

Summary
Newborn screening (NBS) dramatically improves outcomes in severe childhood disorders by treatment before symptom onset. In many genetic diseases, however, outcomes remain poor because NBS has lagged behind drug development. Rapid whole-genome sequencing (rWGS) is attractive for comprehensive NBS because it concomitantly examines almost all genetic diseases and is gaining acceptance for genetic disease diagnosis in ill newborns. We describe prototypic methods for scalable, parentally consented, feedback-informed NBS and diagnosis of genetic diseases by rWGS and virtual, acute management guidance (NBS-rWGS). Using established criteria and the Delphi method, we reviewed 457 genetic diseases for NBS-rWGS, retaining 388 (85%) with effective treatments. Simulated NBS-rWGS in 454,707 UK Biobank subjects with 29,865 pathogenic or likely pathogenic variants associated with 388 disorders had a true negative rate (specificity) of 99.7% following root cause analysis. In 2,208 critically ill children with suspected genetic disorders and 2,168 of their parents, simulated NBS-rWGS for 388 disorders identified 104 (87%) of 119 diagnoses previously made by rWGS and 15 findings not previously reported (NBS-rWGS negative predictive value 99.6%, true positive rate [sensitivity] 88.8%). Retrospective NBS-rWGS diagnosed 15 children with disorders that had been undetected by conventional NBS. In 43 of the 104 children, had NBS-rWGS-based interventions been started on day of life 5, the Delphi consensus was that symptoms could have been avoided completely in seven critically ill children, mostly in 21, and partially in 13. We invite groups worldwide to refine these NBS-rWGS conditions and join us to prospectively examine clinical utility and cost effectiveness.
Keywords: newborn screening, rapid whole-genome sequencing, genetic disease, virtual management guidance, UK Biobank, sensitivity, specificity, clinical utility, diagnostic odyssey, gene therapy, orphan drug, clinical decision support, diagnosis
Graphical abstract
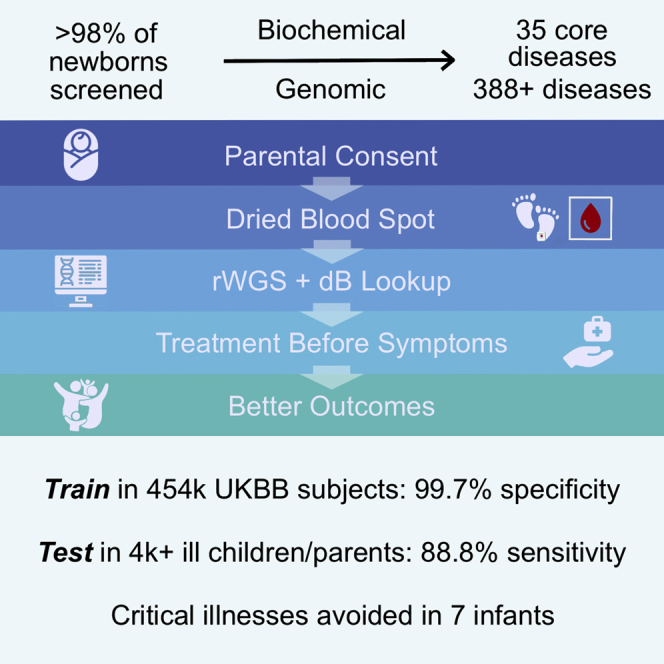
Because highly successful NBS has not kept pace with genome or therapeutic innovation, we adapted rWGS for comprehensive NBS. NBS-rWGS for 388 disorders had 99.7% specificity, 88.8% sensitivity, and could have avoided symptoms completely in seven of 2,208 critically ill infants, mostly in 21, and partially in 13.
