

Abstract
Background
We investigated the causal associations between the genetic liability to cardiovascular and lifestyle risk factors and peripheral artery disease (PAD), using a Mendelian randomization approach.
Methods and Results
We performed a 2‐sample inverse‐variance weighted Mendelian randomization analysis, multiple sensitivity analyses to assess pleiotropy and multivariate Mendelian randomization analyses to assess mediating/confounding factors. European‐ancestry genomic summary data (P<5×10−8) for type 2 diabetes, lipid‐fractions, smoking, alcohol and coffee consumption, physical activity, sleep, and education level were selected. Genetic associations with PAD were extracted from the Million‐Veteran‐Program genome‐wide association studies (cases=31 307, controls=211 753, 72% European‐ancestry) and the GoLEAD‐SUMMIT genome‐wide association studies (11 independent genome‐wide association studies, European‐ancestry, cases=12 086, controls=449 548). Associations were categorized as robust (Bonferroni‐significant (P<0.00294), consistent over PAD‐cohorts/sensitivity analyses), suggestive (P value: 0.00294–0.05, associations in 1 PAD‐cohort/inconsistent sensitivity analyses) or not present. Robust evidence for genetic liability to type 2 diabetes, smoking, insomnia, and inverse associations for higher education level with PAD were found. Suggestive evidence for the genetic liability to higher low‐density lipoprotein cholesterol, triglyceride‐levels, alcohol consumption, and inverse associations for high‐density lipoprotein cholesterol, and increased sleep duration were found. No associations were found for physical activity and coffee consumption. However, effects fully attenuated for low‐density lipoprotein cholesterol and triglycerides after correcting for apoB, and for insomnia after correcting for body mass index and lipid‐fractions. Nonsignificant attenuation by potential mediators was observed for education level and type 2 diabetes.
Conclusions
Detrimental effects of smoking and type 2 diabetes, but not of low‐density lipoprotein cholesterol and triglycerides, on PAD were confirmed. Lower education level and insomnia were identified as novel risk factors for PAD; however, complete mediation for insomnia and incomplete mediation for education level by downstream risk factors was observed.
Keywords: cardiometabolic risk factors, cigarette smoking, education, health risk behaviors, hypercholesterolemia, mendelian randomization analysis, peripheral artery disease
Subject Categories: Peripheral Vascular Disease, Epidemiology, Cardiovascular Disease, Lifestyle, Atherosclerosis
Nonstandard Abbreviations and Acronyms
- IV
instrumental variable
- IVW
inverse variance weighted
- MR
Mendelian randomization
- MR‐PRESSO
Mendelian Randomization Pleiotropy Residual Sum and Outlier
- MVP
Million Veteran Program
- MVPA
moderate‐to‐vigorous physical activity
Clinical Perspective.
What Is New?
Our study strengthened the causality claim on the harmful relationship between smoking, type 2 diabetes, and peripheral artery disease.
Our findings further strengthened the suspicion that apoB, rather than low‐density lipoprotein cholesterol and triglycerides, is responsible for causing peripheral artery disease in people with hyperlipidemia.
Our study identified lower education level and insomnia as upstream risk factors in the development of peripheral artery disease.
What Are the Clinical Implications?
The results of our study contribute to a more targeted prevention strategy for peripheral artery disease, by providing a better understanding of causal risk factors in the development of peripheral artery disease.
Peripheral artery disease (PAD) is characterized by atherosclerosis of the large arteries of the lower extremities. 1 It is estimated to affect >200 million people worldwide with a prevalence rising sharply with age, therefore making it one of the most common manifestations of atherosclerotic disease. 2 Furthermore, individuals with PAD have an increased risk of both major cardiovascular events and cardiovascular mortality. 3
To date, there is no cure for PAD, therefore clinical management mainly focuses on prevention of disease progression by stimulating physical activity and improving cardiovascular risk factor management. 4 However, since executing a randomized controlled trial to assess the causal association between risk factors and PAD remains challenging because of practical and ethical concerns, 4 recommendations given in current guidelines are largely based on observational studies. The negative effects of smoking, diabetes, hypertension, and hypercholesterolemia on PAD have been extensively reported, and corroborated in a meta‐analysis. 2 Furthermore, the impact of cholesterol lowering drugs and glucose lowering therapy on PAD has been investigated in a number of randomized controlled trials. 5 , 6 , 7 , 8 Nevertheless, the evidence from studies investigating the effects of behavioral risk factors and education status on PAD is less convincing. In summary, harmful effects for obesity, lower physical activity, insomnia, and lower education level on PAD, and inverse effects for alcohol use and coffee consumption on PAD 2 , 9 , 10 , 11 , 12 , 13 have been reported. However, these risk factors were investigated in observational studies containing small sample sizes or using cross‐sectional designs, making them vulnerable to bias such as confounding and reverse causation. 14 Therefore, no conclusions on causality can be made. Given the difficulties in evaluating the causal relationship between lifestyle factors and PAD using conventional epidemiological frameworks, Mendelian randomization (MR) studies can function as an alternative way to assess causality and therefore fill the observed evidence gap.
MR is an instrumental variable (IV) analysis which uses genetic variants that are robustly associated with risk factors as IVs. Since genetic variants are randomly allocated at meiosis, they mimic a randomized controlled setting in which all other variables except the exposure are distributed equally between subgroups, this makes the design less vulnerable to confounding and reverse causation bias. 15 Previously executed MR studies observed significant associations between genetic liability to insomnia, alcohol consumption, low‐density lipoprotein cholesterol (LDL‐C), type 2 diabetes and PAD, 16 , 17 , 18 , 19 , 20 however the data used to assess PAD in these studies were derived from smaller genome‐wide association studies (GWASs) and therefore potentially lacks power. Recently, 2 GWASs on PAD with larger sample sizes were published; The Million Veteran Program (MVP) GWAS, consisting of predominantly males of multi‐ancestry origin (72% European‐ancestry), 21 and the GoLEAD‐SUMMIT GWAS consisting of males and females of European‐ancestry. 22 A number of recently published MR studies using the MVP GWAS reported significant associations between hyperlipidemia, 23 hypertension, 24 overall smoking 25 and obesity 26 with PAD. However, to our knowledge, no MR studies have investigated the causal effects of the genetic liability to impaired glucose tolerance, smoking cessation, coffee consumption, increased sleep duration, short and long sleep duration, physical activity, and education status on PAD. Furthermore, to validate prior studies, analyses on the genetic liability to insomnia, alcohol consumption, type 2 diabetes, and lipid subfractions on PAD should be repeated in cohorts consisting of larger sample sizes or cohorts consisting of participants of European ancestry to overcome bias.
In this study, we aim to give a comprehensive overview of putative cardiovascular and lifestyle related risk factors for PAD by investigating the causal associations of the genetic liability to these risk factors on PAD using an MR design. Our aim is 2‐fold; first we aim to investigate risk factors for PAD that have not yet been investigated in MR studies, and second we aim to update research on previously investigated risk factors, using genetic summary data of the 2 largest, most recently published GWAS available of both European and multi‐ancestry origin.
METHODS
Data Availability and Institutional Review Board Approval
All GWAS exposure data used in this MR study, and data from the GoLEAD‐SUMMIT consortium GWAS are publicly available. Information on how to access these data can be found in the below cited GWAS articles. Million Veteran Program PAD GWAS summary statistics are available on dbGAP (Accession phs001672.v6.p1). All used GWASs were approved by their respective institutional review committees, and all participants provided written informed consent. Information on institutional review board approval can be found in the below cited articles.
