

Abstract
Background
Sarcoidosis is an inflammatory, noncaseating, granulomatous disorder of unknown cause that can affect any body system and is associated with cardiovascular disease including sudden cardiac death (SCD). Cardiac involvement in sarcoidosis is associated with higher risk of SCD, but the SCD risk in the general sarcoidosis population is unknown. We aimed to determine the risk of SCD in people with sarcoidosis versus the matched general population.
Methods and Results
A population‐based cohort of sarcoidosis and age‐ and sex‐matched comparators from January 1, 1976 to December 31, 2013 was used; presence of other comorbidities in the comparator group was not an exclusion criterion. Mortality, including time, place, and cause of death were measured and manually adjudicated for SCD events. Incidence rates are reported per 100 000 person‐years, and Cox models were used for group comparisons. Of the 345 incident cases of sarcoidosis (171 men; 50%) there were 58 reported deaths; 10 were definite/probable SCD versus 57 all‐cause and 9 SCDs in comparators. Median follow‐up was 12.9 years (interquartile range, 6.0–23.4 years) . Incidence rate of SCD in sarcoidosis was 192 (95% CI, 92–352) versus 155 (95% CI, 71–294) in comparators (hazard ratio [HR], 1.28 (95% CI, 0.52–3.17). Nocturnal deaths were more frequent in sarcoidosis 57 (95% CI, 12–168) versus 17 (95% CI, 0.4–95) (HR, 3.76 [95% CI, 0.39–36.47]). No significant differences were detected between the groups by sex, age, calendar year of diagnosis, or disease duration.
Conclusions
In a population‐based cohort of patients with sarcoidosis, the risk for SCD compared with matched comparators was not increased. There were more nocturnal deaths among patients with sarcoidosis, yet this was statistically insignificant.
Keywords: arrhythmia, cardiac sarcoidosis, inflammatory cardiomyopathy, sudden death
Subject Categories: Sudden Cardiac Death, Epidemiology, Arrhythmias
Nonstandard Abbreviations and Acronyms
- CS
cardiac sarcoidosis
- SCD
sudden cardiac death
Clinical Perspective.
What Is New?
An epidemiologically matched cohort of sarcoidosis affecting any organ has similar sudden cardiac death risk to the nonsarcoid population.
The sarcoidosis cohort was more likely to have nocturnal deaths compared with the matched cohort.
Most published data on sudden cardiac death risk in sarcoidosis are highly prone to tertiary center referral bias, with more severe and advanced forms of cardiac sarcoidosis. These are the first population‐based data addressing sudden cardiac death risk showing no difference.
What Are the Clinical Implications?
The majority of patients have extracardiac sarcoidosis and want to know the risk of sudden cardiac death.
Patients can be counseled that the risk is similar to age‐ and sex‐matched controls without sarcoidosis.
Screening for cardiac involvement should continue.
Sarcoidosis is a noncaseating granulomatous disorder that can affect any organ, with a wide spectrum of disease presentation and long‐term sequelae. The cause is unknown but thought to involve an unknown antigenic trigger (possible inorganic molecule) in genetically predisposed individuals mounting an immune response. 1 , 2 This results in granuloma formation, consisting of macrophages predominantly, as well as CD4 (cluster of differentiation 4) and CD8 (cluster of differentiation 8) cells. The primary organ systems involved are the lungs and lymphatic system, but also include skin, liver, and neurological systems. Rarely, the disease can affect the heart in isolation, or more frequently with pulmonary, lymph node, and skin involvement, and when present is called cardiac sarcoidosis (CS). Moreover, CS can occur at the index presentation or involve the heart at a later stage. Involvement of the heart can result in an inflammatory cardiomyopathy presenting as: (1) myocarditis, with arrhythmias (brady and tachy) leading to sudden cardiac death (SCD) events; (2) morphological changes similar to idiopathic dilated cardiomyopathy and arrhythmogenic ventricular cardiomyopathy; (3) coronary artery dissection; (4) chronic forms with heart failure syndrome; and (5) no clinical consequences. 3
Although it is believed that people with isolated or multiorgan CS are at higher risk of SCD events, when this occurs during disease progression is unknown. 4 Furthermore, the sentinel event can be SCD, thus making detection of CS and risk stratification challenging. Most data reporting a higher SCD risk with CS are observational, with tertiary referral bias representing a more advanced disease process and with short follow‐up durations. 3 A common clinical and unknown pertinent question asked by clinicians and patients is: What is the incidence of SCD in patients with sarcoidosis with any organ involvement? Furthermore, it is unknown whether this risk is higher than the general population, different between sexes, has day–night variation, and whether this risk changes with disease trajectory. The aims of this study were to determine the incident rates of SCD events in a population‐based cohort of sarcoidosis with long follow‐up duration.
METHODS
Data will be made available on a case‐by‐case basis to investigators following a written request to the corresponding author.