Introduction
Newborn screening (NBS) is performed worldwide in ∼140 million newborns annually to identify severe congenital disorders and initiate treatments at or before onset of symptoms.1 While NBS can greatly improve health outcomes, the number of genetic disorders screened has not kept pace with genomic or therapeutic innovation.2, 3, 4, 5 Between 2006 and 2022, the number of core disorders that were recommended for NBS of dried blood spots (DBSs) in the United States—the Recommended Uniform Screening Panel (RUSP)—increased from 27 to 35, and the number of affected infants identified increased from 6,439 to 6,466.4,5 However, there are ∼7,200 known genetic diseases and hundreds of targeted treatments that have been approved or are in clinical trials.3,6 Over the past decade, rapid whole-genome sequencing (rWGS) has developed into an effective diagnostic test (Dx-rWGS) for almost all heritable diseases and is gaining acceptance as a first-tier test for critically ill newborns with suspected genetic diseases.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 rWGS is attractive for comprehensive NBS because it concomitantly examines almost all genetic diseases with similar time to result as biochemical NBS of DBSs by mass spectrometry (NBS-MS).21, 22, 23, 24, 25 Here, we describe adaptation of Dx-rWGS methods for comprehensive NBS (NBS-rWGS). We detail scalable, feedback-informed methods for newborn screening, diagnosis, and virtual, acute management guidance for 388 diseases with effective treatments and report analytic performance and clinical utility in large retrospective datasets.
Subjects and methods
Research participants
De-identified UK Biobank (UKBB) participants and exomes were queried through the UKBB Research Analysis Platform under application number 82213. Retrospective analysis of genomes and phenotypes of critically ill newborns and children and their parents who had received rWGS for molecular diagnosis of a suspected genetic disorder at Rady Children’s Institute for Genomic Medicine (RCIGM) was approved by the Institutional Review Board of Rady Children’s Hospital/University of California – San Diego.
Selection of disorders and interventions for NBS-rWGS
Disorder and intervention curation for the Genome-to-Treatment (GTRx) management guidance system has been described in detail.20 Briefly, we examined the efficacy of therapeutic interventions available for 563 childhood-onset, single-locus genetic disorders that met the following criteria: acute, childhood presentations that were likely to lead to neonatal, pediatric, or cardiovascular ICU admission; having somewhat effective treatments; high likelihood of rapid progression without treatment; and diagnosable by rWGS. They were identified by a survey of our Dx-rWGS experience in ∼4,000 critically ill newborns and children and from expanded NBS disorder lists developed by several groups.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20,26, 27, 28, 29, 30 Publications relating to ∼10,000 interventions associated with these disorders were extracted with custom scripts (Genomenon Rancho Biosciences, Epam) and curated manually for relevance.20 The interventions were adjudicated by six pediatric clinical and biochemical geneticists with a modified Delphi technique and electronic data capture (RedCap). Five of the six panel members were retained for the entire project. Consensus was required for inclusion of interventions and disorders regarding: (1) age groups in which the intervention was indicated; (2) optimal time of intervention initiation after NBS or diagnosis; (3) contraindications; (4) efficacy category (curative, effective, ameliorative); and (5) level of evidence supporting efficacy.20 A web resource integrated the GTRx information resources and the adjudicated interventions of 457 retained disorders associated with 352 genes and 1,527 interventions (https://gtrx.rbsapp.net/).
We then evaluated the suitability of the 457 genetic diseases retained in GTRx for NBS-rWGS by using established criteria31, 32, 33 and the same expert panel, electronic data capture system, and modified Delphi methods. The panel comprised six pediatric clinical and biochemical geneticists representing hospitals in four states. They met weekly for 1 year. Each week, prior to meeting, they reviewed a set of disorders in a RedCap electronic data capture system. To reach consensus regarding inclusion of each GTRx disorder in NBS-rWGS, the panel considered six questions and clarifying sub-questions (Figure 1).
-
1Is the natural history of this genetic disease well understood?
-
aIs there at least one well-established gene-phenotype association?
-
bIs there significant variation in expressivity?
-
cIs there reduced penetrance?
-
dIs inheritance (autosomal dominant, autosomal recessive, X-linked, mitochondrial) well understood?
-
eIs pathogenicity of at least a subset of DNA variants well understood (gain versus loss of function)?
