

Abstract
Background
Arteriovenous fistula (AVF) maturation failure is a main limitation of vascular access. Maturation is determined by the intricate balance between outward remodeling and intimal hyperplasia, whereby endothelial cell dysfunction, platelet aggregation, and vascular smooth muscle cell (VSMC) proliferation play a crucial role. von Willebrand Factor (vWF) is an endothelial cell–derived protein involved in platelet aggregation and VSMC proliferation. We investigated AVF vascular remodeling in vWF‐deficient mice and vWF expression in failed and matured human AVFs.
Methods and Results
Jugular‐carotid AVFs were created in wild‐type and vWF−/− mice. AVF flow was determined longitudinally using ultrasonography, whereupon AVFs were harvested 14 days after surgery. VSMCs were isolated from vena cavae to study the effect of vWF on VSMC proliferation. Patient‐matched samples of the basilic vein were obtained before brachio‐basilic AVF construction and during superficialization or salvage procedure 6 weeks after AVF creation. vWF deficiency reduced VSMC proliferation and macrophage infiltration in the intimal hyperplasia. vWF−/− mice showed reduced outward remodeling (1.5‐fold, P=0.002) and intimal hyperplasia (10.2‐fold, P<0.0001). AVF flow in wild‐type mice was incremental over 2 weeks, whereas flow in vWF−/− mice did not increase, resulting in a two‐fold lower flow at 14 days compared with wild‐type mice (P=0.016). Outward remodeling in matured patient AVFs coincided with increased local vWF expression in the media of the venous outflow tract. Absence of vWF in the intimal layer correlated with an increase in the intima‐media ratio.
Conclusions
vWF enhances AVF maturation because its positive effect on outward remodeling outweighs its stimulating effect on intimal hyperplasia.
Keywords: AVF maturation, intimal hyperplasia, outward remodeling, von Willebrand Factor, VSMC
Subject Categories: Smooth Muscle Proliferation and Differentiation, Vascular Biology, Ultrasound, Remodeling
Nonstandard Abbreviations and Acronyms
- αSMA
α‐smooth muscle actin
- AVF
arteriovenous fistula
- CCA
common carotid artery
- EC
endothelial cell
- IEL
internal elastic lamina
- IH
intimal hyperplasia
- I/M ratio
intima/media ratio
- OR
outward remodeling
- VSMC
vascular smooth muscle cell
- vWF
von Willebrand Factor
Clinical Perspective.
What Is New?
The von Willebrand Factor is an important mediator of the vascular response in arteriovenous fistulas (AVFs) in the early phase after AVF surgery.
von Willebrand Factor deficiency results in reduced vascular smooth muscle cell proliferation and a decrease in macrophage infiltration, leading to impaired intimal hyperplasia and outward remodeling, hindering AVF maturation in mice.
In patients undergoing hemodialysis, von Willebrand Factor expression in the medial layer of the venous outflow tract is higher in matured AVFs and accompanied by increased wall thickening and outward remodeling, when compared with nonmatured AVFs.
What Are the Clinical Implications?
Nonmaturation of AVFs remains a significant clinical problem for patients with end‐stage kidney disease treated with hemodialysis.
Our observations suggest that local von Willebrand Factor–induced vascular smooth muscle cell proliferation might be a valuable therapeutic strategy aimed at improving AVF maturation.
The arteriovenous fistula (AVF) is considered the criterion standard of vascular access for hemodialysis. 1 Compared with central venous catheters and arteriovenous grafts, AVFs have better longevity and fewer complications. 2 , 3 , 4 However, unassisted maturation rates range from 60% to 79%. 5 This results in frequent vascular access–related interventions and hospitalization in order to achieve, maintain, or restore patency. Therefore, there is an urgent need to improve AVF maturation.
AVF maturation depends on the ability of the venous outflow tract to adapt to the rapid increase in pressure and flow upon AVF creation. Concordantly, the luminal diameter of the vein needs to expand after AVF placement in order to facilitate the increase in blood flow created at the arteriovenous anastomosis. This process is determined by the intricate balance between outward remodeling (OR) of the vessel and the degree of obstruction of the lumen by intimal hyperplasia (IH). One key player in both OR and IH is the vascular smooth muscle cell (VSMC). 6 , 7 , 8 , 9 However, the interplay between favorable VSMC proliferation that enables OR, and detrimental VSMC proliferation that causes IH, is an unknown process.
von Willebrand Factor (vWF) is a multimeric glycoprotein that is essential for blood clotting by binding platelets and as carrier protein of circulating Factor VIII. 10 vWF is synthesized by megakaryocytes located in the bone marrow and by endothelial cells (ECs) lining the intima, but upon vascular damage, vWF can localize subintimally in the vicinity of VSMCs. 11 , 12 vWF−/− mice have reduced angiogenesis, arteriogenesis, wound healing, and leukocyte infiltration, 13 , 14 and it has been established in several animal models that vWF is involved in intimal thickening. 11 , 15 , 16 Moreover, previous studies revealed that vWF is a potent inducer of VSMC proliferation 15 , 17 and inhibits gene expression of mature nonmitotic VSMCs. 18 Hence, vWF might play an important role in the proliferation of VSMCs in AVFs and influence the degree of OR and IH. We hypothesized that vWF‐deficient mice will present with reduced OR and IH, affecting vascular remodeling and thereby AVF maturation. In the present study we investigated the effect of vWF deficiency on murine AVF vascular remodeling and vWF expression levels in the intima and medial layer of native veins and patient‐matched AVFs obtained during two‐stage brachio‐basilic surgery. We hereby assessed whether vWF deficiency tips the scale in the intricate balance between OR and IH to achieve AVF maturation.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals and Study Design
This study was performed in compliance with Dutch government guidelines, the Directive 2010/63/EU of the European Parliament, and was approved by the Institutional Committee for Animal Welfare at the Leiden University Medical Centre. Adult C57BL/6J and B6.129S2‐Vwf tm1Wgr (vWF knockout 19 ) mice, aged 8 to 12 weeks and bred in our own facility, were used for the in vivo studies. For histological analysis, 9 vWF−/− mice (4 females) and 9 wild‐type (WT) mice (4 females) were included. Ultrasound analysis was performed on 9 vWF−/− and 6 WT male mice, because female vWF−/− mice had a higher exsanguination rate during surgery and were less likely to survive subsequent anesthesia during ultrasound analysis. Before the surgery, the mice were anesthetized via intraperitoneal injection of midazolam (5 mg/kg, Roche), medetomidine (0.5 mg/kg, Orion), and fentanyl (0.05 mg/kg, Janssen) and received unilateral AVFs, as previously described 20 between the dorsomedial branch of the external jugular vein and the common carotid artery (CCA). To prevent massive blood loss, human plasma–derived vWF (Wilfactin 150 IU/kg, Sanquin) was administered intravenously to the AVF during surgery. After surgery, anesthesia was antagonized with atipamezole (2.5 mg/kg, Orion) and flumazenil (0.5 mg/kg, Fresenius Kabi). Buprenorphine (0.1 mg/kg, MSD Animal Health) was given after surgery to relieve pain.
Ultrasound Analysis
The Vevo 3100 LAZR‐X Imaging System (FUJIFILM Visual Sonics) coupled to a 50‐MHz linear‐frequency transducer was used to longitudinally follow blood velocity and vessel diameter of the CCA of the AVF. The mice were anesthetized with 4% isoflurane and placed on the animal imaging platform where temperature, heart rate, and respiration rate were monitored in real time. During the experiments, anesthesia was maintained using a vaporized isoflurane gas system (1 L/min of oxygen, 0.3 L/min air, and 1.5% to 2.5% isoflurane). The concentration of isoflurane was adjusted according to the pedal reflex and respiration rate to ensure adequate anesthesia. Measurements were performed at baseline 1 day before surgery, at t=0 immediately following surgery, 7 and 14 days. The number of animals included varied per time point because of swelling and wound crust formation hindering ultrasound measurement (minimum n=3).
Data were obtained using B‐mode to identify the anatomical region, EKV (ECG‐gated Kilohertz Visualization) to visualize vessel wall characteristics such as diameter of the CCA, and pulsed wave color mode to quantify the pulse‐wave velocity. Quantification was performed with measurements that were obtained in between breathing cycles. The data were analyzed using Vevo lab software. Flow rate was calculated from pulse‐wave velocity measurements and vessel diameter of the CCA afferent to the anastomosis.
