

Abstract

Herein, we report the first catalytic one-step synthesis of cyclopropenium cations (CPCs) with readily available alkynes and hypervalent iodine reagents as carbyne sources. Key to the process is the catalytic generation of a novel Rh-carbynoid that formally transfers monovalent cationic carbynes (:+C-R) to alkynes via an oxidative [2+1] cycloaddition. Our process is able to synthesize a new type of CPC substituted with an ester group that underpins the regioselective attack of a broad range of carbon and heteroatomic nucleophiles, thus providing a new platform for the synthesis of valuable cyclopropenes difficult or not possible to make by current methodologies.
Cyclopropenium cations (CPCs), discovered by Prof. Ronald Breslow in the late 1950s,1−3 are the smallest member of the Hückel aromatic systems. These aromatic cations with two π-electrons delocalized over three 2p orbitals are known to have considerable thermodynamic stability and molecular strain (Figure 1A). The highly stable tris(dialkylamino)CPC derivatives have recently found applications as electrophotocatalysts,4 gene delivery agents,5 catholytes for nonaqueous redox batteries,6 or liquid crystals.7 However, CPCs have been largely underappreciated in organic synthesis, despite their potential as three-carbon building blocks in the construction of complex architectures.
Figure 1.

Catalytic synthesis of cyclopropenium cations.
One of the main reasons is that the majority of synthetic strategies toward CPCs, developed between the 1950s and 1980s, rely on multistep sequences and show limitations in efficiency. Those methods require the synthesis of cyclopropene derivatives that lead to the CPC upon (pseudo)halide, nitrile, carbonyl, or hydride abstraction with strong Lewis/Brønsted acids.1,3,8 Alternative approaches based on functionalizations of alkynes with chlorocarbenes generated from chlorodiazirines under UV light irradiation9 or with a cationic metal-carbyne [(η5-C5H5)(CO)2Mn≡CPh]+ show very limited scope (only two examples are described in the latter case).10 In addition to this, nonsymmetrical CPCs are likely to react with poor regioselectivity in nucleophilic events. In fact, pioneering work by Padwa showed that reactions between CPCs and Grignard reagents provided mixtures of regioisomers.11
Herein, we would like to disclose the invention of a one-step Rh-catalyzed process for the preparing of CPCs that combines readily available alkynes and hypervalent iodine reagents as formal cationic carbyne sources (Figure 1B).12 The process showed a broad scope of a new class of CPCs substituted with an ester group that, upon treatment with a diverse range of nucleophiles, provided access to valuable and elusive classes of cyclopropene derivatives.13
Our research group is focused on the development of a carbyne transfer platform in organic synthesis using tailored hypervalent iodine reagents14 as carbyne synthons.15 The catalytic activation with dirhodium carboxylate catalysts16 provides access to Rh-carbynoids (int-1) (Figure 2),15b,15d a novel class of Rh-carbene species substituted with an ester group and a hypervalent iodine moiety as outstanding nucleofuge.17 Such species have the ability to emulate the carbene/carbocation behavior of a monovalent cationic carbyne (:+C–R) and provide a transient cyclopropyl-I(III) intermediate upon alkene cyclopropanation. Recently, we questioned whether we could exploit our Rh-catalyzed carbyne transfer platform for the discovery and development of the first one-step and catalytic synthesis of cyclopropenium cations (Figure 2). We hypothesized that the catalytically generated Rh-carbynoid (int-1) could intercept alkynes and provide cyclopropenyl-I(III) intermediates (int-2) that, upon an ionization process that occurs with the departure of the I(III) leaving group, would lead to a new class of CPC substituted with an ester group.18
Figure 2.

Mechanistic hypothesis.
With this mechanistic proposal in mind, we evaluated the feasibility of this idea using 1-phenyl-1-propyne, a broad range of commercial Rh2 catalysts, and diazomethyl-based hypervalent iodine reagents 2 (see Supporting Information for full optimization studies). Taking into account the possible instability and reactivity of the desired CPC, we employed 1,3,5-trimethoxybenzene as external nucleophile to quantify the efficiency of the transformation. To our delight, after extensive optimization studies, we were able to find that pseudocyclic reagent 2a and the Du Bois catalyst19 Rh2(esp)2 (1 mol %) led to cyclopropene 3a* with excellent efficiency (94% yield, >20:1 r.r.) (Table 1). The only regioisomer observed comes from the selective attack of the nucleophile to the cyclopropenium carbon atom substituted with the ester group.
Table 1. Optimization Studiesa,b.

| entry | deviation of reaction conditions | yieldb3a*, % |
|---|---|---|
| 1 | Rh2(OPiv)4 instead of Rh2(esp)2 | 93 |
| 2 | Rh2(OAc)4 instead of Rh2(esp)2 | 4 |
| 3 | Rh2(TFA)4 instead of Rh2(esp)2 | 0 |
| 4 | 2b instead of 2a | 50 |
| 5 | 2c,d instead of 2a | 0 |
| 6 | 2d used with Zn(OTf)2 instead of 2a | 60c |
| 7 | reaction carried out at –50 °C | 81 |
Reactions performed with alkyne 1a (0.2 mmol, 2.0 equiv), reagents 2 (0.1 mmol, 1 equiv), Rh catalyst (1 mol %), and 1,3,5-trimethoxybenzene (4 equiv) in CH2Cl2 (0.067 M).
Yields are reported on the basis of 1H NMR analysis using CH2Br2 as the internal standard.
0.5 equiv of Zn(OTf)2 was used.
During the optimization of the reaction, we observed that other sterically demanding catalysts worked well (Table 1, entry 1); however, Rh2(OAc)4 or the more electrophilic Rh2(TFA)4 was not competent (entries 2, 3). The nature of the hypervalent iodine reagent played also a key role in the efficiency of the transformation. The use of triflate as counterion provided a moderate yield of 3a* (entry 4). However, although full consumption of linear reagent 2c was observed, no conversion to the desired product was detected (entry 5). On the other hand, cyclic reagent 2d was inert to catalytic diazo activation with Rh2(esp)2 (entry 5). Only upon addition of Zn(OTf)2 as the additive, a well-known activator of cyclic I(III) reagents,14 was a moderate yield of 3a* observed (entry 6). We also found that higher reaction temperatures could give product 3a* but with lower efficiency (entry 7).

