

Abstract
After ischemic stroke, there is a significant burden of cardiovascular complications, both in the acute and chronic phase. Severe adverse cardiac events occur in 10% to 20% of patients within the first few days after stroke and comprise a continuum of cardiac changes ranging from acute myocardial injury and coronary syndromes to heart failure or arrhythmia. Recently, the term stroke–heart syndrome was introduced to provide an integrated conceptual framework that summarizes neurocardiogenic mechanisms that lead to these cardiac events after stroke. New findings from experimental and clinical studies have further refined our understanding of the clinical manifestations, pathophysiology, and potential long‐term consequences of the stroke–heart syndrome. Local cerebral and systemic mediators, which mainly involve autonomic dysfunction and increased inflammation, may lead to altered cardiomyocyte metabolism, dysregulation of (tissue‐resident) leukocyte populations, and (micro‐) vascular changes. However, at the individual patient level, it remains challenging to differentiate between comorbid cardiovascular conditions and stroke‐induced heart injury. Therefore, further research activities led by joint teams of basic and clinical researchers with backgrounds in both cardiology and neurology are needed to identify the most relevant therapeutic targets that can be tested in clinical trials.
Keywords: brain–heart axis, heart, inflammation, outcomes, stroke
Subject Categories: Ischemic Stroke, Complications, Mortality/Survival, Biomarkers, Heart Failure
Nonstandard Abbreviations and Acronyms
- AFDAS
AF detected after stroke
- BDNF
brain‐derived neurotrophic factor
- CaMKII
Ca2+/calmodulin‐dependent kinase II
- CAN
central autonomic network
- CORONA‐IS
Cardiomyocyte Injury Following Acute Ischemic Stroke
- MACE
major adverse cardiovascular events
- MCAo
middle cerebral artery occlusion
- NLRP3
NLR family pyrin domain containing 3
- Nr4a1
nuclear receptor subfamily 4 group a member 1
- PPARα
peroxisome proliferator‐activated receptor α
- PRAISE
Prediction of Acute Coronary Syndrome in Acute Ischemic Stroke
- SICFAIL
Stroke‐Induced Cardiac Failure in Mice and Men
- TTS
Takotsubo syndrome
Ischemic stroke patients are at substantial risk of heart disease, and, vice versa, heart disease increases the risk of ischemic stroke. Randomized controlled trials and observational studies reported severe adverse cardiac events in ≈10% to 20% of patients with acute ischemic stroke. 1 , 2 , 3 , 4 , 5 , 6 , 7 These clinically severe cardiac complications are more common in patients with severe stroke and may even occur in patients without known comorbid cardiac disease. 1 , 2 , 4 , 6 Stroke patients with early severe cardiac complications are at a 2‐ to 3‐fold increased risk of short‐term mortality. 1 , 8 Beyond these acute neurocardiogenic alterations, the occurrence of cardiovascular events and cardiac death are the main drivers of long‐term prognosis after stroke. 9 , 10 Nearly one‐third of deaths in stroke survivors can be attributed to a cardiac cause. 10 , 11 Both short‐term and long‐term cardiac events after stroke happen despite contemporary secondary prevention measures. Thus, there is a medical need to reduce the burden of both acute and long‐term cardiovascular complications after stroke.
Within the past years, it is increasingly recognized that the interactions between heart disease and ischemic stroke are more than a mere consequence of traditional vascular risk factors that affect both organs. Based upon the evidence from preclinical and clinical studies that acute ischemic stroke can have immediate deleterious effects on the heart, the concept of a distinct stroke–heart syndrome was introduced in 2018 (see Box and Figure 1 for definition). 1 , 3 , 12 , 13 , 14 , 15 The intention of the concept was to provide an integrated view of stroke‐related cardiac complications to increase the clinical awareness, generate a common mechanistic framework, and facilitate the development of research collaborations and clinical management pathways. The stroke–heart syndrome concept summarizes the full spectrum of cardiac changes newly observed within the first 30 days after an acute ischemic stroke. 3 , 7 Its pathophysiological model views ischemic stroke as a specific event triggering neurocardiogenic heart injury and is based on both preclinical and clinical data suggesting a strong overlap between the continuum of clinical phenotypes and underlying mechanisms. 1 , 3 The biggest controversy around this concept relates to the underlying pathophysiologic mechanisms: Is cardiac dysfunction following ischemic stroke caused by vascular comorbidity along the cardiovascular risk continuum, is it a specific event triggered by the stroke, or both? On an individual patient level, physicians are challenged by the clinical problem whether acute cardiac injury in a patient with stroke represents a critical event requiring urgent cardiac diagnostics and potentially harmful intervention.
Box 1. Updated Criteria and Features of the Stroke–Heart Syndrome* .
|
Clinical definition
Clinical phenotype
†
Time course
Risk factors
Differential diagnoses
Recent key advances
|
Figure 1. Summary of key criteria and of the stroke–heart syndrome.
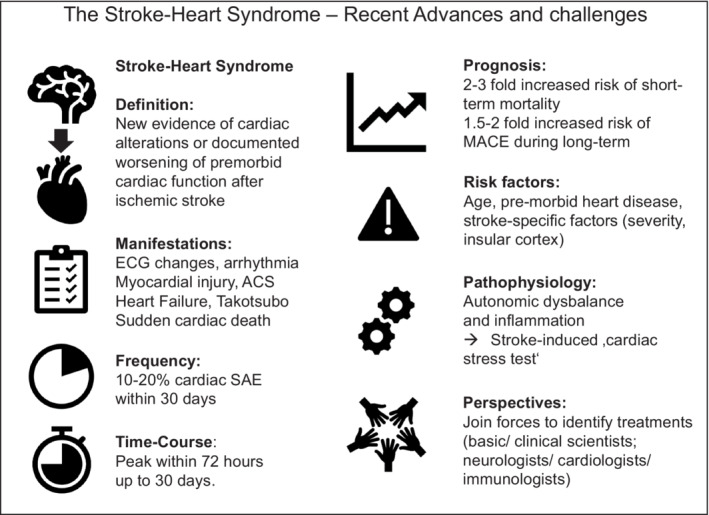
ACS indicates acute coronary syndrome; MACE, major adverse cardiovascular events; and SAE, severe adverse events.
In this review, we aimed to provide an updated overview of the experimental phenotype and clinical manifestations of the stroke–heart syndrome, and to extend the existing pathophysiological model, focusing on the most relevant stroke characteristics, key mediators, as well as downstream cardiac signaling pathways. Finally, we aimed to derive the most challenging questions and promising directions for future research.
MANIFESTATIONS OF STROKE–HEART SYNDROME
Evidence From Animal Models
Rodent models of experimental stroke provide a unique opportunity to study both the immediate short‐term and longer‐term effects of ischemic stroke on the heart. In the late 1980s and early 1990s, there was an initial period of flourishing animal research on deleterious heart–brain interactions following stroke. Ground‐breaking experiments in cats and rats demonstrated that stroke can impair physiological autonomic cardiac function and provoke proarrhythmogenic electrocardiographic changes. 16 , 17 In recent years, several well‐designed experimental studies have helped to refine our knowledge of the stroke–heart syndrome phenotype in rodent models (Figure 2). 18 , 19 , 20 , 21 , 22 , 23 , 24 To study the acute cardiac effects of stroke, Veltkamp and colleagues induced an experimental ischemic stroke via transient (60 minutes) filament occlusion of the left middle cerebral artery (MCAo) in mice. 18 This paradigm resulted in large cerebral infarcts and was paralleled by early cardiac dysfunction and damage. Serial echocardiographic measurements showed that MCAo reduced left ventricular (LV) ejection fraction and LV fractional shortening by 10% to 15% as compared with sham‐operated animals. These effects began on the first day after experimental stroke and persisted for up to 14 days. These findings were accompanied by myocardial injury as reflected by transiently 3‐ to 5‐fold increase in levels of high‐sensitivity cardiac troponin (hs‐cTn), bradycardia, rapid cardiomyocyte atrophy, and cardiac weight loss. 18 Vornholz and colleagues were able to reproduce this phenotype of acute cardiac dysfunction also after right‐sided MCAo. 19 A similar study in rats demonstrated that embolic strokes resulting in large brain infarcts and severe neurologic deficits induced early and persistent reduction in LV fractional shortening. 23
Figure 2. Phenotypes and time course of the stroke–heart syndrome in rodent models and clinical practice.
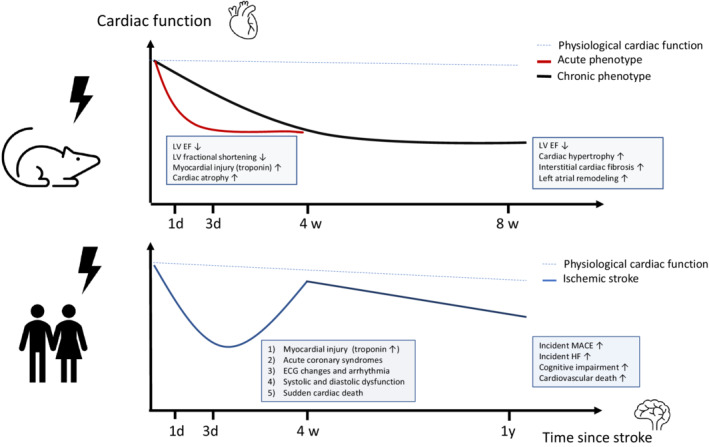
(Top) Phenotype observed in rodent ischemic stroke models. The dashed blue line indicates physiological cardiac function. The red line indicates the severity of acute cardiac dysfunction that peaks within 24 to 72 hours and persists up to 14 days after experimental stroke. Note that this phenotype was most consistently inducible by severe brain ischemia. The black line indicates a phenotype of chronic cardiac dysfunction starting 4 to 8 weeks after experimental brain ischemia. Note that this phenotype was most consistently inducible by mild, right‐sided brain ischemia. (Bottom) Spectrum and time‐course of stroke–heart syndrome observed in human clinical studies. The dashed blue line indicates physiological trajectory of cardiac function during aging. The blue line indicates the trajectory of cardiac alterations after stroke observed in observational clinical studies. Note that there is large individual variability, and that further studies are needed to predict the incidence of long‐term cardiovascular outcomes and heart failure. EF indicates ejection fraction; HF, heart failure; LV, left ventricular; and MACE, major adverse cardiovascular events.
Beyond these immediate cardiac effects, there is evidence that ischemic stroke produces a chronic heart failure phenotype. Bieber and colleagues performed short‐term, right‐sided MCAo in mice resulting in small ischemic lesions in the basal ganglia and the rodent analogue of the insular cortex. 21 Eight weeks after surgery, MCAo resulted in a significant reduction of LV ejection fraction of about 15% compared with sham‐operated and control animals. This was accompanied by a significantly higher heart rate, increased LV end‐systolic and end‐diastolic volumes, cardiac hypertrophy, and adverse cardiac remodeling as measured by increased LV collagen content. 21 Similarly, in several murine stroke models that induced right‐sided small to intermediate cortical lesions, a progressive 10% to 25% reduction in LV ejection fraction was found over a 3‐day to 4‐week period along with cardiac interstitial fibrosis. 20 , 22 , 24 There is evidence that cardiac alterations are not limited to the left ventricle, but also include the left atrium. In a rat model with selective ischemic lesions of the left and right insular cortex stroke by local endothelin‐1 injection, stroke resulted in left atrial remodeling and fibrosis, particularly in atrial regions with rich autonomic innervation. 25
A potential drawback of animal research is that experimental stroke models usually use young, male, and healthy animals. 18 , 25 , 26 This may hamper translation into clinical practice, because ischemic stroke is typically a disease of the elderly with a high burden of chronic cardiovascular comorbidities. 27 , 28 To improve clinical translation, international guidelines recommend repeating experiments in aged and animals of both sexes as well as animals with comorbidities such as diabetes or hypertension. 29 Of note, there is evidence that the stroke–heart syndrome is more pronounced in diabetic mice and similar in young and aged mice. 20 , 21 , 30 This underscores the notion that modeling the stroke–heart syndrome in rodents can be achieved under clinically relevant conditions. If these aspects are further considered in the future, streamlined rodent models will remain highly valuable tools to probe neurocardiogenic mechanisms of stroke–heart syndrome, identify causal mediators, and test promising interventions.