-
fIs genotype-phenotype correlation sufficient for those variants to predict disease course?
-
gCan variability in outcome or disease severity be clarified by additional investigation (such as an analyte, enzyme, biomarker, or functional test)?
-
a
-
2Is this genetic disease a significant risk for morbidity and mortality in infants or young children?
-
aIs penetrance high enough such that identification of clinically insignificant disease is minimal or causes minimal harm?
-
a
-
3Is a treatment or intervention available that is effective and accepted?
-
aIs a treatment available that can affect outcome?
-
bIs a treatment effective for all affected individuals?
-
cIs response to treatment consistent for a given recognized pathogenic variant(s)?
-
dIs treatment effective for all symptoms of a disorder?
-
eIf no specific treatment is available, would making a diagnosis change management some other way?
-
fIs a treatment widely available and are there sufficient providers, facilities, and resources to accommodate all identified individuals?
-
gIs a treatment acceptable to the majority population? Considerations include cost, morbidity of the treatment, and religious or political beliefs. For example, does this intervention require use of fetal-derived tissue?
-
a
-
4Does early treatment improve outcome?
-
aIs there a latent phase during which initiation of treatment leads to improved outcome or prevents complications?
-
bDoes delayed diagnosis lead to poorer outcome or serious complications?
-
cDoes early diagnosis and treatment lead to improved outcome over reactive care following symptom onset?
-
a
-
5Do the benefits of early intervention clearly outweigh the risks?
-
aAre false positives problematic with this gene?
-
bMight NBS adoption of this condition have a negative net benefit? Considerations include the proband, family, and the general population.
-
cDo concerns exist regarding identification of carriers?
-
a
-
6
For genes with more than one associated disorder, do their treatments differ, and can they be distinguished by rWGS or additional testing?
Figure 1.

Flowchart of the modified Delphi technique for ongoing selection of disorders for NBS-rWGS after they have been included in the Genome-to-Treatment virtual management guidance system (GTRx)20
Abbreviations: ICU, intensive care unit; rWGS, rapid whole-genome sequencing.
At the meeting, the individual classification for each disorder was presented by each member. Retained conditions were divided into two groups. Group A was conditions for which there were not major gaps in the evidence, high likelihood of benefit, and low risk of harm. Group B was conditions for which there were gaps in the evidence or uncertainty regarding net benefit that required further assessment by NBS-rWGS research. If there was not initial consensus, discussion regarding differences in opinion ensued. The members then decided whether to change their classification. Decisions required at least two-thirds of panelists to agree. The time taken to review a disorder, the extent of initial agreement, and number of rounds of discussion changed as familiarity with the process increased. For most disorders, a majority of members initially agreed about classification. Few disorders had initial unanimity. In addition to the panel members, a software applications specialist audited RedCap entries and refined the electronic data capture methods. The first author provided feedback to the panel regarding all other pertinent aspects of the project, such as the analytic performance of disorders in test datasets, as needed to help facilitate decision making. The opinions of other pediatric subspecialists at Rady Children’s Hospital, a very large quaternary referral center, were sought if consensus was elusive or if specific domain expertise was required. Four of the panel members had bridging expertise in NBS-MS and Dx-rWGS. We retained NBS-MS RUSP disorders and included American College of Medical Genetics and Genomics (ACMG)-recommended incidental finding disorders with infant onset.34 It should be noted that the consensus to retain a disorder did not imply that the evidence was sufficient for inclusion in a clinical NBS-rWGS product or public health system. Rather, it indicated that the benefit-to-harm ratio for NBS-rWGS was sufficient for inclusion in NBS-rWGS research studies. Not all disorders have been evaluated for inclusion in NBS-rWGS. We encourage submission of recommendations for review and anticipate updating the NBS-rWGS panel twice a year.