Two‐Sample MR Design
A 2‐sample MR design was used to investigate the relationship between the genetic liability to cardiovascular and lifestyle related risk factors and PAD. Single nucleotide polymorphisms (SNPs), genetic variants that are robustly associated with risk factors, were used as IVs. Since SNPs are randomly allocated at meiosis, they are largely free of the classic forms of bias that are observed in observational research (eg, confounding, reverse causation, measurement error) and can therefore be used to explore causal effects on an outcome, providing certain assumptions are met. These assumptions include the following: (1) the SNPs are associated with the exposure; (2) the SNPs are independent of confounders of the risk factor– outcome association; and (3) the SNPs influence the outcome only via the exposure (Figure S1). 15
Data Sources for and Selection of Genetic Instruments
We used GWASs conducted primarily among individuals of European ancestry as data sources for the genetic instruments of the investigated risk factors. For each risk factor the largest and most recently published GWASs were identified (Table). We selected SNPs as IVs if associated with the risk factors at the genome‐wide significance level (P<5×10−8). If SNPs were in linkage disequilibrium (r2>0.1), we included the SNP with the strongest correlation with the risk factor. If SNPs were not available (n=103 [MVP], n=22 [GoLEAD‐SUMMIT]) in the PAD GWAS, the SNPs were excluded from the analyses. Because of the minimal number of unavailable SNPs, no linkage disequilibrium proxies were used.
Table 1.
Overview of Genome‐Wide Association Studies Used as Instrumental Variables
| Risk factor (reference) | Cohort(s) | Measurement unit* | Sample size | Ancestry |
No. SNPs (MVP/GoLEAD‐SUMMIT) of total identified |
% Variance explained | F‐statistic |
Sample overlap MVP |
Sample overlap GoLEAD‐SUMMIT |
|---|---|---|---|---|---|---|---|---|---|
| Glucose metabolism | |||||||||
| Type 2 diabetes 27 | Meta‐analysis >30 cohorts | Odds of type 2 diabetes | 898 130 | European | 280/281 of 403 | 16.3% for 403 SNPs | 165 | none | 96% |
| Fasting glucose 28 | MAGIC Consortium (new data meta‐analyzed with 4 existing GWASs) | mmol/L | 133 010 | European | 35/35 of 36 | 4.8% For 36 SNPs | 350 | none | 0.5% |
| Lipid metabolism | |||||||||
| LDL‐C 29 | Meta‐analysis >40 cohorts | 1 SD increase in LDL‐C | 188 577 | Predominantly European | 53/54 of 58 | 14.6% for 58 SNPs | 784 | none | 5% |
| HDL‐C 29 | Meta‐analysis >40 cohorts | 1 SD increase in HDL‐C | 188 577 | Predominantly European | 65/65 of 71 | 13.7 for 71 SNPs | 593 | none | 5% |
| Triglycerides 29 | Meta‐analysis >40 cohorts | 1 SD increase in triglycerides | 188 577 | Predominantly European | 35/35 of 40 | 11.7% for 40 SNPs | 920 | none | 5% |
| Smoking | |||||||||
| Smoking initiation 30 | GSCAN Consortium (>30 cohorts) | Ever smoked regularly compared with never smoked | 1 232 091 | European | 346/360 of 378 | 2.3% for 378 SNPs | 17 | none | 31% |
| Smoking cessation 30 | GSCAN Consortium (>30 cohorts) | Current compared with former smokers | 547 219 | European | 21/21 of 24 | 0.1% For 24 SNPs | 11 | none | 42% |
| Smoking heaviness/cigarettes per day 30 |
GSCAN Consortium (>30 cohorts) |
SD increase in number of cigarettes smoked daily | 337 334 | European | 45/47 of 55 | ≈1% for 55 SNPs | 55 | none | 88% |
| Diet | |||||||||
| Alcohol consumption 30 | GSCAN Consortium (>30 cohorts) | SD of log‐transformed alcoholic drinks/week | 941 280 | European | 84/90 of 99 | ≈0.2% for 99 SNPs | 5.8 | none | 75% |
| Coffee consumption 31 | UK Biobank; Nurses' Health Study; Health Professionals Follow‐Up Study; Women's Genome Health Study | 50% increase | 375 833 (374 046 for rs117692895) | European | 14/14 of 15 | 0.48% for 15 SNPs | 84 | none | 90% |
| Physical activity | |||||||||
| Physical activity, MVPA 32 | UK Biobank | SD increase in MET‐minutes /wk of MVPA | 377 234 | European | 7/7 of 9 | 0.073% for 9 SNPs† | 25 | none | 82% |
| Sedentary behavior 33 | UK Biobank | SD increase in sedentary time | 91 105 | European | 4/4 of 4 | 0.08% for 4 SNPs | 48 | none | 20% |
| Sleep | |||||||||
| Insomnia 34 | UK Biobank; 23 and Me | Odds of Insomnia | 1 331 010 | European | 229/238 of 248 | 2.6% for 248 SNPs | 28 | none | 84% |
| Sleep duration 35 | UK Biobank | H/d | 446 118 | European | 75/78 of 78 | 0.69% for 78 SNPs | 23 | none | 90% |
| Long sleep duration 35 | UK Biobank | ≥9 h compared with 7–8 h/d | 446 118 | European | 7/7 of 8 | Not available | Not available | none | 74% |
| Short sleep duration 35 | UK Biobank | <7 h compared with 7–8 h /d | 446 118 | European | 27/26 of 27 | Not available | Not available | none | 89% |
| Education | |||||||||
| Education level 36 | Meta‐analysis >70 cohorts | 1 SD increase in years of educational attainment | 1 131 881 | European | 1155/1201 of 1271 | 11–13% for 1271 SNPS | 29 | none | 90% |
GSCAN indicates GWAS and Sequencing Consortium of Alcohol and Nicotine Use; GWAS, Genome‐wide association study; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; MAGIC, Meta‐Analysis of Glucose and Insulin related traits Consortium; MET, metabolic equivalent of task; MVP, Million Veteran Program; MVPA, moderate to vigorous physical activity; and SNP, single nucleotide polymorphism.
Units as used in this Mendelian randomization study.
Not reported, calculated using the formula: r2=2×Minor Allele Frequency×(1 – Minor Allele Frequency)×(beta/SD)2.
Overall we included 280/281 SNPs (MVP/GoLEAD‐SUMMIT) for type 2 diabetes, 27 35/35 SNPs for fasting glucose, 28 53/54 SNPs for LDL‐C, 29 65/65 SNPs for high‐density lipoprotein cholesterol (HDL‐C), 29 35/35 SNPs for triglycerides, 29 346/360 SNPs for smoking initiation, 30 21/21 SNPs for smoking cessation, 30 45/47 SNPs for smoking heaviness, 30 84/90 SNPs for alcohol consumption, 30 14/14 SNPs for coffee consumption, 31 7/7 SNPs for moderate‐to‐vigorous physical activity (MVPA), 32 4/4 SNPs for sedentary behavior, 33 229/238 SNPs for insomnia, 34 75/78 SNPs for overall sleep duration, 35 27/26 SNPs for short sleep duration (<7 versus 7–8 h/day), 35 7/7 SNPs for long sleep duration (≥9 versus 7–8 h/day) 35 and 1155/1201 SNPs for education level 36 (Figure S2). The phenotypic variance explained by the genetic instruments varied from 0.073% for MVPA to 16.3% for type 2 diabetes (Table). No sample overlap was present between the IVs and the MVP GWAS, however sample overlap was present between the IVs and the GoLEAD‐SUMMIT GWAS (ranging from 0.5% for the genetic liability to a higher fasting glucose to 96% for the genetic liability to type 2 diabetes) (Table).
Data Source for PAD
We extracted the genetic associations of the instrumental variables with PAD from 2 cohorts: the 2019 MVP GWAS and the 2021 GoLEAD‐SUMMIT GWAS.
The 2019 Million Veteran Program GWAS of PAD by Klarin et al 21 is a predominantly male (85.2–97.9%) multiethnic (72% European‐ancestry) GWAS of 31 307 cases and 211 753 controls in which 19 PAD loci were identified, 18 of which have not been previously reported. The results were replicated in an independent sample of 5117 PAD cases and 389 291 controls (UK Biobank). Cases and controls were defined based on electronic health record phenotyping, in which individuals were defined as having PAD based on International Classification of Diseases, Ninth and Tenth Revision (ICD‐9 and ICD‐10) codes and/or Current Procedural Terminology coded associated with PAD and/or having at least 2 visits to a vascular surgeon within a 14‐month period. Individuals were defined as controls if they had no diagnosis/procedure codes suggesting a diagnosis of PAD. The diagnosis of PAD was validated against ankle brachial index measurement and manual chart review. The GWAS was adjusted for age, sex, and 5 principal components of ancestry.