Murine Tissue Harvesting and Processing
Fourteen days after AVF surgery, the mice were anesthetized using an anesthetic mixture containing midazolam (5 mg/kg, Roche), medetomidine (0.5 mg/kg, Orion), and fentanyl (0.05 mg/kg, Janssen) through intraperitoneal injection. The thorax was opened and flushed with 4% formalin through intracardiac perfusion, whereafter the AVF was dissected and submerged in 4% formalin overnight, then embedded in paraffin. Tissue sections of 5 μm perpendicular to the venous outflow tract of the AVF were collected with an interval of 150 μm as described before. 20
Plasma and the superior vena cava were collected from mice 6 to 8 weeks of age; they were anesthetized as described above and were euthanized by cervical dislocation after tissue collection. Retro‐orbital blood was collected in EDTA tubes for the luminex assay and in Eppendorfs supplemented with 9:1 sodium citrate for cell culture purposes. The tubes were tumbled to mix the blood and anticoagulant and spun down for 20 minutes at 2000g, after which the plasma was collected and frozen until further use. The superior vena cava was flushed with PBS and connective tissue was removed in vivo, after which the vein was dissected and placed in PBS on ice.
Histology of Murine Tissue
Because most AVF occlusions occur in the venous outflow tract of the AVF, the first 3 venous cross sections downstream of the anastomosis with 150‐μm interval were used for morphological and immunohistochemical analysis. Morphometric analysis of the murine AVFs was performed using Weigert's elastin staining and αSMA (DAKO M0851) as a VSMC marker. Outward remodeling was studied by measuring the length of the internal elastic lamina (IEL) in Caseviewer (3DHISTECH) in Weigert's Elastin stained samples. Tissue within the IEL was defined as IH and determined using histoquant software (3DHISTECH). Luminal area was calculated by subtracting IH from the area within the IEL. αSMA‐positive tissue within the IH was determined using histoquant software. Thrombi were defined as anuclear αSMA‐negative tissue within the IH and calculated by manual tracing using annotations in Caseviewer.
Nuclei of Mac3+ cells (BD Pharmingen, 550 292) were counted manually in 3 random fields of view (80× magnification) from which the mean was calculated. Proliferative VSMCs were detected by counting Ki67 (BD Pharmingen, 550 609) and αSMA‐positive cells within the IEL and normalized to the αSMA‐positive area, which was defined using histoquant.
Luminex Assay
To verify the effect of vWF deficiency on Weibel‐Palade body (WPB)–stored proteins angiopoietin‐2 (Ang‐2), IL‐6, osteoprotegerin, and P‐selectin, we performed a magnetic luminex assay (BioTechne) on EDTA plasma obtained from 9 WT and 5 vWF−/− mice according to manufacturer's protocol. Dilutions of 1:3, 1:6, and 1:9 were analyzed in duplicate on a Bio‐Rad Bio‐Plex analyzer using the Bioplex Manager 6.0 software. Analyte concentrations (pg/mL) were calculated using a 5‐PL logistic regression model based on a 6‐point standard calibration curve.
Murine VSMC Cell Culture and Proliferation Assay
Primary venous VSMCs were isolated from the superior vena cava of WT and vWF−/− mice without an AVF. In a sterile flow cabinet, 12‐well plates were coated with 1% gelatin/PBS. The vena cava was cut longitudinally and the endothelial monolayer scraped off using sterile surgical forceps. Subsequently, the tissue was dissected into explants of ≈1 to 3 mm. By placing a sterile coverslip on top of the explants a vacuum was created, keeping the explants in close contact with the culture surface.
Cells were cultured in DMEM (Gibco) supplemented with 20% fetal calf serum, 2 mmol/L l‐glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin (penicillin/streptomycin, Gibco) and new medium was added after 7 days. Around 2 weeks after explant isolation, the explants were removed and the cells were trypsinized, replated, and cultured until 80% to 90% confluence, whereafter the cells were cultured overnight in DMEM supplemented with 1% fetal calf serum and used to measure proliferative capacity.
Murine VSMC Proliferation Assay
Venous VSMCs were trypsinized, counted, and resuspended in DMEM. Ten thousand cells were plated per well in a 96‐well plate in a total volume of 100 μL DMEM supplemented with 10% murine plasma of either WT or vWF−/− mice. To verify the direct effect of vWF on VSMCs, 10% vWF−/− plasma supplemented with 100 ng/mL Wilfactin (Sanquin, The Netherlands) was added to vWF−/− and WT VSMCs. After 40 hours incubation at 37 °C with 5% CO2, proliferation was quantified using the CyQuant Direct Cell Proliferation Assay according to the manufacturer's protocol (Thermo Fisher Scientific). The experiment was performed in biological n=3 to 6, in technical duplicates.
Patient Samples: Pre‐Access Native Vein and AVF Venous Outflow Tract and Plasma
Five micrometer paraffin‐embedded cross‐sections of matched pre‐access veins and AVF tissue pairs of patients with end‐stage renal disease (ESRD) were obtained from the University of Miami Human Vascular Biobank. The study was performed according to the ethical principles of the Declaration of Helsinki and regulatory requirements at Jackson Memorial Hospital and University of Miami. The ethics committee and University of Miami Institutional Review Board approved the study. Thirty‐eight patients who underwent 2‐stage AVF creation at Jackson Memorial Hospital or the University of Miami Hospital (19 with successful maturation and 19 with anatomic maturation failure) were selected from a total of 273 patients with tissue pairs in the biorepository (189 matured, 84 failed). 21 , 22 Anatomic maturation failure was defined as an AVF with an internal cross‐sectional luminal diameter ≤6 mm as determined intraoperatively using intravascular probes. Random selection was performed using propensity score matching while adjusting for age, sex, ethnicity, diabetes, and predialysis status. Fifteen mature AVFs and 16 failed AVFs were available for matched‐pair analysis. Patient characteristics can be found in Table 1.
Table 1.
Characteristics of Patients With End‐Stage Renal Disease Who Underwent 2‐Stage Brachio‐basilic Arteriovenous Fistula Surgery and Were Included for Histological Analysis
| Average age (years±SD) | Sex (% female) | Ethnicity (% Black) | Diabetes (%) | ASCVD (%) | Pre‐dialysis (%) | Previous AVF (%) | Smoking (%) | |
|---|---|---|---|---|---|---|---|---|
| Failed (N=19) | 57.45±11.95 | 40 | 60 | 50 | 50 | 35 | 15 | 40 |
| Mature (N=19) | 58.80±15.72 | 40 | 40 | 70 | 45 | 35 | 30 | 25 |
ASCVD indicates atherosclerotic cardiovascular disease; and AVF, arteriovenous fistula.
Human samples were stained with vWF (DAKO A0082), CD31 (DAKO M0823), and αSMA (DAKO M0851). The medial layer was defined as the area between the IEL and external elastic lamina and determined using Caseviewer. Positively stained tissue was defined using Caseviewer's Histoquant. Matched pair analysis of native veins and AVFs (15 mature and 16 failed) was performed for IH area, OR (IEL perimeter), area of the medial layer, and luminal area. Intima/media (I/M) area ratio was calculated by dividing the IH by the medial layer. The I/M ratio of 25 of 67 samples was previously analyzed. 22 vWF and CD31 co‐staining was performed on 5 patient‐matched native veins and AVFs of both outcome groups—20 samples in total. Plasma samples from 8 patients with AVF failure and 10 with AVF maturation were collected during surgery at the time of AVF creation and at superficialization through transposition. vWF antigen levels in patient plasma were determined as previously described. 23
Statistical Analysis
Graphpad Prism 8 was used to perform statistical analysis. Normally distributed data are presented as mean±SD. Nonparametric data are presented as the median±interquartile range. Unpaired t test, 1‐way ANOVA, Wilcoxon matched‐pairs signed rank test, and Mann–Whitney U test (2‐tailed) were used when applicable. P<0.05 is considered significant (*), **P<0.01, ***P<0.001, ****P<0.0001.
