

SUMMARY
Heterochromatin functions as a scaffold for factors responsible for gene silencing and chromosome segregation. Heterochromatin can be assembled by multiple pathways, including RNAi and RNA surveillance. We identified factors that form heterochromatin using dense profiles of transposable element integration in Schizosaccharomyces pombe. The candidates include a large number of essential proteins such as four canonical mRNA cleavage and polyadenylation factors. We find that Iss1, a subunit of the poly(A) polymerase module, plays a role in forming heterochromatin in centromere repeats that is independent of RNAi. Genome-wide maps reveal that Iss1 accumulates at genes regulated by RNA surveillance. Iss1 interacts with RNA surveillance factors Mmi1 and Rrp6, and importantly, Iss1 contributes to RNA elimination that forms heterochromatin at meiosis genes. Our profile of transposable element integration supports the model that a network of mRNA cleavage and polyadenylation factors coordinates RNA surveillance, including the mechanism that forms heterochromatin at meiotic genes.
In Brief
Lee et al. use dense profiles of transposon integration to identify genes important for the formation of heterochromatin. Among many candidates, Iss1 is a canonical mRNA cleavage and polyadenylation factor found to be important for heterochromatin at meiotic genes by recruiting the nuclear exosome.
Graphical Abstract
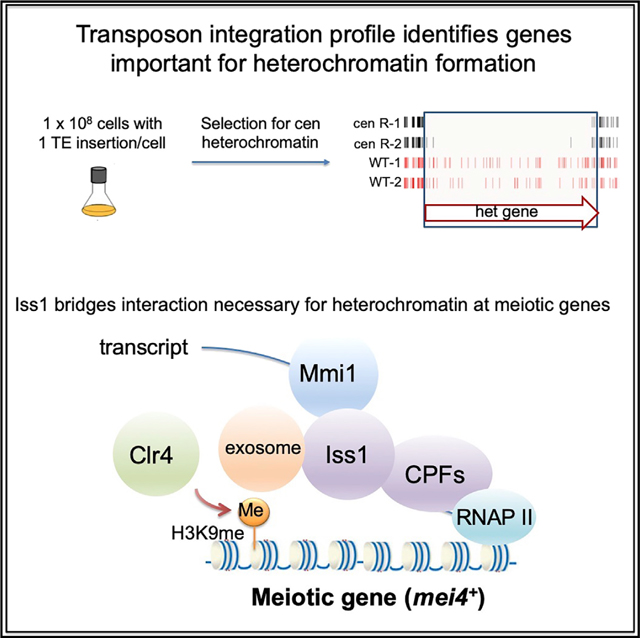
INTRODUCTION
In eukaryotes, the assembly of DNA into highly condensed heterochromatin is critical for a broad range of functions related to genome integrity (Allshire and Madhani, 2018). The methylation of histone H3 on lysine 9 (H3K9me) is central to the formation of heterochromatin by creating binding sites for a range of chromatin proteins important for silencing transposable elements, chromosome segregation, and the epigenetic inheritance that maintains chromatin with H3K9me from one generation to the next. Schizosaccharomyces pombe is an excellent model for studying the molecular mechanisms that generate and regulate heterochromatin (Allshire and Ekwall, 2015; Martienssen and Moazed, 2015; Wang et al., 2016). A single histone methyltransferase, Clr4, is responsible for H3K9me wherever it occurs in the genome. Centromeres, subtelomeres, and the mating-type region are packaged into constitutive heterochromatin, while meiosis genes are silenced by facultative heterochromatin that dissociates when cells are limited for nitrogen (Cam et al., 2005; Tashiro et al., 2013; Zofall et al., 2012). Importantly, Clr4 is recruited to heterochromatin regions by multiple mechanisms.
Constitutive heterochromatin results from RNAi factors that include Ago1-containing RNA-induced transcriptional silencing complex (RITS) (Verdel et al., 2004). The Clr4-containing complex (CLRC) is recruited to chromatin by RITS through a bridging factor, Stc1 (Bayne et al., 2010; Hong et al., 2005; Horn et al., 2005; Noma et al., 2004; Zhang et al., 2008). RNAi-independent mechanisms for producing heterochromatin include a process mediated by the nuclear exosome that produces constitutive heterochromatin in centromeric repeats (Reyes-Turcu et al., 2011). In addition, facultative heterochromatin at meiosis genes is independent of RNAi and relies on RNA elimination factors Red1, Mmi1, the exosome (Tashiro et al., 2013; Zofall et al., 2012), and conserved exoribonuclease Dhp1 that couples pre-mRNA 3′-end processing to transcription termination (Chalamcharla et al., 2015). Competition between the exosome pathway and RNAi factors can be observed in the absence of Rrp6, as cells readily form RNAi-dependent heterochromatin in discreet sets of genes (HOODs) (Yamanaka et al., 2013). These parallel pathways for generating H3K9me provide a key framework for understanding how heterochromatin forms at different types of sequences and growth conditions. However, gaps exist in understanding how different RNA processing activities such as RNAi and the exosome function to target heterochromatin and how these pathways compete.
One limitation in understanding which factors control levels of H3K9me is that most proteins were identified in screens of deletion strains that cannot include essential genes. An alternative approach for identifying gene function is high-throughput sequencing of integration profiles, also known as Tn-seq, which identifies genes important for growth in a wide range of selective conditions (Kwon et al., 2016; van Opijnen and Camilli, 2013). Genes necessary to sustain growth under one condition but not others do not tolerate insertions in the selective condition. Tn-seq has been applied specifically to characterize gene function in bacteria. However, this method has recently been validated for identifying essential genes in a number of yeast species (Guo et al., 2013; Michel et al., 2017; Sanchez et al., 2019; Segal et al., 2018; Zhu et al., 2018). The great potential of this approach is that with the increasing throughput of sequencing technology, functions of individual domains can be observed, including in proteins required for viability (Michel et al., 2017).
With the goal of identifying factors important for forming heterochromatin, we produced dense profiles of integrations using a silencing reporter positioned in centromere heterochromatin. We identified a total of 199 candidate factors involved in silencing expression in the centromere, a full 65 of which are essential for viability. Although many of these candidates have established roles in forming heterochromatin, the majority have no previous association. Importantly, four of the essential proteins are components of the canonical mRNA cleavage and polyadenylation complex. One candidate, Iss1, is implicated by homology to Saccharomyces cerevisiae as tethering the poly(A) polymerase Pla1 to the cleavage and polyadenylation factor (CPF) subcomplex (Preker et al., 1995). In S. pombe, iss1 was identified in a genetic screen for factors that participate in RNA elimination that inhibits the expression of meiotic genes (Yamashita et al., 2013). Our results show that Iss1 is important for forming heterochromatin in centromeric repeats and that this function does not depend on RNAi. Importantly, we found that Iss1 interacts with the exosome factor Rrp6 and limits expression of genes under surveillance of the nuclear exosome, including meiosis genes and iron-regulated genes. In addition, we found that Iss1 interacts with RNA elimination factor Mmi1 and contributes to forming heterochromatin at meiosis genes. Genome-wide chromatin immunoprecipitation sequencing (ChIP-seq) maps revealed Iss1 accumulates at meiosis genes and genes under surveillance by Rrp6.
RESULTS
To identify pathways and factors that promote heterochromatin structure, we generated high-density profiles of transposable element integration in S. pombe cells that contained a heterochromatin reporter. Integration of the Hermes transposable element that disrupted genes required for heterochromatin structure resulted in expression of a silenced ura4 reporter positioned in the outer centromere repeats of chromosome 1 (otr1R::ura4). Passaging of cells in medium containing 5-fluoroorotic acid (FOA) restricted growth when ura4 was expressed, causing integrations in heterochromatin genes to be depleted. Candidate genes important for heterochromatin were identified from passages with FOA on the basis of having lower integration densities in cells that contained otr1R::ura4 compared with the densities in control cells that lacked otr1R::ura4 (Figure 1A). Cultures were passaged for five generations in FOA to identify genes with large contributions to heterochromatin, while 80 generations were used to provide a broad list of candidates. Integration sites were determined in two biologically independent cultures each for cells with and without otr1R::ura4, and combining the replicas provided an average of 954,000 independent insertions for each strain and condition. The total integration in each of the eight cultures passaged in FOA averaged one insertion for every 29 bp of the genome (Table S1). Genes with well-established roles in heterochromatin assembly, such as mit1 and sir2, had significantly fewer insertions in the cells with the centromere reporter otr1R::ura4 compared with cells lacking the reporter (Shankaranarayana et al., 2003; Sugiyama et al., 2007; Figure 1B).
Figure 1. Dense Maps of Transposable Element Integration Identify Genes Important for Heterochromatin at Centromere Repeats.

(A) Single insertions of the transposable element Hermes were generated in cells with WT cen1 and cen1 otr1R::ura4. Cultures were passaged in FOA for 5 or 80 generations. Cells with insertions in heterochromatin genes (het1) express ura4 and cannot grow in FOA. After growth on FOA, fewer insertions were detected in het genes in cells with cen1 otr1R::ura4.
(B) Genes involved in forming centromere heterochromatin, such as mit1 and sir2, had fewer inserts in cells with the cen1 otr1R::ura4 (black, duplicate libraries) relative to cells with WT cen1 (red, duplicate libraries).
(C) ORFs of candidate heterochromatin genes (blue) had lower relative integration densities in cells with cen1 otr1R::ura4 compared with cells with WT cen1.
We assessed the potential contribution to heterochromatin structure of all open reading frames (ORFs) by comparing their insert densities in cells lacking the otr1R::ura4 reporter relative to densities in cells with the centromere reporter. To generate a conservative list of candidates likely to promote heterochromatin, we selected for each ORF the control replicate with the lowest integration density and the replicate of the centromere reporter with the highest densities. When these integration densities were plotted, the vast majority of ORFs fell above the diagonal, indicating little contribution to centromere heterochromatin (Figure 1C). ORFs were chosen to be candidates with potential roles in heterochromatin if the selected control replicate had a density 2-fold or higher than the replicate of the strain with the centromere reporter. The list of candidates was a total of 199 genes (Table S2); 28 were identified in cultures grown five generations in FOA, and 184 resulted from cultures grown for 80 generations in FOA (Figure 2A). We were surprised to find that 65 of the candidates were essential for viability (Figure 2B). This was unexpected, because integration densities in essential ORFs are low in strains lacking the ura4 reporter (Guo et al., 2013). Upon closer inspection, we found that these essential genes were candidates because they tolerated many insertions in their 3′ sequences that reduced heterochromatin function but not viability. As an example, Iss1 is shown in Figure S1.
Figure 2. Candidates with Heterochromatin Function Include Five Essential Factors Important for mRNA Cleavage and Polyadenylation.

