most common presentation of BAV is typical-presentation valvulo-aortopathy with progressive BAV dysfunction and/or aorta dilatation (the clinical manifestation of aortopathy). This typical-presentation valvulo-aortopathy commonly is diagnosed in young adults and adults, requires long-term surveillance, and commonly necessitates surgical treatment, particularly for aortic valve dysfunction (>50% of patients need aortic valve replacement within 25 years after BAV diagnosis).1 The 24-year-old patient seemed to present in a typical manner, with a known nondysfunctional BAV with predominance of aortopathy manifested as a root of 48 mm measured by ECG-gated CTA, the gold standard for measuring the diameter of the aorta.2 There are 2 general BAV aortopathy phenotypes depending on which aortic segment is predominantly dilated: the ascending phenotype and the root phenotype, affecting ≈70% and ≈20% of patients with BAV aortopathy, respectively.1,2 In patients with BAV without risk factors for aortic complications, elective surgical aorta repair is recommended for a 55-mm root or ascending aorta diameter.2 If the patient exhibits any risk factor, such as root phenotype (preferential root dilatation), uncontrolled hypertension, family history of aortic dissection or early sudden cardiac death, substantial aortic regurgitation, or aortic diameter increase >3 mm/year, then surgery should be considered at a lower threshold of 50 mm.2 Although transthoracic echocardiogram single-plane (parasternal long-axis) assessment may underestimate root measurements in BAV,2 our patient’s root increased from 42 mm (by transthoracic echocardiogram) to 48 mm (by CTA) in 3 years—about 2 mm/year—which represents progressive dilatation at a higher rate than the commonly described 0.4 to 0.6 mm/year.1,2 Although BAV with aortopathy seemed to run in the patient’s family, there was no family history of dissection or early unexplained sudden cardiac death, and no syndromic features described on examination to suggest the need for genetic testing. Familial BAV is not an indication for genetic testing because available genetic panels do not include genes specific for BAV aortopathy in the absence of syndromic features or family history of dissection. Our patient exhibited a progressively enlarging root phenotype, which is suspected to carry a higher risk of complications, including aortic dissection in adults.1,2 Planning elective future surgical repair in our patient approaching a 50-mm root seemed appropriate.
Patient presentation (continued): One week later (before the cardiology consultation), the patient presented again to the emergency department with severe frontal headache and blurred vision in the left eye. Brain magnetic resonance angiography demonstrated a 6-mm saccular left supraclinoid internal carotid artery aneurysm (Figure 1) and a tortuous basilar artery, with no evidence of subarachnoid hemorrhage. The patient underwent successful treatment of the intracranial aneurysm with endovascular deployment of a pipeline stent. The patient was discharged without neurologic deficits and started on dual antiplatelet therapy (aspirin and clopidogrel) for 6 months’ duration. Outpatient transthoracic echocardiogram revealed root dilatation (Figure 2 and Movie I in the Data Supplement) of 48 mm, borderline proximal ascending aorta dilatation of 37 mm, right–left cusp fusion BAV with trivial aortic regurgitation, and no stenosis (Figure 2 and Movie II in the Data Supplement). The ventricles were normal in size and function. There was mild mitral valve posterior leaflet prolapse with trivial late-systolic mitral valve regurgitation and persistent left superior vena cava discovered incidentally.
Figure 2. Transthoracic echocardiography and intraoperative photographs.