Results
vWF Enhances Outward Remodeling and Intimal Hyperplasia
To investigate how the vWF influences AVF maturation, we created an end‐to‐side carotid‐jugular AVF in vWF‐deficient (vWF−/−) and WT mice. Two weeks postsurgery, the AVFs were collected and morphometric analysis of the murine AVF venous outflow tract was performed using Weigert's elastin staining for vessel circumference (left Figure 1B and 1C) and αSMA as a VSMC marker (right Figure 1B and 1C). αSMA‐negative and anuclear lesions were defined as thrombi. The IEL perimeter of unoperated external jugular veins was comparable between WT and vWF−/− mice at ±1200 μm (Figure 1A). At 14 days after AVF surgery, vWF−/− mice had a 1.5‐fold smaller IEL perimeter compared with WT mice (Figure 1D, P=0.002). Total IH surface area in vWF−/− mice was reduced 10.2‐fold compared with WT mice (P<0.0001). Within the IEL, the αSMA‐positive area in vWF−/− mice was 8.1‐fold smaller than in WT mice (P=0.0002). Almost no thrombi were present in the vWF−/− AVF, with a mean area of 4 μm2, whereas thrombi in WT mice occupied 2868 μm2 (P<0.0001). There was a 1.4‐fold reduction in luminal area of vWF−/− mice, although this finding did not reach statistical significance (P=0.10).
Figure 1. vWF deficiency leads to reduced outward remodeling and intimal hyperplasia following AVF surgery.
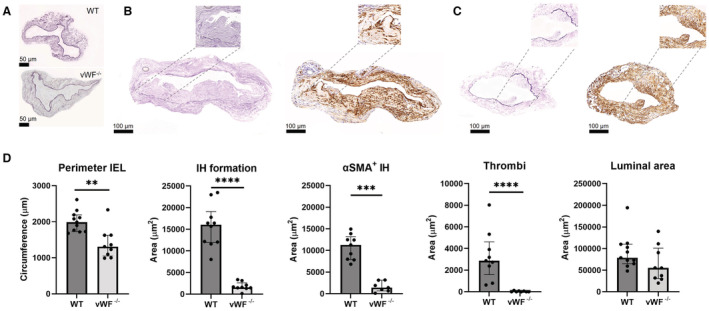
Representative images of baseline vena jugularis, scale bar=50 μm (A) and AVFs stained using Weigert's Elastin (left) and αSMA staining (right) from a WT (B) and vWF−/− (C) mouse AVF. Inlays illustrate IH in closer detail, including a thrombus in Figure 1B. The perimeter was measured by lining the internal elastic lamina (IEL); thrombi were defined as αSMA‐negative anuclear tissue. The stainings were quantified and presented as median±IQR (interquartile range) (D). OR (outward remodeling) was calculated using the IEL perimeter in Weigert's Elastin stained samples, IH was defined as the tissue within the IEL. αSMA‐positive tissue within the IH was determined using histoquant software. Thrombi were manually traced. Luminal area was calculated by subtracting the IH from the area within the IEL. Scale bar is 100 μm. N=9 per group. Statistical significance between groups was determined using the Mann–Whitney U test **P=0.002, ***P<0.001, ****P<0.0001. AVF indicates arteriovenous fistula; IEL, internal elastic lamina; IH, intimal hyperplasia; IQR, interquartile range; OR, outward remodeling; αSMA, α‐smooth muscle actin; vWF, von Willebrand Factor; and WT, wild type.
vWF‐Deficient Mice Do Not Show Incremental AVF Flow
Ultrasonography was performed on the AVFs at baseline, postsurgery at 0, 7, and 14 days post AVF creation to determine AVF functionality in terms of blood flow in the CCA, proximal to the site of anastomosis (Figure 2A). WT and vWF−/− mice had comparable flow at baseline. Flow rate in the WT mice increased over 2 weeks post‐AVF creation, with a 1.3‐fold increase at 7 days (P=0.057) and 2.55‐fold at 14 days (P=0.016) compared with the AVF flow volume at baseline (Figure 2B). AVF flow remained constant in vWF−/− mice over 2 weeks post‐AVF creation, congruent with the low degree of OR. At t=14 days, flow in the AVFs of vWF−/− mice was two‐fold lower than in WT mice (P=0.008).
Figure 2. vWF deficiency leads to reduced AVF flow 14 days post‐AVF creation.
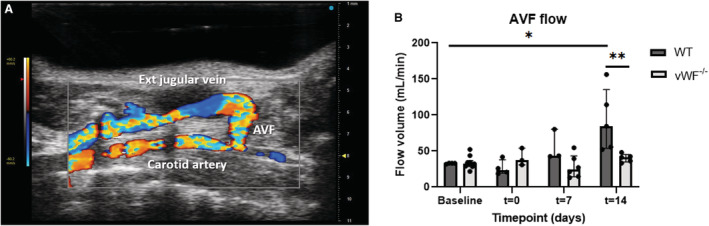
Effect of vWF deficiency on AVF blood flow was determined in the common carotid artery (CCA) afferent to the anastomosis (A). AVF blood flow in the CCA was calculated with the diameter of the CCA using EKV obtained data and blood velocity obtained in PW color mode (B). Ultrasound measurements were performed 1 day before surgery (baseline), immediately postsurgery (t=0), and at t=7 and t=14 days. Data are presented as median±interquartile range, n=6 WT and 9 vWF−/− mice. Statistical significance between groups was determined at timepoint t=14 days, and between baseline and t=14 days in WT mice using the Mann–Whitney U test. *P=0.016, **P=0.008. AVF indicates arteriovenous fistula; EKV, ECG‐gated Kilohertz Visualization; PW, pulsed wave; vWF, von Willebrand Factor; and WT, wild type.
vWF Deficiency Affects Weibel Palade Body Proteins
WPBs are the storage granules of ECs, of which vWF is a pivotal component. To verify whether deficiency of vWF affects the systemic release of WPB‐stored proteins angiopoietin‐2 (Ang‐2), IL‐6, osteoprotegerin and P‐selectin, we performed a luminex assay on plasma obtained from unoperated WT and vWF−/− mice. IL‐6 was undetectable in the plasma of both groups (data not shown) and no difference was observed in Ang‐2 levels (Figure 3A). Systemic levels of P‐selectin (3‐fold, P=0.007) and osteoprotegerin (2.9‐fold, P=0.001) (Figure 3B and 3C) expression were significantly enhanced in vWF−/− mouse plasma.
Figure 3. Luminex analysis of systemic WPB protein expression.
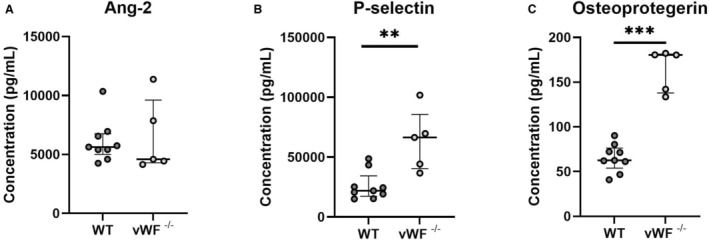
The effect of vWF deficiency on Weibel‐Palade body (WPB)‐proteins angiopoeitin‐2 (A), P‐selectin (B), and osteoprotegerin (C) was determined in 1:3 diluted EDTA plasma from n=9 WT and 5 vWF−/− mice using a 5‐PL logistic regression model. Data are presented as median±interquartile range. Statistical significance between groups was determined using the Mann–Whitney U test. **P=0.007, ***P=0.001. vWF indicates von Willebrand Factor; and WT, wild type.
vWF −/− Mice Exhibit Less Mac3+ Cells in the Intimal Hyperplasia
Similar to vWF, transmembrane protein P‐selectin is involved in leukocyte recruitment and adhesion. To investigate the effect of vWF deficiency combined with increased systemic levels of P‐selectin on local recruitment of inflammatory cells and IH formation, we performed a double‐staining for macrophage marker Mac3 and αSMA. Compared with WT mice (Figure 4A), AVFs of vWF−/− mice (Figure 4B) have a 37% reduction in expression of Mac3+ cells in the IH (Figure 4C), P=0.006.