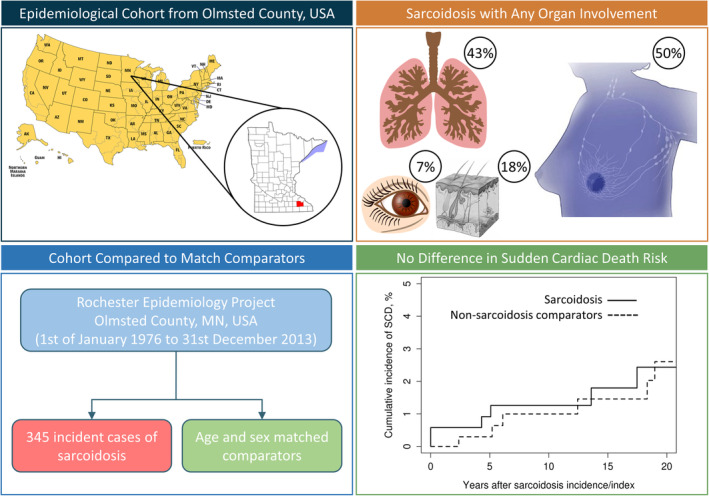
This study was approved by the institutional review boards of both the Mayo Clinic and Olmsted Medical Center. Patients provided written permission for review of their medical records. A population‐based cohort of incident cases of sarcoidosis and 345 age‐ and sex‐matched comparators in Olmsted County, Minnesota over January 1, 1976 to December 31, 2013 was identified through the resources of the Rochester Epidemiology Project. 5 In brief, a combination of validated International Classification of Diseases, Ninth Revision (ICD‐9), Hospital International Classification of Disease Adaptation, and pre–ICD‐9 Mayo Berkson codes to identify possible sarcoidosis and noncaseating granulomatous disease that underwent manual adjudication were used. This strategy included cases diagnosed at postmortem and cases with sudden death. The Rochester Epidemiology Project provides access to the comprehensive and complete medical records of all residents of Olmsted County, which includes the city of Rochester, seeking medical care through all of the health care providers (including Mayo Clinic, Olmsted Medical Center and its affiliated hospitals, nursing homes, and private providers) for over 6 decades. A record linkage system unifies these records as part of the Rochester Epidemiology Project, facilitating robust epidemiological research, and through multiple‐source ascertainment ensures virtually all clinical cases of sarcoidosis diagnosed in vivo and postmortem are captured. 6 In addition to census data, mortality and cause of death are collected regularly using Minnesota state records and the National Death Index.
Each case was manually reviewed, 3 , 7 and clinical variables abstracted using a predefined data dictionary and case note abstraction form set up and piloted in REDCap (Research Electronic Data Capture; Vanderbilt University, Nashville, TN). Date of diagnosis, age, sex, self‐reported ethnicity, smoking status, intrathoracic and extrathoracic disease, symptoms, radiological findings, serum angiotensin‐converting enzyme levels, and calcium levels were recorded. For living subjects, the last date of follow‐up and clinical status were also recorded. Mortality, including time, place, and cause of death were individually reviewed for decedents and categorized into cardiovascular‐related deaths and SCD events. A death was considered an SCD event in accordance with accepted definition by the American College of Cardiology, American Heart Association, and Heart Rhythm Society: death occurring within 60 minutes of the onset of symptoms or within 24‐hours if the death was unexpected, unwitnessed, and the subject was well in the preceding days, or an autopsy confirmed no other competing cause. 8
Comparator Group
Nonsarcoid (referent control) comparators matched by age and sex in the year of sarcoid diagnosis were randomly selected from the Rochester Epidemiology Project by computer (1 case:1 comparator). Each comparator was manually reviewed to ensure there was no prior diagnosis of sarcoidosis (either CS or extra‐CS) but could have any other disease (ie, the comparator group is not disease‐free healthy individuals). Baseline variables were manually abstracted for each comparator as described above for the cases. The presence of other comorbidities was not an exclusion criterion, because the aim was to determine prevalence of comorbidities and outcomes including SCD in the general nonsarcoid population.
Internal Validity and Accuracy
Review of mortality was conducted by the first author (C.A.A.C.). Case verification and abstraction of variables were conducted by the senior author (P.U.), and to ensure validity and accuracy, 10% were randomly selected and reviewed by the coauthors (E.L.M. and P.U.). 5
Statistical Analysis
Categorical variables are reported as frequencies and continuous variables with either means and SD or where skewed medians with 25th and 75th percentiles. Characteristics were compared between cohorts using χ2 and rank sum tests. Incidence rates of SCD are reported per 100 000 person‐years, and 95% CIs were obtained by assuming that the number of SCD events followed the Poisson distribution. Poisson regression models with smoothing splines were used to examine trends over time to allow for nonlinear effects. Overall mortality rates were estimated using the Kaplan‐Meier method. The cumulative incidence of SCD was estimated with adjustment for competing risk of death by other causes. 9 These methods are similar to the Kaplan‐Meier method, with censoring of patients who are still alive at last follow‐up. Patients who die without SCD events are appropriately accounted for to avoid overestimation of the rate of occurrence of SCD, which can occur if such subjects are simply censored at death. Cox proportional hazards models were used to compare the rate of SCD between cases and comparators. The proportional hazards assumptions were checked using Schoenfeld residuals, and no violations were found. A P value of <0.05 was considered statistically significant for all analyses. Analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC) and R version 3.4.2 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
The final study sample included 345 incident cases of sarcoidosis from January 1, 1976 to December 31, 2013 and 345 comparators matched by age, sex, and year of diagnosis. Baseline characteristics are described in Table 1. The body mass index was slightly higher in the sarcoidosis cohort versus the comparator group (30.1±7.5 versus 27.4±5.6, P<0.001). Cardiac involvement was only seen in 10 (2.9%) cases, none of which had isolated CS.
