

Abstract
Bispecific T-cell engagers (BiTEs) preferentially targeting tumour-associated antigens and stimulating CD3-mediated signalling are being used in patients to treat acute B-cell lymphoblastic leukaemia. However, the potency of BiTEs in solid tumours is limited by their short half-life and by their severe toxicity at relevant therapeutic doses. Here we report the design and in vivo performance of a bispecific antibody that simultaneously targets the murine T-cell co-receptor CD3ε and the murine immune checkpoint programmed-death ligand 1 (PD-L1). In multiple syngeneic tumour models, the bispecific antibody generated higher antitumour immune responses than conventional BiTEs targeting tumour-associated antigens and CD3ε. We found that the durable antigen-specific T-cell responses resulted from the rejuvenation of CD8 T cells, owing to the blockade of PD-L1 on dendritic cells, but not on tumour cells, and to co-stimulation by B7–1/2 (a peripheral membrane protein on dendritic cells). Bispecific T-cell engagers targeting dendritic cells rather than tumour cells may represent a general means of T-cell rejuvenation for durable cancer immunotherapy.
One-sentence editorial summary:
A bispecific antibody targeting the T-cell co-receptor CD3ε and the immune checkpoint programmed-death ligand 1 on dendritic cells rejuvenates tumour-specific CD8 T cells, leading to durable antitumour responses in murine models of cancer.
Therapeutic strategies aiming to redirect T cells in the tumor have been increasingly studied in multiple cancer types over the past decades1–4. The bispecific T-cell engager (BiTE), which simultaneously binds tumor-associated antigen (TAA) and CD3ε, is one of the most potent technologies that can redirect T cells in the tumor tissue to cancer cells regardless of their intrinsic T-cell receptors (TCRs). Blinatumomab is the first U.S. Food and Drug Administration (FDA) approved BiTE for patients with B-cell precursor acute lymphoblastic leukemia (ALL) who are in remission but still have minimal residual disease (MRD)5,6. Despite the successful application of blinatumomab in hematopoietic malignancies, the application of BiTEs in solid tumors has been hampered, presumably by limited tumor tissue penetration, immunosuppressive tumor microenvironment (TME) and severe side effects7–9. The in vivo efficacy of the BiTEs mainly depend on the specificity of the TAAs, which are usually expressed on noncancerous tissue as well. Extensive efforts have been made to discover appropriate targets on tumor cells, such as the EGFRxCD3, EpCAMxCD3 and Her2xCD310–12. However, it is clear that the CD3 complex still gets cross-linked by such TAAs in peripheral noncancerous tissue, causing “on-target off-tumor” distractions and severe toxicity in the form of cytokine storm and tissue damage13. In addition, it is still unclear whether such BiTEs can generate tumor specific memory responses. Generally, current BiTEs are evaluated in xenograft mouse models, where human tumors and peripheral blood mononuclear cells (PBMCs) interact in immune-deficient hosts. However, such xenograft models fail to recapitulate “on-target off-tumor” distractions and severe toxicity because human antigens are not presented on the non-tumor mouse cells. It is also difficult to evaluate memory responses due to the nature of Graft-Versus-Host Disease effects by PBMCs. Therefore, the efficacy of BiTEs can be overestimated while toxicity is far underestimated. Severe side effects have been observed in clinical trials of BiTEs despite the promising efficacy evaluated in xenograft mouse models. Optimized strategies or targets are needed to overcome this primary barrier14,15.
The TCR signaling threshold can determine the fate of T cell activation 16. Insufficient TCR stimulation and lack of co-stimulatory engagement with professional antigen presenting cells (APCs) can lead to T cell exhaustion17,18. In addition, TCR stimulation alone can cause activation-induced cell death (AICD) 19. During natural T cell priming, APCs especially dendritic cells (DCs) provide three signals for proper T cell activation and survival. The TCR engagement of peptide-MHC (signal 1), co-stimulation between B7 and CD28 (signal 2), and inflammatory cytokines interleukin-2 (IL-2), IL-12 or type I IFN (signal 3) 20. To mimic this natural interaction, chimeric antigen receptor T cells (CAR-T) are designed to provide both signal 1 (via a portion of the CD3ζ cytodomain) and signal 2 (via a portion of the CD28 or 4–1BB cytodomain)21,22. BiTEs have shown the capacity to induce a potent T cell mediated tumor cell killing in the presence of signal 1 alone23–25. However, a lack of co-stimulatory signaling will induce T cell apoptosis after activation26. Approaches have been implemented to address this issue by providing anti-CD28 simultaneously with the BiTE, but it remains unknown whether anti-CD3 and anti-CD28 signaling can efficiently rescue tumor specific T cells27,28. Moreover, anti-CD28 signaling can activate a broader range of T cells, leading to acute cytokine storm29. Thus, treatments that can provide all three signals for T cell reactivation in the tumor tissue become an important criteria for bispecific antibody design30.
Since BiTEs engage CD3 signaling on T cells, any T cell can be activated regardless of their functional properties. CD4 T cells, CD8 T cells, regulatory T cells (Tregs) and Natural killer T (NKT) cells are highly enriched in the TME, and contribute to the heterogeneity of potentially activated T cells31. Among all T cell populations, antigen specific T cells play an indispensable role in establishing proper anti-tumor immunity, but the percentage of antigen specific T cells is limited in the tumor tissue32. Besides the potent activation of all tumor infiltrated T cells by BiTE treatment, bispecific antibodies that favor antigen specific T cell stimulation may be an alternative strategy to generate anti-tumor immunity inside the TME. Furthermore, studies have shown that CD8 T cells within the tumor consist of distinct populations of terminally differentiated and stem-like cells, the latter of which have an effector-molecule secretion potential and reside in APC niches33. Thus, targeting APCs to engage T cells is a potential strategy to rejuvenate specific anti-tumor T cell immunity.
Meanwhile, many types of immune cells (such as myeloid-derived suppressor cells (MDSCs), macrophages and Tregs) and tumor cells create an immune-resistant tumor microenvironment by providing co-inhibitory signals such as PD-L1. PD-L1 can inhibit the function of CD8 T cells at either the cytotoxic stage or re-activation stage34–36. Effector molecules released after T cell engagement, like Interferon gamma (IFNγ), also upregulate the expression of PD-L1, which further promotes adaptive resistance to BiTE treatment37,38. Thus the therapeutic effect of BiTE treatment can be improved in combination with checkpoint blockade 39.
To improve the anti-tumor effect of current tumor cell targeting BiTE therapy, we designed a bispecific antibody that targets the immune checkpoint PD-L1 to redirect T cells to APCs. We observed that PD-L1xCD3 generates superior anti-tumor immunity compared with the combination of anti-PD-L1 and anti-CD3. We also revealed a potential therapeutic strategy that allows APCs to rejuvenate T cells by enhancing B7/CD28 co-stimulation. Lastly, we highlighted the indispensable role of DCs over tumor cells in PD-L1-targeting T-cell engager treatment. Thus, an immune cell targeting bispecific antibody (BsAb) can be a complementary strategy to tumor cell targeting BiTE, which could generate synergistic anti-tumor effects. This study raises novel insights and overcomes major hurdles encountered in the current concept of BiTE therapies.
Results
PD-L1xCD3 targets PD-L1 to activate T cells in vitro.
In order to evaluate the anti-tumor efficacy of PD-L1-targeting T cell engager, we generated two different bispecific antibodies. One targets the human epidermal growth factor receptor (EGFR) and murine CD3ε, while the other targets PD-L1 and murine CD3ε. Both antibodies consist of two single-chain variable fragments (ScFv, anti-EGFR from Cetuximab, anti-PD-L1 from Atezolizumab, anti-CD3 from clone 17A2) and an Fc domain of human IgG1 that prolongs protein half-life in vivo. The CH3 domains of the antibodies were engineered with ‘Knobs-into-holes’ mutants to form heterodimers and the CH2 domains were engineered with ‘LALA-PG’ mutants to reduce Fc γ receptor (FcγR) binding (Fig. 1a and Supplementary Fig. 1a)40,41. We first confirmed the purity and molecular weight of the bispecific antibodies by gel electrophoresis under reducing and non-reducing conditions (Supplementary Fig. 1b). We then used high performance size-exclusion chromatography (HP-SEC) to confirm there is less than 0.5% aggregates and fragments in the purified proteins, with an endotoxin level less than 0.02 EU/ml (Supplementary Fig. 1c,d). In order to compare the binding affinity and therapeutic effects of each bispecific antibody, we derived a target cell line from the murine colorectal cancer cell line MC3842. This cell line, termed MC38E5, expresses a chimeric mouse EGFR with six amino acids mutated to allow binding of the anti-human EGFR antibody Cetuximab. The PD-L1 targeting BsAb (PD-L1xCD3) can specifically bind to PD-L1+ tumor cells, whereas the EGFR targeting BsAb (ErbxCD3) preferentially binds to EGFR+ tumor cells (Fig. 1b left and Supplementary Fig. 2a). Furthermore, both antibodies have similar affinity to CD3ε on naïve CD8 T cells (Fig. 1b right, Supplementary Fig. 2b and Supplementary Table 1).
Fig. 1 |. PD-L1xCD3 targets PD-L1 to activate T cells in vitro.

