

Key Points
Question
Are different CHEK2 pathogenic variants (PVs) associated with different cancer phenotypes?
Findings
In this retrospective cohort study of 36 817 participants tested for cancer predisposition, 3783 had 1 or more CHEK2 PVs; after excluding the lower-risk variants, p.I157T, p.S428F, and p.T476M, other CHEK2 PVs were associated with similar cancer phenotypes to c.1100del.
Meaning
CHEK2 PVs were not associated with colorectal cancer and were associated with breast, kidney, and thyroid cancers; the p.I157T, p.S428F, and p.T476M variants—42% of PVs in this cohort—had a tenuous association with breast cancer only. These findings may guide cancer surveillance of individuals with CHEK2 PVs.
This retrospective cohort study examines the association of different CHEK2 pathogenic variants with different cancer phenotypes.
Abstract
Importance
Germline CHEK2 pathogenic variants (PVs) are frequently detected by multigene cancer panel testing (MGPT), but our understanding of PVs beyond c.1100del has been limited.
Objective
To compare cancer phenotypes of frequent CHEK2 PVs individually and collectively by variant type.
Design, Setting, and Participants
This retrospective cohort study was carried out in a single diagnostic testing laboratory from 2012 to 2019. Overall, 3783 participants with CHEK2 PVs identified via MGPT were included. Medical histories of cancer in participants with frequent PVs, negative MGPT (wild type), loss-of-function (LOF), and missense were compared.
Main Outcomes and Measures
Participants were stratified by CHEK2 PV type. Descriptive statistics were summarized including median (IQR) for continuous variables and proportions for categorical characteristics. Differences in age and proportions were assessed with Wilcoxon rank sum and Fisher exact tests, respectively. Frequencies, odds ratios (ORs), 95% confidence intervals were calculated, and P values were corrected for multiple comparisons where appropriate.
Results
Of the 3783 participants with CHEK2 PVs, 3473 (92%) were female and most reported White race. Breast cancer was less frequent in participants with p.I157T (OR, 0.66; 95% CI, 0.56-0.78; P<.001), p.S428F (OR, 0.59; 95% CI. 0.46-0.76; P<.001), and p.T476M (OR, 0.74; 95% CI, 0.56-0.98; P = .04) PVs compared with other PVs and an association with nonbreast cancers was not found. Following the exclusion of p.I157T, p.S428F, and p.T476M, participants with monoallelic CHEK2 PV had a younger age at first cancer diagnosis (P < .001) and were more likely to have breast (OR, 1.83; 95% CI, 1.66-2.02; P < .001), thyroid (OR, 1.63; 95% CI, 1.26-2.08; P < .001), and kidney cancer (OR, 2.57; 95% CI, 1.75-3.68; P < .001) than the wild-type cohort. Participants with a CHEK2 PV were less likely to have a diagnosis of colorectal cancer (OR, 0.62; 95% CI, 0.51-0.76; P < .001) compared with those in the wild-type cohort. There were no significant differences between frequent CHEK2 PVs and c.1100del and no differences between CHEK2 missense and LOF PVs.
Conclusions and Relevance
CHEK2 PVs, with few exceptions (p.I157T, p.S428F, and p.T476M), were associated with similar cancer phenotypes irrespective of variant type. CHEK2 PVs were not associated with colorectal cancer, but were associated with breast, kidney, and thyroid cancers. Compared with other CHEK2 PVs, the frequent p.I157T, p.S428F, and p.T476M alleles have an attenuated association with breast cancer and were not associated with nonbreast cancers. These data may inform the genetic counseling and care of individuals with CHEK2 PVs.
Introduction
The CHEK2 gene codes a protein kinase (CHK2) that acts as a tumor suppressor and plays a role in DNA damage repair.1,2,3,4 CHEK2 variants were first described among families who met clinical criteria for Li-Fraumeni syndrome (LFS) but were TP53 negative.5 CHEK2 pathogenic and likely pathogenic variants (PVs) have been associated with breast cancer (BC). Although their association with LFS has been negated,6,7 the association with other cancers remains controversial.8,9,10 There is an urgent need to delineate the cancer phenotypes associated with CHEK2 PVs because they are frequently identified on cancer panel testing.11,12,13
The best-studied CHEK2 variant, c.1100del, is a loss-of-function (LOF) variant that has been well characterized in European populations.14,15,16 The cumulative risk of BC with CHEK2 c.1100del was estimated to be 37% in a meta-analysis of patients with BC.17 In the Copenhagen General Population Study,18 the c.1100del variant was associated with breast and stomach cancers and enriched in kidney cancers and sarcomas, but this was not significant after correcting for multiple comparisons. CHEK2 PVs have been associated with colorectal, kidney, prostate, and thyroid cancers,11,19,20,21,22,23,24 but these findings are limited by small sample sizes and high genetic homogeneity.
Many questions about CHEK2 and its associated cancer risks remain. In general, LOF variants tend to be PVs.25 In contrast, missense variants have more variable effects and depend on whether a critical protein domain is affected.25 A few missense variants, p.I157T, p.S428F, and p.T476M, are associated with attenuated BC risk compared with LOF PVs in CHEK2.12,26,27 Muranen et al26 compared p.I157T with c.1100del, and among patients with BC, p.I157T had a more favorable prognosis compared with those with c.1100del. Others have excluded both p.I157T and p.S428F, from analyses owing to the lower association with BC (odds ratio [OR], <1.5).12 The high population frequency of p.I157T, p.S428F, and p.T476M and the seemingly attenuated cancer risks have made it challenging to delineate CHEK2-associated cancer risks by variant type.
We aimed to compare cancer phenotypes among participants with CHEK2 PVs by variant and variant type, and to compare the cancer phenotypes of CHEK2 monoallelic and biallelic PVs.
Methods
This retrospective cohort study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guidelines and was conducted to evaluate participants with CHEK2 PVs identified by genetic testing ordered by health care professionals between July 2012 through September 2019 at a single diagnostic testing laboratory (Ambry Genetics). Individuals with a concurrent PV in CHEK2 and another gene were excluded. The CHEK2 PV cohort underwent 8 to 25 gene targeted breast/ovarian cancer panel testing (n = 2199), 49 to 67 gene panel testing (n = 1857), or testing with a customizable panel of 1 to 75 genes (n = 119). The CHEK2 wild-type (WT) cohort included 33 034 participants without any PVs on a pancancer panel (49-67 genes). Individuals with variants of uncertain significance in any gene were not excluded uniformly. The Western institutional review board provided an exemption from review and a waiver of consent for the deidentified data.
Clinical characteristics, including sex, race and ethnicity, cancer history, BC hormone receptor subtype, age at genetic testing, and first cancer diagnosis were obtained from clinician-completed requisition forms and from clinical documentation (pedigrees and clinic notes) when provided. Diverse cancer types were examined including adrenal, brain, colorectal, endometrial, gastric, kidney, melanoma, ovarian, pancreatic, prostate, thyroid, and sarcoma.
