

Abstract
Diabetic bladder dysfunction (DBD) is the most common complication in diabetes. Myogenic abnormalities are common in DBD; however, the underlying mechanisms leading to these remain unclear. To understand the importance of smooth muscle insulin receptor (IR)-mediated signaling in the pathogenesis of DBD, we conditionally deleted it to achieve either heterozygous (SMIR+/−) or homozygous (SMIR−/−) deletion in smooth muscle cells. Despite impaired glucose and insulin tolerance seen with SMIR−/− mice, both SMIR+/− and SMIR−/− mice exhibited normal blood glucose and plasma insulin levels. Interestingly, these mice had abnormal voiding phenotypes, that included urinary frequency and small voids, and bladder smooth muscle (BSM) had significantly diminished contraction force. Morphology revealed a dilated bladder with thinner BSM layer, and BSM bundles were disorganized with penetrating interstitial tissue. Deletion of IR elevated FoxO and decreased mTOR protein expression, which further decreased the expression of Chrm3, P2x1, Sm22, and Cav1.2, crucial functional proteins for BSM contraction. Furthermore, we determined the expression of adiponectin in BSM, and deletion of IR in BSM inhibited adiponectin-mediated signaling. In summary, disruption of IR-mediated signaling in BSM caused abnormalities in proliferation and differentiation, leading to diminished BSM contractility and a voiding dysfunction phenotype that recapitulates human DBD.
Introduction
Insulin signaling is crucial for glucose metabolism and protein/lipid synthesis, and recent progress indicates that it is also important for gene transcription, cell proliferation, and cell differentiation, in regulating multiple pathways, including mTOR and FoxO signaling (1–8). Studies in several organs suggest that insulin signaling plays differential roles in different tissues by regulating a variety of downstream effectors. For example, specific IR deletion in skeletal muscle results in increased serum triglycerides and decreased muscle growth (4,9), and liver-specific deletion creates severe insulin resistance associated with glucose intolerance (4,10), while in adipose tissue, deletion of IR markedly reduced fat mass and protected the animal from age-related obesity and glucose intolerance (11).
Diabetic bladder dysfunction (DBD) is the most common complication of diabetes, affecting 40–100% of patients with diabetes (12). DBD is commonly associated with myogenic and neurogenic alterations. Patients often exhibit decreased sensation, polyuria with increased bladder volume, poor emptying with increased postvoid residual urine volume, and bladder instability and overactivity, as well as urinary urgency and incontinence (13–20). These debilitating symptoms of DBD significantly impact quality of life.
The control of bladder smooth muscle (BSM) contractility is central to whether the bladder functions properly. In general, micturition is mediated by neurotransmitters ATP and acetylcholine (Ach), which are coreleased by parasympathetic nerves. ATP binds to P2x1 receptor and elicits a rapid contraction to initiate voiding, while Ach binds to muscarinic M2/M3 receptors to induce a slower cholinergic contraction to expel urine (21–25). Indeed, alterations in BSM contractility have consistently been observed in DBD of both people and animal models (13–20). With induction of diabetes with streptozotocin in animal DBD studies and a Zucker diabetic fatty (ZDF) rat model, diabetes caused significantly altered BSM contractility in response to electric field stimulation (EFS), KCl, or agonists like carbachol and α,β-methyleneadenosine 5′-triphosphate trisodium (α,β-meATP) (20,26).
The molecular mechanisms underlying DBD are not fully understood. Hyperglycemia-induced polyuria might play a role in bladder hypertrophy and overactivity, and oxidative stress due to hyperglycemia might induce molecular and cellular damage of bladder tissue (27). As mentioned, insulin signaling might play a central role in the pathogenesis of DBD, and injection of insulin has been shown to reverse streptozotocin-induced DBD in animal models (17). A recent study has shown that deletion of insulin receptor in arterial smooth muscle significantly reduces intimal hyperplasia in a high-fat-fed mouse model, suggesting insulin signaling is an important pathway during the pathogenesis of restenosis in diabetes (28). How insulin receptor signaling in bladder tissue regulates bladder function and whether the disruption of insulin signaling in bladder tissue itself contributes to the pathogenesis of DBD are, however, completely unknown. To address this question, we generated both heterozygous and homozygous smooth muscle–specific IR knockout mouse models to mimic the disrupted insulin signaling in diabetes. These mice exhibited altered voiding function, abnormal bladder morphology, impaired BSM contractility, and dysregulated IR-mediated signaling in bladder, which suggests a pathogenesis recapitulating DBD in human patients with diabetes.
Research Design and Methods
Reagents
Unless otherwise specified, all chemicals were obtained from MilliporeSigma and were of reagent grade or better. P2x1 agonists α,β-meATP and carbachol were purchased from R&D Systems.
Animals
All mice used in this study were on the C57BL/6J background and 12–16 weeks old with matching conditions for age and sex. Both male and female mice were used unless otherwise specified. The mice were housed in standard polycarbonate cages with free access to normal food and water. Wild-type C57BL/6J mice were purchased from The Jackson laboratory (Bar Harbor, ME). We generated both heterozygous and homozygous smooth muscle–specific insulin receptor–deficient mice (SMIR+/− and SMIR−/−) by mating INSRf/f mice (generously donated by Dr. C. Ronald Kahn’s Laboratory at Harvard Medical School) with Sm22α-cre mice (B6.Cg-Tg(Tagln-cre)1Her/J; The Jackson Laboratory). All animal studies were performed in adherence with U.S. National Institutes of Health guidelines for animal care and use and with the approval of the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Glucose Tolerance Test and Insulin Tolerance Test
Glucose tolerance tests (GTTs) were performed on mice fasted overnight (14 h), and then glucose (2 mg/g body wt i.p.) was administered. Insulin tolerance rests (ITTs) were performed on mice fasted for 6 h, and then insulin (0.75 mIU/g body wt i.p.) was administered. Blood glucose was measured from tail nicks at 0, 15, 30, 45, 60, and 120 min after glucose or insulin injection by Contour glucometer (Bayer, Mishawaka, IN).
