

Summary
It is known that the human cellular models of Alzheimer’s disease (AD) and tauopathy can only recapitulate the very early stage of the disease. To overcome these limitations, we developed a technology to make forebrain organoids (FBOs) from feeder-free induced pluripotent stem cells (iPSC)s by regulating a FGF2 concentration and applied this method to generate FBOs from patients with familial AD (fAD FBOs). The obtained fAD FBOs recapitulated the amyloid-β pathology and increased tau phosphorylation but not tau aggregates. To fully induce the tau pathology, FBOs were injected with adeno-associated virus (AAV)-expressing P301L mutant tau. In these Tau-P301L FBOs, tau fibrils were observed in the neuronal cell body and neurites with immunoelectron microscopy, in addition to the sarkosyl-insoluble and thioflavin S-positive phospho-tau aggregates. Collectively, this model can be used as a platform for investigating pathogenetic mechanisms and evaluation of target molecules for drug discovery for tauopathy.
Keywords: feeder-free iPSCs, brain organoids, FGF2, tauopathy, Alzheimer's disease, tau, AAV
Graphical abstract
Highlights
-
•
An efficient method to generate neuropeithelia from feeder-free iPSCs
-
•
Forebrain organoids from fAD-iPSCs exhibit amyloid-β pathology but no tau aggregation
-
•
AAV delivery of Tau-P301L causes tau aggregation throughout the FBOs
-
•
AAV injection at later time points enhances tau phenotypes
Motivation
One of the reasons why drug development for Alzheimer’s disease (AD) and tauopathy has been difficult to date is the lack of appropriate models. Rodent models cannot fully recapitulate the disease pathology due to species differences between rodents and humans. Due to the low accessibility of patient brain for experimental use, human induced pluripotent stem cell (iPSC) technology is a promising approach for the generation of human cell models as an alternative research tool. However, even using human iPSCs, most cellular models of AD and tauopathy can only recapitulate the early stage of the disease, and it is challenging to display protein aggregation, such as amyloid-β plaques and tau fibrils, which are hallmarks of AD. To overcome these limitations, we made an improvement to increase the efficiency of neuroepithelia formation, leading to an increase in the efficiency of brain organoid generation. Furthermore, we established an efficient system of gene transfer by adeno-associated virus (AAV) injection into brain organoids and applied this to generate brain organoids with pronounced tau pathology, including tau fibrils.
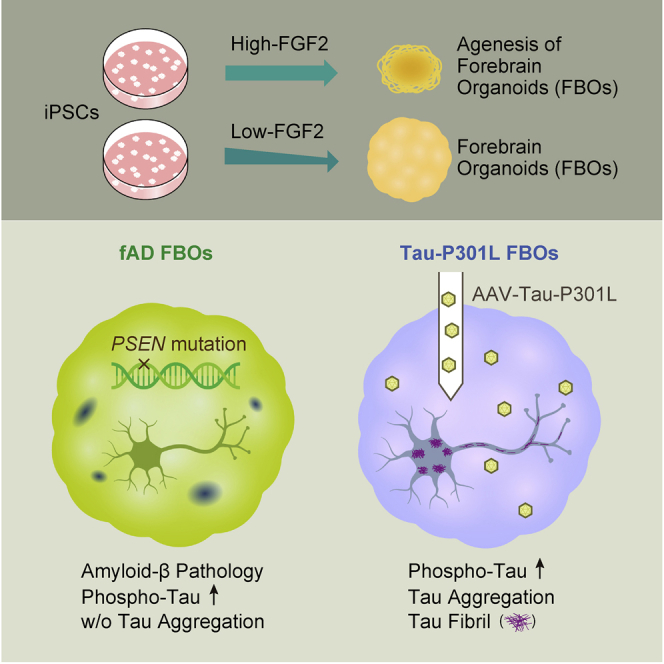
Shimada et al. develop a method to create forebrain organoids (FBOs) by regulating a FGF2 concentration in the medium of feeder-free iPSCs. AAV injection of mutant tau to FBOs induces pronounced tau aggregates containing tau fibrils, suggesting the utility of this approach for modelling tauopathy.
Introduction
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disease and is associated with progressive memory impairments and cognitive decline. The major hallmarks of AD are amyloid-β and tau protein aggregates, which lead to senile plaques and neurofibrillary tangles (NFTs), respectively. Mouse models with familial AD (fAD)-related mutations have allowed researchers to obtain extensive knowledge about AD pathogenesis; however, despite this extensive research, most candidate molecules developed using mouse models have failed in late clinical trials. Because the use of patient brain tissues is limited, human induced pluripotent stem cell (iPSC) technology is a promising approach for the generation of in vitro models of AD in humans (Klimmt et al., 2020; Okano and Yamanaka, 2014; Sandoe and Eggan, 2013; Takahashi et al., 2007; Takahashi and Yamanaka, 2006). A number of research groups have reported that neurons derived from human iPSCs from patients with AD exhibit AD phenotypes. Most of these groups have reported higher secretion levels of Aβ40 and Aβ42 or a higher Aβ42/Aβ40 ratio and increased phosphorylated tau (phospho-tau) in neurons obtained from iPSCs derived from patients with AD (AD-iPSCs); however, amyloid-β plaque deposition, tau aggregation, and neurodegeneration have seldom been reported (Arber et al., 2017; Klimmt et al., 2020). Three-dimensional (3D) culture strategies allow more complex cell-cell interactions, cell-extracellular matrix (ECM) interactions, and spatial organization than conventional 2D culture approaches; therefore, 3D brain organoids are expected to exhibit disease phenotypes such as extracellular amyloid-β aggregation (Centeno et al., 2018) (Di Lullo and Kriegstein, 2017). Raja et al. were the first to generate cerebral organoids from AD-iPSCs and observed increased extracellular accumulation of amyloid-β puncta but not amyloid-β plaque deposition (Raja et al., 2016). Gonzalez et al. reported that thioflavin derivative-positive amyloid-β plaques are formed in iPSC-cerebral organoids derived from a patient with AD (Gonzalez et al., 2018). It is surprising that they also detected NFT-like inclusions that are positive for Gallyas silver staining in their cerebral organoids. In fact, this is the only previous report that detected both amyloid-β plaques and NFT-like inclusions in AD-iPSC-derived brain organoids. However, in this report, while they showed that there is an increased amount of phospho-tau by immunoblot analysis, it remains unclear whether Gallyas-positive inclusions are composed of tau fibrils. Thus, the recapitulation of amyloid-β and tau aggregation in human iPSC-derived brain organoids remains a challenging issue to overcome.
In the present study, we developed next-generation brain organoid technologies for recapitulating late-onset phenotypes of tauopathy such as frontotemporal lobar degeneration (FTLD) in vitro. For this purpose, we developed a method for efficient production of forebrain organoids (FBOs) from feeder-free iPSCs and adeno-associated virus (AAV)-mediated gene effective transduction into FBOs through the microinjection of viral particles into the individual FBOs. Through these optimizations, we could achieve technological breakthroughs to recapitulate the late hallmarks of tau pathology, such as tau aggregation in control and fAD iPSC-derived FBOs with AAV-expressing Tau-P301L, which is the mutation of MAPT gene that causes a form of FTLD (FTLD-Tau/FTDP-17).
Results
Efficient generation of 3D FBOs from control and AD-iPSCs
It has been reported that neurons in 3D culture are more mature than those in 2D culture and that brain organoids are more appropriate cellular models than 2D models (Centeno et al., 2018; Trujillo et al., 2019). In addition, since secreted amyloid-β is expected to be retained within brain organoids, brain organoids are considered to be suitable for detecting amyloid-β deposition. To create 3D FBOs, we modified Ming et al.’s protocol (Qian et al., 2016, 2018) to generate 3D FBOs from feeder-free iPSCs. Feeder-free iPSCs were routinely cultured in StemFit Basic 02 medium (Ajinomoto) with a high concentration (100 ng/mL) of FGF2. However, it was difficult to generate embryoid bodies (EBs) with neuroepithelium-like structure reproducibly; rather, small aggregates with many dead and unhealthy cells around the aggregates were induced (Figure 1B). To improve the protocol, we focused on the FGF2 concentration because feeder-dependent iPSCs, from which brain organoids are successfully generated, are usually maintained with much a lower concentration (4 ng/mL) of FGF2 than feeder-free iPSCs. FGF2 has been known to be a key factor for maintaining the pluripotency but also functions as an inhibitor for neural induction (Greber et al., 2011). Therefore, we hypothesized that reducing FGF2 concentration in feeder-free iPSC cultures enables the reproducible generation of brain organoids. The concentration of FGF2 was reduced stepwise from 100 to 30 ng/mL and finally to 10 ng/mL immediately before differentiation since the FGF2 concentration may affect the undifferentiated state of cells and the efficiency of their differentiation into brain organoids (Figure 1A). iPSCs cultured with 10 ng/mL FGF2 (low-FGF2 iPSCs) formed EBs (low-FGF2 EBs) that were more stable in size and morphology than those (high-FGF2 EBs) generated from iPSCs cultured with 100 ng/mL FGF2 (high-FGF2 iPSCs) (Figures 1B and 1C). These low-FGF2 EBs formed multiple thick neuroepithelium-like structures, although most high-FGF2 EBs became cystic and possessed fewer neuroepithelium-like structures during Matrigel embedding from days 7 to 14 (Figure 1B).