a, Schematic structure of PD-L1xCD3 bispecific antibody. PD-L1xCD3 is composed of a single-chain variable fragment (ScFv) to PD-L1 and a ScFv to murine CD3ε, fused to a mutant human IgG1. b, Binding of PD-L1xCD3 to PD-L1 on MC38 cells overexpressing PD-L1. Cells were incubated with serial dilutions of PD-L1xCD3 or human IgG control, followed by a fluorophore-conjugated anti-human IgG secondary antibody. Flow cytometry measured specific fluorescence index (SFI) using the mean fluorescence intensity (MFI). Binding of PD-L1xCD3 to CD3ε on CD8 T cells purified from mouse spleen. Cells were incubated with serial dilutions of PD-L1xCD3 or human IgG control, followed by a fluorophore-conjugated anti-human IgG secondary antibody. Flow cytometry measured SFI. c, Binding of PD-L1xCD3 to FcγR on RAW 264.7 cells. Cells were incubated with serial dilutions of WT IgG BsAbs, mutant IgG BsAbs, or WT IgG BsAbs with anti- FcγR, followed by a fluorophore-conjugated anti-human IgG secondary antibody. Flow cytometry measured SFI. d-f, MC38-GFP cells (3×104) and purified splenic CD8 T cells (3×105) were co-cultured with serial dilutions of PD-L1xCD3 or human IgG control for 48 hours. IFNγ in the supernatant was detected by cytokine beads array (CBA) (d). CD25 and CD69 expression on T cells were detected by flow cytometry (e). GFP+ 7AAD- tumor cells (viability) were detected by flow cytometry (f). g-h, MC38 tumor cells (WT or PD-L1−/−, 3×104) and purified splenic CD8 T cells (3×105) were co-cultured with PD-L1xCD3 or human IgG control for 48 hours, T cell activation (g) and IFNγ in the supernatant (h) were detected respectively. Data were presented as means ± SEM from a representative experiment (n=3 (b-f), 4 (h-i) biological replicates) of two independent experiments. Data were analyzed using non-linear best fits for (b-f) and two-tailed unpaired Student’s t-test for (g-h). ****P ≤ 0.0001.
The Fc domain plays multiple roles in bispecific antibody function. On one hand, it prolongs in vivo half-life; on the other hand, it also cross-links CD3 complex non-specifically. We observed that antibody-dependent cellular cytotoxicity (ADCC) also reduces rather than increases T cell percentages in the spleen (Supplementary Fig. 3a,b). Thus, we re-engineered the Fc domain so that neonatal Fc receptor (FcRn) binding affinity is preserved but FcγR binding affinity is reduced. Antibodies with a wild type CH2 domain can bind to an FcγR+ murine macrophage cell line. In contrast, antibodies with this re-engineered mutant CH2 domain exhibit a reduced binding affinity, which was similar to using anti-CD16/CD32 to block FcγR binding (Fig. 1c).
We next tested whether T cells can be activated by bispecific antibodies to kill tumor cells. When antibodies were applied to co-cultured tumor cells and CD8 T cells, naïve T cells rapidly upregulated the expression of CD25 and CD69 on the cell surface with increased secretion of IFNγ in a dose dependent manner (Fig. 1d,e and Supplementary Fig. 2c,d). Meanwhile, tumor cells were also efficiently killed, indicating a fully functional activation of the T cells (Fig. 1f and Supplementary Fig. 2e). Even though ErbxCD3 and PD-L1xCD3 have similar EC50 in T cell activation markers (1.67 vs 3.218 nM) and IC50 in tumor cell killing (5.498 vs 4.186 nM), IFNγ was more potently induced by PD-L1xCD3 compared to ErbxCD3 (Supplementary Table 1). Furthermore, when PD-L1 was knocked out on tumor cells, PD-L1xCD3 completely lost the ability to activate T cells (Fig. 1g,h). These results demonstrate that PD-L1xCD3 can activate T cells to kill tumor cells in a PD-L1 dependent manner in vitro.
PD-L1xCD3 generates superior anti-tumor effects to combination treatment in vivo.
Since PD-L1xCD3 generated potent anti-tumor effects in vitro, we next investigated whether it can also induce anti-tumor immune responses in syngeneic mouse models. When PD-L1xCD3 was administrated intraperitoneally to MC38 bearing mice, tumors were completely eradicated after the second infusion. Even though non-specific engagement of CD3 signaling by anti-CD3 displayed similar anti-tumor effects with PD-L1xCD3 at early stage, treated tumor eventually relapsed after second infusion. Compared with PD-L1xCD3, neither anti-PD-L1 nor anti-PD-L1 + anti-CD3 combination treatment was able to generate comparable anti-tumor effects, indicating that the anti-tumor effect of PD-L1xCD3 is not due to the synergistic effect of stimulating CD3 signal and block the checkpoint simultaneously (Fig. 2a and Supplementary Fig. 4a). In addition, PD-L1xCD3 treatment dramatically improved the overall survival rate compared with combination treatment (Fig. 2b). More importantly, PD-L1xCD3 treatment also induced OT-I-specific IFNγ producing cells in the spleen of MC38OVA bearing mice, further demonstrating the efficient generation of antigen specific T cell responses (Fig. 2c,d). Memory T cell responses play a critical role in establishing protective immunity against cancer, but the ability of BiTEs to induce immunological memory has not been demonstrated in vivo43. However, when we re-challenged PD-L1xCD3 cured mice with 10-fold higher number of tumor cells on day 50 after treatment, no tumors were able to grow out, indicating that PD-L1xCD3 treatment efficiently generated memory immune responses after eradicating tumors (Fig. 2e and Supplementary Fig. 4b). To further prove that PD-L1xCD3 treatment generated memory T cells, splenocytes from cured mice were adoptively transferred to Rag1−/− mice. Transferred splenocytes not only prevented tumorigenesis (Fig. 2f and Supplementary Fig. 4c) but also effectively rejected established tumors (Fig. 2g and Supplementary Fig. 4d) in vivo. Taken together, we hypothesized that PD-L1xCD3 may provide distinct signals to T cells compared to combination treatment, which triggers a specific immune response against the tumor.
Fig. 2 |. PD-L1xCD3 generates superior anti-tumor effect than combination treatment in vivo.

a-b, C57BL/6J mice were subcutaneously inoculated with 1×106 MC38 tumor cells and treated with 0.25 mg/kg of anti-CD3, anti-PD-L1, anti-CD3 plus anti-PD-L1 or PD-L1xCD3 twice on day 10 and 15. Tumor volume (a) and percentage of survival (b) was shown. c-d, C57BL/6J mice were subcutaneously inoculated with 1×106 MC38OVA tumor cells and treated as in panel a. 25 days after treatment, splenocytes from different treatment groups were isolated and stimulated with either OT-I peptide or irradiated MC38 tumor cell (IR MC38). Antigen specific T cells were detected by Elispot assay. e-g, MC38 bearing C57BL/6J mice were treated with PD-L1xCD3 twice on day 10 and day 15 after tumor inoculation. 50 days after treatment, cured mice were re-challenged with 1×107 MC38 tumor cell (e), 2×107 splenocytes from cured mice were adoptively transferred to Rag1−/− mice two days before MC38 tumor cell inoculation (f), 2×107 splenocytes from cured mice were adoptively transferred to MC38 bearing Rag1−/− mice 10 days after tumor inoculation (g). Data were presented as mean ± SEM from a representative experiment (n=5 (a-e), 4 (f-g) biological replicates) of two independent experiments. Statistical analysis was performed by two-way ANOVA with Tukey’s multiple comparisons test (a, c), Sidak’s multiple comparisons test (e-g) or Log-rank (Mantel-Cox) test (b). ***P ≤ 0.001, and ****P ≤ 0.0001.
We next investigated the systemic toxicity of PD-L1xCD3 treatment. Mice treated with the combination of anti-CD3 and anti-PD-L1 lost about 10% of their initial body weight and had a transient increase of serum alanine transaminase (ALT) or aspartate aminotransferase (AST) at 12 hours post injection. Surprisingly, treatment with PD-L1xCD3 dramatically reduced this systemic toxicity (Supplementary Fig. 5a-c). Moreover, when serum cytokines were detected at different time points after treatment, IFNγ, tumor necrosis factor-α (TNF-α) and IL-6 levels transiently peaked within 24 hours in combination treatment but not after PD-L1xCD3 treatment (Supplementary Fig. 5d-f). To further evaluate the on-target off-tumor side effect of our BsAb treatment, we analyzed the histology of tissues including liver, spleen, heart, kidney, and lung from mice that received PD-L1xCD3 treatment. Compared with human IgG (hIgG) control, treatment with PD-L1xCD3 did not induce visible tissue damage or inflammation in H&E staining (Supplementary Fig. 6). Thus, PD-L1xCD3 treatment did not induce severe systemic toxicity in vivo.