The 2021 GoLEAD‐SUMMIT GWAS by van Zuydam et al, 22 combined 11 independent GWASs of individuals of European ancestry (n‐cases: 12086, n‐controls: 499548). The majority of PAD cases (n=7172) were identified using clinical parameters (eg, ankle‐brachial index, a clinical diagnosis, procedures specific to PAD and treatment for claudication). A comparatively smaller subset of the PAD cases was identified based on a mixture of self‐reported PAD in patients with clinical evidence of vascular disease and hospital admission codes related to PAD (n=4914). Summary statistics across the 11 GWASs were combined in a fixed‐effects meta‐analysis, under an inverse‐variance weighting scheme. Detailed information on the combined and individual GWASs can be found elsewhere. 22
Main Analysis
For the main analysis a 2‐sample MR analysis was performed in each cohort separately, using the inverse variance weighted (IVW) method which combines the Wald estimates for each SNP (SNP outcome estimates/SNP exposure estimate, where the intercept is constrained to zero) into 1 causal estimate of each risk factor. Afterwards, the SNPs were combined using an IVW multiplicative random‐effect meta‐analysis to obtain 1 causal estimate for each risk factor. The IVW method provides an unbiased estimate in the absence of horizontal pleiotropy or when horizontal pleiotropy is uncorrelated with SNP‐confounder associations and is balanced. 15
Validation of the MR Assumptions
We assessed adherence to the first MR assumption, meaning that genetic subgroups defined by the variant have different average levels of exposure, or in other words; there is a systematic difference between the different group, by only including SNPs that were at the genome‐wide significance level (P<5×10−8). The strength of the IVs was evaluated by the proportion of variance explained (Table), which was reported in the original papers or calculated using the formula 2×minor allele frequency×(1–minor allele frequency)×(β estimate in SD units). 2 Furthermore the F statistic was calculated as a measurement for instrument strength, 37 a strength of F>10 was considered sufficient 38 (Table). Lastly, post hoc‐power calculations were performed for the main IVW analyses using an online power calculation tool (https://sb452.shinyapps.io/power/) (Table S1). 39
The second MR assumption ensures that all other variables are distributed equally between subgroups and the third assumption ensures that the only causal pathway from the genetic variant to the outcome is via the exposure. 15 By adhering to these assumptions the genetic variant is not directly associated with the outcome, nor is there any alternative pathway by which the variant is associated with the outcome other than through the exposure. Pleiotropy might be present if these assumptions are not met. The presence of pleiotropy was assessed for each cohort separately by plotting the beta coefficients and corresponding standard errors of the SNP‐exposure association against the beta coefficients and standard errors of the SNP‐outcome association to get a first impression about the presence of unbalanced pleiotropy and the direction of the causal effects. 40 Furthermore, a number of robust sensitivity analyses were performed, in which the causal estimates from the various methods were compared with test for potential directional pleiotropy. First, we performed complementary analyses using the weighted median and MR‐Egger regression methods which are relatively robust under different assumptions about pleiotropy, although at the cost of statistical power. 41 , 42 Deviation of the MR‐Egger intercept from zero was used as another tool to assess pleiotropy and the Q statistics were used to assess heterogeneity. Second, the MR Pleiotropy Residual Sum and Outlier (MR‐PRESSO) test was used to identify outlying SNPs reflecting likely pleotropic biases, in which we checked whether exclusion of the outlying SNP changed the causal estimate. 43 Third, the contamination mixture method, which is a robust method if the largest group of SNPs estimating the same quantity is the group of valid instruments, was performed. 44
For the IVs MVPA, sedentary behavior and long sleep duration, only a limited number of SNPs was available that met the threshold of genome‐wide significance. For these exposures the IVW analyses were repeated using a more liberal threshold (P<5x10−6) for selecting SNPs. Furthermore, possible pleiotropic associations between the instrumental SNPs and potential confounders for the lifestyle factors with <20 independent SNPs were identified using Phenoscanner Database Version 2 and were excluded from the analysis in a conservative sensitivity analysis.
Lastly, a number of multivariable MR analyses were performed: The genetic instruments for LDL‐C, HDL‐C, triglycerides, and the lipid subfractions (apo A1 and apoB) are partly overlapping. 45 Therefore, we performed multivariable MR as a sensitivity analysis to obtain causal estimates adjusted for these genetic correlations. For LDL‐C, HDL‐C, and triglycerides summary data from a GWAS by Willer et al were used, 29 for the lipid subfractions summary data from a GWAS by Sinnot‐Armstrong et al were used. 46 Furthermore, the relationship between genetically predicted education level, insomnia, and type 2 diabetes is likely to be mediated by other cardiometabolic and lifestyle‐related risk factors. To assess potential mediation by these risk factors, the analyses were adjusted for the genetic liability to hyperlipidemia, 29 hypertension, 47 type 2 diabetes, 27 smoking initiation, 30 and body mass index 48 by adding these risk factors to the analyses.
All analyses were conducted using RStudio version 4.0.3, with the R packages MR 49 and MRPRESSO. 45 Results were reported as odds ratios (OR) with corresponding 95% CIs. To account for multiple testing in our principal analyses, we used a Bonferroni corrected significance level of P<0.00294 (0.05 divided by 17 risk factors). P values between 0.00294 and 0.05 were considered as potential causal associations.
RESULTS
We identified 2824 SNPs that could serve as IVs for the 17 selected exposures. A total of 239 SNPs in the exposure GWASs were in linkage disequilibrium or were not genome‐wide significant and were therefore excluded from the analyses; 103 SNPs were not available in the MVP outcome GWAS, and 22 SNPs were not available in the GoLEAD‐SUMMIT GWASs. Overall, 2482 and 2563 SNPs were used in the MVP GWAS and the GoLEAD‐SUMMIT GWAS analyses, respectively (Figure S2). Results of the IVW analyses in both cohorts are depicted in Figure 1. Figure 2 shows a graphic overview of the outcomes of the main and sensitivity analyses. A complete overview of results including sensitivity analyses, and parameters for the assessment of pleiotropy can be found in Table S2, S3 and Figure S3 through S6. A post hoc analysis of instrument strength can be found in Table S1.
Figure 1. The association between cardiovascular risk factors and lifestyle behaviors with peripheral artery disease using the inverse variance‐weighted Mendelian randomization method.
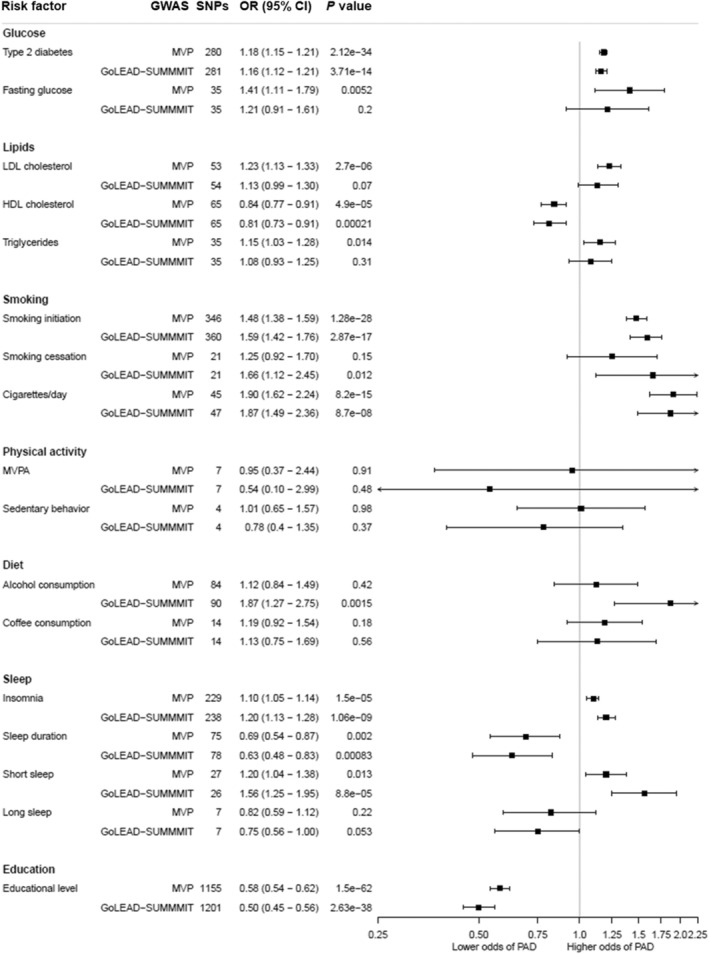
Odds ratios represent the associations of peripheral artery disease with listed risk factors. GoLEAD‐SUMMIT indicates genome‐wide association study, executed by the GoLEAD‐SUMMIT consortium; GWAS, Genome wide association study; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; MVP, Genome wide association study on peripheral artery disease, executed by the Million veteran program; MVPA, moderate‐to‐vigorous physical activity; OR, odds ratio; PAD, peripheral artery disease; and SNP, single nucleotide polymorphism.