Variant interpretation was performed according to a model based on the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines.28 Pathogenic and likely pathogenic variants were both denoted as PV. CHEK2 PVs were categorized as follows: biallelic, monoallelic, LOF (includes truncating, splice site variants, initiation codons, gross deletions, and duplications), missense, and more or less frequent (using an approximate 100 participant threshold). The p.I157T, p.S428F, and p.T476M variants have conflicting interpretations between laboratories in ClinVar29,30,31,32 or functional studies have led to questions about their pathogenicity.7,12,26,32,33,34,35,36 Therefore, the BC phenotypes of these groups were first compared with the other PV and WT CHEK2 cohorts to determine how they should be categorized for subsequent analyses.10,34
Descriptive statistics for participants stratified by variant category are summarized as median (IQR) for continuous and proportions for categorical characteristics. Differences in ages were assessed with Wilcoxon rank sum test and proportions across variant categories were analyzed with Fisher exact tests. Frequencies of specific tumor types and ORs with 95% CIs were summarized by variant category. All statistical tests were 2-sided, and a P <.05 was considered statistically significant. Adjustments for multiple tests were made according to the Bonferroni method where applicable and P values of less than 0.00278 are significant. All analyses were conducted with R statistical software (version 4.0.4; R Foundation, Inc).
Results
Of 36 817 evaluable participants who underwent multigene cancer panel testing, 3783 CHEK2 PVs were identified (3734 monoallelic and 49 biallelic, Figure 1). Of the 3783 individuals carrying a PV, 3473 participants (91.8%) (95% CI, 90.9%-92.7%) were female, 2818 (74.5%) had cancer (95% CI, 73.1%-75.9%), and, of the female participants, 2202 (63.4%) (95% CI, 61.7%-65.0%) had BC. The most frequent monoallelic PV was c.1100del (n = 1252) followed by p.I157T (n = 992), p.S428F (n = 324), p.T476M (n = 250), p.R117G (n = 125), exon8_9 deletion (ex8_9del, n = 113), and c.444 + 1G>A (n = 92) (Figure 1). The ex8_9del variant is a recurrent 5395 base pair deletion that spans coding exons 8 and 9.37
Figure 1. Strengthening the Reporting of Observational Studies in Epidemiology Diagram.
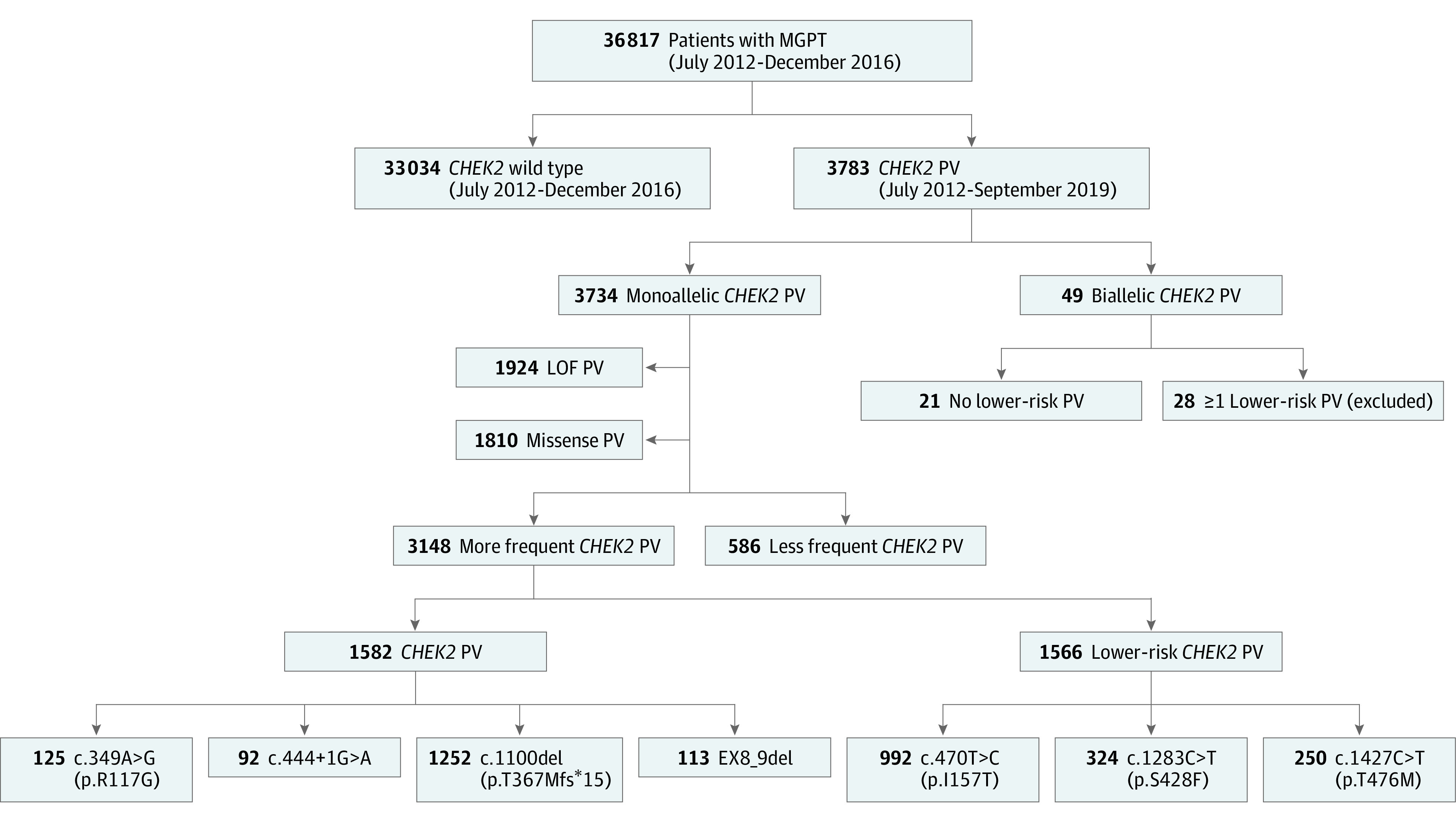
LOF indicates loss of function; MGPT, multigene cancer panel testing; PV, pathogenic or likely pathogenic variant.
Breast Cancer Among Frequent PVs
We compared the BC prevalence among participants with the 7 more frequent PVs to all other CHEK2 PVs and to CHEK2 WT to inform our classification of the proposed lower-risk variants in our cohort. The BC prevalence was highest among participants with c.444 + 1G>A (OR, 2.63; 95% CI, 1.59-4.35; P < .001), ex8_9del variants (OR, 2.36; 95% CI, 1.53-3.64; P < .001), followed by p.R117G (OR, 1.65; 95% CI, 1.12-2.44; P = .01) and c.1100del (OR, 1.76; 95% CI, 1.55-2.00; P < .001; Figure 2, eTable 8 in the Supplement) compared with WT.
Figure 2. Breast Cancer Prevalence by CHEK2 Variant.