During the optimization process, we observed that the addition of 2a to 1a and Rh2(esp)2 at −60 °C provided the formation of a slurry, which quickly turned into a clear solution after the addition of 1,3,5-trimethoxybenzene. We believed that this observation suggested the potential formation of either intermediate int-2 or CPC 3a. Initial experiments performed to isolate the intermediate at room temperature were unsuccessful; however, slow addition of dry hexane at −60 °C and quick filtration provided a white solid, which was subjected to spectroscopic analysis using mono- and bidimensional nuclear magnetic resonance (1H, 13C, 19F, 31P, 1H–13C NMR, CD3NO2 as solvent), IR, and HRMS. The three deshielded signals at 171.5, 166.7, and 156.0 ppm observed in the 13C NMR spectra clearly referred to the cyclopropenium carbon atoms, thus confirming the formation of 3a (86% yield) (Table 2). 3a could be stored at −20 °C under argon for at least 1 month without detectable decomposition and can be handled at room temperature without the need of a glovebox.
Table 2. Scope of the Catalytic Synthesis of Cyclopropenium Cations 3a.
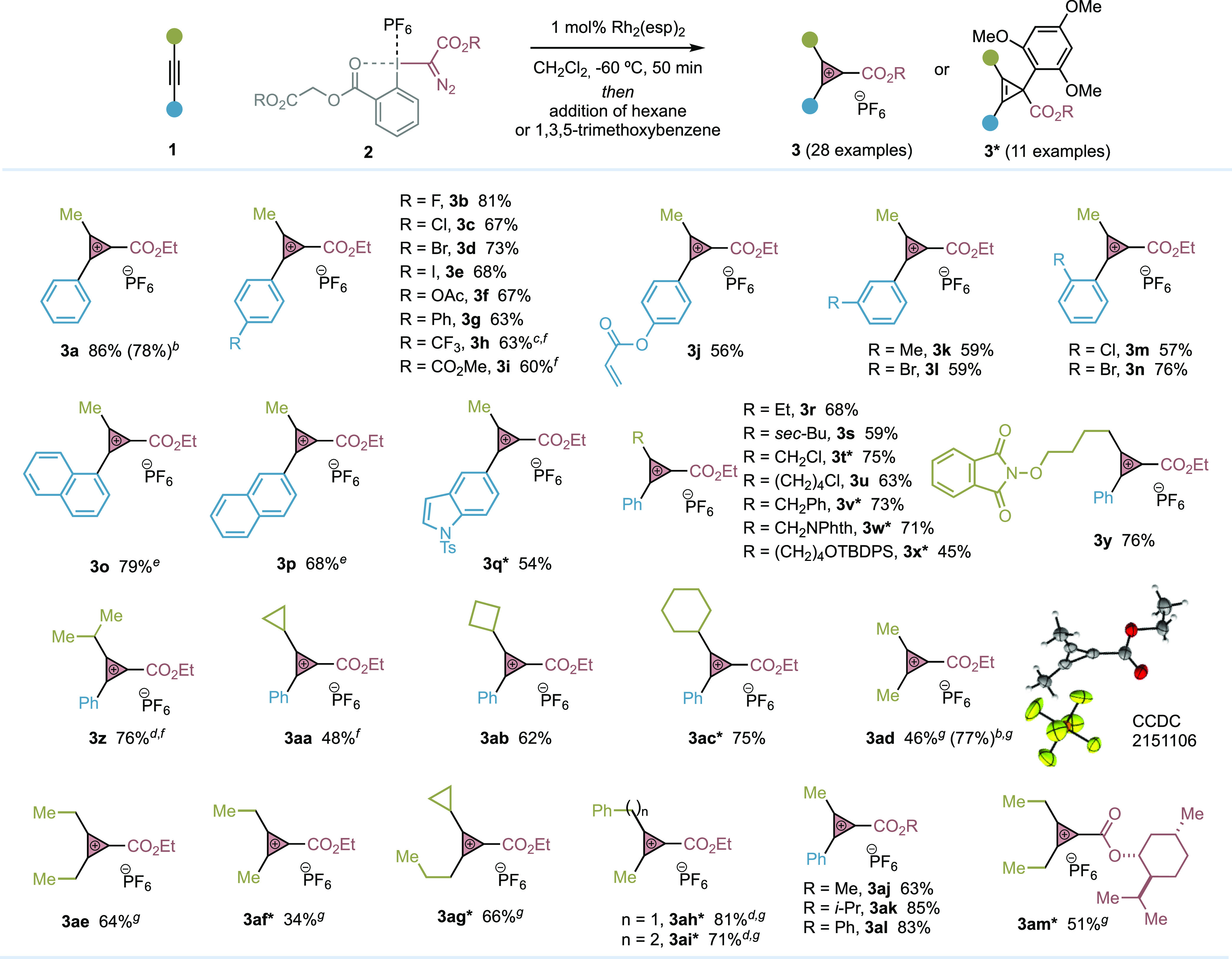
Performed with alkyne 1 (0.26 mmol, 1.3 equiv), hypervalent iodine reagents 2 (0.2 mmol, 1.0 equiv), CH2Cl2, −60 °C, 50 min, 1,3,5-trimethoxybenzene (0.8 mmol, 4.0 equiv) was added for the synthesis of 3*.
Yield of the reaction to give 1.08 g of 3a and 1.10 g of 3ad.
1.5 mol % catalyst.
2 mol % catalyst.
Reaction carried out at −63 °C.
1.5 equiv of alkyne.
2.0 equiv of alkyne.
Encouraged by the results, we next aimed to develop the scope of our catalytic protocol for the synthesis of CPCs 3 by examining a wide range of alkynes (Table 2). In case the CPC was difficult to isolate, it was transformed to the corresponding cyclopropene 3* with 1,3,5-trimethoxybenzene. We were delighted to observe that our process worked well for alkynes with aryl rings substituted with synthetically useful functionalities such as halogens (3b–e, 3l–n), acetoxy (3f), phenyl (3g), CF3 (3h), ester (3i), alkene (3j), and methyl (3k) in para, meta, and ortho positions as well as naphthalene (3o,p) and heterocycle cores (3q*).
Alternative primary alkyl groups such as ethyl (3r), sec-butyl (3s), chloroalkyls (3t*, 3u), benzyl (3v*), and alkyl groups functionalized with protected amines (3w*) or alcohols (3x*, 3y) were well tolerated. Secondary alkyl substituents like isopropyl required an increment on the catalyst loading (2 mol %) and alkyne (1.5 equiv) (3z); however, cyclic derivatives such as cyclopropyl (3aa), cyclobutyl (3ab), and cyclohexyl (3ac*) worked well under the standard conditions. Alkynes substituted with tertiary groups like tert-butyl or trimethylsilyl provided traces of the desired CPCs. On the other hand, while alkynes substituted with two alkyl groups were well tolerated (3ad–ai*), diphenylacetylene was unreactive and terminal alkynes such as phenylacetylene provided mixtures of products difficult to identify.20 We also demonstrated that alternative ester substituents at the hypervalent iodine reagents were possible (3aj–al, 3am*). Finally, it is worth highlighting that CPC 3a and 3ad were prepared in >1 g without compromising the efficiency of the process.