Figure 1.

A FGF2 concentration in feeder-free iPSCs contributes to forebrain identity acquisition in EBs
(A) Schematic illustration of generating EBs and neuroepithelia from feeder-free iPSCs with high (100 ng/mL, high-FGF2 iPSCs) or low (10 ng/mL, low-FGF2 iPSCs) concentrations of FGF2.
(B) Representative images of high- or low-FGF2 iPSCs (201B7), EBs, and neuroepithelia. n > 30. Scale bar, 300 μm.
(C) Diameter of high- or low-FGF2 EBs generated from high- or low-FGF2 iPSCs cultured for 3, 4, and 10 weeks. Greater than 6 EBs from three different batches.
(D) Volcano plot of differential gene expression in high- versus low-FGF2 iPSCs by DEG analysis. The expression difference is considered significant for a log2 fold change of >1 (outer pink dash vertical line) and a p value of <0.00124 corresponding to 0.05 in adjusted p value (pink dash horizontal line). See also Table S1.
(E) Volcano plot of differential gene expression in high- versus low-FGF2 EBs by DEG analysis. The expression difference is considered significant for a log2 fold change of >1 (outer pink dash vertical line) and a p value of <0.00242 corresponding to 0.05 in adjusted p value (pink dash horizontal line). See also Table S1.
(F) Enrichment analysis for GO biological process of significant up- or down-regulated genes between high- and low-FGF2 iPSCs. Six top terms are shown. See also Figure S1A and Table S1.
(G) Enrichment analysis for GO biological process of significant up- or down-regulated genes between high- and low-FGF2 EBs. Six top terms are shown. See also Figure S1B and Table S1.
(H) Heatmap of neural stem/progenitor cell (NS/PC), forebrain, midbrain, hindbrain, and spinal cord marker gene expression in high- and low-FGF2 iPSCs (n = 3), and high- and low-FGF2 EBs on day 7 (n = 5). See also Figure S2A.
(I) PCA map showing PC1 and PC3 of high- and low-FGF2 iPSCs and high- and low-FGF2 EBs (pink triangle: high-FGF2 iPSCs; green circle: low-FGF2 iPSCs; purple triangle: high-FGF2 EBs; blue circle: low-FGF2 EBs). See also Figure S2B.
(J) Immunohistochemical staining of the forebrain marker FOXG1 and the neural progenitor marker SOX2 in high- (left panel) or low-FGF2 (right panel) neuroepithelia derived from 201B7 iPSC line on day 14 (n = 3). The nuclei were stained with Hoechst 33258. Scale bar, 50 μm. See also Figures S2C–S2F.
To assess differences in EB formation between high- and low-FGF2 EBs, differentially expressed genes (DEGs) were analyzed. Among 22,633 genes, 205 genes in low-FGF2 iPSCs and 160 genes in high-FGF2 iPSCs were significantly up-regulated, while 334 genes in low-FGF2 EBs and 400 genes in high-FGF2 EBs were significantly up-regulated (Figures 1D and 1E; Table S1). Enrichment analysis of Gene Ontology (GO) terms in biological process in revealed enrichment of regionalization (green) and regulation of cellular response to growth factor stimulus pathways (blue) in iPSCs (Figures 1D, 1F, and S1A; Table S1). Both low- and high-FGF2 iPSCs expressed PSC markers, such as POU5F1, LIN28A, SOX2, and NANOG (Figure S2A). On the other hand, pattern specification process (green), regionalization (green), and forebrain development (red) pathways were enriched in EBs (Figures 1E, 1G, and S1B; Table S1). The regionalization pathway was enriched in both iPSCs and EBs, though the forebrain development pathway was enriched only in EBs. Among those DEGs, we noted that some forebrain markers, such as FOXG1, SIX3, LHX2, and RAX, were more highly expressed in low-FGF2 EBs than in high-FGF2 EBs, while several mid-brain and mid-hindbrain markers (ZIC1, EN1, LMX1B, WNT1) were more highly expressed in high-FGF2 EBs (Figure 1H). The principal-component analysis (PCA) of mRNA sequencing (mRNA-seq) results, including expression data of 22,633 genes in low- and high-FGF2-iPSCs and EBs, showed that the PC1 component, in which the contribution ratio is 64.63%, represented the variation in gene expressions between iPSCs and EBs, while the PC3 component, in which the contribution ratio is 5.14%, was considered to represent the difference in gene expressions between low- and high-FGF2-EBs (Figures 1I and S2B). Notably, both low- and high-FGF2 iPSCs did not separate along the PC2 components as well as PC3 components, suggesting that the concentration of FGF2 largely affects gene expressions in EBs after initiation of neural induction. Moreover, immunohistochemical analysis showed that SOX2+ neural progenitor cells positive for the forebrain marker FOXG1 were observed in low-FGF2 EBs, while FOXG1+ cells were not found in high-FGF2 neuroepithelia on day 14 (Figure 1J). These data suggest that low-FGF2 EBs acquired forebrain identity.
Low-FGF2 EBs with neuroepithelium-like structures were selected on day 14 and cultured in a 30 mL bioreactor (Figure S2C). Although it depends on the iPSC lines, more than 50% of low-FGF EBs can then be cultured in a bioreactor. Immunohistochemical analysis showed that most cells were positive for the neural progenitor markers Pax6 and Nestin on day 14 (Figure S2D). On day 28, βIII tubulin+/COUP-TF-interacting protein 2 (CTIP2)+ deep layers were observed around SOX2+ ventricular zone (VZ)-like structures. The FBOs were cultured with 40% O2 from day 35 to increase oxygen penetration. By day 56, T-box brain protein 1 (TBR1)+ and CTIP2+ deep layers had formed above the distinct VZ-like structures. On day 81, special AT-rich sequence-binding protein 2 (SATB2)+ upper-layer neurons were found near the surface, and glial fibrillary acidic protein (GFAP)+ astrocytes were observed (Figure S2D). Also, puncta positive for the presynaptic marker synaptophysin were observed in FBOs on day 84 (Figure S2E). Furthermore, myelin basic protein (MBP)+ oligodendrocytes were spontaneously produced on day 126 FBOs (Figure S2F). We successfully generated FBOs from several control (KhES1, 414C2, 201B7, RPC771) cell lines and AD-iPSC (PS1-2, PS2-2) lines. These results indicate that FBOs could be efficiently generated from feeder-free iPSCs by simply reducing the concentration of FGF2 in the iPSC culture medium in a reproducible manner, suggesting that the initial state of iPSCs is an important deciding factor for subsequent differentiation.
Recapitulation of amyloid-β pathology in fAD FBOs
To investigate whether amyloid-β pathology was observed in 3D culture, FBOs were generated from fAD-iPSC lines (PS1-2, PS2-2) (Yagi et al., 2011). Immunohistochemistry was performed using two antibodies, 6E10 and 4G8, which recognize different peptides within amyloid-β. While amyloid-β plaque-like structures were not observed in fAD FBOs on day 84 (Figure S3A), 6E10+ or 4G8+ amyloid-β plaque-like structures, which were similar to amyloid-β plaque depositions found in the brains of patients with AD in size and morphology, were detected in fAD FBOs on day 120 (Figures 2A and S3B). To measure secreted Aβ40 and Aβ42 levels, the culture medium of day 120 FBOs was subjected to ELISA. As exhibited by a previous study (Yagi et al., 2011), the ratio of secreted levels of Aβ42 to Aβ40 was significantly higher in the medium of fAD FBOs than in that of control FBOs, 414C2 iPSCs, and KhES1 embryonic stem cells (ESCs) (Figure 2B). Day 120 FBOs were also stained with BTA-1, a thioflavin-T derivative with high affinity for amyloid deposits. BTA-1-positive aggregates observed in the fAD FBOs resembled those found in the AD brain, whereas BTA-1-positive staining was not detected in control FBOs or the control healthy brain (Figure S3C). These data demonstrate that amyloid-β pathology occurs spontaneously in fAD FBOs in a time-dependent manner.
Figure 2.

Amyloid-β and tau pathology in day 120 fAD FBOs
(A) Immunohistochemical staining for amyloid-β (4G8: red), MAP2 (green), and GFAP (magenta) in day 120 414C2 (control) FBOs and PS1-2 (fAD) FBOs. Images are representative of 414C2 (control) FBOs and PS1-2 (fAD) FBOs that were imaged with these markers (n = 3). The nuclei were stained with Hoechst 33258 (blue). Scale bar, 50 μm. See also Figures S3A–S3C.
(B) The Aβ42/Aβ40 ratio was significantly different between control FBOs (iPSCs: 414C2, ESCs: KhES1) and fAD-FBOs (PS1-2, PS2-2). The concentrations of Aβ42 and Aβ40 in the culture medium of day 120 control FBOs and fAD FBOs with 2–3 mm in diameter were measured by ELISA (n = 3). Error bars represent ± SEM of three organoids. Statistical significance was determined using unpaired Student’s t test (∗p < 0.05).