Furthermore, we also explored whether PD-L1xCD3 could generate superior anti-tumor effect compared to conventional TAAxCD3. We treated MC38E5 bearing mice with either ErbxCD3 or PD-L1xCD3. Even though PD-L1xCD3 has similar tumor-killing ability to ErbxCD3 in vitro, it displays a much stronger anti-tumor effect in vivo (Extended Data Fig. 1a). Consistent results were also observed in chimeric EGFR expressing tumor models, including cervical cancer model TC-1 (TC1E5) (Extended Data Fig. 1c), melanoma model B16 (B16E5) (Extended Data Fig. 1e), and breast cancer model TUBO (TuBoE5) (Extended Data. 1f). In addition, tumor-free mice from PD-L1xCD3 treated groups also obtained memory immunity to reject re-challenged tumor cells (Extended Data Fig. 1b,d). To investigate whether ErbxCD3 treatment requires a higher therapeutic dose to generate similar anti-tumor effect with PD-L1xCD3 treatment, we treated MC38E5 bearing mice with ErbxCD3 intratumorally. Local injection considerably improved the anti-tumor effect of ErbxCD3 compared with systemic injection, but the overall anti-tumor effect was still weaker than PD-L1xCD3 (Extended Data Fig. 1g). Therefore, other mechanisms besides dose effects may contribute to the resistance to ErbxCD3 in vivo. Taken together, we demonstrated that PD-L1xCD3 generates superior anti-tumor effects compared to ErbxCD3 at the therapeutic dose we used (0.25 mg/kg, twice) in multiple mouse tumor models. These data also raises the possibility that PD-L1xCD3 creates a unique microenvironment by engaging different signaling pathways or inducing different cell-cell interactions.
Pre-existing CD8 T cells are sufficient for the anti-tumor effect of PD-L1xCD3.
Next, we investigated the mechanisms underlying the therapeutic effects of PD-L1xCD3. PD-L1xCD3 showed no effect on MC38 bearing Rag1−/− mice, which confirms that adaptive immunity is essential for the therapeutic effect of PD-L1xCD3 (Fig. 3a and Supplementary Fig. 7a). Moreover, depleting different T cell subsets with anti-CD4 or anti-CD8 (Extended Data Fig. 2a,b) confirmed that CD8 T cells but not CD4 T cells contribute to the therapeutic effect (Fig. 3b and Supplementary Fig. 7b). To further determine whether PD-L1xCD3 treatment depends on pre-existing T cells in the tumor microenvironment or recruitment of T cells from peripheral tissue, we used FTY720 (a S1P receptor agonist) to block T cell trafficking to tumor tissue during PD-L1xCD3 treatment. Additional FTY720 blocking did not affect the therapeutic effect of PD-L1xCD3, indicating the sufficient role of pre-existing CD8 T cells in the TME for this treatment (Fig. 3c and Supplementary Fig. 7c). To further clarify whether PD-L1xCD3 is targeting tumor tissue to induce the anti-tumor effect, we treated mice intratumorally with the BsAb. As expected, local treatment was also sufficient to achieve a similar anti-tumor effect compared to systemic treatment (Extended Data Fig. 2e). We also tested the in vivo distribution of the bispecific antibody at different time points post treatment. The antibody was preferentially enriched in the tumor tissue starting at 24 hours post intraperitoneal injection and detectable levels were sustained for up to 5 days (Extended Data Fig. 2f). Hence, PD-L1xCD3 can target tumor tissue to rejuvenate CD8 T cell immunity. Besides CD8 T cells, many other types of immune cell were enriched in the TME in response to treatment. Even though they were not the primary effector cells in tumor killing, they may still play important roles by interacting with CD8 T cells. To identify key components that contribute to the anti-tumor effects during treatment, we applied a series of depletion experiments (Extended Data Fig. 2c,d). NK cell depletion by anti-NK1.1 or macrophage depletion by anti-CSF1R did not affect tumor growth control by PD-L1xCD3 (Fig. 3d and Supplementary Fig. 7d). Since the uniqueness of PD-L1xCD3 to ErbxCD3 mainly exists through the targets by which CD3 signaling engages (PD-L1 vs EGFR), we proposed that PD-L1+ cells in the tumor microenvironment might contribute to the CD8 dependent anti-tumor immunity.
Fig. 3 |. Pre-existing CD8 T cells are sufficient for the anti-tumor effect of PD-L1xCD3.

a, Rag1−/− mice were inoculated with 1×106 MC38 tumor cells and treated with PD-L1xCD3 (0.25 mg/kg on day 10 and 15). b, C57BL/6 mice were inoculated with 1×106 MC38 tumor cells and treated with PD-L1xCD3 (0.25 mg/kg on day 10 and 15). 200μg anti-CD8 or anti-CD4 was administrated one day before treatment initiation and then twice a week for 2 weeks. c, C57BL/6 mice were inoculated with 1×106 MC38 tumor cells and treated with PD-L1xCD3 (0.25 mg/kg on day 14 and 18). 20μg FTY720 was administrated one day before treatment initiation and then 10μg every other day for 2 weeks. d, C57BL/6 mice were inoculated with 1×106 MC38 tumor cells and treated with PD-L1xCD3 (0.25 mg/kg on day 10 and 15). 200 μg anti-NK1.1 or 500 μg anti-CSF1R was administrated one day before treatment initiation and then twice a week for 2 weeks. Data were presented as mean ± SEM from a representative experiment (n=5 biologically independent animals) of two independent experiments. Statistical analysis was performed by two-way ANOVA with Sidak’s multiple comparisons test (a) or Dunnett’s multiple comparisons test (b-d). ****P ≤ 0.0001.
PD-L1 on dendritic cells is essential for the anti-tumor effect of PD-L1xCD3.
PD-L1 is widely expressed by a variety of cell types in multiple tissues including lymphocytes, myeloid cells, and tumor cells. To elucidate the role of PD-L1 on different cells in contributing to PD-L1xCD3 treatment, we performed multiple knockout (KO) studies. We first tested the therapeutic effect of PD-L1xCD3 on PD-L1 KO tumors. Surprisingly, PD-L1xCD3 still generated an effective anti-tumor response in mice bearing PD-L1 KO MC38 (Fig. 4a left and Supplementary Fig. 8a) or B16 (Fig. 4a right and Supplementary Fig. 8b) tumors. However, its therapeutic effect was completely abolished in PD-L1 deficient mice bearing WT MC38, indicating that PD-L1 on host cells but not tumor cells plays a critical role in the anti-tumor effect of PD-L1xCD3 (Fig. 4b and Supplementary Fig. 8c). Since myeloid cells are the dominant host cells that are PD-L1+ in the tumor microenvironment, we next used conditional KO mice to study which PD-L1 expressing cells are essential for the anti-tumor effects of PD-L1xCD3. Our results showed that, PD-L1xCD3 is able to control tumor growth in Lyz2CrePD-L1f/f mice (macrophage specific PD-L1 knockout) but not Zbtb46CrePD-L1f/f mice (dendritic cell specific PD-L1 knockout). This indicates that PD-L1 on dendritic cells but not macrophages is required for PD-L1xCD3 treatment (Fig. 4c and Supplementary Fig. 8d,e). We have also measured the expression level of PD-L1 on different cells in the tumor. DCs, especially CD103+ DCs expressed the highest level of PD-L1 in vivo, which may contribute to DC targeting of PD-L1xCD3 (Supplementary Fig. 9a,b). To determine if those DCs are essential, we administered PD-L1xCD3 treatment to tumor bearing Batf3−/− mice that lack of CD103+ cDC1. Strikingly, the bispecific antibody completely lost anti-tumor efficacy in these mice, despite few cells are CD103+ in both tumor and draining lymph node. These results indicate that our treatment may improve CD8 T cell function through a unique subset of DCs (Fig. 4d and Supplementary Fig. 8f). Taken together, PD-L1 on CD103+ cDC1 plays a critical role in facilitating the anti-tumor effect of PD-L1xCD3 treatment.
Fig. 4 |. PD-L1 on dendritic cells is essential for the anti-tumor effect of PD-L1xCD3.

a, C57BL/6J mice were subcutaneously inoculated with 1×106 MC38-PD-L1−/− tumor cells (a, left) or 5×105 B16-PD-L1−/− tumor cells (a, right) and treated with 0.25 mg/kg of PD-L1xCD3 or human IgG control twice on day 10 and 15 after tumor inoculation. b, PD-L1−/− mice were subcutaneously inoculated with 1×106 MC38 tumor cells and treated with 0.25 mg/kg of PD-L1xCD3 or human IgG control twice on day 10 and 15 after tumor inoculation. c, Zbtb46CrePD-L1f/f mice (c, left) or Lyz2CrePD-L1f/f mice (c, right) were subcutaneously inoculated with 1×106 MC38 tumor cells and treated with 0.25 mg/kg of PD-L1xCD3 or human IgG control twice on day 10 and 15 after tumor inoculation. d, Batf3−/− mice were subcutaneously inoculated with 1×106 MC38 tumor cells and treated with PD-L1xCD3 or human IgG control (0.25 mg/kg on day 10 and 15). Data were presented as mean ± SEM from a representative experiment (n=5 biologically independent animals) of two independent experiments. Statistical analysis was performed by two-way ANOVA with Sidak’s multiple comparisons test. ****P ≤ 0.0001.
PD-L1xCD3 reshapes a distinct immunophenotypic signature in tumor-bearing mice.
Since PD-L1xCD3 targets PD-L1 on DCs to facilitate a superior immune response to ErbxCD3, we further investigated how the TME is reshaped to promote CD8 T cell responses. Lymphocytes and myeloid cell populations in the spleen and tumor were analyzed by flow cytometry at 48 hours post treatment (Supplementary Fig. 10). The percentage of CD8 T cells, CD4 T cells and NKT cells in the tumor dramatically increased in PD-L1xCD3 but not ErbxCD3 treated group (Supplementary Fig. 11a-c). In contrast, the percentage of NK cell and B cell remains unchanged (Supplementary Fig. 11d,e). We further analyzed the immunophenotype of CD8 T cells in the tumor, as they play an essential role during treatment. PD-L1xCD3 treatment increased not only CD69 but also Ki-67 expression on CD8 T cells, indicating that CD8 T cells were activated and expanded after treatment (Fig. 5a). The percentage of PD-1 and TIM-3 double positive terminally exhausted CD8 T cells was significantly reduced after treatment (Fig. 5a left). Meanwhile, the percentage of TCF1+ and CD28+ stem-like CD8 T cells increased (Fig. 5b), indicating that the phenotype of intra-tumoral CD8 T cells have been reshaped by PD-L1xCD3 treatment.