Figure 2. Overview of the associations of cardiovascular risk factors and lifestyle behaviors with peripheral artery disease.

All results can be found in Figure 1 and Table S2 and S3. *Bonferroni corrected significance level of P<0.00294 (0.05 divided by 17 risk factors). IWV indicates Inverse variance‐weighted method; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; MVPA, moderate‐to‐vigorous physical activity; MR‐PRESSO, Mendelian Randomization Pleiotropy Residual Sum and Outlier; NA, not applicable; and PAD, peripheral artery disease.
Diabetes
Genetic liability to having type 2 diabetes was strongly associated with a higher odds of PAD in both cohorts (MVP: OR, 1.18 [CI, 1.15–1.21]; GoLead‐SUMMIT: OR, 1.16 [CI, 1.12–1.21]). The observed effect remained significant in all sensitivity analyses, with exception of the MR‐Egger analysis in the GoLEAD‐SUMMIT cohort, and the intercept of the MR‐Egger analyses in both cohorts which significantly deviated from 0 (0.004; P value: 0.012 [MVP], 0.01; P: 3.37E‐4 [GoLEAD‐SUMMIT]). Further exploration of pleiotropy by excluding outlying SNPs in the MR‐PRESSO analysis did not change the estimates (distortion test P value: 0.93). Overall, the robustness of the results in nearly all sensitivity analyses suggest little to no interference by pleiotropy. Furthermore, adjusting for the genetic correlation between possible mediators (genetic liability to smoking initiation, body mass index, LDL‐C, HDL‐C, triglycerides, systolic and diastolic blood pressure) via multivariable MR analyses did not significantly change the association between type 2 diabetes and PAD (Figure S4).
A complementary analysis investigating the genetic liability to having a higher fasting glucose (increase of 1‐mmol L−1) showed a suggestive causal association with higher odds of PAD (OR, 1.41 [CI, 1.11–1.79]) in the MVP cohort, but lacked statistical power when corrected for multiple testing. A similar, albeit nonsignificant, trend was seen in the IVW results from the GoLEAD SUMMIT cohort (OR, 1.21 [CI, 0.91–1.61]). The suspicion of pleotropic inference was strengthened by the results of the MR‐PRESSO analysis, in which the estimates changed significantly after exclusion of outliers (distortion test P value: 0.05), and the corrected estimate did not reach statistical significance when corrected for multiple testing (OR, 1.26 [CI, 1.05–1.51], P value: 0.02). Given the results of the main and sensitivity analyses in both cohorts, pleiotropic inference cannot be ruled out.
Lipid Metabolism
Genetic liability to having higher LDL‐C (1‐SD increase in LDL‐C) was associated with higher odds of PAD in the IVW (OR, 1.23 [CI, 1.13–1.33]) and sensitivity analyses of the MVP cohort. However, even though a similar trend was observed, the IVW failed to reach statistical significance in the GoLEAD‐SUMMIT cohort (OR, 1.13 [CI, 0.99–1.30]) and most sensitivity analyses except for the contamination mixture analysis (OR, 1.18 [CI, 1.08–1.29]). Furthermore, adjusting for the genetic correlation between the different lipids and lipid subfractions via multivariable MR analyses showed full attenuation of the effect, when adjusted for apoB (Figure S5) in both cohorts.
An association was found for the genetic liability to having a higher HDL‐C (1‐SD increase in HDL‐C) with a lower odds of PAD in both cohorts (OR, 0.84 [CI, 0.77–0.91] MVP; OR, 0.81 [CI, 0.73–0.91] GoLEAD‐SUMMIT), and a suggestive causal association was found for the genetic liability to having higher triglyceride levels (1‐SD increase in triglycerides) with higher odds of PAD (OR, 1.15 [CI, 1.03–1.28]) in the MVP cohort, but no associations were found in the GoLEAD‐SUMMIT cohort (OR, 1.08 [CI, 0.93–1.25]). For both HDL‐C and triglycerides, the observed effects could not be confirmed in all sensitivity analyses, therefore pleiotropic inference cannot be ruled out. Furthermore, adjusting for the genetic correlation between the different lipids and lipid subfractions via multivariable MR analyses led to a partial attenuation of the association between HDL‐C and PAD after correcting for LDL‐C, triglycerides, and apoB, and a full attenuation of the association between triglycerides and PAD after correction for LDL‐C, HDL‐C, and apoB (Figure S6).
Smoking
Genetic predisposition to smoking initiation (ever smoked regularly versus never smoked) and to smoking an increased number of cigarettes per day (1‐SD increase) were associated with higher odds of PAD in both cohorts (OR, 1.48 [CI, 1.38–1.59] and OR 1.90 [CI, 1.62–2.24], respectively in the MVP cohort and OR, 1.59 [CI, 1.42–1.76] and OR, 1.87 [CI, 1.49–2.36], respectively in the GoLEAD‐SUMMIT cohort). These results were robust to sensitivity analyses, suggesting the absence of directional pleiotropy.
The genetic predisposition to smoking cessation (former versus current smoker) was found to be associated with higher odds of PAD in the GoLEAD‐SUMMIT GWAS IVW analysis (OR, 1.66 [CI, 1.12–2.45]), and showed a stable trend in sensitivity analyses, however results but not meet the Bonferroni‐corrected significance level. Null results were reported for the genetic liability to smoking cessation in the MVP cohort, even though a trend towards a harmful association is visible (OR, 1.25 [CI, 0.92–1.70]). Results are likely to be influenced by pleiotropy, as well as weak instrument (explained variance 0.1%), and insufficient power (3.1% and 9.9% in the MVP and GoLEAD‐SUMMIT cohorts, respectively, Table S1).
Education
Genetically predicted higher education status (1‐SD increase in years of education attainment) was associated with lower odds of PAD in both cohorts (MVP: OR, 0.58 [CI, 0.54–0.62]; GoLEAD‐SUMMIT: OR, 0.50 [CI, 0.45–0.56]). The observed effect remained significant in all sensitivity analyses. Overall making a compelling case for a causal relationship with minimal to no interference of directional pleiotropy.
Adjusting for the genetic correlation between possible mediating factors (genetic liability to smoking initiation, body mass index, type 2 diabetes, LDL‐C, HDL‐C, triglycerides, and systolic and diastolic blood pressure) via multivariable MR analyses did not change the association between education diabetes and PAD. Nevertheless, effects sizes did slightly attenuate after correcting for the genetic liability to smoking initiation, body mass index, and lipid fractions (Figure S6).
Diet
In the GoLEAD‐SUMMIT cohort, genetically predicted alcohol consumption (1‐SD increase in log‐transformed alcohol drinks/week) was associated with higher odds of PAD (OR, 1.87 [CI, 1.27–2.75]). Effect sizes remained significant in the weighted median and MR‐Egger analyses, though the MR egger intercept significantly deviated from 0 (−0.01, P: 0.02). No effects were found for genetically predicted alcohol consumption and PAD in the MVP cohort (OR, 1.12 [CI, 0.84–1.49]), but these analyses were significantly underpowered (0.4% for the MVP cohort compared with 50.5% for the GoLEAD‐SUMMIT cohort, Table S1).