(C) Immunostaining for phospho-tau (AT8: magenta) and total tau (cyan) in day 120 414C2 (control) FBOs and PS1-2 (fAD) FBOs. Images are representative of 414C2 (control) FBOs and PS1-2 (fAD) FBOs that were imaged with these markers (n = 3). The nuclei were stained with Hoechst 33258. Scale bar, 50 μm.
(D) Western blot analysis of RIPA buffer-soluble fractions using tau antibody and AT8 for phospho-tau. Quantification of ratio of phospho-tau/total tau protein revealed a significant difference between 414C2 (control) and PS1-2 (fAD) FBOs. Error bars represent ± SEM of three organoids. Statistical significance was determined using an unpaired Student’s t test (∗p < 0.05) (n = 3).
(E) Immunostaining for aggregated tau (MC1: magenta) and total tau (cyan) in day 120 414C2 (control) FBOs and PS1-2 (fAD) FBOs. Images are representative of 414C2 (control) FBOs and PS1-2 (fAD) FBOs that were imaged with these markers (n = 3). The nuclei were stained with Hoechst 33258. Scale bar, 50 μm.
(F) Schematic representation of phenotypes in fAD FBOs. Amyloid-β pathology and phospho-tau were observed, but tau aggregation was not developed in fAD FBOs.
(G) Representative images of PS1-2 (fAD) FBOs overexpressing GFP (left panels) or Tau-P301L (right panels) by AAV injection immunostained for aggregated tau (MC1: magenta) and total tau (RTM38: cyan). The nuclei were stained with Hoechst 33258 (blue). The FBOs were analyzed 5 weeks after AAV injection. Images are representative of PS1-2 (fAD) FBOs overexpressing GFP or Tau-P301L that were imaged with these markers (n = 2, GFP: n = 3, Tau-P301L). Scale bar, 200 μm.
Developing tau pathologies in FBOs: AAVs-Tau-P301L transduction into FBOs results in detergent-insoluble tau and filamentous structure of tau aggregates
The accumulation of tau fibrils are observed in various neurodegenerative diseases including AD and FTLD (Soeda and Takashima, 2020). Binding of tau protein to microtubules is regulated by its phosphorylation. Phospho-tau (recognized by an AT8 antibody) detached from microtubules and formed oligomer in pathological condition (recognized by T22 and MC1 antibodies). Further promotion of conformational changes to forms β-sheet structures leads to the formation of tau fibrils (also recognized by a MC1 antibody) (Soeda and Takashima, 2020).
Based on these backgrounds, we first investigated whether fAD FBOs (with PS1 mutations) exhibited tau pathology by immunohistochemical method using AT8 and MC1 antibodies. AT8+-phospho-tau was detectable in fAD FBOs, but not in control FBOs, on day 120 (Figure 2C); however, MC1+-aggregated tau (presumably tau oligomer) was not detected in either control FBOs or fAD FBOs (Figure 2E). To examine the presence of phospho-tau in fAD FBOs, immunoblotting of radioimmunoprecipitation assay (RIPA)-soluble fractions was performed. The ratio of phospho-tau/total tau protein was significantly higher in fAD FBOs than in control FBOs (Figure 2D). These data suggest that while AT8+ phospho-tau, which is an early hallmark of AD pathology, is present in fAD FBOs carrying PS1 or PS2 mutations, it does not lead to pathological structural changes recognized by a MC1 antibody (Figure 2F).
As a next step, to fully induce the tau pathology in FBOs, we transduced fAD FBOs (with PS1 mutation) with AAV-expressing Tau-P301L (1N4R), encoded by mutant MAPT gene causing frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), a subtype of FTLD, which has a higher aggregation ability than wild-type tau in vitro (Aoyagi et al., 2007). As a result, MC1-positive aggregated tau was observed prominently throughout the FBOs when Tau-P301L was overexpressed compared with the control where GFP was overexpressed (Figure 2G).
To investigate the appropriate timing of AAV injection, AAV was injected into 5-, 8-, and 13-week control iPSC-derived FBOs, and the Tau-P301L-overexpressing FBOs (Tau-P301L FBOs) were analyzed 5 weeks after AAV injection (Figures 3A and 3B; Video S1). Immunofluorescence revealed that AT8+ phospho-tau and MC1+ aggregated tau levels were dramatically increased in Tau-P301L FBOs compared with GFP-transduced controls in an injection time point-dependent manner (Figures 3C–3E, S4A, and S4B). Moreover, T22+ tau oligomers were observed in Tau-P301L FBOs (Figures S5A and S5B). Remarkably, a marked degree of tau pathology was observed in 13-week Tau-P301L FBOs. A presynaptic marker, synaptophysin+ puncta were prominently observed in the 13-week control FBOs, suggesting that the presence of more mature neurons compared with young FBOs (Figures S6A and S6B). We then analyzed protein levels in Tau-P301L FBOs. Western blot analysis confirmed that the level of 4-repeat tau protein was increased in the soluble fractions of 8- and 13-week Tau-P301L FBOs (Figure 4A). To explore the presence of insoluble phospho-tau aggregates, which are observed in patients with FTLD (FTDP-17), the sarkosyl-insoluble fractions of Tau-P301L FBOs were assessed by immunoblotting using an HT7 antibody that detects both phosphorylated and unphosphorylated tau and an antibody targeting tau phosphorylated at the S396 residue specifically (Figure 4B). Insoluble phospho-tau were detected in the sarkosyl-resistant fractions of 8- and 13-week Tau-P301L FBOs but not in GFP-expressing control FBOs (Figure 4B). Next, 13-week Tau-P301L FBOs were subjected to thioflavin-S staining. Colocalization of thioflavin-S-positive deposits with AT8 staining was observed in 13-week Tau-P301L FBOs as well as the AD brain, which shows a tau pathology as a positive control (Figure 4C).
Figure 3.

Tau-P301L overexpression in FBOs by AAV injection
(A) Schematic of AAV injection into FBOs. AAV expressing GFP or Tau-P301L was injected into FBOs at 5, 8, and 13 weeks. Five weeks after AAV injection, the FBOs were processed for immunohistochemistry.
(B) A representative image of AAV injection into FBOs. A needle stick in an FBO.
(C) Immunofluorescence analysis of phospho-tau (AT8: magenta) and total tau (RTM38: cyan) in 5-, 8-, and 13-week 201B7 (control) FBOs. Images are representative of 201B7 (control) FBOs overexpressing GFP or Tau-P301L that were imaged with these markers (n = 3 or 4). The nuclei were stained with Hoechst 33258 (blue). Scale bar, 50 μm. See also Figures S4A, S5, and S6.
(D) Immunofluorescence analysis of aggregated tau (MC1: magenta) and total tau (RTM38: cyan) in 5-, 8-, and 13-week 201B7 (control) FBOs. Images are representative of 201B7 (control) FBOs overexpressing GFP or Tau-P301L that were imaged with these markers (n = 3 or 4). The nuclei were stained with Hoechst 33258 (blue). Scale bar, 50 μm. See also Figures S4B, S5, S6, and S7.
(E) The values shown in the graph are the areas of AT8- (left) or MC1-positive (right) area normalized to the total area. The data are expressed as the mean values ± SEM. For statistical analysis, two-way ANOVA was used (∗p < 0.04, ∗∗p < 0.006, ∗∗∗p < 0.001) (n = 3 or 4). See also Table S2.
Figure 4.

Detection of aggregated phospho-tau in the sarkosyl-insoluble fractions of Tau-P301L FBOs
(A) Total tau, phospho-tau, and tau isoform levels in the soluble fractions of 8- and 13-week 201B7 (control) FBOs overexpressing GFP or Tau-P301L. The white arrowheads indicate 4R-tau. The black arrowhead indicates 3R-tau. The data are expressed as the mean values ± SEM. For statistical analysis, two-way ANOVA was used (∗p < 0.05, ∗∗p < 0.004, ∗∗∗p < 0.0008) (n = 3). See also Table S2.
(B) Total tau and phospho-tau levels in the sarkosyl-insoluble fractions of 8- and 13-week 201B7 (control) FBOs overexpressing GFP or Tau-P301L. The arrowheads indicate 4R-tau. The data are expressed as the mean values ± SEM. For statistical analysis, two-way ANOVA was used (∗p = 0.0436) (n = 3). See also Table S2.
(C) Tau inclusions in 13-week 201B7 (control) FBOs overexpressing GFP or Tau-P301L, and the brains of a control and a patient with AD were stained with thioflavin S and AT8. Images are representative of 201B7 (control) FBOs overexpressing GFP or Tau-P301L that were imaged with these markers (n = 3). Scale bar, 10 μm.