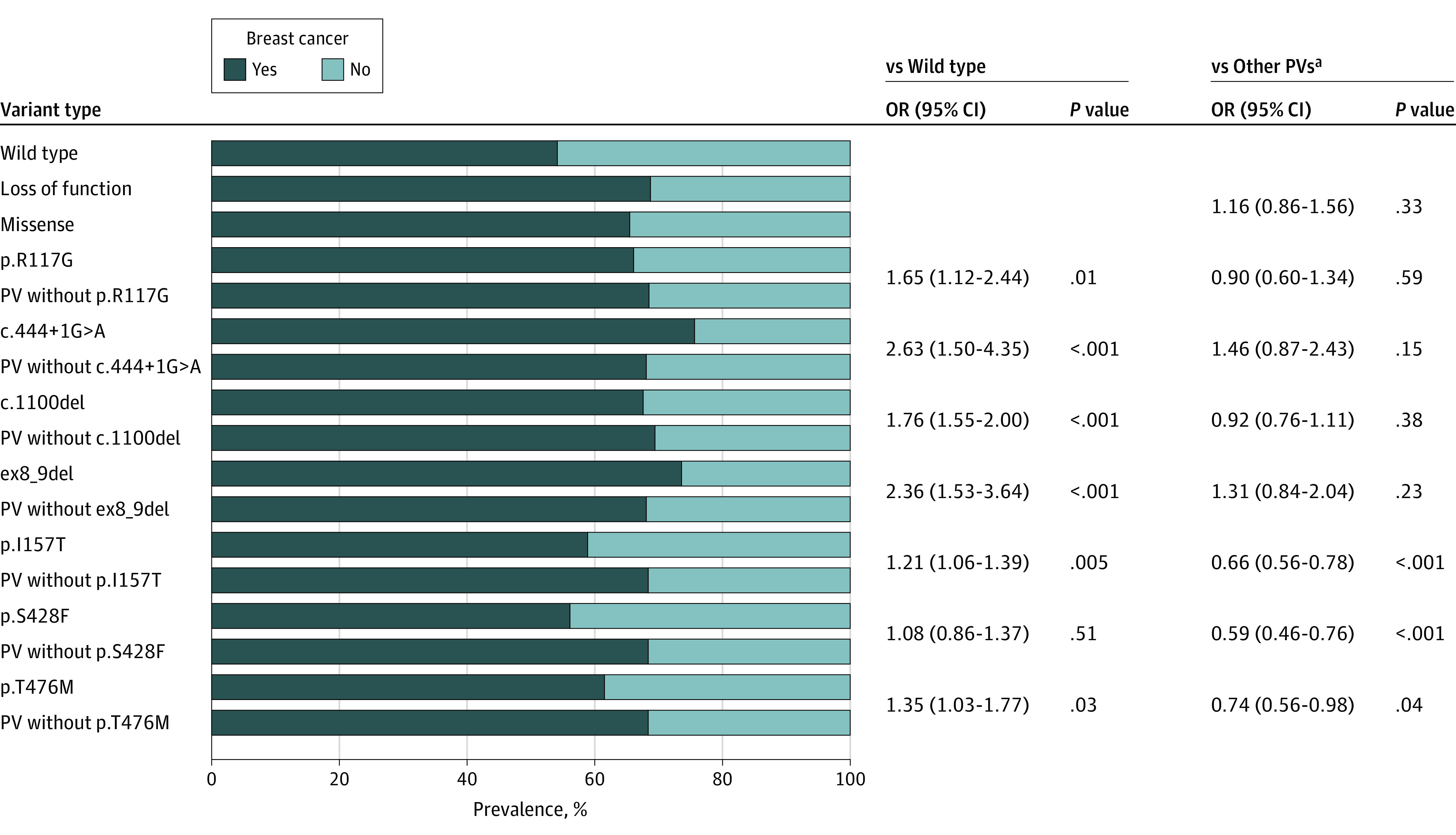
ap.I157F, p.S428F, and p.T476M were not included in other PVs. OR indicates odds ratio; PV, pathogenic or likely pathogenic variant.
The BC prevalence was lower with the p.I157T, p.S428F, and p.T476M variants compared with other PVs (P < .001, P < .001, and P = .04, respectively) and the ORs were fewer than 1.4 compared with WT (Figure 2; eTables 1-3 in the Supplement). Owing to these marked differences from other PVs, p.I157T, p.S428F and p.T476M were deemed lower risk and analyzed and reported separately.
Cancer Phenotypes of Participants With CHEK2 PVs and WT
More participants reported White race (1707 [78.7%]) among CHEK2 PVs compared with the WT cohort (21 907 [66.3%]) (Table; eTable 2 in the Supplement). Cancer characteristics were compared between the PV cohort (n = 2168 after excluding p.I157T, p.S428F, and p.T476M) and the WT cohort. Participants with PVs were more likely to be diagnosed with a cancer (1664 [76.8%]) compared with WT (23 065 [69.8%]) (OR, 1.43; 95% CI, 1.29-1.58; P < .001, Table). Median (IQR) age at first cancer diagnosis was younger among participants with PVs: 47 (40-56) vs 49 (42-58) years for WT (Table). A history of multiple primary tumors, defined as a primary cancer of more than 1 site (eg, breast and colon), was more frequent among the PV cohort than WT (OR, 1.37; 95% CI, 1.18-1.58; P < .001, Table).
Table. Cohort Characteristics and Cancer Associations Stratified by Wild-type vs Monoallelic Pathogenic Variants.
| Characteristic | No. (%) | CHEK2 monoallelic PV vs WT | ||
|---|---|---|---|---|
| WT | CHEK2 monoallelic PVa | OR (95% CI) | P value | |
| Sexb | ||||
| Female | 30 429 (92.1) | 1995 (92.0) | 0.99 (0.85-1.17) | .93 |
| Male | 2605 (7.9) | 172 (7.9) | ||
| Age, median (IQR), y | ||||
| At genetic testing | 53 (44-63) | 53 (43-62) | NA | .005 |
| At first breast cancer | 50 (43-59) | 47 (41-56) | NA | <.001 |
| At first cancer | 49 (42-58) | 47 (40-56) | NA | <.001 |
| Race and ethnicity, ancestry | ||||
| African American/Black | 1815 (5.5) | 37 (1.7) | 0.26 (0.18-0.36) | <.001 |
| Ashkenazi Jewish | 2247 (6.8) | 92 (4.2) | 0.53 (0.42-0.65) | <.001 |
| Asian | 1071 (3.2) | 20 (0.9) | 0.24 (0.15-0.37) | <.001 |
| Hispanic | 1582 (4.8) | 66 (3.0) | 0.54 (0.41-0.69) | <.001 |
| White | 21 907 (66.3) | 1707 (78.7) | 0.72 (0.62-0.82) | <.001 |
| Other | 4409 (13.4) | 246 (11.4) | 1.43 (1.29-1.58) | <.001 |
| Any cancer | ||||
| Yes | 23 065 (69.8) | 1664 (76.8) | NA | NA |
| No | 9969 (30.2) | 504 (23.3) | NA | NA |
| Primary cancers, No. | ||||
| 0 | 9969 (30.2) | 504 (23.3) | NA | <.001 |
| 1 | 18 498 (56.0) | 1348 (62.2) | NA | |
| >1 | 4567 (13.8) | 316 (14.6) | NA | |
| Breast cancer | ||||
| Yes | 16 029 (52.7) | 1339 (67.1) | 1.83 (1.66-2.02) | <.001 |
| No | 13 584 (44.6) | 620 (31.1) | ||
| Bilateral breast cancer | ||||
| Yes | 2228 (7.3) | 273 (13.7) | 1.99 (1.74-2.28) | <.001 |
| No | 27 345 (89.9) | 1683 (84.4) | ||
| First breast cancer ER/PR positivec | ||||
| Yes | 8355 (78.0) | 837 (92.8) | 3.64 (2.81-4.78) | <.001 |
| No | 2362 (22.0) | 65 (7.2) | ||
| First breast cancer ERBB2 positive | ||||
| Yes | 1492 (19.0) | 168 (25.9) | 1.49 (1.23-1.79) | <.001 |
| No | 6362 (81.0) | 482 (74.2) | ||
| Colorectal cancer | ||||
| Yes | 2575 (7.8) | 109 (5.0) | 0.62 (0.51-0.76) | <.001 |
| No | 29 550 (89.5) | 2015 (92.9) | ||
| Kidney cancer | ||||
| Yes | 214 (0.7) | 36 (1.7) | 2.57 (1.75-3.68) | <.001 |
| No | 31 896 (96.6) | 2088 (96.3) | ||
| Melanoma | ||||
| Yes | 1259 (3.8) | 60 (2.8) | 0.71 (0.54-0.93) | .01d |
| No | 30 854 (93.4) | 2064 (95.2) | ||
| Pancreatic cancer | ||||
| Yes | 597 (1.8) | 20 (0.9) | 0.5 (0.3-0.78) | <.001 |
| No | 31 517 (95.4) | 2104 (97.1) | ||
| Thyroid cancer | ||||
| Yes | 696 (2.1) | 74 (3.4) | 1.63 (1.26-2.08) | <.001 |
| No | 31 416 (95.1) | 2050 (94.6) | ||
Abbreviations: ER, estrogen receptor; ERBB2, formerly HER2 (human epidermal growth factor receptor 2); NA, not applicable; PR, progesterone receptor; PV, pathogenic or likely pathogenic variant; WT, wild type.