Figure 4. Intimal hyperplasia in the AVF of vWF−/− mice displays fewer Mac3+ cells.
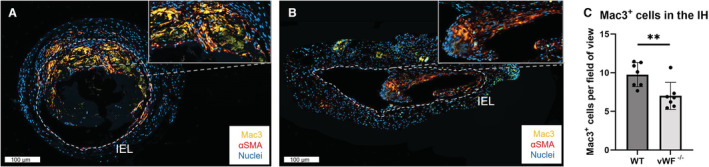
Immunofluorescent staining for nuclei (blue), αSMA (red), and Mac3 (yellow) of an AVF of a WT (A) and vWF−/− (B) mouse, with a higher magnification inlay of part of the IH. The IEL is traced in white, scale bar=100 μm. Nuclei of Mac3+ cells were counted manually per field of fiew and quantified for n=7 per group (C). Data are presented as mean±SD, **P=0.006, statistical significance between groups was determined using unpaired t test. αSMA indicates α‐smooth muscle actin; AVF, arteriovenous fistula; IEL, internal elastic lamina; IH, intimal hyperplasia; vWF, von Willebrand Factor; and WT, wild type.
vWF Enhances VSMC Proliferation
To assess the effect of vWF expression on VSMC proliferation, sections from the venous outflow tract of AVFs from WT (Figure 5A) and vWF−/− mice (Figure 5B) were stained for Ki67 and αSMA. Double‐positive cells within the IH were counted and normalized to the αSMA‐positive area. Compared with WT mice, vWF−/− mice showed an 82% decrease in proliferating Ki67+ VSMCs in the IH (Figure 5C, P=0.0002).
Figure 5. Characterization of proliferating VSMCs in the IH at 2 weeks post AVF creation and in vitro.
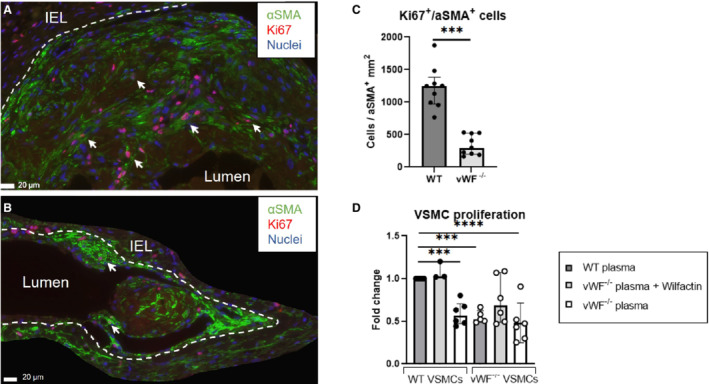
Immunofluorescent staining for nuclei (blue), Ki67 (red), and αSMA (green) in a WT (A) and vWF−/− mouse (B). The IEL is traced in white. Ki67+/αSMA+ cells, examples indicated with an arrow, within the IH were counted manually and normalized to the αSMA‐positive area within the IH. Scale bar=20 μm. Data are analyzed by the Mann–Whitney U test and presented as median±interquartile range, n=9 per group (C). VSMCs from WT (left 3 bars) and vWF−/− (right 3 bars) mice were stimulated with 10% murine plasma of WT or vWF−/− mice ±100 ng/mL Wilfactin (see box legend) for 40 hours, after which proliferation was measured. Data are presented as mean±SD, n=3 to 6. Statistical significance between WT VSMCs stimulated with WT plasma (control) and other experimental conditions was determined using 1‐way ANOVA and Dunnett's multiple comparisons test ***P<0.001, ****P=0.0001 (D). IEL indicates internal elastic lamina; IH, intimal hyperplasia; αSMA, α‐smooth muscle actin; VSMCs, vascular smooth muscle cells; vWF, von Willebrand Factor; and WT, wild type.
To verify the direct effect of vWF on VSMC proliferation, VSMCs were grown from explants of the superior vena cava and stimulated with murine plasma to simulate in vivo conditions. Compared with WT VSMCs exposed to WT plasma, vWF‐deficient plasma induced less proliferation in both WT and vWF−/− VSMCs (0.6‐fold and 0.5‐fold, Figure 5D), which was rescued by addition of human plasma–derived vWF (Wilfactin). A nonsignificant 1.5‐fold upregulation was observed when vWF−/− VSMCs were stimulated with Wilfactin in addition to vWF‐deficient plasma alone (P=0.09), indicating a trend of VSMC proliferation because of rescue of vWF deficiency.
vWF Resides in the IH and Vasa Vasorum of Patients With ESRD
Human pre‐access native veins and pair‐matched venous AVF samples were obtained from patients with ESRD undergoing 2‐stage brachio‐basilic AVF surgery. We observed vWF expression in the IH of both murine (Figure 6A) and human AVFs (Figure 6B). Human AVFs show vWF and CD31 co‐expression in the vasa vasorum in the tunica media (medial layer) and tunica adventitia (asterisk, Figure 6B). Expression of vWF not in the vicinity of CD31‐positive ECs was also observed in the tunica media of a mature AVF (arrow in Figure 6B). No difference in systemic VWF antigen levels were observed in samples obtained during the first procedure—AVF surgery—(1.31±0.69 IU/mL in patients with an AVF that would mature; 1.58±0.69 IU/mL in patients with an AVF that would fail) and during the second procedure—superficialization of the basilic vein—(mature AVF: 1.36±0.55 IU/mL; failed AVF: 1.35±0.55 IU/mL).
Figure 6. vWF resides in the intima and medial layer.
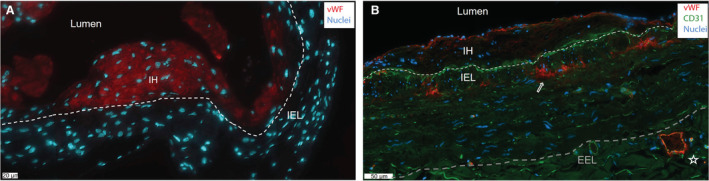
Representative immunofluorescent staining for vWF (red) in a wild‐type murine AVF (A) and in a matured AVF of a patient with end‐stage renal disease at superficialization (B). Endothelial cell marker CD31 is stained in the human AVF in green, nuclei in blue. The white dashed line signifies the IEL (internal elastic lamina), and the gray line indicates the EEL (external elastic lamina). Scale bar=20 μm in (A) and 50 μm in (B). AVF indicates arteriovenous fistula; IH, intimal hyperplasia; and vWF, von Willebrand Factor.
Matured AVFs Show Increased Expression of vWF in the Venous Tunica Media
Matured AVFs (Figure 7A) have a thicker tunica media compared with failed AVFs (Figure 7B). vWF colocalized with CD31+ cells in the vasa vasorum of both failed and mature AVFs (Figure 7A, indicated with an asterisk), or with αSMA, indicated by arrowheads in a mature AVF sample (Figure 7A). When studying maturation outcome parameters, the median increase in intimal area was not significantly different in the pair‐matched samples of patients with matured versus failed AVFs (Figure 7C; IH 2.1 versus 1.7 mm2, respectively, P=0.67).The increase in IEL perimeter, medial area, as well as luminal area in patients with matured AVFs was significantly larger when compared with patients with failed AVFs (median perimeter 5.4 versus 2.7 mm, P=0.022; medial area: 2.7 versus 0.6 mm2, P=0.009; luminal area: 1.8 versus 0.7 mm2, P=0.016). In addition, the increase in I/M ratio was smaller in patients with matured AVFs (0.2) when compared with patients with failed AVFs (0.7, P=0.01), because of reduced thickening of the medial area in failed AVFs, indicating the importance of OR in human AVF maturation. The wall thickening and OR in matured AVFs coincided with 167% (P=0.03) increase in vWF expression in the medial layer of pre‐access veins to mature venous AVF samples, but not in patient samples of failed AVFs (Figure 7D).
Figure 7. Morphometric and vWF expression analysis of samples from patient with ESRD.