(A and B) Venn diagrams of candidates identified after 5 and 80 generations of growth in FOA (A). Among 199 candidate genes, 65 are required for viability (B).
(C) GO analysis of all candidate factors showed significant enrichment for functions of centromere heterochromatin and siRNA. p values corrected for false discovery rates; *p < 0.025, **p < 3.5 × 10−3, ***p < 2.5 × 10−4, and ****p < 3.5 × 10−6.
(D) GO analysis of the 40 RNA-processing genes in (C) determined relative to genome frequency.
(E) Canonical complexes cleavage factor 1A (CF1A; green), cleavage and polyadenylation factor (CPF; blue), nuclease module (Ysh1 and Cft2), and cleavage factor 1B (CF1B; gray).
Initial Evaluation of Heterochromatin Candidates
In an initial evaluation of the candidates for a role in heterochromatin formation, we examined multi-subunit complexes reported to contribute to heterochromatin formation. FACT, Lid2, Mediator, the spliceosome, TFIID, SAGA, and CPF are complexes with at least one subunit that contributes to heterochromatin formation. Twenty-five of the candidates from our screen not previously associated with heterochromatin are subunits of these complexes (Table 1). These multi-subunit complexes provide strong evidence that a large number of our candidates are indeed involved in heterochromatin formation.
Table 1.
Candidates Not Previously Associated with Heterochromatin Formation Are Components of Complexes with Reported Roles in Heterochromatin
| Complexes | Reported Role in Heterochromatin Formation | Identified Candidates |
|---|---|---|
| FACT complex | Pob3 | Pob3, Spt16a,b, Nhp6b |
| Lid2 complex | Lid2 | Lid2a, Jmj3a,b, Snt2b |
| Mediator | Med8, Med18, Med20, Hrp1 | Med1b, Med20, Srb8b |
| Splicing factors | Prp5, Prp8, Prp10, Prp12, Prp16, Cwf10, Prp39 | Cwf2a,b, Cwf11b, Prp11a,b, Prp31a,b, Saf3a,b, Sap49a,b, Smd2a,b, SPAC30D11.14cb, Srp2a,b, Uaf2a,b |
| TFIID complex | TBP (Drosophila) | Taf1a,b, Taf4a,b, Taf7a,b |
| SAGA complex | Gcn5, Ada3, Tra1 | Sgf11b, Ubp8b |
| Cleavage and polyadenylation | Pla1 | Ysh1a,b, Iss1a,b, Rna14a,b, Msi2a,b |
Essential genes.
Factors not previously associated with heterochromatin formation.
In another approach to validate the candidates from our screen, we tested a representative sample of five candidates for a role in centromere heterochromatin by measuring silencing of the otr1R::ura4 reporter. Deletion of these five candidate genes (hpt1, dus3, pub2, mak2, and cat1) resulted in increased growth on media lacking uracil, indicating that they play a role in centromere silencing (Figure S2). Mak2 is a histidine kinase in a pathway that responds to oxidative stress (Buck et al., 2001), and Cat1 is a plasma membrane amino acid transporter (Aspuria and Tamanoi, 2008). These candidates suggest the possibility that centromere heterochromatin may be regulated by environmental conditions.
Gene Ontology (GO) analysis of the 199 candidates showed strong enrichments for functions in chromatin silencing at centromere outer repeats, production of small interfering RNA (siRNA) involved in chromatin silencing, regulation of histone methylation, and chromatin silencing at mating-type cassette (Figure 2C). These enrichments were strongly significant and demonstrated that the integration profile had succeeded in identifying factors important for heterochromatin structure. The candidate list included all four factors that produce siRNA involved in silencing and 11 of 26 (42%) of the proteins that silence the outer centromere repeats (Allshire and Ekwall, 2015; Allshire and Madhani, 2018; Allshire et al., 1995). Although 42% represents a considerable fraction of factors known to promote heterochromatin at centromere repeats, it is not comprehensive, as mutations in these genes can result in slow growth in the strain that lacks the otr1R::ura4 reporter, which masks the contributions to heterochromatin in the strain with the reporter. Although our screen did not identify all 26 factors linked to silencing of the outer centromere repeats, it is important to point out these previously determined factors are all nonessential for viability. The advantage of our screen is that it identified 65 essential gene candidates and also had the capability to identify 133 nonessential gene candidates.
Heterochromatin Candidates Included a Module of Polyadenylation and Cleavage Factors
Although the enriched ontologies were consistent with previously identified contributions to heterochromatin, RNA processing stood out because it contains a substantial number of genes not previously linked to heterochromatin structure. The candidates classified as RNA-processing factors were evaluated for subsets of function by determining enrichment for just these 40 genes relative to genome frequency (Figure 2D). Among the RNA-processing factor candidates, a substantial number are involved in mRNA cleavage, polyadenylation, and 3′-end processing. Strikingly, four of the RNA-processing candidates form an interaction module of canonical mRNA polyadenylation and cleavage factors as predicted from highly homologous proteins in Saccharomyces cerevisiae (Casañal et al., 2017; Ezeokonkwo et al., 2011; Vanoosthuyse et al., 2014; Wood et al., 2012; Yang and Doublié, 2011). These factors are Msi2, Rna14, Iss1, and Ysh1 (Figure 2E, black dashed outlines). A fifth candidate, Ssu72, is a catalytic subunit of the phosphatase module that associates with CPF and promotes 3′ mRNA cleavage (He et al., 2003; Vanoosthuyse et al., 2014). We were particularly interested in testing polyadenylation and cleavage candidates for function in promoting heterochromatin in centromere repeats because Pla1, the poly(A) polymerase, has previously been shown to be involved in heterochromatin structure of sexual differentiation genes and retrotransposons (Yamanaka et al., 2013).
Iss1 Promotes Heterochromatin Structure in Outer Centromere Repeats
To determine whether polyadenylation and cleavage contribute to heterochromatin structure at the centromere repeats, we focused on the function of Iss1, a subunit of CPF (Vanoosthuyse et al., 2014; Yang and Doublié, 2011). We generated a C-terminal truncation of Iss1 removing 38 amino acids that, on the basis of the Hermes insertions, were important for silencing the otr1R::ura4 reporter. Two independently generated truncations of Iss1 (Iss1-ΔC) showed no growth restriction on non-selective medium (Figure 3A). However, the Iss1-ΔC mutation caused a silencing defect as demonstrated by growth in the absence of uracil and reduced growth with FOA. Although the defects in silencing were apparent, they were not as strong as in cells lacking the sole histone H3K9 methyltransferase, Clr4. The defects in silencing resulting from the Iss1-ΔC mutation coincided with increased expression of the otr1R::ura4 reporter and reduced levels of H3K9 dimethylation (H3K9me2) (Figure 3B). These results demonstrate the Hermes screen correctly identified Iss1 as important for heterochromatin structure at the otr1R::ura4 reporter.
Figure 3. Iss1 Is Important for RNAi-Independent Heterochromatin Formation at Centromere Repeats.

(A) Growth phenotypes of cells with the silencing reporter (otr1R:ura4+) using medium lacking uracil (−URA) and medium containing FOA. clr4Δ lacks H3K9me and illustrates phenotypes when heterochromatin is absent.
(B) ura4 mRNA levels measured by qRT-PCR and H3K9me2 enrichment associated with otr1R:ura4+ measured by ChIP relative to adh1 (p ≤ 0.05).
(C and D) TBZ sensitivity of cells lacking the silencing reporter (otr1R:ura4+). Strains with clr4Δ and swi6Δ are controls that illustrate sensitivity (C). Ectopic expression of GFP-iss1+ demonstrates complementation of the chromosomal allele iss1-ΔC (D).
(E) qRT-PCR measures of cen-dg transcripts relative to adh1 (p ≤ 0.01).
(F) H3K9me2 associated with cen-dg measured by ChIP relative to adh1 (**p ≤ 0.01, ***p ≤ 0.001).
We next explored whether Iss1 contributes to heterochromatin of centromere repeats in cells that lack the otr1R::ura4 reporter. Factors that affect centromeric heterochromatin assembly are in general defective in chromosome segregation and show sensitivity to spindle-poisoning drug thiabendazole (TBZ). We therefore tested whether cells carrying the iss1 mutant allele also display TBZ sensitivity. Consistent with a role in centromere structure, independent Iss1-ΔC truncations showed significant sensitivity to TBZ equivalent to cells lacking the Hp1 protein Swi6 (Figure 3C). Resistance to TBZ was rescued when we expressed a GFP-Iss1 fusion, indicating the mutant phenotype was due to loss of Iss1 function (Figure 3D). However, in these cells lacking the reporter, the iss1-ΔC mutation caused little or no change in the expression and H3K9me2 level of the outer centromere dg repeats (Figures 3E and 3F). In experiments to test for a role in heterochromatin structure that is independent of RNAi, we added the iss1-ΔC mutation to cells lacking Ago1. Importantly, in cells lacking Ago1, the iss1-ΔC mutation caused a significant increase in expression of the centromere dg repeats and a substantial reduction of dg H3K9me2 (Figures 3E and 3F). These data demonstrate Iss1 makes a significant contribution to heterochromatin structure at centromere repeats when the RNAi system is compromised.
Iss1 Restricts Expression of Genes Regulated by the Nuclear Exosome
Truncation of an essential polyadenylation and cleavage factor could potentially cause defects in transcription termination, resulting in considerable changes in expression genome-wide. In addition, genes with altered expression could include factors important for heterochromatin structure, which could similarly manifest as defects in heterochromatin formation. To address these questions, we analyzed wild-type and iss1-ΔC cells by strand-specific RNA sequencing (RNA-seq) using three biological replicates to validate statistical significance. Truncation of Iss1 resulted in 73 genes with increased expression and 86 with reduced levels based on changes that were two-fold or greater (Figure 4A). Importantly, these lists do not include genes that have an established role in forming heterochromatin structure (Table S3).
Figure 4. Iss1 Is Involved in 3′ Processing and RNA Elimination.

(A) RNA isolated from three biological replicas of iss1+ and iss1-ΔC cells was characterized using RNA-seq. Listed are the number of genes with expression altered 2-fold or more.
(B) RNA-seq reads mapped to annotated transcripts divided into 15 equal segments. Reads that aligned to intergenic sequences upstream or downstream were mapped in 100 bp bins by bp number from the nearest annotated transcript.
(C) RNA-seq reads that mapped downstream of annotated transcripts as in (B) shown with an expanded y axis.
(D) Maps of RNA-seq reads for highly expressed genes. Red vertical lines bracket reads that mapped downstream of annotated transcripts.
(E) Gene Ontology enrichment for genes that had increased expression in cells with iss1-ΔC. *Pyruvate metabolic process, false discovery rate (FDR)-corrected p ≤ 0.022; *iron assimilation, FDR-corrected p ≤ 0.011.