Genetically predicted coffee consumption (50% increase in coffee consumption) showed no association with PAD in both cohorts (MVP: OR, 1.19 [CI, 0.92–1.54]; GoLEAD‐SUMMIT: OR, 1.13 [CI, 0.75–1.69]). Exclusion of outlying SNPs, identified by MR‐PRESSO analysis, did not alter the results significantly. Furthermore, exclusion of potentially pleiotropic SNPs correlated with body mass index and LDL‐C (rs574367, rs66723169, rs1260326, rs10865548, rs34060476) did not alter the results for coffee consumption (Figure S3).
Physical Activity
Genetically predicted MVPA (1‐SD increment in metabolic equivalent of task min/week) and genetic liability to sedentary behavior (1 SD increase) were not associated with PAD in both cohorts (MVP: OR, 0.95 [CI, 0.37–2.44], OR, 1.01 [CI, 0.65–1.57], respectively; and GoLEAD‐SUMMIT: OR, 0.54 [CI, 0.10–2.99], OR, 0.78 [CI, 0.4–1.35], respectively) in the main and sensitivity analyses. Excluding outlying SNPs via MR‐PRESSO analysis did not change the outcomes. Exclusion of rs2035562 for MVPA and rs25981 for sedentary behavior, both correlated with coronary artery disease, higher cholesterol, and diabetes, and additional analyses using a more liberal cut‐off to select instrumental SNPs (P<5x10−6) did not alter the results (Figure S3). However, even though instrument strength based on the F‐statistic appears sufficient, the low variance explained for both IVs (0.073% and 0.08%, respectively), and lack of power (0.6–11.2% for MVPA and 1.2–1.3% for sedentary behavior), should be considered when interpreting the results (Table S1).
Sleep
Genetically predicted insomnia was associated with higher odds of PAD in both cohorts (MVP: OR, 1.10 [CI, 1.05–1.14]; GoLEAD‐SUMMIT: OR, 1.20 [CI, 1.13–1.28]). Consistent estimates were obtained by the weighted median, contamination mixture and MR‐PRESSO analyses. However, the MR‐Egger showed attenuation of the effect sizes (MVP: OR, 0.99 [CI, 0.84–1.17]; GoLEAD‐SUMMIT: OR, 1.28 [CI, 1.02–1.63]). Overall the robustness of the results in nearly all sensitivity analyses suggested little interference by pleiotropy. Similarly, genetically predicted sleep duration (increase of 1 hour sleep per day) was associated with lower odds of PAD (MVP: OR, 0.69 [CI, 0.54–0.87]; GoLEAD‐SUMMIT: OR, 0.63 [CI, 0.48–0.83]), and persisted in most sensitivity analyses, although with insufficient statistical strength when corrected for multiple testing. Exclusion of outlying SNPs identified by MR‐PRESSO analysis did not alter the results.
Adjusting for the genetic correlation between genetic liability to smoking initiation, higher body mass index, type 2 diabetes, LDL‐C, HDL‐C, systolic and diastolic blood pressure via multivariable MR analyses showed that effect sizes significantly attenuated when corrected for body mass index, type 2 diabetes, and lipid fractions in both cohorts. No attenuation was observed after correcting for smoking initiation and markers of blood pressure (Figure S4).
Complementary analyses revealed a harmful effect of the genetic liability to short sleep duration (<7 h/d) on PAD (MVP: OR, 1.20 [CI, 1.04–1.38]; GoLEAD‐SUMMIT: OR, 1.56 [CI, 1.25–1.95]). This trend persisted in most sensitivity analyses. However, the Q‐statistic indicated heterogeneity. Furthermore, a nonsignificant trend towards beneficial effects of genetic liability to long sleep duration (≥9 h/d) (MVP: OR, 0.82 [CI, 0.59–1.12]; GoLEAD‐SUMMIT: OR, 0.75 [CI, 0.56–1.00]) was observed. Nevertheless, the MR Egger intercept significantly deviated from 0 in the MVP cohort. After exclusion of outlying SNPs by MR‐PRESSO analysis, the estimates did not change significantly. Exclusion of potentially pleiotropic SNPs (rs17817288 and rs17688916) both associated with body mass index attenuated the protective trend of long sleep duration on PAD to a null association (Figure S3). Nevertheless, additional analyses with a more liberal cut‐off (5x10−6) emphasized the protective trend of the genetic liability to long sleep duration (MVP: OR, 0.81 [CI, 0.64–1.03]; GoLEAD‐SUMMIT: OR, 0.80 [CI, 0.62–0.98]) (Figure S3) but did not meet statistical significance.
DISCUSSION
In this MR study, we found robust evidence (present in both cohorts, and consistent over most sensitivity analyses) for a causal relationship between genetically predicted type 2 diabetes, smoking initiation, smoking heaviness, insomnia and lower education status with increased risk of PAD. However effects found for insomnia significantly attenuated after correcting for potential mediating risk factors, and a similar but non‐significant effect was observed for education level. Furthermore, we found strong evidence (eg, consistent over most sensitivity analyses) for a causal relationship between genetically predicted increased sleep duration and higher levels of HDL‐C with a decreased risk of PAD. Finally, suggestive evidence of causality was found for genetically predicted increased fasting glucose, higher alcohol consumption, smoking cessation and short sleep duration on PAD, but statistical strength was lacking when corrected for multiple testing and/or effects were only found in one of the 2 investigated outcome cohorts. Even though in the main analyses a harmful trend on PAD was observed for the genetic liability to increased LDL‐C and triglycerides, these effects fully attenuated after correcting for apoB. No evidence was found for a causal relationship between MVPA, sedentary behavior, long sleep duration and coffee consumption and PAD.
Classic Cardiovascular Risk Factors
The harmful effects of smoking initiation, smoking heaviness, and type 2 diabetes on PAD found in our study align with results found in observational studies 2 and previously executed MR studies. 17 , 20 , 25 , 50 Furthermore, for type 2 diabetes these results were corroborated in a short term randomized controlled trial in which positive effects on PAD were observed after glucose lowering therapies. 8 Additionally, we found suggestive evidence for a causal relationship between the genetic liability to having higher fasting glucose levels and PAD, which to our knowledge, has not yet been investigated via an MR design, even though a certain influence of pleiotropy cannot be excluded based on our analyses. Combined, the evidence of multiple studies using different analyses techniques, together with a strong pathophysiological rationale, a compelling case for the presence of a causal relationship between these classic cardiovascular risk factors and PAD can be made. In our analysis on the GoLEAD‐SUMMIT cohort, genetic liability for smoking cessation was found to be detrimental for the development of PAD, no effects were observed in the MVP cohort. In observational research a lower risk was found for former smoking compared with current smoking. 2 The discrepancy between our findings and findings in observational studies could be due to the genetic instrument being defined as current versus former smoker, therefore a correlation with smoking initiation can be expected.
In observational studies and short‐term randomized controlled trials, hypercholesterolemia has been associated with higher risk of PAD. 2 , 5 , 6 , 7 However, we found the effects of HDL‐C on PAD was partially attenuated, and the association between triglycerides and PAD was fully attenuated when corrected for LDL‐C. Furthermore the effects of LDL‐C and triglycerides fully attenuated after correction for apoB, insinuating that in accordance with findings of comparable MR studies 23 , 51 apoB, rather than LDL‐C or triglycerides is likely to be the causal subfraction in PAD.
Education Status
In observational studies lower education level has been linked to PAD 13 , 52 and the results from our MR analysis fully corroborate this association. The strength and robustness of the observed association and the consistency with observational studies suggest a causal relationship. Nevertheless, the results from the multivariate MR analysis showed non‐significant attenuation after correcting for the most common mediators suggesting that education level is a upstream determinant of health; a higher education level likely leads to healthier life choices, thereby lowering the chance of getting exposed to other risk.