Spectrum, Severity, and Time Course of Clinical Manifestations
The stroke–heart syndrome encompasses a broad clinical spectrum of cardiovascular changes including (1) acute myocardial injury (as evidenced by acute elevation of hs‐cTn), (2) acute coronary syndromes (ACS; including type 1 and type 2 myocardial infarction [MI]), (3) systolic and diastolic LV dysfunction including Takotsubo syndrome (TTS), (4) cardiac arrhythmias including atrial fibrillation (AF) and relevant ECG changes, and (5) neurogenic sudden cardiac death (Box, Figure 2). 1 , 3 Importantly, these categories are not mutually exclusive, and there is substantial overlap between these manifestations. 1 , 31 , 32 Established risk factors include older age, premorbid cardiac disease, and stroke characteristics such as stroke severity, infarct size, and lesion site in the insular cortex. 1 , 2 The stroke–heart syndrome apparently affects women and men equally, although certain phenotypes, such as TTS secondary to stroke, mainly affect women. 33 , 34 , 35 In the original description of the stroke–heart syndrome, we proposed the differentiation of stroke–heart syndrome from myocardial ischemia because of coronary plaque rupture or plaque thrombosis (ie, type 1 MI). Recent evidence from experimental studies in humans and mice, however, suggest that acute mental stress and neuroinflammation may lead to destabilization of atherosclerotic plaques via activation of the neuroendocrine axis and subsequent uptake of inflammatory leucocyte populations. 36 , 37 Thus, we now propose that also type 1 MI may occur as part of the spectrum of the stroke–heart syndrome. Given the high frequency of atypical clinical presentation of ACS in the elderly, especially those with neurological deficits (such as aphasia), the accurate diagnosis of ACS has to rely on strict and systematic clinical examination, typical ECG findings, and cardiac imaging showing regional wall‐motion abnormalities or coronary lesions. 3 , 38
With the introduction of increasingly sensitive and specific cardiac diagnostic tests within the past decades, it became clear that the manifestations of the stroke–heart syndrome range from oligo‐ or even asymptomatic laboratory or ECG findings to severe clinical manifestations requiring urgent cardiac diagnostics and treatments. The most common cardiac changes include myocardial injury (ie, elevated hs‐cTn levels above assay‐specific reference limits), ECG alterations such as QTc prolongation, and LV diastolic dysfunction, which are observed in up to 60% of patients with acute ischemic stroke, typically during the initial in‐hospital workup. 8 , 31 , 38 These patients are at increased risk of death and more likely to leave the hospital with more severe stroke‐related disabilities. 8 , 39 Because the individual premorbid cardiac status is usually unknown, thorough clinical assessment and serial diagnostic tests are required to distinguish comorbid and clinically stable heart disease from stroke–heart syndrome. Several observational studies documented that myocardial injury and prolonged QTc time are associated with the incidence of more severe forms of stroke–heart syndrome. 2 , 4 , 32 , 39 , 40 , 41 Therefore, it is reasonable and also recommended by current guidelines to measure hs‐cTn and perform a standard 12‐lead ECG to screen for subclinical early cardiac involvement after ischemic stroke. 42 In case of pathological findings, prolonged ECG monitoring, thorough clinical cardiac assessment, and serial measurement of hs‐cTn are warranted to identify patients at higher risk of developing severe or fatal cardiovascular complications. Data from systematic evaluations of adverse events documented in high‐quality randomized controlled trial settings have shown that 10% to 20% of patients with acute ischemic stroke suffer a severe cardiac adverse event requiring urgent evaluation and treatment. 2 , 5 , 6 , 7 These mainly include acute myocardial injury (characterized by a distinctive rise or fall pattern of hs‐cTn levels), 8 , 38 , 43 LV systolic dysfunction, 31 , 40 , 44 , 45 and arrhythmia including atrial fibrillation. 4 , 46 In 1% to 5% of patients, even more severe manifestations, such as myocardial infarction, 5 , 47 , 48 overt heart failure including TTS, 1 , 3 , 7 , 34 , 45 and malignant tachycardia or cardiac arrest, have to be expected. 4 , 5 , 7 The spectrum of severe forms of stroke–heart syndrome are associated with a 2‐ to 3‐fold increased risk of mortality or poor functional outcome. 1 , 5 , 48
In regard to the time course, there is evidence that the acute manifestations of the stroke–heart syndrome peak within the first 72 hours after brain ischemia (Figures 1 and 2) 1 , 2 , 4 but may not be limited to this early phase. A recent Canadian population‐based study of adults aged ≥66 years, 9.1% of 21 931 patients with first‐ever ischemic stroke had incident major adverse cardiovascular events (MACE) at 1 year (including ACS and MI, coronary interventions, incident congestive heart failure, or cardiovascular death). 9 Compared with 71 696 matched individuals without stroke or heart disease, ischemic stroke was associated with a 4.5‐times higher risk of MACE. Of note, patients with known heart disease at baseline were excluded from this study, and the increased risk was strongly time dependent. 9 , 33 Although adjusted risk of MACE was increased 25‐fold in the first 30 days, it remained >2 times higher during the first year after ischemic stroke. 9 Based on this time‐dependent trajectory, it can be assumed that MACE occurring within the first 30 days after stroke may be directly attributable to the stroke–heart syndrome. 3 After this phase, incident MACE and heart failure can be considered as possible long‐term consequences of the initial cardiac injury attributable to stroke–heart syndrome.
Current Challenges
It remains challenging to differentiate whether poststroke cardiac events are manifestations of stroke–heart syndrome, an expression of an underlying cardiac condition, or both. This is because the cardiac status before the stroke is usually not known when the patient presents to the emergency department and because systematic cardiac evaluations by performing serial echocardiography or cardiovascular magnetic resonance (CMR) imaging during follow‐up of stroke are lacking. As outlined above, however, there is strong evidence supporting a potential causal relationship between stroke and subsequent cardiac complications. In addition to the wealth of animal data that show experimental stroke can induce immediate and chronic cardiac dysfunction, 13 , 18 , 22 clinical data provide evidence that there is a time‐dependent occurrence of cardiac complications with a clear peak during the first few days after stroke. 1 , 2 , 4 , 9 , 35 Major adverse cardiac events are also observed even when patients with known history of heart disease are excluded. 9 , 49 Further evidence comes from observational studies that applied coronary angiography in stroke patients with elevation of hs‐cTn, showing that the majority of these patients had no underlying obstructive coronary artery disease. 47 , 50 Serial measurements of hs‐cTn demonstrate a rise or fall pattern in 15% to 30% of patients, 8 , 43 which means that myocardial injury is acute and not chronic in these patients. Finally, specific stroke characteristics like stroke severity and stroke lesion site promote the occurrence of poststroke cardiac events even when statistically accounting for premorbid cardiac conditions. 32 , 41 , 51 , 52 These stroke characteristics linked to stroke–heart syndrome are discussed in more detail below.
To date, it remains also challenging to determine whether long‐term vascular events and post‐stroke cognitive dysfunction are prompted by stroke–heart syndrome or whether premorbid subclinical heart disease, or both, accounts for the observed associations. However, there is increasing evidence that this might be the case. As described earlier, a phenotype of chronic cardiac dysfunction with myocardial fibrosis can be induced 4 to 8 weeks after experimental stroke. 21 , 24 Moreover, ex vivo experiments using rat hearts derived from animals that underwent an experimental stroke paradigm were more susceptible to second ischemic hits to the heart. 23 This suggests an increased susceptibility to secondary cardiac events poststroke. This presumption is supported by a recent retrospective analysis using a global (primarily US based) network of 365 383 health care records of patients with stroke and 5‐year follow‐up that compared the long‐term cardiovascular outcomes of stroke patients with and without manifestations of stroke–heart syndrome (defined as new‐onset cardiovascular complications within 30 days after stroke). 7 Patients with stroke–heart syndrome had a 1.5‐ to 2‐fold higher odds of 5‐year mortality and MACE depending on the respective manifestation of stroke–heart syndrome. 7 Although this study focused on substantial manifestations of stroke–heart syndrome diagnosed based on International Classification of Diseases (ICD‐10) codes, there is also evidence that more subtle manifestations may entail a higher cardiovascular risk. In a clinical study of 201 ischemic stroke patients, those who had evidence of autonomic imbalance (ie, lower high‐frequency power in heart‐rate variability analysis) within 48 hours after stroke had a higher risk of secondary ischemic events within 90 days. 53 However, most of the events detected in this study occurred early after stroke, highlighting the need for more confirmative data with regard to the longer term. Of 220 patients with first‐ever, mild‐to‐moderate ischemic stroke and myocardial injury measured with hs‐cTn within a median of 4 days after the event, but without clinical diagnosis of concurrent ACS, 27.3% experienced a cumulative end point of recurrent stroke, MI, or death within 3 years of follow‐up. 54 This was significantly higher than in the control group of 342 patients with normal hs‐cTn levels during the acute poststroke phase (10.2%; adjusted hazard ratio, 2.0). 54 Beyond the occurrence of cardiovascular events, there is also evidence that patients with (subclinical) myocardial injury in the acute phase after the ischemic stroke have a higher burden of cerebral small vessel disease and cognitive impairment than those without. 55 , 56 Among 555 patients with first‐ever ischemic stroke, global cognitive impairment was present in 43% of patients with hs‐cTn values in the highest quartile. 56 This proportion was significantly higher than in the group of patients with hs‐cTn in the lowest quartile (15%) and remained statistically significant after adjusting for potential confounders. Cognitive function remained worse in this group during the 3 years of follow‐up. 56
Another challenging field of research includes the entity of AF detected after stroke (AFDAS), which may be considered a particular expression of the stroke–heart syndrome. According to a meta‐analysis, previously unknown AF can be found in ≈10% of patients with ischemic stroke during the initial in‐hospital workup and in ≈25% by sequentially combining cardiac monitoring methods during long‐term follow‐up. 46 The theoretical construct of AFDAS has evolved in recent years and addresses its distinctive characteristics when compared with AF known before stroke. 57 , 58 Conceptually, AFDAS results from the interplay of underlying cardiogenic mechanisms and stroke‐related neurogenic phenomena. 58 AFDAS seems to have a lower prevalence of cardiovascular comorbidities and less severe structural heart disease than AF known before stroke occurrence. 58 , 59 These findings may explain why patients with AFDAS seem to have a lower risk of stroke recurrence than those with a known history of AF before stroke onset. 60 , 61 AFDAS probably encompasses higher and lower risk phenotypes based on the severity of preexisting atrial cardiopathy and the prevalence of risk factors. 58 Within the spectrum of AFDAS risk, patients with severe atrial cardiopathy (eg, dilated left atrium) are those who may bear the higher risk, whereas those with structurally healthy hearts and lower‐burden AFDAS triggered by self‐limited and short‐lasted neurogenic mechanisms may have a lower embolic risk. 3 , 62 , 63 Studies combining biomarkers, neuroimaging patterns, and cardiovascular comorbidities are needed to further characterize higher‐ and lower‐risk AFDAS phenotypes. Characterizing these phenotypes may help improve the selection of ischemic stroke and transient ischemic attack patients who could benefit more from anticoagulation if AFDAS is found on prolonged cardiac monitoring.