Variant selection
We evaluated 29,865 rare (gnomAD allele frequency < 0.5%), germline, pathogenic (P), or likely pathogenic (LP) ClinVar nucleotide variants that mapped to 388 NBS-rWGS gene-disorder dyads (317 genes and 381 disorders). They included variants with conflicting assertions of pathogenicity and where the associated condition was not specified. Variants of uncertain significance, likely benign, and benign variants were excluded. Well established disease-causing variants with gnomAD allele frequency > 0.5% were retained.35 Following training, 94 “block-listed” variants were removed, leaving a reconciled set of 29,771 variants. Thirteen of these ClinVar variants were associated with more than one gene. These were manually associated with a single gene (Table S1). Ten variants were located in variant call file (VCF) regions overlapping both the hemoglobin α1 (HBA1 [MIM: 141800]) and α2 (HBA2 [MIM: 141850]) loci. These were manually corrected to the ClinVar gene association (HBA2). Additional P or LP variants not found in ClinVar were identified with Mastermind followed by curation of evidence and variant interpretation according to standard ACMG clinical guidelines (Genomenon).
Rapid diagnostic whole-genome sequencing
Clinical Dx-rWGS methods from EDTA blood samples and DBS have been described in detail (Figure 2).36 Briefly, genomic DNA (gDNA) was isolated from blood with the EZ1 DSP DNA Blood Kit (Qiagen). gDNA was isolated from five 3 mm2 DBS punches (Nucleic card, Thermo Fisher or Protein Saver 903 Card, GE Healthcare) with either the DNA Flex Lysis Reagent Kit (Illumina) or Proteinase K (QIAGEN). gDNA quality was assessed with the Quant-iT Picogreen dsDNA assay, Nanodrop A260/A280 assay, and by electrophoresis on 0.8% agarose gels (Thermo Fisher). Sequencing libraries were prepared with either DNA PCR-free prep kits (Illumina) or KAPA Hyper-Plus PCR-free library kits (Roche). Libraries with concentration >3 nM and acceptable fragment size were sequenced (2 × 101 nucleotide [nt]) on NovaSeq 6000 instruments (Illumina). Quality controls for rWGS included Q30 ≥ 80%, error rate ≤ 3%, and >120 Gb sequence generated per sample after top-off sequencing. rWGS were aligned to human genome assembly GRCh37 (hg19) and variants identified and genotyped with the DRAGEN platform (Illumina). Structural variants were filtered to retain those affecting coding regions of genes associated with genetic diseases and with allele frequencies < 2% in the RCIGM database. rWGS variant quality controls included (1) identity tracking by CODIS short tandem repeats (STR) by capillary electrophoresis (Thermo Fisher) and in silico STR from rWGS; (2) <15% duplicates; (3) >98% aligned reads; (4) Ti/Tv ratio 2.0–2.2); (5) Hom/Het variant ratio 0.40–0.61); (6) >90% of OMIM genes with >10-fold coverage of all coding nucleotides; (7) sex match; and (8) coverage uniformity by GC bias, standard deviation of coverage normalized to average coverage, and the total length of the reference genome with read coverage.
Figure 2.

Workflow diagrams of diagnostic rWGS and newborn screening by rWGS
Shown are comparisons of the workflow for Dx-rWGS (A) with that for NBS-rWGS (B) and for a secondary use of data generated by NBS-rWGS (C). The interpretation burden of NBS-rWGS is approximately 1,000-fold less than that of Dx-rWGS. The light blue shading indicates the activities occurring in places of care for newborns or older children, while the darker blue shading indicates activities occurring in clinical laboratories. The dashed green arrows ① and ② in NBS-rWGS indicate feedback loops. Abbreviations: dB, database; EDTA, ethylene diamine tetra-acetic acid; ICU, intensive care unit; EHR, electronic health record; CLIA, clinical laboratory improvements act; GEM AI, a genome interpretation tool that employs artificial intelligence15; GTRx, Genome-to-Treatment virtual management guidance system.20
Comprehensive variant interpretation was performed according to standard guidelines by clinical molecular geneticists with GEM and Enterprise software (Fabric Genomics) with the variant call file (VCF), list of observed human phenotype ontology terms, and individual metadata (coded identifier, name, electronic health record [EHR] number, ordering physician, date of birth, location, relationship to proband).15,20 Variants of each type and inheritance mode were ranked according to phenotypic match with the associated genetic disease and locus, pathogenicity classification, and rarity in population databases. Reported variants were confirmed by Sanger sequencing, multiplex-ligation-dependent probe amplification, or chromosomal microarray, as appropriate. Secondary findings were not systematically sought, but medically actionable incidental findings were reported if families requested this information. Using the consensus recommendations of the ACMG, a diagnosis was considered made if pathogenic or likely pathogenic variants were identified in a genomic locus that providers agreed led to the disease causing the critical illness.