Serum Insulin Response
Mice fasted for 14 h were injected with glucose (2 mg/g body wt i.p.). Blood was sampled from the tail at 0, 15, 30, 45, 60, and 120 min after glucose injection. Plasma insulin levels were measured with a mouse insulin ELISA kit (Ultrasensitive Mouse Insulin ELISA kit, cat. no. 90080; Crystal Chem) on an xMark Microplate Spectrophotometer (Bio-Rad Laboratories) according to the manufacturer’s instructions.
Void Spot Assay
Void spot assay (VSA) was performed during daytime with light from ∼9:00 a.m. to 1:00 p.m. in female mice. We did not perform VSA on male mice because male voiding data are often complicated by their territorial marking behavior. Individual mice were gently placed in a standard mouse cage with BLICK Cosmos Blotting Paper (cat. no. 10422-1005) placed at the bottom, during which time water was withheld and standard dry mouse chow was available. Recovered filters were imaged as previously described (29,30). Overlapping voiding spots were visually examined and manually separated by outlining and copying and then pasting to a nearby empty space in Fiji software for analysis. A volume:area standard curve showed that a 1 mm2 voiding spot represents 0.283 μL urine. Urine spots with an area ≥80 mm2 were considered primary voiding spots (PVS) based on a cutoff established from the voiding spot patterns analyzed from hundreds of mice (29–31).
Cystometrogram
Cystometrogram (CMG) was performed with PBS infusion (25 μL/min) as previously described in female mice, which gives a consistent regular voiding pattern in wild-type animals (32,33). With urethane (1.4 g/kg body wt) and continuous-flow isoflurane (3% induction, 1.0% maintenance) anesthetization, a 1 cm midline abdominal incision was performed. PE50 tubing was then implanted through the dome of the bladder. The catheter was connected to a pressure transducer and syringe pump coupled to data-acquisition devices (WPI Transbridge and ADInstruments PowerLab 4/35) and a computerized recording system (ADInstruments LabChart software). Isoflurane was withdrawn right after surgery, and CMG was performed under urethane anesthetization, which spares voiding reflex. CMG was performed for at least >1 h of stable and regular voiding cycles, and minimum five cycles of filling and voiding traces were assessed for the change in mice urodynamics. Bladder compliance was determined as follows:
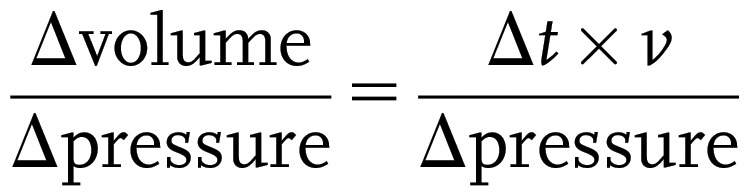 |
where v is 25 μL/min.
BSM Myography
BSM myography was performed on male mice. Male have larger bladders (∼30 mg) compared with female mice, which is easier for dissecting away the epithelial layer and yields four muscle strips per bladder consistently. Mucosa-dissected BSM strips were cut longitudinally (∼2 mm wide and ∼7 mm long). Muscle strips were mounted in an SI-MB4 tissue bath system (World Precision Instruments, Sarasota, FL). Force sensors were connected to TBM4M Transbridge (WPI), and the signal was amplified by PowerLab (ADInstruments) and monitored with LabChart software (ADInstruments). BSM strips were gently prestretched to optimize contraction force and then pre-equilibrated for at least 1 h. Contraction force was sampled at 2,000/s with use of LabChart software. EFS was carried out with a Grass S48 field stimulator (Grass Technologies, West Warwick, RI) according to previously described standard protocols: voltage 50 V, duration 0.05 ms, trains of stimuli 3 s, and frequencies 1, 2, 5, 10, 20, and 50 Hz (32,33).
Histological and Immunofluorescence Staining
Excised bladders were fixed in 4% formaldehyde, embedded in paraffin blocks, and the middle equatorial region of the bladder was sectioned prior to staining with Masson trichrome for bladder wall thickness evaluation. A minimum of six points (shortest distance) where infolding urothelial layers were lacking were averaged on each section and three or more sections per bladder, and the final mean is bladder wall thickness. Muscle layer thickness is measured on six or more randomly chosen points on each section, and three or more sections per bladder. For immunofluorescence staining, excised bladders were fixed in 4% (w/v) paraformaldehyde and then cryoprotected, frozen, sectioned, and incubated with antibodies (1:100) (Supplementary Table 1) overnight at 4°C. The sections were then incubated with an Alexa Fluor 488–conjugated secondary antibody (diluted 1:100), and nuclei were stained with DAPI. Imaging was performed on an Olympus BX60 fluorescence microscope with a 40×/0.75 objective or on a Leica THUNDER imaging system with a 63× objective (33).
Western Blot
Whole bladder tissues from female mice were lysed with radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS). The protein concentration was determined with use of BCA protein assay reagent (Thermo Fisher Scientific, Rockford, IL). Proteins (25 µg) were loaded into each well and separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes. Membranes were incubated with primary antibodies (Supplementary Table 1) at 4°C for 2 h. Protein bands were visualized with Amersham ECL reagent (Arlington Heights, IL). The membranes were incubated with Restore PLUS Western Blot Stripping Buffer (Thermo Fisher Scientific) for 5 min to remove primary and secondary antibodies for reblotting with new antibodies, and each membrane was stripped and reprobed no more than three times. The intensity of protein bands was scanned and then quantitated with Fiji software (33). Targeted protein in each lane was normalized to β-actin intensity in the same lane. The average protein expression level in wild-type mice was designed as 1, and the fold change of protein expression level in knockout mice was reported.