Finally, we investigated whether aggregated tau formed filamentous structures by immunoelectron microscopy (iEM). In 5- and 8-week Tau-P301L FBOs, filamentous structures labeled with a MC1 antibody, which recognizes conformational changes in tau protein, were detected only in neurites (Figure 5A); however, in 13-week Tau-P301L FBOs and fAD FBOs (with PS2 mutation) overexpressing Tau-P301L, MC1-labeled filamentous structures were observed in neurites (Figure 5A) and were randomly distributed in the cytoplasm (Figure 5B), similar to the tau filaments found in patients with FTDP-17 (Spillantini et al., 1998). These results indicate that 13-week Tau-P301L FBOs exhibited the later hallmarks of tau pathology, such as tau fibrils (Figure 5C). Although astrocytes generated as FBOs were developing in culture, tau aggregations occurred in neuronal cells in 13-week Tau-P301L FBOs (Figure 5D).
Figure 5.

iEM revealed the presence of tau fibrils labeled with a MC1 antibody in neurites and cell bodies
(A) Filamentous assemblies labeled with a MC1 antibody were detected in neurites in 5-, 8-, and 13-week Tau-P301L FBOs (201B7 [control]). The bottom panels show enlarged images of the corresponding top panels. The pictures on the right show 13-week Tau-P301L fAD FBO (PS2-2). The black dots indicate a MC1 antibody bound with nanogold-conjugated anti-mouse secondary antibodies. Images are representative of 201B7 (control) and PS2-2 (fAD) FBOs overexpressing Tau-P301L. n = 2, 5- and 8-week 201B7; n = 3, 13-week 201B7; n = 1, 13-week PS2-2. The nanogold signals were enhanced with silver enhancement solution.
(B) MC1-labeled tau fibrils were randomly distributed in cell bodies only in 13-week 201B7 (control) and PS2-2 (fAD) FBOs overexpressing Tau-P301L. Images are representative of 201B7 and PS2-2 FBOs overexpressing Tau-P301L. n = 3, 13-week 201B7, n = 1, 13-week PS2-2. N, nucleus; M, mitochondria.
(C) Schematic representation of phenotypes in fAD FBOs and Tau-P301L FBOs. Amyloid-β pathology and phospho-tau were observed in fAD FBOs, and tau pathology such as tau fibrils recognized by iEM was found in Tau-P301L FBOs.
(D) Immunostaining for markers of aggregated tau (MC1: magenta), neurons (ELAVL3/4: cyan), and astrocytes (GFAP: green) in 13-week Tau-P301L 201B7 (control) FBOs to identify cells with tau accumulation. The top panels show the outer part of the FBOs, and the bottom panels show the inner part of the FBOs. The arrowheads indicate MC1 and ELAVL3/4 double-positive cells. Images are representative of 201B7 (control) FBOs overexpressing Tau-P301L (n = 3). The nuclei were stained with Hoechst 33258 (blue). Scale bar, 50 μm.
Discussion
We were able to efficiently produce FBOs from feeder-free iPSCs by controlling the concentration of FGF2 in the culture medium of iPSCs. Notably, FGF2 concentration did not affect the undifferentiated state of iPSCs but affected gene expressions that control forebrain identity once neural differentiation was initiated. There is a major obstacle hindering the precise recapitulation of disease phenotypes by brain organoid technology: a low reproducibility. Compared with on-feeder iPSCs, feeder-free iPSCs are not only easier to handle but also more reproducible, as they are not affected by lot differences in feeder cells. Therefore, it has been eagerly awaited to develop reproducible methods for brain organoids from feeder-free iPSCs. Thus, this breakthrough may further develop the research of brain organoids using iPSCs.
To generate tauopathy models in FBOs, we used AAVs for overexpressing Tau-P301L in FBOs. Several groups have succeeded in delivering fluorescence reporters in brain organoids by simply adding AAVs to the medium (Fischer et al., 2019). Modest tau aggregation was observed after AAV-Tau-P301L was just added to the medium of FBOs; however, tau aggregation was markedly enhanced in whole FBOs after injecting AAV-Tau-P301L, indicating that high expression of tau protein is necessary to induce tau pathology in an experimental time frame. Injection of virus particles into the FBOs, rather than simply adding AAV to the culture medium, may have resulted in high expression of tau protein across the FBOs, leading to recapitulate pathology of tauopathy. This is also suggested by the fact that tau pathology was not developed in Tau-P301L knockin mouse (Gilley et al., 2012), while transgenic mouse models that overexpress Tau-P301L displayed features of human tauopathies (Lewis et al., 2000; Ramsden et al., 2005). This AAV injection technology in FBOs allows us to acquire high levels of disease-associated proteins in brain organoids, which could be applied to other neurodegenerative diseases.
Although early tau pathology has been observed when neurons are differentiated from iPSCs with mutations observed in patients with FTD and FTLD in 2D cultures (Imamura et al., 2016; Silva et al., 2016), it is still challenging to reproduce pronounced tau aggregation observed in patients with tauopathy. In fact, overexpression of mutant tau was not enough to provoke tau aggregation in 2D culture. When exogenous tau fibrils are exposed to cells stably expressing tau, tau inclusions form (Matsumoto et al., 2018; Sanders et al., 2014). Here, we demonstrated that overexpression of mutated tau alone was sufficient to cause drastic tau aggregations in FBOs. The 13-week Tau-P301L FBOs, where the most pronounced tau pathology was observed, seemed to be more mature than 5- and 8-week Tau-P301L FBOs, suggesting that tau aggregations are not caused in immature FBOs. The difficulty in causing tau aggregates in 2D culture may be due to the immaturity of 2D neurons compared with 3D neurons (Centeno et al., 2018; Trujillo et al., 2019). Amyloid-β plaques and NFT-like structures were also observed in a 3D neural culture system in which immortalized human neural progenitor cells overexpressing APP and PS1 mutations were embedded in a Matrigel scaffold (Choi et al., 2014, 2016). Therefore, 3D models are more appropriate than 2D models for recapitulating the protein accumulation observed in patients with AD and tauopathy.
As FBOs increase in size, hypoxic cell death occurs in the interior since vascularization is required for oxygen penetration and nutrient supply (Qian et al., 2020). Additionally, it has been reported that stress-related genes such as the glycolysis-related gene PGK1 and ER stress-related genes ARCN1 and GORASP2 are activated in cortical organoids (Bhaduri et al., 2020). The stressful environment of over 13-week FBOs may be one of the factors that caused the late hallmarks of AD and tauopathy such as amyloid-β and tau aggregation, although transcriptome analysis showed that brain organoids related to embryonic human neocortex (Qian et al., 2016). In fact, tau accumulation was observed in neurons in the interior of fAD FBOs on day 84 (Figures S7A and S7B), although aggregated tau was not detected in the sarkosyl-insoluble fractions, suggesting that a hypoxic environment enhances tau accumulation. Neurons showing tau accumulation were positive for BNIP3, a hypoxia marker related to mitophagy (Figure S7C).
We successfully induced significant tau pathology, such as sarkosyl-insoluble phospho-tau and tau fibrils recognized by a MC1 antibody using iEM, in Tau-P301L FBOs by AAV microinjection. The obtained model may be useful for exploring tau pathology and evaluating candidate molecules and target genes for tauopathy treatment. This FBO model of tauopathy will pave the way for new approaches for a better understanding tau pathology and the development of therapeutic agents for patients with tauopathy worldwide.