PATHOPHYSIOLOGY OF STROKE–HEART SYNDROME
The pathophysiological model of stroke–heart syndrome considers the spectrum of clinical manifestations to be the result of neurocardiogenic mechanisms leading to stroke‐induced heart injury (stroke‐induced cardiac stress‐test). 1 , 3 Of note, neurocardiogenic injury is not restricted to an ischemic stroke cause and can be observed in other acute brain disorders such as seizures, traumatic brain injury, and intracranial hemorrhage (reviewed elsewhere). 64 Within the past few years, further advances have been made that have helped to further elucidate the complex interplay of ischemic stroke‐lesion characteristics, local cerebral and systemic mediators, and downstream cardiac mechanisms leading to the stroke–heart syndrome (Figure 3). The next paragraphs summarize these advances and describe promising future directions of research.
Figure 3. Overview about stroke‐specific characteristics, mediators, downstream cardiac mechanisms, and outcomes of stroke–heart syndrome.

The 4 columns describe the most promising and well‐studied stroke characteristics, mediators, cardiac mechanisms, and short‐term and long‐term outcomes of stroke–heart syndrome.
Impact of Stroke Characteristics
There is evidence that specific localizations of the ischemic lesion favor the occurrence of stroke–heart syndrome. A well‐studied example is the association between ischemic lesions in the insular cortex and the occurrence of acute myocardial injury, TTS, and arrhythmia. 8 , 34 , 41 , 51 , 52 In experimental studies, the extent of histological damage in the correlate of the insula in the mouse brain damage correlated with the severity of cardiac dysfunction and troponin elevation. 19 , 26 The insular cortex is an integral part of central autonomic network (CAN) and is involved in both cardiac interoception and efferent cardiovascular response to emotional experience. 65 , 66 Under physiological conditions, the CAN regulates sympathetic and parasympathetic neuronal outflow to the heart. 3 , 66 Ischemic stroke may lead to abrupt changes in the physiological organization of the CAN, resulting in autonomic dysfunction. Importantly, the occurrence of the stroke–heart syndrome is not limited to ischemia within the insular cortex. Both in clinical and experimental settings, a phenotype of poststroke cardiac dysfunction can also be observed when the insular cortex itself is not affected. 22 , 24 , 67 Other relevant regions of the CAN with documented impact on cardiac function include the amygdala, anterior cingulate cortex, ventromedial prefrontal cortex, hypothalamus, mediodorsal thalamus, hippocampus, and brainstem regions. 3 , 66 , 68 , 69 , 70 With regard to the latter, QTc prolongation on ECG upon hospital admission was found in 9 out of 12 patients with acute medullary infarction. 71 Lesion mapping revealed that QTc prolongation correlated with lesions in the right or left dorsal vagal nucleus. Remarkably, QTc time partially normalized during the in‐hospital stay, suggesting a causal relationship with the stroke event. In addition, lateral medullary infarction can occasionally result in central sleep‐related hypoventilation, which may lead to secondary cardiac injury (eg, via endothelial dysfunction) or coronary demand ischemia. 72 , 73 Of note, stroke lesion sites in the insula and brain stem are associated with sleep‐disordered breathing during the early poststroke phase. 74 In turn, sleep‐disordered breathing after stroke was linked to impaired cardiac autonomic dynamics, endothelial dysfunction, ECG alterations like sinus brady‐ or tachycardia, and reduced LV function. 73 , 74 , 75 Therefore, the interplay between the CAN and sleep‐disordered breathing and its potential impact on the occurrence of the stroke–heart syndrome deserves further study.
Recent evidence from patients with TTS (synonymous with stress cardiomyopathy) highlights the role of the CAN in the occurrence of cardiac dysfunction. Compared with healthy controls, a surface‐ and voxel‐based morphometry study revealed that patients with TTS related to emotional stress but unrelated to acute stroke have reduced cortical thickness in both insulae and reduced amygdala volume. 76 Moreover, a functional magnetic resonance imaging study demonstrated altered parasympathetic and sympathetic network activity in TTS patients versus matched controls. 77 In a case–control study of 104 individuals undergoing positron emission tomography–computed tomography imaging, higher activity in the amygdala was associated with the occurrence of TTS during a median follow‐up of 2.5 years. 78 Interestingly, individuals with the highest activity within the amygdala developed TTS up to 2 years earlier than those with relatively lower activity. 78 Of note, higher resting activity of the amygdala and higher perceived stress was associated with arterial inflammation, bone marrow activity, and incident cardiovascular events in patients without known cardiovascular or cerebrovascular disease. 79 However, it remains challenging to determine whether inherited or acquired (eg, by epigenetic mechanisms) disturbance of stress response and function of the CAN well before the stroke increase the individual's susceptibility to develop stroke–heart syndrome. Moreover, further clinical studies (eg, using lesion‐network mapping approaches) are needed to determine the specificity of CAN dysfunction (beyond the insula and brain stem) for the occurrence of the stroke–heart syndrome.
Beyond specific lesion sites, observational clinical data provide compelling evidence that stroke severity is associated with the occurrence of virtually all manifestations of the stroke–heart syndrome. 1 This association has been shown most consistently in relation to the occurrence of myocardial injury, cardiac arrhythmia, and impaired LV function after stroke. 4 , 8 , 60 , 67 In line with this data, animal studies provided evidence that the severity of acute and chronic cardiac dysfunction correlates with the size of brain ischemia. 18 , 21 , 26 Larger ischemic brain injury not only increases the likelihood of CAN dysfunction, but also leads to more profound disruption of the blood–brain barrier and damage to the neurovascular unit. As a result, danger signals and extracellular vesicles enter the bloodstream, which results in a systemic activation of the immune system. 80 This is in line with the notion that the extent of cerebral injury and inflammation correlate with detrimental effects on the heart. Whether this can be targeted by therapeutic interventions remains to be proven.
Key Mediators
The knowledge about critical mediators of the stroke–heart syndrome is rather scarce. However, the way toward mechanistic studies is paved by combining experimental animal models such as MCAo with deeper cardiac phenotyping. Sympathetic overdrive and reduced parasympathetic activity as a cause for cardiac dysfunction in the stroke–heart syndrome are among the most consistently discussed mediators. 1 , 3 , 81 MCAo‐induced cardiac dysfunction in mice was associated with higher levels of norepinephrine in serum and the heart and linked to peripheral sympathetic overactivity. 21 , 26 Seemingly contradicting, cardiac norepinephrine stores were enhanced 3 days after MCAo in the study of Veltkamp and colleagues, which could be interpreted as a decrease in sympathetic activity 3 days after stroke. 18 However, unbiased RNA microarray analyses showed massive changes in expression of catecholamine‐driven genes in the latter experimental setup. This suggests an early sympathetic overactivation in the heart persisting for at least 1 but not for 3 days after MCAo. In line with these considerations, it has been shown that already 2 hours after cerebral embolism in rats, circulating catecholamines were increased along with a reduction in cardiac function. 23 Therefore, local cardiac catecholamine homoeostasis should, in particular, be examined and targeted in the first hours after ischemia in the brain. Of note, MCAo‐induced cardiac dysfunction could be attenuated by β‐blockers. 21 Moreover, different experiments found that monoamine oxidase inhibition resulted in both reduced local cerebral neuroinflammation and reduced catecholamine‐induced cardiac dysfunction. 82 , 83 This points to the potential of neuro‐humoral blockade as therapeutic target. The role of autonomic dysfunction is supported by clinical evidence of impaired autonomic reflexes, and variability of blood pressure and heart rate in patients with ischemic stroke. 84 , 85 , 86 High blood pressure variation, especially within the first 3 hours in the early critical phase of autonomic disbalance is associated with increased mortality at 90 days. 87 Both tachycardia and hypertensive blood pressure levels are established triggers of coronary demand ischemia, which can cause myocardial injury. 88 In addition, pressure overload can lead to reversible LV dysfunction with troponin release and myocyte apoptosis even without necrosis. 89
Recent experimental studies support an important role of inflammatory responses in the occurrence of stroke–heart syndrome. 19 , 24 Stroke‐induced cardiac dysfunction in mice was accompanied by a systemic inflammatory response, upregulation of proinflammatory cytokines within the myocardial tissue, and macrophage infiltration into the heart. 19 , 24 Thus, besides sympathetic overactivation, proinflammatory pathways represent independent or perhaps interdependent mediators of the stroke–heart syndrome. In regard to the latter, sympathetic activation leads to the mobilization of inflammatory cells from the bone marrow or spleen. 90 , 91 In a photothrombotic stroke model, splenectomy attenuated cardiac dysfunction along with interstitial cardiac fibrosis at 4 weeks. 24 This was preceded by an attenuation of proinflammatory cytokines and cardiac macrophage recruitment, suggesting that the spleen is critically involved in the development of subacute and long‐term consequences of stroke–heart syndrome, eventually leading to chronic remodeling. Beyond the brain–bone marrow–spleen axis, further interorgan cross talks that involve the kidney and gut may be relevant drivers of inflammation and promote poststroke cardiac injury. 92 , 93
The directed recruitment of CD45+ leukocytes to the heart in response to injury has been shown to depend on cardiomyocyte‐born cardiokines that serve as chemoattractant signaling molecules. 94 , 95 Although the specific role of these cardiomyocyte‐born signals in stroke–heart syndrome remains to be shown, similar pathophysiological roles have already been described for CC‐chemokine ligands 2 (monocyte chemotactic protein 1) and 3 (macrophage inflammatory protein) in ischemia–reperfusion injury of the heart. 94 , 95 Importantly, this cascade was mediated by a catecholamine‐sensitive protein kinase, namely CaMKII (Ca2+/calmodulin‐dependent kinase II) in cardiomyocytes, pointing to a model in which both peripheral sympathetic activation along with local cardiac sympathetic activation collectively orchestrate myocardial inflammation. Another candidate mediator in this particular context is BDNF (brain‐derived neurotrophic factor), a neurotrophin involved in the development of (autonomic) neurons, angiogenesis, and maintenance of endothelial function. 96 , 97 It has been demonstrated that BDNF improves cardiac muscle contraction and relaxation via the before‐mentioned CaMKII pathway. 97 Circulating levels of BDNF are reduced after stroke, especially after severe stroke. 98 Therefore, BDNF signaling may represent a pathway mediating the interplay between autonomic tone, endothelial integrity, and myocardial dysfunction. Further experimental stroke studies in this direction are warranted in the future.
Both systemic inflammation and sympathetic overdrive can result in activation of coagulation, platelet hyperactivity, and endothelial dysfunction. 99 , 100 , 101 This process of thromboinflammation has received scientific attention during the COVID‐19 pandemic and has been proposed as an important mediator of myocardial injury, coronary microcirculatory dysfunction, and other extrapulmonary manifestations of COVID‐19. 101 , 102 The impact of thromboinflammation in the occurrence of the stroke–heart syndrome, however, has yet to be proven.