Re-pipelining of WGS, TileDB development, and queries
To serve as a reference and test set for NBS-rWGS, we created CSI and TBL files for 3,202 One Thousand Genome Project (1KGP) subjects, Genome in a Bottle reference samples, and 4,376 critically ill children and their parents who received rWGS at RCIGM for diagnosis of suspected genetic disorders.9,11, 12, 13, 14, 15, 16, 17, 18, 19, 20,37,38 We re-aligned 3,202 (30 × 2 × 150 nt) 1KGP WGS and 4,376 (>40 × 2 × 100 nt) RCIGM rWGS to the GRCh38 reference genome by using DRAGEN (v3.8 and v3.9, respectively) on Illumina Connected Analytics (ICA). We developed array-based data models for genomic variants and metadata extracted from Fabric Enterprise, Ensembl, gnomAD, Clinvar, and variant effect prediction (VEP). The resultant 7,578 single sample VCFs were ingested into a TileDB array (v2.8) on AWS S3 with TileDB-VCF (v0.15). TileDB-VCF is a specialized application that parses VCF files in a sparse, 3-dimensional array in which records are indexed by their chromosome, chromosomal position, and sample of origin.39 During ingestion, every VCF is read and converted into the TileDB-VCF on disk format. While the VCF record is in memory, the genotype for each variant is inspected to determine the frequencies of each allele, which are stored in an additional grouped, variant-centric, TileDB array. Metadata fields from prior RCIGM diagnostic rWGS were extracted from Fabric Enterprise and interpretation reports were de-identified, lifted from GRCH37 to GRCH38 coordinates, and ingested into TileDB-Cloud (v0.7.41), together with Ensembl (v104), gnomAD (v3.1.1), Clinvar (downloaded 2022-5-20), and VEP (v105) metadata for each variant. We parsed 317 NBS-rWGS genes and queried the 4,376 RCIGM VCFs with ClinVar P and LP variants mapping to these genes on the basis of positions and alleles. Multi-allelic variant rows were flattened. We retained high quality variants and annotated the query results with gene information, project-specific subject codes, gender, and disorder pattern of inheritance. We used custom scripts to calculate variant zygosity and to determine whether genotypes represented NBS-rWGS positives on the basis of diplotypes and disorder pattern of inheritance. Completeness of query results was assessed by comparison with results of prior Dx-rWGS interpretation. Queries were performed repeatedly and debugged until reproducibility was assured. Among individuals who had been diagnosed with an NBS-rWGS disorder, additional NBS-rWGS positive individuals were sought by analysis of VCFs with the automated interpretation tool, GEM in Fabric Enterprise.15 GEM was performed with a Bayes factor-based cutoff of >0.1 and a generic phenotype (phenotypic abnormality, HP: 0000118).
UK Biobank queries
The 454,707 UK Biobank (UKBB) subjects were 208,120 (46%) male and 246,587 (54%) female.40, 41, 42 NBS-rWGS gene regions were extracted from UKBB pVCFs. We split multiallelic rows, normalized indels, and filtered out low-quality variants as described.42 We retrieved ClinVar variants with clinical significance (CLNSIG) of “likely_pathogenic” or “pathogenic” that mapped to the NBS-rWGS gene regions. We intersected the two variant sets and identified positive individuals on the basis of pattern of inheritance and individual zygosity (heterozygous for dominant disorders and compound heterozygous, hemizygous, or homozygous for recessive disorders). Where Mendelian Inheritance in Man (MIM) indicated the pattern of inheritance to be mixed dominant and recessive, we retained only individuals exhibiting recessive patterns of inheritance. We used the aggregated International Statistical Classification of Diseases and Related Health Problems (ICD)-9/10 codes, read v2 medication codes, death register codes, and self-reported medical condition data provided for UKBB subjects to identify those affected by specific conditions, including hemophilia A.