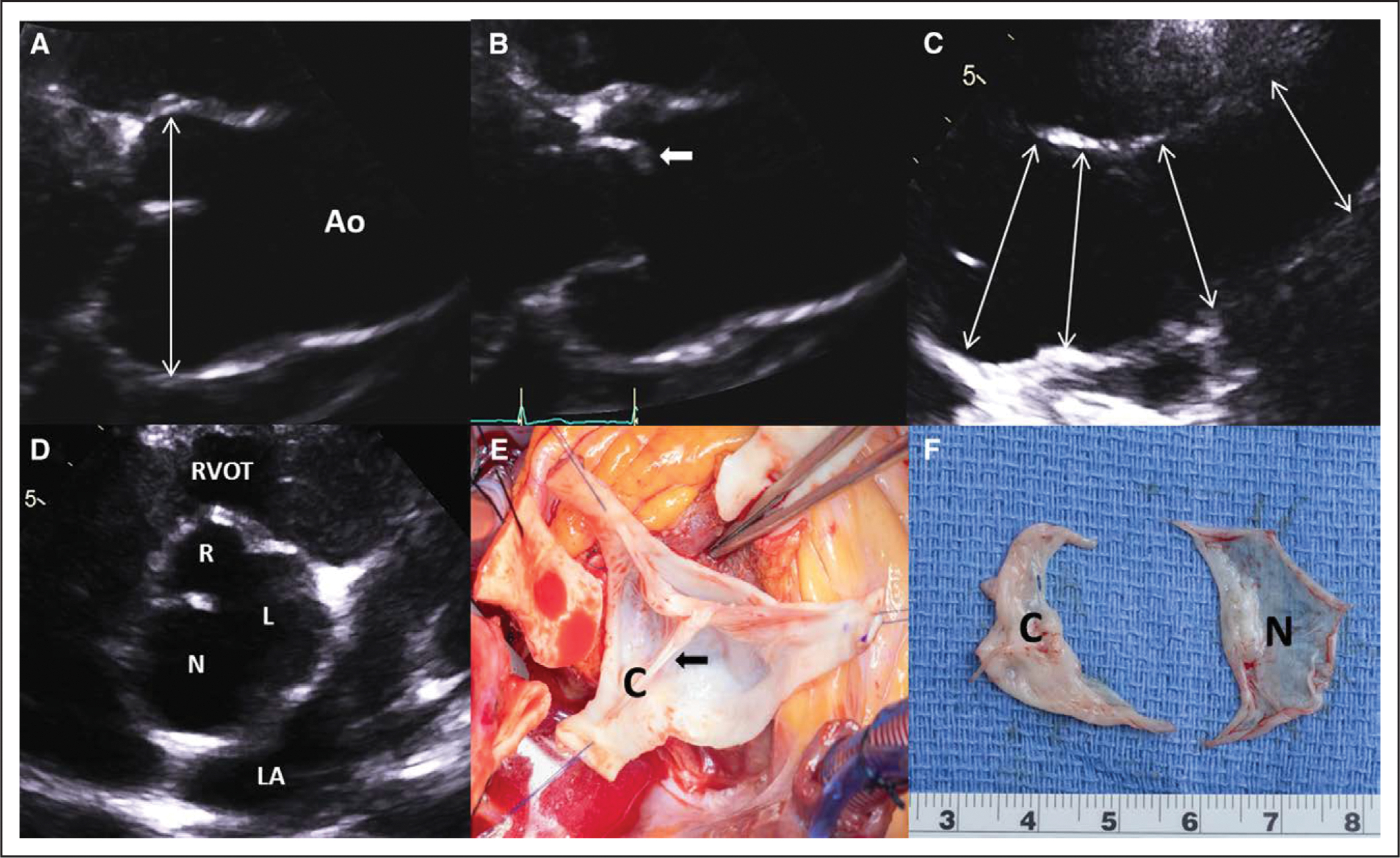
A, Transthoracic echocardiography demonstrates significant dilatation of the aortic root in the left parasternal long-axis diastolic zoomed view (double-headed arrow; see Movie I in the Data Supplement. B, Systolic doming of the conjoined cusp (anterior cusp; arrow; see Movie I in the Data Supplement. C, High left parasternal view of the ascending aorta (Ao) showing progressive tapering of the aortic size from proximal to middle sections (double-headed arrows). D, Left parasternal short-axis diastolic view with zoom showing right (R)–left (L) fusion bicuspid aortic valve with dominant noncoronary cusp (N)/sinus (see Movie II in the Data Supplement. E, Intraoperative photograph with the root already excised shows the conjoined cusp (C) with prominent raphe (arrow). F, The excised bicuspid valve with a thickened conjoined cusp (C) and large, translucent noncoronary cusp (N). LA indicates left atrium; and RVOT, right ventricular outflow tract.