Table 1.
Baseline Clinical Characteristics
| Variable | Cohort with sarcoidosis, n=345 | Comparator group, n=345 | P value |
|---|---|---|---|
| Age at diagnosis of sarcoid, y, mean±SD | 45.6±13.6 | 45.4±13.7 | 0.87 |
| Male sex, n (%) | 174 (50) | 174 (50) | 1.0 |
| BMI, kg/m2, mean±SD | 30.1±7.5 | 27.4±5.6 | <0.001* |
| Organ involvement, n (%) | |||
| Intrathoracic | 335 (97%) | … | … |
| Intrathoracic lymphadenopathy | 169 (50%) | … | … |
| Lung parenchymal involvement | 145 (43%) | … | … |
| Skin | 63 (18%) | … | … |
| Arthralgia | 43 (20%) | … | … |
| Ophthalmological | 24 (7%) | … | … |
| Hepatic | 20 (6%) | … | … |
| Splenic | 13 (4%) | … | … |
| Renal | 12 (3%) | … | … |
| Neurological | 12 (3%) | … | … |
| Exocrine gland | 7 (2%) | … | … |
| Upper respiratory tract | 6 (2%) | … | … |
| Cardiac involvement | 10 (2.9%) | … | … |
| Bone | 1 | … | … |
| Smoking status at diagnosis, n (%) | |||
| Never | 198 (60) | 132 (42%) | <0.001* |
| Former smoker | 71 (21) | 70 (22%) | |
| Current smoker | 63 (19) | 115 (36%) | |
| Unknown | 13 | 28 | |
| Laboratory values, n positive/n tested (%) | |||
| Elevated serum ACE | 121/287 (42%) | … | … |
| Elevated serum calcium level | 32/295 (16%) | … | … |
Continuous variables are presented as mean±SD and categorical variables as number (percent). Asterisk (*) refers to being significant. ACE indicates angiotensin‐converting enzyme; and BMI, body mass index.
Follow‐Up and Survival
The median length of follow‐up was 12.9 years (interquartile range, 6.0–23.4 years), with a total of 5220 person‐years of follow‐up. In the matched comparator group, median follow‐up was 15.8 years (interquartile range, 6.8–25.5 years), with a total of 5819 person‐years follow‐up. The overall all‐cause mortality was similar in the cohort of incident sarcoidosis cases (58 deaths) and the comparator group (57 deaths; hazard ratio [HR], 1.14 [95% CI, 0.79–1.64]).
The number of SCD events was similar, with 10 in the sarcoid cohort and 9 in the comparator group (Table 2). The incidence rate of SCD in sarcoidosis was 192 (95% CI, 92–352) versus 155 (95% CI, 71–294) per 100 000 person‐years in comparators (HR, 1.28 [95% CI, 0.52–3.17]; Figure 1). There were no significant differences between the groups by sex, disease duration, age, or calendar year of incidence/index (Figure 2). There was no difference in cumulative incidence of any SCD events adjusted for competing risk of death from other causes in patients with sarcoidosis versus nonsarcoidosis comparators (Figure 3). Nocturnal deaths were more frequent in sarcoidosis, with an incidence rate of 57 (95% CI, 12–168) versus 17 (95% CI, 0.4–95) per 100 000 per son‐years (HR, 3.76 [95% CI, 0.39–36.47]; P=0.25; Figure 4). The nocturnal frequency was higher for both women and men, though it did not reach significance. Overall design and results are summarized in Figure 5. Cox proportional hazards ratios for univariate variables of age, male sex, and absence of intrathoracic disease are included in Table S1.
Table 2.
SCD Among Patients With Sarcoidosis Compared With Age‐ and Sex‐Matched Comparators Without Sarcoidosis
| Cause category | No. of deaths in sarcoidosis/nonsarcoidosis | Rate* in sarcoidosis (95% CI) | Rate* in nonsarcoidosis (95% CI) | Hazard ratio† (95% CI) |
|---|---|---|---|---|
| Any SCD, A, B, C, D, E | 10/9 | 192 (92–352) | 155 (71–294) | 1.28 (0.52–3.17) |
| Any definite SCD, A, B, C | 3/1 | 57 (12–168) | 17 (0.4–95) | 3.35 (0.35–32.24) |
| Any SCD at night, 22:00–06:00 | 3/1 | 57 (12–168) | 17 (0.4–95) | 3.76 (0.39–36.47) |
| Any SCD at day | 6/6 | 115 (42–250) | 103 (38–224) | 1.16 (0.37–3.60) |
| Any SCD at unknown time | ½ | … | … | |
| Women: any SCD | 6/6 | 217 (79–471) | 205 (75–447) | 1.12 (0.36–3.47) |
| Men: any SCD | 4/3 | 163 (44–418) | 103 (21–302) | 1.49 (0.33–6.69) |
A indicates definite fatal myocardial infarction; B, definite sudden death because of coronary heart disease; C, definite fatal coronary heart disease; D, possible fatal coronary heart disease; E, non–coronary heart disease (ie, arrhythmic SCD); and SCD, sudden cardiac death.
Rate per 100 000 person‐years.
Unadjusted because of small number of events.
Figure 1. Incidence rates of sudden cardiac death events in patients with sarcoidosis (‐‐‐‐‐‐) and nonsarcoidosis comparators (‐‐‐‐‐) based on day–night variation and sex as a biological variable.