Fig. 5 |. PD-L1xCD3 reshapes a distinct immunophenotypic signature in tumor-bearing mice.

a, C57BL/6J mice were subcutaneously inoculated with 1×106 MC38-OVA tumor cells and treated with 0.25 mg/kg of PD-L1xCD3 or human IgG control. Flow cytometry analysis was performed with splenocytes and dissociated tumor samples for the percentage of PD-1high TIM-3+ CD8 T cells (a, left), Ki-67+ CD8 T cells (a, middle), CD69+CD8+ T cells (a, right). b, C57BL/6J mice were subcutaneously inoculated with 1×106 MC38-OVA tumor cells and treated with 0.25 mg/kg of PD-L1xCD3 or human IgG control. Flow cytometry analysis was performed with splenocytes and dissociated tumor samples for the percentage of TCF1+ CD8 T cells (b, left), CD28+ CD8 T cells (b, middle), and tetramer+ cells (b, right). c, MC38 bearing mice were treated with PD-L1xCD3 or human IgG. 48 hours after treatment, the percentage of macrophage (c, left), MDSC (c, middle), and dendritic cell (c, right) were detected by flow cytometry with splenocytes and dissociated tumor samples. Representative result from two independent experiments were shown as mean ± SEM (n=5 biological replicates). Statistical analysis was performed by two-tailed unpaired Student’s t-test (a, left and middle; b, left and middle) or two-way ANOVA with Sidak’s multiple comparisons test (a, right; b, right and c). ***P ≤ 0.001, ****P ≤ 0.0001.
As previous studies have shown, antigen presenting cells maintain the stem-like CD8 T cell niche in the TME33,44. We have also observed that our bispecific antibody targets PD-L1 on DCs to reactivate T cells. Thus, it is possible that stem-like CD8 T cells but not terminally exhausted CD8 T cells were preferentially activated by PD-L1xCD3, due to their physiological co-localization with DCs. More importantly, the percentage of antigen specific CD8 T cell in the tumor also increased after PD-L1xCD3 treatment (Fig. 5b right). Thus, PD-L1xCD3 may preferentially activate a ‘DC-interacting’ population of CD8 T cells to establish specific anti-tumor immunity. In addition, we also examined the dynamics of myeloid cells in the TME. The percentage of both macrophages and MDSCs dramatically decreased after treatment since they are considered as ‘PD-L1+ targets’ (Fig. 5c). Meanwhile, the percentage of DCs also decreased despite their critical role in initiation of the anti-tumor effect (Fig. 5c right). Notably, the percentage of Tregs was also decreased despite an increase in the percentage of total CD4 T cells (Supplementary Fig. 11f). Even though the mechanism is still unknown, the expression of PD-L1 on Tregs has been reported, which may be an explanation for this phenomenon45. As we observed a notable reduction in myeloid cell percentages in the tumor tissue, we further investigated how the immune response was affected by these changes. First of all, the percentage of macrophages, MDSCs and dendritic cells in the draining lymph node and spleen remained unchanged after treatment (Fig. 5c and Supplementary Fig. 12a). Second, we investigated how the dynamic changes of myeloid cells in the tumor tissue regulates the anti-tumor immunity using PD-L1 knockout mice. When PD-L1 was knocked out from macrophages, PD-L1xCD3 could still generate effective anti-tumor response (Fig. 4c middle). However, the percentage of macrophages and MDSCs remains unchanged (Supplementary Fig. 12b-d). Thus, PD-L1 expression on DCs but not the reduction of inhibitory cells is essential to ignite the TME. The reduction of macrophages may contribute to initiate T cell activation but is not sufficient, because PD-L1xCD3 treatment also reduced the percentage of macrophages in Zbtb46CrePD-L1f/f mice (Supplementary Fig. 12b), but an anti-tumor effect was not observed (Fig. 4c left). Finally, the dynamics of all other immune populations was restricted to the tumor. PD-L1xCD3 treatment did not substantially alter splenic immune cell populations compared to the control group, indicating that the anti-tumor effect was mainly generated in the tumor. Taken together, PD-L1xCD3 reshapes the TME to provoke specific anti-tumor immunity.
Co-stimulatory signaling is required for PD-L1xCD3 mediated anti-tumor effects.
The generation of protective T cell immunity is one of the most desired goals in cancer immunotherapy. However, it remains the major hurdle of current tumor cell targeting BiTE therapy. Our data has shown that PD-L1xCD3 can target DCs to promote antigen specific T cell immunity. Therefore, we further investigated the underlying mechanisms participating in this process. Previous studies have shown that the therapeutic effect of anti-PD-1 treatment is CD28 dependent which highlights the importance of co-stimulatory signaling in generating and maintaining T cell immunity46,47. Therefore, we hypothesized that the therapeutic effect of PD-L1xCD3 may depend on T cell co-stimulation. To test this hypothesis, we combined anti-CD80/86 antibodies together with PD-L1xCD3 treatment (Fig. 6a). To our surprise, blocking B7-CD28 co-stimulation completely abolished the therapeutic effect of PD-L1xCD3 (Fig. 6a and Supplementary Fig. 13). Similar results were also observed when using CTLA4-Ig, a fusion protein that competitively binds to CD80/86 to block its signaling (Extended Data Fig. 3a,b). Blocking B7-CD28 interactions also inhibited the generation of antigen specific T cells in the tumor (Fig. 6a right).
Fig. 6 |. Co-stimulatory signaling is required for PD-L1xCD3 mediated anti-tumor effects.

a, C57BL/6J mice were inoculated with 1×106 MC38 tumor cells and treated with PD-L1xCD3 (0.25 mg/kg on day 10 and 15), 200 μg anti-B7–1 and anti-B7–2 were administrated on day 10, 13 and 15. Experimental design (a, left), tumor growth curve (a, middle) and IFNγ-producing antigen specific CD8 T cells (a, right) were shown. b-e, CD8 T cells were co-cultured with either tumor cells or dendritic cells in the presence of bispecific antibodies. T cell activation (b), supernatant IFNγ (c), apoptotic T cells (d) and supernatant IL-2 (e) were measured by flow cytometry and CBA. f-g, Cumulative survival in colorectal adenocarcinoma (f) and liver hepatocellular carcinoma (g) patients according to CD8 infiltration and CD28 level in TCGA database (top 10% vs bottom 10%). Representative result from two independent experiments were shown as mean ± SEM (n=5 biological replicates). Statistical analysis was performed by one-way ANOVA (a, right and b-e), two-way ANOVA (a, middle) with Tukey’s multiple comparisons test and Log-rank test (f-g). ****P ≤ 0.0001.
To further determine how co-stimulatory signaling helps DCs generate proper T cell response in the presence of PD-L1xCD3, we co-cultured tumor cells or splenic DCs with naïve CD8 T cells in the presence of either ErbxCD3 or PD-L1xCD3. PD-L1xCD3 but not ErbxCD3 induced similar level of CD25+CD69+ expression on T cells and IFNγ in the supernatant in both tumor-T and DC-T cell co-cultures (Fig. 6b,c and Extended Data Fig. 3c,e). Yet surprisingly, the percentage of live T cells in tumor-T cell group was much lower than that of DC-T cell group. AICD occurred in tumor-cell-activated T cells treated with either ErbxCD3 or PD-L1xCD3, but was greatly reduced in dendritic-cell-activated T cells treated with PD-L1xCD3 (Fig. 6d and Extended Data Fig. 3d). Thus, targeting tumor cell to reactivate T cells may only have transient anti-tumor effect and cannot generate long-lasting effects due to a lack of co-stimulation, increased T cell death and no memory response. Our in vivo data also showed that ErbxCD3 treatment group had a lower frequency of intratumoral CD8 T cell compared with the control group, which was consistent with our in vitro results shown here (Supplementary Fig.11a and Extended Data Fig. 3d). Moreover, when anti-CD80/86 blocking antibody was administered together with PD-L1xCD3 in our DC-T cell co-culture, T cell activation was reduced, and T cell apoptosis was increased to levels similar to those of tumor-T cell co-cultures (Fig. 6b,d and Extended Data Fig. 3c,d). This further confirmed that PD-L1xCD3 treatment promotes T cell survival through enhancing CD28 costimulation. When cytokines in the supernatant was detected by Cytometric Bead Array (CBA), we observed that the DC-T cell co-culture induced the highest level of IL-2, which is known to support T cell survival and proliferation48. However, IL-2 is undetectable in ErbxCD3 group. As expected, the production of IL-2 is also B7-CD28 dependent (Fig. 6e and Extended Data Fig. 3e). Finally, when TCGA database was analyzed, patients with high level of CD28 expression but not CD8 T cell infiltration have better cumulative survival rate (Fig. 6f,g and Extended Data Fig. 4a). The level of dendritic cell infiltration and CD80/86 expression correlated with the level of CD28 significantly, which indicates the potential importance of dendritic cell mediated T cell costimulation in anti-cancer immunity (Extended Data Fig. 4b,c). In summary, PD-L1xCD3 targets dendritic cells to activate antigen specific T cells in a B7-CD28 dependent manner and overcomes the major barrier of conventional BiTE therapy (Fig. 7).