Dr Michelena: The combination of a progressively dilating proximal ascending aorta (ie, root phenotype), likely symptomatic intracranial aneurysm (extrathoracic medium-sized artery involvement), mitral prolapse, and familial aortopathy (irrespective of basilar artery tortuosity, mitral prolapse) in this 24-year-old patient does not represent a typical-presentation valvulo-aortopathy from the nosology perspective but rather a complex-presentation valvulo-aortopathy (Figure 3), in which concomitant or associated disorders may be clinically and prognostically more serious than the BAV condition per se (ie, Turner syndrome, Loeys-Dietz syndrome, Shone complex, or severe aortic coarctation), or there is early or accelerated valve dysfunction or aortopathy, or both. This complex-presentation valvulo-aortopathy is more commonly diagnosed earlier in the pediatric and young adult population and usually requires close surveillance and early surgical or invasive treatment. Our patient’s complex presentation must make the clinician suspicious for an underlying connective tissue abnormality or genetic disorder that could explain the combination of findings.
Figure 3. Nosologic spectrum of the bicuspid aortic valve (BAV) condition.
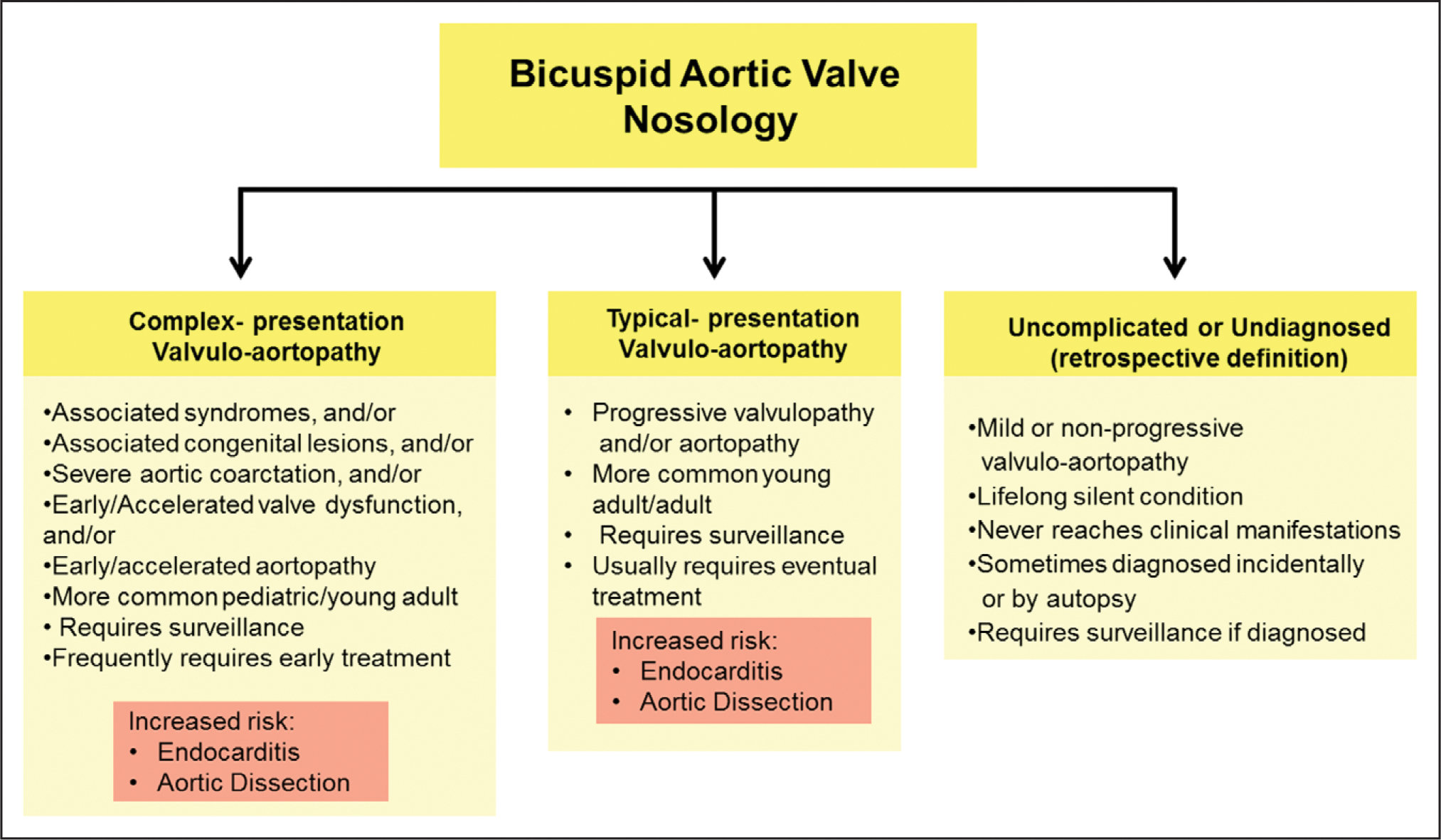
Complex-presentation and typical-presentation valvulo-aortopathies are described in the text and are risk factors for infective endocarditis and aortic dissection. The uncomplicated or undiagnosed category is more of a retrospective definition of cases of BAV in patients who never develop complications and remain undiagnosed through their lifetime, or are incidentally diagnosed by autopsy or imaging.
I evaluated the patient in clinic and found him very anxious regarding the recent events. On detailed interrogation, the patient endorsed a history of right inguinal hernia repair, right shoulder subluxation, easy skin bruising, flat feet, and tooth overcrowding, but his physical examination was normal with the exception of mild pectus excavatum. His height was 183 centimeters, weight was 71 kilograms, and