We next turned our attention to evaluate the reactivity of our cyclopropenium cations with a broad range of carbon and heteroatomic nucleophiles aiming to develop a novel synthetic route to cyclopropenes (Table 3). These highly strained, three-membered unsaturated carbocycles are well known for their unique potential as versatile building blocks in organic synthesis that can undergo nucleophilic or electrophilic additions, substitutions, rearrangements, cycloadditions, and ring-opening reactions, delivering pharmaceutical-relevant scaffolds like cyclopropanes or complex structures,21 but also as biorthogonal reagents for chemical biology applications.22
Table 3. Synthesis of Tri- and Tetrasubstituted Cyclopropenes by Nucleophilic Attacka.
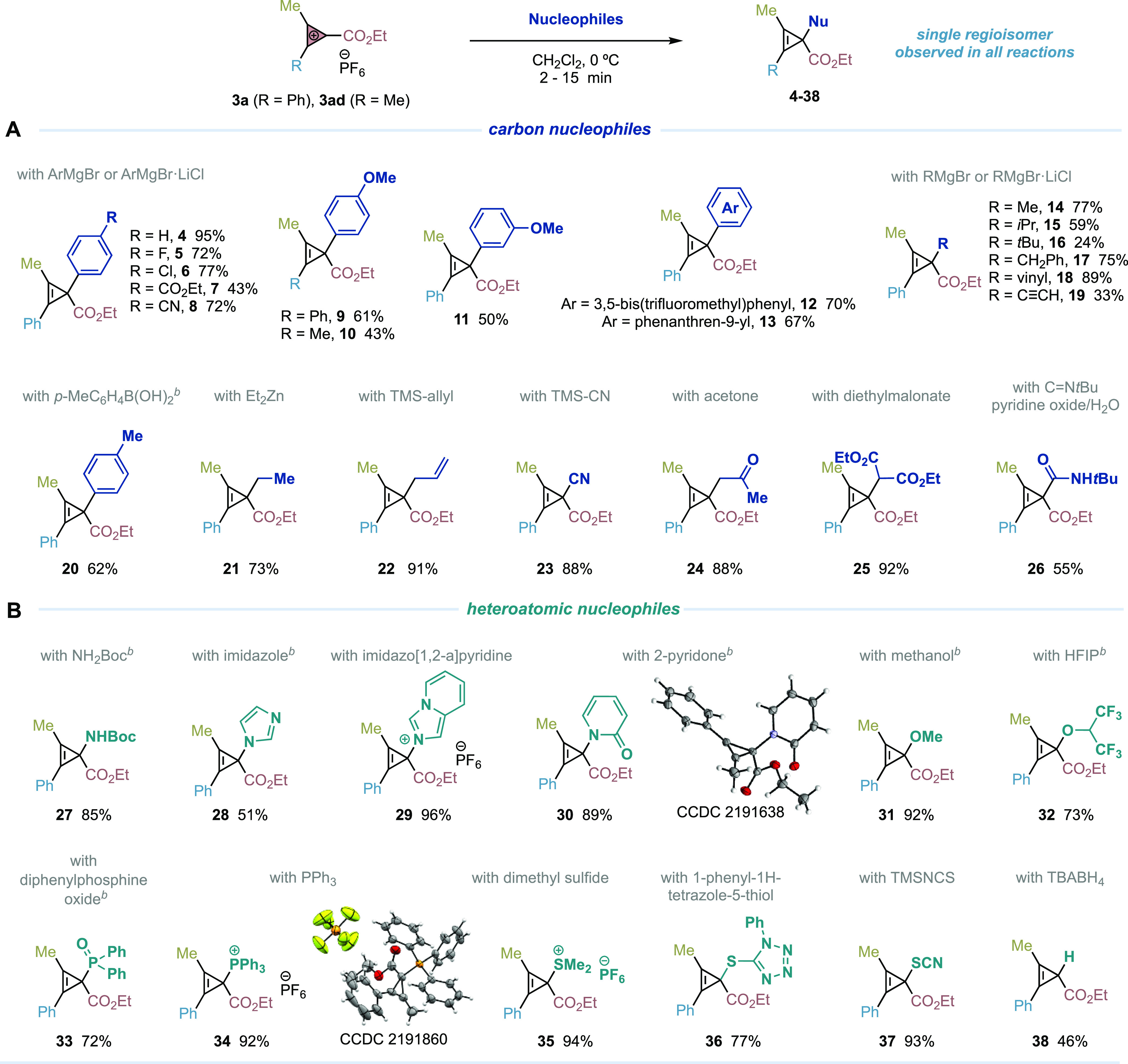
Performed with 3 (0.1 mmol), carbon or heteroatomic nucleophiles (0.15–0.2 mmol), CH2Cl2, 0 °C, 2–15 min.
Cs2CO3 (0.1 mmol) was added as base. Yields are reported on the basis of isolated pure product.
Considering that the nucleophilic attack of 1,3,5-trimethoxybenzene to 3a proceeds with outstanding regioselectivity, we hoped that alternative nucleophiles could behave analogously. We were delighted to observe that a variety of commercial or readily available aryl, alkyl, vinyl, and alkynyl Grignard reagents provided instantaneous access to cyclopropenes 4–19 as single regioisomers and in high efficiency with simple reaction conditions (CH2Cl2, 0 °C, 2–15 min). Moreover, alternative nucleophiles such as boronic acid (20), organozinc (21), organosilicon (22, 23), carbonyl (24, 25) and isocyanide nucleophiles (26) performed well, and in many cases, no chromatographic column was needed to obtain the corresponding cyclopropene product.
It is worth highlighting that our methodology with cyclopropenium cations provides a new approach to the synthesis of complex cyclopropenes23 and solutions to challenges observed in metal-catalyzed carbene transfer with diazo acetates to internal alkynes. Catalytically generated metal-carbenes substituted with alkyl or allyl groups undergo faster β-hydride migration24 or intramolecular cyclopropanation,25 respectively, before the intermolecular cyclopropenation of the internal alkyne takes place.