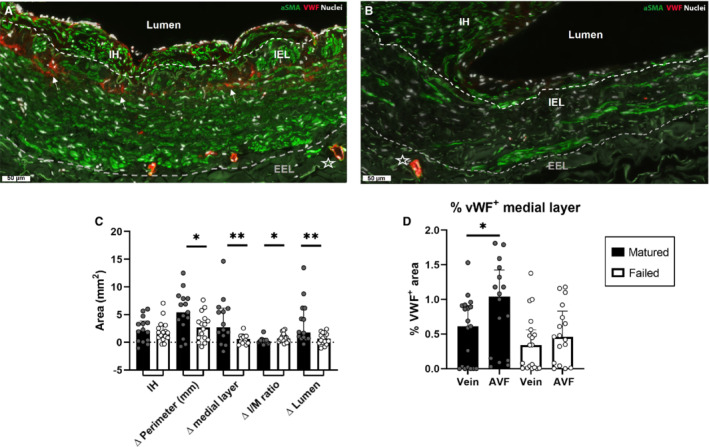
Analysis of ESRD pair‐matched samples of pre‐access native veins and venous AVF outflow tracts obtained during 2‐stage brachio‐basilic AVF surgery. AVF failure was defined as having an internal cross‐sectional luminal diameter ≤6 mm. Representative sample of a mature (A) and failed AVF (B). Scale bar=50 μm, star indicates a vessel in the vasa vasorum, αSMA+/vWF+ staining is indicated with arrowheads. The inner white line signifies the IEL, and the outer gray line signifies the external elastic lamina (EEL). Pair‐matched analysis of increase in IH area, delta (Δ) OR (perimeter in mm), area of the medial layer, the intima/media (I/M) ratio, and luminal size was calculated by subtracting the pre‐access venous parameters from the patient‐matched AVF. N=15 pair‐matched samples from matured AVFs and 16 pair‐matched failed AVFs. Statistical significance between groups was determined using the Mann–Whitney U test (C). vWF+ tissue in the medial layer was quantified in immunofluorescently stained samples and normalized to the area of the medial layer. Statistical significance was determined using Wilcoxon matched‐pairs signed rank test (D). Data are presented as median±interquartile range. *P<0.05, **P<0.01. AVF indicates arteriovenous fistula; ESRD, end‐stage renal disease; IEL, internal elastic lamina; IH, intimal hyperplasia; OR, outward remodeling; αSMA, α‐smooth muscle actin; and vWF, von Willebrand Factor.
Failed AVFs Express Less vWF in the Intimal Layer
In both samples from mature and failed AVFs, absence of vWF lining the IH was observed. This was often at sites with increased IH (Figure 8A), whereas little IH was observed at sites with vWF lining the intima (Figure 8B). We observed a reduction of vWF+ intima from the native vein to the venous AVF outflow tract in both patient groups (Figure 8D), with a significant 3.6‐fold reduction in failed AVF samples compared with mature AVFs (P=0.04). The intimal hyperplasia to medial layer (I/M) ratio increased from the native vein to the venous AVF sample (Figure 8C) with 2.6‐fold in mature AVFs (P=0.02) and 4.0‐fold in failed AVFs (P=0.0001). With no significant difference in IH between the 2 groups, and significant wall thickening in matured AVFs (Figure 7C), the high fold increase in I/M ratio in native veins to failed AVFs is caused by inefficient wall thickening. We hypothesized that vWF lining the intima might correlate with decreased I/M ratio because of vWF production to induce thickening of the tunica media. The I/M and vWF+ intima of AVFs were negatively correlated using linear regression (Figure 8E) and showed that when the vWF+ intimal layer is disrupted, the I/M ratio increases (P=0.0017 for matured AVFs and P=0.0264 for failed AVFs).
Figure 8. Relationship between disruption of vWF lining the intima and an enhanced I/M ratio.
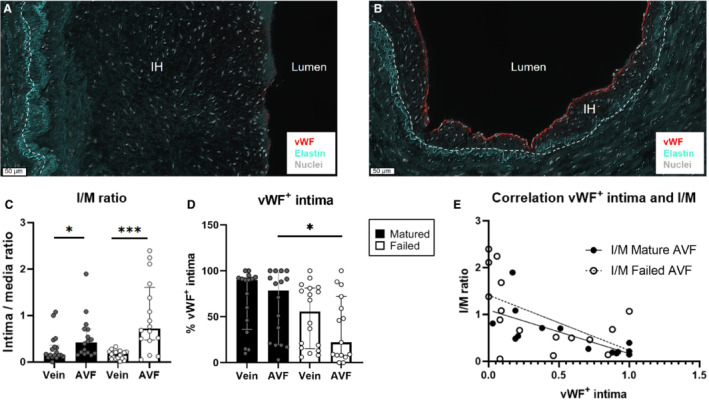
Analysis of ESRD samples of pre‐access native veins and venous AVF outflow tracts obtained during 2‐stage brachio‐basilic AVF surgery. Absence of vWF lining the intimal layer was observed at sites with increased IH (A) while an intact vWF+ intimal layer was observed at sites with little IH (B). The dashed line signifies the IEL, vWF is stained in red, elastin in turquoise, and nuclei in white. Scale bar=50 μm. The ratio between intimal area and the medial layer (I/M) (C) and the percentage of vWF+ intimal layer in native veins and AVFs were quantified (D) and correlated using linear regression (E). N=15 pair‐matched samples from matured AVFs and 16 pair‐matched failed AVFs; data are represented as median±interquartile range. Statistical significance between groups was determined using the Mann–Whitney U test; *P<0.05, ***P=0.0001. AVF indicates arteriovenous fistula; ESRD, end‐stage renal disease; IEL, internal elastic lamina; IH, intimal hyperplasia; I/M, intima/media ratio; and vWF, von Willebrand Factor.
Discussion
In this study we investigated the role of vWF in the process of AVF maturation and observed that vWF deficiency in mice impaired VSMC proliferation, macrophage infiltration, and both OR as well as IH, which resulted in the inability to increase AVF flow. Human AVF samples showed no difference in systemic vWF plasma levels, but we did observe a local increase in vWF+ tissue in the medial layer of matured AVFs, which did not occur in failed AVFs. Secondly, we observed a correlation between disruption of a vWF expression in the intimal layer and increase in the I/M ratio, signifying the importance of vWF production to promote OR. This suggests a potential role for vWF in AVF maturation in patients with ESRD as well.
Our study is the first to directly investigate the effect of vWF depletion on murine AVF maturation. vWF is well known for its function in platelet aggregation and venous thrombosis. 24 , 25 , 26 Indeed, previous studies illustrated that platelet inhibition causes reduction of IH in mice. 27 We observed that in our vWF‐deficient mouse model, systemic plasma levels of WPB‐proteins P‐selectin and osteoprotegerin were significantly higher than in WT mice. Osteoprotegerin is known to be atheroprotective and anticalcific by inhibiting calcium deposition in VSMCs, 28 possibly positively affecting AVF function in vWF−/− mice by reducing AVF stiffness. vWF‐deficient mice are known to exhibit altered local regulation of P‐selectin through a reduction in both intracellular storage and expression at the EC surface, along with impaired leukocyte recruitment. 14 , 29 This reduction in inflammatory cells might also be because of the reduction in vascular permeability and leukocyte docking after loss of vWF and thereby vWF–platelet interaction. 30 , 31 We indeed observed a reduction of thrombi and Mac3+ cells in the IH of vWF−/− mice, possibly affecting production of matrix metalloproteinases 32 and AVF wall thickening. 33
Besides inflammation and thrombus formation, vWF is also involved in the development of αSMA+ IH and OR of the venous outflow tract of murine AVFs, with an increase in expression. To verify that vWF promotes VSMC proliferation in our murine model, we performed an αSMA/Ki67 co‐staining, which showed vWF−/− mice to have a significant 82% decrease in proliferating αSMA+ cells in the IH. This is reinforced by recent work showing that vWF induces proliferation and migration of human arterial VSMCs through binding via its A2 domain to the LRP4‐receptor, causing αVβ3 integrin signaling. 17 αVβ3 integrin expression is upregulated in the rabbit AVF, compared with control vessels, 34 and inhibition of αVβ3 reduced SMC invasion of a collagen type I lattice, 35 similar to migration through vessel layers. These findings suggest that αVβ3 inhibition results in a reduction of IH and possibly thickening of the medial layer. Furthermore, because vWF can bind to several growth factors through its A1 heparin binding domain, vWF−/− mice present with decreased proliferation of αSMA+/CD45− cells. 13 These growth factors include vascular endothelial growth factor‐A, which promotes VSMC proliferation, 36 , 37 platelet‐derived growth factor‐BB, which induces VSMC phenotypic switching, proliferation, migration, and IH, 38 , 39 and C‐X‐C motif chemokine 12, which helps recruit VSMCs. 40 Furthermore, arteries from patients with CADASIL (Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) show vWF expression extending into the arterial wall, where vWF inhibits expression of mature contractile smooth muscle cell markers such as SM22, SM‐actin, calponin, and smooth muscle myosin heavy chain, indicating that vWF causes phenotypic VSMC switching towards a proliferative profile. 18 , 41 This provides support for the role of vWF in inducing VSMC proliferation in the tunica intima and tunica media of the AVF.