Excluding the lower risk CHEK2 variants: p.I157T, p.S428F, and p.T476M.
Sex was not available for 1 participant.
ER/PR positive breast cancer included tumors that were estrogen receptor positive and/or progesterone receptor positive.
After Bonferroni correction, this P value was not significant.
The CHEK2 PV cohort was more likely to have a personal history of BC (n = 1339, 67.1%) than WT (n = 16 029, 52.7%) with an OR of 1.83 (95% CI, 1.66-2.02; P < .001). The median (IQR) age at first BC diagnosis was slightly younger among the PV cohort (47 [41-56] years) compared with WT (50 [43-59] years, Table). Other significant differences in BC were noted among the PV cohort vs the WT cohort, with the former including more bilateral BCs (OR, 1.99; 95% CI, 1.74-2.28; P < .001), more estrogen and/or progesterone receptor (ER/PR)-positive BCs (OR, 3.64; 95% CI, 2.81-4.78; P < .001), and more ERBB2 (formerly HER2)-positive BC (OR, 1.49; 95% CI, 1.23-1.79; P < .001; Table 1).
Nonbreast Cancers Among Participants With CHEK2 PVs and WT
In addition to BC, the CHEK2 PV cohort had more kidney (OR, 2.57; 95% CI, 1.75-3.68; P < .001) and thyroid cancers (OR, 1.63; 95% CI, 1.26-2.08; P < .001) compared with WT (Table 1). The PV cohort had less colorectal cancer (CRC) than those in the WT group, 5.0% vs 7.8% (OR, 0.62; 95% CI, 0.51-0.76; P < .001; Table 1).
Both endometrial cancer (2.1% vs 4.2%, P < .001) and pancreatic cancer (0.9% vs 1.8%, P = .001) were less frequent in the PV cohort than those in the WT cohort (Table 1; eTable 4 in the Supplement). The frequencies of brain, gastric, melanoma, sarcoma, prostate, and ovarian cancer were not different between the 2 cohorts (Table 1; eTable 4 in the Supplement).
Frequent CHEK2 PVs and Cancer Phenotype
The individual frequent PVs, p.R117G, c.444 + 1G>A, c.1100del, and ex8_9del were compared with the other CHEK2 PVs and WT by cancer type. Participant BC frequencies were overlapping for different CHEK2 PVs and higher than those in the WT cohort: p.R117G (OR, 1.65; 95% CI, 1.1-2.44; P = .01; not significant after correcting for multiple comparisons), c.444 + 1G>A (OR, 2.63; 95% CI, 1.59-4.35; P < .001), c.1100del (OR, 1.76; 95% CI, 1.55-2.00; P < .001), ex8_9del (OR, 2.36, 95% CI, 1.53-3.64; P < .001; eTable 8 in the Supplement). Bilateral BCs were associated with the p.R117G (2.20; 95% CI, 1.31-3.69; P = .002) and c.1100del (OR, 1.97; 95% CI, 1.65-2.34; P<.001) PVs compared with those in the WT group (eTable 8 in the Supplement).
Compared with WT, the c.1100del PV was associated with kidney (OR, 3.11; 95% CI, 1.96-4.74; P < .001) and thyroid cancer (OR, 1.68; 95% CI, 1.21-2.3; P = .002; eTable 8 in the Supplement). In contrast, CRC (OR, 0.69; 95% CI, 0.53-0.88; P = .002; eTable 8 in the Supplement) and endometrial cancer (OR, 0.46; 95% CI, 0.29-0.69; P < .001; eTable 5 in the Supplement) were less frequent in participants with CHEK2 c.1100del than those in the WT cohort. Other examined cancers (adrenal, brain, gastric, sarcoma, prostate, and ovarian cancer) were not associated with CHEK2 PV (eTable 5 in the Supplement).
Biallelic CHEK2, LOF, Missense, and c.1100del
Comparisons were conducted for biallelic (n = 21) vs monoallelic (n = 2168) (eTable 6 in the Supplement); LOF (n = 1924) vs missense (n = 244) variants (eTable 7 in the Supplement); and c.1100del (n = 1252) vs other PVs (n = 916) for any cancers and BC (eTable 8 in the Supplement, Figure 3).
Figure 3. Box Plot of Any Cancer and Breast Cancer by CHEK2 Pathogenic Variant Type and Age at Diagnosis.