Sleep Status
Despite the sparse observational epidemiological evidence of impaired sleeping on PAD, a number of studies support the hypothesis that increased levels of inflammation markers facilitate the process of atherosclerosis in individuals suffering from lack of sleep. 53 , 54 We observed a robust association between genetically predicted insomnia, an inverse association between genetically predicted increased sleep duration and a suggestive association between short sleep duration and PAD. For insomnia, our results corroborate the results of a recently published MR study. 16 The larger sample size of our study, and the addition of other not prior investigated IVs that represent sleep duration (eg, increased sleep duration, short and long sleep duration), which all show detrimental effects of increased sleep duration on PAD, further strengthen the causality claim. However, since the GWASs used in our, and previously executed MR studies, are based on self‐reported sleep data, bias cannot be completely eliminated. Furthermore, the results from our multivariate MR study indicate that insomnia functions as a precursor of other risk factors, and should rather be considered an upstream determinant of health.
Alcohol consumption
Analyses carried out in the GoLEAD‐SUMMIT cohort reported a harmful effect of alcohol consumption on PAD, which is in accordance with 2 other recently published MR studies. 17 , 18 Such associations were not found in the MVP cohort; however, the MVP analysis was significantly underpowered (Table S1) and overall instrument strength should be considered weak (F: 5.8). When interpreting these results, it should be taken into account that based on observational research a “J” shaped association curve is expected, indicating a reduced risk for PAD incidence for within‐guideline drinkers compared with either abstainers or excessive drinkers. 9 , 55 Neither our nor the previously published MR studies were designed to assess non‐linear relationships which could have resulted in a distortion of results.
Physical activity and coffee consumption
No effects on PAD were found for physical activity and coffee consumption. However, when looking at evidence from prior research there seems to be a strong rationale for a beneficial effect of physical activity on PAD, since the majority of observational studies and a number of small trials reporting associations between higher physical activity and risk of the development of PAD. 56 , 57 , 58 , 59 , 60 For coffee consumption, prior evidence is inconsistent, but just as for alcohol consumption, a “J” shaped association is expected. 9 , 61 Overall, it is important to notice that the analyses for physical activity, alcohol‐ and coffee consumption in our study are all limited by a low variance explained for the used IVs (Table) and a lack of power (Table S1), leading to weak instrument bias by violation of the first assumption of MR studies. Until stronger genetic instruments are identified by GWAS studies, MR studies are most likely not to be able to draw conclusions on causality.
Clinical Implications
With the expected continuation of the global aging process, a considerable increase in the prevalence of PAD is expected. In order to alleviate the societal and economic burden of PAD, efforts should be made to improve prevention, early diagnosis and treatment of PAD. In the western world smoking, hypertension, type 2 diabetes and hypercholesterolemia are known to be the most abundant risk factors in the development of PAD, and preventive strategies to reduce those risk factors have been implemented. However, globally these risk factors are projected to increase substantially the next 10 years, 62 , 63 therefore worldwide awareness of these risk factors, especially in low income countries, is needed. 2 Our study confirmed and strengthened the causality claim on the harmful relationship between the classic cardiovascular risk factors; smoking, type 2 diabetes and hypercholesterolemia and PAD, thereby reinforcing the importance of optimizing cardiovascular risk factor control. However, our results and the results from recently published MRs indicate that lipid subfractions such as apoB are likely to be the harmful components in the development of PAD, even though more in‐depth research is needed to confirm this association. Furthermore, with the improved prevention and management of classic risk factors the importance of discovering lifestyle related risk factors in the development of PAD is pressing. In this study we identified lower education level and insomnia as other, upstream, risk factors in the development of PAD. By adapting policies that target upstream determinants of health, multiple downstream risk factors can be affected simultaneously, thereby significantly lowering the risk of PAD. We believe these findings could contribute to a more targeted prevention strategy for PAD, both on national level as well as in a daily clinical setting by providing a better understanding of risk factors in the development of PAD.
Strengths and Limitations
This study has several strengths. First, the use of an MR design, which is for a large part independent of forms of bias that are inherent to observational research, enabled us to make causal claims provided all MR assumptions are met. To satisfy these assumptions we incorporated several parameters in our study. In short, by using only SNPs that are associated with the risk factor at genome‐wide significance level in our main analysis, we avoided weak instrument bias and by adding a vast amount of sensitivity analyses we diminished the likelihood of horizontal pleiotropy. Second, the 2 GWASs included in this study used data from large genome‐wide studies, thereby strengthening statistical power. Third, sample overlap (eg, overlap of study sample between IV and outcome GWAS) and population stratification (eg, the IVs and the outcome GWAS not consisting of the same population) can bias the results of MR studies. In this current study, we used 2 GWASs of which the MVP had no sample overlap between IVs and outcomes, thereby keeping the type 1 error rate as low as possible and the GoLEAD‐SUMMIT consisting of participants of European‐ancestry only thereby keeping the presence of population stratification as low as possible. By comparing associations found in both GWASs and by executing a vast amount of sensitivity analyses, we aimed to keep the impact of both forms of bias as low as possible.
However, a number of limitations to our study should be considered. First, PAD outcomes were studied among primarily male participants of the Veterans Affairs MVP. Second, for smoking cessation, MVPA and sedentary behavior, we were not able to draw conclusions on causality because of limited precision as a result of a small explained variance. Third, post‐hoc power analyses showed the analyses for smoking cessation, alcohol, and coffee consumption, MVPA and sedentary behavior are significantly underpowered, potentially distorting results. Furthermore, during the selection of SNPs for the IVs, we used a relatively liberal cut‐off of r2>0.1 for linkage disequilibrium, potentially introducing bias in our instruments. Finally, our analyses did not enable to investigate potential non‐linear relationships even though several lifestyle variables such as alcohol and coffee consumption are suspected to have a nonlinear relationship with cardiovascular disease.
CONCLUSIONS
Our study strengthened the causality claim on the harmful relationship between smoking, type 2 diabetes and PAD. Furthermore, our findings further strengthened the suspicion that apoB, rather that LDL‐C and triglycerides, functions as the harmful component causing PAD in hyperlipidemia. Lower education level and insomnia are identified as upstream risk factors in the development of PAD, and suggestive harmful associations for alcohol consumption and impaired sleeping and PAD were found. No associations between physical activity and coffee consumption and PAD were observed, but these analyses were underpowered, limited by weak instrument bias and the inability to investigate non‐linear relationships. The results of our study contribute to a more targeted prevention strategy for PAD, by providing a better understanding of causal risk factors in the development of PAD.
Sources of Funding
J.W.J. Beulens is supported by a ZorgOnderzoek Nederland (ZON) en het gebied Medische Wetenschappen (NW), Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) Vidi grant (91 718 304).
Disclosures
None.
Supporting information
Tables S1–S3
Figures S1–S6
Acknowledgments
We gratefully acknowledge the authors and participants of all GWAS from which we used summary statistics data. The authors especially thank the Million Veteran Program (MVP) staff, researchers, and volunteers, who have contributed to MVP, and participants who previously served their country in the military and now generously agreed to enroll in the study. (See https://www.research.va.gov/mvp/ for more details). This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by the Veterans Administration (VA) Cooperative Studies Program (CSP) award #G002.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.025644
For Sources of Funding and Disclosures, see page 12 and 13.