Besides apical secretion into the circulation, ECs can also secrete vWF basally into the subendothelial matrix. 42 We observed vWF expression in αSMA+ tissue in the tunica media, indicating that vWF might directly influence VSMCs and switch them to a proliferative state to enhance OR and wall thickening. This illustrates the importance of the interplay between vWF produced by the endothelial layer and VSMCs in AVF maturation. Indeed, impaired endothelial functioning pre‐AVF creation is associated with decreased AVF remodeling and function in patients with ESRD. 43 , 44 This corresponds to our observed correlation between loss of vWF expression in the AVF endothelial intima and an increase in the I/M ratio in ESRD patient samples.
A previous study in mice 6 observed high numbers of dedifferentiated Ki67+ VSMCs in the IH 4 weeks after AVF creation. Furthermore, matured MYH11+/Ki67− VSMCs were mainly detected in the vascular wall, provoking the statement that VSMCs have a dual function in AVF remodeling: proliferative VSMCs aggravate IH, while mature VSMCs facilitate wall thickening. We postulate, however, that VSMC proliferation is also required in the medial layer to facilitate OR, but occurs primarily in the first 2 weeks after murine AVF creation, whereafter proliferation of VSMCs in the intima is still ongoing. 45 With both IH and OR reduction in our vWF−/− mice resulting in a worse outcome in AVF functionality, this reinforces the current hypothesis that the degree of OR is of greater importance than minimizing the degree of IH. Indeed, patient‐matched data did not indicate differential increase in IH between matured and failed AVFs, while matured AVFs showed more OR and wall thickening than failed AVFs. This was accompanied by a significant local upregulation of vWF expression in the medial layer of mature AVFs, signifying the clinical translation of our murine findings that vWF might enhance OR in patients with ESRD.
Some aspects of our study, however, require further discussion. Unlike the murine AVFs, the patient samples have not been perfused with formalin as the murine AVFs were, possibly affecting the analysis of OR. Secondly, human plasma–derived vWF was administered during AVF surgery to rescue vWF deficiency and prevent excessive blood loss. Human plasma–derived vWF has a mean residence time of ≈2.2 hours in mice, thus affecting our model temporarily. 46 Moreover, we used a constitutive vWF knockout model, which does not reflect biological variations in humans, affects storage of WPB proteins, and could have a broader impact on EC function. Lastly, since the surgical procedure and ultrasound analysis combined with vWF deficiency puts a strain on the mice, our experiments have been performed in a model without renal failure. Previous studies revealed that chronic kidney disease significantly accelerates wall thickening and IH in mice, 47 , 48 but the composition of the stenotic lesions in these mice seems comparable with mice with normal kidney function. 47 However, it is unknown to which degree the murine IH process exactly resembles human IH formation in the early phase after AVF surgery.
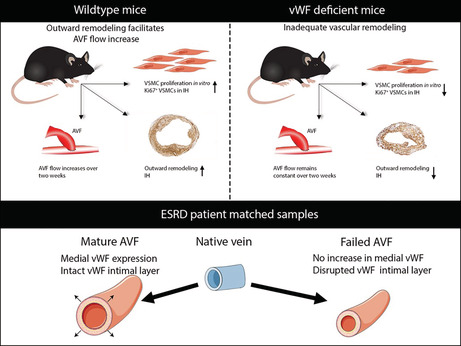
In conclusion, we have shown the importance of vWF in VSMC proliferation and the AVF maturation process in mice, with human data reinforcing the importance of an intact vWF+ intima and medial vWF expression to facilitate OR and wall thickening (Figure 9).
Figure 9. Overview of the findings.
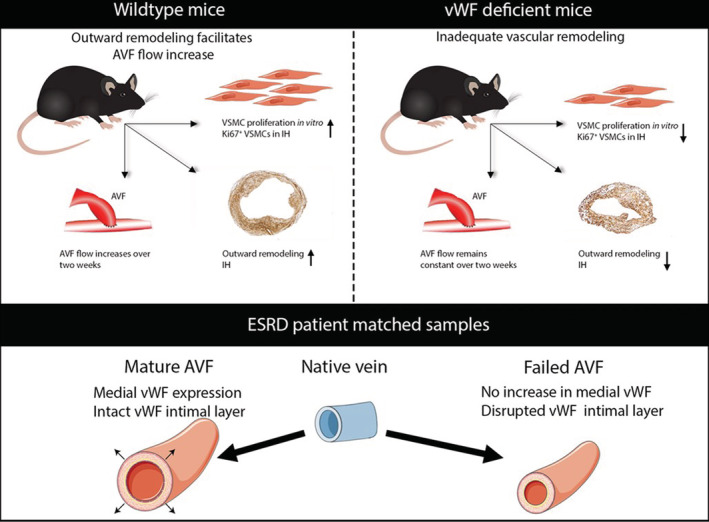
AVF indicates arteriovenous fistula; ESRD, end‐stage renal disease; IH, intimal hyperplasia; VSMC, vascular smooth muscle cell; and vWF, von Willebrand Factor.
To apply this knowledge clinically, further research should be conducted on the origin of VSMCs that cause OR, IH, and wall thickening, because currently there is no consensus about where the VSMCs populating the IH originate from. 7 , 49 , 50 , 51 Secondly, future studies with inducible vWF expression in a vWF‐knockout model would enhance the understanding of the direct effect of vWF on vascular remodeling in AVF, besides its effect on EC function. Moreover, because systemic vWF will greatly enhance the risk of thrombosis, local delivery such as a slow‐releasing gel or targeted nanoparticles should be further investigated. Most importantly, the time frame of VSMC proliferation that is involved in OR and wall thickening in ESRD mice and patients should be studied to determine the optimal treatment delivery. Knowledge of timing and delivery of vWF will provide a solid background to develop a promising vWF therapy to enhance AVF maturation, thereby reducing the complication‐ and intervention burden on the already fragile group of patients with ESRD.
Sources of Funding
This work was supported by a VIDI grant (NWO, Nederlandse Organisatie voor Wetenschappelijk Onderzoek; 91715328 to J.I.R.) from ZonMw (The Netherlands Organization for Health Research and Development: ZorgOnderzoek Nederland (ZON) en het gebied Medische Wetenschappen (MW)).
Disclosures
None.
Acknowledgments
The authors thank Anouk Spruit for her help during the luminex assay, Erna Peters for her assistance during the setting up of the murine VSMCs culture, and Jeroen Eikenboom for scientific input.
Author contributions: M.R.d.V. and J.I.R. conceived the idea and directed the work. S.L.L. performed the experiments, data analysis, and wrote the manuscript. M.R.d.V. performed the murine AVF surgeries and together with A.d.J. set up the ultrasound analysis protocol and murine sample acquisition. H.C.d.B. provided scientific input on the research model. R.A.L. performed tissue staining and gave scientific input. L.M. and R.I.V.P. provided the patient samples and gave scientific input. All authors revised the manuscript.
For Sources of Funding and Disclosures, see page 12.