Statistical Analyses
All data are presented as box and whisker plots. The centerline is the median, the box represents 75% of the data, and the whiskers indicate minimum to maximum. Data were analyzed with one-way ANOVA for comparison among groups. Bonferroni multiple comparisons post hoc tests were used where necessary between the two groups with GraphPad Prism 8 software, and a P value <0.05 was considered significant.
Data and Resource Availability
Further information, reagents, and other supporting data in this study are available from the corresponding author on request.
Results
SMIR−/− Mice Exhibit Impaired Glucose and Insulin Tolerance
Given that IR plays a crucial role in glucose homeostasis, glucose metabolism in SMIR+/− and SMIR−/− mice was assessed. In comparisons with wild-type mice, there was no detectable difference in blood glucose levels in both nonfasted and fasted female SMIR+/− and SMIR−/− mice (Fig. 1A). Male mice were similar (Supplementary Fig. 1A). However, both male and female SMIR−/− mice exhibited hyperglycemia when challenged with an injection of glucose (2 mg/g body wt i.p.). There was elevated blood glucose in SMIR+/− mice in response to GTT, but the difference was not statistically different compared with wild type (Fig. 1B and C and Supplementary Fig. 1B and C). Consistent with the GTT response, blood glucose level in both male and female SMIR−/− mice in response to ITT was significantly higher than in wild-type animals (Fig. 1D and E and Supplementary Fig. 1D and E), indicating impaired ability of SMIR−/− mice in glucose metabolism during stressed condition. In comparison with wild-type mice, the plasma insulin levels were not different in either SMIR+/− or SMIR−/− female mice. The overall plasma insulin levels in response to GTT were also similar in all three studied groups, though the SMIR−/− mice tend to show mildly elevated insulin levels (Fig. 1F–H).
Figure 1.

Female SMIR−/− mice exhibit impaired glucose and insulin tolerance. A: Blood glucose levels of female mice fasted (n = 15, 14, and 19 for WT, SMIR+/−, and SMIR−/− mice) for 14 h or nonfasted (n = 10, 10, and 16 for wild-type [WT], SMIR+/−, and SMIR−/− mice). B: GTT in female wild-type (n = 10), SMIR+/− (n = 10), and SMIR−/− (n = 16) mice. C: Area under the curve (AUC) for B. D: ITT in female wild-type (n = 9), SMIR+/− (n = 8), and SMIR−/− (n = 10) mice. E: Area under the curve for D. F: Serum insulin levels in female wild-type (n = 14), SMIR+/− (n = 9), and SMIR−/− (n = 10) mice. G: Serum insulin levels in response to GTT in female wild-type (n = 10), SMIR+/− (n = 10), and SMIR−/− (n = 8) mice. H: Area under the curve for G. Data are shown as boxes and whiskers. The centerline is the median of the data set, the box represents 75% of the data, and bars indicate whiskers from minimum to maximum. Data were analyzed with use of Student t test. *P < 0.05 and **P < 0.01 compared with wild-type mice; #P < 0.05 and ##P < 0.01 compared between SMIR+/− and SMIR−/− mice.
SMIR+/− and SMIR−/− Mice Have Altered Voiding Phenotype and Urodynamics
For understanding of whether deficiency or deletion in BSM insulin signaling can directly cause voiding dysfunction, we performed VSA to examine the voiding phenotype in both SMIR+/− and SMIR−/− mice. Compared with wild-type mice, which produce on average 3.03 PVS in a 4-h period, significantly increased voiding frequency was seen in SMIR+/− (5.83 PVS/4 h) and SMIR−/− (5.07 PVS/4 h) mice. Wild-type mice have an average PVS size of ∼434 mm2/spot (∼123 μL/void); however, PVS size is significantly smaller in SMIR+/− (319 mm2/spot, ∼90.33 μL/void) and SMIR−/− (273 mm2/spot, ∼77.29 μL/void) mice (Fig. 2A–F). Voiding spots were further grouped according to their different sizes as shown in the frequency distribution chart in Fig. 2G, and spots showed a distribution pattern similar to a skewed bell curve, with control group peaks at ∼200–300 mm2, while SMIR+/− and SMIR−/− groups peak at ∼80–200 mm2, indicating a significantly shifted voiding pattern. For confirmation of the altered voiding pattern detected in VSA and exploration of potential underlying mechanisms, urodynamic studies were performed with CMG. CMG data consistently indicated that SMIR+/− and SMIR−/− mice exhibited increased voiding frequency with decreased bladder compliance (Fig. 2H–N).
Figure 2.

SMIR+/− and SMIR−/− mice have altered voiding phenotype and urodynamics. A–C: Representative filters show ultraviolet light–illuminated urine spots from female wild-type (WT) (n = 30), SMIR+/− (n = 28), and SMIR−/− (n = 27) mice. D–F: Summarized data of the numbers of PVS, area of PVS, and the total area of voiding spots per filter. G: Summarized frequency distribution chart of varied spot size. H–J: Representative CMG traces from female wild-type (n = 5), SMIR+/− (n = 4), and SMIR−/− (n = 5) mice. K–N: Summarized urodynamic data of voiding interval, peak pressure, basal pressure, and compliance from CMGs. Data are shown as boxes and whiskers. The centerline is the median of the data set, the box represents 75% of the data, and bars indicate whiskers from minimum to maximum. Data were analyzed with use of Student t test. *P < 0.05 and **P < 0.01.