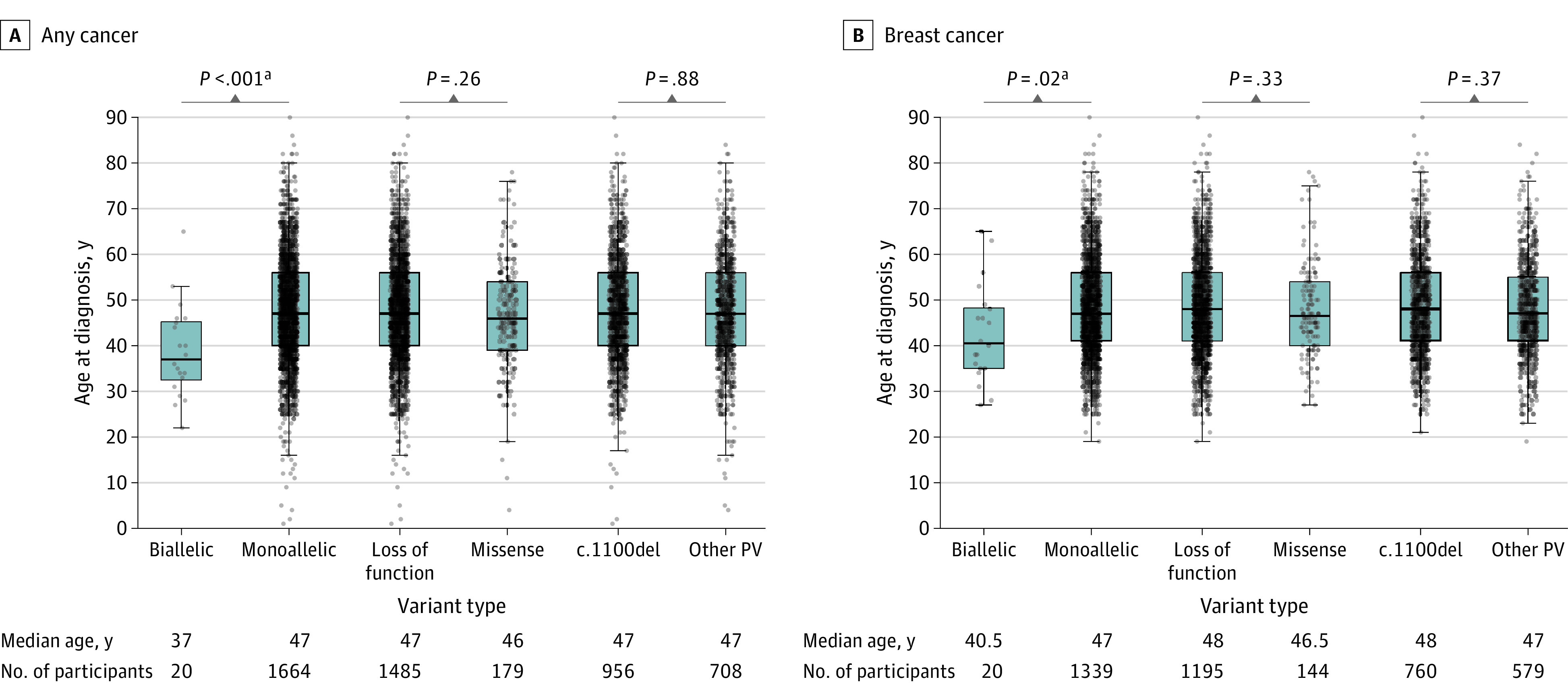
Only affected participants are included. For example, of the 21 participants with biallelic CHEK2 PVs, 20 were known to have cancer. PV indicates pathogenic or likely pathogenic variant; y, years.
aStatistically significant findings.
The biallelic CHEK2 PV cohort had a younger age at any cancer diagnosis (median age, 37 years vs 47 years; P < .001) and at BC diagnosis (median age, 40 years vs 47 years; P = .02) compared with the monoallelic PV cohort (Figure 3; eTable 6 in the Supplement). Breast cancer was more frequent in the biallelic cohort compared to the WT group and this was statistically significant (P < .001).
There were no significant differences in age at cancer diagnosis between LOF vs missense variants and c.1100del vs other PVs for both any cancer types including BC (Figure 3; eTables 7 and 8 in the Supplement).
Frequent Lower-Risk CHEK2 Variants
The lower-risk variants, p.I157T, p.S428F, and p.T476M were examined separately by comparing cancer characteristics of each group with the WT and the PV cohorts (eTables 1-3 in the Supplement).
There were no significant differences in the frequencies of any cancer including BC between the p.I157T and WT cohorts (eTable 1 in the Supplement). The frequency of bilateral BC (OR, 1.52; 95% CI, 1.23-1.89; P < .001) was higher with p.I157T than those in the WT cohort. However, when compared with the other PVs, the p.I157T cohort had a significantly lower frequency of BC (OR, 0.66; 95% CI, 0.56-0.78; P < .001; eTable 1 in the Supplement). When comparing p.I157T with WT and with other PVs, there were no differences in the frequencies of any cancer diagnosis including colorectal, kidney, and thyroid cancer.
Compared with WT, the p.S428F cohort was no different in the frequencies of any cancer diagnosis, BC, and bilateral BCs (eTable 2 in the Supplement). Any cancer (OR, 0.50; 95% CI, 0.39-0.65; P < .001), BC (OR, 0.59; 95% CI, 0.46-0.76; P < .001), and bilateral BC (OR, 0.44; 95% CI, 0.27-0.44; P < .001) were less frequent in the p.S428F cohort compared with other PVs.
The frequency of BC was higher with p.T476M compared with WT (OR, 1.35; 95% CI, 1.03-1.77; P = .03), but lower compared with other PVs (OR, 0.74; 95% CI, 0.56-0.98; P = .04) (eTable 3 in the Supplement), though neither was significant when correcting for multiple comparisons. There were no significant differences for any cancer, bilateral BC, CRC, kidney, and thyroid cancers between p.T476M, other CHEK2 PVs, and WT (eTable 3 in the Supplement).
Discussion
Germline CHEK2 PVs are frequently identified on cancer panel testing and are enriched in female participants with BC.38,39 Except for the frequent lower-risk variants (p.I157T, p.T476M, and p.S428F), we found that most CHEK2 PVs, whether missense or LOF, were associated with similar cancer phenotypes. This has important implications for counseling and care of individuals with lower-risk variants because they accounted for 41.9% of the total CHEK2 monoallelic PV cohort. There were no differences in BC prevalence or age at onset when comparing LOF vs missense CHEK2 PVs.
Collectively, CHEK2 PVs were associated with BC (OR, 1.83), ER/PR-positive BC (OR, 3.60), ERBB2 protein–positive BC (OR, 1.50), and bilateral BC (OR, 2.00) compared with WT. These findings confirm previous associations of CHEK2 c.1100del with BC17,40,41 and bilateral BC.42 As in other studies, we found CHEK2 PVs to be associated with ER/PR-positive and ERBB2 protein–positive BC.12,25,40,42,43,44
Currently, patients with CHEK2 PVs initiate BC screening at age 40 with magnetic resonance imaging (MRI) and mammogram, and earlier if there is a family history of early-onset BC.8 A recent comparative modeling study45 proposed annual breast MRI beginning at age 30 to 35 years with the addition of mammogram at age 40 years for women with CHEK2 PVs. We found the median (IQR) age of first BC diagnosis was 47 (41-50) years, which suggests screening prior to 35 years may be of limited utility.
Biallelic CHEK2 PVs were associated with higher rates of BC and earlier age of BC (median age, 40 years) compared with monoallelic PVs (median age, 47 years), a finding consistent with Rainville et al.46 As compared with monoallelic PVs, biallelic CHEK2 PVs were also associated with an even earlier age of any cancer by a decade (median age, 37 vs 47 years for monoallelic PVs). This finding is noteworthy and supports even earlier screening for patients with biallelic CHEK2 PVs.
CHEK2 PVs were associated with multiple primary (P < .001), kidney (OR, 2.6; P < .001) and thyroid cancers (OR, 1.6; P < .001) compared with WT. An association with kidney cancer was previously suggested for c.1100del (hazard ratio, 3.61; P = .01), although not statistically significant after Bonferroni correction.18 In the present study, the association remains significant even when correcting for multiple comparisons, likely due to the larger sample size. This association is further supported by studies of patients with kidney cancers in which CHEK2 PVs were overrepresented and found in 2%-3.5% of patients.47,48 More data on the prognosis of CHEK2-associated kidney cancers including modeling and cost-effectiveness studies are needed to inform screening.
An association with thyroid cancer was suggested by Naslund-Koch et al,18 though it, too, was not significant after Bonferroni correction. Here, we found a persistent association supporting the work of Kamihara et al, who found CHEK2 PVs were enriched among patients with thyroid cancer.22 As above, more data are needed on the aggressiveness of CHEK2-associated thyroid cancers to inform surveillance.