Root cause analysis
Root cause analysis was performed manually on all NBS-rWGS positive subjects in the UKBB and RCIGM sets to assess the likelihood that they were true or false positives (Figure 3). We first checked gene names, disorder names, and patterns of inheritance to ensure that each variant matched an NBS-rWGS disorder. We ranked genes by frequency of positive subjects and compared observed frequencies with known incidences of those disorders. Genes with more positive subjects than the population incidence underwent detailed variant analysis. We also ranked variants by frequency of positive subjects and compared observed frequencies with the proportion of affected subjects expected to harbor those variants, where known, and their population incidence. Outlier variants identified by these searches underwent the following. (1) Literature review to assess the quality and quantity of evidence of pathogenicity, including variant effect predictions, the number of citations reporting affected individuals with the NBS-rWGS disorder in ClinVar or PubMed, and the quality of evidence for pathogenicity in PubMed, including quantitative functional evidence, number of affected subjects, and phenotypes in affected subjects. For disorders with well-established locus-specific variant databases, these largely replaced review of the primary literature. (2) Review of putative compound heterogyzotes to remove those that were either known to occur in cis as recurrent haplotypes or novel haplotypes that were identified by inspection of aligned and phased sequencing reads. (3) Review for evidence that they mapped to regions of the genome that are difficult to genotype with short read sequences. Variants for which root cause analysis identified an artifactual reason for high positivity were block listed. Recurrent variants with strong support for pathogenicity were white listed.
Figure 3.

Funnel plots demonstrating the use of step-wise root cause analysis to improve the specificity and sensitivity of newborn screening by genomic sequencing
Funnel plots showing reduction in 2,982 positive individuals in 73 positive NBS-rWGS genes among 454,707 UK Biobank participants by root cause analysis (A) and increase in retrospective NBS-rWGS positives among 4,376 children and their parents (B). Variant identifiers are from ClinVar. Abbreviations: LB, likely benign; B, benign; AR, autosomal recessive; AD, autosomal dominant; ICD, International Statistical Classification of Diseases and Related Health Problems; dB, database; UKBB, United Kingdom Biobank.
Retrospective clinical utility assessment
The potential clinical utility of NBS-rWGS was evaluated retrospectively in 4,376 critically ill children with a suspected genetic disorder and their parents, who had received Dx-rWGS.9,11, 12, 13, 14, 15, 16, 17, 18, 19, 20 In each proband child who had received a molecular diagnosis by rWGS that had been recapitulated by NBS-rWGS, the observed clinical features were compared with those listed in MIM, Genetic and Rare Diseases Information Center, and MEDLINE to determine which were attributable to that molecular diagnosis. Based on the assessed efficacy of each indicated intervention for that disorder in GTRx, one of us compared the impact on the observed, reversible, attributable clinical features of starting those interventions at the actual age of diagnosis by rWGS with that of treatment initiation at the counterfactual age of diagnosis by NBS-rWGS (day of life 5), as previously described.9,16 The extent to which NBS-rWGS could have prevented or avoided the occurrence of each of the attributable clinical features was adjudged on a five-point Likert scale (completely, mostly, partially, none, uncertain).