(F) Gene Ontology enrichment of genes with increased expression in cells with rrp6Δ (Mukherjee et al., 2016). *Hypergeometric distance p ≤ 0.05.
(G) Overlap of genes with increased expression in cells with iss1-ΔC and rrp6Δ (hypergeometric distance p ≤ 2.8 × 10−5).
To determine the impact of the iss1-ΔC mutation on overall transcript levels and lengths, we performed a meta-analysis of RNA levels. Each ORF was divided into 15 equal sized segments, and the RNA reads for all ORFs were divided into these segments and averaged. Maps of RNA reads across all transcripts show that the iss1-ΔC mutation caused little overall change in distribution (Figure 4B). However, there was a noticeable shift extending the 3′ end of transcripts in the RNA from the iss1-ΔC cells. Although the percentage of total reads was relatively small, the iss1-ΔC mutation caused a reproducible and statistically significant extension of the 3′ end of transcripts by about 200 nt (Figure 4C; Figure S3). The 3′ extension of transcripts can be visualized most clearly for genes with high levels of expression, such as ribosomal protein genes (Figure 4D). Together, these results indicate the C-terminal truncation of Iss1 resulted in relatively few changes in genome-wide expression but for some genes a noticeable extension in the 3′ ends of transcripts.
The 73 genes with increased expression in the iss1-ΔC mutant were evaluated for common functions and were strongly enriched for factors important for iron assimilation (Figure 4E; Gene Ontology [GO]: 0033212). Interestingly, a similar increase in iron assimilation genes occurs in cells lacking Rrp6, the 3′−5′ exonuclease subunit of the nuclear exosome (Mukherjee et al., 2016; Figure 4F). The overlap of genes upregulated by iss1-ΔC and rrp6Δ is strongly significant and extends beyond iron assimilation to include meiotic-induced genes and noncoding RNAs (Figure 4G; Table S4). The similarity in their upregulated genes suggests Rrp6 and Iss1 may participate in a common mechanism of regulation.
Iss1 Functions with RNA Elimination Factors and the Exosome to Assemble Meiotic Genes into Heterochromatin Islands
Rrp6, as a key subunit of the nuclear exosome plays an important role in RNA surveillance that includes the degradation of meiotic transcripts expressed during vegetative growth (Yamanaka et al., 2013; Yamanaka et al., 2010). It is significant that Rrp6 is also required at meiotic genes such as mei4 and ssm4 for the facultative heterochromatin that assembles during vegetative growth (Zofall et al., 2012). We therefore asked whether the role Iss1 plays in forming heterochromatin extends to meiotic genes. We found the iss1-ΔC mutation did result in increased accumulation of ssm4 and mei4 mRNA (Figure 5A). Consistent with a role in forming heterochromatin, the iss1-ΔC mutation significantly reduced H3K9me2 at both ssm4 and mei4 (Figure 5B). Rrp6 plays a direct role at ssm4 and mei4 by associating to chromatin in a complex of RNA-binding proteins that recruits the H3K9 histone methyltransferase Clr4 (Zofall et al., 2012). With ChIP assays, we found that Iss1 is also enriched at mei4 indicating Iss1 may play a direct role in forming the heterochromatin at these genes (Figure 5C). The association of Iss1with mei4 is maintained with iss1-ΔC.
Figure 5. Iss1 Functions with the Exosome to Form Heterochromatin Associated with Meiosis Genes.

(A and B) mRNA relative to adh1 measured by qRT-PCR (A) and H3K9me2 measured by ChIP relative to adh1 (B) of ssm4 and mei4. *p ≤ 0.05 and **p ≤ 0.01.
(C) Enrichment of Iss1 and Iss1-ΔC at mei4. **p ≤ 0.01.
(D) Co-immunoprecipitation of Rrp6 associated with Iss1.
(E) Co-immunoprecipitation of Mmi1 associated with Iss1.
(F) Co-immunoprecipitation of Iss1 and Iss1-ΔC associated with Pla1.
(G) Network of RNA elimination factors that form heterochromatin at meiosis genes.
(H) Immunofluorescence microscopy of S. pombe expressing Pla1-GFP and mCherry-Iss1.
The elimination of meiotic mRNAs during vegetative growth depends on the RNA-binding protein Mmi1 to bind the determinant of selective removal (DSR) sequence in order to recruit the exosome (Harigaya et al., 2006; Yamanaka et al., 2010). Factors important for Mmi1-dependent mRNA elimination include Rrp6, poly(A)-binding protein Pab2, and polyadenylation and cleavage factors Pla1 and Rna15 (Egan et al., 2014; Lee et al., 2013; St-André et al., 2010; Yamanaka et al., 2010). Co-immunoprecipitation (coIP) experiments, affinity purifications, and two-hybrid assays demonstrate that Mmi1 forms a network of interactions with Rrp6, Pab2, Pla1, and Rna15 (Egan et al., 2014; Harigaya et al., 2006; Yamanaka et al., 2010). Our coIP experiments revealed Iss1 interacts with Rrp6, Mmi1, and Pla1, indicating that Iss1 is associated with this network of elimination factors (Figures 5D–5G), and the association with Pla1 is independent of the C-terminal residues deleted in iss1-ΔC (Figure S5). Importantly, we found that the iss1-ΔC truncation did disrupt its interaction with Mmi1 (Figure 5F). However, the truncation did not reduce Iss1 interaction with Rrp6 (Figure S5).
Additional evidence that the RNA elimination factors Mmi1, Rrp6, Pab2, and Pla1 interact in vivo is that they co-localize into sub-nuclear dot structures (Harigaya et al., 2006; Lemay et al., 2010; Sugiyama and Sugioka-Sugiyama, 2011; Yamanaka et al., 2010). We found that Iss1 assembles into nuclear dots, and these co-localized with Pla1, indicating that Iss1 also assembles in vivo with RNA elimination factors (Figure 5H).
Iss1 Associates with Genes Regulated by Mmi1 and Rrp6 Including Heterochromatin Islands
Iss1 associates physically with Rrp6 and Mmi1, and it contributes to the heterochromatin structure at meiotic genes mei4 and ssm4. These data suggest that Iss1 may play a direct role in RNA elimination and the formation of heterochromatin islands across the genome. We explored this possibility by mapping the chromatin interactions of Iss1 genome-wide. By mapping ChIP-seq reads relative to input, we identified a total of 886 Iss1 peaks with p values less than 0.05 (Table S5). The ChIP-seq data confirmed Iss1 binding at meiotic-regulated genes mei4 and ssm4 (Figure 6A). Importantly, Iss1 binding formed a peak in the center of mei4 that correlates with the DSR, the position at which Mmi1 binding triggers RNA elimination (Harigaya et al., 2006; Yamanaka et al., 2010). The binding of Iss1 at ssm4 was positioned at the 3′ end of the gene, which is also the location of the DSR (Harigaya et al., 2006). However, this position at the 3′ end also contains cleavage and adenylation signals (Chalamcharla et al., 2015). The ChIP-seq results demonstrate that Iss1 also forms binding peaks at a number of other heterochromatin islands (Figure 6A).
Figure 6. Iss1 Is Enriched at Genes that Are Regulated by the RNA Elimination and at Genes Transcribed by RNA Pol I and Pol III.

(A) ChIP-seq results for Iss1 and H3K9me2 are mapped to regions of meiotic Islands.
(B) ChIP-seq peak intensity of Iss1 accumulation mapped to ORFs divided into 15 equal segments. ChIP-seq peaks of Iss1 were mapped to all annotated ORFs divided into 15 equal segments. Peaks that aligned to intergenic sequences upstream or downstream were mapped in 100 bp bins by bp number from the nearest annotated ORF.
(C) Accumulation of Iss1 ChIP-seq reads relative to the 3′ end of annotated transcripts. Iss1-binding peaks relative to all annotated transcription termination sites were averaged.
(D) Number of Iss1 peaks overlapping genes sorted into ten groups ranked by expression.
In an effort to delineate the roles of Iss1 in RNA elimination versus the cleavage and polyadenylation of mRNAs, we determined the distribution of Iss1 binding by averaging the ChIP-seq peaks across all gene bodies divided into 15 equal-sized segments. The majority of Iss1 binding clustered 400 bp 3′ of coding sequences of genes, although a modest amount of Iss1 also clustered upstream of ORFs (Figure S4). By excluding ChIP-seq data of Iss1 bound to tandem intragenic sequences that could be associated with the 3′ or 5′ ends of genes, we found that Iss1 interacts primarily with the 3′ ends of genes, consistent with its role as a subunit of the canonical cleavage and polyadenylation complex CPF (Figure 6B). Mapping Iss1-binding peaks relative to all annotated transcription termination sites (TTSs) showed that maximum interaction occurred approximately 200 bp downstream of the TTSs (Figure 6C). The position of this peak suggests that the bulk of Iss1 detected was involved in canonical mRNA cleavage and adenylation. To explore the functional relevance of Iss1 binding at the 3′ end of genes, we tested the possibility that genes with high levels of transcription would require more Iss1 to perform cleavage and polyadenylation. When we organized the 6,732 genes of S. pombe into ten equal-sized groups ranked by levels of expression, the group with the highest RNA levels had significantly more genes with overlapping peaks of Iss1 binding (Figure 6D). Of all the 675 genes that contain an overlapping peak of Iss1, 215 (30.3%) are in the group of the most highly expressed genes. Thus, the genes in the group of highest expression have a 3.0-fold enrichment of Iss1 overlapping peaks relative to all genes (p = 1.8 × 1−59). The relatively similar amount of Iss1 binding to genes in the first nine groups and the sharp increase of Iss1 binding in the group with the greatest expression suggest that Iss1 accumulation increases significantly when transcription levels exceed a threshold amount.
Iss1 Regulates Genes that Are Controlled by RNA Elimination
The interaction of Iss1 with Rrp6 and Mmi1 and the finding that the heterochromatin islands mei4 and ssm4 were silenced by Iss1 led us to ask whether other genes controlled by RNA elimination are regulated by Iss1. Although a subset of Iss1-bound genes are highly expressed and are associated with the function of Iss1 in mRNA processing, most Iss1-bound genes have lower expression. By surveying the Iss1-bound genes with lower levels of expression (Figure 6D), we found that many have previously been shown to lie in heterochromatin islands, regulated by Mmi1, or controlled by Rrp6 (Figure 6A; Zofall et al., 2012). Importantly, we found that the iss1-ΔC mutation caused significant increases in RNA levels of these genes. To determine whether Iss1 enrichment at genes plays a significant role in RNA elimination, we tested for significant associations between genes with Iss1 enrichment and groups of genes known to be controlled by Rrp6 or Mmi1. Of 261 genes that are upregulated in the absence of Rrp6 (Mukherjee et al., 2016), a significantly greater number than expected by chance (19%, p = 0.04) contained an Iss1-binding site (Table S6). With RNA immunoprecipitation (RNA-IP), 30 RNAs were found to bind specifically to Mmi1 (Touat-Todeschini et al., 2017). Here too we found a significant enrichment of associated genes that had Iss1 peaks (43%, p = 1.4 × 10−4). Of the 36 genes assembled into heterochromatin islands (Zofall et al., 2012), there were significantly more than expected by chance that had Iss1 enrichment (28%, p = 0.03). Taken together, chromatin-bound Iss1 showed a strong correlation with genes regulated by mechanisms of RNA elimination and genes assembled into heterochromatin.