Limitations of the study
Differences between lines and batches of iPSCs have always been a problem in the generation of brain organoids. Although our low-FGF2 method has improved this problem, further improvements are needed to reduce line-to-line and batch-to-batch differences, especially for disease-specific iPSCs. In addition, the poor environment inside the organoids may have been a factor in causing the tauopathy in the Tau-P301L FBOs, but on the other hand, the cells in the center of the organoids are easily dying, making it difficult to analyze. The numbers of organoids evaluated in the present study were sufficient to demonstrate the statistical significance of key tau phenotypes; however, future investigations would be required to examine the large-scale heterogeneity of this model and its application to high-throughput screening assays for tauopathy therapeutics. Also, it is desirable to develop brain organoids that more closely resemble the human brain and to generate disease models that approach the real pathogenesis, and this study may contribute to these goals.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FOXG1 | Abcam | Cat#ab18259; RRID: AB_732415 |
| βIII tubulin | Sigma-Aldrich | Cat#T8660; RRID: AB_477590 |
| MAP2 | Sigma-Aldrich | Cat#M4403; RRID: AB_477193 |
| ELAVL3/4 (HuC/D) | Molecular Probes | Cat#A21271; RRID: AB_221448 |
| SATB2 | Abcam | Cat#ab92446; RRID: AB_10563678 |
| CTIP2 | Abcam | Cat#ab18465; RRID: AB_2064130 |
| TBR1 | Abcam | Cat#ab31940; RRID: AB_2200219 |
| GFAP | Thermo Fisher Scientific | Cat#13-0300; RRID: AB_2532994 |
| Nestin | Sigma-Aldrich | Cat#MAB5326; RRID: AB_2251134 |
| Pax6 | MBL | Cat#PD022; RRID: AB_1520876 |
| Sox2 | Santa Cruz Biotechnology | Cat#sc-365823; RRID: AB_10842165 |
| MBP | Aveslabs | Cat#MBP-0020 |
| 6E10 | BioLegend | Cat#803001; RRID: AB_2564653 |
| 4G8 | BioLegend | Cat#800701; RRID: AB_2564633 |
| Tau | Dako | Cat#A0024; RRID: AB_10013724 |
| HT7 | Thermo Fisher Scientific | Cat#MN1000; RRID: AB_2314654 |
| RTM38 | FUJI Wako Chemical | Cat#017-26893 |
| MC1 | Gift from Dr. Peter Davies | N/A |
| AT8 | Thermo Fisher Scientific | Cat#MN1020; RRID: AB_223647 |
| T22 | Merck Millipore | Cat#ABN454; RRID: AB_2888681 |
| Synaptophysin | Sigma-Aldrich | Cat#S5768; RRID: AB_477523 |
| Phospho-Tau (Ser 396) | Thermo Fisher Scientific | Cat#44-752G; RRID: AB_2533745 |
| RD3 | Sigma-Aldrich | Cat#05-803; RRID: AB_310013 |
| RD4 | Sigma-Aldrich | Cat#05-804; RRID: AB_310014 |
| Alexa Fluor 488 goat anti-rabbit IgG | Thermo Fisher Scientific | Cat#A11034; RRID: AB_2576217 |
| Alexa Fluor 555 goat anti-rabbit IgG | Thermo Fisher Scientific | Cat#A21429; RRID: AB_2535850 |
| Alexa Fluor 488 goat anti-chicken IgG | Thermo Fisher Scientific | Cat#A11039; RRID: AB_2534096 |
| Alexa Fluor 488 goat anti-rat IgG | Thermo Fisher Scientific | Cat#A11006; RRID: AB_141373 |
| Alexa Fluor 555 goat anti-rat IgG | Thermo Fisher Scientific | Cat#A21434; RRID: AB_141733 |
| Alexa Fluor 647 goat anti-rat IgG | Thermo Fisher Scientific | Cat#A21247; RRID: AB_141778 |
| Alexa Fluor 488 goat anti-mouse IgG | Thermo Fisher Scientific | Cat#A11029; RRID: AB_2534088 |
| Alexa Fluor 555 goat anti-mouse IgG | Thermo Fisher Scientific | Cat#A21424; RRID: AB_141780 |
| Alexa Fluor 555 goat anti-mouse IgG1 | Thermo Fisher Scientific | Cat#A21127; RRID: AB_141596 |
| Alexa Fluor 647 goat anti-mouse IgG1 | Thermo Fisher Scientific | Cat#A21240; RRID: AB_141658 |
| Alexa Fluor 488 goat anti-mouse IgG2b | Thermo Fisher Scientific | Cat#A21141; RRID: AB_141626 |
| Alexa Fluor 555 goat anti-mouse IgG2b | Thermo Fisher Scientific | Cat#A21147; RRID: AB_1500897 |
| Alexa Fluor 647 goat anti-mouse IgG2b | Thermo Fisher Scientific | Cat#A21242; RRID: AB_1500900 |
| Alexa Fluor 488-FluoroNanogold-conjugated goat anti-mouse secondary antibody | Thermo Fisher Scientific | Cat# A24920; RRID: AB_1500792 |
| Amersham ECL Rabbit IgG, HRP-linked whole Ab (from donkey) | Cytiva | Cat#NA934; RRID: AB_772206 |
| Amersham ECL Mouse IgG, HRP-linked whole Ab (from donkey) | Cytiva | Cat#NA931; RRID: AB_772210 |
| Bacterial and virus strains | ||
| AAV-PHP.B-CAG-Tau(1N4R)-P301L | This paper | N/A |
| AAV-PHP.B-CAG-eGFP | This paper | N/A |
| Biological samples | ||
| Frozen Tissue Section - Alzheimer's Disease: Brain: Temporal Lobe | BioChain | T1236078ALZ |
| Frozen Tissue Section - Human Adult Normal: Brain: Temporal Lobe | BioChain | T1234078 |
| Chemicals, peptides, media and recombinant proteins | ||
| Recombinant Human FGF-basic (154a.a.) | Peprotech | Cat#100-18B |
| Y27632 | FUJIFILM Wako Chemicals | Cat#034-24024 |
| iMatrix | Nippi | Cat#892012 |
| StemFit Basic02 | Ajinomoto | Cat#SFB-503 |
| TrypLE Select | Thermo Fisher Scientific | Cat#12563011 |
| 0.5 mmol/L-EDTA /PBS Solution | Nacalai tesque | Cat#13567-84 |
| Phosphate Buffered Saline (10X) | Nacalai tesque | Cat#27575-31 |
| Dorsomorphin | Sigma-Aldrich | P5499 |
| A83-01 | Tocris | Cat#2939 |
| DMEM/F-12, GlutaMAX | Thermo Fisher | Cat#10565018 |
| SB431542 | Cellagen Technology | Cat#C7243 |
| CHIR99021 | Cellagen Technology | Cat#C2447 |
| N-2 supplement | Thermo Fisher Scientific | Cat#17502001 |
| MEM Nonessential Amino Acids Solution (100X) | Nacalai tesque | Cat#06344-56 |
| Matrigel Matrix Basement Membrane Growth Factor Reduced | Corning | Cat#354230 |
| B27 supplement | Thermo Fisher Scientific | Cat#17504044 |
| Insulin, Human, recombinant | FUJIFILM Wako Chemicals | Cat#093-06476 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat#M3148 |
| Penicillin-Streptomycin Mixed Solution | Nacalai tesque | Cat#09367-34 |
| B-27 Plus Neuronal Culture System | Thermo Fisher Scientific | Cat#A3653401 |
| BDNF | Peprotech | Cat#450-02 |
| GDNF | Peprotech | Cat#450-10 |
| cAMP | Sigma-Aldrich | Cat#A6885 |
| Leukemia inhibitory factor human | Sigma-Aldrich | Cat#LIF1010 |
| Antibiotic-Antimycotic (100X) | Thermo Fisher Scientific | Cat#15240062 |
| Trypan Blue Stain (0.4%) | Thermo Fisher Scientific | Cat#15250-061 |
| Critical commercial assays | ||
| 16% Formaldehyde Solution | Thermo Fisher Scientific | Cat#28906 |
| Phosphate Buffered Saline (10x) (pH7.4) | Nacalai tesque | Cat#27575-31 |
| Tissue-Tek O.C.T. Compound | Sakura Finetek | N/A |
| Triton-X | Sigma-Aldrich | Cat#X100 |
| Dimethyl Sulfoxide | Nacalai tesque | Cat#13445-74 |
| Ethanol | Nacalai tesque | Cat#14713-53 |
| Methanol | FUJIFILM Wako Chemicals | Cat#131-01826 |
| 1M Tris-HCl Buffer, pH7.5 | Thermo Fisher Scientific | Cat#15567027 |
| NP40 Alternative | Millipore | Cat#492018 |
| NaCl (5M) | Thermo Fisher Scientific | Cat#AM9759 |
| Sodium Deoxycholate 10% | bioWORLD | Cat#40430018 |
| 0.5M EDTA (pH8.0) | Nippon Gene | Cat#311-90075 |
| 0.1M EGTA | Nacalai tesque | Cat#37346-05 |
| Sucrose | FUJIFILM Wako Chemicals | Cat#193-09545 |
| N-Lauroylsarcosine sodium salt solution | Sigma-Aldrich | Cat#61747 |
| PMSF | Sigma-Aldrich | Cat# P7626 |
| PhosSTOP | Roche | Cat# 4906837001 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693132001 |
| Ultrapure Water | FUJIFILM Wako Chemicals | Cat# 217-01031 |
| NuPAGE LDS Sample Buffer (4x) | Thermo Fisher Scientific | Cat#NP0008 |
| NuPAGE Sample Reducing Agent (10x) | Thermo Fisher Scientific | Cat# NP0009 |
| PBS pH7.4 | Thermo Fisher Scientific | Cat# 10010-023 |
| Skim Milk | Megmilk Snow Brand | N/A |
| Tween 20 | Nacalai tesque | Cat#35624-15 |
| Tris Buffered Saline (TBS) Tablets, pH7.6 | Takara | Cat#T9141 |
| Albumin, Bovine Serum | Nacalai tesque | Cat#01860-07 |
| Normal Donor Goat Serum | MBL | Cat#EQ-102 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23227 |
| QIAzol Lysis Reagent | Qiagen | Cat#79306 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| TruSeq Stranded mRNA LT Sample Prep Kit | Illumina | Part# 15031047 |
| Block Ace | KAC | Cat#UK-B80 |
| BTA-1 | Sigma-Aldrich | Cat#B9934 |
| Thioflavin S | Sigma-Aldrich | Cat#T1892 |
| Human Amyloid (1–40) ELISA Kit II | FUJIFILM Wako Chemicals | Cat#298–64601 |