SMIR+/− and SMIR−/− Mice Display a Dilated Bladder With Abnormal BSM Layer
To understand how deficiency in BSM insulin signaling contributes to abnormal voiding function exhibited by VSA and CMG, we examined SMIR+/− and SMIR−/− mice morphologically. As shown in Figure 3H, SMIR+/− and SMIR−/− mice bladders are lighter compared with those of wild-type mice. Masson trichrome staining of tissue sections from middle part of bladders from SMIR+/− and SMIR−/− mice revealed significantly altered morphology, with bladder wall thickness and muscle layer thickness significantly thinner compared with wild type (Fig. 3). Furthermore, SMIR+/− and SMIR−/− bladder muscle bundles are disorganized with enlarged spaces among the bundles, and, particularly in SMIR−/− bladders, significant interstitial tissue penetrates and segregates the muscle bundles into looser smaller strips (Fig. 3F). The increased proportion of interstitial tissue is obvious in both SMIR+/− and SMIR−/− mice bladders, indicating a decompensatory response. Consistent with dilated bladders, the vasculature in SMIR+/− and SMIR−/− mice bladder lamina propria layer also dilated (Fig. 3D–F), suggesting a potential significant oxidative stress due to abnormal vascular function, which could contribute to the DBD as well.
Figure 3.

SMIR+/− and SMIR−/− mice display a dilated bladder with abnormal BSM layer. A–C: Representative images of wild-type (WT), SMIR+/−, and SMIR−/− mice bladders. D–F: Enlarged images of bladder wall from panels A–C. G–J: Summarized data of body weight (n = 12, 12, and 8 for wild-type, SMIR+/−, and SMIR−/− including both male and female mice), bladder weight (n = 12, 12, and 8 for wild-type, SMIR+/−, and SMIR−/− including both male and female mice), bladder wall thickness (n = 10, 10, and 8 for wild-type, SMIR+/−, and SMIR−/− female mice), and muscle layer thickness (n = 10, 10, and 8 for wild-type, SMIR+/−, and SMIR−/− female mice). Data are shown as boxes and whiskers. The centerline is the median of the data set, the box represents 75% of the data, and bars indicate whiskers from minimum to maximum. L, lamina propria layer; M, smooth muscle layer; U, urothelium layer. White arrows indicate blood vessels in lamina propria. Data were analyzed with use of Student t test. *P < 0.05 and **P < 0.01.
SMIR+/− and SMIR−/− Mice Have Diminished Smooth Muscle Contraction Force
Our morphological evidence suggested that IR deletion could impact BSM contractility, thereby leading to altered bladder voiding function. For determination of whether this was true, myography was performed on isolated BSM strips. As shown in Figure 4A–D, the contractile force of BSM strips increases in response to increased EFS frequencies, which mimics the in vivo BSM contraction in response to neurotransmitter release. In comparisons with wild-type mice, the contraction force from SMIR+/− and SMIR−/− mice BSM strips was significantly smaller. For confirmation that the diminished contraction force is due to BSM itself, and not due to altered neurotransmitter release, KCl, which depolarizes BSM membrane potential and therefore causes BSM contraction, was used to test BSM response. Results consistently indicated significantly smaller contraction force in BSM strips from SMIR+/− and SMIR−/− bladders (Fig. 4E). As noted, bladder contraction is mainly mediated by parasympathetic ATP and Ach co-release, which induce P2x1 receptor– and Chrm3 receptor–mediated signaling cascades leading to BSM contraction. For determination of changes of these signaling pathways in IR-deleted BSM, α,β-meATP, an agonist for P2x1 receptor, and carbachol, an agonist for Chrm3 receptor, were tested, and our data showed significantly impaired contractile force in response to both agonists, suggesting significant change of these pathways in SMIR+/− and SMIR−/− mice bladders (Fig. 4F and G).
Figure 4.

SMIR+/− and SMIR−/− mice have diminished smooth muscle contraction force. A–C: Representative traces of BSM contraction from male wild-type (WT) (n = 16 strips from 4 mice), SMIR+/− (n = 16 strips from 4 mice), and SMIR−/− (n = 12 strips from 3 mice) mice in response to EFS. D: Summarized data. E–G: Summarized data of BSM contraction force in response to KCl (n = 16, 16, and 12 BSM strips for wild-type, SMIR+/−, and SMIR−/− mice), carbachol (n = 20, 16, and 12 BSM strips for wild-type, SMIR+/−, and SMIR−/− mice), and α,β-meATP (n = 16, 16, and 12 BSM strips for wild-type, SMIR+/−, and SMIR−/− mice) stimuli. Data are shown as boxes and whiskers, the centerline is the median of the data set, the box represents 75% of the data, and bars indicate whiskers from minimum to maximum. Data were analyzed with use of Student t test. *P < 0.05 and **P < 0.01.
Altered Molecular Expression Is Responsible for the Diminished BSM Contraction Force in IR-Deficient Bladders
To further understand the mechanism of impaired smooth muscle contractile function in SMIR+/− and SMIR−/− mice, we performed Western blot studies on potential pathways for BSM contraction machinery. Consistent with our functional data showing diminished BSM contraction force (Fig. 4), the expression levels of both Chrm3 receptor and P2x1 receptor were significantly downregulated (Fig. 5A–C). Interestingly, integrin β1, which we have previously shown is an important cell adhesion protein mediating BSM mechanotransduction and voiding function (34), remains unchanged when BSM is IR-null (Fig. 5A and D). However, sm22α, a smooth muscle–specific protein involved in cell differentiation and cytoskeletal organization, is significantly reduced in both SMIR+/− and SMIR−/− bladders, suggesting reduced BSM cell proliferation/differentiation. Cav1.2, a voltage-gated ion channel mediating calcium influx, is an essential protein for BSM cell contractile function and gene regulation, and smooth muscle null for Cav1.2 causes a lethal phenotype (33,35). Interestingly, in our SMIR+/− and SMIR−/− mice, Cav1.2 is significantly downregulated (Fig. 5A and F), which might also contribute to the diminished contraction force and altered voiding phenotype in these mice.