the aortic root cross-sectional area-to-height ratio (r2π [cm2]/height [m]) was 9.8 cm2/m, which was close to the proposed cutoff of 10 cm2/m that is associated with worse aortic outcomes.2 His young age, aortic diameter, and substantial associated abnormalities prompted consideration of urgent aortic surgery and genetic testing and counseling. Although no data are available for patients with BAV, I started him on low-dose β-blockade for his aorta and agreed for him to continue noncompetitive exercise at a comfortable level without weight lifting or other isometric exercise that would require straining. I recommended echocardiographic evaluations in his siblings (the patient had no children), formal psychiatric evaluation for anxiety and depression management, abdomen–pelvis CTA, and medical genetics testing and counseling. In discussion with interventional radiology, we concluded that it was relatively safe to stop the Plavix at 3 months after intracranial aneurysm treatment to allow for prompt surgical root-ascending aorta repair.
Patient presentation (continued): Abdomen–pelvis CTA showed no evidence of any other aorta segment dilatation, no dural ectasia, and mild scoliosis. Genetic testing revealed a heterozygous pathogenic variant in the transforming growth factor β receptor 1 gene (TGFBR1), a specific variant called c.1459C>T (p.R487W), consistent with Loeys-Dietz syndrome (LDS) type I, although the patient had only the TGFBR1 pathogenic variant without typical syndromic features. This prompted genetic testing of his family; his mother was positive for the same genetic pathogenic variant without syndromic features and his uncle was negative. The patient underwent successful root ascending hemiarch repair without preservation of the aortic valve (because of prominent fenestrations and frail tissue; Figure 2) and received a mechanical valve in the aortic position. Surgical pathology revealed a congenital BAV with fenestrated raphe and aorta with focal moderate medial degeneration. The patient was discharged from the hospital without complications.