| Human Amyloid (1–42) ELISA Kit High Sensitivity | FUJIFILM Wako Chemicals | Cat#296–64401 |
| Hoechst 33258 solution | Sigma-Aldrich | Cat#94403 |
| bisBenzimide H33258 | Sigma-Aldrich | Cat#B2883 |
| Midi Format, 0.2 μm PVDF | Bio-Rad | Cat#1704157 |
| Immobilon Western Chemiluminescent HRP Substrate | Merck Millipore | Cat#WBKLS00500 |
| XV pantera MP Gel | DRC | Cat#NXV-336P |
| Intercept Blocking Buffer | LI-COR | Part#927-60001 |
| 0.1M Phosphate Buffer pH7.4 | Muto Pure Chemicals | Cat#8038-2 |
| HEPES, minimum 99.5%(titration) | Sigma-Aldrich | Cat#3375 |
| Osmium tetroxide, high purify crystal | TAAB Laboratories | Cat#O001 |
| Acetone | Sigma-Aldrich | Cat#179124 |
| n-butyl glycidyl ether (QY-1) | Nisshin EM. | Cat#310-1 |
| Epoxy resin (Oken EPOK812 set) | Okenshoji | Cat#02-1001 |
| Deposited data | ||
| The BioProject | This paper | PRJNA792673 |
| Experimental models: Cell lines | ||
| Human: Passage 27-45 201B7 iPSCs | Takahashi et al. (2007) | RRID:CVCL_A324 |
| Human: Passage 33-45 414C2 iPSCs | Okita et al. (2011) | RRID:CVCL_DP60 |
| Human: Passage 26-37 RPC771 iPSCs | ReproCELL | Cat#RCRP001N |
| Human: Passage 37-45 PS1-2 iPSCs | Yagi et al. (2011) | RRID:CVCL_T873 |
| Human: Passage 17-45 PS2-2 iPSCs | Yagi et al. (2011) | RRID:CVCL_T876 |
| Human: Passage 32-45 KhES1 ES cells | Suemori et al. (2006) | RRID:CVCL_B231 |
| Software and algorithms | ||
| DESeq2 package version 1.28.1 | Love et al., 2014 | https://bioconductor.riken.jp/packages/3.11/bioc/html/DESeq2.html |
| ClusterProfiler package version 3.16.1 | Wu et al., 2021 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| FeatureCounts version 1.5.2 of the Subread package | Liao et al., 2014 | http://subread.sourceforge.net |
| Hisat2 version 2.2.1 | Kim et al., 2019 | http://daehwankimlab.github.io/hisat2/download/ |
| ImageMaster software | Vilber | https://www.vilber.com/fusion-fx/ |
| Odyssey CLx imaging system | LI-COR | https://www.licor.com/bio/odyssey-dlx/ |
| Other | ||
| Tungsten Needles | Fine Science Tools | Cat#10130-10 |
| Moria Nickel Plated Pin Holder-12cm | Fine Science Tools | Cat#26016-12 |
| 6ch Magnetic Stirrer for 100 mL, 30mL | ABLE Biott | Cat#BWS-S03N0S-6 |
| Single Use Bioreactor 30mL | ABLE Biott | Cat#BWV-S03A |
| Patch-Clamp System | Muromachi Kikai | Model#MD-1 |
| Electric Microinjector | BEX | Cat#BJ-120 |
| Calibrated Pipets | Drummond Scientific Company | Cat#2-000-001-90 |
| Branson Digital Sonifier | Emerson Electric | SFX250 |
| Ultrasonic homogenizer | TITEC | VP-050N |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hideyuki Okano (hidokano@a2.keio.jp).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human PSCs
We previously generated iPSCs from two patients with fAD carrying missense mutations, i.e., A246E in PS1 (PS1–2) and N141I in PS2 (PS2–2) (Yagi et al., 2011). The human ESC line KhES1 (Suemori et al., 2006) and the human iPSC lines 414C2 (Okita et al., 2011), 201B7 (Takahashi et al., 2007), and RPC771 (ReproCELL) were used as healthy controls.
Methods details
Human PSCs culture
Feeder-free iPSCs were passaged every 7 days and seeded at a density of 1.0–1.5 × 105 cells/well in StemFit Basic 02 medium (Ajinomoto) with 100 ng/mL FGF-2 (PeproTech) and 10 μM Y27632 (FUJIFILM Wako Chemicals) in a six-well tissue culture plate coated with iMatrix-511 (Nippi). The culture medium was replaced with fresh medium without Y27632 on days 1, 3, 5 and 6 after passage. For differentiation, iPSCs between passages 20 and 50 were used. Before differentiation, the iPSCs were maintained in StemFit Basic 02 medium containing 30 ng/mL FGF-2 for one week, and then, the concentration of FGF-2 was reduced to 10 ng/mL for more than one week (low-FGF2 iPSCs). The culture medium of low-FGF2 iPSCs was refreshed every day.
Generation of FBOs from iPSCs
FBOs were generated according to a protocol published by Qian et al., 2016, 2018 with some modifications. On day 0, iPSCs were dissociated into single cells with 0.5x TrypLE Select (Thermo Fisher Scientific) diluted in phosphate-buffered saline (PBS) containing 1 mM EDTA, and 9,000 cells were plated in StemFit Basic 02 medium containing 10 μM Y27632 in each well of a PrimeSurface 96V plate (Sumitomo Bakelite Co, Ltd, cat. no. MS-9096V). Dual SMAD inhibitors (2 μM dorsomorphin (Sigma-Aldrich) and 2 μM A-83-01 (Tocris)) were added on either day 0 or day 1 depending on the cell line. Half of the medium was replaced with fresh StemFit Basic 02 containing the dual SMAD inhibitors on days 1 and 3. On day 5, half the medium was exchanged with induction medium consisting of DMEM/F-12, Glutamax (Thermo Fisher Scientific), 1 μM SB431542 (Cellagentech), 1 μM CHIR99021 (Cellagentech), 1x N-2 supplement (Thermo Fisher Scientific), 1x MEM-NEAA (Nacalai tesque), and 1x penicillin-streptomycin (Nacalai tesque). On day 7, the obtained EBs were embedded in Matrigel (Corning) in induction medium to form a neuroepithelium, and the medium was changed every two days. On day 14, the EBs were detached from the Matrigel by pipetting several times, and the remaining Matrigel was removed from each EB with needles under a microscope. EBs with neuroepithelium were selected, transferred to a 30 mL bioreactor (ABLE Biott) in differentiation medium consisting of DMEM/F-12, GlutaMAX, 1x N-2 supplement, 1x B27 supplement (Thermo Fisher Scientific), 1x MEM-NEAA, 2.5 μg/mL insulin (FUJIFILM Wako Chemicals), 0.1 mM 2-mercaptoethanol (Sigma-Aldrich) and 1x penicillin-streptomycin (Nacalai tesque) and cultured on a magnetic stir system (ABLE Biott) at a speed of 50 rpm at 37°C and 5% CO2. Two to three times per week, 2/3 of the medium was replaced with fresh medium. FBOs were cultured in an incubator at 37°C and 40% O2 and 5% CO2 from day 35. On day 71, the medium was replaced with maturation medium comprising, the B-27 Plus Neuronal Culture System (Thermo Fisher Scientific), 1x MEM-NEAA, 0.2 mM ascorbic acid, 20 ng/mL BDNF (PeproTech), 20 ng/mL GDNF (PeproTech), 0.5 mM cAMP (Sigma-Aldrich) and 1x antibiotic-antimycotic (Thermo Fisher Scientific). Two to three times per week, 2/3 of the medium was replaced with fresh medium. For STAT3 activation, 10 ng/mL leukaemia inhibitory factor (Sigma-Aldrich) was added from day 14. For example, the 201B7 control iPSC line could generate about 50 brain organoids from 1 × 106 cells using the low-FGF protocol.
Transcriptome analysis
Total RNA was extracted from iPSCs or EBs and purified using the QIAzol Lysis Reagent or RNeasy Mini Kit (Qiagen). Subsequently, the total RNA was used to prepare an RNA-seq library using the TruSeq Stranded mRNA LT Sample Prep Kit according to the TruSeq Stranded mRNA Sample Preparation Guide, part #15031047 Rev. E (Illumina). The libraries were sequenced for 100 cycles (paired ends) using a NovaSeq 6000 instrument (Illumina). The sequence reads from each RNA-seq library were mapped onto the human genome (build hg19) using Hisat2 version 2.2.1 (Kim et al., 2019), and the number of sequence reads for each gene was calculated using featureCounts version 1.5.2 of the Subread package (Liao et al., 2014). DEG, heatmap, and PCA was analyzed using DESeq2 package version 1.28.1 (Love et al., 2014). Enrichment of GO biological process and correlation network of enriched GO terms was used clusterProfiler package version 3.16.1 (Wu et al., 2021) with 0.01 and 0.05 as p value and q-value (BH methods) cut-off values.
AAV injection
FBOs of similar size and morphology were selected. Then, 2 μL of AAV vector (AAV-PHP.B-CAG-Tau(1N4R)-P301L, 1.3 × 1013 vg/mL; AAV-PHP.B-CAG-eGFP, 2.0 × 1013 vg/mL) was injected into the center of each FBO at 5, 8 and 13 w. A FBO was placed in a dish containing PBS, and the plate was put in a patch-clamp system (Muromachi Kikai). A glass pipette connected to a gas injector (BEX) was lowered until the desired depth (450–700 μm) was reached, and the AAV was injected at a speed of 100 nL/min. The needle was retracted at a rate of 100 μm/min 5 min after completion of AAV injection (Video S1).