Fig. 7 |. Schematic of hypothesized working model.

PD-L1xCD3 will predominantly distribute and accumulate in the tumor tissue, targeting PD-L1 positive cells to reactivate the T cells in close proximity with them. As dendritic cells express the highest level of PD-L1 in the TME, PD-L1xCD3 mainly targets dendritic cells to not only crosslink the CD3 complex for the 1st signal activation but also interrupt PD1-PD-L1 mediated inhibition. Meanwhile, PD-L1xCD3 mediated binding to PD-L1 on DCs will release CD80 from the PD-L1-CD80 heterodimer, which can then bind to CD28 on T cells to provide the 2nd signal for T cell activation. With both the 1st and 2nd signals activated, tumor reactivate T cells will overcome activation induced cell death (AICD) and be expanded via IL-2. Thus, T cells in the tumor tissue will be reactivated for effective anti-tumor immunity and memory T cell differentiation. This schematic was created with BioRender.
Discussion
The implementation of bispecific T-cell engagers in solid tumors has been hampered presumably by not only immunosuppressive TME but also the lack of an appropriate target. In the presented study, we designed and evaluated the efficacy and safety of a PD-L1 targeting bispecific antibody in syngeneic mouse models. Compared with conventional BiTE treatment, PD-L1xCD3 treatment targets and reactivates pre-existing tumor-infiltrating lymphocytes (TILs) in vivo, which can sufficiently eradicate tumors and establish protective immunity. Mechanistically, targeting a subset of DCs instead of tumor cells with PD-L1xCD3 can not only enhance B7-CD28 interactions but also simultaneously block the PD-1/PD-L1 checkpoint to establish a proper antigen specific CD8 T cell response to control tumor progression. Taken together, our study highlighted the indispensable role of Batf3+ DC instead of tumor cell in PD-L1 targeting bi-specific antibody therapy to rejuvenate and maintain a durable immune response against cancer.
Even though T cell-redirecting therapies have received advances in patients with hematopoietic malignancies, their safety and efficacy in patients with solid tumors remain very limited. Several anti-CD3 bispecific antibodies have been evaluated in preclinical models, targeting tumor associated antigens like EGFR, Her2 and EpCAM for years49,50. Studies using a murine melanoma model have shown that targeting TAAs to redirect T cells to tumor cells fails to generate memory immune responses, and tumors eventually relapse despite initial control43. In our syngeneic mouse study, the TAA-targeting BsAb (ErbxCD3) also activated T cells efficiently to kill tumor cells in vitro but had very limited anti-tumor efficacy in vivo. T cell frequency in tumor decreased after treatment, which indicates that the TME initiates an immune resistance to evade killing. However, PD-L1xCD3 treatment could generate a strong anti-tumor effect both in vitro and in vivo. Thus, these results indicated that targeting PD-L1 to rejuvenate T cells can induce more effective anti-tumor immunity than bridging T cells to tumor cells directly.
One conceptual issue is whether engagement of T cells by tumor cells can sufficiently rejuvenate exhausted T cells. The lack of proper co-stimulatory molecules on tumor cells may result in sustained T cell dysfunction51. Bispecific antibodies engineered with additional anti-CD28 activity have been reported recently in either a trispecific format or in two separate bispecific antibody combination28,30. With additional anti-CD28 signaling, the therapeutic effect is better than BiTE alone indicating the indispensable role of T cell co-stimulation. However, tumor cells may produce various suppressive factors to restrain T cell re-activation. Therefore, targeting DCs may be a better strategy to rescue those T cells. Furthermore, the cross-linking of either CD3 or CD28 complex by anti-TAA still depends on the specificity of the tumor associated antigens, which are shared by many normal tissues. Thus, this anti-CD28 strategy also encounters the same ‘on-target off-tumor’ adverse effects as conventional BiTE therapy. Even though PD-L1xCD3 can bind PD-L1+ tumor cells to activate T cells in vitro, it mainly targets DCs instead of tumor cells in vivo52. Thus, endogenous B7–1/2 from DCs can provide co-stimulatory signaling for effective T cell activation, which is rarely expressed by solid tumors. Studies have also shown that the therapeutic effect of anti-PD-L1 treatment also depends on dendritic cells and B7 co-stimulation53. This is consistent with a recent study showing that anti-PD-L1 can release CD80 from the CD80&PD-L1 heterodimer, which provides a potential explanation for the mechanism of anti-PD-L1 treatment54. In fact, CAR-T and BsAb treatments that target CD19 for B cell leukemia consistently have better therapeutic effect than for other tumors. This may be due to B cell lymphoma cells potentially serve as APCs in lymphoid tissues to provide co-stimulation and a local milieu that favors T cell activation.
Recent studies have shown that the APC niche in the tumor microenvironment maintains a specific subset of stem-like T cells that express TCF1 and CD2833,44. Analyses of the TCGA database revealed that CD28 expression highly correlates with DC infiltration in multiple cancers. In addition, our results have also shown that CD103+ cDC1 and pre-existing CD8 T cells in the tumor are required for PD-L1xCD3 treatment. The percentage of CD28+ and TCF1+ T cells increased after PD-L1xCD3 treatment, thus indicating that PD-L1xCD3 can activate T cells that are interacting with DCs. The DC-interacting T cells have some unique therapeutic potentials such as better activation due to CD28 expression and interacting with DCs to receive B7 ligation. More importantly, DC-interacting T cells are likely to be antigen specific since DC is the dominant tumor antigen presenting APC. Our results also showed that antigen-specific T cells increased after treatment. Thus, targeting DCs to rejuvenate T cells for tumor killing may be a better strategy than directly linking T cells against tumor cells.
Immune checkpoints are another factor that may limit the anti-tumor effect of BsAb treatment. Studies have demonstrated that blocking the PD-1 pathway can enhance the therapeutic effect of anti-CD19 CAR-T55,56. There are also several ongoing clinical trials testing the combination of BsAbs with checkpoint blockade 4. PD-L1xCD3 treatment achieved this goal by simultaneously blocking negative signaling (PD-L1) and engaging positive signaling (CD3) for sustained T cell activation. PD-L1 may play a dual role for PD-L1xCD3 treatment. First, it may act as a target to redirect T cells since tumor tissues have higher level of PD-L1 than other tissues. Our results showed that intraperitoneally injected PD-L1xCD3 preferentially distributes to the tumor. It is known that multiple cells in the TME have high levels of PD-L1 expression including tumor cells, stromal cells, T cells and myeloid cells driven by abundant IFN signaling. Thus, PD-L1 may serve as a potent target for local rejuvenation of T cells in the tumor. Second, the anti-PD-L1 arm of PD-L1xCD3 also blocks the PD-L1/PD-1 pathway to prevent cytotoxic T lymphocyte (CTL) exhaustion in close proximity during T cell activation57. By conditional knocking out PD-L1 on different cells, we demonstrate that PD-L1 on DCs plays an essential role in eliciting the therapeutic effect. Intriguingly, Batf3+ DCs are the most efficient APCs in cross-presenting tumor antigens to T cells because of their highly professional ability to process antigens58. In addition, Batf3+ DCs also express higher levels of PD-L1 than other DCs or tumor cells, thus they are preferentially targeted by our bispecific antibody. Since Batf3+ DCs are essential for the efficacy of PD-L1xCD3, it is possible that PD-L1xCD3 brings T cells to this rare but potent APC for their re-activation.
As shown in our data, redirecting T cells to tumor cells for killing only induces a limited immune response. T cells that are activated by CD3 engagement also undergo AICD due to lack of CD28 co-stimulation. IFNγ will not only kill tumor cells but also induce T cell apoptosis 59. Thus, whether T cells can survive after activation becomes a key factor in determining the therapeutic effect of a BsAbs in vivo. Treatment with PD-L1xCD3, predominantly rejuvenates the T cells interacting with DCs. B7 from DCs may stimulate CD28 signaling for Bcl-XL production to abrogate AICD26. We also observed B7 dependent IL-2 production after PD-L1xCD3 treatment, which may contribute to T cell expansion and survival. Taken together, these data highlight the importance of targeting DCs to activate T cells. Despite the presented results, we acknowledge limitations of current study. In vivo efficacy should also be tested on humanized mouse models in multiple cancers to validate our major conclusions. Other DC-targeting BsAbs should also be compared to PD-L1xCD3 like CD103xCD3 or CD40xCD3, to potentially achieve similar or better anti-tumor immune responses. The combination of PD-L1xCD3 with either radiation or anti-CTLA4 should also be tested for the synergistic effect in the future.
In summary, our study not only demonstrates a novel tumor immunological concept but also proposes a strategy for BsAb based therapy, which highlights the indispensable role of targeting DCs instead of tumor cells for cancer immunotherapy.