CHEK2 PVs were not associated with colorectal cancer compared with WT (OR, 0.62). These results are consistent with a meta-analysis49 on CHEK2 and CRC, and the Copenhagen study,18 where the control group was population-based and negative for c.1100del. In totality, these findings suggest intensive CRC screening may not be indicated and present guidelines for the care of patients with CHEK2 PVs should be refined.
Consistent with prior literature, the p.I157T and p.S428F variants were less frequently associated with BC compared with other CHEK2 PVs.12,25,50 When corrected for multiple comparisons, BC frequency with p.T476M was not different from other PVs or from WT. Moreover, p.I157T, p.T476M, and p.S428F were associated with lower rates of non-BCs compared with WT. Classification of these variants is inconsistent,29,30,31,32,51 and in a previous study of bioinformaticists and clinical geneticists, Agaoglu et al52 showed that the p.T476M alteration was classified as a variant of unknown significance, likely pathogenic or pathogenic by different interpreters. Although BC frequency with the p.I157T variant was not significantly different from WT, the frequency of bilateral BCs was (OR, 1.52; P < .001), indicating this variant may act in concert with other genetic modifiers influencing BC risk.
In 2019 the American Society of Breast Surgeons recommended all women with BC consider germline genetic testing.53 As testing expands, more patients will be identified with the frequent, lower-risk CHEK2 PVs (p.I157T, p.S428F, and p.T447M), hastening the need for health care professionals to be knowledgeable and to counsel patients about CHEK2 cancer associations (or lack thereof) prior to making BC surgical decisions.
Limitations
Limitations of this study include a homogeneous (predominately White female participants) study population, most with a history of BC. Recognizing that men are less likely to undergo genetic testing,54,55,56,57,58,59 there are ascertainment biases inherent in a cohort selected for genetic testing for cancer predisposition. Ordinarily, these ascertainment biases would lead to overestimations in the ORs for cancers. However, our WT cohort was subject to the same testing bias, which may lower ORs for cancer for CHEK2 PVs. Therefore, these data were not used to estimate cancer risk by genotype as this cohort was not population based. Conversely, some tumor associations may not be apparent, such as those previously reported for cancers of the colorectum and prostate, owing to this bias. In a recent phenome-wide association study,60 CHEK2 was associated with leukemia and plasma cell neoplasms; however, this was not evaluable in our study because peripheral blood is not the optimal specimen for germline testing of patients with hematologic malignant diseases. We did not evaluate the frequency of nonmelanoma skin cancer despite evidence for an association.61 Single-nucleotide variation testing and polygenic risk scores were unavailable to further refine genotype and analyze phenotype. The large number of participants with the lower-risk alleles, p.I157T, p.S428F, and p.T476M, allowed us to examine these separately. However, it is possible that some less frequent alleles included in the PV category may also be associated with attenuated phenotypes.
These data provide keen insights into genotype-phenotype associations for CHEK2. CHEK2 PVs were associated with a younger age at any cancer diagnosis, with multiple primary cancers; ER/PR-positive, ERBB2 protein–positive BC, bilateral BC, and kidney and thyroid cancers. Other previously reported associations, including CRC, were not identified. This large study discerns cancer phenotype by genotype. Genetic modifiers affect CHEK2 penetrance and in the future will likely aid in cancer risk stratification of patients with CHEK2 PVs.62,63,64 Although additional studies in population-based cohorts are needed, these data help to refine cancer associations of CHEK2 PVs and lower-risk alleles.
Conclusions
In this cohort study CHEK2 PVs, except the low-penetrance alleles of p.I157T, p.S428F, and p.T476M, were associated with similar cancer phenotypes irrespective of variant type (missense or LOF). Associations with ERBB2-positive BC, kidney, and thyroid cancers were noted while associations with CRC and other cancers were not found. These data may inform genetic counseling and risk assessment of patients with CHEK2 PVs.
eTable 1. Comparison of CHEK2 p.I157T to WT and CHEK2 monoallelic pathogenic variants
eTable 2. Comparison of CHEK2 p.S428F to WT and CHEK2 monoallelic pathogenic variants
eTable 3. Comparison of CHEK2 p.T476M to WT and CHEK2 monoallelic pathogenic variants
eTable 4. Cancer Associations by WT vs. CHEK2 Monoallelic pathogenic variants
eTable 5. Cancer Phenotype of Individual CHEK2 PVs: R117G, c.1100del, ex8-9del, and c.444+1G>A compared to Wildtype and Other CHEK2 PVs
eTable 6. Cohort Characteristics and Cancer Associations comparing CHEK2 Monoallelic PV versus Biallelic PV
eTable 7. Cohort Characteristics and Cancer Associations of LOF versus missense CHEK2 pathogenic variants.
eTable 8. Cancer Phenotype of Individual CHEK2 PVs: R117G, c.1100del, ex8-9del, and c.444 + 1G>A compared With Wildtype (WT) and Other CHEK2 PVs
References
- 1.Lukas C, Bartkova J, Latella L, et al. DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res. 2001;61(13):4990-4993. [PubMed] [Google Scholar]
- 2.Hirao A, Kong YY, Matsuoka S, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287(5459):1824-1827. doi: 10.1126/science.287.5459.1824 [DOI] [PubMed] [Google Scholar]
- 3.Bartek J, Falck J, Lukas J. CHK2 kinase—a busy messenger. Nat Rev Mol Cell Biol. 2001;2(12):877-886. doi: 10.1038/35103059 [DOI] [PubMed] [Google Scholar]
- 4.Cai Z, Chehab NH, Pavletich NP. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Mol Cell. 2009;35(6):818-829. doi: 10.1016/j.molcel.2009.09.007 [DOI] [PubMed] [Google Scholar]
- 5.Bell DW, Varley JM, Szydlo TE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286(5449):2528-2531. doi: 10.1126/science.286.5449.2528 [DOI] [PubMed] [Google Scholar]
- 6.Batalini F, Peacock EG, Stobie L, et al. Li-Fraumeni syndrome: not a straightforward diagnosis anymore-the interpretation of pathogenic variants of low allele frequency and the differences between germline PVs, mosaicism, and clonal hematopoiesis. Breast Cancer Res. 2019;21(1):107. doi: 10.1186/s13058-019-1193-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SB, Kim SH, Bell DW, et al. Destabilization of CHK2 by a missense mutation associated with Li-Fraumeni Syndrome. Cancer Res. 2001;61(22):8062-8067. [PubMed] [Google Scholar]
- 8.National Comprehensive Cancer Network (NCCN) . Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 1.2022. Accessed February 14, 2022. www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
- 9.National Comprehensive Cancer Network (NCCN) . Genetic/Familial High-Risk Assessment: Colorectal. Version 1.2021. Accessed February 14, 2022. www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf
- 10.Roeb W, Higgins J, King MC. Response to DNA damage of CHEK2 missense mutations in familial breast cancer. Hum Mol Genet. 2012;21(12):2738-2744. doi: 10.1093/hmg/dds101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leedom TP, LaDuca H, McFarland R, Li S, Dolinsky JS, Chao EC. Breast cancer risk is similar for CHEK2 founder and non-founder mutation carriers. Cancer Genet. 2016;209(9):403-407. doi: 10.1016/j.cancergen.2016.08.005 [DOI] [PubMed] [Google Scholar]
- 12.Hu C, Hart SN, Gnanaolivu R, et al. A population-based study of genes previously implicated in breast cancer. N Engl J Med. 2021;384(5):440-451. doi: 10.1056/NEJMoa2005936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neben CL, Zimmer AD, Stedden W, et al. Multi-gene panel testing of 23,179 individuals for hereditary cancer risk identifies pathogenic variant carriers missed by current genetic testing guidelines. J Mol Diagn. 2019;21(4):646-657. doi: 10.1016/j.jmoldx.2019.03.001 [DOI] [PubMed] [Google Scholar]
- 14.Vahteristo P, Bartkova J, Eerola H, et al. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet. 2002;71(2):432-438. doi: 10.1086/341943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meijers-Heijboer H, Wijnen J, Vasen H, et al. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am J Hum Genet. 2003;72(5):1308-1314. doi: 10.1086/375121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.CHEK2 Breast Cancer Case-Control Consortium . CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet. 2004;74(6):1175-1182. doi: 10.1086/421251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weischer M, Bojesen SE, Ellervik C, Tybjaerg-Hansen A, Nordestgaard BG. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. J Clin Oncol. 2008;26(4):542-548. doi: 10.1200/JCO.2007.12.5922 [DOI] [PubMed] [Google Scholar]
- 18.Näslund-Koch C, Nordestgaard BG, Bojesen SE. Increased risk for other cancers in addition to breast cancer for CHEK2*1100delC heterozygotes estimated from the Copenhagen General Population Study. J Clin Oncol. 2016;34(11):1208-1216. doi: 10.1200/JCO.2015.63.3594 [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Dai B, Ye D. CHEK2 mutation and risk of prostate cancer: a systematic review and meta-analysis. Int J Clin Exp Med. 2015;8(9):15708-15715. [PMC free article] [PubMed] [Google Scholar]
- 20.Cybulski C, Górski B, Huzarski T, et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet. 2004;75(6):1131-1135. doi: 10.1086/426403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siołek M, Cybulski C, Gąsior-Perczak D, et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int J Cancer. 2015;137(3):548-552. doi: 10.1002/ijc.29426 [DOI] [PubMed] [Google Scholar]
- 22.Kamihara J, Zhou J, LaDuca H, et al. Germline pathogenic variants in cancer risk genes among patients with thyroid cancer and suspected predisposition. Cancer Med. 2022;11(8):1745-1752. doi: 10.1002/cam4.4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truong H, Sheikh R, Kotecha R, et al. Germline variants identified in patients with early-onset renal cell carcinoma referred for germline genetic testing. Eur Urol Oncol. 2021;4(6):993-1000. doi: 10.1016/j.euo.2021.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cybulski C, Wokołorczyk D, Kładny J, et al. Germline CHEK2 mutations and colorectal cancer risk: different effects of a missense and truncating mutations? Eur J Hum Genet. 2007;15(2):237-241. doi: 10.1038/sj.ejhg.5201734 [DOI] [PubMed] [Google Scholar]
- 25.Dorling L, Carvalho S, Allen J, et al. ; Breast Cancer Association Consortium . Breast cancer risk genes—association analysis in more than 113,000 women. N Engl J Med. 2021;384(5):428-439. doi: 10.1056/NEJMoa1913948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muranen TA, Blomqvist C, Dörk T, et al. Patient survival and tumor characteristics associated with CHEK2:p.I157T—findings from the Breast Cancer Association Consortium. Breast Cancer Res. 2016;18(1):98. doi: 10.1186/s13058-016-0758-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akcay IM, Celik E, Agaoglu NB, et al. Germline pathogenic variant spectrum in 25 cancer susceptibility genes in Turkish breast and colorectal cancer patients and elderly controls. Int J Cancer. 2021;148(2):285-295. doi: 10.1002/ijc.33199 [DOI] [PubMed] [Google Scholar]
- 28.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ClinVar . NM_007194.4(CHEK2):c.1427C>T (p.Thr476Met). Accessed March 3, 2022. www.ncbi.nlm.nih.gov/clinvar/variation/128060/?new_evidence=false
- 30.ClinVar . NM_007194.4(CHEK2):c.1283C>T (p.Ser428Phe). Accessed March 3, 2022. www.ncbi.nlm.nih.gov/clinvar/variation/5603/?new_evidence=false
- 31.ClinVar . NM_007194.4(CHEK2):c.470T>C (p.Ile157Thr). Accessed March 3, 2022. https://www.ncbi.nlm.nih.gov/clinvar/variation/5591/?new_evidence=false
- 32.Mundt E, Nix P, Brown K, Bowles KR, Manley S. Complexities of variant classification in clinical hereditary cancer genetic testing. J Clin Oncol. 2017;35(34):3796-3799. doi: 10.1200/JCO.2017.74.5182 [DOI] [PubMed] [Google Scholar]
- 33.Shaag A, Walsh T, Renbaum P, et al. Functional and genomic approaches reveal an ancient CHEK2 allele associated with breast cancer in the Ashkenazi Jewish population. Hum Mol Genet. 2005;14(4):555-563. doi: 10.1093/hmg/ddi052 [DOI] [PubMed] [Google Scholar]
- 34.Kleiblova P, Stolarova L, Krizova K, et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int J Cancer. 2019;145(7):1782-1797. doi: 10.1002/ijc.32385 [DOI] [PubMed] [Google Scholar]
- 35.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877-885. doi: 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boonen RACM, Wiegant WW, Celosse N, et al. Functional analysis identifies damaging CHEK2 missense variants associated with increased cancer risk. Cancer Res. 2022;82(4):615-631. doi: 10.1158/0008-5472.CAN-21-1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cybulski C, Wokołorczyk D, Huzarski T, et al. A large germline deletion in the Chek2 kinase gene is associated with an increased risk of prostate cancer. J Med Genet. 2006;43(11):863-866. doi: 10.1136/jmg.2006.044974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dominguez-Valentin M, Nakken S, Tubeuf H, et al. Results of multigene panel testing in familial cancer cases without genetic cause demonstrated by single gene testing. Sci Rep. 2019;9(1):18555. doi: 10.1038/s41598-019-54517-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurian AW, Ward KC, Howlader N, et al. Genetic testing and results in a population-based cohort of breast cancer patients and ovarian cancer patients. J Clin Oncol. 2019;37(15):1305-1315. doi: 10.1200/JCO.18.01854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weischer M, Nordestgaard BG, Pharoah P, et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer-specific death, and increased risk of a second breast cancer. J Clin Oncol. 2012;30(35):4308-4316. doi: 10.1200/JCO.2012.42.7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y, Zhang F, Wang Y, Liu SC. CHEK2 1100delC variant and breast cancer risk in Caucasians: a meta-analysis based on 25 studies with 29,154 cases and 37,064 controls. Asian Pac J Cancer Prev. 2012;13(7):3501-3505. doi: 10.7314/APJCP.2012.13.7.3501 [DOI] [PubMed] [Google Scholar]