A potential contribution of cell–cell communication in combination with proinflammatory signals have recently been shown in experimental stroke models. Stroke‐induced cardiac fibrosis and LV dysfunction was accompanied by decreased levels of the microRNA miR‐126 in the serum and the heart. 22 Mice specifically lacking miR‐126 in endothelial cells showed an even more severe phenotype. This study demonstrates how a stroke‐associated reduction in miR‐126 and miR126‐target genes may mediate the stroke–heart syndrome via the involvement of endothelial cells. Thus, it is possible that vascular injury after stroke might lead to long‐distance cell–cell communication independent of the surrounding tissue. In this regard, metabolites or extracellular vesicles should also be considered. 103 Of note, administration of exosomes derived from cluster of differentiation 133+ umbilical cord blood cells attenuated cardiac dysfunction in a photothrombotic stroke model. 20 This was accompanied by reduced oxidative stress, reduced transforming growth factor‐beta expression, and reduced recruitment of proinflammatory macrophages. cluster of differentiation 133+ umbilical cells express miR‐126, thereby providing a potential link to the aforementioned endothelial mechanism. The vasculature, with its enormous surface, may thereby serve as an amplifier of deleterious signals as was recently demonstrated in tumor biology. 104
Cardiac Downstream Pathways
The above‐mentioned mediators affect various cardiac cell populations via induction of coronary demand ischemia (eg, via coronary vasoconstriction), microvascular dysfunction, metabolic switches, and cardiac inflammation (Figure 2). Within the past years, it is increasingly recognized that cardiomyocytes constitute less than half of the total cardiac cell population. 105 In response to acute injury, other cardiac‐resident cells, including fibroblasts as well as the vasculature and immune cell compartments, also undergo phenotype changes that can trigger maladaptive cardiac remodeling. Interestingly, it has been shown that tissue‐resident cardiac macrophages can be activated by remote injury processes including stroke. 106 Several downstream cardiac pathways have been established to mediate cardiac dysfunction. 107 However, signaling pathways to be potentially targeted by pharmacologic interventions that mediate acute cardiac dysfunction after stroke are largely elusive. Previous approaches to use calcium sensitizers in stroke models failed, 108 indicating that the underlying pathways may be more complex. Evidence for an important contribution of pro‐inflammatory pathways was provided by a recent study in which an inhibitor of NLRP3 (NLR family pyrin domain containing 3), a key component of the inflammasome, restored cardiac function in mice after MCAo. 30 In a rat stroke model, increased oxidative stress and altered NO signaling were measured in the left atria. This was accompanied by the recruitment of neutrophils, T and B lymphocytes into the left atrium, as well as atrial fibrosis. 25 A detailed analysis of the recruitment of proinflammatory cells, receptor‐ligand binding studies in combination with single cell, or even single nucleus sequencing may add to a better mechanistic understanding of these processes and their role in reducing the burden of stroke–heart syndrome.
In addition, recent discoveries highlight the role of the interface between metabolic and epigenetic processes. Nr4a1 (nuclear receptor subfamily 4 group a member 1) is a catecholamine‐sensitive nuclear receptor that integrates cytokine signaling in inflammatory disorders by acting in nonmyocytes. 109 Nr4a1 was found to be upregulated 1 day after MCAo in the heart. 18 Interestingly, Nr4a1 was also enhanced in cardiomyocytes derived from induced pluripotent stem cells of TTS patients, 110 suggesting an overlapping molecular signature of these 2 brain‐dependent forms of acute cardiac dysfunction. Interestingly, Nr4a1 was shown to be a key target of an epigenetic axis affecting cardiomyocyte calcium handling in response to stress. 111 Mechanistic experiments revealed that this process was causative for exercise‐induced cardiac fatigue, which is reminiscent of the stroke–heart syndrome. Thus, it will be interesting to explore the potential contribution of these epigenetic‐metabolic signaling pathways to the stroke–heart syndrome. Supporting this notion, many of the genes acutely regulated after experimental stroke were found to depend on the activity of the transcription factor PPARα (peroxisome proliferator‐activated receptor α) that regulates fatty acid storage and glucose metabolism. 18
Besides upregulation of metabolic pathways, Veltkamp and colleagues also reported that pathways that mediate muscle wasting were regulated following experimental stroke. 18 These expression changes persisted over >3 days, pointing to more long‐term consequences. Likewise, heart weight was reduced in the first days after MCAo, suggesting that these changes might impact the heart, resulting in pathological atrophy, a process that is also seen in muscle wasting diseases such as cancer‐related cachexia. However, data that investigate the potential causal involvement are missing but may represent another new research direction.
OPEN QUESTIONS, ONGOING STUDIES, AND DIRECTIONS FOR FUTURE RESEARCH
Currently, there are many unanswered questions on the pathophysiology, long‐term outcomes, and specific therapeutic approaches to stroke–heart syndrome, with many of them being addressed in ongoing studies (Table). 40 , 67 , 112 , 113 , 114 , 115 , 116 , 117 , 118 , 119 , 120 Because elevation of serum markers of myocardial injury is associated with all other clinical categories of stroke–heart syndrome, 4 , 32 , 40 , 41 it seems reasonable to measure hs‐cTn in all patients to screen for early cardiac involvement after ischemic stroke and identify patients who are at risk for a more severe clinical course and further specific cardiac diagnostics. As described above, it is currently not clear how to differentiate causes of myocardial injury at the individual patient level and identify patients at high risk of ACS requiring timely coronary revascularization. It is therefore crucial to refine diagnostic algorithms to identify the corresponding underlying pathophysiology in the respective patient, in the future ideally without invasive diagnostics. This question is currently being answered by the PRAISE (Prediction of Acute Coronary Syndrome in Acute Ischemic Stroke) study, in which stroke patients with relevant hs‐cTn elevation undergo coronary angiography in addition to standardized electrocardiography and echocardiographic assessment. 119 In the future, other suitable diagnostic measures may routinely involve contrast‐enhanced multislice cardiac computed tomography and/or comprehensive CMR to differentiate cardiac pathologies. 121 , 122 In particular, CMR offers opportunities to distinguish causes of myocardial injury after stroke and investigate long‐term cardiac remodeling. The localization, pattern, and estimated reversibility of myocardial tissue changes enables the differentiation of ischemic and nonischemic injury. Focal fibrosis, as detected by CMR, may reflect unrecognized MI and has a prognostic impact in suspected ischemic heart disease. 123 Beyond focal fibrosis, diffuse fibrosis can be quantified by CMR and strongly correlates with histopathological findings of collagen content in the myocardium. 124 Diffuse fibrosis on CMR is associated with ventricular remodeling and event‐free survival in patients aortic stenosis. 125 The role of CMR to differentiate mechanisms of myocardial injury after stroke is being addressed in the CORONA‐IS (Cardiomyocyte Injury Following Acute Ischemic Stroke) study. 116
Table 1.
Ongoing Clinical Studies Exploring the Pathophysiology or Targeting of Long‐Term Consequences of Stroke–Heart Syndrome
| Study | Title of study (registration) | Design | City, country | Target population | Target no. of patients | Outcomes of interest | Measures | Progress |
|---|---|---|---|---|---|---|---|---|
| 1 | Atrial Cardiomyopathy in Patients With Stroke of Undetected Mechanism 112 (NCT03830983) | Prospective, observational, case–control‐study (healthy age‐ and sex‐matched controls) | Copenhagen, Denmark | Ischemic stroke (≤30 d before inclusion) without atrial fibrillation | 150 (originally estimated enrolment=225) | Extent of LA fibrosis incidence of silent brain lesions, LA volume, LAEF | Gadolinium‐enhanced CMR imaging | Active, recruiting |
| 2 | BEHABIS (The Bern Heart and Brain Interaction Study) 67 (NCT03720522) | Prospective, observational, single‐center cohort study | Bern, Switzerland | Acute ischemic stroke (<12 h after symptom onset) without severe renal failure (GFR <40) | 220 | TTS (prevalence of neurogenic stunned myocardium), subacute MI | CMR (gadolinium‐enhanced with or without perfusion) | Active, recruiting |
| 3 |
BeLOVE (Berlin Long‐Term Observation of Vascular Events) 113 (DRKS00016852) |
Prospective, observational, multicenter cohort study | Berlin, Germany | Hospitalization for: acute ischemic stroke; acute coronary syndrome, acute heart failure, acute kidney injury (4 study arms) | 10 000, 2000 each study arm | MACE (cardiovascular mortality, nonfatal stroke, nonfatal myocardial infarction, hospitalization because of heart failure) | Clinical assessment, CMR, MRI (head), ECG, 3‐dimensional echocardiography, ocular coherence tomography | Active, recruiting |
| 4 | CONVINCE (Colchicine for Prevention of Vascular Inflammation in Non‐CardioEmbolic Stroke) 115 (NCT02898610) | Randomized controlled trial (colchicine vs placebo) | Several countries in Europe | Noncardioembolic ischemic stroke without major disability | 2623 | Recurrence of nonfatal ischemic stroke, nonfatal major cardiac event, vascular death | Clinical assessment | Active recruiting |
| 5 | CORONA‐IS (Cardiomyocyte Injury Following Acute Ischemic Stroke) 116 (NCT03892226) | Prospective, observational, single‐center cohort study | Berlin, Germany | Acute ischemic stroke with hospital admission <48 h after symptom onset | 300 | Quantify autonomic dysfunction and decipher downstream cardiac mechanisms leading to myocardial injury | Multimodal CMR, echocardiography, autonomic ECG markers, biobanking | Active, recruiting |
| 6 | Heart and Brain Study–Substudy of Whitehall II Imaging cohort 117 (NCT03335696) | Prospective, observational, cohort study | Oxford, United Kingdom | Retired British civil servants | 775 | Brain atrophy, cognitive decline | Vascular ultrasound, MRI (head) | Recruitment completed, extended follow‐up |
| 7 | InsuCor (Insular–Noninsular Stroke Underlying Cardiac Failure (DRKS00012454) | Prospective, observational, case–control study | Würzburg, Germany | Acute ischemic stroke (onset <3 d; with and without involvement of the insular lobe) | 180 | (New) systolic cardiac dysfunction, stroke, vascular events within 3 mo | Echocardiography, blood biomarker | Active, recruiting |
| 8 | MIRACLE (MR Evidence of Cardiac Inflammation Post‐Stroke Study) | Prospective, observational, cohort study | London, Ontario, Canada | Acute ischemic embolic stroke of undetermined source | 44 | NT‐proBNP, systemic inflammation, myocardial infarction, LV function, LA fibrosis | NT‐proBNP, inflammatory markers, gadolinium enhanced CMR imaging | Active, recruiting |
| 9 |
Multifactorial Risk Stratification in Patients With Ischemic Stroke or Transient Ischemic Attack and Structural, Inflammatory, or Arrhythmogenic Cardiac Disease 118 |
Prospective, observational, single‐center cohort study | Tübingen, Germany | Ischemic stroke or TIA admitted to hospital | 878 | Any stroke, mortality, ischemic stroke, TIA, systemic embolism, myocardial infarction, intracranial hemorrhage, major bleeding | Clinical follow‐up | Active, not recruiting |
| 10 |
PRAISE (Prediction of Acute Coronary Syndrome After Acute Ischemic Stroke) 119 (NCT3609385) |
Prospective, observational, multicenter cohort study | Multicenter, Germany | Acute ischemic stroke (<72 h) with troponin elevation | 251 | Presence of acute coronary syndrome, deaths, functional outcome, cardiovascular events | Coronary angiography, echocardiography, ECG | Recruitment completed. Follow‐up ongoing |
| 11 | Predicting the Development of Myocardial Depression in Acute Neurological Patients (NCT03801694) | Prospective, observational single‐center cohort study | Columbus, Ohio, USA | Female patients with acute ischemic stroke or patients with SAH, >50 y, predicted to be on norepinephrine infusion for at least 48 h | 10 | Stress‐induced cardiomyopathy | ST‐T changes on ECG, echocardiography and measurement of catecholamines and troponin | Active and recruiting |
| 12 |
PROSCIS (Prospective Cohort With Incident Stroke) 120 |
Prospective, observational, hospital‐based cohort study | Berlin (PROSCIS‐B); Munich (PROSCIS‐M), Germany | First ever acute stroke (including intracerebral hemorrhage in Berlin) | 627 with first‐ever ischemic stroke (Berlin), 850 (Munich) | Composite of stroke, myocardial infarction, and vascular death (within 3 y) | Clinical follow‐up including cerebral MRI, cognitive testing | Recruitment completed (Berlin); active (Munich). |
| 13 |
RIC‐ACS (Protective Effects of Remote Ischemic Conditioning in Elderly With Acute Ischemic Stroke Complicating Acute Coronary Syndrome) |
Randomized, controlled, double‐blind, trial (sham procedure) | Beijing, China | Acute ischemic stroke (onset <24 h) plus acute coronary syndrome (onset <24 h), elderly patients (≤60 y) | 80 | Any death and recurrence of cardiac and cerebrovascular ischemic events within 3 mo | Remote ischemic conditioning (brief and transient limb ischemia) | Active and recruiting |
| 14 |
SICFAIL (Stroke‐Induced Cardiac Failure in Mice and Men) 40 (DRKS00011615) |
Prospective, observational, single‐center cohort study | Würzburg, Germany | Acute ischemic stroke with treatment on stroke unit | 696 | Heart failure, manifestation of cardiovascular disease | Follow‐up by mail or telephone, echocardiography and ECG | Recruitment completed. First results published. |
CMR indicates cardiovascular MRI; LA, left atrium; LAEF, left atrial ejection fraction; LV, left ventricle; MACE, major adverse cardiovascular events; MI, myocardial infarction; MR, magnetic resonance; MRI, magnetic resonance imaging; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; SAH, subarachnoid hemorrhage; TIA, transient ischemic attack; and TTS, Takotsubo syndrome.