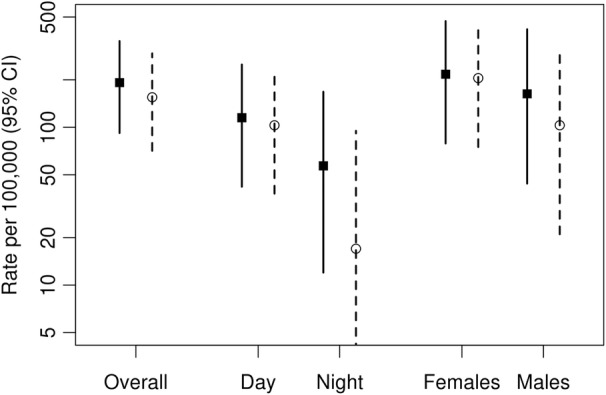
Figure 2. Rate of sudden cardiac death (SCD) in patients with sarcoidosis (‐‐‐‐‐‐) and nonsarcoidosis comparators (‐‐‐‐‐) according to disease duration (top), age (middle), and calendar year of sarcoidosis incidence/index date (bottom).
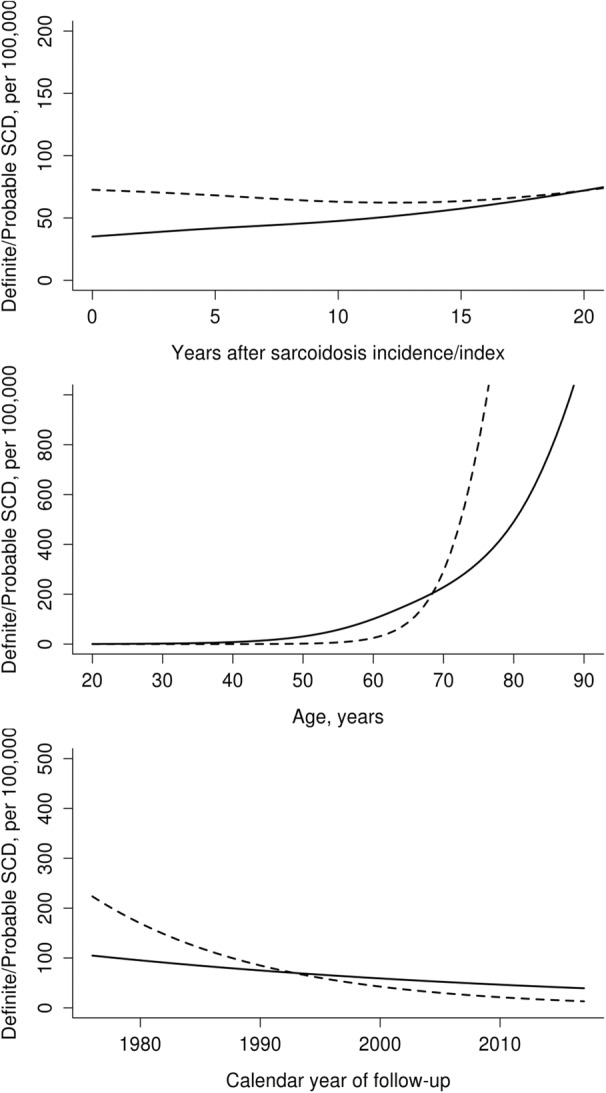
Figure 3. Cumulative incidence of any sudden cardiac death (SCD) adjusted for competing risk of death from other causes in patients with sarcoidosis (‐‐‐‐‐‐) and nonsarcoidosis comparators (‐‐‐‐‐).
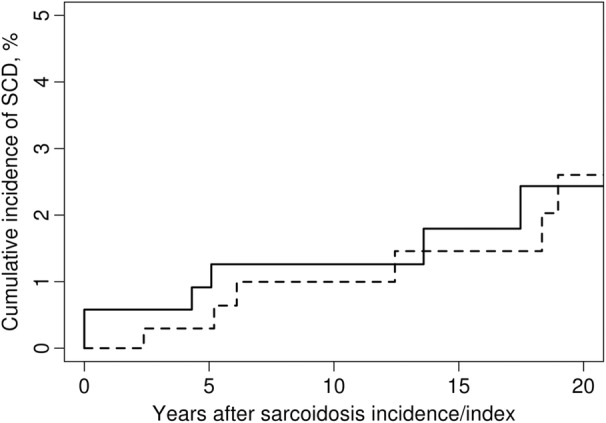
Figure 4. Incidence rates of sudden cardiac death events in sarcoidosis vs nonsarcoidosis comparators based on day–night variation and sex as a biological variable.