- 42.Kriege M, Hollestelle A, Jager A, et al. Survival and contralateral breast cancer in CHEK2 1100delC breast cancer patients: impact of adjuvant chemotherapy. Br J Cancer. 2014;111(5):1004-1013. doi: 10.1038/bjc.2014.306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt MK, Hogervorst F, van Hien R, et al. Age- and tumor subtype-specific breast cancer risk estimates for CHEK2*1100delC Carriers. J Clin Oncol. 2016;34(23):2750-2760. doi: 10.1200/JCO.2016.66.5844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mavaddat N, Dorling L, Carvalho S, et al. ; Breast Cancer Association Consortium . Pathology of tumors associated with pathogenic germline variants in 9 breast cancer susceptibility genes. JAMA Oncol. 2022;8(3):e216744. doi: 10.1001/jamaoncol.2021.6744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lowry KP, Geuzinge HA, Stout NK, et al. ; Breast Working Group of the Cancer Intervention and Surveillance Modeling Network (CISNET), in collaboration with the Breast Cancer Surveillance Consortium (BCSC), and the Cancer Risk Estimates Related to Susceptibility (CARRIERS) Consortium . Breast cancer screening strategies for women with ATM, CHEK2, and PALB2 pathogenic variants: a comparative modeling analysis. JAMA Oncol. 2022;8(4):587-596. doi: 10.1001/jamaoncol.2021.6204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rainville I, Hatcher S, Rosenthal E, et al. High risk of breast cancer in women with biallelic pathogenic variants in CHEK2. Breast Cancer Res Treat. 2020;180(2):503-509. doi: 10.1007/s10549-020-05543-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carlo MI, Mukherjee S, Mandelker D, et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol. 2018;4(9):1228-1235. doi: 10.1001/jamaoncol.2018.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yngvadottir B, Andreou A, Bassaganyas L, et al. Frequency of pathogenic germline variants in cancer susceptibility genes in 1336 renal cell carcinoma cases. Hum Mol Genet. 2022;ddac089. doi: 10.1093/hmg/ddac089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiang HP, Geng XP, Ge WW, Li H. Meta-analysis of CHEK2 1100delC variant and colorectal cancer susceptibility. Eur J Cancer. 2011;47(17):2546-2551. doi: 10.1016/j.ejca.2011.03.025 [DOI] [PubMed] [Google Scholar]
- 50.Liu C, Wang Y, Wang QS, Wang YJ. The CHEK2 I157T variant and breast cancer susceptibility: a systematic review and meta-analysis. Asian Pac J Cancer Prev. 2012;13(4):1355-1360. doi: 10.7314/APJCP.2012.13.4.1355 [DOI] [PubMed] [Google Scholar]
- 51.Ivanov M, Sharova M, Olsen A, et al. Letter to the Editor: CHEK2 I157T—pluto among numerous low-risk genetic factors requiring discharge from a range of pathogenic variants? J Natl Compr Canc Netw. 2022;20(2):xxv. doi: 10.6004/jnccn.2021.7103 [DOI] [PubMed] [Google Scholar]
- 52.Agaoglu NB, Unal B, Akgun Dogan O, et al. Consistency of variant interpretations among bioinformaticians and clinical geneticists in hereditary cancer panels. Eur J Hum Genet. 2022;30(3):378-383. doi: 10.1038/s41431-022-01060-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The American Society of Breast Surgeons . Consensus Guideline on Genetic Testing for Hereditary Breast Cancer. Approved on Feb 10, 2019. Accessed April 22, 2022. www.breastsurgeons.org/docs/statements/Consensus-Guideline-on-Genetic-Testing-for-Hereditary-Breast-Cancer.pdf
- 54.Collins V, Halliday J, Warren R, Williamson R. Assessment of education and counselling offered by a familial colorectal cancer clinic. Clin Genet. 2000;57(1):48-55. doi: 10.1034/j.1399-0004.2000.570107.x [DOI] [PubMed] [Google Scholar]
- 55.Holloway SM, Bernhard B, Campbell H, Cetnarskyj R, Lam WW. Inequality of use of cancer genetics services by members of breast, ovarian and colorectal cancer families in South East Scotland. Fam Cancer. 2008;7(3):259-264. doi: 10.1007/s10689-008-9184-x [DOI] [PubMed] [Google Scholar]
- 56.Taylor S. Gender differences in attitudes among those at risk for Huntington’s disease. Genet Test. 2005;9(2):152-157. doi: 10.1089/gte.2005.9.152 [DOI] [PubMed] [Google Scholar]
- 57.Srinivasan S, Won NY, Dotson WD, Wright ST, Roberts MC. Barriers and facilitators for cascade testing in genetic conditions: a systematic review. Eur J Hum Genet. 2020;28(12):1631-1644. doi: 10.1038/s41431-020-00725-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanz J, Ramón y Cajal T, Torres A, et al. Uptake of predictive testing among relatives of BRCA1 and BRCA2 families: a multicenter study in northeastern Spain. Fam Cancer. 2010;9(3):297-304. doi: 10.1007/s10689-009-9313-1 [DOI] [PubMed] [Google Scholar]
- 59.Jeong GW, Shin W, Lee DO, et al. Uptake of family-specific mutation genetic testing among relatives of patients with ovarian cancer with BRCA1 or BRCA2 mutation. Cancer Res Treat. 2021;53(1):207-211. doi: 10.4143/crt.2020.364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeng C, Bastarache LA, Tao R, et al. Association of pathogenic variants in hereditary cancer genes with multiple diseases. JAMA Oncol. 2022;8(6):835-844. doi: 10.1001/jamaoncol.2022.0373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bui AN, LeBoeuf NR, Nambudiri VE. Skin cancer risk in CHEK2 mutation carriers. J Eur Acad Dermatol Venereol. 2021;35(2):353-359. doi: 10.1111/jdv.16729 [DOI] [PubMed] [Google Scholar]
- 62.Gallagher S, Hughes E, Kurian AW, et al. Comprehensive breast cancer risk assessment for CHEK2 and ATM pathogenic variant carriers incorporating a polygenic risk score and the Tyrer-Cuzick model. JCO Precis Oncol. 2021;5:PO.20.00484. doi: 10.1200/PO.20.00484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muranen TA, Greco D, Blomqvist C, et al. ; NBCS Investigators; kConFab/AOCS Investigators . Genetic modifiers of CHEK2*1100delC-associated breast cancer risk. Genet Med. 2017;19(5):599-603. doi: 10.1038/gim.2016.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wendt C, Muranen TA, Mielikäinen L, et al. A search for modifying genetic factors in CHEK2:c.1100delC breast cancer patients. Sci Rep. 2021;11(1):14763. doi: 10.1038/s41598-021-93926-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Comparison of CHEK2 p.I157T to WT and CHEK2 monoallelic pathogenic variants
eTable 2. Comparison of CHEK2 p.S428F to WT and CHEK2 monoallelic pathogenic variants
eTable 3. Comparison of CHEK2 p.T476M to WT and CHEK2 monoallelic pathogenic variants
eTable 4. Cancer Associations by WT vs. CHEK2 Monoallelic pathogenic variants
eTable 5. Cancer Phenotype of Individual CHEK2 PVs: R117G, c.1100del, ex8-9del, and c.444+1G>A compared to Wildtype and Other CHEK2 PVs
eTable 6. Cohort Characteristics and Cancer Associations comparing CHEK2 Monoallelic PV versus Biallelic PV
eTable 7. Cohort Characteristics and Cancer Associations of LOF versus missense CHEK2 pathogenic variants.
eTable 8. Cancer Phenotype of Individual CHEK2 PVs: R117G, c.1100del, ex8-9del, and c.444 + 1G>A compared With Wildtype (WT) and Other CHEK2 PVs