The remarkable promiscuity observed of our CPCs to react with carbon nucleophiles encouraged us to question whether heteroatomic nucleophiles could work. If successful, such reactions would provide access to a type of tetrasubstituted cyclopropenes not possible to make by any reaction currently available, because of the lack of heteroatom-substituted diazoacetates or alternative carbene sources.26 We were delighted to observe that a selection of commercial nitrogen, oxygen, phosphorus, and sulfur nucleophiles provided cyclopropenes 27–37 with high efficiency. In addition, a reaction carried out with tetrabutylammonium borohydride provided trisubstituted cyclopropene derivative 38 by the regioselective hydride attack to 3a.27,28
Notably, all kinds of heteroatomic nucleophiles underwent regioselective attack to the cyclopropenium carbon atom substituted with the ester group. The reactions provide access to a plethora of novel cyclopropene derivatives with unknown reactivity, which promise applications in reaction discovery and in the construction of complex skeletons.21
Finally, in order to provide an explanation of the outstanding and intriguing regiocontrol observed in the nucleophilic addition to CPCs 3, we calculated the geometry optimization and LUMO map using SPARTAN 20 at the ωb97xd/6-31G(d) level. In Figure 3, it can clearly be appreciated that the carbon atom substituted with the ester group has the highest LUMO coefficient among the cyclopropenium carbon sites, thus suggesting that nucleophilic attack occurs under orbital control.
Figure 3.
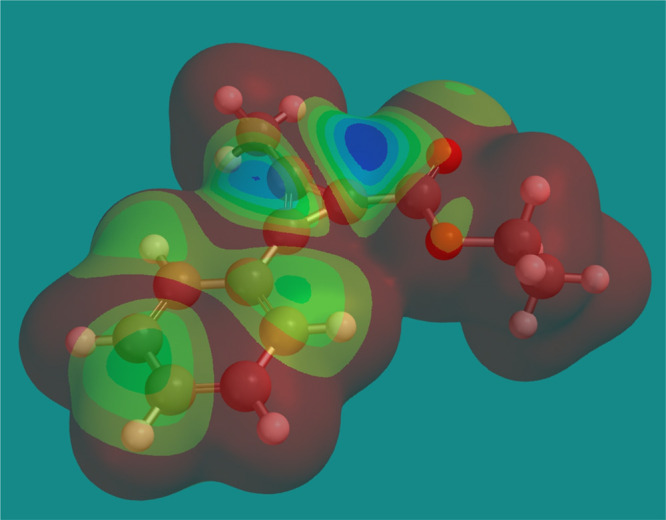
LUMO map of 3a (method: ωB97X-D, basis set: 6-31G(D)).
In summary, we have discovered and developed the first catalytic synthesis of cyclopropenium cations based on the formal transfer of monovalent cationic carbynes (:+C–R) to readily available alkynes from a catalytically generated Rh-carbynoid. This type of group transfer process is uncommon for metal-carbyne complexes, which mainly evolve via [2+2] cycloadditions with alkynes.29,10 Our process accesses previously unknown ester-substituted CPCs, which can be handled outside of a glovebox, from a broad range of internal aryl- and alkyl-substituted alkynes. The synthetic utility of our CPCs has been demonstrated by the regioselective attack of a broad range of carbon and heteroatomic nucleophiles that provided valuable cyclopropenes. Several of those compounds cannot be made by current approaches due to the lack of appropriate diazoacetate reagents as carbene sources or limitations in current methodologies. Current studies are focused on exploiting novel cyclopropenium reactivity for asymmetric synthesis and other applications.30
Acknowledgments
European Research Council (ERC-CoG 2019, 865554), Agencia Estatal de Investigación (AEI) of the Ministerio de Ciencia e Innovación (EUR2019-103814, PID2019-104101GB-I00, Severo Ochoa Excellence Accreditation 2020–2023 -CEX2019-000925-S), ICIQ Foundation, and the CERCA Programme (Generalitat de Catalunya) are gratefully acknowledged for financial support. We thank the European Union for a Marie Skłodowska-Curie Individual Fellowship (101032589).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c07769.
Experimental procedures and spectral data (PDF)
The authors declare the following competing financial interest(s): A patent application has been filed through the Fundaci Institut Catal D'Investigaci Qumica (ICIQ) based on the results presented here.
Supplementary Material
References
- Breslow R. Synthesis of the s-Triphenylcyclopropenyl Cation. J. Am. Chem. Soc. 1957, 79, 5318. 10.1021/ja01576a067. [DOI] [Google Scholar]
- Komatsu K.; Kitagawa T. Cyclopropenylium Cations, Cyclopropenones, and Heteroanalogues - Recent Advances. Chem. Rev. 2003, 103, 1371–1427. 10.1021/cr010011q. [DOI] [PubMed] [Google Scholar]
- a Breslow R.; Yuan C. The sym-Triphenylcyclopropenyl Cation, a Novel Aromatic System. J. Am. Chem. Soc. 1958, 80, 5991–5994. 10.1021/ja01555a026. [DOI] [Google Scholar]; b Breslow R.; Hover H.; Chang H. W. The Synthesis and Stability of Some Cyclopropenyl Cations with Alkyl Substituents. J. Am. Chem. Soc. 1962, 84, 3168–3174. 10.1021/ja00875a027. [DOI] [Google Scholar]; c Sundaralingam M.; Jensen L. H. The Structure of a Carbonium Ion. Refinement of the Crystal and Molecular Structure of sym-Triphenylcyclopropenium Perchlorate. J. Am. Chem. Soc. 1966, 88, 198–204. 10.1021/ja00954a002. [DOI] [Google Scholar]; d Breslow R.; Groves J. T.; Ryan G. Cyclopropenyl Cation. J. Am. Chem. Soc. 1967, 89, 5048. 10.1021/ja00995a042. [DOI] [PubMed] [Google Scholar]