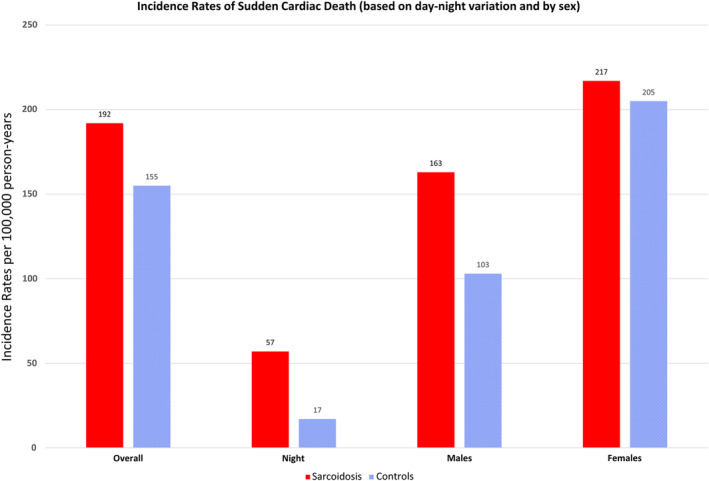
Day vs night incidence rates 57 (95% CI, 12–168) vs 17 (95% CI, 0.4–95). Cox proportional hazards regression hazard ratio 3.76 (95% CI, 0.39–36.47; P=0.25).
Figure 5. Overall study design and results.
SCD indicates sudden cardiac death.
Of the 345 sarcoid cases, 18 patients received a cardiac implantable electronic device. This included 11 with a permanent pacemaker, 6 with an implantable cardioverter‐defibrillator (ICD), and 1 with a biventricular ICD. Of these device recipients, 8 had confirmed CS (ICD, n=4; permanent pacemaker, n=3; and biventricular ICD, n=1). Of the remaining 10 patients who received a cardiac implantable electronic device and did not have evidence of CS, 1 received an ICD for primary prevention with hypertrophic cardiomyopathy and a remote history of biopsy‐proven isolated pulmonary sarcoidosis. The other 9 patients received a permanent pacemaker when in the seventh and eighth decades of life. These cardiac implantable electronic device implants occurred several decades after a diagnosis of pulmonary sarcoidosis, when sarcoidosis was quiescent or burnt out and the cause of conduction system disease most likely to be age‐related fibrosis and sclerosis. 10
DISCUSSION
This epidemiological study has several important and clinically relevant findings. First, in this cohort of sarcoidosis in a geographically and temporally well‐defined North Central US population with 5819 person‐years of follow‐up, the incidence of overall mortality and SCD events was similar, with no significant difference to the general population matched by age and sex. 11 Second, the major clinical concern of SCD being the first manifestation of CS in patients with non‐CS was not observed. Third, there were no differences in lifetime all‐cause mortality or SCD using sex as a biological variable. Fourth, there was a trend to differential day–night variation of SCD events, with more nocturnal events in patients with sarcoidosis versus the general population, but this did not reach statistical significance.
Incidence and Organ Involvement in Sarcoidosis
A wide‐range of estimated incidence rates of sarcoidosis have been previously reported with differences between countries, 12 sexes, as well as differences between racial groups within the same country. 12 , 13 , 14 The incidence in this study cohort is similar to other predominantly White populations estimated as 10.0 (95% CI, 9.0–10.9) per 100 000 population after adjusting for age and sex to the 2010 US White population. 5 , 15 The majority of studies report primary intrathoracic involvement; isolated nonpulmonary involvement is rare. 15 In our cohort, intrathoracic involvement (defined as parenchymal involvement and/or lymphadenopathy) was present in 335 (97%) cases. Of the intrathoracic cases, 293 (87%) had lymphadenopathy and 169 (50%) had radiographic and/or histological evidence of interstitial lung infiltration. Only 145 (43%) reported respiratory symptoms (dry cough, shortness of breath, and chest pain). The most frequent extrathoracic involvement included skin (63 cases, 18%), arthralgia (43 cases, 12%), and ophthalmologic involvement (24 cases, 7%). Cardiac involvement was seen in 10 (2.9%) cases, none of which had isolated CS. Isolated extrapulmonary sarcoidosis was rare and was seen in only 10 (3%) patients.
Mortality and SCD
Mortality estimates for patients with sarcoidosis have varied, with some studies suggesting no difference compared with the general population and others suggesting differences based on the presence of primary organ involvement, the degree of organ dysfunction and multiorgan involvement, sex, and race. 16 , 17 A French death‐certificate based study reported a higher age‐adjusted standardized mortality ratio of 3.6 per million population in sarcoidosis versus nonsarcoidosis, younger age at diagnosis, and male sex as highest risk, as well as a marked north–south gradient of mortality. 16 However, this is prone to reporting and causes of death, dependent on the seniority and expertise of the certifier. There was no denominator of actual sarcoid cases, no manual review of each case, granularity such as organ involvement, and the authors conclude this is an overestimate and not applicable to the general population. A Korean study of electronic health records reported an incidence of 1.3 per 100 000 (markedly lower than in our data), yet a standardized mortality ratio of 1.7 (95% CI, 1.5–1.8). 17 This study was based entirely on single diagnostic codes, no manual validation or case note review, unknown causes of death, and no assessment of tuberculosis status, which is known to be high in Korea. The authors acknowledge that these data likely include many false‐positive cases, excluding sarcoidosis as the primary diagnosis code.