Further open questions concern in particular the longer‐term outcomes of stroke patients with stroke–heart syndrome, especially with regard to chronic cardiac function and vascular end points. At present, it is not clear if the high frequency of systolic and diastolic dysfunction detected in stroke patients is transient and recovers over time or whether this will result in clinically manifest heart failure. Approximately one‐third of patients with early poststroke cardiac dysfunction have known heart failure before the stroke. 45 More longitudinal clinical studies are needed to determine the time‐course, reversibility, and progression of heart failure in stroke patients. Among others, the SICFAIL (Stroke‐Induced Cardiac Failure in Mice and Men) study aims to characterize and provide mechanistic understanding for long‐term cardiac dysfunction after stroke. 40
The biggest task for the future will be to prove how to specifically treat stroke–heart syndrome and its long‐term consequences. From our point of view, it seems conclusive that stroke patients with ACS should primarily be treated with coronary revascularization. 126 , 127 In contrast, patients with other manifestations of stroke–heart syndrome may benefit from a pathophysiologically oriented drug therapy. This could consist of targeting the sympathetic nervous system (eg, with β‐blockers), heart rate control, and improvement of blood pressure variability (eg, β‐blockers, ivabradine, renin–angiotensin system inhibitors), anti‐inflammatory approaches (eg, inhibition of the inflammasome or inflammatory cytokines like interleukin‐1β with colchicine or canakinumab), improvement of vascular endothelial function, and reduction of oxidative stress (eg, with statins, renin–angiotensin system inhibitors, antioxidants, new generation monoamine oxidase inhibitors), a combined antithrombotic medication (eg, temporary use of dual antiplatelets or adding low‐dose anticoagulation), and avoiding proarrhythmic drugs (QTc prolongation). Until now, however, none of these candidates have formally been tested in appropriate randomized and blinded therapy studies. Joint research initiatives led by interdisciplinary teams of cardiologists and neurologists from both preclinical and clinical perspectives are needed to reduce the knowledge gap and improve the clinical management of stroke‐associated cardiac complications.
Sources of Funding
M.E. received funding from DFG under Germany’s Excellence Strategy – EXC‐2049 ‐ 390688087, Collaborative Research Center ReTune TRR 295‐ 424778381, BMBF, DZNE, DZHK, EU, Corona Foundation, and Fondation Leducq.
Disclosures
Dr Scheitz reports a research grant from the Corona‐Stiftung, outside the submitted work. Dr Sposato reports personal fees from Boehringer Ingelheim and Pfizer, and grants from Bayer, outside the submitted work. Dr Backs reports personal fees from Lead Discovery Center (LDC) Dortmund, outside the submitted work. Dr Endres reports grants from Bayer and fees paid to the Charité from Abbot, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, Daiichi Sankyo, Sanofi, Novartis, and Pfizer, all outside the submitted work. The remaining authors have no disclosures to report.
For Sources of Funding and Disclosures, see page 14.
References
- 1. Scheitz JF, Nolte CH, Doehner W, Hachinski V, Endres M. Stroke‐heart syndrome: clinical presentation and underlying mechanisms. Lancet Neurol. 2018;17:1109–1120. doi: 10.1016/S1474-4422(18)30336-3 [DOI] [PubMed] [Google Scholar]
- 2. Prosser J, MacGregor L, Lees KR, Diener HC, Hacke W, Davis S; Investigators V . Predictors of early cardiac morbidity and mortality after ischemic stroke. Stroke. 2007;38:2295–2302. doi: 10.1161/STROKEAHA.106.471813 [DOI] [PubMed] [Google Scholar]
- 3. Sposato LA, Hilz MJ, Aspberg S, Murthy SB, Bahit MC, Hsieh CY, Sheppard MN, Scheitz JF. Post‐stroke cardiovascular complications and neurogenic cardiac injury: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2020;76:2768–2785. doi: 10.1016/j.jacc.2020.10.009 [DOI] [PubMed] [Google Scholar]
- 4. Kallmunzer B, Breuer L, Kahl N, Bobinger T, Raaz‐Schrauder D, Huttner HB, Schwab S, Kohrmann M. Serious cardiac arrhythmias after stroke: incidence, time course, and predictors—a systematic, prospective analysis. Stroke. 2012;43:2892–2897. doi: 10.1161/STROKEAHA.112.664318 [DOI] [PubMed] [Google Scholar]
- 5. Lettow I, Jensen M, Schlemm E, Boutitie F, Quandt F, Cheng B, Ebinger M, Endres M, Fiebach JB, Thijs V, et al. Serious adverse events and their impact on functional outcome in acute ischemic stroke in the WAKE‐UP trial. Stroke. 2021;52:3768–3776. doi: 10.1161/STROKEAHA.120.033425 [DOI] [PubMed] [Google Scholar]
- 6. Johnston KC, Li JY, Lyden PD, Hanson SK, Feasby TE, Adams RJ, Faught RE Jr, Haley EC Jr. Medical and neurological complications of ischemic stroke: experience from the RANTTAS trial. RANTTAS Investigators. Stroke. 1998;29:447–453. doi: 10.1161/01.str.29.2.447 [DOI] [PubMed] [Google Scholar]
- 7. Buckley BJR, Harrison SL, Hill A, Underhill P, Lane DA, Lip GYH. Stroke‐heart syndrome: incidence and clinical outcomes of cardiac complications following stroke. Stroke. 2022;53:1759–1763. doi: 10.1161/STROKEAHA.121.037316 [DOI] [PubMed] [Google Scholar]
- 8. Scheitz JF, Mochmann HC, Erdur H, Tutuncu S, Haeusler KG, Grittner U, Laufs U, Endres M, Nolte CH. Prognostic relevance of cardiac troponin T levels and their dynamic changes measured with a high‐sensitivity assay in acute ischaemic stroke: analyses from the TRELAS cohort. Int J Cardiol. 2014;177:886–893. doi: 10.1016/j.ijcard.2014.10.036 [DOI] [PubMed] [Google Scholar]
- 9. Sposato LA, Lam M, Allen B, Richard L, Shariff SZ, Saposnik G. First‐ever ischemic stroke and increased risk of incident heart disease in older adults. Neurology. 2020;94(15):e1559–e1570. doi: 10.1212/WNL.0000000000009234 [DOI] [PubMed] [Google Scholar]
- 10. Hankey GJ, Jamrozik K, Broadhurst RJ, Forbes S, Burvill PW, Anderson CS, Stewart‐Wynne EG. Five‐year survival after first‐ever stroke and related prognostic factors in the Perth Community Stroke Study. Stroke. 2000;31:2080–2086. doi: 10.1161/01.str.31.9.2080 [DOI] [PubMed] [Google Scholar]
- 11. Rutten‐Jacobs LC, Arntz RM, Maaijwee NA, Schoonderwaldt HC, Dorresteijn LD, van Dijk EJ, de Leeuw FE. Long‐term mortality after stroke among adults aged 18 to 50 years. JAMA. 2013;309:1136–1144. doi: 10.1001/jama.2013.842 [DOI] [PubMed] [Google Scholar]
- 12. Samuels MA. Neurogenic heart disease: a unifying hypothesis. Am J Cardiol. 1987;60:15J–19J. doi: 10.1016/0002-9149(87)90678-3 [DOI] [PubMed] [Google Scholar]
- 13. Samuels MA. The brain‐heart connection. Circulation. 2007;116:77–84. doi: 10.1161/CIRCULATIONAHA.106.678995 [DOI] [PubMed] [Google Scholar]
- 14. Norris JW, Froggatt GM, Hachinski VC. Cardiac arrhythmias in acute stroke. Stroke. 1978;9:392–396. doi: 10.1161/01.str.9.4.392 [DOI] [PubMed] [Google Scholar]
- 15. Oppenheimer SM, Cechetto DF, Hachinski VC. Cerebrogenic cardiac arrhythmias. Cerebral electrocardiographic influences and their role in sudden death. Arch Neurol. 1990;47:513–519. doi: 10.1001/archneur.1990.00530050029008 [DOI] [PubMed] [Google Scholar]
- 16. Smith KE, Hachinski VC, Gibson CJ, Ciriello J. Changes in plasma catecholamine levels after insula damage in experimental stroke. Brain Res. 1986;375:182–185. doi: 10.1016/0006-8993(86)90973-x [DOI] [PubMed] [Google Scholar]
- 17. Hachinski VC, Wilson JX, Smith KE, Cechetto DF. Effect of age on autonomic and cardiac responses in a rat stroke model. Arch Neurol. 1992;49:690–696. doi: 10.1001/archneur.1992.00530310032009 [DOI] [PubMed] [Google Scholar]
- 18. Veltkamp R, Uhlmann S, Marinescu M, Sticht C, Finke D, Gretz N, Grone HJ, Katus HA, Backs J, Lehmann LH. Experimental ischaemic stroke induces transient cardiac atrophy and dysfunction. J Cachexia Sarcopenia Muscle. 2019;10:54–62. doi: 10.1002/jcsm.12335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vornholz L, Nienhaus F, Gliem M, Alter C, Henning C, Lang A, Ezzahoini H, Wolff G, Clasen L, Rassaf T, et al. Acute heart failure after Reperfused ischemic stroke: association with systemic and cardiac inflammatory responses. Front Physiol. 2021;12:782760. doi: 10.3389/fphys.2021.782760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Venkat P, Cui C, Chen Z, Chopp M, Zacharek A, Landschoot‐Ward J, Culmone L, Yang XP, Xu J, Chen J. CD133+ exosome treatment improves cardiac function after stroke in type 2 diabetic mice. Transl Stroke Res. 2021;12:112–124. doi: 10.1007/s12975-020-00807-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bieber M, Werner RA, Tanai E, Hofmann U, Higuchi T, Schuh K, Heuschmann PU, Frantz S, Ritter O, Kraft P, et al. Stroke‐induced chronic systolic dysfunction driven by sympathetic overactivity. Ann Neurol. 2017;82:729–743. doi: 10.1002/ana.25073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen J, Cui C, Yang X, Xu J, Venkat P, Zacharek A, Yu P, Chopp M. MiR‐126 affects brain‐heart interaction after cerebral ischemic stroke. Transl Stroke Res. 2017;8:374–385. doi: 10.1007/s12975-017-0520-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meloux A, Rigal E, Rochette L, Cottin Y, Bejot Y, Vergely C. Ischemic stroke increases heart vulnerability to ischemia‐reperfusion and alters myocardial cardioprotective pathways. Stroke. 2018;49:2752–2760. doi: 10.1161/strokeaha.118.022207 [DOI] [PubMed] [Google Scholar]
- 24. Yan T, Chen Z, Chopp M, Venkat P, Zacharek A, Li W, Shen Y, Wu R, Li L, Landschoot‐Ward J, et al. Inflammatory responses mediate brain‐heart interaction after ischemic stroke in adult mice. J Cereb Blood Flow Metab. 2020;40:1213–1229. doi: 10.1177/0271678X18813317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balint B, Jaremek V, Thorburn V, Whitehead SN, Sposato LA. Left atrial microvascular endothelial dysfunction, myocardial inflammation and fibrosis after selective insular cortex ischemic stroke. Int J Cardiol. 2019;292:148–155. doi: 10.1016/j.ijcard.2019.06.004 [DOI] [PubMed] [Google Scholar]