Dr Prakash: Most BAV cases are sporadic, with <10% being familial and only 1% associated with genetic syndromes such as Turner syndrome (≈30% with associated BAV) or LDS (≈10% with associated BAV)3 (Figure 4). We refer patients with BAV for genetic testing and counseling if they have syndromic features (suggesting a specific syndrome), systemic features suggestive of nonspecific connective tissue disease (especially if associated with proximal aorta dilatation, such as in our patient), complex congenital cardiac lesions (truncus arteriosus, tetralogy of Fallot, transposition), family history of aortic dissection or early unexplained sudden cardiac death (ie, <55 years), or arterial aneurysms or dissections involving medium-sized arteries beyond the thoracic aorta, such as our patient’s intracranial aneurysm (Figures 1 and 4). Whereas syndromic features are increased among patients with LDS who experience aortic events, <50% of carriers of the TGFBR1 pathogenic variant present with recognizable syndromic features; thus it is not surprising that our patient had no specific LDS syndromic features. Male sex and increased arterial tortuosity are more highly predictive of dissection and death than are syndromic features in patients with TGFBR1 pathogenic variants4; our male patient had basilar artery tortuosity. The persistent superior vena cava is not associated with LDS. Whereas there are no specific anatomic features that distinguish BAV in LDS from BAV in sporadic cases, valvular disease tends to manifest at younger ages in patients with LDS or TGFBR1 pathogenic variants, who frequently present with aortic regurgitation attributable to proximal (ie, root) aortic dilatation.
Figure 4. Genetic syndromes associated with bicuspid aortic valve (BAV) and clinical indications for genetic testing in patients with BAV.
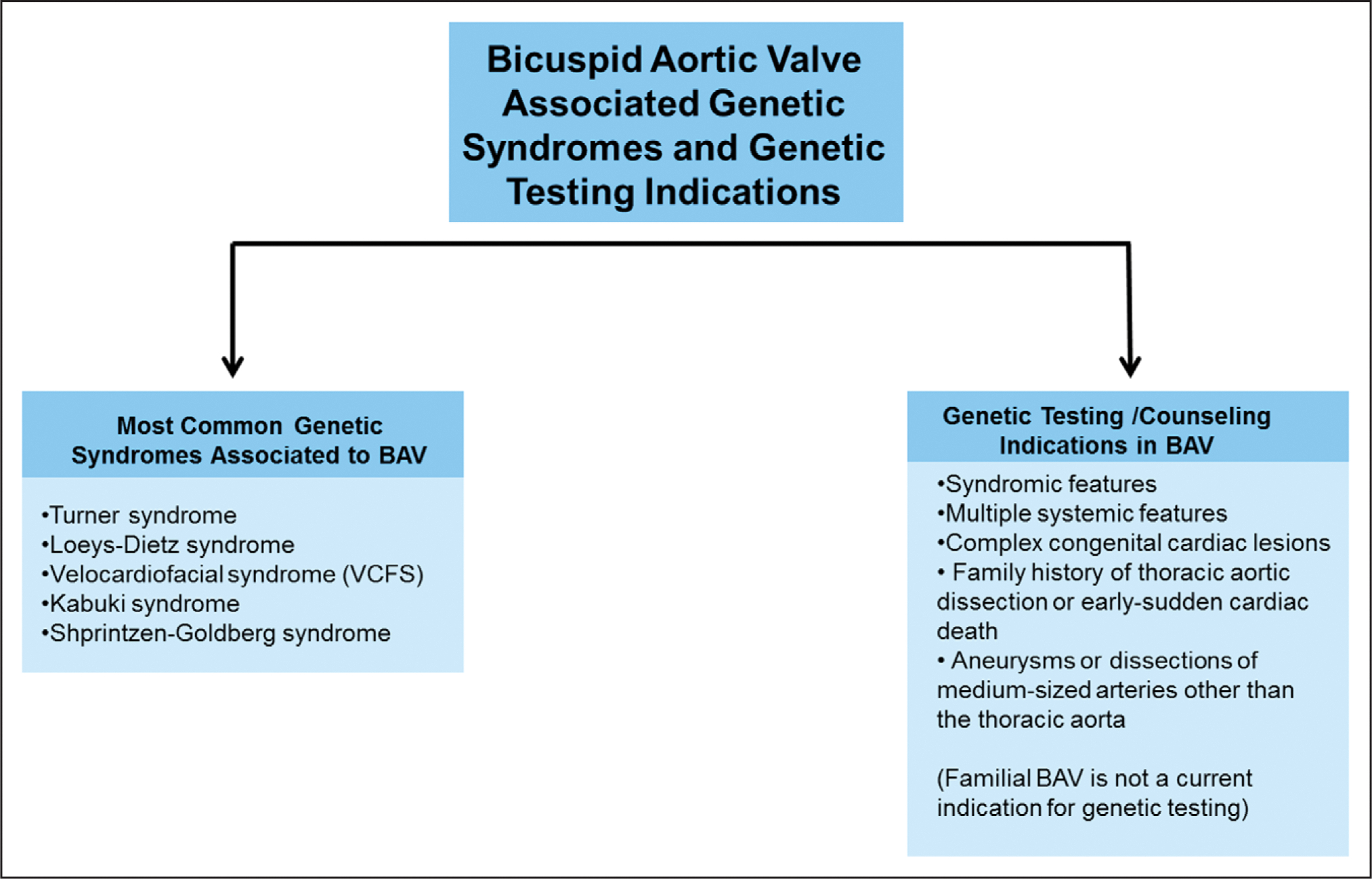
Guidelines recommend gene-specific personalized management of LDS or TGFBR pathogenic variant–related aortic disease. The frequency of aortic dissection varies substantially by causal gene, with carriers of TGFBR1 and TGFBR2 pathogenic variants experiencing earlier and more highly penetrant aortic events (median age at first event, 28 to 34 years) than carriers of the SMAD3 pathogenic variant (median age at first event, 45 years). Dissections occurred at larger aortic root diameters (mean, 68 mm) in patients with TGFBR1 pathogenic variants than in those with TGFBR2 pathogenic variants (mean, 52 mm), and no dissections were observed in patients with TGFBR1 pathogenic variants when the root diameter was <45 mm.4 The recommended threshold for surgical intervention (45 mm for male patients, closer to 40 mm for small female patients),4 as implemented in our case, reflects the somewhat milder course of TGFBR1-related aortic disease.
DISCUSSION