References
- 1. Schmidli J, Widmer MK, Basile C, de Donato G, Gallieni M, Gibbons CP, Haage P, Hamilton G, Hedin U, Kamper L, et al. Vascular access: 2018 clinical practice guidelines of the European Society for Vascular Surgery (ESVS). Eur J Vasc Endovasc Surg. 2018;55:757–818. doi: 10.1016/j.ejvs.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 2. Astor BC, Eustace JA, Powe NR, Klag MJ, Fink NE, Coresh J, Study ftC . Type of vascular access and survival among incident hemodialysis patients: the choices for healthy outcomes in caring for ESRD (CHOICE) study. J Am Soc Nephrol. 2005;16:1449–1455. doi: 10.1681/ASN.2004090748 [DOI] [PubMed] [Google Scholar]
- 3. Banerjee T, Kim SJ, Astor B, Shafi T, Coresh J, Powe NR. Vascular access type, inflammatory markers, and mortality in incident hemodialysis patients: the Choices for Healthy Outcomes in Caring for End‐Stage Renal Disease (CHOICE) Study. Am J Kidney Dis. 2014;64:954–961. doi: 10.1053/j.ajkd.2014.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dhingra RK, Young EW, Hulbert‐Shearon TE, Leavey SF, Port FK. Type of vascular access and mortality in U.S. hemodialysis patients. Kidney Int. 2001;60:1443–1451. doi: 10.1046/j.1523-1755.2001.00947.x [DOI] [PubMed] [Google Scholar]
- 5. Voorzaat BM, Janmaat CJ, van der Bogt KEA, Dekker FW, Rotmans JI. Patency outcomes of arteriovenous fistulas and grafts for hemodialysis access: a trade‐off between nonmaturation and long‐term complications. Kidney360. 2020;1:916–924. doi: 10.34067/KID.0000462020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao J, Jourd'heuil FL, Xue M, Conti D, Lopez‐Soler RI, Ginnan R, Asif A, Singer HA, Jourd'heuil D, Long X. Dual function for mature vascular smooth muscle cells during arteriovenous fistula remodeling. J Am Heart Assoc. 2017;6. doi: 10.1161/JAHA.116.004891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liang M, Wang Y, Liang A, Mitch WE, Roy‐Chaudhury P, Han G, Cheng J. Migration of smooth muscle cells from the arterial anastomosis of arteriovenous fistulas requires notch activation to form neointima. Kidney Int. 2015;88:490–502. doi: 10.1038/ki.2015.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roy‐Chaudhury P, Wang Y, Krishnamoorthy M, Zhang J, Banerjee R, Munda R, Heffelfinger S, Arend L. Cellular phenotypes in human stenotic lesions from haemodialysis vascular access. Nephrol Dial Transplant. 2009;24:2786–2791. doi: 10.1093/ndt/gfn708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Mey JG, Schiffers PM, Hilgers RH, Sanders MM. Toward functional genomics of flow‐induced outward remodeling of resistance arteries. Am J Physiol Heart Circ Physiol. 2005;288:H1022–H1027. doi: 10.1152/ajpheart.00800.2004 [DOI] [PubMed] [Google Scholar]
- 10. Ruggeri ZM, Ware J. von Willebrand factor. FASEB J. 1993;7:308–316. doi: 10.1096/fasebj.7.2.8440408 [DOI] [PubMed] [Google Scholar]
- 11. Giddings JC, Banning AP, Ralis H, Lewis MJ. Redistribution of von Willebrand factor in porcine carotid arteries after balloon angioplasty. Arterioscler Thromb Vasc Biol. 1997;17:1872–1878. doi: 10.1161/01.atv.17.10.1872 [DOI] [PubMed] [Google Scholar]
- 12. Bosmans JM, Kockx MM, Vrints CJ, Bult H, De Meyer GR, Herman AG. Fibrin(ogen) and von Willebrand factor deposition are associated with intimal thickening after balloon angioplasty of the rabbit carotid artery. Arterioscler Thromb Vasc Biol. 1997;17:634–645. doi: 10.1161/01.atv.17.4.634 [DOI] [PubMed] [Google Scholar]
- 13. Ishihara J, Ishihara A, Starke RD, Peghaire CR, Smith KE, McKinnon TAJ, Tabata Y, Sasaki K, White MJV, Fukunaga K, et al. The heparin binding domain of von Willebrand factor binds to growth factors and promotes angiogenesis in wound healing. Blood. 2019;133:2559–2569. doi: 10.1182/blood.2019000510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Vries MR, Peters EAB, Quax PHA, Nossent AY. von Willebrand factor deficiency leads to impaired blood flow recovery after ischaemia in mice. Thromb Haemost. 2017;117:1412–1419. doi: 10.1160/TH16-12-0957 [DOI] [PubMed] [Google Scholar]
- 15. Qin F, Impeduglia T, Schaffer P, Dardik H. Overexpression of von Willebrand factor is an independent risk factor for pathogenesis of intimal hyperplasia: preliminary studies. J Vasc Surg. 2003;37:433–439. doi: 10.1067/mva.2003.63 [DOI] [PubMed] [Google Scholar]
- 16. De Meyer GR, Hoylaerts MF, Kockx MM, Yamamoto H, Herman AG, Bult H. Intimal deposition of functional von Willebrand factor in atherogenesis. Arterioscler Thromb Vasc Biol. 1999;19:2524–2534. doi: 10.1161/01.atv.19.10.2524 [DOI] [PubMed] [Google Scholar]
- 17. Lagrange J, Worou ME, Michel JB, Raoul A, Didelot M, Muczynski V, Legendre P, Plénat F, Gauchotte G, Lourenco‐Rodrigues MD, et al. The VWF/LRP4/αVβ3‐axis represents a novel pathway regulating proliferation of human vascular smooth muscle cells. Cardiovasc Res. 2022;118:622–637. doi: 10.1093/cvr/cvab042 [DOI] [PubMed] [Google Scholar]
- 18. Meng H, Zhang X, Lee SJ, Wang MM. von Willebrand factor inhibits mature smooth muscle gene expression through impairment of notch signaling. PLoS One. 2013;8:e75808. doi: 10.1371/journal.pone.0075808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Denis C, Methia N, Frenette PS, Rayburn H, Ullman‐Cullere M, Hynes RO, Wagner DD. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci USA. 1998;95:9524–9529. doi: 10.1073/pnas.95.16.9524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wong CY, de Vries MR, Wang Y, van der Vorst JR, Vahrmeijer AL, van Zonneveld A‐J, Hamming JF, Roy‐Chaudhury P, Rabelink TJ, Quax PHA, et al. A novel murine model of arteriovenous fistula failure: the surgical procedure in detail. J Vis Exp. 2016;e53294. doi: 10.3791/53294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tabbara M, Duque JC, Martinez L, Escobar LA, Wu W, Pan Y, Fernandez N, Velazquez OC, Jaimes EA, Salman LH, et al. Pre‐existing and postoperative intimal hyperplasia and arteriovenous fistula outcomes. Am J Kidney Dis. 2016;68:455–464. doi: 10.1053/j.ajkd.2016.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martinez L, Duque JC, Tabbara M, Paez A, Selman G, Hernandez DR, Sundberg CA, Tey JCS, Shiu YT, Cheung AK, et al. Fibrotic venous remodeling and nonmaturation of arteriovenous fistulas. J Am Soc Nephrol. 2018;29:1030–1040. doi: 10.1681/ASN.2017050559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Jong A, Dirven RJ, Oud JA, Tio D, van Vlijmen BJM, Eikenboom J. Correction of a dominant‐negative von Willebrand factor multimerization defect by small interfering rna‐mediated allele‐specific inhibition of mutant von Willebrand factor. J Thromb Haemost. 2018;16:1357–1368. doi: 10.1111/jth.14140 [DOI] [PubMed] [Google Scholar]