Figure 5.

Altered molecular expression is responsible for the diminished BSM contraction force in SMIR+/− and SMIR−/− mice bladder. A: Western blot of Chrm3, P2x1, Itgβ1, Sm22, and Cav1.2 proteins from wild-type (WT), SMIR+/−, and SMIR−/− mice bladders (n = 4). β-Actin is used as loading control for normalization and quantitated data are shown as B–F. Data are shown as boxes and whiskers, the centerline is the median of the data set, the box represents 75% of the data, and bars indicate whiskers from minimum to maximum. Data were analyzed with use of Student t test. *P < 0.05 and **P < 0.01.
Insulin Signaling Abnormalities in SMIR+/− and SMIR−/− Mice Bladders
As noted, insulin signaling is crucial for glucose metabolism, protein/lipid synthesis, and cell proliferation/differentiation in regulating gene transcription. Insulin signaling is mediated by insulin receptor (IR), which belongs to receptor tyrosine kinase (RTK) family and contains the intrinsic tyrosine kinase domain. On activation, IR phosphorylates multiple substrate proteins and modulates many downstream signal pathways. Among them, Akt/protein kinase B, a serine/threonine protein kinase, plays crucial roles by inhibiting Forkhead Box O (FoxO)-mediated protein degradation, autophagy, and apoptosis or by promoting target of rapamycin (mTOR)-mediated metabolism, protein synthesis, and cell proliferation. As expected, IR expression levels in SMIR+/− and SMIR−/− mice bladders are significantly reduced (Fig. 6A and B), and the remaining messenger and protein signaling reflects the IR expression from other cell types in bladder wall such as urothelial and interstitial cells. To our surprise, Akt and p-Akt (Akt phosphorylated at Ser473) levels were not changed (Fig. 6A, C, and D), and this could be due to multiple factors. (See discussion.) Interestingly, multiple FoxO proteins are upregulated in SMIR+/− and SMIR−/− mice bladders, including FoxO1, FoxO3, and FoxO6 (Fig. 6A and E–G), and correspondingly, mTOR and p-mTOR (mTOR phosphorylated at Ser2448) are significantly decreased in SMIR+/− and SMIR−/− mice bladders (Fig. 6A, H, and I). These data indicate an increased catabolism-like protein degradation and autophagy in these mice, which is consistent with our morphological (Fig. 2), functional (Figs. 2 and 4), and molecular (Fig. 5) data shown above.
Figure 6.

Insulin signaling abnormalities in SMIR+/− and SMIR−/− mice bladders. A: Western blot of Insr, Akt, p-Akt, FoxO1, FoxO3, Fox6, mTOR, and p-mTOR proteins from wild-type (WT), SMIR+/−, and SMIR−/− mice bladders (n = 4). β-Actin is used as loading control for normalization, and quantitated data are shown in B–I. Data are shown as boxes and whiskers. The centerline is the median of the data set, the box represents 75% of the data, and bars indicate whiskers from minimum to maximum. Data were analyzed with use of Student t test. *P < 0.05 and **P < 0.01.
Insulin Signaling Is Associated With Adiponectin Signaling in IR-Deficient Bladders
Adiponectin is a soluble protein secreted mainly by adipocytes, and recent studies have shown that adiponectin-mediated signaling is important in regulating insulin sensitivity, glucose uptake, and lipid metabolism (36,37). Insulin signaling cross talks with adiponectin signaling by interacting with adaptor proteins such as APPL1, and knockdown of IR in adipocytes has been shown to suppress adiponectin production (38). Interestingly, recent studies indicated that vascular smooth muscle cells (VSMC) also express adiponectin, and adiponectin-mediated signaling in VSMC regulates VSMC differentiation and contractile function (39,40). Using immunofluorescent staining and imaging, we determined that adiponectin is also expressed in BSM and in vasculature elements in bladder wall (Fig. 7A and Supplementary Fig. 2). Further Western blot studies indicate that adiponectin protein levels are significantly downregulated in SMIR+/− and SMIR−/− mice bladders, suggesting a role of adiponectin signaling in BSM function (Fig. 7B and C). AMPK is a key downstream molecule mediating adiponectin signaling. AMPK plays an important role in cellular energy homeostasis by regulating glucose and fatty acid uptake and oxidation. While there were no detectable changes in AMPK expression levels in SMIR+/− and SMIR−/− bladders, p-AMPK (phosphorylated AMPK-α subunit Thr172) levels are inhibited (Fig. 7B, D, and E), suggesting that downregulated adiponectin resulted in reduced AMPK phosphorylation as a consequence of IR deletion.
Figure 7.

Inhibition of adiponectin signaling in SMIR+/− and SMIR−/− mice bladders. A: Immunostaining and imaging of adiponectin (green) in BSM cell membrane. Nuclei are stained with DAPI (blue). B: Western blot of Adpn, Ampk, and p-Ampk proteins from wild-type (WT), SMIR+/−, and SMIR−/− mice bladders (n = 4). β-Actin is used as loading control for normalization, and quantitated data are shown in C–E. Data are shown as boxes and whiskers. The centerline is the median of the data set, the box represents 75% of the data, and bars indicate whiskers from minimum to maximum. Data were analyzed with use of Student t test. *P < 0.05 and **P < 0.01.