We tested the function of Iss1 bound to regions of heterochromatin at meiotic genes by measuring changes in H3K9me2 levels resulting from the iss1-ΔC mutation. ChIP-seq results demonstrate that the levels of H3K9me2 associated with Iss1 peaks were significantly reduced by the iss1-ΔC mutation (Figure 6A). These results demonstrate that Iss1 plays an important role in heterochromatin formation at meiotic islands.
DISCUSSION
Transposable Element Profiles
Dense integration profiles (Tn-seq) of transposable elements in bacteria are now widely used to pinpoint pathogenic genes or virulence factors that play major roles in a variety of infections. With this innovation, virulence and antibiotic response genes have been defined for pathogens responsible for tuberculosis (Carey et al., 2018), malaria (Zhang et al., 2018), urinary tract infections (Armbruster et al., 2017), and dysentery (Li et al., 2017), to name a few. This recent innovation has also identified fitness phenotypes for thousands of unannotated genes in diverse bacteria (Price et al., 2018). Until recently, the insights resulting from transposable element profiles were strictly limited to studies of bacteria. The discovery of transposable elements with high integration efficiency in yeast species has now opened the door to profiling gene function in cells that possess eukaryotic features that are widely conserved in vertebrates, such as organelles, cytoskeletal structures, and heterochromatin. Initial profiles in yeast used the transposable element Hermes to reveal which genes of S. pombe are required for growth (Guo et al., 2013). The maize Ac/Ds system has strong mobility in S. cerevisiae and was used to detect genes required for growth and resistance to rapamycin (Michel et al., 2017). Hermes profiles of S. cerevisiae have also been used to identify genes that provide resistance to prions (Edskes et al., 2018). Our application of Hermes profiles to identify genes important for heterochromatin formation demonstrates the versatility and universal significance of the approach, especially given that we were able to identify large numbers of essential genes with our screen. In particular, the simultaneous identification of four essential components of the mRNA cleavage and polyadenylation complex by our Hermes profile illustrates a valuable feature of integration profiles that is not obtainable with other high-throughput genetic screens, including CRISPR. The validation of our screen by the identification of many known silencing and heterochromatin factors and our experiments that confirm a role of Iss1 in promoting H3K9me2 demonstrate that our list of candidates is a valuable resource for future studies on heterochromatin.
Redundancy of Machinery that Forms Centromere Heterochromatin
We found that Iss1 contributed significantly to the silencing function and heterochromatin structure of the centromere, but the majority of this function is redundant with RNAi. However, in the absence of RNAi factors such as Ago1, the iss1-ΔC mutation resulted in sharp increases in cen-dg RNA and decreases in cen-dg H3K9me2 (Figures 3E and 3F). RNAi-independent centromere heterochromatin has been observed previously and was found to require the nuclear exosome, as demonstrated in cells lacking rrp6 (Reyes-Turcu et al., 2011). Heterochromatin at the environmentally regulated meiosis genes in islands also relies on rrp6 and RNA elimination (St-André et al., 2010; Yamanaka et al., 2010; Zofall et al., 2012). Importantly, RNA elimination-mediated silencing of meiotic genes requires 3′ processing machinery such as CPFs Rna15 and Pcf11, the poly(A)-binding protein Pab2, and pre-mRNA 3′-end processing transcription termination factor Dhp1 (Chalamcharla et al., 2015; St-André et al., 2010; Yamanaka et al., 2010; Zofall et al., 2012). It is therefore reasonable to propose that the function of Iss1 in cleavage and polyadenylation promotes the formation of centromere heterochromatin mediated by Rrp6 and the exosome.
Iss1 Is a Component of the RNA Elimination Machinery that Forms Heterochromatin
RNA surveillance and elimination play key roles in suppressing a broad range of genes, including many activated during meiosis. For example, nuclear exosome factor Rrp6 selectively eliminates mRNAs from more than 100 genes (Mukherjee et al., 2016; Zhang et al., 2011). We found that Iss1 reduces amounts of many of the same mRNAs, including those of meiotic genes. This overlap with Rrp6-regulated mRNAs, and the interaction we detected between Iss1and Rrp6 indicates that Iss1 is a component of the RNA elimination pathway. Our finding that Iss1 participates in the RNA elimination of meiotic genes is independently supported by the identification of Iss1 in a genetic screen for mutations that reduced meiotic RNA elimination (Yamashita et al., 2013).
Meiosis-regulated genes, including mei4 and ssm4, are silenced by RNA elimination and the exosome by a mechanism that requires the RNA surveillance factor Red1 and CPFs Rna15 and Pcf11 (St-André et al., 2010; Yamanaka et al., 2010; Zofall et al., 2012). As the result of an interaction between Red1 and Clr4, RNA elimination is also responsible for the RNAi-independent formation of H3K9me heterochromatin that assembles on mei4, ssm4, and a number of other meiotic genes (Zofall et al., 2012). Our measures of H3K9me2 and the interactions between Iss1 and Mmi1 and Rrp6 demonstrate that Iss1 plays a direct role in the formation of heterochromatin at the meiotic genes. The role of Iss1 in forming heterochromatin at these genes likely functions together with Rna15, Pcf11, and Pla1. Given the cluster of polyadenylation and cleavage factors identified by our screen, it is also likely that Rna15, Pcf11, and Pla1 function together with Iss1 in this RNAi-independent formation of heterochromatin.
Other clusters of genes upregulated during sexual differentiation assemble into regions of heterochromatin in certain growth conditions or in an exosome mutant. These regions, termed HOODs, form heterochromatin that is dependent on the RNAi pathway (Yamanaka et al., 2013). Importantly, the formation of heterochromatin in HOODs relies on the poly(A) polymerase Pla1, the poly(A)-binding protein Pab2, and Red1 (Yamanaka et al., 2013). The activities of Pla1 and Pab2 are required for H3K9me2 (Yamanaka et al., 2013). This indicates that Pla1 and Pab2 serve as core machinery along with other factors to create H3K9me2-modified heterochromatin that is RNAi dependent at HOODs and possibly RNAi independent at islands. Our finding that Iss1 and potentially three other CPFs (Figure 2E) contribute to H3K9me2 of centromere repeats and meiosis genes indicates that multiple cleavage and adenylation factors serve as core machinery in producing heterochromatin.
Structural studies of CPFs demonstrate that within the poly(A) polymerase module, the S. cerevisiae homolog of Pla1 (Pap1) is tethered to the core factor of the module Cft1 by the S. cerevisiae homolog of Iss1, Fip1 (Casañal et al., 2017; Ezeokonkwo et al., 2011; Ghazy et al., 2009; Preker et al., 1995). In addition, Fip1 forms an interaction that tethers the poly(A) polymerase module to the CF IA complex through a contact with Rna14 (Casañal et al., 2017; Preker et al., 1995). Fip1 also plays a key role in the integrity of the cleavage and polyadenylation complex through its interaction with Yth1, which associates Pap1 to the ribonuclease Ysh1 (modeled in Figure 2E with S. pombe homologs). The multiple contacts of Fip1 indicate that it likely plays a key role as a hub in bringing together the polymerase Pap1, CF I1, substrate RNA, and Ysh1 (Figure 2E). This organization is also supported by the nuclear dot structure formed by these proteins (Figure 5H; Harigaya et al., 2006; Lemay et al., 2010; Sugiyama and Sugioka-Sugiyama, 2011; Yamanaka et al., 2010). The multiple contacts of Fip1 and that factors in all three complexes were identified in our screen suggest that the full cleavage and polyadenylation complex must be intact for H3K9me-modified heterochromatin to form. Our finding that the C-terminal truncation mutation iss1-ΔC disrupts the Iss1 interaction with Mmi1 indicates that the role of Iss1 in forming heterochromatin is to promote the key interaction of the CPFs with Mmi1, the sequence-specific RNA-binding factor that recruits the nuclear exosome (Figure 5G). This suggests that the cleavage and polyadenylation complex plays a central role in recruiting the exosome to the Mmi1-bound RNA targets. Consistent with this model is the result that the C-terminal truncation of Iss1 does not compromise the Iss1 interaction with the exosome factor Rrp6 (Figure S5B).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Henry Levin (henry_levin@nih.gov). Plasmids and strains generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. No other reagents were generated.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. pombe cells were grown at 32°C. Strains of S. pombe used to generate the integration libraries are listed in Table S1. The full list of strains and oligonucleotides are listed in the Key Resource Table. The rich media used was YES (yeast extract plus supplements) which consists of yeast extract and nutritional supplements (Rai et al., 2018). Minimal growth media for S. pombe consisted of PMG supplemented with all amino acids (Rai et al., 2018). When indicated, Vitamin B1 (Sigma) was added to a final concentration of 10 μM to repress the promoter nmt1, 5-fluoroorotic acid (5-FOA) (US Biologicals) to a final concentration of 1 mg/ml, and antibiotic Geneticin (G418, GIBCO by Life Technologies) to a final concentration of 500 μg/ml. Strains with the truncated allele of iss1 and with C-terminal epitope tags were created by homologous recombination using a PCR product templated by versions of pFa6a (Bähler et al., 1998). PCR products and plasmids were introduced into S. pombe by lithium acetate transformation (Rai et al., 2018).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-dimethyl-histone H3-K9 antibody | Abcam | ab1220; RRID:AB_449854 |