Tissue preparation and immunohistochemistry
FBOs were fixed in 4% paraformaldehyde (Thermo Fisher Scientific) in PBS (Nacalai tesque) for 30–60 min at room temperature or overnight at 4°C, washed three times with PBS, and then cryoprotected in 30% sucrose solution (Fujifilm Wako Chemicals) overnight. The FBOs were embedded in OCT compound (Sakura Finetek), and 10–12 μm thick sections were obtained for immunohistochemistry. The sections were hydrated in TBS (Takara) for 10 min and incubated in blocking solution containing 10% (v/v) normal goat serum (MBL) and 1% (w/v) bovine serum albumin (Nacalai tesque) in TBS for 1 h. The sections were incubated with primary antibodies diluted in TBS containing 1% bovine serum albumin overnight at 4°C. The primary antibodies used in this study were diluted as follows: FOXG1 (1:100, Abcam), βIII tubulin (1:250, Sigma-Aldrich), MAP2 (1:200, Sigma-Aldrich), ELAVL3/4 (1:200, Molecular Probes), SATB2 (1:100, Abcam), CTIP2 (1:100, Abcam), TBR1 (1:300, Abcam), GFAP (1:500, Thermo Fisher Scientific), Nestin (1:250, Sigma-Aldrich), Pax6 (1:200, MBL), Sox2 (1:200, Santa Cruz), MBP (1:200, Aveslabs) 6E10 (1:100, BioLegend), 4G8 (1:100, BioLegend), Tau (1:1000, Dako), HT7 (1:500, Thermo Fisher Scientific), RTM38 (1:500, FUJIFILM Wako Chemical), MC1 (1:500, gift from Dr. Peter Davies), AT8 (1:300, Thermo Fisher Scientific), T22 (1:200, Merk Millipore), Synaptophysin (1:100, Sigma-Aldrich), The sections were washed three times for 10 min each with 0.05% Triton-X (Sigma-Aldrich) in TBS, incubated with Alexa Fluor® 488-, 555-, 647-conjugated secondary antibody solutions (1:1000, Thermo Fisher Scientific) for 1.5 h at room temperature and washed three times with 0.05% Triton-X in TBS. Hoechst 33258 (Sigma-Aldrich) was used to stain the nuclei. Immunofluorescence images were obtained with an LSM 700 or 780 laser scanning microscope (Zeiss).
BTA-1 and thioflavin S staining
The sections were hydrated in TBS for 10 min and permeabilized with 80% MeOH for 20 min at −20°C. The sections were stained with 500 μM BTA-1 (Sigma-Aldrich) or 0.1% thioflavin S (Sigma-Aldrich) diluted in DMSO for 30 min at room temperature in the dark and then washed three times with 50% ethanol for 5 min each. The BTA-1 and thioflavin S signals were captured using an LSM 700 or 780 laser scanning microscope (ZEISS).
Preparation of soluble and sarkosyl-insoluble fractions for western blot analysis
FBOs were homogenized in RIPA buffer containing 50 mM Tris-HCl (pH 7.5, Thermo Fisher Scientific), 5 mM EDTA (Nippon Gene), 1 mM EGTA (Nacalai Tesque), 1% NP40 (Millipore), 0.25% sodium deoxycholate (bioWORLD), protease inhibitor mixture (cOmplete EDTA-free, Roche), phosphatase inhibitor cocktail (PhosSTOP, Roche), 0.5 mM phenylmethylsulfonyl fluoride (PMSF, Sigma-Aldrich) and 0.1 M NaCl (Thermo Fisher Scientific) by using a homogenizer (Emerson Electric). The homogenates were centrifuged for 20 min at 163,000 × g at 4°C, and the supernatants were kept as the soluble fractions. The RIPA-insoluble pellets were then resuspended in buffer A68 containing 10 mM Tris-HCl (pH 7.5), 0.5 M NaCl, 1 mM EGTA, 10% sucrose and 1% sarkosyl (Sigma-Aldrich) and homogenized by using a homogenizer (TITEC). The samples were incubated at 37°C for 1 h and then centrifuged for 20 min at 163,000 × g. The sarkosyl-insoluble pellets were washed with PBS and centrifuged for 20 min at 163,000 × g. The pellets were homogenized in PBS to yield the sarkosyl-insoluble fractions.
Western blot analysis
Protein samples (10 μg) were resolved by 12.5% SDS-PAGE (DRC), and the proteins were transferred to PVDF membranes (Bio-Rad). The membranes were blocked with 2.5% skim milk (Megmilk Snow Brand) in 0.005% Tween 20 (Nacalai tesque) in TBS and incubated with the following primary antibodies diluted as indicated: tau (1:2000; DAKO), AT8 (1:500; Thermo Fisher Scientific), HT7 (1:2,000, Thermo Fisher Scientific), phospho-tau (Ser 396) (1:1,000, Thermo Fisher Scientific), RD3 (1:1,000, Sigma-Aldrich), and RD4 (1:1,000, Sigma-Aldrich). The membranes were washed and incubated with IRDye 680T- and IRDye 800CW-conjugated secondary antibodies (LI-COR). Proteins were detected and quantified by an Odyssey CLx imaging system (LI-COR). Or the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:2,000; Cytiva). The western blot images were visualized using Immobilon Western Chemiluminescent HRP Substrate (Merck Millipore), and images were captured with a Fusion FX system (Vilber) and quantified with ImageMaster software (Vilber).
ELISA
FBOs were transferred from the bioreactor to an ultralow attachment 48-well plate (one FBO/well) and cultured in fresh maturation media for 48 h at 37°C and 40% O2. The culture medium was harvested after 48 h and centrifuged at 15,000 rpm for 5 min to remove the debris. The supernatants were collected and stored at −80°C. The concentrations of Aβ 40 and Aβ42 in the supernatants were measured using a commercially available Human Amyloid (1–40) ELISA Kit II (Fujifilm Wako Chemicals) and a Human Amyloid (1–42) ELISA Kit High Sensitivity (Fujifilm Wako Chemicals) according to the manufacturer’s instructions. The absorbance of the samples was measured at 450 nm with an iMark microplate reader (Bio-Rad).
Electron microscopy (EM)
The detailed pre-embedding iEM protocol was described previously (Shibata et al., 2019). Briefly, frozen sections of FBOs for iEM were placed on glass slides, dried with a cool air dryer, washed in 0.1 M phosphate buffer (PB, pH 7.4, Muto Pure Chemicals) 3 times, and incubated with blocking solution containing 5.0% Block Ace (KAC) and 0.01% saponin in 0.1 M PB for one hour at 25°C. The sections were incubated with a mouse anti-AT8 antibody (1:150) and a mouse anti-MC1 antibody (1:250) in blocking solution for 72 h at 4°C and then incubated with Hoechst 33258 (1:1000, Sigma-Aldrich) and Alexa Fluor 488/FluoroNanogold-conjugated goat anti-mouse secondary antibody (1:100, Thermo Fisher Scientific) for 24 h at 4°C. The specific area of interest was identified as the area exhibiting Alexa Fluor 488 signals under a fluorescence microscope (LSM880, Carl Zeiss). After the sections were fixed in 2.5% glutaraldehyde in 0.1 M PB, washed in 0.1 M PB and 50 mM HEPES (pH 5.8, Sigma-Aldrich) 3 times for 10 min each, and washed with diluted water for 1 min, the nanogold signals were enhanced with silver enhancement solution for 5–7 min at 25°C. The gold-labelled sections were postfixed with 1.0% osmium tetroxide (TAAB Laboratories) for 1.5 h at 4°C; en bloc stained with uranyl acetate for 20 min at 25°C; dehydrated in a series of ethanol solutions (50, 70, 80, 90, 100% EtOH (twice in each solution)) for 5 min each, 100% acetone (Sigma-Aldrich) for 5 min, and n-butyl glycidyl ether (QY1, Nisshin EM.) twice for 5 min each; incubated with epoxy resin (Okenshoji) with QY1 (1:1) for 1 h and 100% epoxy resin for 48 h at 4°C; and embedded in 100% epoxy resin. After polymerization for 72 h at 60°C, the resin blocks were trimmed and sectioned at a thickness of 80 nm with an ultramicrotome (UC7, Leica) with a diamond knife (Ulutra, DiATOME). The ultrathin sections were collected on copper grids and stained with uranyl acetate and lead citrate. The ultrathin sections were imaged with a transmission electron microscope (JEM-1400 plus, JEOL) at 100 keV.
Quantification and statistical analysis
Statistical analyses were performed using t tests (Figures 2B and 2D), and one-way or two-way ANOVA followed by Tukey’s post hoc test (Figures 3E, 4A, 4B, S4B, and S5B) with GraphPad Prism 8 software. F and p values of two-way ANOVA analyses were presented in Table S2. The data are expressed as the mean values ± s.e.m.