- 26. Min J, Farooq MU, Greenberg E, Aloka F, Bhatt A, Kassab M, Morgan JP, Majid A. Cardiac dysfunction after left permanent cerebral focal ischemia: the brain and heart connection. Stroke. 2009;40:2560–2563. doi: 10.1161/STROKEAHA.108.536086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gallacher KI, Jani BD, Hanlon P, Nicholl BI, Mair FS. Multimorbidity in stroke. Stroke. 2019;50:1919–1926. doi: 10.1161/STROKEAHA.118.020376 [DOI] [PubMed] [Google Scholar]
- 28. Elamy AH, Shuaib A, Carriere KC, Jeerakathil T. Common comorbidities of stroke in the Canadian population. Can J Neurol Sci. 2020;47:314–319. doi: 10.1017/cjn.2020.17 [DOI] [PubMed] [Google Scholar]
- 29. Savitz SI, Baron JC, Fisher M; Consortium SX . Stroke treatment academic industry roundtable X: brain cytoprotection therapies in the reperfusion era. Stroke. 2019;50:1026–1031. doi: 10.1161/STROKEAHA.118.023927 [DOI] [PubMed] [Google Scholar]
- 30. Lin HB, Wei GS, Li FX, Guo WJ, Hong P, Weng YQ, Zhang QQ, Xu SY, Liang WB, You ZJ, et al. Macrophage‐NLRP3 inflammasome activation exacerbates cardiac dysfunction after ischemic stroke in a mouse model of diabetes. Neurosci Bull. 2020;36:1035–1045. doi: 10.1007/s12264-020-00544-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wrigley P, Khoury J, Eckerle B, Alwell K, Moomaw CJ, Woo D, Flaherty ML, Rios DL, la Rosa F, Mackey J, et al. Prevalence of positive troponin and echocardiogram findings and association with mortality in acute ischemic stroke. Stroke. 2017;48:1226–1232. doi: 10.1161/STROKEAHA.116.014561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ahn SH, Kim YH, Shin CH, Lee JS, Kim BJ, Kim YJ, Noh SM, Kim SM, Kang HG, Kang DW, et al. Cardiac vulnerability to cerebrogenic stress as a possible cause of troponin elevation in stroke. J Am Heart Assoc. 2016;5:e004135. doi: 10.1161/JAHA.116.004135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sposato LA, Lam M, Allen B, Shariff SZ, Saposnik G. First‐ever ischemic stroke and incident major adverse cardiovascular events in 93 627 older women and men. Stroke. 2020;51:11–19. doi: 10.1161/STROKEAHA.119.028066 [DOI] [PubMed] [Google Scholar]
- 34. Jung JM, Kim JG, Kim JB, Cho KH, Yu S, Oh K, Kim YH, Choi JY, Seo WK. Takotsubo‐like myocardial dysfunction in ischemic stroke: a hospital‐based registry and systematic literature review. Stroke. 2016;47:2729–2736. doi: 10.1161/STROKEAHA.116.014304 [DOI] [PubMed] [Google Scholar]
- 35. Cammann VL, Scheitz JF, von Rennenberg R, Jancke L, Nolte CH, Szawan KA, Stengl H, Wurdinger M, Endres M, Templin C, et al. Clinical correlates and prognostic impact of neurologic disorders in Takotsubo syndrome. Sci Rep. 2021;11:23555. doi: 10.1038/s41598-021-01496-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roth S, Singh V, Tiedt S, Schindler L, Huber G, Geerlof A, Antoine DJ, Anfray A, Orset C, Gauberti M, et al. Brain‐released alarmins and stress response synergize in accelerating atherosclerosis progression after stroke. Sci Transl Med. 2018;10:eaao1313. doi: 10.1126/scitranslmed.aao1313 [DOI] [PubMed] [Google Scholar]
- 37. Hinterdobler J, Schott S, Jin H, Meesmann A, Steinsiek AL, Zimmermann AS, Wobst J, Muller P, Mauersberger C, Vilne B, et al. Acute mental stress drives vascular inflammation and promotes plaque destabilization in mouse atherosclerosis. Eur Heart J. 2021;42:4077–4088. doi: 10.1093/eurheartj/ehab371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scheitz JF, Stengl H, Nolte CH, Landmesser U, Endres M. Neurological update: use of cardiac troponin in patients with stroke. J Neurol. 2021;268:2284–2292. doi: 10.1007/s00415-020-10349-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fure B, Bruun WT, Thommessen B. Electrocardiographic and troponin T changes in acute ischaemic stroke. J Intern Med. 2006;259:592–597. doi: 10.1111/j.1365-2796.2006.01639.x [DOI] [PubMed] [Google Scholar]
- 40. Heuschmann PU, Montellano FA, Ungethüm K, Rücker V, Wiedmann S, Mackenrodt D, Quilitzsch A, Ludwig T, Kraft P, Albert J, et al. Prevalence and determinants of systolic and diastolic cardiac dysfunction and heart failure in acute ischemic stroke patients: the SICFAIL study. ESC Heart Fail. 2021;8:1117–1129. doi: 10.1002/ehf2.13145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheitz JF, Erdur H, Haeusler KG, Audebert HJ, Roser M, Laufs U, Endres M, Nolte CH. Insular cortex lesions, cardiac troponin, and detection of previously unknown atrial fibrillation in acute ischemic stroke: insights from the troponin elevation in acute ischemic stroke study. Stroke. 2015;46:1196–1201. doi: 10.1161/STROKEAHA.115.008681 [DOI] [PubMed] [Google Scholar]
- 42. Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, Biller J, Brown M, Demaerschalk BM, Hoh B, et al. 2018 guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2018;49:e46–e110. doi: 10.1161/STR.0000000000000158 [DOI] [PubMed] [Google Scholar]
- 43. Faiz KW, Einvik G, Brekke PH, Omland T. Cardiac troponin T increase in patients with acute ischemic stroke with and without cancer. Clin Chem. 2018;64:404–406. doi: 10.1373/clinchem.2017.280016 [DOI] [PubMed] [Google Scholar]
- 44. Hellwig S, Grittner U, Elgeti M, Wyschkon S, Nagel SN, Fiebach JB, Krause T, Herm J, Scheitz JF, Endres M, et al. Evaluation of left ventricular function in patients with acute ischaemic stroke using cine cardiovascular magnetic resonance imaging. ESC Heart Fail. 2020;7:2572–2580. doi: 10.1002/ehf2.12833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Siedler G, Sommer K, Macha K, Marsch A, Breuer L, Stoll S, Engelhorn T, Dorfler A, Arnold M, Schwab S, et al. Heart failure in ischemic stroke: relevance for acute care and outcome. Stroke. 2019;50:3051–3056. doi: 10.1161/STROKEAHA.119.026139 [DOI] [PubMed] [Google Scholar]
- 46. Sposato LA, Cipriano LE, Saposnik G, Ruiz Vargas E, Riccio PM, Hachinski V. Diagnosis of atrial fibrillation after stroke and transient ischaemic attack: a systematic review and meta‐analysis. Lancet Neurol. 2015;14:377–387. doi: 10.1016/S1474-4422(15)70027-X [DOI] [PubMed] [Google Scholar]
- 47. Mochmann HC, Scheitz JF, Petzold GC, Haeusler KG, Audebert HJ, Laufs U, Schneider C, Landmesser U, Werner N, Endres M, et al. Coronary angiographic findings in acute ischemic stroke patients with elevated cardiac troponin: the troponin elevation in acute ischemic stroke (TRELAS) study. Circulation. 2016;133:1264–1271. doi: 10.1161/CIRCULATIONAHA.115.018547 [DOI] [PubMed] [Google Scholar]
- 48. Alqahtani F, Aljohani S, Tarabishy A, Busu T, Adcock A, Alkhouli M. Incidence and outcomes of myocardial infarction in patients admitted with acute ischemic stroke. Stroke. 2017;48:2931–2938. doi: 10.1161/STROKEAHA.117.018408 [DOI] [PubMed] [Google Scholar]
- 49. Scheitz JF, Endres M, Mochmann HC, Audebert HJ, Nolte CH. Frequency, determinants and outcome of elevated troponin in acute ischemic stroke patients. Int J Cardiol. 2012;157:239–242. doi: 10.1016/j.ijcard.2012.01.055 [DOI] [PubMed] [Google Scholar]
- 50. Kruska M, Kolb A, Fastner C, Mildenberger I, Hetjens S, Kittel M, Bail K, Behnes M, Akin I, Borggrefe M, et al. Coronary artery disease in patients presenting with acute ischemic stroke or transient ischemic attack and elevated troponin levels. Front Neurol. 2021;12:781553. doi: 10.3389/fneur.2021.781553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Krause T, Werner K, Fiebach JB, Villringer K, Piper SK, Haeusler KG, Endres M, Scheitz JF, Nolte CH. Stroke in right dorsal anterior insular cortex is related to myocardial injury. Ann Neurol. 2017;81:502–511. doi: 10.1002/ana.24906 [DOI] [PubMed] [Google Scholar]
- 52. Seifert F, Kallmunzer B, Gutjahr I, Breuer L, Winder K, Kaschka I, Kloska S, Doerfler A, Hilz MJ, Schwab S, et al. Neuroanatomical correlates of severe cardiac arrhythmias in acute ischemic stroke. J Neurol. 2015;262:1182–1190. doi: 10.1007/s00415-015-7684-9 [DOI] [PubMed] [Google Scholar]
- 53. Guan L, Wang Y, Claydon VE, Mazowita G, Wang Y, Brant R, Collet JP. Autonomic parameter and stress profile predict secondary ischemic events after transient ischemic attack or minor stroke. Stroke. 2019;50:2007–2015. doi: 10.1161/STROKEAHA.118.022844 [DOI] [PubMed] [Google Scholar]
- 54. Scheitz JF, Lim J, Broersen LHA, Ganeshan R, Huo S, Sperber PS, Piper SK, Heuschmann PU, Audebert HJ, Nolte CH, et al. High‐sensitivity cardiac troponin T and recurrent vascular events after first ischemic stroke. J Am Heart Assoc. 2021;10:e018326. doi: 10.1161/JAHA.120.018326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. von Rennenberg R, Siegerink B, Ganeshan R, Villringer K, Doehner W, Audebert HJ, Endres M, Nolte CH, Scheitz JF. High‐sensitivity cardiac troponin T and severity of cerebral white matter lesions in patients with acute ischemic stroke. J Neurol. 2019;266:37–45. doi: 10.1007/s00415-018-9085-3 [DOI] [PubMed] [Google Scholar]
- 56. Broersen LHA, Siegerink B, Sperber PS, von Rennenberg R, Piper SK, Nolte CH, Heuschmann PU, Endres M, Scheitz JF, Liman TG. High‐sensitivity cardiac troponin T and cognitive function in patients with ischemic stroke. Stroke. 2020;51:1604–1607. doi: 10.1161/STROKEAHA.119.028410 [DOI] [PubMed] [Google Scholar]
- 57. Sposato LA, Fridman S, Whitehead SN, Lopes RD. Linking stroke‐induced heart injury and neurogenic atrial fibrillation: a hypothesis to be proven. J Electrocardiol. 2018;51:430–432. doi: 10.1016/j.jelectrocard.2018.02.006 [DOI] [PubMed] [Google Scholar]