| Mouse monoclonal anti-c-Myc antibody | SIGMA | M4439; RRID:AB_439694 |
| Mouse monoclonal anti-FLAG M2 antibody | SIGMA | F1804; RRID:AB_262044 |
| Mouse anti-green fluorescent protein | Roche | 11814460001; RRID:AB_390913 |
| Mouse anti-HA monoclonal antibody | Sigma-Aldrich | 12CA5; RRID:AB_514505 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| 5-fluoroorotic Acid (FOA) | USBiological | N/A |
| Thiabendazole | SIGMA | T8904 |
| EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Ribo-Zero Gold rRNA Removal Kit | Illumina | MRZY1324 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | 20020594 |
| Agencourt AMPure XP beads | Beckman Coulter | A63880 |
| RNeasy MinElute Cleanup Kit | QIAGEN | 74204 |
|
| ||
| Deposited Data | ||
|
| ||
| RNaseq of iss1+ and isslΔC cells | SRA#PRJNA520927 | N/A |
| Hermes integration profiles | SRA#PRJNA517661 | N/A |
| ChIPseq of Iss1-3XFLAG | SRA#PRJNA517678 | N/A |
| ChIPseq of H3K9me2 | SRR10758214 SRR10758215 |
N/A |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| S. pombe YHL10155: mat1Msmt0 leu1-32 ade6-210 his2 ura4DS/E otr1R(Sph1)::ura4+ | (Chalamcharla et al., 2015) | N/A |
| S. pombe YHL12148: mat1Msmt0 leu1-32 ade6-216 his2 ura4 otr1R(Sph1)::ura4+ clr4Δ::KanMX | This paper | N/A |
| S. pombe: h+ leu1-32 ura4-D18 ade6-M216 clr4::KanMX | This paper | N/A |
| S. pombe YHL11414: h+ leu1-32 ura4-D18 ade6-M216 swi6::ura4 | This paper | N/A |
| S. pombe: h+ ade6-M216 ura4-D18 leu1-32 | Bioneer Corporation | ED668 |
| S. pombe YHL12159:h+ ade6-M216 ura4-D18 leu1-32 iss1ΔC-3xflag::KanMX | This study | N/A |
| S. pombe YHL12215:h+ ade6-M216 ura4-D18 leu1-32 iss1-3xflag::KanMX | This study | N/A |
| S. pombe YHL12160:mat1Msmt0 leu1-32 ade6-216 his2 ura4 otr1R(Sph1)::ura4+ iss1ΔC-3xflag::KanMX | This study | N/A |
| S. pombe YHL12266:h+ ade6 m210 his3-da leu1-32 ura4-d18 iss1-13xmyc::kanMX rrp6-3xflag::hyg | This study | N/A |
| S. pombe YHL12237:h+ leu1-32 ura4-D18 ade6-M216 rrp6-3xFlag::Hyg | This study | N/A |
| S. pombe YHL12783:YHL12237 transformed with pREP41-Iss1-HA | This study | N/A |
| S. pombe YHL12786:YHL12237 transformed with pREP41-Iss1ΔC HA | This study | N/A |
| S. pombe YHL12789:YHL12237 transformed with pREP41-GFP-HA | This study | N/A |
| S. pombe YHL12282:h+ ade6 m210 his3-da leu1-32 ura4-d18 mmi1-3xflag::hyg | This study | N/A |
| S. pombe YHL12792.YHL12282 transformed with pREP41-Iss1-HA | This study | N/A |
| S. pombe YHL12795.YHL12282 transformed with pREP41-Iss1ΔC HA | This study | N/A |
| S. pombe YHL12798.YHL12282 transformed with pREP41-GFP-HA | This study | N/A |
| S. pombe SR282 Paul Russell:h90 leu1-32 ade6-216 his2- ago1::KanMX6 | This study | N/A |
| S. pombe YHL12167:h90 leu1-32 ade6-216 his2- ago1::KanMX6 iss1ΔC-3xflag::KanMX | This study | N/A |
| S. pombe YHL12164:h+ leu1-32 ura4-D18 iss1 ΔC-3xflag::KanMX pRep41-N-GFP | This study | N/A |
| S. pombe YHL12162:h+ leu1-32 ura4-D18 iss1 ΔC-3xflag::KanMX pRep41-N-GFP-iss1 | This study | N/A |
| S. pombe YHL12179:h+ leu1-32 ura4-D18 pRep41-N-GFP | This study | N/A |
| S. pombe YHL12165:h+ leu1-32 ura4-D18 pRep41-N-GFP-iss1 | This study | N/A |
| S. pombe YHL12195:h+ ade6 m210 his3-da leu1-32 ura4-d18 iss1-GFP::KanMX | This study | N/A |
| S. pombe YHL12284 JT3 A. Yamashita:h90 pla1-GFP-kanR ade6-M216 leu1 | This study | N/A |
| S. pombe YHL12301:h90 pla1-GFP-kanR ade6-M216 leu1 pREP41-iss1 ΔC-3HA | This study | N/A |
| h90 pla1-GFP-kanR ade6-M216 leu1 pREP41-mCherry-iss1 | This study | N/A |
| S. pombe YHL12214:h+ ade6 m210 his3-da leu1-32 ura4-d18 iss1-13xmyc::kanMX | This study | N/A |
| S. pombe YHL12295:h+ ade6 m210 his3-da leu1-32 ura4-d18 iss1-13xmyc::kanMX mmi1-3xflag::hyg | This study | N/A |
| S. pombe YHL9451A:h- leu1-32, ura4-294 + pHL2577, pHL2578 | This study | N/A |
| S. pombe YHL9451C:h- leu1-32, ura4-294 + pHL2577, pHL2578 | This study | N/A |
| S. pombe YHL10161A:mat1Msmt0 leu1-32 ade6-210 his2 ura4DS/E otr1R(Sph1)::ura4+ pHL2577, pHL2578 | This study | N/A |
| S. pombe YHL10161B:mat1Msmt0 leu1-32 ade6-210 his2 ura4DS/E otr1R(Sph1)::ura4+ pHL2577, pHL2578 | This study | N/A |
| S. pombe YHL9451:YHL912 with pHL2577 | This study | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| HL3835, forward primer for deletion of 38 aa from C-terminal of iss1, ATGCACAACCTACTCATAATCCTACATCGAGTTATGGGAATGGAGCCTCTACGAACTATAATGCCTCAAGACCTCCTAGTCGGATCCCCGGGTTAATTAA | This study | N/A |
| HL3836, reverse primer for deletion of 38 aa from C-terminal of iss1, TAGAAGTAGTCGACACCCCATAGCATACAATTAAAACGATTCTCCTAAGATTTGCATACACAAACGTTATTGTATTTATCGAATTCGAGCTCGTTTAAAC | This study | N/A |
| HL5086, forward primer for N-terminal tagging
of iss1, AAGGAATCCATAATTTCAGGTGCGAGACTCAGTCAATGA
ATGGACTTGCCCCATATACATTTTAGAAGTGG
GTTTTTACTGAATTCGAGCTCGTTTAAAC |
This study | N/A |
| HL5087, reverse primer for N-terminal tagging of iss1, ACTCCATCATTCCGTTCCTCTATGGTTTCTCCATATAAATATTCATCCTCATCAACATCCATGTGCTGATCAGCATTTGACATGATTTAACAAAGCGACTATA | This study | N/A |
| HL5041, forward primer for N-terminal tagging of mmi1, TGGAAATGATGTATGATGAAGGCAGCAGACTTTGTACGTTGATTAACTATGCTATAATGAAGCGAATTGGAAGAGACCGTCGGATCCCCGGGTTAATTAA | This study | N/A |
| HL5042, reverse primer for N-terminal tagging of mmi1, CCGCTCCAATAGTATATAGTTTCATGAATTTTGTTAAAGGAAGCATCCAACATTCTTGAAAATCCAGCAAAAGGGAGATAGAATTCGAGCTCGTTTAAAC | This study | N/A |
| HL4200, forward primer for N-terminal tagging of rrp6, TTAACAAGGTAAGTTTACCAAAGCGAAATACTAGGGAACTCAAAAAAAGGAAGGTTTCAGACGGGAAGAGTACATCTTATCGGATCCCCGGGTTAATTAA | This study | N/A |
| HL4197, reverse primer for N-terminal tagging of rrp6, TTTTCAATTTCTACAATGTATTTAAAAGTAAATTTTCATTTTGTCAAAATTGTTTCGTTTATTTAAAATTAAAACTGATAGAATTCGAGCTCGTTTAAAC | This study | N/A |
| HL1870, linker (adaptor) that ligates to MseI-digested end of genomic DNA fragment, GTAATACGACTCACTATAGGGCTCCGCTTAAGGGAC | (Guo et al., 2013) | N/A |
| HL1871, 5 Phos, 3 AmMO linker (adaptor) that ligates to MseI-digested end of genomic DNA fragment 5Phos/TAGTCCCTTAAGCGGAG/3AmMO/ | (Guo et al., 2013) | N/A |
| HL3504, The primer specific for the TIR end of Hermes with 5 bp barcode (ACGTC) at upstream, AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTACGTCCTATGTGGCTTACGTTTGCCTG | This study | N/A |
| HL3505, primer specific for the TIR end of Hermes with 5 bp barcode (TGCAG) at upstream, AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTGCA GCTATGTGGCTTACGTTTGCCTG | This study | N/A |
| HL3506, primer specific for the TIR end of Hermes with 5 bp barcode (GATCA) at upstream, AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATC ACTATGTGGCTTACGTTTGCCTG | This study | N/A |
| HL3507, primer specific for the TIR end of Hermes with 5 bp barcode (CTAGT) at upstream, AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCTAGTCTATGTGGCTTACGTTTGCCTG | This study | N/A |
| HL3508, primer specific for the TIR end of Hermes with 5 bp barcode (ACTGC) at upstream, AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTACTGCCTATGTGGCTTACGTTTGCCTG | This study | N/A |
| HL3509, primer specific to the adaptor used to amplify all Hermes libraries, CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTGTAATACGACTCACTATAGG GC | This study’ | N/A |
| HL3643, primer specific for the TIR end of Hermes with 5 bp barcode (GTACG) at upstream, AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCGTACGTCTATGTGGCTTACGTTTGCCTG | This study | N/A |
| HL3644, primer specific for the TIR end of Hermes with | This study | N/A |
| HL3645, primer specific for the TIR end of Hermes with 5 bp barcode (CAGAT) at upstream, AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCCAGATTCTATGTGGCTTACGTTTGCCTG | This study | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pREP41-mCherry-iss1 | This study | N/A |
| pREP41-iss1ΔC-3HA, Akira Yamashita | This study | N/A |
| pREP41-iss1-3HA, Akira Yamashita | This study | N/A |
| pRep41-N-GFP-iss1 | This study | N/A |
| pRep41-N-GFP | This study | N/A |
| pHL2577: PSP2 containing kanMX6 flanked by Hermes left, right | (Guo et al., 2013) | N/A |
| pHL2578: Rep81×1 transposase (Hermes) express w/low nmt | (Guo et al., 2013) | N/A |
| pHL1768: Rep81X, Empty vector (serves as control, as there is no Hermes transposase) | (Guo et al., 2013) | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| HTtools | (Esnault et al., 2019) | N/A |
| CLCbio Workbench v11 | QIAGEN | (https://digitalinsights.qiagen.com/) |
| Deeptools | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/en/develop/content/list_of_tools.html |
| Lewis-Sigler Institute for Integrative Genomics web tool | (Boyle et al., 2004) | https://lsi.princeton.edu/research/facilities |
METHOD DETAILS
Spotting Assays
Growth assays were performed by spotting 5- or 10-fold serial dilutions of exponentially growing cells onto yeast extract with glucose and supplements (YES) plates in the absence or presence of the indicated agents. Plates were incubated at 30°C and photographed after 2 to 7 days of growth.