Acknowledgments
We would like to thank Peter Davies at Albert Einstein College of Medicine for providing a MC1 antibody. We also thank Drs. Kazuhiro Sakurada, Jay W. Shin, Satoru Morimoto, Masaki Takao, Junko Hayashi, Mitsuru Ishikawa, Tomoko Andoh-Noda, Hirotaka Watanabe, Mie Furuhashi, Sumihiro Maeda, Mika Terumitsu, and Takuya Shimazaki (Keio University) for their helpful comments and support. We appreciate Profs. Hirofumi Suemori, Shinya Yamanaka, and Keisuke Okita (Kyoto University) for kindly providing hPSC lines KhES1, 201B7, and 414C2 respectively. This work was supported by the program for Regenerative Medicine (the Program for Intractable Disease Research Utilizing Disease-specific iPSCs and the Acceleration Program for Intractable Diseases Research Utilizing Disease-specific iPSCs), Practical Research Project for Rare/Intractable Diseases (H.O.) (JP16ek0109013, JP16ek0109158, JP16bm0609003, JP17bm0804003, JP18bm0804003, JP19bm0804003, JP20bm0804003, and JP21bm0804003), no. 17pc0101006 from the Japan Agency for Medical Research and Development, and research foundation from Eisai Co., Ltd.
Author contributions
Overall conceptualization, H.S. and H.O.; primary experimentation, H.S.; AAV injections, H.S., Y.S., A.S., K.K., T.K., and H.A.; immunohistochemistry, H.S. and S.M.; biochemistry, H.S., A.S., and H.A.; electron microscopy, T.S. and S.S.; gene-expression analysis, T.S. and K.I.; methodology, discussion, and investigation, H.S., A.S., H.A., S.I., J.K., and H.O.; writing – original draft, H.S.; writing – review & editing, all authors; funding acquisition, H.S. and H.O.; supervision and project administration, H.S., D.I., and H.O.
Declaration of interests
H.O. is a scientific consultant for SanBio, Co., Ltd., and K Pharma, Inc. H.S. declares non-financial competing interests. H.A. and J.K. are full-time employees of Eisai, a pharmaceutical company listed on the Tokyo Stock Exchange (TYO:4523) for the period in which the data reported in this study were generated. Y.S. and T.K. are the founders of ALAN, Inc., and held shares in ALAN, Inc. There are two international patent applications related to this work, and these application numbers are PCT/JP2020/025605 (H.O. and H.S.) and PCT/JP2022/12502 (H.O., H.S., Y.S., and T.K.).
Published: September 8, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100289.
Contributor Information
Hiroko Shimada, Email: hiroko.z5@keio.jp.
Hideyuki Okano, Email: hidokano@keio.jp.
Supplemental information
Data and code availability
-
•
The BioProject accession number for RNA sequencing data reported in this paper is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aoyagi H., Hasegawa M., Tamaoka A. Fibrillogenic nuclei composed of P301L mutant tau induce elongation of P301L tau but not wild-type tau. J. Biol. Chem. 2007;282:20309–20318. doi: 10.1074/jbc.M611876200. [DOI] [PubMed] [Google Scholar]
- Arber C., Lovejoy C., Wray S. Stem cell models of Alzheimer's disease: progress and challenges. Alzheimer's Res. Ther. 2017;9:42. doi: 10.1186/s13195-017-0268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaduri A., Andrews M.G., Mancia Leon W., Jung D., Shin D., Allen D., Jung D., Schmunk G., Haeussler M., Salma J., et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature. 2020;578:142–148. doi: 10.1038/s41586-020-1962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centeno E.G.Z., Cimarosti H., Bithell A. 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener. 2018;13:27. doi: 10.1186/s13024-018-0258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.H., Kim Y.H., Hebisch M., Sliwinski C., Lee S., D'Avanzo C., Chen H., Hooli B., Asselin C., Muffat J., et al. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014;515:274–278. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.H., Kim Y.H., Quinti L., Tanzi R.E., Kim D.Y. 3D culture models of Alzheimer's disease: a road map to a “cure-in-a-dish”. Mol. Neurodegener. 2016;11:75. doi: 10.1186/s13024-016-0139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lullo E., Kriegstein A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017;18:573–584. doi: 10.1038/nrn.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J., Heide M., Huttner W.B. Genetic modification of brain organoids. Front. Cell. Neurosci. 2019;13:558. doi: 10.3389/fncel.2019.00558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Seereeram A., Ando K., Mosely S., Andrews S., Kerschensteiner M., Misgeld T., Brion J.P., Anderton B., Hanger D.P., Coleman M.P. Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a "P301L" tau knockin mouse. Neurobiol. Aging. 2012;33:621.e1–621.e15. doi: 10.1016/j.neurobiolaging.2011.02.014. [DOI] [PubMed] [Google Scholar]
- Gonzalez C., Armijo E., Bravo-Alegria J., Becerra-Calixto A., Mays C.E., Soto C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry. 2018;23:2363–2374. doi: 10.1038/s41380-018-0229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber B., Coulon P., Zhang M., Moritz S., Frank S., Müller-Molina A.J., Araúzo-Bravo M.J., Han D.W., Pape H.C., Schöler H.R. FGF signalling inhibits neural induction in human embryonic stem cells. Embo J. 2011;30:4874–4884. doi: 10.1038/emboj.2011.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K., Sahara N., Kanaan N.M., Tsukita K., Kondo T., Kutoku Y., Ohsawa Y., Sunada Y., Kawakami K., Hotta A., et al. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep. 2016;6:34904. doi: 10.1038/srep34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimmt J., Dannert A., Paquet D. Neurodegeneration in a dish: advancing human stem-cell-based models of Alzheimer's disease. Curr. Opin. Neurobiol. 2020;61:96–104. doi: 10.1016/j.conb.2020.01.008. [DOI] [PubMed] [Google Scholar]
- Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X., et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G., Matsumoto K., Kimura T., Suhara T., Higuchi M., Sahara N., Mori N. Tau fibril formation in cultured cells compatible with a mouse model of tauopathy. Int. J. Mol. Sci. 2018;19:E1497. doi: 10.3390/ijms19051497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano H., Yamanaka S. iPS cell technologies- significance and applications to CNS regeneration and disease. Mol. Brain. 2014;7:22. doi: 10.1186/1756-6606-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K.i., et al. A more efficient method to generate integration-free human iPs cells.pdf. Nat. Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Qian X., Jacob F., Song M.M., Nguyen H.N., Song H., Ming G.L. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 2018;13:565–580. doi: 10.1038/nprot.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X., Nguyen H.N., Song M.M., Hadiono C., Ogden S.C., Hammack C., Yao B., Hamersky G.R., Jacob F., Zhong C., et al. Brain-Region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 2016;165:1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X., Su Y., Adam C.D., Deutschmann A.U., Pather S.R., Goldberg E.M., Su K., Li S., Lu L., Jacob F., et al. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell. 2020;26:766–781.e9. doi: 10.1016/j.stem.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja W.K., Mungenast A.E., Lin Y.T., Ko T., Abdurrob F., Seo J., Tsai L.H. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate alzheimer's disease phenotypes. PLoS One. 2016;11:e0161969. doi: 10.1371/journal.pone.0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M., Kotilinek L., Forster C., Paulson J., McGowan E., SantaCruz K., Guimaraes A., Yue M., Lewis J., Carlson G., et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J. Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders D.W., Kaufman S.K., DeVos S.L., Sharma A.M., Mirbaha H., Li A., Barker S.J., Foley A.C., Thorpe J.R., Serpell L.C., et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoe J., Eggan K. Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat. Neurosci. 2013;16:780–789. doi: 10.1038/nn.3425. [DOI] [PubMed] [Google Scholar]
- Shibata S., Iseda T., Mitsuhashi T., Oka A., Shindo T., Moritoki N., Nagai T., Otsubo S., Inoue T., Sasaki E., et al. Large-area fluorescence and electron microscopic correlative imaging with multibeam scanning electron microscopy. Front. Neural Circuits. 2019;13:29. doi: 10.3389/fncir.2019.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M.C., Cheng C., Mair W., Almeida S., Fong H., Biswas M.H.U., Zhang Z., Huang Y., Temple S., Coppola G., et al. Human iPSC-derived neuronal model of tau-A152T frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell Rep. 2016;7:325–340. doi: 10.1016/j.stemcr.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeda Y., Takashima A. New insights into drug discovery targeting tau protein. Front. Mol. Neurosci. 2020;13:590896. doi: 10.3389/fnmol.2020.590896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M.G., Bird T.D., Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8:387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suemori H., Yasuchika K., Hasegawa K., Fujioka T., Tsuneyoshi N., Nakatsuji N. Efficient establishment of human embryonic stem cell lines and long-term maintenance with stable karyotype by enzymatic bulk passage. Biochem. Biophys. Res. Commun. 2006;345:926–932. doi: 10.1016/j.bbrc.2006.04.135. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Trujillo C.A., Gao R., Negraes P.D., Gu J., Buchanan J., Preissl S., Wang A., Wu W., Haddad G.G., Chaim I.A., et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell Stem Cell. 2019;25:558–569.e7. doi: 10.1016/j.stem.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T., Ito D., Okada Y., Akamatsu W., Nihei Y., Yoshizaki T., Yamanaka S., Okano H., Suzuki N. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The BioProject accession number for RNA sequencing data reported in this paper is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.