Discussion
Insulin signaling is crucial for metabolic homeostasis, and tissue-specific deletion of IR can prompt distinct metabolic disorders (9–11). In a recent report, feeding a high-fat diet for 8 weeks to smooth muscle–specific IR-deleted mice induced obesity in both wild-type and SMIR−/− mice to reach body weights of ∼40 g. In that case, there was not a significant difference detected between wild-type and SMIR−/− mice with regard to GTT, ITT, and triglyceride cholesterol levels. The plasma insulin level was somewhat higher in SMIR−/− mice but did not reach significance (28). However, in that study they did not report on the metabolic condition of SMIR−/− mice fed on a normal diet. It is possible the high-fat diet could mask abnormal metabolic conditions in these mice. In this study, SMIR+/− and SMIR−/− mice fed regular food were smaller compared with wild-type control mice (mostly ranging from 18 to 30 g), and our results indicate that SMIR−/− mice exhibit normal blood glucose but have impaired GTT and ITT responses (Fig. 1). These data suggest that smooth muscle tissue could contribute to overall metabolic syndromes during stressed condition due to local consumption and storage of glucose.
DBD is an extremely common complication in human patients with diabetes, and they can present with a spectrum of symptoms and urodynamics due to different diabetic cystopathies, including neuronal impairment, BSM damage and areflexia, and urothelial dysfunction (15,41,42). Neuronal impairment and urothelial dysfunction might cause sensory abnormalities resulting in insensate bladder, which leads to increased bladder capacity and overdistended bladder. BSM damage and areflexia might cause diminished BSM contractility and increased postvoid residual volume (PVR). Therefore, patients with diabetes often manifest with urinary frequency, urgency, incontinence, hesitancy, dribbling and weak stream, nocturia, and urinary retention. In our SMIR+/− and SMIR−/− mice, we were able to recapitulate many of the symptoms and urodynamic changes observed in human DBD patients, such as frequency (Fig. 2), diminished BSM contractility (Fig. 4), dilated bladder indicating increased bladder capacity (Fig. 3), and small voids (Fig. 2) with dilated bladder (Fig. 3) suggesting urinary retention and increased PVR. However, our conventional CMG study did not generate PVR data in these SMIR+/− and SMIR−/− mice, which needs to be further investigated in future with other novel methods such as X-ray videocystometry (43).
Hyperglycemia is often blamed for DBD pathogenesis and polyuria (27). It is believed that hyperglycemia results in oxidative stress and causes cellular damage, and polyuria leads to diuresis and bladder remodeling. In our study, blood glucose levels in both SMIR+/− and SMIR−/− mice were normal, and only SMIR−/− mice exhibited abnormal GTT and ITT responses (Fig. 1 and Supplementary Fig. 1); however, as shown above, significant voiding dysfunctions and morphological changes mimicking human DBD were observed in both homozygous and heterozygous models. This observation implies several important ideas with regard to the pathogenesis of DBD: 1) while hyperglycemia might play some role in the pathogenesis of DBD, it may not be the most essential or major contributing factor. 2) Instead, IR-mediated signaling in bladder itself, which not only regulates bladder tissue metabolic homeostasis but also modulates tissue phenotype and function through regulation of gene expression, could be the major contributing factor leading to DBD pathogenesis. 3) SMIR+/− and SMIR−/− mice do not exhibit hyperglycemia (Fig. 1A and Supplementary Fig. 1A) and diuresis and do not exhibit any polyuria (Fig. 2F), both of which are often invoked as causative for DBD. These findings suggest that a major shift may be required in how we think about the etiology of DBD.
In addition to disrupted homeostasis of energy metabolism, what other underlying mechanism(s) might contribute to DBD? In high-fat-fed SMIR−/− mice, VSMC proliferation in femoral artery at 7 days after wire injury was decreased by 51% compared with wild-type mice, and the intimal hyperplasia upon wire injury was decreased by 37%, with a much thinner femoral arterial wall thickness exhibited (28). These data are fully consistent with our findings that BSM layer is much thinner in SMIR+/− and SMIR−/− mice, and the vasculature in the bladder wall is also dilated like the bladder wall (Fig. 3), indicating decreased proliferation in BSM cells due to disrupted insulin signaling. Furthermore, we showed that FoxO1, FoxO3, and FoxO6 protein levels are significantly upregulated, while mTOR and p-mTOR protein levels are significantly downregulated, in SMIR+/− and SMIR−/− mice bladder (Fig. 6). Further study is required regarding the exact role of these proteins in BSM function; however, it is generally believed that an increase of FoxO transcription factors and a corresponding decrease of mTOR favors pathways for cell cycle arrest, protein degradation, inflammation, and apoptosis (44). Therefore, IR deletion not only decreases BSM proliferation but also might increase BSM protein degradation and cell apoptosis and thereby change both BSM phenotype and function. This idea is further supported by decreased expression levels of key functional proteins in IR-deficient BSM, including Chm3, P2x1, Cav1.2, and sm22 (Fig. 5), which eventually leads to diminished BSM contraction force (Fig. 4) and voiding dysfunction (Fig. 2).
While only high-fat-fed SMIR−/− mice were studied in arterial intimal hyperplasia (28), we included both SMIR+/− and SMIR−/− mice in this study because we believe the heterozygotes might better mimic the diminished insulin signaling cascades in BSM cells of type 2 diabetes due to decreased insulin release or insulin resistance. Conversely, SMIR−/− mice might better mimic type 1 diabetes completely lacking in insulin signaling. Interestingly, both mice exhibit significant DBD phenotypes, with SMIR−/− mice showing relatively more severe symptoms and molecular changes in most cases, and this is in good agreement with the human type 1 and type 2 diabetes situations.
Hyperglycemia, diminished β-cell insulin release, and insulin resistance are key features in human patients with diabetes. Hyperglycemia is a direct consequence of either diminished β-cell insulin release or insulin resistance, which reflects the disruption of normal hepatic regulation of blood glucose by IR-mediated pathway, and in liver-specific IR knockout mice severe hyperglycemia and insulin resistance have been reported, while in other tissue-specific IR knockout studies, including fat, skeletal muscle, and smooth muscle, hyperglycemia has not been shown. However, abnormal tissue/organ function mimicking patients with diabetes has been consistently reported in these studies. Therefore, it seems rational to conclude that disrupted IR signaling in the bladder tissue of human patients with type 1 diabetes and type 2 diabetes is a major pathological mechanism for the development of DBD.