- 58. Sposato LA, Chaturvedi S, Hsieh CY, Morillo CA, Kamel H. Atrial fibrillation detected after stroke and transient ischemic attack: a novel clinical concept challenging current views. Stroke. 2022;53:e94–e103. doi: 10.1161/STROKEAHA.121.034777 [DOI] [PubMed] [Google Scholar]
- 59. Fridman S, Jimenez‐Ruiz A, Vargas‐Gonzalez JC, Sposato LA. Differences between atrial fibrillation detected before and after stroke and TIA: a systematic review and meta‐analysis. Cerebrovasc Dis. 2022;51:152–157. doi: 10.1159/000520101 [DOI] [PubMed] [Google Scholar]
- 60. Sposato LA, Cerasuolo JO, Cipriano LE, Fang J, Fridman S, Paquet M, Saposnik G; Group PS . Atrial fibrillation detected after stroke is related to a low risk of ischemic stroke recurrence. Neurology. 2018;90:e924–e931. doi: 10.1212/WNL.0000000000005126 [DOI] [PubMed] [Google Scholar]
- 61. Yang XM, Rao ZZ, Gu HQ, Zhao XQ, Wang CJ, Liu LP, Liu C, Wang YL, Li ZX, Xiao RP, et al. Atrial fibrillation known before or detected after stroke share similar risk of ischemic stroke recurrence and death. Stroke. 2019;50:1124–1129. doi: 10.1161/strokeaha.118.024176 [DOI] [PubMed] [Google Scholar]
- 62. Yaghi S, Kamel H. Stratifying stroke risk in atrial fibrillation: beyond clinical risk scores. Stroke. 2017;48:2665–2670. doi: 10.1161/STROKEAHA.117.017084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sposato LA, Cipriano LE, Riccio PM, Hachinski V, Saposnik G. Very short paroxysms account for more than half of the cases of atrial fibrillation detected after stroke and TIA: a systematic review and meta‐analysis. Int J Stroke. 2015;10:801–807. doi: 10.1111/ijs.12555 [DOI] [PubMed] [Google Scholar]
- 64. Tahsili‐Fahadan P, Geocadin RG. Heart‐brain axis: effects of neurologic injury on cardiovascular function. Circ Res. 2017;120:559–572. doi: 10.1161/CIRCRESAHA.116.308446 [DOI] [PubMed] [Google Scholar]
- 65. Craig AD. How do you feel—now? The anterior insula and human awareness. Nat Rev Neurosci. 2009;10:59–70. doi: 10.1038/nrn2555 [DOI] [PubMed] [Google Scholar]
- 66. Beissner F, Meissner K, Bar KJ, Napadow V. The autonomic brain: an activation likelihood estimation meta‐analysis for central processing of autonomic function. J Neurosci. 2013;33:10503–10511. doi: 10.1523/JNEUROSCI.1103-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liesirova K, Abela E, Pilgrim T, Bickel L, Meinel T, Meisterernst J, Rajeev V, Sarikaya H, Heldner MR, Dobrocky T, et al. Baseline troponin T level in stroke and its association with stress cardiomyopathy. PLoS One. 2018;13:e0209764. doi: 10.1371/journal.pone.0209764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kumral D, Schaare HL, Beyer F, Reinelt J, Uhlig M, Liem F, Lampe L, Babayan A, Reiter A, Erbey M, et al. The age‐dependent relationship between resting heart rate variability and functional brain connectivity. Neuroimage. 2019;185:521–533. doi: 10.1016/j.neuroimage.2018.10.027 [DOI] [PubMed] [Google Scholar]
- 69. Thayer JF, Ahs F, Fredrikson M, Sollers JJ III, Wager TD. A meta‐analysis of heart rate variability and neuroimaging studies: implications for heart rate variability as a marker of stress and health. Neurosci Biobehav Rev. 2012;36:747–756. doi: 10.1016/j.neubiorev.2011.11.009 [DOI] [PubMed] [Google Scholar]
- 70. Mueller K, Thiel F, Beutner F, Teren A, Frisch S, Ballarini T, Moller HE, Ihle K, Thiery J, Schuler G, et al. Brain damage with heart failure: cardiac biomarker alterations and gray matter decline. Circ Res. 2020;126:750–764. doi: 10.1161/CIRCRESAHA.119.315813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Je G, Sun Y, Keyhanian K, Yaghi S, Henninger N. Dorsal vagal nucleus involvement relates to QTc‐prolongation after acute medullary infarction. Acta Neurol Scand. 2021;144:283–287. doi: 10.1111/ane.13445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lassman AB, Mayer SA. Paroxysmal apnea and vasomotor instability following medullary infarction. Arch Neurol. 2005;62:1286–1288. doi: 10.1001/archneur.62.8.1286 [DOI] [PubMed] [Google Scholar]
- 73. Cereda CW, Tamisier R, Manconi M, Andreotti J, Frangi J, Pifferini V, Bassetti CL. Endothelial dysfunction and arterial stiffness in ischemic stroke: the role of sleep‐disordered breathing. Stroke. 2013;44:1175–1178. doi: 10.1161/STROKEAHA.111.000112 [DOI] [PubMed] [Google Scholar]
- 74. Siccoli MM, Valko PO, Hermann DM, Bassetti CL. Central periodic breathing during sleep in 74 patients with acute ischemic stroke—neurogenic and cardiogenic factors. J Neurol. 2008;255:1687–1692. doi: 10.1007/s00415-008-0981-9 [DOI] [PubMed] [Google Scholar]
- 75. Tobaldini E, Proserpio P, Oppo V, Figorilli M, Fiorelli EM, Manconi M, Agostoni EC, Nobili L, Montano N, Horvath T, et al. Cardiac autonomic dynamics during sleep are lost in patients with TIA and stroke. J Sleep Res. 2020;29:e12878. doi: 10.1111/jsr.12878 [DOI] [PubMed] [Google Scholar]
- 76. Hiestand T, Hanggi J, Klein C, Topka MS, Jaguszewski M, Ghadri JR, Luscher TF, Jancke L, Templin C. Takotsubo syndrome associated with structural brain alterations of the limbic system. J Am Coll Cardiol. 2018;71:809–811. doi: 10.1016/j.jacc.2017.12.022 [DOI] [PubMed] [Google Scholar]
- 77. Templin C, Hanggi J, Klein C, Topka MS, Hiestand T, Levinson RA, Jurisic S, Luscher TF, Ghadri JR, Jancke L. Altered limbic and autonomic processing supports brain‐heart axis in Takotsubo syndrome. Eur Heart J. 2019;40:1183–1187. doi: 10.1093/eurheartj/ehz068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Radfar A, Abohashem S, Osborne MT, Wang Y, Dar T, Hassan MZO, Ghoneem A, Naddaf N, Patrich T, Abbasi T, et al. Stress‐associated neurobiological activity associates with the risk for and timing of subsequent Takotsubo syndrome. Eur Heart J. 2021;42:1898–1908. doi: 10.1093/eurheartj/ehab029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tawakol A, Ishai A, Takx RA, Figueroa AL, Ali A, Kaiser Y, Truong QA, Solomon CJ, Calcagno C, Mani V, et al. Relation between resting amygdalar activity and cardiovascular events: a longitudinal and cohort study. Lancet. 2017;389:834–845. doi: 10.1016/S0140-6736(16)31714-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gulke E, Gelderblom M, Magnus T. Danger signals in stroke and their role on microglia activation after ischemia. Ther Adv Neurol Disord. 2018;11:1756286418774254. doi: 10.1177/1756286418774254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dorrance AM, Fink G. Effects of stroke on the autonomic nervous system. Compr Physiol. 2015;5:1241–1263. doi: 10.1002/cphy.c140016 [DOI] [PubMed] [Google Scholar]
- 82. Liu Y, Feng S, Subedi K, Wang H. Attenuation of ischemic stroke‐caused brain injury by a monoamine oxidase inhibitor involves improved proteostasis and reduced neuroinflammation. Mol Neurobiol. 2020;57:937–948. doi: 10.1007/s12035-019-01788-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, Chen K, Gabrielson KL, Blakely RD, Shih JC, et al. Monoamine oxidase A‐mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. 2010;106:193–202. doi: 10.1161/CIRCRESAHA.109.198366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tang S, Xiong L, Fan Y, Mok VCT, Wong KS, Leung TW. Stroke outcome prediction by blood pressure variability, heart rate variability, and baroreflex sensitivity. Stroke. 2020;51:1317–1320. doi: 10.1161/STROKEAHA.119.027981 [DOI] [PubMed] [Google Scholar]
- 85. Jimenez‐Ruiz A, Racosta JM, Kimpinski K, Hilz MJ, Sposato LA. Cardiovascular autonomic dysfunction after stroke. Neurol Sci. 2021;42:1751–1758. doi: 10.1007/s10072-021-05128-y [DOI] [PubMed] [Google Scholar]
- 86. Tian G, Xiong L, Leung H, Soo Y, Leung T, Wong LK. Beat‐to‐beat blood pressure variability and heart rate variability in relation to autonomic dysregulation in patients with acute mild‐moderate ischemic stroke. J Clin Neurosci. 2019;64:187–193. doi: 10.1016/j.jocn.2019.03.003 [DOI] [PubMed] [Google Scholar]
- 87. Stead LG, Gilmore RM, Vedula KC, Weaver AL, Decker WW, Brown RD Jr. Impact of acute blood pressure variability on ischemic stroke outcome. Neurology. 2006;66:1878–1881. doi: 10.1212/01.wnl.0000219628.78513.b5 [DOI] [PubMed] [Google Scholar]
- 88. Saaby L, Poulsen TS, Hosbond S, Larsen TB, Pyndt Diederichsen AC, Hallas J, Thygesen K, Mickley H. Classification of myocardial infarction: frequency and features of type 2 myocardial infarction. Am J Med. 2013;126:789–797. doi: 10.1016/j.amjmed.2013.02.029 [DOI] [PubMed] [Google Scholar]