Results
The starting points for development of a system for genomic NBS were the existing state-run NBS by mass spectrometry (NBS-MS) systems and 10 years of experience with rapid, diagnostic whole-genome sequencing (Dx-rWGS) and precision treatment in critically ill newborns (Figure 2A).9,11, 12, 13, 14, 15, 16, 17, 18, 19, 20 Dx-rWGS was modified to achieve NBS-rWGS during the first week of life and with a scope of parentally consented, feedback-informed screening and diagnosis of hundreds of genetic diseases, together with immediate treatment informed by virtual, acute management guidance (Figure 2B).20 The DBS that are collected in the first 24–48 h of life and universally used for NBS-MS were validated for clinical-grade rWGS (Figure 2B).36 Of over 260 archived California NBS DBS, collected between 2005 and 2018, none failed genomic DNA extraction and WGS quality control (Table S2). The average library yield was 15.3 nM (range 5.1–24.2 nM), average WGS yield was 156 GB (range 107.8–310 GB), average proportion of nucleotide variants passing quality control was 98.9 (range 98.6–99.2%), average number of nucleotide variants was 5,018,555 (range 4,714,850–5,884,774), average mitochondrial genome coverage was 4,524-fold (range 1,256–18,248), and average proportion of Mendelian Inheritance in Man genes with all coding nucleotides with at least 10-fold coverage was 96.1% (range 90.1%–98.2%, Table S2).
NBS-rWGS required adaptation of Dx-rWGS to a much lower pre-test probability of genetic disease. Among critically ill newborns in intensive care units (ICUs) with suspected genetic diseases, the pre-test probability is ∼40% (Figure 2A).7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 Available data suggested the probability to be 10%–15% among all newborns in ICUs and 1%–2% in ostensibly healthy newborns, the populations who would receive NBS-rWGS (Figure 2B).14,43,44 The analytic performance desired for NBS-rWGS was based on that of NBS by mass spectrometry (NBS-MS). 20 years ago, NBS-MS had low positive predictive value (2% PPV).45 Low PPV is unacceptable to parents, pediatricians, ethicists, and payors.46 Methodologic improvements have increased the PPV of NBS-MS to ∼50% in term births (for 48 disorders with a combined true positive rate of 0.03%, Figure S1A).47 We developed NBS-rWGS with a similar target PPV to current NBS-MS. Unlike NBS-MS, however, NBS-rWGS will not have a lower PPV in premature newborns. NBS-rWGS required variant interpretation without guiding clinical features (Figure 2B). Dx-rWGS interpretation, in contrast, is predicated on a rank ordered differential diagnosis based on goodness of fit of the newborn’s clinical features to those of all genetic diseases (Figure 2A). For both of these reasons, NBS-rWGS was developed to query a set of variants that were well established to be causal in genetic diseases known to cause severe morbidity in young children and with effective treatments31, 32, 33 (Figure 2B).
Selection of disorders for the primary use (NBS-rWGS) started by evaluating the 457 childhood-onset genetic diseases with effective treatments that are included in GTRx, a virtual management guidance system for pediatricians caring for critically ill, newly diagnosed children in ICUs (Figure 1, phase i).20 To develop GTRx, we evaluated the efficacy, evidence of efficacy, indications, contraindications, and urgency of initiation of ∼10,000 interventions for 563 genetic diseases that are diagnosed by rWGS in critically ill children. 457 disease-gene dyads (446 disorders associated with 346 genes) and 1,527 drugs, dietary modifications, devices, surgeries, and other interventions with adequate evidence of efficacy were retained.20 GTRx functions similarly to the ACT sheets developed by the ACMG to guide confirmatory testing and management at time of receipt of a positive result from traditional NBS.48 Since medical and genome science are evolving rapidly, we wished to develop auditable methods for ongoing, annual selection of disease-gene dyads appropriate for screening in all newborns. While well-established criteria for selection of disorders for NBS exist, they predate the genomic era, and most genetic diseases have not been evaluated in this regard.31, 32, 33 The suitability of the genetic diseases in GTRx for NBS-rWGS was assessed by a national panel of six pediatric geneticists using the electronic survey database (REDCap v10.6.3) and modified Delphi technique that were effective for development of GTRx (Figure 1, phases iii–vi).20 To reach consensus