- a Huang H.; Strater Z. M.; Lambert T. H. Electrophotocatalytic C–H Functionalization of Ethers with High Regioselectivity. J. Am. Chem. Soc. 2020, 142, 1698–1703. 10.1021/jacs.9b11472. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shen T.; Lambert T. H. Electrophotocatalytic Diamination of Vicinal C–H Bonds. Science 2021, 371, 620–626. 10.1126/science.abf2798. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Huang H.; Lambert T. H. Electrophotocatalytic C–H Heterofunctionalization of Arenes. Angew. Chem., Int. Ed. 2021, 60, 11163–11167. 10.1002/anie.202100222. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Huang H.; Lambert T. H. Electrophotocatalytic Acetoxyhydroxylation of Aryl Olefins. J. Am. Chem. Soc. 2021, 143, 7247–7252. 10.1021/jacs.1c01967. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Shen T.; Lambert T. H. C–H Amination via Electrophotocatalytic Ritter-type Reaction. J. Am. Chem. Soc. 2021, 143, 8597–8602. 10.1021/jacs.1c03718. [DOI] [PMC free article] [PubMed] [Google Scholar]; For other type of catalysis:; f Bandar J. S.; Tanaset A.; Lambert T. H. Phase-Transfer and Other Types of Catalysis with Cyclopropenium Ions. Chem.—Eur. J. 2015, 21, 7365–7368. 10.1002/chem.201500124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucks S. D.; Freyer J. L.; Lambert T. H.; Campos L. M. Influence of Substituent Chain Branching on the Transfection Efficacy of Cyclopropenium-Based Polymers. Polymers 2017, 9, 79. 10.3390/polym9030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sevov C. S.; Samaroo S. K.; Sanford M. S. Cyclopropenium Salts as Cyclable, High-Potential Catholytes in Nonaqueous Media. Adv. Energy Mater. 2017, 7, 1602027. 10.1002/aenm.201602027. [DOI] [Google Scholar]; b Ji W.; Huang H.; Huang X.; Zhang X.; Zheng D.; Ding T.; Chen J.; Lambert T. H.; Qu D. A Redox-active Organic Cation for Safer High Energy Density Li-ion Batteries. J. Mater. Chem. A 2020, 8, 17156–17162. 10.1039/D0TA06133F. [DOI] [Google Scholar]; c Yan Y.; Vaid T. P.; Sanford M. S. Bis(diisopropylamino)cyclopropenium-arene Cations as High Oxidation Potential and High Stability Catholytes for Non-aqueous Redox Flow Batteries. J. Am. Chem. Soc. 2020, 142, 17564–17571. 10.1021/jacs.0c07464. [DOI] [PubMed] [Google Scholar]; d Yan Y.; Vogt D. B.; Vaid T. P.; Sigman M. S.; Sanford M. S. Development of High Energy Density Diaminocyclopropenium-Phenothiazine Hybrid Catholytes for Non-Aqueous Redox Flow Batteries. Angew. Chem., Int. Ed. 2021, 60, 27039–27045. 10.1002/anie.202111939. [DOI] [PubMed] [Google Scholar]
- a Freyer J. L.; Brucks S. D.; Gobieski G. S.; Russell S. T.; Yozwiak C. E.; Sun M.; Chen Z.; Jiang Y.; Bandar J. S.; Stockwell B. R.; Lambert T. H.; Campos L. M. Clickable Poly(ionic liquids): A Materials Platform for Transfection. Angew. Chem., Int. Ed. 2016, 55, 12382–12386. 10.1002/anie.201605214. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Litterscheidt J.; Judge P.; Bühlmeyer A.; Bader K.; Bandar J. S.; Lambert T. H.; Laschat S. When Size Matters: Exploring the Potential of Aminocyclopropenium Cations as Head Groups in Triphenylene-derived Ionic Liquid Crystals in Comparison with Guanidinium and Ammonium Units. Liq. Cryst. 2018, 45, 1250–1258. 10.1080/02678292.2018.1427811. [DOI] [Google Scholar]
- a Tobey S. W.; West R. The Trichlorocyclopropenium Ion. J. Am. Chem. Soc. 1964, 86, 1459. 10.1021/ja01061a055. [DOI] [Google Scholar]; b West R.; Sadô A.; Tobey S. W. Synthesis of Trihalocyclopropenium Salts and Normal Coordinate Analysis of C3Cl3+. J. Am. Chem. Soc. 1966, 88, 2488–2494. 10.1021/ja00963a024. [DOI] [Google Scholar]; c Yoshida Z.; Tawara Y. Aminocyclopropenium Ion. J. Am. Chem. Soc. 1971, 93, 2573–2574. 10.1021/ja00739a057. [DOI] [Google Scholar]; d Buchholz H. A.; Prakash G. K. S.; Deffieux D.; Olah G. A. Electrochemical Preparation of Tris(tert-butyldimethylsilyl)cyclopropene and Its Hydride Abstraction to Tris(tert-butyldimethylsilyl)cyclopropenium tetrafluoroborate. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 10003–10005. 10.1073/pnas.96.18.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gilbertson R. D.; Weakley T. J. R.; Haley M. M. Preparation, X-ray Crystal Structures, and Reactivity of Alkynylcyclopropenylium Salts. J. Org. Chem. 2000, 65, 1422–1430. 10.1021/jo9915372. [DOI] [PubMed] [Google Scholar]; f Lavallo V.; Canac Y.; Donnadieu B.; Schoeller W. W.; Bertrand G. Cyclopropenylidenes: From Interstellar Space to an Isolated Derivative in the Laboratory. Science 2006, 312, 722–724. 10.1126/science.1126675. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Riu M.-L. Y.; Jones R. L.; Transue W. J.; Müller P.; Cummins C. C. Isolation of an Elusive Phosphatetrahedrane. Sci. Adv. 2020, 6, eaaz3168 10.1126/sciadv.aaz3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Komatsu K.; Tomioka I.; Okamoto K. The Tricyclopropylcyclopropenium Ion. Tetrahedron Lett. 1980, 21, 947–950. 10.1016/S0040-4039(00)77747-1. [DOI] [Google Scholar]; b Moss R. A.; Shen S.; Krogh-Jespersen K.; Potenza J. A.; Schugar H. J.; Munjal R. C. Cyclopropyl/Phenylcyclopropenyl Cations: Studies in Stabilization. J. Am. Chem. Soc. 1986, 108, 134–140. 10.1021/ja00261a022. [DOI] [Google Scholar]
- Fischer H.; Troll C. Transfer of the Carbyne Ligand to the C≡C Bond versus Alkyne–Carbyne–CO Coupling in the Reactions of [(η5-C5H5)(CO)2Mn≡CPh]+ with Electron-Rich Alkynes. J. Chem. Soc., Chem. Commun. 1994, 457–458. 10.1039/C39940000457. [DOI] [Google Scholar]