Methods
Mice
Female C57BL/6J, BALB/c, FcγR−/−, Batf3−/−, Zbtb46-Cre and Lyz2-Cre mice were purchased from The Jackson Laboratory. Rag1−/− mice on C57BL/6 background were purchased from UT southwestern mouse breeding core. Pdl1f/f mice were generated in the UT southwestern mouse breeding core. PD-L1−/− mice were provided by L. Chen (Yale University, New Haven, Connecticut, USA). All mice were maintained under specific pathogen-free conditions. Animal care and experiments were carried out under institutional and National Institutes of Health protocol and guidelines. Animal work described in this manuscript has been approved and conducted under the oversight of the UT Southwestern Institutional Animal Care and Use Committee.
Cell lines and reagents
B16, MC38 cell lines were purchased from American Type Culture Collection (ATCC). TC-1 cells were kindly provided by Dr. T. C. Wu at John Hopkins University. TUBO was derived from a spontaneous mammary tumor in a BALB/c Neu-Tg mouse. MC38-OVA cells were made by lenti-viral transduction of OVA gene. B16E5, TC1E5, MC38E5 were sorted and sub-cloned after being transduced by lentivirus expressing murine-human chimeric EGFR (full-length of the murine EGFR with six mutated amino acids that are critical for human EGFR binding to Cetuximab). PD-L1 deficient MC38 or B16 cells were generated by CRISPR/Cas9 technology as described by previous study. All cell lines were routinely tested using mycoplasma con-tamination kit (R&D) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin under 5% CO2 at 37 °C. Anti-CD4 (GK1.5), anti-NK1.1 (PK136), anti-CD8 (53–5.8), anti-CSF1R (AFS98), anti-CD80 (16–10A1), anti-CD86 (GL-1), and CTLA-Ig mAbs were purchased from BioXCell. FTY720 were purchased from Selleckchem. A list of key resouces was pvovided in Supplementary Table.
Flow Cytometry Analysis
Single cell suspensions from either spleen, tumor or in vitro co-cultured cells were incubated with anti-FcγIII/II receptor (clone 2.4G2) for 15 minutes to block non-specific binding before staining with the conjugated antibodies. 7-AAD Viability Staining Solution or Fixable Viability Dye eFluor™ 506 was used to exclude dead cells. Foxp3, Ki-67 and TCF1 were stained intracellularly by using True-Nuclear transcription factor buffer set (BioLegend) following the manufacturer’s instructions. To assess the EGFR, PD-L1 binding affinity, EGFR and PD-L1 expressing cells were firstly stained with ErbxCD3, PD-L1xCD3 or Control IgG, then PE conjugated donkey anti-human IgG was used as a secondary antibody. To assess the FcγR binding affinity, RAW264.7 cells were first stained with bispecific antibodies with WT or mutant Fc, then PE conjugated donkey anti-human IgG was used as a secondary antibody. All staining steps were conducted at 4 °C in the dark. BD™ Cytometric Bead Array (CBA) Mouse Th1/Th2/Th17 Kit was used to measure the cytokines in the supernatants from in vitro cell culture or mice serum according to the manufacturer’s protocol (BD Biosciences). Data were collected on CytoFLEX flow cytometer (Beckman Coulter, Inc) and analyzed by using CytExpert (Beckman Coulter, Inc) or FlowJo (Tree Star Inc., Ashland, OR) software.
Enzyme-Linked ImmunoSorbent Assay (ELISA)
Microtiter plates (Corning Costar) were coated with 2 μg/mL (100 μl/well) capture antibody (AffiniPure Goat Anti-Human IgG, Fcγ fragment specific) overnight at 4 °C. After washing and blocking, diluted tissue lysate from PD-L1xCD3 treated mice were added and incubated at 37 °C for 1 hr. After washing, Horseradish Peroxidase (HRP) conjugated Goat Anti-Human IgG (H+L) was added and incubated at 37 °C for 30 minutes. Finally, the plates were visualized by adding 100 μl TMB solution plus 50 μl H2SO4 and read at 450 nm using the SPECTROstar Nano (BMG LABTECH).
IFN-γ Enzyme-Linked Immunosorbent Spot Assay (ELISPOT)
MC38-OVA (1×106) tumors were injected subcutaneously on the right flank of C57BL/6. For PD-L1xCD3 single treatment, 0.25 mg/kg PD-L1xCD3 was intraperitoneally given twice on days 10 and 15. 25 days after second treatment, splenocytes from PD-L1xCD3 treated and control mice were collected for single-cell suspension preparation. 3×105 splenocytes was seeded in each well with either irradiated MC38-OVA tumor cells (3×104) or 5 μg/mL SIINFEKL peptide (OVA257–264) to stimulate the tumor-specific T cells. After 48 hrs culture, the ELISPOT assay was performed using the IFN-γ ELISPOT kit (BD Bioscience) according to the manufacturer’s instructions. IFN-γ spots were enumerated with the CTL-ImmunoSpot® S6 Analyzer (Cellular Technology Limited). For anti-B7–1&2 blocking treatment, 0.25mg/kg PD-L1xCD3 was intraperitoneally given twice on days 10 and 15, 200 μg of anti-B7–1&2 was given intraperitoneally on day 10, 13 and 15. CD45+ cells in the tumor were enriched by EasySep™ Biotin Positive Selection Kit II. ELISPOT assay was performed by described above.
Tumor growth and treatment
A total of 1×106 MC38, 3×105 MC38E5, 1×106 MC38OVA, 1×106 TC1E5, 3×105 B16E5, 5×105 TUBOE5, 1×106 MC38-PDL1KO or 5×105 B16-PDL1KO cells were inoculated subcutaneously into right dorsal flanks of the mice in 100 μl phosphate buffered saline (PBS). Tumor-bearing mice were randomly grouped into treatment groups when tumors grew to around 80–100mm3. For PD-L1xCD3 treatment, two doses of 0.25mg/kg antibody was intraperitoneally given starting from day 8–10 with 3–4 days interval. For CSF1R, NK1.1, CD4+ and CD8+ T cell depletion, 200 µg of antibodies were intraperitoneally injected 1 day before treatment initiation and then twice a week for 2 weeks. For FTY720 treatment, 20 μg FTY720 was intraperitoneally administrated one day before treatment initiation and then 10 μg every other day for 2 weeks. For anti-B7–1&2 and CTLA-4-Ig treatment, 200μg anti-B7–1, anti-B7–2 or CTLA-4-Ig was administrated on day 10, 13 and 15. Tumor volumes were measured by the length (a), width (b) and height (h) and calculated as tumor volume = abh/2.
Production of Bispecific and Monoclonal antibodies
Based on the heterodimeric Fc variant KiHss-AkKh technology as previously described41, the ScFv fragment of anti-PD-L1 or anti-EGFR was fused with knob variant Fc region, and the anti-CD3 ScFv was fused with hole variant Fc region. PD-L1xCD3 and ErbxCD3 was generated by transient co-transfection of two arms of plasmids into FreeStyle™ 293-F cells. The supernatant containing bispecific antibodies was purified using Protein A affinity chromatography according to the manufacturer’s protocol. Protein A purified proteins were first desalted into PBS with 50 KDa ultrafiltration discs. Then, proteins were centrifuged at 12000 rpm for 10 minutes to remove aggregates. Finally, gel permeate chromatography (GPC) using Superdex200pg (GE Healthcare) was performed to remove high molecular weight species. The heterogeneity and purity were confirmed by SDS-PAGE (unprocessed SDS-PAGE is provided in Supplementary Fig. 14). The anti-PD-L1 (Atezolizumab) and anti-CD3 (17A2) monoclonal control antibodies, which have the same isotype (hIgG1) with PD-L1xCD3 were generated and purified in same procedure as described above.
Biophysical characterization of Bispecific Antibodies
The purify of bispecific antibody was analyzed by high performance size-exclusion chromatography (HP-SEC). The molecular weight was calculated with gel filtration calibration kit (HMW). The aggregation was detected by PROTEOSTAT® Protein aggregation assay (Enzo Life Sciences) with Flow cytometry. The endotoxin level was detected by chromogenic LAL assay (GenScript).
Hematoxylin and Eosin (H&E) Staining
Formalin fixed paraffin embedded slides (5 μm) were deparaffinized with xylene, ethanol, and deionized water. Slides were then stained with hematoxylin (Fisher Chemical, Cat# SH26500D) and destained excess signal with acid ethanol. Eosin (Poly Scientific, Cat# s176) was applied to slides and washed with 95% ethanol. Stained slides were dehydrated with ethanol and xylene. Slides were scanned by Hamamatsu Nanozoomer 2.0HT.
Tissue homogenate preparation
Spleen, kidney, heart, liver and tumor were excised on day 0.5, 1, 3, 5 after PD-L1xCD3 treatment and homogenized in the Cell Lysis Kit (Bio-Rad Laboratories) with the FastPrep-24 5G Homogenizer. Then centrifuge for 20 minutes at 12000 rpm. Supernatant was collected and stored at −80 °C for ELISA.
Tumor digestion
Tumor tissues were excised and digested with 2 mg/mL Collagenase I (Sigma) and 0.5 mg/mL DNase I (Roche) in the 37°C for 30mins, tumor was then passed through a 70-μm cell strainer to remove large pieces of undigested tumor. Tumor infiltrating cells were washed twice with PBS containing 2 mM EDTA.
Immune cell isolation
CD8+ T cells were isolated from lymph nodes and spleens of naïve C57BL/6J mice with a negative CD8 isolation kit (STEMCELL Technologies) following the manufacturer’s instructions. DCs in the spleen and lymph nodes were stained with CD11c and sorted by BD FACSMelody™.