The usual age of onset for sarcoidosis is between the ages of 25 and 45 years, with onset rare in those aged <15 years and those aged >70 years. A prior study from Olmsted County, Minnesota determined incident rates of SCD in subjects aged 20 to 40 years as 44.9 per 100 000 (61 per 1000 person‐years) over the periods from 1960 to 1989. 18 In that study, none of the deaths were because of sarcoidosis, although 4 were attributed to myocarditis histologically, in the absence of granulomata. In the current study, the overall SCD rate for the matched comparator group is 155 per 100 000 person‐years, which is 2.5‐fold higher than the prior study from Olmsted County. 18 This could be explained by age, the referent‐comparator group age range is 20 to 87 years versus 20 to 40 years for the group reported by Shen et al., and also better recognition of SCD with greater awareness and dissemination of knowledge. The incidence rate of SCD for the general adult US population is 141 (95% CI, 138–143) per 100 000 population, which is similar in our comparator group. 11 Thus, these findings suggest the risk of SCD in patients with sarcoidosis is similar to that of the general population, and special surveillance programs and/or preemptive interventions for prevention of SCD in a general sarcoid population would not be recommended based on these observations alone.
However, our data do not include subclinical CS, because in line with prior clinical standards, our patients were not routinely screened for cardiac involvement (with the use of ECG, Holter, and cardiac imaging). Importantly, given that we included cases diagnosed at postmortem with sarcoidosis (with or without heart involvement) including sudden death cases, the lack of regular screening for CS and consequent prognostic effects are minimal. In other words, including decedent sudden death cases in our geographical location, where all postmortems are exclusively conducted at our institution, ensures prognostically significant sarcoidosis is captured.
The risk of SCD in this population cohort is lower than that observed for people with CS (whether isolated or with systemic sarcoidosis). 3 , 4 However, these are not epidemiological cohorts and reflect strong referral bias and a more severe form of cardiac involvement. Our data directly answer the question of SCD risk in an epidemiological cohort with sarcoid involving any organ. The primary basis for regular screening of CS is that the sentinel event can be SCD, which our data show to be no different than the general population. Although our data show SCD risk in the general sarcoidosis population is low over a 30‐year follow‐up period, we still advocate that it is prudent to evaluate these patients for manifestations of CS including arrhythmia, impairment of right or left ventricular function, and heart failure syndrome, because these can be intervened on in a timely manner, for example, for implantation of a permanent pacemaker for brady‐arrhythmias. Isolated CS is rare and may represent a more aggressive form of the disease; a multicenter tertiary electrophysiology study of 235 cases of CS with an ICD reported only 13 (5%) with definite or suspected isolated CS, which was associated with 61.5% appropriate ICD shocks versus 26.6% in the remaining multisystem sarcoid cases (P=0.010) over a mean 4.2‐year follow‐up. 4 The authors acknowledge this is a highly biased population group and not reflective of typical sarcoid cases. Our study directly addresses the general sarcoid population risk of SCD.
Sex‐Based Differences in Mortality and SCD
Sex‐based differences have been consistently reported in sarcoidosis with higher incidence in women versus men, and mortality greatest in non‐Hispanic Black women, attributed to the higher prevalence of autoimmune diseases in women. 13 However, mortality and SCD have age‐ and sex‐dependent incidence in the general population, and are higher in men compared to premenopausal women. In our study, we found no differences in mortality or SCD when considering sex as a biological variable. Our study population is 90% White, with only 18 (5%) Black individuals and 6 (2%) Asian individuals. Thus, our data of no sex‐based differences are not generalizable to the non‐White population and may explain our reported differences.
Day–Night Variation With SCD
On day–night variation, our data trended toward a difference with higher nocturnal mortality, which did not reach significance, likely attributable to being underpowered. This could have important implications on other coexisting nocturnal disorders, such as undiagnosed sleep apnea, which is itself an independent risk factor for SCD. Our data showed a difference in body mass index between patients with sarcoidosis and the referent‐control group, a well‐recognized risk factor for sleep apnea. Whether sleep disorders and sarcoid synergistically increase the risk of nocturnal SCD remains unknown and deserving of further study. The other possibilities include coexisting obstructive lung disorders, which if present together with pulmonary sarcoid, may increase the propensity for SCD.
Screening for Cardiac Involvement
Symptomatic CS usually occurs in association with pulmonary or systemic organ involvement. The reported prevalence varies across studies, ranging from <1% to almost 5%. 3 , 5 , 19 , 20 Similarly, asymptomatic involvement and incidental findings are highly variable, from 25% in autopsy‐based studies and from 3.7% to 54.9% in imaging studies of extrathoracic sarcoidosis. This range reflects both sampling (referral) bias and selection bias (inconsistent definitions of what constitutes cardiac involvement) and use of different imaging modalities that may not detect inflammation with the same degree of sensitivity and specificity. Without consistently applied definitions of cardiac involvement, these discrepancies will persist, which influence recommendations of the Heart Rhythm Society on diagnosis and management of arrhythmia in sarcoidosis. 3 These guidelines acknowledge the lack of evidence for management recommendations, pointing out that most of the recommendations made by the panel of 14 experts are based on observational data. The recommendations of the Heart Rhythm Society are closely aligned to the World Association of Sarcoidosis and Other Granulomatous Disorders 2014 guidelines.
In our epidemiological cohort, 2.9% of patients had cardiac involvement, based on symptoms, ECG criteria, imaging evidence, and biopsy. All cases of CS were diagnosed in living patients and none were diagnosed postmortem (despite comprehensively searching). In A Case Control Etiologic Study of Sarcoidosis–control study from the United States, based on predefined criteria for organ involvement, 19 only 2.3% of patients with sarcoidosis had cardiac involvement. 21 In this study, 95% of patients had pulmonary involvement, followed by 15.9% with skin, 15.2% with lymph node, 11.8% with eye, and 11.5% with liver involvement.