References
- 1. Criqui MH, Aboyans V. Epidemiology of peripheral artery disease. Circ Res. 2015;116:1509–1526. doi: 10.1161/CIRCRESAHA.116.303849 [DOI] [PubMed] [Google Scholar]
- 2. Song P, Rudan D, Zhu Y, Fowkes FJI, Rahimi K, Fowkes FGR, Rudan I. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: an updated systematic review and analysis. Lancet Glob Health. 2019;7:e1020–e1030. doi: 10.1016/S2214-109X(19)30255-4 [DOI] [PubMed] [Google Scholar]
- 3. Ankle Brachial Index C, Fowkes FG, Murray GD, Butcher I, Heald CL, Lee RJ, Chambless LE, Folsom AR, Hirsch AT, Dramaix M, et al. Ankle brachial index combined with Framingham risk score to predict cardiovascular events and mortality: a meta‐analysis. JAMA. 2008;300:197–208. doi: 10.1001/jama.300.2.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gerhard‐Herman MD, Gornik HL, Barrett C, Barshes NR, Corriere MA, Drachman DE, Fleisher LA, Fowkes FG, Hamburg NM, Kinlay S, et al. 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2017;135:e726–e779. doi: 10.1161/CIR.0000000000000471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bonaca MP, Nault P, Giugliano RP, Keech AC, Pineda AL, Kanevsky E, Kuder J, Murphy SA, Jukema JW, Lewis BS, et al. Low‐density lipoprotein cholesterol lowering with evolocumab and outcomes in patients with peripheral artery disease: insights from the fourier trial (further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk). Circulation. 2018;137:338–350. doi: 10.1161/CIRCULATIONAHA.117.032235 [DOI] [PubMed] [Google Scholar]
- 6. Heart Protection Study Collaborative G . Randomized trial of the effects of cholesterol‐lowering with simvastatin on peripheral vascular and other major vascular outcomes in 20,536 people with peripheral arterial disease and other high‐risk conditions. J Vasc Surg. 2007;45:645–654; discussion 653‐644 [DOI] [PubMed] [Google Scholar]
- 7. Pedersen TR, Kjekshus J, Pyorala K, Olsson AG, Cook TJ, Musliner TA, Tobert JA, Haghfelt T. Effect of simvastatin on ischemic signs and symptoms in the scandinavian simvastatin survival study (4s). Am J Cardiol. 1998;81:333–335. doi: 10.1016/S0002-9149(97)00904-1 [DOI] [PubMed] [Google Scholar]
- 8. Sardar P, Udell JA, Chatterjee S, Bansilal S, Mukherjee D, Farkouh ME. Effect of intensive versus standard blood glucose control in patients with type 2 diabetes mellitus in different regions of the world: systematic review and meta‐analysis of randomized controlled trials. J Am Heart Assoc. 2015;4:e001577. doi: 10.1161/JAHA.114.001577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ogilvie RP, Lutsey PL, Heiss G, Folsom AR, Steffen LM. Dietary intake and peripheral arterial disease incidence in middle‐aged adults: the atherosclerosis risk in communities (ARIC) study. Am J Clin Nutr. 2017;105:651–659. doi: 10.3945/ajcn.116.137497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heikkila K, Coughlin PA, Pentti J, Kivimaki M, Halonen JI. Physical activity and peripheral artery disease: two prospective cohort studies and a systematic review. Atherosclerosis. 2019;286:114–120. doi: 10.1016/j.atherosclerosis.2019.05.008 [DOI] [PubMed] [Google Scholar]
- 11. Aziz M, Ali SS, Das S, Younus A, Malik R, Latif MA, Humayun C, Anugula D, Abbas G, Salami J, et al. Association of subjective and objective sleep duration as well as sleep quality with non‐invasive markers of sub‐clinical cardiovascular disease (CVD): a systematic review. J Atheroscler Thromb. 2017;24:208–226. doi: 10.5551/jat.36194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nagayoshi M, Lutsey PL, Benkeser D, Wassel CL, Folsom AR, Shahar E, Iso H, Allison MA, Criqui MH, Redline S. Association of sleep apnea and sleep duration with peripheral artery disease: the Multi‐Ethnic Study Of Atherosclerosis (MESA). Atherosclerosis. 2016;251:467–475. doi: 10.1016/j.atherosclerosis.2016.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pande RL, Creager MA. Socioeconomic inequality and peripheral artery disease prevalence in us adults. Circ Cardiovasc Qual Outcomes. 2014;7:532–539. doi: 10.1161/CIRCOUTCOMES.113.000618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grimes DA, Schulz KF. Bias and causal associations in observational research. Lancet. 2002;359:248–252. doi: 10.1016/S0140-6736(02)07451-2 [DOI] [PubMed] [Google Scholar]
- 15. Burgess S, Thompson SG. Mendelian randomization: methods for using genetic variants in causal estimation. Boca Raton, FL: Chapman and Hall/CRC; 2015. [Google Scholar]
- 16. Yuan S, Mason AM, Burgess S, Larsson SC. Genetic liability to insomnia in relation to cardiovascular diseases: a Mendelian randomisation study. Eur J Epidemiol. 2021;36:393–400. doi: 10.1007/s10654-021-00737-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rosoff DB, Davey Smith G, Mehta N, Clarke TK, Lohoff FW. Evaluating the relationship between alcohol consumption, tobacco use, and cardiovascular disease: a multivariable Mendelian randomization study. PLoS Med. 2020;17:e1003410. doi: 10.1371/journal.pmed.1003410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Larsson SC, Burgess S, Mason AM, Michaelsson K. Alcohol consumption and cardiovascular disease: a Mendelian randomization study. Circ Genom Precis Med. 2020;13:e002814. doi: 10.1161/CIRCGEN.119.002814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emanuelsson F, Nordestgaard BG, Tybjaerg‐Hansen A, Benn M. Impact of LDL cholesterol on microvascular versus macrovascular disease: a Mendelian randomization study. J Am Coll Cardiol. 2019;74:1465–1476. [DOI] [PubMed] [Google Scholar]
- 20. Liu B, Mason AM, Sun L, Di Angelantonio E, Gill D, Burgess S. Genetically predicted type 2 diabetes mellitus liability, glycated hemoglobin and cardiovascular diseases: a wide‐angled Mendelian randomization study. Genes (Basel). 2021;12:1644. doi: 10.3390/genes12101644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klarin D, Lynch J, Aragam K, Chaffin M, Assimes TL, Huang J, Lee KM, Shao Q, Huffman JE, Natarajan P, et al. Genome‐wide association study of peripheral artery disease in the million veteran program. Nat Med. 2019;25:1274–1279. doi: 10.1038/s41591-019-0492-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Zuydam NR, Stiby A, Abdalla M, Austin E, Dahlstrom EH, McLachlan S, Vlachopoulou E, Ahlqvist E, Di Liao C, Sandholm N, et al. Genome‐wide association study of peripheral artery disease. Circ Genom Precis Med. 2021;14:e002862. doi: 10.1161/CIRCGEN.119.002862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levin MG, Zuber V, Walker VM, Klarin D, Lynch J, Malik R, Aday AW, Bottolo L, Pradhan AD, Dichgans M, et al. Prioritizing the role of major lipoproteins and subfractions as risk factors for peripheral artery disease. Circulation. 2021;144:353–364. doi: 10.1161/CIRCULATIONAHA.121.053797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Levin MG, Klarin D, Walker VM, Gill D, Lynch J, Hellwege JN, Keaton JM, Lee KM, Assimes TL, Natarajan P, et al. Association between genetic variation in blood pressure and increased lifetime risk of peripheral artery disease. Arterioscler Thromb Vasc Biol. 2021;41:2027–2034. doi: 10.1161/ATVBAHA.120.315482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levin MG, Klarin D, Assimes TL, Freiberg MS, Ingelsson E, Lynch J, Natarajan P, O'Donnell C, Rader DJ, Tsao PS, et al. Genetics of smoking and risk of atherosclerotic cardiovascular diseases: a Mendelian randomization study. JAMA Netw Open. 2021;4:e2034461. doi: 10.1001/jamanetworkopen.2020.34461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gill D, Zuber V, Dawson J, Pearson‐Stuttard J, Carter AR, Sanderson E, Karhunen V, Levin MG, Wootton RE, Klarin D, et al. Risk factors mediating the effect of body mass index and waist‐to‐hip ratio on cardiovascular outcomes: Mendelian randomization analysis. Int J Obes (Lond). 2021;45:1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. Fine‐mapping type 2 diabetes loci to single‐variant resolution using high‐density imputation and islet‐specific epigenome maps. Nat Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, Luan J, Magi R, Strawbridge RJ, Rehnberg E, Gustafsson S, et al. Large‐scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005. doi: 10.1038/ng.2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, Datta G, Davila‐Velderrain J, McGuire D, Tian C, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–244. doi: 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhong VW, Kuang A, Danning RD, Kraft P, van Dam RM, Chasman DI, Cornelis MC. A genome‐wide association study of bitter and sweet beverage consumption. Hum Mol Genet. 2019;28:2449–2457. doi: 10.1093/hmg/ddz061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klimentidis YC, Raichlen DA, Bea J, Garcia DO, Wineinger NE, Mandarino LJ, Alexander GE, Chen Z, Going SB. Genome‐wide association study of habitual physical activity in over 377,000 UKbiobank participants identifies multiple variants including CADM2 and APOE. Int J Obes (Lond). 2018;42:1161–1176. doi: 10.1038/s41366-018-0120-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Doherty A, Smith‐Byrne K, Ferreira T, Holmes MV, Holmes C, Pulit SL, Lindgren CM. GWAS identifies 14 loci for device‐measured physical activity and sleep duration. Nat Commun. 2018;9:5257. doi: 10.1038/s41467-018-07743-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jansen PR, Watanabe K, Stringer S, Skene N, Bryois J, Hammerschlag AR, de Leeuw CA, Benjamins JS, Munoz‐Manchado AB, Nagel M, et al. Genome‐wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat Genet. 2019;51:394–403. doi: 10.1038/s41588-018-0333-3 [DOI] [PubMed] [Google Scholar]