In Vitro tumor cell killing, T cell activation, and cytokine analysis
3×104 MC38E5-GFP tumor cells and 3×105 naïve CD8 T cells were seeds in 96-well plate with 200 μl of RPMI-1640. A series dilutions of bispecific antibodies were added to the supernatant. T cell activation, T cell and tumor cell viability and serum cytokines was measured at 24, 48 or 72 hrs after incubation.
TCGA database analyze
Cumulative survival rate in patient with different level of CD28 expression and correlation of CD28 expression with CD80&CD86 expression, DCs infiltration were analyzed using TIMER: Tumor IMmune Estimation Resource (https://cistrome.shinyapps.io/timer/).
Statistical analysis
All the data analyses were performed with GraphPad Prism statistical software and shown as mean ± SEM. P value was determined by two-way ANOVA for tumor growth followed by multiple comparison test as indicated in the figure captions. Log-rank test for survival, Spearman’s rho correlation test for correlation or unpaired two-tailed t-tests for other analysis were used respectively. A value of p < 0.05 was considered statistically significant. The experiments were repeated multiple times as independent experiments.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request. Source data for the figures are available from figshare with the identifier https://doi.org/10.6084/m9.figshare.14984793.
Extended Data
Extended Data Fig. 1 |. PD-L1xCD3 generates superior antitumour effects than TAA-targeting BiTE in vivo.
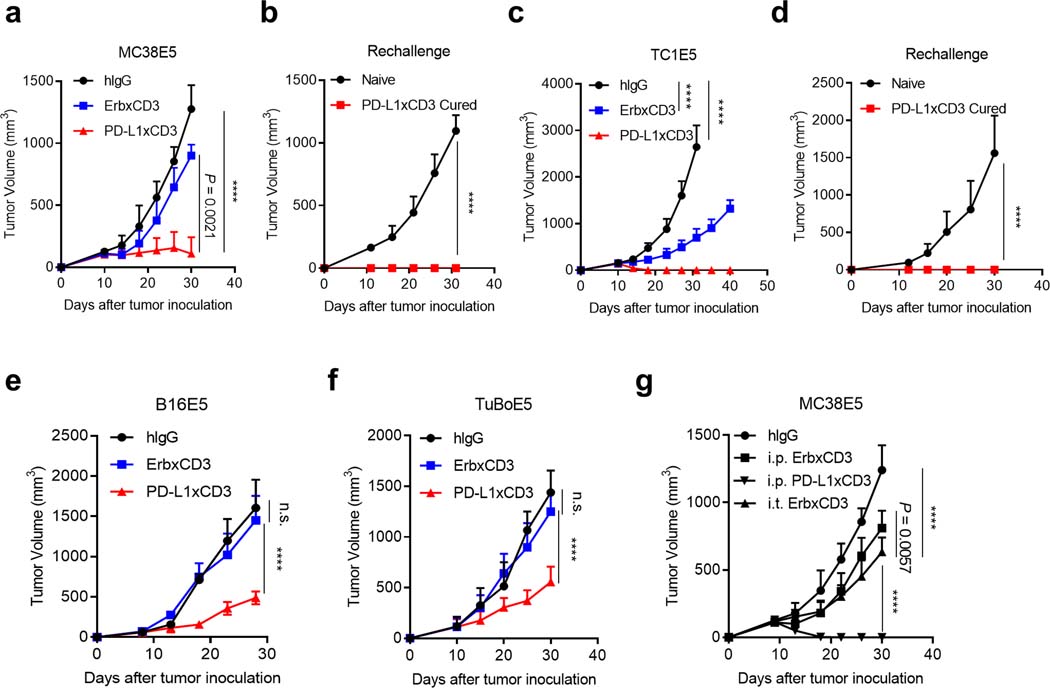
a-b, C57BL/6 J mice were subcutaneously inoculated with 3 × 105 MC38E5 tumor cells and treated with 0.25 mg kg−1 of bispecific antibodies twice on day 10 and 15. Tumor volume was measured twice per week (a). 60 days post treatment, tumor free mice were re-challenged with 3 × 106 tumor cells (b). c-d, C57BL/6 J mice were subcutaneously inoculated with 1 × 106 TC1E5 tumor cells and treated with 0.25 mg kg−1 of bispecific antibodies twice on day 10 and 15. Tumor volume was measured twice per week (c). 60 days post treatment, tumor free mice were re-challenged with 1 × 107 tumor cells (d). e, C57BL/6 J mice were subcutaneously inoculated with 3 × 105 B16E5 tumor cells and treated with 0.25 mg kg−1 of bispecific antibodies intraperitoneally twice on day 8 and 12. f, BALB/c mice were subcutaneously inoculated with 5 × 105 TuBoE5 tumor cells and treated with 0.25 mg kg−1 of fusion proteins intraperitoneally twice on day 10 and 14. g, C57BL/6 J mice were subcutaneously inoculated with 3 × 105 MC38E5 tumor cells and treated with 0.25 mg kg−1 of fusion proteins either intratumorally or intraperitoneally twice on day 9 and 13. Data were presented as mean ± s.e.m from a representative experiment (n = 5 (a, b, f, g), 4 (c-e) biologically independent animals) of two independent experiments. Statistical analysis was performed by two-way ANOVA with Tukey’s multiple comparisons test. ****P ≤ 0.0001.
Extended Data Fig. 2 |. PD-L1xCD3 targets pre-existing CD8 T cells in the tumour tissue to initiate the antitumour immune response.
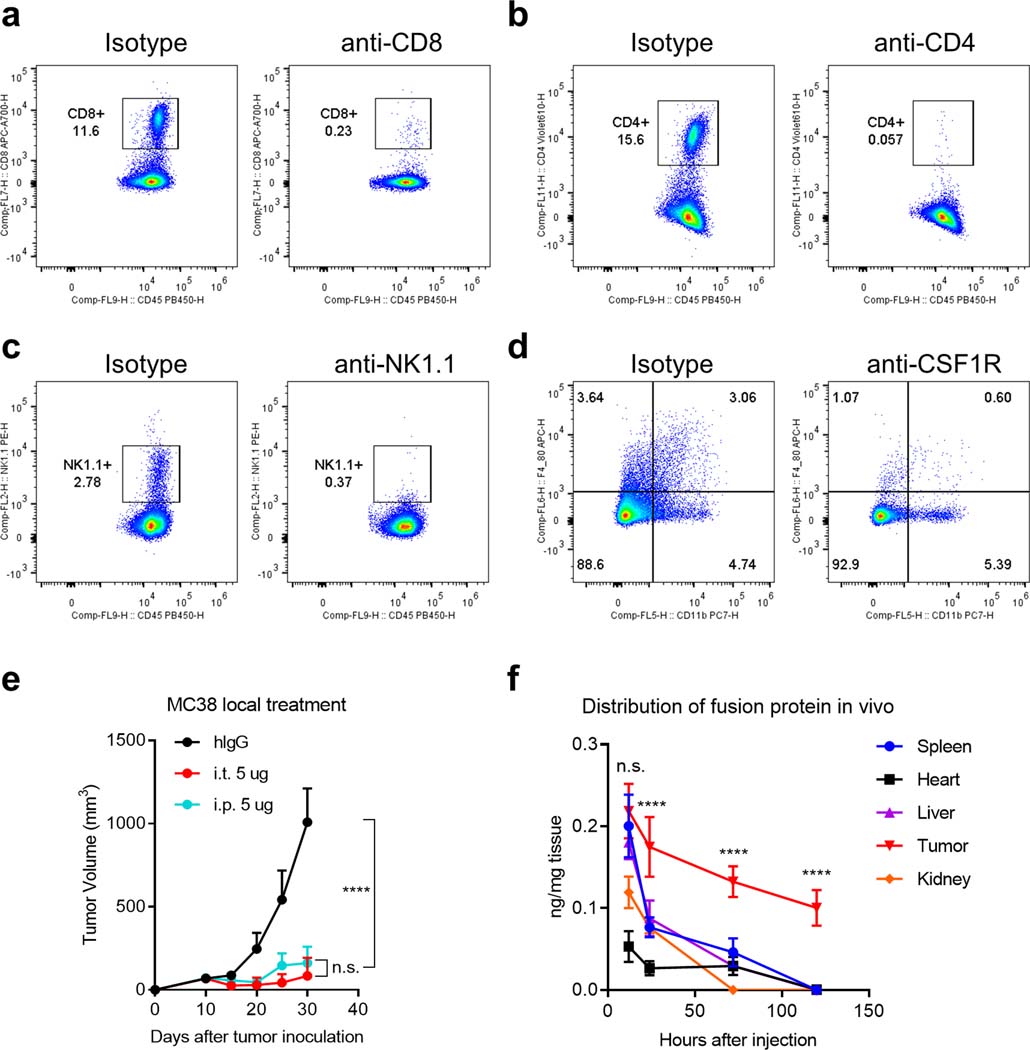
a-d, C57BL/6 J mice were inoculated with 1 × 106 MC38 tumor cells and treated with PD-L1xCD3 (0.25 mg kg−1 on day 10 and 15). 200 μg of anti-CD8, anti-CD4, anti-NK1.1 or 500 μg of anti-CSF1R was administrated respectively one day before treatment initiation and then twice a week for 2 weeks. The percentage of CD8 +cells (a), CD4 +cells (b), NK1.1+ cells (c) and CD11b +F4/80+ cells (d) in the spleen were detected by flow cytometry. e, C57BL/6 J mice (n = 5 biologically independent animals) were subcutaneously inoculated with 1 × 106 MC38 tumor cells and treated with 0.25 mg kg−1 of PD-L1xCD3 either intratumorally or intraperitoneally twice on day 10 and 15. f, C57BL/6 J mice (n = 3 biological replicates) were subcutaneously inoculated with 1 × 106 MC38 tumor cells and intraperitoneally treated with 0.25 mg kg−1 of PD-L1xCD3. Concentration of fusion protein in different tissues were measured by hIgG ELISA at indicated time point. Data were shown as mean ± s.e.m from a representative experiment of two independent experiments. Statistical analysis was performed by two-way ANOVA with Tukey’s multiple comparisons test. ****P ≤ 0.0001.