Despite the interesting and novel findings uncovered in these experiments, there are also a number of emerging questions that need be answered in future studies. For example, insulin-like growth factor 1 (IGF1), a growth hormone with a molecular structure similar to that of insulin, can also activate IR signaling. IGF1 receptor (IGF1R) and IR not only form homodimers by themselves but also can form heterodimers with each other. In femoral artery, IGF1R has been shown to be a negative regulator of insulin signaling (28). Elevated IGF1 signaling is suggested to be important in BSM hypertrophy and bladder remodeling (45,46); however, further investigation is needed for detailed understanding of roles of IR- and IGF1R-mediated signaling in bladder physiology and pathogenesis.
Activation of both IR and IGF1R share many common downstream effectors, such as Akt. How different receptors induce differential effects by modulating a common effector is not clear. To our surprise, we observed that Akt and p-Akt expression levels are not affected in SMIR+/− and SMIR−/− bladders; however, the downstream FoxO proteins are elevated (Fig. 6). This could be a confounding effect of IGF1 signaling in BSM, or IR signaling in other tissue in bladder wall as well. In wild-type VSMCs, a dose of insulin activation can boost the p-Akt dramatically, while in SMIR−/− VSMCs diminished p-Akt signaling has been observed (28).
Further insight is also needed regarding the role of adiponectin in BSM, which has never been explored in DBD. Due to the important role of adiponectin in obesity, glucose, and fat metabolism, and its interaction with insulin signaling, we expect that the determination of adiponectin expression in BSM will lead to more discoveries on the details of adiponectin-mediated DBD pathogenesis.
In summary, we have discovered that the bladder phenotype displayed in SMIR+/− and SMIR−/− mice recapitulates symptoms and urodynamics observed in human DBD patients, and disruption of IR-mediated proliferation and protein degradation pathways might be the major mechanisms in the pathogenesis of DBD.
Article Information
Acknowledgments. The authors thank Dr. C. Ronald Kahn’s Laboratory at Harvard Medical School for providing insulin gene floxed mice and Dr. Anne Brunet’s laboratory at Standard University for providing FoxO6 antibody. The authors also thank Dr. Warren Hill (Beth Israel Deaconess Medical Center) for helpful comments on the manuscript.
Funding. The authors acknowledge funding received from the National Institute on Aging/National Institutes of Health (grant R21AG064633 to W.Y.) and National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health (grant R01DK126674 to W.Y.) and financial support provided by the National Institute of Diabetes and Digestive and Kidney Diseases Diabetic Complications Consortium (DiaComp) (https://www.diacomp.org) (grant DK076169 to W.Y.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. H.C. performed metabolic measurements, ELISA, VSA, CMG, myography, histological and immunostaining and imaging, and Western blot and analyzed data. A.W. performed Western blot and immunostaining and imaging. W.Y. performed immunostaining and imaging and analyzed data. M.L.Z. contributed to study conception and interpretation of data. W.Y. conceived and supervised the project. H.C. and W.Y. wrote the manuscript, and all authors critically reviewed the manuscript, discussed idea and results, and contributed to the manuscript. W.Y. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.20349528.
H.C. is currently affiliated with The School of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan, China.
References
- 1. Yoon MS. The role of mammalian target of rapamycin (mTOR) in insulin signaling. Nutrients 2017;9:1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilcox G. Insulin and insulin resistance. Clin Biochem Rev 2005;26:19–39 [PMC free article] [PubMed] [Google Scholar]
- 3. Kitamura T, Kahn CR, Accili D. Insulin receptor knockout mice. Annu Rev Physiol 2003;65:313–332 [DOI] [PubMed] [Google Scholar]
- 4. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 2014;6:a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kadowaki T. Insights into insulin resistance and type 2 diabetes from knockout mouse models. J Clin Invest 2000;106:459–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol 2018;19:31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol 2014;220:T1–T23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mackenzie RW, Elliott BT. Akt/PKB activation and insulin signaling: a novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes Metab Syndr Obes 2014;7:55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brüning JC, Michael MD, Winnay JN, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 1998;2:559–569 [DOI] [PubMed] [Google Scholar]
- 10. Michael MD, Kulkarni RN, Postic C, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 2000;6:87–97 [PubMed] [Google Scholar]
- 11. Blüher M, Michael MD, Peroni OD, et al. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell 2002;3:25–38 [DOI] [PubMed] [Google Scholar]
- 12. Mumtaz FH, Thompson CS, Khan MA, et al. Alterations in the formation of cyclic nucleotides and prostaglandins in the lower urinary tract of the diabetic rabbit. Urol Res 1999;27:470–475 [DOI] [PubMed] [Google Scholar]
- 13. Brown JS, Wessells H, Chancellor MB, et al. Urologic complications of diabetes. Diabetes Care 2005;28:177–185 [DOI] [PubMed] [Google Scholar]
- 14. Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S. Diabetic bladder dysfunction: current translational knowledge. J Urol 2009;182(Suppl.):S18–S26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Golbidi S, Laher I. Bladder dysfunction in diabetes mellitus. Front Pharmacol 2010;1:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Daneshgari F, Leiter EH, Liu G, Reeder J. Animal models of diabetic uropathy. J Urol 2009;182(Suppl.):S8–S13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daneshgari F, Huang X, Liu G, Bena J, Saffore L, Powell CT. Temporal differences in bladder dysfunction caused by diabetes, diuresis, and treated diabetes in mice. Am J Physiol Regul Integr Comp Physiol 2006;290:R1728–R1735 [DOI] [PubMed] [Google Scholar]