- 89. Weil BR, Suzuki G, Young RF, Iyer V, Canty JM Jr. Troponin release and reversible left ventricular dysfunction after transient pressure overload. J Am Coll Cardiol. 2018;71:2906–2916. doi: 10.1016/j.jacc.2018.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Vasamsetti SB, Florentin J, Coppin E, Stiekema LCA, Zheng KH, Nisar MU, Sembrat J, Levinthal DJ, Rojas M, Stroes ESG, et al. Sympathetic neuronal activation triggers myeloid progenitor proliferation and differentiation. Immunity. 2018;49:93–106.e7. doi: 10.1016/j.immuni.2018.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Monteiro S, Pinho AG, Macieira M, Serre‐Miranda C, Cibrao JR, Lima R, Soares‐Cunha C, Vasconcelos NL, Lentilhas‐Graca J, Duarte‐Silva S, et al. Splenic sympathetic signaling contributes to acute neutrophil infiltration of the injured spinal cord. J Neuroinflammation. 2020;17:282. doi: 10.1186/s12974-020-01945-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhao Q, Yan T, Chopp M, Venkat P, Chen J. Brain‐kidney interaction: renal dysfunction following ischemic stroke. J Cereb Blood Flow Metab. 2020;40:246–262. doi: 10.1177/0271678X19890931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Singh V, Roth S, Llovera G, Sadler R, Garzetti D, Stecher B, Dichgans M, Liesz A. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J Neurosci. 2016;36:7428–7440. doi: 10.1523/JNEUROSCI.1114-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Weinreuter M, Kreusser MM, Beckendorf J, Schreiter FC, Leuschner F, Lehmann LH, Hofmann KP, Rostosky JS, Diemert N, Xu C, et al. CaM kinase II mediates maladaptive post‐infarct remodeling and pro‐inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol Med. 2014;6:1231–1245. doi: 10.15252/emmm.201403848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J. CaMKIIdelta‐mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight. 2018;3:e97054. doi: 10.1172/jci.insight.97054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jin H, Chen Y, Wang B, Zhu Y, Chen L, Han X, Ma G, Liu N. Association between brain‐derived neurotrophic factor and von Willebrand factor levels in patients with stable coronary artery disease. BMC Cardiovasc Disord. 2018;18:23. doi: 10.1186/s12872-018-0762-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Feng N, Huke S, Zhu G, Tocchetti CG, Shi S, Aiba T, Kaludercic N, Hoover DB, Beck SE, Mankowski JL, et al. Constitutive BDNF/TrkB signaling is required for normal cardiac contraction and relaxation. Proc Natl Acad Sci USA. 2015;112:1880–1885. doi: 10.1073/pnas.1417949112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mojtabavi H, Shaka Z, Momtazmanesh S, Ajdari A, Rezaei N. Circulating brain‐derived neurotrophic factor as a potential biomarker in stroke: a systematic review and meta‐analysis. J Transl Med. 2022;20:126. doi: 10.1186/s12967-022-03312-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Connors JM, Levy JH. Thromboinflammation and the hypercoagulability of COVID‐19. J Thromb Haemost. 2020;18:1559–1561. doi: 10.1111/jth.14849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Austin AW, Wissmann T, von Kanel R. Stress and hemostasis: an update. Semin Thromb Hemost. 2013;39:902–912. doi: 10.1055/s-0033-1357487 [DOI] [PubMed] [Google Scholar]
- 101. Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, Bikdeli B, Ahluwalia N, Ausiello JC, Wan EY, et al. Extrapulmonary manifestations of COVID‐19. Nat Med. 2020;26:1017–1032. doi: 10.1038/s41591-020-0968-3 [DOI] [PubMed] [Google Scholar]
- 102. Sandoval Y, Januzzi JL Jr, Jaffe AS. Cardiac troponin for assessment of myocardial injury in COVID‐19: JACC review topic of the week. J Am Coll Cardiol. 2020;76:1244–1258. doi: 10.1016/j.jacc.2020.06.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ramirez SH, Andrews AM, Paul D, Pachter JS. Extracellular vesicles: mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS. 2018;15:19. doi: 10.1186/s12987-018-0104-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Singhal M, Gengenbacher N, Abdul Pari AA, Kamiyama M, Hai L, Kuhn BJ, Kallenberg DM, Kulkarni SR, Camilli C, Preuss SF, et al. Temporal multi‐omics identifies LRG1 as a vascular niche instructor of metastasis. Sci Transl Med. 2021;13:eabe6805. doi: 10.1126/scitranslmed.abe6805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Litvinukova M, Talavera‐Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, et al. Cells of the adult human heart. Nature. 2020;588:466–472. doi: 10.1038/s41586-020-2797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hoyer FF, Naxerova K, Schloss MJ, Hulsmans M, Nair AV, Dutta P, Calcagno DM, Herisson F, Anzai A, Sun Y, et al. Tissue‐specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity. 2019;51:899–914.e7. doi: 10.1016/j.immuni.2019.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bleilevens C, Roehl AB, Zoremba N, Tolba R, Rossaint R, Hein M. Insular infarct size but not levosimendan influenced myocardial injury triggered by cerebral ischemia in rats. Exp Brain Res. 2015;233:149–156. doi: 10.1007/s00221-014-4096-5 [DOI] [PubMed] [Google Scholar]
- 109. Murphy EP, Crean D. Molecular interactions between NR4A orphan nuclear receptors and NF‐kappaB are required for appropriate inflammatory responses and immune cell homeostasis. Biomolecules. 2015;5:1302–1318. doi: 10.3390/biom5031302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Borchert T, Hubscher D, Guessoum CI, Lam TD, Ghadri JR, Schellinger IN, Tiburcy M, Liaw NY, Li Y, Haas J, et al. Catecholamine‐dependent beta‐adrenergic signaling in a pluripotent stem cell model of Takotsubo cardiomyopathy. J Am Coll Cardiol. 2017;70:975–991. doi: 10.1016/j.jacc.2017.06.061 [DOI] [PubMed] [Google Scholar]
- 111. Lehmann LH, Jebessa ZH, Kreusser MM, Horsch A, He T, Kronlage M, Dewenter M, Sramek V, Oehl U, Krebs‐Haupenthal J, et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat Med. 2018;24:62–72. doi: 10.1038/nm.4452 [DOI] [PubMed] [Google Scholar]
- 112. Larsen BS, Aplin M, Host N, Dominguez H, Christensen H, Christensen LM, Havsteen I, Prescott E, Jensen GB, Vejlstrup N, et al. Atrial cardiomyopathy in patients with ischaemic stroke: a cross‐sectional and prospective cohort study‐the COAST study. BMJ Open. 2022;12:e061018. doi: 10.1136/bmjopen-2022-061018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Siegerink B, Weber J, Ahmadi M, Eckardt KU, Edelmann F, Endres M, Gerhardt H, Haubold K, Hübner N, Landmesser U, et al. Disease overarching mechanisms that explain and predict outcome of patients with high cardiovascular risk: rationale and design of the Berlin long‐term observation of vascular events (BeLOVE) study. medRxiv. 2019.
- 114. Kelly PJ, Murphy S, Coveney S, Purroy F, Lemmens R, Tsivgoulis G, Price C. Anti‐inflammatory approaches to ischaemic stroke prevention. J Neurol Neurosurg Psychiatry. 2018;89:211–218. doi: 10.1136/jnnp-2016-314817 [DOI] [PubMed] [Google Scholar]
- 115. Kelly P, Weimar C, Lemmens R, Murphy S, Purroy F, Arsovska A, Bornstein NM, Czlonkowska A, Fischer U, Fonseca AC, et al. Colchicine for prevention of vascular inflammation in non‐CardioEmbolic stroke (CONVINCE)—study protocol for a randomised controlled trial. Eur Stroke J. 2021;6:222–228. doi: 10.1177/2396987320972566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Stengl H, Ganeshan R, Hellwig S, Blaszczyk E, Fiebach JB, Nolte CH, Bauer A, Schulz‐Menger J, Endres M, Scheitz JF. Cardiomyocyte injury following acute ischemic stroke: protocol for a prospective observational cohort study. JMIR Res Protoc. 2021;10:e24186. doi: 10.2196/24186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Suri S, Bulte D, Chiesa ST, Ebmeier KP, Jezzard P, Rieger SW, Pitt JE, Griffanti L, Okell TW, Craig M, et al. Study protocol: the Heart and Brain Study. Front Physiol. 2021;12:643725. doi: 10.3389/fphys.2021.643725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Poli S, Siebert E, Mbroh J, Poli K, Krumbholz M, Mengel A, Greulich S, Hartig F, Muller KAL, Bocksch W, et al. Closure or medical therapy of patent foramen ovale in cryptogenic stroke: prospective case series. Neurol Res Pract. 2021;3:16. doi: 10.1186/s42466-021-00114-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Nolte CH, von Rennenberg R, Litmeier S, Scheitz JF, Leistner DM, Blankenberg S, Dichgans M, Katus H, Petzold GC, Pieske B, et al. PRediction of acute coronary syndrome in acute ischemic StrokE (PRAISE)—protocol of a prospective, multicenter trial with central reading and predefined endpoints. BMC Neurol. 2020;20:318. doi: 10.1186/s12883-020-01903-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Liman TG, Zietemann V, Wiedmann S, Jungehuelsing GJ, Endres M, Wollenweber FA, Wellwood I, Dichgans M, Heuschmann PU. Prediction of vascular risk after stroke—protocol and pilot data of the Prospective Cohort with Incident Stroke (PROSCIS). Int J Stroke. 2013;8:484–490. doi: 10.1111/j.1747-4949.2012.00871.x [DOI] [PubMed] [Google Scholar]
- 121. Linde JJ, Kelbaek H, Hansen TF, Sigvardsen PE, Torp‐Pedersen C, Bech J, Heitmann M, Nielsen OW, Hofsten D, Kuhl JT, et al. Coronary CT angiography in patients with non‐ST‐segment elevation acute coronary syndrome. J Am Coll Cardiol. 2020;75:453–463. doi: 10.1016/j.jacc.2019.12.012 [DOI] [PubMed] [Google Scholar]
- 122. Sorensson P, Ekenback C, Lundin M, Agewall S, Bacsovics Brolin E, Caidahl K, Cederlund K, Collste O, Daniel M, Jensen J, et al. Early comprehensive cardiovascular magnetic resonance imaging in patients with myocardial infarction with nonobstructive coronary arteries. JACC Cardiovasc Imaging. 2021;14:1774–1783. doi: 10.1016/j.jcmg.2021.02.021 [DOI] [PubMed] [Google Scholar]
- 123. Antiochos P, Ge Y, Steel K, Bingham S, Abdullah S, Mikolich JR, Arai AE, Bandettini WP, Patel AR, Farzaneh‐Far A, et al. Imaging of clinically unrecognized myocardial fibrosis in patients with suspected coronary artery disease. J Am Coll Cardiol. 2020;76:945–957. doi: 10.1016/j.jacc.2020.06.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Faragli A, Merz S, Muzio FPL, Doeblin P, Tanacli R, Kolp C, Abawi D, Otvos J, Stehning C, Schnackenburg B, et al. Estimation of total collagen volume: a T1 mapping versus histological comparison study in healthy Landrace pigs. Int J Cardiovasc Imaging. 2020;36:1761–1769. doi: 10.1007/s10554-020-01881-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Everett RJ, Treibel TA, Fukui M, Lee H, Rigolli M, Singh A, Bijsterveld P, Tastet L, Musa TA, Dobson L, et al. Extracellular myocardial volume in patients with aortic stenosis. J Am Coll Cardiol. 2020;75:304–316. doi: 10.1016/j.jacc.2019.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Neumann FJ, Sousa‐Uva M, Ahlsson A, Alfonso F, Banning AP, Benedetto U, Byrne RA, Collet JP, Falk V, Head SJ, et al. 2018 ESC/EACTS guidelines on myocardial revascularization. Eur Heart J. 2019;40:87–165. doi: 10.1093/eurheartj/ehy394 [DOI] [PubMed] [Google Scholar]
- 127. Collet JP, Thiele H, Barbato E, Barthelemy O, Bauersachs J, Bhatt DL, Dendale P, Dorobantu M, Edvardsen T, Folliguet T, et al. 2020 ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST‐segment elevation. Eur Heart J. 2021;42:1289–1367. doi: 10.1093/eurheartj/ehaa575 [DOI] [PubMed] [Google Scholar]