Extended Data Fig. 3 |. Co-stimulatory signaling blockade abolished PD-L1xCD3 mediated antitumour effects and the in vitro activation of T cells by TAA-targeting BiTE.

a-b, C57BL/6 J mice were inoculated with 1 × 106 MC38 tumor cells and treated with PD-L1xCD3 (0.25 mg kg−1 on day 10 and 15), 200 μg CTLA4-Ig was administrated on day 10, 13 and 15. Experimental design (a) and tumor growth curve (b) are shown. c-e, CD8 T cells were co-cultured with either tumor cells or dendritic cells in the presence of ErbxCD3. T cell activation (c), apoptotic T cells (d), supernatant IL-2 and IFN-γ (e) were measured by flow cytometry. Data were shown as mean ± s.e.m from a representative experiment of two independent experiments (n = 5 biologically independent animals). Statistical analyses were performed by two-way ANOVA with Dunnett’s multiple comparisons test (b), two-tailed unpaired Student’s t-test (c-e). ***P ≤ 0.001, ****P ≤ 0.0001.
Extended Data Fig. 4 |. Correlation analysis of CD28 expression with CD80/86 expression, dendritic cell infiltration and patient survival.

TCGA database was analyzed for cumulative survival according to CD28 expression (a), correlation of CD28 level with CD80 and CD86 level (b) and correlation of CD28 level with dendritic cell infiltration (c). Skin cutaneous melanoma (SKCM), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), lung adenocarcinoma (LUAD), colon adenocarcinoma (COAD), breast invasive carcinoma (BRCA). Statistical analyses were performed by log-rank test (a), and Spearman’s rho correlation test (b-c).
Supplementary Material
Acknowledgements
We thank the UT Southwestern Institutional Animal Care & Use Committee (IACUC) and Animal Resources Center (ARC). Y.-X.F. holds the Mary Nell and Ralph B. Rogers Professorship in Immunology. This work was supported by Cancer Prevention and Research Institute of Texas (CPRIT) grant RR150072 given to Y.-X.F and National Institutes of Health (NIH) grant 1U54CA244719-01 to J.Q. We also thank Yong Liang, Xuezhi Cao, Zhenhua Ren, Anli Zhang, Chunbo Dong, Zhida Liu, Changzheng Lu and Benjamin Moon for providing experiment materials and helpful discussions.
Peer review information
Nature Biomedical Engineering thanks Reviewer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information [please do not modify this section]
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-021-00800-2.
References
- 1.Staerz UD, Kanagawa O & Bevan MJ Hybrid antibodies can target sites for attack by T cells. Nature 314, 628–631 (1985). [DOI] [PubMed] [Google Scholar]
- 2.Garber K Bispecific antibodies rise again. Nature reviews. Drug discovery 13, 799–801 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Baeuerle PA & Reinhardt C Bispecific T-cell engaging antibodies for cancer therapy. Cancer research 69, 4941–4944 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Trabolsi A, Arumov A & Schatz JH T Cell-Activating Bispecific Antibodies in Cancer Therapy. Journal of immunology 203, 585–592 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Bargou R et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321, 974–977 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Topp MS et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. The Lancet. Oncology 16, 57–66 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Maude SL, Barrett D, Teachey DT & Grupp SA Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer journal 20, 119–122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topp MS et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 32, 4134–4140 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Li J et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Science translational medicine 11 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Reusch U et al. Anti-CD3 x anti-epidermal growth factor receptor (EGFR) bispecific antibody redirects T-cell cytolytic activity to EGFR-positive cancers in vitro and in an animal model. Clinical cancer research : an official journal of the American Association for Cancer Research 12, 183–190 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Cioffi M, Dorado J, Baeuerle PA & Heeschen C EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. CClinical cancer research : an official journal of the American Association for Cancer Research 18, 465–474 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Han H et al. Bispecific anti-CD3 x anti-HER2 antibody mediates T cell cytolytic activity to HER2-positive colorectal cancer in vitro and in vivo. International journal of oncology 45, 2446–2454 (2014). [DOI] [PubMed] [Google Scholar]
- 13.of I et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology 7, e1450710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kebenko M et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology 7, e1450710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lutterbuese R et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proceedings of the National Academy of Sciences of the United States of America 107, 12605–12610 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Panhuys N TCR Signal Strength Alters T-DC Activation and Interaction Times and Directs the Outcome of Differentiation. Frontiers in immunology 7, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chai JG & Lechler RI Immobilized anti-CD3 mAb induces anergy in murine naive and memory CD4+ T cells in vitro. International immunology 9, 935–944 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Harding FA, McArthur JG, Gross JA, Raulet DH & Allison JP CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 356, 607–609 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Green DR, Droin N & Pinkoski M Activation-induced cell death in T cells. Immunological reviews 193, 70–81 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Curtsinger JM & Mescher MF Inflammatory cytokines as a third signal for T cell activation. Current opinion in immunology 22, 333–340 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalos M et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science translational medicine 3, 95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKay M et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nature biotechnology 38, 233–244 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Labrijn AF, Janmaat ML, Reichert JM & Parren P Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov 18, 585–608 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Seckinger A et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 31, 396–410 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Rius Ruiz I et al. p95HER2-T cell bispecific antibody for breast cancer treatment. Sci Transl Med 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boise LH et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3, 87–98 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Garfall AL & June CH Trispecific antibodies offer a third way forward for anticancer immunotherapy. Nature 575, 450–451 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Skokos D et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Science translational medicine 12 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Suntharalingam G et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. The New England journal of medicine 355, 1018–1028 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Wu L et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nature Cancer 1, 86–98 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Binnewies M et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature medicine 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knutson KL & Disis ML Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer immunology, immunotherapy : CII 54, 721–728 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jansen CS et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou W, Wolchok JD & Chen L PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Science translational medicine 8, 328rv324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin H et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. The Journal of clinical investigation 128, 805–815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang H et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. The Journal of clinical investigation 128, 580–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Diaz A et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell reports 19, 1189–1201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohnke T, Krupka C, Tischer J, Knosel T & Subklewe M Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. Journal of hematology & oncology 8, 111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobold S, Pantelyushin S, Rataj F & Vom Berg J Rationale for Combining Bispecific T Cell Activating Antibodies With Checkpoint Blockade for Cancer Therapy. Frontiers in oncology 8, 285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlothauer T et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein engineering, design & selection : PEDS 29, 457–466 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Wei H et al. Structural basis of a novel heterodimeric Fc for bispecific antibody production. Oncotarget 8, 51037–51049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiao J et al. Targeting Tumors with IL-10 Prevents Dendritic Cell-Mediated CD8(+) T Cell Apoptosis. Cancer cell 35, 901–915 e904 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Benonisson H et al. CD3-Bispecific Antibody Therapy Turns Solid Tumors into Inflammatory Sites but Does Not Install Protective Memory. Molecular cancer therapeutics 18, 312–322 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Sade-Feldman M et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013 e1020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diskin B et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nature immunology 21, 442–454 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Hui E et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamphorst AO et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 355, 1423–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelly E, Won A, Refaeli Y & Van Parijs L IL-2 and related cytokines can promote T cell survival by activating AKT. Journal of immunology 168, 597–603 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Heiss MM et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. International journal of cancer 127, 2209–2221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiewe P et al. Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 12, 3085–3091 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Schildberg FA, Klein SR, Freeman GJ & Sharpe AH Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity 44, 955–972 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Horn LA et al. CD3xPDL1 bi-specific T cell engager (BiTE) simultaneously activates T cells and NKT cells, kills PDL1(+) tumor cells, and extends the survival of tumor-bearing humanized mice. Oncotarget 8, 57964–57980 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayoux M et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Science translational medicine 12 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 51, 1059–1073 e1059 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rafiq S et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nature biotechnology 36, 847–856 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hill BT, Roberts ZJ, Xue A, Rossi JM & Smith MR Rapid tumor regression from PD-1 inhibition after anti-CD19 chimeric antigen receptor T-cell therapy in refractory diffuse large B-cell lymphoma. Bone marrow transplantation (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wallberg M et al. Anti-CD3 treatment up-regulates programmed cell death protein-1 expression on activated effector T cells and severely impairs their inflammatory capacity. Immunology 151, 248–260 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.del Rio ML, Bernhardt G, Rodriguez-Barbosa JI & Forster R Development and functional specialization of CD103+ dendritic cells. Immunological reviews 234, 268–281 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Refaeli Y, Van Parijs L, Alexander SI & Abbas AK Interferon gamma is required for activation-induced death of T lymphocytes. The Journal of experimental medicine 196, 999–1005 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request. Source data for the figures are available from figshare with the identifier https://doi.org/10.6084/m9.figshare.14984793.