Our case illustrates the complex presentation BAV valvulo-aortopathy and highlights that younger patients with BAV with proximal thoracic aorta dilatation (ie, root phenotype) require thorough family history-taking, careful physical examination including search for syndromic features, thorough interrogation to elicit connective tissue disease features, and genetic testing and counseling, when associated with systemic features and extrathoracic vascular complications, as was the case in our patient (Figure 4).
Typical Presentation BAV Valvulo-Aortopathy
There is no indication for preventive ruling out of intracranial aneurysms in patients with the typical valvulo-aortopathy if they do not have aortic coarctation2 or family history of intracranial aneurysms. Whereas patients with BAV may have a higher incidence of small intracranial aneurysms compared with patients without BAV, patients with BAV do not appear to incur subarachnoid hemorrhage at a higher rate compared to the general population,5 with no evidence of aneurysmal growth or de novo aneurysms noted on follow-up.5 Genetic testing is not recommended for the typical valvulo-aortopathy presentation of the BAV condition even if familial (Figure 4) unless there is a family history of aortic dissection or early or unexplained sudden cardiac death or syndromic features (Figure 4). Typical BAV valvulo-aortopathy does not require imaging beyond the thoracic aorta.2
Complex Presentation BAV Valvulo-Aortopathy
Our patient represents the complex presentation valvulo-aortopathy of the BAV spectrum (Figure 3) with an associated disorder (TGFBR1 pathogenic variant), which is clinically and prognostically worse than the BAV condition per se, and manifested with an intracranial aneurysm (the risk of aneurysms or dissections of medium-sized arteries including intracranial arteries is increased in patients with the TGFBR pathogenic variant) and progressive proximal aorta dilatation (ie, root phenotype) at a relatively early age, which prompted urgent surgery (highlighting the importance of not considering the aortic diameter in isolation but within the clinical context of each patient). Had an intracranial aneurysm not been discovered in our patient, his TGFBR1 pathogenic variant may not have been uncovered, particularly because many patients with TGFBR1 pathogenic variants do not exhibit recognizable syndromic features.4 However, the combination of his young age, root phenotype, and nonspecific connective tissue disease features (ie, right inguinal hernia repair, right shoulder subluxation, easy skin bruising, flat feet, tooth overcrowding, pectus excavatum, scoliosis, and mitral prolapse) should have prompted genetic evaluation irrespective of the intracranial aneurysm (Figure 4). The genetic finding changed the patient’s management, because it prompted CTA evaluation of the abdominopelvic aorta and confirmed the need for urgent aorta surgery; will modify his future interval clinical and imaging follow-up; will serve a critical role for his future offspring; and identified the same pathogenic variant in his mother, which prompted further investigations and early aorta surgery in her as well.