Hermes integration profiles
Due to the highly mutagenic nature of Hermes in S. pombe, the strains with the donor plasmid (pHL2577) are propagated without the plasmid that expresses the Hermes transposase (pHL2578). Once pHL2578 is introduced single colonies are directly activated for transposition in 50 mL cultures of PMG containing uracil and G418 but lacked Vitamin B1 and leucine. Table S7 describes the media used for each stage of the integration profile procedure. Each day the 50 mL cultures were passaged by inoculating fresh medium at 0.05 OD. The proportion of cells with integration events was measured by determining the ratio of colonies that form on plates with PMG + FOA + G418 divided by the number of colonies on PMG + FOA (Guo et al., 2013). Once the transposition frequency reaches 20%, the cells from the PMG (− leu − B1 + uracil + G418) culture were used to inoculate a 500 ml, PMG (complete + FOA) culture starting at 0.5 OD to remove the donor plasmid. After 24 hours of growth at 32°C, these cells were used to inoculate a 500 ml, PMG (complete + FOA + G418) culture starting at 0.5 OD to select for the library of cells with integration events. After 24 hours cells from these saturated cultures were harvested and resuspended in 15% glycerol at OD 5.0 and aliquots were frozen at −80°C. Having grown for approximately 5 generations in FOA genomic DNA was extracted from 200 OD units of cells for sequencing insertion profiles. These cells were also used to inoculate further passages of 100 mL cultures (PMG complete +U + leucine +B1 +FOA) inoculated at 0.05 OD. Daily passages were continued by inoculating at 0.05 OD until a total of 80 generations were reached. Genomic DNA was isolated from 200 OD units of these cells and used for sequencing the integration profiles.
Library preparation for deep sequencing of integration profiles
Genomic DNA was prepared as discussed previously (Guo and Levin, 2010). Pellets containing 200 OD units of cells were resuspended in 5 mL of SP1 buffer (1.2M Sorbitol, 50mM Citric Acid monohydrate, 50mM Na2HPO4*7H20, 40mM EDTA, SP1 adjusted to pH 5.6 with NaOH) containing 15mg zymolyase100T (Seikagaku Kogyo). The cells were incubated at 65°C for 2 hours or until they darken as observed with a phase contract microscope. The spheroblasts were pelleted for five minutes at RCF 1,855, resuspended in 15 mL 5X TE (50 mM Tris, 5 mM EDTA) and 1.5 mL of 10% SDS was added. The cells were then incubated at 65°C for 5 minutes, transferred to a 50 mL screwtop tube, 5 mL of KOAc was added followed by an incubation at room temperature for 30 minutes. The sample was centrifuged at 4,000 rpm (Beckman JS4.2 rotor, RCF = 3,300) for 15 min to remove debris. The supernatant was pipetted into a 40 mL high speed centrifuge tube. 20 mL ice-cold isopropanol was added, and the sample was incubated on ice for 5 minutes and centrifuge at 8000 rpm (Sorval GSA rotor) for 10min to pellet the nucleic acids. The isopropanol was decanted, and the pellet was air-dried. The pellet was resuspended in 6ml 5X TE, RNase A was added to a final concentration of 100 μg/ml and the mixture was incubated at 37°C for 1 hour. Then 0.2ml of 10% SDS was added and Proteinase K was added to final concentration of 50 μg/ml. The sample was incubated for 2 hours at 50°C. An equal volume of equilibrated phenol was added followed by vigorous mixing. The mixture was centrifuged at 5000 (Sorval GSA rotor) rpm for 5 min. The top aqueous layer was added to a fresh tube and extracted at least twice more. The supernatant was extracted with an equal volume of phenol/CHCl3/isoamylalcohol and centrifuged at 5000 rpm (Sorval GSA rotor) for 5 min. The top aqueous layer was extracted with phenol/CHCl3/isoamylalcohol at least twice or until interface between aqueous and organic layer was clear. The DNA was EtOH precipitated by adding 1/10 volume 5M NaCl and 2.5 volumes 100% ethanol. The bulk solution was transferred to microcentrifuge tubes and centrifuged for 20 minutes to pellet DNA. The pellets were washed with 70% ethanol and air-dried. The pellets were resuspended in 200 μl of TE and the concentration determined.
For each library seven micrograms of genomic DNA was digested with MseI at 37 deg. overnight. The digested DNA was purified with magnetic beads using AMPure XP (Beckman Coulter) and the final concentration determined.
The linker oligos HL1870 and HL1871 were annealed in PCR buffer at a final concentration of 10 μM in a 50-μl volume by heating at 94°C for 2 minutes and gradually cooling to 4°C. For each library five micrograms of AMPure XP digested DNA was ligated to the annealed linker in a series of 20 μl reactions each containing 400 nanograms of DNA, 1X ligation buffer (50 mM Tris Buffer pH 7.4, 10 mM MgCl2, 10 mM dithiothreitol, 0.1 mg/ml BSA), 1 mM ATP, and 200 units New England Biolabs T4 ligase. The ligation reactions were incubated at 16°C for 16 hours. Successful linker ligation was evaluated on a 1.7% agarose gel by observing the genomic DNA ligated to linker did not greatly increase in molecular weight compared to a control ligation of genomic DNA that lacked linker.
To optimize the diversity of each library 95 PCR reactions were performed plus one reaction lacking DNA template. Each reaction was 20 μl and contained two microliters of 10X clonetech Titanium buffer and 0.4 μl each of Clonetech Titanium Taq, 10 μM linker primer (HL3509), 10 μM Hermes TIR primer (barcoded primers listed in the Key Resources Table), and 10 mM dNTP mix. The PCR program used was:
94°C for 4 min
94°C for 15 s
65°C for 30 s
72°C for 45 s
go to step 2 for 6 cycles total
94°C for 15 s
60°C for 30 s
72°C for 45 s
go to step 6 for 24 cycles total
68°C for 10 min
4°C until sample retrieved
The PCR products from the 95 wells were concentrated on 6 QIAGEN PCR purification columns loading DNA-buffer PI mixture twice. Also, 50 μl of EB buffer was loaded onto each column twice to increase elution. The products were run on a 2% preparative agarose gel and fragments between 200 bp and 500 bp were purified with a QIAGEN gel purification kit. In preparation for Illumina sequencing the DNA concentrations were determined with the KAPA library quantification kit. The primers used to amplify the integration products contained different barcodes for each library and the Illumina P5 and P7 sequences which allowed them to be sequenced directly on a Illumina HiSeq2000 by the University of California Irvine Genomic High-Throughput Facility.
Analysis of Tf1 integration profiles
The mapping of Tf1 integration sites to the S. pombe genome (unmasked EMBL release ASM294v2.21 from September 2009, ftp://ftp.ensemblgenomes.org/pub/fungi/release-21/fasta/schizosaccharomyces_pombe/dna/Schizosaccharomyces_pombe.ASM294v2.21.dna.genome.fa.gz) was performed as previously described using the custom scripts suite HTtools (Guo and Levin, 2010), available online as supplementary material (Esnault et al., 2019).
Gene Ontology analysis
Gene ontology enrichment calculations were performed on April 30, 2018 using the Princeton University Lewis-Sigler Institute for Integrative Genomics web tool that relies on established algorithms (Boyle et al., 2004). P values were corrected for false discovery rates with Bonferroni correction. The fold enrichment calculations performed on the web tool were based on the proportion of genes in an ontology class included in our candidate list divided by the proportion of all S. pombe genes in the candidate list.
Overlap of gene sets independently found to be involved in RNA elimination
Enrichment of Iss1 bound genes in three independent studies of genes regulated by RNA elimination were calculated with hypergeometric distance including false rates of discovery.
Co-IP
To test for an interaction between Iss1 and Pla1, the S. pombe strain YHL12284 (h90 pla1-GFP-kanR, ade6-M216, leu1) was transformed with plasmids expressing Iss1–3HA or Iss1-ΔC-3HA (lacking the C-terminal 39 amino acids). For other Co-IP experiments we used strains containing 13X-Myc or 3X-FLAG at the C-termini of Iss1, Mmi1, and Rrp6. Harvested cells were lysed by vortexing vigorously with glass beads in HB buffer [25 mM MOPS (pH7.2), 5 mM ethylene glycol bis-(2-aminoehylether) tetraacetic acid (EGTA) (pH7.2), 15 mM MgCl2, 150 mM KCl, 50 mM beta-glycerophosphate, 15 mM p-nitrophenylphosphate, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.1 mM Na3VO4, 0.2% NP-40, protease inhibitor cocktail (Complete Mini EDTA-free, Roche)]. Native cell extracts were incubated with magnetic beads conjugated to anti-HA monoclonal antibody (16B12, Abcam) for 40 min at 4°C. The beads were then washed three times with HB buffer. Anti-HA monoclonal antibody (12CA5, Sigma-Aldrich) and full-length GFP polyclonal antibody (Clonetech) were used to detect target proteins.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (Rozenzhak et al., 2010) using anti-dimethyl-histone H3-K9 antibody (Abcam. Ab1220), anti-c-Myc antibody (Sigma, M4439), or anti–FLAG (Sigma, F1804). Quantitative PCR was performed on input and ChIP samples. The enrichment was calculated as E = (target_IP/normalization_IP)/(target_WCE/normalization_WCE). Statistical significance was determined using p values calculated with the Student’s t test.
ChIP-Seq
ChIP-seq experiments for H3k9me2 were performed in iss1+ and iss1-ΔC S. pombe cells. Cells (OD600 = 0.6) were fixed for 15 min in a 1% formaldehyde solution at room temperature. Crosslinked samples were lysed in FA-lysis buffer (50mM HEPES-KOH, pH 7.5, 140mM NaCl, 1mM EDTA, 0.1% Na-deoxycholate, 1% Triton X-100, 1mM PMSF, 1 μg/ml Leupeptin, 1 μg/ml Pepstatin A and 10 μg/ml Aprotinin) and fragmented to average size 200–500bp using a water bath sonicator (BioRuptor Pico, Diagenode). Five μg of chromatin was immuno-precipitated with 5 μg of H3k9me2 antibody (Abcam. Ab1220) conjugated Protein A magnetic beads and beads were washed 2 times with each of three wash buffers (FA-lysis buffer: 50mM HEPES-KOH, pH 7.5, 140mM NaCl, 1mM EDTA, 0.1% Na-deoxycholate, 1% Triton X-100, 1mM PMSF, 1 μg/ml Leupeptin, 1 μg/ml Pepstatin A and 10 μg/ml Aprotinin. Wash Buffer II: 50mM HEPES-KOH, pH 7.5, 500mM NaCl, 1mM EDTA, 0.1% Na-deoxycholate and 1% Triton X-100. Wash Buffer III: 10mM Tris-Cl, pH8, 250mM LiCl, 1mM EDTA, 0.5% Na-deoxycholate and 1% NP-40). Reverse-crosslinked ChIP and input DNA were purified by using a ChIP DNA Clean & concentrator kit (Zymo Research # D5205).