A prospective single‐center study from the United States evaluated 62 consecutive patients by symptoms, 12‐lead ECG, and transthoracic echocardiography, comparing these with the working gold standard of either abnormal computerized tomography‐positron emission tomography or magnetic resonance imaging, detecting a frequency of CS of 39%. 20 A significant limitation of that study was the exclusion of 32 patients who may or may not have had CS, thus affecting the reported frequency. There have been no published reports reproducing findings with screening using computerized tomography‐positron emission tomography and magnetic resonance imaging from the United States, raising concerns over selection bias. Importantly, despite the higher reported frequency of CS over a 2‐year follow‐up period, no patients developed heart failure and none had arrhythmias, SCD events, or appropriate therapies for ventricular arrhythmias. None of the parameters had high sensitivity alone when compared with computerized tomography‐positron emission tomography and magnetic resonance imaging to detect inflammation and evidence of CS. However, specificity was high for individual tests and increased when used in combination. Thus, the reported frequency is not an accurate estimate of prevalence and includes a significant proportion with subclinical disease, which does not impact mortality. Our data with longer follow‐up are consistent with no impact on SCD prognosis. The role of cardiac blood biomarkers including brain‐type natriuretic peptides, atrial natriuretic peptides, and troponins in detecting cardiac involvement is uncertain. Currently, no blood biomarkers are recommended or in routine use; these were not routinely collected in our cohort. However, with greater awareness of CS, falling costs of the tests, and increased use of electronic health records for following patients, this may be changing, with some centers now routinely monitoring these parameters despite lack of recommendations. 3
Strengths and Limitations
Strengths of the current study include the robust epidemiological design, which minimizes referral bias intrinsic to most studies from single centers and assures complete case ascertainment. The >30‐year follow‐up period is the longest reported in the literature and permits assessment of the disease course over time, particularly because medical visits and mortality are reliably captured.
Limitations include those inherent to the retrospective cohort design, and that these data represent minimal estimates, because undiagnosed or subclinical disease may not have been captured. Of note, any case identified at postmortem was included in this study, and thus should have identified local sudden death cases with sarcoidosis. The results of this study may not be generalizable to more diverse populations because the population of Olmsted County is predominately of Northern European ancestry and less diverse than other US populations. Indigenous Northern European populations have a higher incidence of sarcoid, which may in part be attributable to a genetic predisposition.
CONCLUSIONS
There were no significant differences in all‐cause mortality or SCD events between people with sarcoidosis and the general population. SCD event rates were similar in the matched comparator group, as well as that of the general US population, and no sex‐based differences were observed. Although there were more nocturnal deaths among patients with sarcoidosis, this did not reach statistical significance.
Sources of Funding
This publication was made possible by the Rochester Epidemiology Project (R01 AG034676 from the National Institutes of Health) and Clinical and Translational Science Award, grant number UL1 TR002377 RR024150‐01 from the National Center for Advancing Translational Sciences, a component of the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of National Institutes of Health. C.A.A.C. and V.K.S. are supported by the National Institutes of Health [grant numbers HL65176 and HL134885]. C.A.A.C. is supported by the American Heart Association [grant number 17POST33400211].
Disclosures
V.K.S. has served as a consultant for ResMed, Phillips, GlaxoSmithKline, Respicardia, Ronda Gray, Biosense Webster, Dane Garvin, Bayer, Itamar, and U‐Health. He works with Mayo Health Solutions and their industry partners on intellectual property related to sleep and obesity. However, none of these entities were involved in this study in any way. The remaining authors have no disclosures to report.
Supporting information
Table S1
Acknowledgments
The authors acknowledge the secretarial support from E.M. Emanuel, Department of Cardiology, Mayo Clinic with article preparation and submission.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.025479
For Sources of Funding and Disclosures, see page 9.
Contributor Information
C. Anwar A. Chahal, Email: chahal.anwar@mayo.edu.
Patompong Ungprasert, Email: p.ungprasert@gmail.com.