- 35. Dashti HS, Jones SE, Wood AR, Lane JM, van Hees VT, Wang H, Rhodes JA, Song Y, Patel K, Anderson SG, et al. Genome‐wide association study identifies genetic loci for self‐reported habitual sleep duration supported by accelerometer‐derived estimates. Nat Commun. 2019;10:1100. doi: 10.1038/s41467-019-08917-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, Nguyen‐Viet TA, Bowers P, Sidorenko J, Karlsson Linner R, et al. Gene discovery and polygenic prediction from a genome‐wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50:1112–1121. doi: 10.1038/s41588-018-0147-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35:1880–1906. doi: 10.1002/sim.6835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–665. doi: 10.1002/gepi.21758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burgess S. Sample size and power calculations in mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol. 2014;43:922–929. doi: 10.1093/ije/dyu005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burgess S, Thompson SG. Bias in causal estimates from mendelian randomization studies with weak instruments. Stat Med. 2011;30:1312–1323. doi: 10.1002/sim.4197 [DOI] [PubMed] [Google Scholar]
- 41. Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, Hartwig FP, Holmes MV, Minelli C, Relton CL, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2019;4:186. doi: 10.12688/wellcomeopenres.15555.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28:30–42. doi: 10.1097/EDE.0000000000000559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burgess S, Foley CN, Allara E, Staley JR, Howson JMM. A robust and efficient method for mendelian randomization with hundreds of genetic variants. Nat Commun. 2020;11:376. doi: 10.1038/s41467-019-14156-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–698. doi: 10.1038/s41588-018-0099-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sinnott‐Armstrong N, Tanigawa Y, Amar D, Mars N, Benner C, Aguirre M, Venkataraman GR, Wainberg M, Ollila HM, Kiiskinen T, et al. Genetics of 35 blood and urine biomarkers in the UKbiobank. Nat Genet. 2021;53:185–194. doi: 10.1038/s41588-020-00757-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Evangelou E, Warren HR, Mosen‐Ansorena D, Mifsud B, Pazoki R, Gao H, Ntritsos G, Dimou N, Cabrera CP, Karaman I, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50:1412–1425. doi: 10.1038/s41588-018-0205-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM, et al. Meta‐analysis of genome‐wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet. 2018;27:3641–3649. doi: 10.1093/hmg/ddy271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yavorska OO, Burgess S. Mendelianrandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–1739. doi: 10.1093/ije/dyx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Larsson SC, Mason AM, Back M, Klarin D, Damrauer SM, Million Veteran P, Michaelsson K, Burgess S. Genetic predisposition to smoking in relation to 14 cardiovascular diseases. Eur Heart J. 2020;41:3304–3310. doi: 10.1093/eurheartj/ehaa193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Richardson TG, Sanderson E, Palmer TM, Ala‐Korpela M, Ference BA, Davey Smith G, Holmes MV. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med. 2020;17:e1003062. doi: 10.1371/journal.pmed.1003062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fowkes FG, Aboyans V, Fowkes FJ, McDermott MM, Sampson UK, Criqui MH. Peripheral artery disease: epidemiology and global perspectives. Nat Rev Cardiol. 2017;14:156–170. doi: 10.1038/nrcardio.2016.179 [DOI] [PubMed] [Google Scholar]
- 53. Miller MA, Kandala NB, Kumari M, Marmot MG, Cappuccio FP. Relationships between sleep duration and von willebrand factor, factor VII, and fibrinogen: Whitehall II study. Arterioscler Thromb Vasc Biol. 2010;30:2032–2038. doi: 10.1161/ATVBAHA.110.206987 [DOI] [PubMed] [Google Scholar]
- 54. Meier‐Ewert HK, Ridker PM, Rifai N, Regan MM, Price NJ, Dinges DF, Mullington JM. Effect of sleep loss on c‐reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol. 2004;43:678–683. doi: 10.1016/j.jacc.2003.07.050 [DOI] [PubMed] [Google Scholar]
- 55. Bell S, Daskalopoulou M, Rapsomaniki E, George J, Britton A, Bobak M, Casas JP, Dale CE, Denaxas S, Shah AD, et al. Association between clinically recorded alcohol consumption and initial presentation of 12 cardiovascular diseases: population based cohort study using linked health records. BMJ. 2017;356:j909. doi: 10.1136/bmj.j909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stein RA, Rockman CB, Guo Y, Adelman MA, Riles T, Hiatt WR, Berger JS. Association between physical activity and peripheral artery disease and carotid artery stenosis in a self‐referred population of 3 million adults. Arterioscler Thromb Vasc Biol. 2015;35:206–212. doi: 10.1161/ATVBAHA.114.304161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hamer M, O'Donovan G, Stamatakis E. Association between physical activity and sub‐types of cardiovascular disease death causes in a general population cohort. Eur J Epidemiol. 2019;34:483–487. doi: 10.1007/s10654-018-0460-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parsons TJ, Sartini C, Ellins EA, Halcox JP, Smith KE, Ash S, Lennon LT, Wannamethee SG, Lee IM, Whincup PH, et al. Objectively measured physical activity and sedentary behaviour and ankle brachial index: cross‐sectional and longitudinal associations in older men. Atherosclerosis. 2016;247:28–34. doi: 10.1016/j.atherosclerosis.2016.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nardi Gomes TJ, Martins de Albuquerque I, de Moraes CP, Cardoso DM, de Moraes CG, da Costa Vieira JL. Association between the ankle‐brachial index, intermittent claudication, and physical activity level: what is the influence on the functional capacity of patients with or at high risk of cardiovascular disease? Int J Gen Med. 2015;8:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Housley E, Leng GC, Donnan PT, Fowkes FG. Physical activity and risk of peripheral arterial disease in the general population: Edinburgh Artery Study. J Epidemiol Community Health. 1993;47:475–480. doi: 10.1136/jech.47.6.475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ding M, Bhupathiraju SN, Satija A, van Dam RM, Hu FB. Long‐term coffee consumption and risk of cardiovascular disease: a systematic review and a dose‐response meta‐analysis of prospective cohort studies. Circulation. 2014;129:643–659. doi: 10.1161/CIRCULATIONAHA.113.005925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mendez D, Alshanqeety O, Warner KE. The potential impact of smoking control policies on future global smoking trends. Tob Control. 2013;22:46–51. doi: 10.1136/tobaccocontrol-2011-050147 [DOI] [PubMed] [Google Scholar]
- 63. Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figures S1–S6
Data Availability Statement
All GWAS exposure data used in this MR study, and data from the GoLEAD‐SUMMIT consortium GWAS are publicly available. Information on how to access these data can be found in the below cited GWAS articles. Million Veteran Program PAD GWAS summary statistics are available on dbGAP (Accession phs001672.v6.p1). All used GWASs were approved by their respective institutional review committees, and all participants provided written informed consent. Information on institutional review board approval can be found in the below cited articles.