- a Padwa A.; Blacklock T. J.; Chou C. S.; Hatanaka N. Photochemical Transformations of Small Ring Compounds. 107. Studies Dealing with the Intramolecular Hydrogen Atom Transfer Reaction of Tetrasubstituted Cyclopropenes. J. Am. Chem. Soc. 1979, 101, 5743–5759. 10.1021/ja00513a047. [DOI] [Google Scholar]; b Padwa A.; Rieker W. F.; Rosenthal R. J. Synthesis of the Benzotricyclo[3.2.0.02,7]heptene Ring System via the Intramolecular [2 + 2]-Cycloaddition Reaction of Some Cyclopropene Derivatives. J. Am. Chem. Soc. 1983, 105, 4446–4456. 10.1021/ja00351a051. [DOI] [Google Scholar]; c Padwa A.; Rieker W. F.; Rosenthal R. J. Intramolecular Ring Closure Reactions of Cyclopropene Derivatives as a Method for Synthesizing Novel Tricyclic Ring Compounds. J. Org. Chem. 1984, 49, 1353–1360. 10.1021/jo00182a008. [DOI] [Google Scholar]; d Padwa A.; Rieker W. F.; Rosenthal R. J. Studies Dealing with the Intramolecular Ene Reaction of Cyclopropene Derivatives. J. Am. Chem. Soc. 1985, 107, 1710–1717. 10.1021/ja00292a042. [DOI] [Google Scholar]; e Padwa A.; Goldstein S. I.; Rosenthal R. J. Single-Electron-Transfer Pathway in the Coupling of Cyclopropenyl Cations with Organometallic Reagents. J. Org. Chem. 1987, 52, 3278–3285. 10.1021/jo00391a018. [DOI] [Google Scholar]
- a For literature on free carbyne generation:DoMinh T.; Gunning H. E.; Strausz O. P. The Formation and Reactions of Monovalent Carbon Intermediates. I. Photolysis of Diethyl Mercurybisdiazoacetate. J. Am. Chem. Soc. 1967, 89, 6785–6787. 10.1021/ja01001a084. [DOI] [Google Scholar]; b Strausz O. P.; DoMinh T.; Font J. The Formation and Reactions of Monovalent Carbon Intermediates. II. Further Studies on the Decomposition of Diethyl Mercurybisdiazoacetate. J. Am. Chem. Soc. 1968, 90, 1930–1931. 10.1021/ja01009a063. [DOI] [Google Scholar]; c Strausz O. P.; Kennepohl G. J. A.; Garneau F. X.; DoMinh T.; Kim B.; Valenty S.; Skell P. S. The Formation and Reactions of Monovalent Carbon Intermediates. III. The Reaction of Carbethoxymethyne with Olefins. J. Am. Chem. Soc. 1974, 96, 5723–5732. 10.1021/ja00825a007. [DOI] [Google Scholar]; d Patrick T. B.; Kovitch G. H. Photolysis of Diethyl Mercurybisdiazoacetate and Ethyl Diazoacetate in Chloroalkanes. J. Org. Chem. 1975, 40, 1527–1528. 10.1021/jo00898a043. [DOI] [Google Scholar]; e Patrick T. B.; Wu T.-T. Photodecomposition of Diethyl Mercurybis(Diazoacetate) in Several Heterocyclic Systems. J. Org. Chem. 1978, 43, 1506–1509. 10.1021/jo00402a004. [DOI] [Google Scholar]; f Bino A.; Ardon M.; Shirman E. Formation of a Carbon-Carbon Triple Bond by Coupling Reactions In Aqueous Solution. Science 2005, 308, 234–235. 10.1126/science.1109965. [DOI] [PubMed] [Google Scholar]; g Bejot R.; He A.; Falck J. R.; Mioskowski C. Chromium–Carbyne Complexes: Intermediates for Organic Synthesis. Angew. Chem., Int. Ed. 2007, 46, 1719–1722. 10.1002/anie.200604015. [DOI] [PubMed] [Google Scholar]; h Bogoslavsky B.; Levy O.; Kotlyar A.; Salem M.; Gelman F.; Bino A. Do Carbyne Radicals Really Exist in Aqueous Solution?. Angew. Chem., Int. Ed. 2012, 51, 90–94. 10.1002/anie.201103652. [DOI] [PubMed] [Google Scholar]; i Levy O.; Bino A. Metal Ions Do Not Play a Direct Role in the Formation of Carbon-Carbon Triple Bonds during Reduction of Trihaloalkyls by CrII or VII. Chem. Eur. J. 2012, 18, 15944–15947. 10.1002/chem.201202918. [DOI] [PubMed] [Google Scholar]; j Zeng T.; Wang H.; Lu Y.; Xie Y.; Wang H.; Schaefer H. F.; Ananth N.; Hoffmann R. Tuning Spin-States of Carbynes and Silylynes: A Long Jump with One Leg. J. Am. Chem. Soc. 2014, 136, 13388–13398. 10.1021/ja5073993. [DOI] [PubMed] [Google Scholar]
- In a recent work reported by Wang and co-workers, a cyclopropenium intermediate was proposed to explain the formation of alkenyl ketones generated from a photoredox coupling of alkynes, α-diazo sulfonium triflate, and water:Wang X.; Tong W.-Y.; Huang B.; Cao S.; Li Y.; Jiao J.; Huang H.; Yi Q.; Qu S.; Wang X. Convergent Synthesis of 1,4-Dicarbonyl Z-Alkenes through Three-Component Coupling of Alkynes, α-Diazo Sulfonium Triflate, and Water. J. Am. Chem. Soc. 2022, 144, 4952–4965. 10.1021/jacs.1c12874. [DOI] [PubMed] [Google Scholar]
- a For reviews in hypervalent iodine chemistry:Zhdankin V. V.; Stang P. J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yoshimura A.; Zhdankin V. V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. 10.1021/acs.chemrev.5b00547. [DOI] [PubMed] [Google Scholar]; c Li Y.; Hari D. P.; Vita M. V.; Waser J. Cyclic Hypervalent Iodine Reagents for Atom-Transfer Reactions: Beyond Trifluoromethylation. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. 10.1002/anie.201509073. [DOI] [PubMed] [Google Scholar]; d Zhao R.; Shi L. Reactions between Diazo Compounds and Hypervalent Iodine(III) Reagents. Angew. Chem., Int. Ed. 2020, 59, 12282–12292. 10.1002/anie.202003081. [DOI] [PubMed] [Google Scholar]
- a Wang Z.; Herraiz A. G.; del Hoyo A. M.; Suero M. G. Generating Carbyne Equivalents with Photoredox Catalysis. Nature 2018, 554, 86–91. 10.1038/nature25185. [DOI] [PubMed] [Google Scholar]; b Wang Z.; Jiang L.; Sarró P.; Suero M. G. Catalytic Cleavage of C(sp2)–C(sp2) Bonds with Rh-Carbynoids. J. Am. Chem. Soc. 2019, 141, 15509–15514. 10.1021/jacs.9b08632. [DOI] [PubMed] [Google Scholar]; c Jiang L.; Wang Z.; Armstrong M.; Suero M. G. β-Diazocarbonyl Compounds: Synthesis and their Rh(II)-catalyzed 1,3 C–H Insertions. Angew. Chem., Int. 2021, 60, 6177–6184. 10.1002/anie.202015077. [DOI] [PubMed] [Google Scholar]; d Jiang L.; Sarró P.; Teo W. J.; Llop J.; Suero M. G. Catalytic Alkene Skeletal Modification for the Construction of Fluorinated Tertiary Stereocenters. Chem. Sci. 2022, 13, 4327–4333. 10.1039/D2SC00968D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Doyle M. P.; Forbes D. C. Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev. 1998, 98, 911–936. 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]; b Davies H. M. L.; Beckwith R. E. J. Catalytic Enantioselective C-H Activation by Means of Metal-Carbenoid-Induced C-H Insertion. Chem. Rev. 2003, 103, 2861–2904. 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; c Davies H. M. L.; Manning J. R. Catalytic C–H Functionalization by Metal Carbenoid and Nitrenoid Insertion. Nature 2008, 451, 417–424. 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ford A.; Miel H.; Ring A.; Slattery C. N.; Maguire A. R.; McKervey M. A. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. 10.1021/acs.chemrev.5b00121. [DOI] [PubMed] [Google Scholar]; e Davies H. M. L.; Liao K. Dirhodium Tetracarboxylates as Catalysts for Selective Intermolecular C–H Functionalization. Nat. Rev. Chem. 2019, 3, 347–360. 10.1038/s41570-019-0099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuyama T.; Takino T.; Sueda T.; Ochiai M. Solvolysis of Cyclohexenyliodonium Salt, a New Precursor for the Vinyl Cation: Remarkable Nucleofugality of the Phenyliodonio Group and Evidence for Internal Return from an Intimate Ion–Molecule Pair. J. Am. Chem. Soc. 1995, 117, 3360–3367. 10.1021/ja00117a006. [DOI] [Google Scholar]
- a For alternative applications using the diazo-substituted hypervalent iodine reagents, see:Weiss R.; Seubert D.-C. J.; Hampel F. α-Aryliodonio Diazo Compounds: SN Reactions at the α-C Atom as a Novel Reaction Type for Diazo Compounds. Angew. Chem., Int. Ed. Engl. 1994, 33, 1952–1953. 10.1002/anie.199419521. [DOI] [Google Scholar]; b Schnaars C.; Hennum M.; Bonge-Hansen T. Nucleophilic Halogenations of Diazo Compounds, a Complementary Principle for the Synthesis of Halodiazo Compounds: Experimental and Theoretical Studies. J. Org. Chem. 2013, 78, 7488–7497. 10.1021/jo401050c. [DOI] [PubMed] [Google Scholar]; c Taylor M. T.; Nelson J. E.; Suero M. G.; Gaunt M. J. A Protein Functionalization Platform Based on Selective Reactions at Methionine Residues. Nature 2018, 562, 563–568. 10.1038/s41586-018-0608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gao Z.-Z.; Xu Y.-Y.; Wang Z.-K.; Wang H.; Zhang D.-W.; Li Z.-T. Porous [Ru(Bpy)3]2+-Cored Metallosupramolecular Polymers: Preparation and Recyclable Photocatalysis for the Formation of Amides and 2-Diazo-2-Phenylacetates. ACS Appl. Polym. Mater. 2020, 2, 4885–4892. 10.1021/acsapm.0c00800. [DOI] [Google Scholar]; e Li J.; Lu X.-C.; Xu Y.; Wen J.-X.; Hou G.-Q.; Liu L. Photoredox Catalysis Enables Decarboxylative Cyclization with Hypervalent Iodine(III) Reagents: Access to 2,5-Disubstituted 1,3,4-Oxadiazoles. Org. Lett. 2020, 22, 9621–9626. 10.1021/acs.orglett.0c03663. [DOI] [PubMed] [Google Scholar]; f Dong J.-Y.; Wang H.; Mao S.; Wang X.; Zhou M.-D.; Li L. Visible Light-Induced [3 + 2] Cyclization Reactions of Hydrazones with Hypervalent Iodine Diazo Reagents for the Synthesis of 1-Amino-1,2,3-Triazoles. Adv. Synth. Catal. 2021, 363, 2133–2139. 10.1002/adsc.202001436. [DOI] [Google Scholar]; g Li J.; Wen J.-X.; Lu X.-C.; Hou G.-Q.; Gao X.; Li Y.; Liu L. Catalyst-Free Visible-Light-Promoted Cyclization of Aldehydes: Access to 2,5-Disubstituted 1,3,4-Oxadiazole Derivatives. ACS Omega 2021, 6, 26699–26706. 10.1021/acsomega.1c04098. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Huang H.; Zou X.; Cao S.; Peng Z.; Peng Y.; Wang X. N-Heterocyclic Carbene-Catalyzed Cyclization of Aldehydes with α-Diazo Iodonium Triflate: Facile Access to 2,5-Disubstituted 1,3,4-Oxadiazoles. Org. Lett. 2021, 23, 4185–4190. 10.1021/acs.orglett.1c01128. [DOI] [PubMed] [Google Scholar]; i Yip W.-M.; Yu Q.; Tantipanjaporn A.; Chan W.-C.; Deng J.-R.; Ko B. C.-b.; Wong M.-K. Synthesis of New Quinolizinium-Based Fluorescent Compounds and Studies on Their Applications in Photocatalysis. Org. Biomol. Chem. 2021, 19, 8507–8515. 10.1039/D1OB00716E. [DOI] [PubMed] [Google Scholar]; j Devi L.; Kumar P.; Kant R.; Rastogi N. Exploiting the Umpolung Reactivity of Diazo Groups: Direct Access to Triazolyl-Azaarenes from Azaarenes. Chem. Commun. 2022, 58, 7062–7065. 10.1039/D2CC01897G. [DOI] [PubMed] [Google Scholar]; k Ma X.; Sun A.; Wang K.-K. Unexpected Ester and Phosphonate Radical Generation by Hypervalent Iodine Compounds for Synthesizing 6-Phenanthridine Derivatives. New J. Chem. 2022, 46, 6856–6859. 10.1039/D2NJ01186G. [DOI] [Google Scholar]; l Wen J.; Zhao W.; Gao X.; Ren X.; Dong C.; Wang C.; Liu L.; Li J. Synthesis of [1,2,3]Triazolo-[1,5-a]Quinoxalin-4(5H)-Ones through Photoredox-Catalyzed [3 + 2] Cyclization Reactions with Hypervalent Iodine(III) Reagents. J. Org. Chem. 2022, 87, 4415–4423. 10.1021/acs.joc.2c00135. [DOI] [PubMed] [Google Scholar]
- Espino C. G.; Fiori K. W.; Kim M.; Du Bois J. Expanding the Scope of C-H Amination through Catalyst Design. J. Am. Chem. Soc. 2004, 126, 15378–15379. 10.1021/ja0446294. [DOI] [PubMed] [Google Scholar]
- Strong electron-donating functionalities in the aryl substituent of the alkyne such as methoxy or electron-rich heterocyclic substituents such as thiophene and unprotected indole were not tolerated. Alkynes substituted with heteroatomic functionalities (PhC≡CX, X = Cl, Br, I, NMeTs) did not afford the desired CPC and instead complex reaction mixtures were observed.