ChIP-Seq libraries were constructed from 30 ng of ChIP DNA using the Ovation Ultralow System V2 1–96 (NuGEN). The number of cycles was determined by a test amplification in order to minimize the number of amplification cycles. The final library was twice purified using Ampure XP PCR Purification Beads (Agencourt). Pooled libraries were sequenced on multiple lanes of a HiSeq 2500 in Rapid mode using version 3 chemistry to achieve a minimum of 23 million 51 base reads by the NIH Intramural Sequencing Center, National Human Genome Research Institute. The data was processed by the Bioinformatics Section of NINDs using RTA version 1.18.64 and CASAVA 1.8.2. ChIP enrichment was calculated as the log2 of the ratio of normalized IP reads to normalized input (whole cell extract) reads.
Fastq files were quality inspected by sample using the FASTQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adaptor clipping and quality trimming was achieved using Trimmomatic (http://www.usadellab.org/cms/index.php?page=trimmomatic). Mapping of the post adaptor clipped and low quality trimmed sequences by sample against the Schizosaccharomyces pombe reference genome (“EF2”) was performed in the CLCbio Genomics Workbench v11.0 (https://www.qiagenbioinformatics.com) using the “Map reads to Reference” tool under default parameters. Peak detection was accomplished for each “IP” sample mapping in conjunction with its “Control” pair mapping using the “Transcription Factor ChIP-Seq” tool under default parameters; providing for a table of detected peaks per “IP” sample that includes peak location, peak center, peak length, peak shape score, and peak P value. Annotation of each peak detected was accomplished using the “Annotate with Nearby Gene Information” tool. Fastq files representing the raw preprocessed sequences are available for download from NCBI SRA
Iss1 ChIPSeq data: fastq records (S401ab and S397ab) are aligned to S. pombe genome with BWA software to generate bam files. Visualizaion: Bedgraph files are generated from bam files with deeptools software with CPM normalization and at 20 bp bin size level. Coverage is visualized with IGV genome browser.
RNaseq analysis
RNA libraries were prepared and sequenced by the DNA Sequencing and Computational Biology Core, NHLBI/NIH. Fastq files were quality inspected by sample using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) then adaptor clipped (TruSeq3-PE.fa:2:30:10) and quality trimmed (HEADCROP:15 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:15) using Trimmomatic (http://www.usadellab.org/cms/index.php?page=trimmomatic). Stranded paired-end reference mapping, with no de novo gene nor transcript discovery, was performed against the “EF2” Ensembl instance of the S. pombe genome (https://support.illumina.com/sequencing/sequencing_software/igenome.html) was then performed on surviving intact read pairs only under default parameters using the RNA-Seq tool supported in the CLCbio Workbench v11 (https://digitalinsights.qiagen.com/). Upon completion of mapping, expression in reads per kilobase per million mapped reads (RPKM) units for 7,015 features (i.e., genes) was exported from the Workbench per sample and imported into R (http://www.cran.r-project.org). In R, expression for each sample was organized in matrix form then cross-sample normalized via quantile tact after pedestalling by 2 and taking the Log2 transform. Quality of the quantile normalized expression was next inspected and assured via Tukey boxplot, covariance-based PCA scatterplot, and correlation-based heatmap. To remove noise-biased features, the mean expression and coefficient of variation per feature by sample group was calculated. Then, by sample group, mean expression was modeled by coefficient of variation using locally weighted scatterplot smoothing (lowess). Resulting fits were then over plotted and inspected to identify and select the noise threshold for the data as the minimum expression value at which the linear relationship between mean expression (i.e., signal) and coefficient of variation (i.e., noise) was grossly lost. The noise threshold was Quantile(Log2(RPKM+2)) expression value = 3. Features not having an expression greater than this threshold for at least one sample were discarded with surviving features floored to the threshold if less. To identify those surviving features having differential expression between sample groups, the analysis of variance (ANOVA) test was first employed under Benjamini Hochberg (BH) multiple comparison correction (MCC) condition followed by post hoc testing using the TukeyHSD test. Features having an ANOVA-based BH MCC p < 0.05, a post hoc p < 0.05 for a group comparison, and an absolute linear mean difference > = 2X for the same group comparison were deemed to have differential expression between the sample groups respectively.
The Pearson Correlation coefficients of expression level for the three replicas of each strain were all higher than 0.976. The PCA scatterplot is presented in Figure S5.
RT-qPCR
Five hundred nanograms of the RNAs were reverse transcribed into cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems by Life Technologies). Gene expression was determined by quantitative PCR on a StepOnePlus system, with the following procedure: 6 μL cDNAs previously diluted to 10 ng/μL, 10 μL of SYBR green PCR master mix (Applied Biosystems by Life Technologies), and 4 μL of forward and reverse primers mixed at 1 μM. The samples were analyzed in triplicate. Gene expression was normalized to that of act1. Relative changes in expression were determined by the comparative CT (ΔCT) method (Schmittgen and Livak, 2008). The P values were calculated using Student–s t test.
RNA meta-analysis of all ORFs
Each ORF was divided into 15 equal sized segments and the RNA reads for all ORFs were divided into these segments and averaged. RNA reads upstream and downstream of ORFs were averaged in 100 bp bins. Reads were partitioned upstream or downstream depending on whether they were closer to the 5′ or 3′ end of the ORF. The Y scale axis represents percent of all RNA reads per sample.
Fluorescence microscopy
The Rep41-mCherry-iss1 plasmid was transformed into the strain carrying GFP at the C terminus of Pla1 (YHL12284). Cells were first grown to early log phase in liquid EMM2 with 5 μg/ml of thiamine, and then in thiamine-free EMM2 for 18 h or 20 h at 30°C after 5 washes with thiamine-free EMM2. Cells were then collected and visualized using a Zeiss Axio Observer.Z1 inverted light microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
In Figure 2C the significance of enriched gene ontologies was calculated with the Princeton University Lewis-Sigler Institute for Integrative Genomics web tool which calculates a P value using the hypergeometric distribution (Boyle et al., 2004). P values were corrected for false discovery rates with Bonferroni correction (see Methods GO analysis). The numbers of enriched genes are listed in Figure 2C. In Figures 3B, 3E, 3F, 4E, 4F, and 5A–5C, p values were calculated with the Student’s t test comparing three independent measurements. In Figure 4G the significance of overlapped gene sets was determined with hypergeometric distance including false rates of discovery. The numbers of enriched genes is listed in Figure 4G.
DATA AND CODE AVAILABILITY
The accession numbers for sequencing results are:
Hermes integration profiles-SRA#PRJNA517661
RNaseq of WT cells and with the Iss1 C-terminal truncation-SRA#PRJNA520927
ChIPseq of Iss1–3XFLAG- SRA#PRJNA517678
ChIPseq of H3K9me2- WT iss1 (YNB 97) SRR10758214 for sample SAMN13671684 iss1ρC-FLAG (YHL 22159) SRR10758215 for sample SAMN13671683
This study did not generate code. The suite HTtools (Guo and Levin, 2010), is available as published in supplementary material (Esnault et al., 2019).
Supplementary Material
Highlights.
Dense transposon integration identified genes important for forming heterochromatin
This transposon method identified many candidates that are essential genes
Four candidates are canonical mRNA cleavage and polyadenylation factors
Iss1 is required for heterochromatin assembly by recruiting Mmi1 to the exosome
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Programs of the NIH from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. We thank Shiv Grewal for sharing unpublished results. During the revision of this manuscript, a related study was published that independently demonstrated that heterochromatin formation at meiotic genes involves components of the cleavage and polyadenylation complex. Specifically, Swd22 and ssu72 were shown to promote heterochromatin formation at meiotic genes (Vo et al., 2019).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.094.