REFERENCES
- 1. Statement on sarcoidosis . Joint statement of the american thoracic society (ATS), the european respiratory society (ERS) and the world association of sarcoidosis and other granulomatous disorders (WASOG) adopted by the ats board of directors and by the ers executive committee, february 1999. Am J Respir Crit Care Med. 1999;160:736–755. doi: 10.1164/ajrccm.160.2.ats4-99 [DOI] [PubMed] [Google Scholar]
- 2. Beijer E, Veltkamp M, Meek B, Moller DR. Etiology and immunopathogenesis of sarcoidosis: novel insights. Semin Respir Crit Care Med. 2017;38:404–416. doi: 10.1055/s-0037-1603087 [DOI] [PubMed] [Google Scholar]
- 3. Birnie DH, Sauer WH, Bogun F, Cooper JM, Culver DA, Duvernoy CS, Judson MA, Kron J, Mehta D, Cosedis Nielsen J, et al. Hrs expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm. 2014;11:1305–1323. doi: 10.1016/j.hrthm.2014.03.043 [DOI] [PubMed] [Google Scholar]
- 4. Kron J, Sauer W, Mueller G, Schuller J, Bogun F, Sarsam S, Rosenfeld L, Mitiku TY, Cooper JM, Mehta D, et al. Outcomes of patients with definite and suspected isolated cardiac sarcoidosis treated with an implantable cardiac defibrillator. J Interv Card Electrophysiol. 2015;43:55–64. doi: 10.1007/s10840-015-9978-3 [DOI] [PubMed] [Google Scholar]
- 5. Ungprasert P, Carmona EM, Utz JP, Ryu JH, Crowson CS, Matteson EL. Epidemiology of sarcoidosis 1946‐2013: a population‐based study. Mayo Clin Proc. 2016;91:183–188. doi: 10.1016/j.mayocp.2015.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rocca WA, Yawn BP, St Sauver JL, Grossardt BR, Melton LJ 3rd. History of the Rochester epidemiology project: Half a century of medical records linkage in a us population. Mayo Clin Proc. 2012;87:1202–1213. doi: 10.1016/j.mayocp.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Costabel U, Hunninghake GW. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis statement committee. American Thoracic Society. European Respiratory Society. World Association for Sarcoidosis and other Granulomatous Disorders. Eur Respir J. 1999;14:735–737. doi: 10.1034/j.1399-3003.1999.14d02.x [DOI] [PubMed] [Google Scholar]
- 8. Buxton AE, Calkins H, Callans DJ, DiMarco JP, Fisher JD, Greene HL, Haines DE, Hayes DL, Heidenreich PA, Miller JM, et al. ACC/AHA/HRS 2006 key data elements and definitions for electrophysiological studies and procedures: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (ACC/AHA/HRS Writing Committee to Develop Data Standards on Electrophysiology). Circulation. 2006;114:2534–2570. doi: 10.1161/CIRCULATIONAHA.106.180199 [DOI] [PubMed] [Google Scholar]
- 9. Gooley TA, Leisenring W, Crowley J, Storer BE. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. 1999;18:695–706. doi: [DOI] [PubMed] [Google Scholar]
- 10. Khurshid S, Choi SH, Weng LC, Wang EY, Trinquart L, Benjamin EJ, Ellinor PT, Lubitz SA. Frequency of cardiac rhythm abnormalities in a half million adults. Circulation. 2018;11:e006273. doi: 10.1161/CIRCEP.118.006273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, et al. Heart disease and stroke statistics‐2018 update: a report from the american heart association. Circulation. 2018;137:e67–e492. doi: 10.1161/CIR.0000000000000558 [DOI] [PubMed] [Google Scholar]
- 12. Morimoto T, Azuma A, Abe S, Usuki J, Kudoh S, Sugisaki K, Oritsu M, Nukiwa T. Epidemiology of sarcoidosis in Japan. Eur Respir J. 2008;31:372–379. doi: 10.1183/09031936.00075307 [DOI] [PubMed] [Google Scholar]
- 13. Cozier YC, Berman JS, Palmer JR, Boggs DA, Serlin DM, Rosenberg L. Sarcoidosis in black women in the United States: data from the black women's health study. Chest. 2011;139:144–150. doi: 10.1378/chest.10-0413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hillerdal G, Nou E, Osterman K, Schmekel B. Sarcoidosis: epidemiology and prognosis. A 15‐year european study. Am Rev Respir Dis. 1984;130:29–32. doi: 10.1164/arrd.1984.130.1.29 [DOI] [PubMed] [Google Scholar]
- 15. Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61:980–985. doi: 10.1136/thx.2006.062836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jamilloux Y, Maucort‐Boulch D, Kerever S, Gerfaud‐Valentin M, Broussolle C, Eb M, Valeyre D, Seve P. Sarcoidosis‐related mortality in France: a multiple‐cause‐of‐death analysis. Eur Respir J. 2016;48:1700–1709. doi: 10.1183/13993003.00457-2016 [DOI] [PubMed] [Google Scholar]
- 17. Park JE, Kim YS, Kang MJ, Kim CJ, Han CH, Lee SM, Park SC. Prevalence, incidence, and mortality of sarcoidosis in Korea, 2003‐2015: a nationwide population‐based study. Respir Med. 2018;144:S28–S34. doi: 10.1016/j.rmed.2018.03.028 [DOI] [PubMed] [Google Scholar]
- 18. Shen WK, Edwards WD, Hammill SC, Bailey KR, Ballard DJ, Gersh BJ. Sudden unexpected nontraumatic death in 54 young adults: a 30‐year population‐based study. Am J Cardiol. 1995;76:148–152. doi: 10.1016/S0002-9149(99)80047-2 [DOI] [PubMed] [Google Scholar]
- 19. Judson MA, Baughman RP, Teirstein AS, Terrin ML, Yeager H Jr. Defining organ involvement in sarcoidosis: the access proposed instrument. Access research group. A case control etiologic study of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16:75–86. [PubMed] [Google Scholar]
- 20. Mehta D, Lubitz SA, Frankel Z, Wisnivesky JP, Einstein AJ, Goldman M, Machac J, Teirstein A. Cardiac involvement in patients with sarcoidosis: diagnostic and prognostic value of outpatient testing. Chest. 2008;133:1426–1435. doi: 10.1378/chest.07-2784 [DOI] [PubMed] [Google Scholar]
- 21. Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H Jr, Bresnitz EA, DePalo L, Hunninghake G, Iannuzzi MC, Johns CJ, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care medicine. 2001;164:1885–1889. doi: 10.1164/ajrccm.164.10.2104046 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1