- 24. Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, Wakefield TW, Lammle B, Massberg S, Wagner DD. von Willebrand factor‐mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–1407. doi: 10.1182/blood-2010-05-287623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chauhan AK, Kisucka J, Lamb CB, Bergmeier W, Wagner DD. von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood. 2007;109:2424–2429. doi: 10.1182/blood-2006-06-028241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Receveur N, Nechipurenko D, Knapp Y, Yakusheva A, Maurer E, Denis CV, Lanza F, Panteleev M, Gachet C, Mangin PH. Shear rate gradients promote a bi‐phasic thrombus formation on weak adhesive proteins, such as fibrinogen in a VWF‐dependent manner. Haematologica. 2019;235754. doi: 10.3324/haematol.2019.235754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drosopoulos JHF, Kraemer R, Shen H, Upmacis RK, Marcus AJ, Musi E. Human solCD39 inhibits injury‐induced development of neointimal hyperplasia. Thromb Haemost. 2010;103:426–434. doi: 10.1160/TH09-05-0305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Callegari A, Coons ML, Ricks JL, Rosenfeld ME, Scatena M. Increased calcification in osteoprotegerin‐deficient smooth muscle cells: dependence on receptor activator of NF‐κB ligand and interleukin 6. J Vasc Res. 2014;51:118–131. doi: 10.1159/000358920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Denis CV, André P, Saffaripour S, Wagner DD. Defect in regulated secretion of p‐selectin affects leukocyte recruitment in von Willebrand factor‐deficient mice. Proc Natl Acad Sci. 2001;98:4072–4077. doi: 10.1073/pnas.061307098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, Goerge T, Schneider SW, Jones C, Nieswandt B, et al. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116:4712–4719. doi: 10.1182/blood-2010-03-276311 [DOI] [PubMed] [Google Scholar]
- 31. Gragnano F, Sperlongano S, Golia E, Natale F, Bianchi R, Crisci M, Fimiani F, Pariggiano I, Diana V, Carbone A, et al. The role of von Willebrand factor in vascular inflammation: from pathogenesis to targeted therapy. Mediators Inflamm. 2017;2017:5620314. doi: 10.1155/2017/5620314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hanania R, Song Sun H, Xu K, Pustylnik S, Jeganathan S, Harrison RE. Classically activated macrophages use stable microtubules for matrix metalloproteinase‐9 (MMP‐9) secretion. J Biol Chem. 2012;287:8468–8483. doi: 10.1074/jbc.M111.290676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guo X, Fereydooni A, Isaji T, Gorecka J, Liu S, Hu H, Ono S, Alozie M, Lee SR, Taniguchi R, et al. Inhibition of the Akt1‐mTORC1 axis alters venous remodeling to improve arteriovenous fistula patency. Sci Rep. 2019;9:11046. doi: 10.1038/s41598-019-47542-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cai W‐J, Li MB, Wu X, Wu S, Zhu W, Chen D, Luo M, Eitenmüller I, Kampmann A, Schaper J, et al. Activation of the integrins α5β1 and αvβ3 and focal adhesion kinase (FAK) during arteriogenesis. Mol Cell Biochem. 2009;322:161–169. doi: 10.1007/s11010-008-9953-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanda S, Kuzuya M, Ramos MA, Koike T, Yoshino K, Ikeda S, Iguchi A. Matrix metalloproteinase and alphavbeta3 integrin‐dependent vascular smooth muscle cell invasion through a type I collagen lattice. Arterioscler Thromb Vasc Biol. 2000;20:998–1005. doi: 10.1161/01.atv.20.4.998 [DOI] [PubMed] [Google Scholar]
- 36. Liao XH, Xiang Y, Li H, Zheng DL, Xu Y, Xi YC, Li JP, Zhang XY, Xing WB, Cao DS, et al. VEGF‐A stimulates STAT3 activity via nitrosylation of myocardin to regulate the expression of vascular smooth muscle cell differentiation markers. Sci Rep. 2017;7:2660. doi: 10.1038/s41598-017-02907-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parenti A, Bellik L, Brogelli L, Filippi S, Ledda F. Endogenous VEGF‐A is responsible for mitogenic effects of MCP‐1 on vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2004;286:H1978–H1984. doi: 10.1152/ajpheart.00414.2003 [DOI] [PubMed] [Google Scholar]
- 38. Lu QB, Wan MY, Wang PY, Zhang CX, Xu DY, Liao X, Sun HJ. Chicoric acid prevents PDGF‐BB‐induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFΚB/MTOR/P70S6K signaling cascade. Redox Biol. 2018;14:656–668. doi: 10.1016/j.redox.2017.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kingsley K, Huff JL, Rust WL, Carroll K, Martinez AM, Fitchmun M, Plopper GE. ERK1/2 mediates PDGF‐BB stimulated vascular smooth muscle cell proliferation and migration on laminin‐5. Biochem Biophys Res Commun. 2002;293:1000–1006. doi: 10.1016/S0006-291X(02)00331-5 [DOI] [PubMed] [Google Scholar]
- 40. Stratman AN, Burns MC, Farrelly OM, Davis AE, Li W, Pham VN, Castranova D, Yano JJ, Goddard LM, Nguyen O, et al. Chemokine mediated signalling within arteries promotes vascular smooth muscle cell recruitment. Commun Biol. 2020;3:734. doi: 10.1038/s42003-020-01462-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang X, Meng H, Blaivas M, Rushing EJ, Moore BE, Schwartz J, Lopes MBS, Worrall BB, Wang MMJTSR. von Willebrand factor permeates small vessels in cadasil and inhibits smooth muscle gene expression. Transl Stroke Res. 2012;3:138–145. doi: 10.1007/s12975-011-0112-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lopes da Silva M, Cutler DF. von Willebrand factor multimerization and the polarity of secretory pathways in endothelial cells. Blood. 2016;128:277–285. doi: 10.1182/blood-2015-10-677054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. MacRae JM, Ahmed S, Hemmelgarn B, Sun Y, Martin BJ, Roifman I, Anderson T. Role of vascular function in predicting arteriovenous fistula outcomes: an observational pilot study. Can J Kidney Health Dis. 2015;2:19. doi: 10.1186/s40697-015-0055-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Owens CD, Wake N, Kim JM, Hentschel D, Conte MS, Schanzer A. Endothelial function predicts positive arterial‐venous fistula remodeling in subjects with stage IV and V chronic kidney disease. J Vasc Access. 2010;11:329–334. doi: 10.5301/jva.2010.5833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wong C‐Y, de Vries MR, Wang Y, van der Vorst JR, Vahrmeijer AL, van Zonneveld AJ, Roy‐Chaudhury P, Rabelink TJ, Quax PHA, Rotmans JI. Vascular remodeling and intimal hyperplasia in a novel murine model of arteriovenous fistula failure. J Vasc Surg. 2014;59:192–201.e191. doi: 10.1016/j.jvs.2013.02.242 [DOI] [PubMed] [Google Scholar]
- 46. Groeneveld DJ, van Bekkum T, Cheung KL, Dirven RJ, Castaman G, Reitsma PH, van Vlijmen B, Eikenboom J. No evidence for a direct effect of von Willebrand factor's abh blood group antigens on von Willebrand factor clearance. J Thromb Haemost. 2015;13:592–600. doi: 10.1111/jth.12867 [DOI] [PubMed] [Google Scholar]
- 47. Kang L, Grande JP, Hillestad ML, Croatt AJ, Barry MA, Katusic ZS, Nath KA. A new model of an arteriovenous fistula in chronic kidney disease in the mouse: beneficial effects of upregulated heme oxygenase‐1. Am J Physiol Renal Physiol. 2016;310:F466–F476. doi: 10.1152/ajprenal.00288.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kokubo T, Ishikawa N, Uchida H, Chasnoff SE, Xie X, Mathew S, Hruska KA, Choi ET. CKD accelerates development of neointimal hyperplasia in arteriovenous fistulas. J Am Soc Nephrol. 2009;20:1236–1245. doi: 10.1681/ASN.2007121312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liang M, Liang A, Wang Y, Jiang J, Cheng J. Smooth muscle cells from the anastomosed artery are the major precursors for neointima formation in both artery and vein grafts. Basic Res Cardiol. 2014;109:431. doi: 10.1007/s00395-014-0431-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Skartsis N, Manning E, Wei Y, Velazquez OC, Liu Z‐J, Goldschmidt‐Clermont PJ, Salman LH, Asif A, Vazquez‐Padron RI. Origin of neointimal cells in arteriovenous fistulae: bone marrow, artery, or the vein itself? Semin Dial. 2011;24:242–248. doi: 10.1111/j.1525-139X.2011.00870.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vazquez‐Padron RI, Martinez L, Duque JC, Salman LH, Tabbara M. The anatomical sources of neointimal cells in the arteriovenous fistula. J Vasc Access. 2021;112972982110118. doi: 10.1177/11297298211011875 [DOI] [PMC free article] [PubMed] [Google Scholar]