- 18. Leiria LO, Mónica FZ, Carvalho FD, et al. Functional, morphological and molecular characterization of bladder dysfunction in streptozotocin-induced diabetic mice: evidence of a role for L-type voltage-operated Ca2+ channels. Br J Pharmacol 2011;163:1276–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Munoz A, Boone TB, Smith CP, Somogyi GT. Diabetic plasticity of non-adrenergic non-cholinergic and P2X-mediated rat bladder contractions. Brain Res Bull 2013;95:40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu G, Daneshgari F. Alterations in neurogenically mediated contractile responses of urinary bladder in rats with diabetes. Am J Physiol Renal Physiol 2005;288:F1220–F1226 [DOI] [PubMed] [Google Scholar]
- 21. Andersson KE, Arner A. Urinary bladder contraction and relaxation: physiology and pathophysiology. Physiol Rev 2004;84:935–986 [DOI] [PubMed] [Google Scholar]
- 22. Berridge MJ. Smooth muscle cell calcium activation mechanisms. J Physiol 2008;586:5047–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heppner TJ, Werner ME, Nausch B, Vial C, Evans RJ, Nelson MT. Nerve-evoked purinergic signalling suppresses action potentials, Ca2+ flashes and contractility evoked by muscarinic receptor activation in mouse urinary bladder smooth muscle. J Physiol 2009;587:5275–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sibley GN. A comparison of spontaneous and nerve-mediated activity in bladder muscle from man, pig and rabbit. J Physiol 1984;354:431–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vial C, Evans RJ. P2X receptor expression in mouse urinary bladder and the requirement of P2X(1) receptors for functional P2X receptor responses in the mouse urinary bladder smooth muscle. Br J Pharmacol 2000;131:1489–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kendig DM, Ets HK, Moreland RS: Effect of type II diabetes on male rat bladder contractility. Am J Physiol Renal Physiol 2016:310:F909–F922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xiao N, Wang Z, Huang Y, Daneshgari F, Liu G. Roles of polyuria and hyperglycemia in bladder dysfunction in diabetes. J Urol 2013;189:1130–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Q, Fu J, Xia Y, et al. Homozygous receptors for insulin and not IGF-1 accelerate intimal hyperplasia in insulin resistance and diabetes. Nat Commun 2019;10:4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen H, Zhang L, Hill WG, Yu W. Evaluating the voiding spot assay in mice: a simple method with complex environmental interactions. Am J Physiol Renal Physiol 2017;313:F1274–F1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rajandram R, Ong TA, Razack AH, MacIver B, Zeidel M, Yu W. Intact urothelial barrier function in a mouse model of ketamine-induced voiding dysfunction. Am J Physiol Renal Physiol 2016;310:F885–F894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu W, Ackert-Bicknell C, Larigakis JD, et al. Spontaneous voiding by mice reveals strain-specific lower urinary tract function to be a quantitative genetic trait. Am J Physiol Renal Physiol 2014;306:F1296–F1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hao Y, Wang L, Chen H, et al. Targetable purinergic receptors P2Y12 and A2b antagonistically regulate bladder function. JCI Insight 2019;4:e122112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen H, Vandorpe DH, Xie X, Alper SL, Zeidel ML, Yu W. Disruption of Cav1.2-mediated signaling is a pathway for ketamine-induced pathology. Nat Commun 2020;11:4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie X, Chen H, Zhang L, et al. Molecular mechanisms of voiding dysfunction in a novel mouse model of acute urinary retention. FASEB J 2021;35:e21447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wegener JW, Schulla V, Lee TS, et al. An essential role of Cav1.2 L-type calcium channel for urinary bladder function. FASEB J 2004;18:1159–1161 [DOI] [PubMed] [Google Scholar]
- 36. Choi HM, Doss HM, Kim KS. Multifaceted physiological roles of adiponectin in inflammation and diseases. Int J Mol Sci 2020;21:1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iwabu M, Okada-Iwabu M, Yamauchi T, Kadowaki T. Adiponectin/AdipoR research and its implications for lifestyle-related diseases. Front Cardiovasc Med 2019;6:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Groeneveld MP, Brierley GV, Rocha NM, Siddle K, Semple RK. Acute knockdown of the insulin receptor or its substrates Irs1 and 2 in 3T3-L1 adipocytes suppresses adiponectin production. Sci Rep 2016;6:21105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ding M, Carrão AC, Wagner RJ, et al. Vascular smooth muscle cell-derived adiponectin: a paracrine regulator of contractile phenotype. J Mol Cell Cardiol 2012;52:474–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ding M, Xie Y, Wagner RJ, et al. Adiponectin induces vascular smooth muscle cell differentiation via repression of mammalian target of rapamycin complex 1 and FoxO4. Arterioscler Thromb Vasc Biol 2011;31:1403–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wittig L, Carlson KV, Andrews JM, Crump RT, Baverstock RJ. Diabetic bladder dysfunction: a review. Urology 2019;123:1–6 [DOI] [PubMed] [Google Scholar]
- 42. Agochukwu-Mmonu N, Pop-Busui R, Wessells H, Sarma AV. Autonomic neuropathy and urologic complications in diabetes. Auton Neurosci 2020;229:102736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Franken J, De Bruyn H, Rietjens R, et al. X-ray videocystometry for high-speed monitoring of urinary tract function in mice. Sci Adv 2021;7:eabi6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 2011;1813:1978–1986 [DOI] [PubMed] [Google Scholar]
- 45. Kanno Y, Mitsui T, Kitta T, et al. The inflammatory cytokine IL-1β is involved in bladder remodeling after bladder outlet obstruction in mice. Neurourol Urodyn 2016;35:377–381 [DOI] [PubMed] [Google Scholar]
- 46. Chen Y, Bornfeldt KE, Arner A, et al. Increase in insulin-like growth factor I in hypertrophying smooth muscle. Am J Physiol 1994;266:E224–E229 [DOI] [PubMed] [Google Scholar]