REFERENCES
- Allshire RC, and Ekwall K (2015). Epigenetic regulation of chromatin states in Schizosaccharomyces pombe. Cold Spring Harb. Perspect. Biol. 7, a018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, and Madhani HD (2018). Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 19, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, Nimmo ER, Ekwall K, Javerzat JP, and Cranston G (1995). Mutations derepressing silent centromeric domains in fission yeast disrupt chromosome segregation. Genes Dev. 9, 218–233. [DOI] [PubMed] [Google Scholar]
- Armbruster CE, Forsyth-DeOrnellas V, Johnson AO, Smith SN, Zhao L, Wu W, and Mobley HLT (2017). Genome-wide transposon mutagenesis of Proteus mirabilis: Essential genes, fitness factors for catheter-associated urinary tract infection, and the impact of polymicrobial infection on fitness requirements. PLoS Pathog. 13, e1006434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspuria PJ, and Tamanoi F (2008). The Tsc/Rheb signaling pathway controls basic amino acid uptake via the Cat1 permease in fission yeast. Mol. Genet. Genomics 279, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, Wach A, Philippsen P, and Pringle JR (1998). Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951. [DOI] [PubMed] [Google Scholar]
- Bayne EH, White SA, Kagansky A, Bijos DA, Sanchez-Pulido L, Hoe KL, Kim DU, Park HO, Ponting CP, Rappsilber J, and Allshire RC (2010). Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell 140, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, and Sherlock G (2004). GO:TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20, 3710–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck V, Quinn J, Soto Pino T, Martin H, Saldanha J, Makino K, Morgan BA, and Millar JB (2001). Peroxide sensors for the fission yeast stress-activated mitogen-activated protein kinase pathway. Mol. Biol. Cell 12, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, and Grewal SI (2005). Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet. 37, 809–819. [DOI] [PubMed] [Google Scholar]
- Carey AF, Rock JM, Krieger IV, Chase MR, Fernandez-Suarez M, Gagneux S, Sacchettini JC, Ioerger TR, and Fortune SM (2018). TnSeq of Mycobacterium tuberculosis clinical isolates reveals strain-specific antibiotic liabilities. PLoS Pathog. 14, e1006939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casañal A, Kumar A, Hill CH, Easter AD, Emsley P, Degliesposti G, Gordiyenko Y, Santhanam B, Wolf J, Wiederhold K, et al. (2017). Architecture of eukaryotic mRNA 3′-end processing machinery. Science 358, 1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalamcharla VR, Folco HD, Dhakshnamoorthy J, and Grewal SI (2015). Conserved factor Dhp1/Rat1/Xrn2 triggers premature transcription termination and nucleates heterochromatin to promote gene silencing. Proc. Natl. Acad. Sci. U S A 112, 15548–15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HK, Mukhamedova M, Edskes BK, and Wickner RB (2018). Hermes transposon mutagenesis shows [URE3] prion pathology prevented by a ubiquitin-targeting protein: evidence for carbon/nitrogen assimilation cross talk and a second function for Ure2p in Saccharomyces cerevisiae. Genetics 209, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan ED, Braun CR, Gygi SP, and Moazed D (2014). Post-transcriptional regulation of meiotic genes by a nuclear RNA silencing complex. RNA 20, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C, Lee M, Ham C, and Levin HL (2019). Transposable element insertions in fission yeast drive adaptation to environmental stress. Genome Res. 29, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeokonkwo C, Zhelkovsky A, Lee R, Bohm A, and Moore CL (2011). A flexible linker region in Fip1 is needed for efficient mRNA polyadenylation. RNA 17, 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazy MA, He X, Singh BN, Hampsey M, and Moore C (2009). The essential N terminus of the Pta1 scaffold protein is required for snoRNA transcription termination and Ssu72 function but is dispensable for pre-mRNA 3′-end processing. Mol. Cell. Biol. 29, 2296–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, and Levin HL (2010). High-throughput sequencing of retrotransposon integration provides a saturated profile of target activity in Schizosaccharomyces pombe. Genome Res. 20, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Park JM, Cui B, Humes E, Gangadharan S, Hung S, FitzGerald PC, Hoe KL, Grewal SI, Craig NL, and Levin HL (2013). Integration profiling of gene function with dense maps of transposon integration. Genetics 195, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigaya Y, Tanaka H, Yamanaka S, Tanaka K, Watanabe Y, Tsutsumi C, Chikashige Y, Hiraoka Y, Yamashita A, and Yamamoto M (2006). Selective elimination of messenger RNA prevents an incidence of untimely meiosis. Nature 442, 45–50. [DOI] [PubMed] [Google Scholar]
- He X, Khan AU, Cheng H, Pappas DL Jr., Hampsey M, and Moore CL (2003). Functional interactions between the transcription and mRNA 3′-end processing machineries mediated by Ssu72 and Sub1. Genes Dev. 17, 1030–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, Villén J, Gerace EL, Gygi SP, and Moazed D (2005). A cullin E3 ubiquitin ligase complex associates with Rik1 and the Clr4 histone H3-K9 methyltransferase and is required for RNAi-mediated heterochromatin formation. RNA Biol. 2, 106–111. [DOI] [PubMed] [Google Scholar]
- Horn PJ, Bastie JN, and Peterson CL (2005). A Rik1-associated, cullin-dependent E3 ubiquitin ligase is essential for heterochromatin formation. Genes Dev. 19, 1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YM, Ricke SC, and Mandal RK (2016). Transposon sequencing: methods and expanding applications. Appl. Microbiol. Biotechnol. 100, 31–43. [DOI] [PubMed] [Google Scholar]
- Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, and Grewal SI (2013). Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155, 1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay JF, D’Amours A, Lemieux C, Lackner DH, St-Sauveur, Bähler J, and Bachand F (2010). The nuclear poly(A)-binding protein interacts with the exosome to promote synthesis of noncoding small nucleolar RNAs. Mol. Cell 37, 34–45. [DOI] [PubMed] [Google Scholar]
- Li P, Jiang W, Yu Q, Liu W, Zhou P, Li J, Xu J, Xu B, Wang F, and Shao F (2017). Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature 551, 378–383. [DOI] [PubMed] [Google Scholar]
- Martienssen R, and Moazed D (2015). RNAi and heterochromatin assembly. Cold Spring Harb. Perspect. Biol. 7, a019323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AH, Hatakeyama R, Kimmig P, Arter M, Peter M, Matos J, De Virgilio C, and Kornmann B (2017). Functional mapping of yeast genomes by saturated transposition. eLife 6, e23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee K, Gardin J, Futcher B, and Leatherwood J (2016). Relative contributions of the structural and catalytic roles of Rrp6 in exosomal degradation of individual mRNAs. RNA 22, 1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Sugiyama T, Cam H, Verdel A, Zofall M, Jia S, Moazed D, and Grewal SI (2004). RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat. Genet. 36, 1174–1180. [DOI] [PubMed] [Google Scholar]
- Preker PJ, Lingner J, Minvielle-Sebastia L, and Keller W (1995). The FIP1 gene encodes a component of a yeast pre-mRNA polyadenylation factor that directly interacts with poly(A) polymerase. Cell 81, 379–389. [DOI] [PubMed] [Google Scholar]
- Price MN, Wetmore KM, Waters RJ, Callaghan M, Ray J, Liu H, Kuehl JV, Melnyk RA, Lamson JS, Suh Y, et al. (2018). Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 557, 503–509. [DOI] [PubMed] [Google Scholar]
- Rai SK, Atwood-Moore A, and Levin HL (2018). High-frequency lithium acetate transformation of Schizosaccharomyces pombe. Methods Mol. Biol. 1721, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Zhang K, Zofall M, Chen E, and Grewal SI (2011). Defects in RNA quality control factors reveal RNAi-independent nucleation of heterochromatin. Nat. Struct. Mol. Biol. 18, 1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenzhak S, Mejía-Ramírez E, Williams JS, Schaffer L, Hammond JA, Head SR, and Russell P (2010). Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-Phase in fission yeast. PLoS Genet. 6, e1001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez MR, Payen C, Cheong F, Hovde BT, Bissonnette S, Arkin AP, Skerker JM, Brem RB, Caudy AA, and Dunham MJ (2019). Transposon insertional mutagenesis in Saccharomyces uvarum reveals trans-acting effects influencing species-dependent essential genes. Genome Res. 29, 396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Segal ES, Gritsenko V, Levitan A, Yadav B, Dror N, Steenwyk JL, Silberberg Y, Mielich K, Rokas A, Gow NAR, et al. (2018). Gene essentiality analyzed by in vivo transposon mutagenesis and machine learning in a stable haploid isolate of Candida albicans. MBio 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaranarayana GD, Motamedi MR, Moazed D, and Grewal SI (2003). Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr. Biol. 13, 1240–1246. [DOI] [PubMed] [Google Scholar]
- St-André O, Lemieux C, Perreault A, Lackner DH, Bähler J, and Bachand F (2010). Negative regulation of meiotic gene expression by the nuclear poly(a)-binding protein in fission yeast. J. Biol. Chem. 285, 27859–27868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, and Sugioka-Sugiyama R (2011). Red1 promotes the elimination of meiosis-specific mRNAs in vegetatively growing fission yeast. EMBO J. 30, 1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Cam HP, Sugiyama R, Noma K, Zofall M, Kobayashi R, and Grewal SI (2007). SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128, 491–504. [DOI] [PubMed] [Google Scholar]
- Tashiro S, Asano T, Kanoh J, and Ishikawa F (2013). Transcription-induced chromatin association of RNA surveillance factors mediates facultative heterochromatin formation in fission yeast. Genes Cells 18, 327–339. [DOI] [PubMed] [Google Scholar]
- Touat-Todeschini L, Shichino Y, Dangin M, Thierry-Mieg N, Gilquin B, Hiriart E, Sachidanandam R, Lambert E, Brettschneider J, Reuter M, et al. (2017). Selective termination of lncRNA transcription promotes heterochromatin silencing and cell differentiation. EMBO J. 36, 2626–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, and Camilli A (2013). Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat. Rev. Microbiol. 11, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoosthuyse V, Legros P, van der Sar SJ, Yvert G, Toda K, Le Bihan T, Watanabe Y, Hardwick K, and Bernard P (2014). CPF-associated phosphatase activity opposes condensin-mediated chromosome condensation. PLoS Genet. 10, e1004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, and Moazed D (2004). RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo TV, Dhakshnamoorthy J, Larkin M, Zofall M, Thillainadesan G, Balachandran V, Holla S, Wheeler D, and Grewal SIS (2019). CPF recruitment to non-canonical transcription termination sites triggers heterochromatin assembly and gene silencing. Cell Rep. 28, 267–281.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Jia ST, and Jia S (2016). New insights into the regulation of heterochromatin. Trends Genet. 32, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood V, Harris MA, McDowall MD, Rutherford K, Vaughan BW, Staines DM, Aslett M, Lock A, Bähler J, Kersey PJ, and Oliver SG (2012). PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res. 40, D695–D699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S, Yamashita A, Harigaya Y, Iwata R, and Yamamoto M (2010). Importance of polyadenylation in the selective elimination of meiotic mRNAs in growing S. pombe cells. EMBO J. 29, 2173–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S, Mehta S, Reyes-Turcu FE, Zhuang F, Fuchs RT, Rong Y, Robb GB, and Grewal SI (2013). RNAi triggered by specialized machinery silences developmental genes and retrotransposons. Nature 493, 557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Takayama T, Iwata R, and Yamamoto M (2013). A novel factor Iss10 regulates Mmi1-mediated selective elimination of meiotic transcripts. Nucleic Acids Res. 41, 9680–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, and Doublié S (2011). Structural biology of poly(A) site definition. Wiley Interdiscip. Rev. RNA 2, 732–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Mosch K, Fischle W, and Grewal SI (2008). Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol. 15, 381–388. [DOI] [PubMed] [Google Scholar]
- Zhang K, Fischer T, Porter RL, Dhakshnamoorthy J, Zofall M, Zhou M, Veenstra T, and Grewal SI (2011). Clr4/Suv39 and RNA quality control factors cooperate to trigger RNAi and suppress antisense RNA. Science 331, 1624–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, Udenze K, Bronner IF, Casandra D, Mayho M, et al. (2018). Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360, eaap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Gong R, Zhu Q, He Q, Xu N, Xu Y, Cai M, Zhou X, Zhang Y, and Zhou M (2018). Genome-wide determination of gene essentiality by transposon insertion sequencing in yeast Pichia pastoris. Sci. Rep. 8, 10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Yamanaka S, Reyes-Turcu FE, Zhang K, Rubin C, and Grewal SI (2012). RNA elimination machinery targeting meiotic mRNAs promotes facultative heterochromatin formation. Science 335, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for sequencing results are:
Hermes integration profiles-SRA#PRJNA517661
RNaseq of WT cells and with the Iss1 C-terminal truncation-SRA#PRJNA520927
ChIPseq of Iss1–3XFLAG- SRA#PRJNA517678
ChIPseq of H3K9me2- WT iss1 (YNB 97) SRR10758214 for sample SAMN13671684 iss1ρC-FLAG (YHL 22159) SRR10758215 for sample SAMN13671683
This study did not generate code. The suite HTtools (Guo and Levin, 2010), is available as published in supplementary material (Esnault et al., 2019).