The heterogeneous BAV condition can be reconciled into 3 groups: typical-presentation valvulo-aortopathy, complex-presentation valvulo-aortopathy, and uncomplicated or undiagnosed BAV (Figure 3). Typical-presentation valvulo-aortopathy is the most common and was the initial assumption in our patient, yet was revealed to be a complex-presentation valvulo-aortopathy that prompted genetic testing and urgent aorta surgery. Most patients with TGFBR1 pathogenic variants do not have recognizable syndromic features, but are at risk for sudden death attributable to highly penetrant thoracic aortic disease. Syndromic aortopathies tend to manifest with proximal aorta dilatation (ie, root phenotype), and our case raises an important question: In the absence of other clinical manifestations or features, should young male patients with BAV (ie, <30 years old) with root phenotypes undergo genetic testing? We propose that the answer is “yes” in light of the existence of known genetic abnormalities without syndromic features. More data are needed to determine whether BAV with root dilatation is in fact a genetically distinct subgroup.
Supplementary Material
Footnotes
Disclosures
None.
ARTICLE INFORMATION
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.120.046892.
Contributor Information
Hector I. Michelena, Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN.
Saarwaani Vallabhajosyula, Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN.
Siddharth K. Prakash, Divisions of Cardiovascular Medicine and Medical Genetics, Department of Internal Medicine, University of Texas Health Science Center at Houston.
REFERENCES
- 1.Michelena HI, Prakash SK, Della Corte A, Bissell MM, Anavekar N, Mathieu P, Bosse Y, Limongelli G, Bossone E, Benson DW, et al. Bicuspid aortic valve: identifying knowledge gaps and rising to the challenge from the International Bicuspid Aortic Valve Consortium (BAVCon). Circulation. 2014;129:2691–2704. doi: 10.1161/CIRCULATIONAHA.113.007851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borger MA, Fedak PWM, Stephens EH, Gleason TG, Girdauskas E, Ikonomidis JS, Khoynezhad A, Siu SC, Verma S, Hope MD, et al. The American Association for Thoracic Surgery consensus guidelines on bicuspid aortic valve-related aortopathy: full online-only version. J Thorac Cardiovasc Surg. 2018;156:e41–e74. doi: 10.1016/j.jtcvs.2018.02.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prakash SK, Bosse Y, Muehlschlegel JD, Michelena HI, Limongelli G, Della Corte A, Pluchinotta FR, Russo MG, Evangelista A, Benson DW, et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J Am Coll Cardiol. 2014;64:832–839. doi: 10.1016/j.jacc.2014.04.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jondeau G, Ropers J, Regalado E, Braverman A, Evangelista A, Teixedo G, De Backer J, Muino-Mosquera L, Naudion S, Zordan C, et al. ; for the Montalcino Aortic Consortium. International registry of patients carrying TGFBR1 or TGFBR2 mutations: results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet. 2016;9:548–558. doi: 10.1161/CIRCGENETICS.116.001485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaulov A, Leibowitz D, Rott D. Prevalence of bicuspid aortic valve in patients presenting with subarachnoid hemorrhage related to an intracerebral aneurysm. Int J Cardiol. 2012;157:142–143. doi: 10.1016/j.ijcard.2012.03.031 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.