- a For selected reviews:Rubin M.; Rubina M.; Gevorgyan V. Transition Metal Chemistry of Cyclopropenes and Cyclopropanes. Chem. Rev. 2007, 107, 3117–3179. 10.1021/cr050988l. [DOI] [PubMed] [Google Scholar]; b Zhu Z.-B.; Wei Y.; Shi M. Recent Developments of Cyclopropene Chemistry. Chem. Soc. Rev. 2011, 40, 5534–5563. 10.1039/c1cs15074j. [DOI] [PubMed] [Google Scholar]; c Vicente R. Recent Progresses towards the Strengthening of Cyclopropene Chemistry. Synthesis 2016, 48, 2343–2360. 10.1055/s-0035-1561644. [DOI] [Google Scholar]; d Dian L.; Marek I. Asymmetric Preparation of Polysubstituted Cyclopropanes Based on Direct Functionalization of Achiral Three-Membered Carbocycles. Chem. Rev. 2018, 118, 8415–8434. 10.1021/acs.chemrev.8b00304. [DOI] [PubMed] [Google Scholar]; e Vicente R. C-C Bond Cleavages of Cyclopropenes: Operating for Selective Ring-Opening Reactions. Chem. Rev. 2021, 121, 162–226. 10.1021/acs.chemrev.0c00151. [DOI] [PubMed] [Google Scholar]
- Row R. D.; Prescher J. A. Constructing New Bioorthogonal Reagents and Reactions. Acc. Chem. Res. 2018, 51, 1073–1081. 10.1021/acs.accounts.7b00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liao L.-a.; Yan N.; Fox J. M. Dianion Approach to Chiral Cyclopropene Carboxylic Acids. Org. Lett. 2004, 6, 4937–4939. 10.1021/ol047837+. [DOI] [PubMed] [Google Scholar]; b Chuprakov S.; Rubin M.; Gevorgyan V. Direct Palladium-Catalyzed Arylation of Cyclopropenes. J. Am. Chem. Soc. 2005, 127, 3714–3715. 10.1021/ja042380k. [DOI] [PubMed] [Google Scholar]; c Fordyce E. A. F.; Luebbers T.; Lam H. W. Synthesis and Application of Alkenylstannanes Derived from Base-Sensitive Cyclopropenes. Org. Lett. 2008, 10, 3993–3996. 10.1021/ol801486v. [DOI] [PubMed] [Google Scholar]; d Fisher L. A.; Fox J. M. Studies on the Stability of Cycloprop-2-ene Carboxylate Dianions and Reactions with Electrophiles. J. Org. Chem. 2008, 73, 8474–8478. 10.1021/jo801683n. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Briones J. F.; Davies H. M. L. Gold(I)-Catalyzed Asymmetric Cyclopropenation of Internal Alkynes. J. Am. Chem. Soc. 2012, 134, 11916–11919. 10.1021/ja304506g. [DOI] [PubMed] [Google Scholar]; f Chen L.; Leslie D.; Coleman M. G.; Mack J. Recyclable Heterogeneous Metal Foil-catalyzed Cyclopropenation of Alkynes and Diazoacetates under Solvent-free Mechanochemical Reaction Conditions. Chem. Sci. 2018, 9, 4650–4661. 10.1039/C8SC00443A. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Yang Y.; Antoni P.; Zimmer M.; Sekine K.; Mulks F. F.; Hu L.; Zhang L.; Rudolph M.; Rominger F.; Hashmi A. S. K. Dual Gold/Silver Catalysis Involving Alkynylgold(III) Intermediates Formed by Oxidative Addition and Silver-Catalyzed C-H Activation for the Direct Alkynylation of Cyclopropenes. Angew. Chem., Int. Ed. 2019, 58, 5129–5133. 10.1002/anie.201812577. [DOI] [PubMed] [Google Scholar]; h He F.; Koenigs R. M. Visible Light Mediated, Metal-free Carbene Transfer Reactions of Diazoalkanes with Propargylic Alcohols. Chem. Commun. 2019, 55, 4881–4884. 10.1039/C9CC00927B. [DOI] [PubMed] [Google Scholar]; i Cohen Y.; Augustin A. U.; Levy L.; Jones P. G.; Werz D. B.; Marek I. Regio- and Diastereoselective Copper-Catalyzed Carbomagnesiation for the Synthesis of Penta- and Hexa-Substituted Cyclopropanes. Angew. Chem., Int. Ed. 2021, 60, 11804–11808. 10.1002/anie.202102509. [DOI] [PubMed] [Google Scholar]
- a Panne P.; Fox J. M. Rh-Catalyzed Intermolecular Reactions of Alkynes with α-Diazoesters That Possess β-Hydrogens: Ligand-Based Control over Divergent Pathways. J. Am. Chem. Soc. 2007, 129, 22–23. 10.1021/ja0660195. [DOI] [PubMed] [Google Scholar]; b Briones J. F.; Davies H. M. L. Silver Triflate-Catalyzed Cyclopropenation of Internal Alkynes with Donor-/Acceptor-Substituted Diazo Compounds. Org. Lett. 2011, 13, 3984–3987. 10.1021/ol201503j. [DOI] [PubMed] [Google Scholar]
- a Panish R.; Chintala S. R.; Boruta D. T.; Fang Y.; Taylor M. T.; Fox J. M. Enantioselective Synthesis of Cyclobutanes via Sequential Rh-catalyzed Bicyclobutanation/Cu-catalyzed Homoconjugate Addition. J. Am. Chem. Soc. 2013, 135, 9283–9286. 10.1021/ja403811t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Qin C.; Davies H. M. L. Enantioselective Synthesis of 2-Arylbicyclo[1.1.0]butane Carboxylates. Org. Lett. 2013, 15, 310–313. 10.1021/ol303217s. [DOI] [PubMed] [Google Scholar]
- a Cheng Y.; Meth-Cohn O. Heterocycles Derived from Heteroatom-Substituted Carbenes. Chem. Rev. 2004, 104, 2507–2530. 10.1021/cr030604w. [DOI] [PubMed] [Google Scholar]; b Priebbenow D. L. Silicon-Derived Singlet Nucleophilic Carbene Reagents in Organic Synthesis. Adv. Synth. Catal. 2020, 362, 1927–1946. 10.1002/adsc.202000279. [DOI] [Google Scholar]
- Compounds 4, 31, and 34 can also be synthesized in one pot from the corresponding alkyne using the previous protocol for the synthesis of 3a* with PhMgBr or Ph2Zn, MeOH, and PPh3 as the nucleophile, respectively. See Supporting Information for details.
- Reactions carried out with water as nucleophile provided mixtures of alkenyl ketones, presumably generated from the ring opening of the corresponding cyclopropenol intermediate.
- Fürstner A. Alkyne Metathesis on the Rise. Angew. Chem., Int. Ed. 2013, 52, 2794–2819. 10.1002/anie.201204513. [DOI] [PubMed] [Google Scholar]
- We have never observed any explosion during the preparation or manipulation of reagents 2 at the scales indicated here in our laboratory. For safety precautions, we always use an antiblast shield in the synthesis of reagents 2. See ref (15a) for DSC studies.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.