regarding retention of each GTRx disorder in NBS-rWGS, the panel considered six questions and clarifying sub-questions (Figure 1, phase ii).31, 32, 33 (1) Is the natural history of this disease well understood? This question was particularly important for ultra-rare and recently discovered diseases. (2) Is this disease a significant risk for morbidity and mortality in infants or young children? (3) Is a treatment or intervention available that is effective and accepted? (4) Does early treatment improve outcome? (5) Do the benefits of early intervention clearly outweigh the risks? This question was particularly important for drugs with serious adverse effects and high-risk interventions. (6) For genes with more than one associated disorder, do their treatments differ, and can they be distinguished by rWGS or other tests? The opinions of other pediatric subspecialists at Rady Children’s Hospital were sought if consensus was elusive. We retained federally recommended NBS-MS disorders and included ACMG-recommended incidental finding disorders with infant onset.34 388 (85%) of 457 gene-disorder dyads (317 [92%] of 346 genes associated with 381 [85%] of 446 disorders) were retained for evaluation in retrospective datasets (Table S3). Retained conditions were divided into two groups. Group A (295, 76%) was conditions for which there were not major gaps in the evidence, high likelihood of benefit, and low risk of harm. Group B (93, 24%) was conditions for which there were gaps in the evidence or uncertainty regarding net benefit that required further assessment by NBS-rWGS research. The average agreement in disorder classification among panel members was 89.9%. The cumulative incidence of these disorders in the US is approximately 0.8% (Table S4). These methods will allow the number of NBS-rWGS disorders to grow with time as further effective interventions are developed and approved.3
The initial variants evaluated by in silico NBS-rWGS were all 29,865 rare (gnomAD allele frequency < 0.5%), germline, pathogenic (P), or likely pathogenic (LP) ClinVar nucleotide (nt) variants that mapped to the 388 NBS-rWGS disease-gene dyads. These included variants where the associated condition was not specified. Variants of uncertain significance were excluded. From this set, we wished to remove variants, disorders, and associated genes with unacceptably high false positive rates. We examined these variants in whole-exome sequences of 454,707 UK Biobank (UKBB) subjects enrolled at age 40–69 years between 2006 and 2010.40,42 This cohort was significantly healthier than the UK population as a whole.41 NBS started in the UK in 1969 and encompasses nine conditions (sickle cell disease [MIM: 603903], cystic fibrosis [MIM: 219700], congenital hypothyroidism [genetically heterogeneous], phenylketonuria [MIM: 261600], medium-chain acyl-CoA dehydrogenase deficiency [MIM: 201450], maple syrup urine disease [MIM: 248600], isovaleric acidemia [MIM: 243500], glutaric aciduria type 1 [MIM: 231670, and homocystinuria [genetically heterogeneous]). We expected the prevalence of other severe, childhood onset disorders to be very low in this population. Screening 29,865 variants identified 147,533 genotypes for 5,348 (18%) variants mapping to 281 (89%) genes (Table S5). When converted to diplotypes and restricted to the patterns of inheritance of the 388 dyads, however, only 2,982 (0.66%) subjects remained positive for 523 (1.8%) variants (Figures 3A and S1B, Table S6). Remarkably, 244 (77%) of 317 NBS-rWGS genes were associated with no false positive participants. However, prior exploratory studies of NBS by WES for small panels of genes found the analytic performance to be inferior to NBS-MS.43,49, 50, 51, 52, 53 Therefore, we examined whether feedback loop learning, implemented as root cause analysis (Figure 2B, indicated by green arrows ① and ②), would reduce false positives. First, we examined variants located within regions of the genome that are known to be difficult to genotype with short-read sequencing (Figure 3A.ii).54 This removed 38 subjects with artifactual homozygous genotypes in cystathionine β-synthase (CBS [MIM: 613381], associated with homocystinuria [MIM: 236200], Table S6). Second, we removed 172 likely true positive subjects, 111 based on concordant, albeit limited, UK Biobank phenotypes, and 61 subjects with variants associated with mild or late onset disease (Table S6, Figures 3A.iii and S1B). An informative example was X-linked hemophilia A (HEMA [MIM: 306700]), which has an incidence ∼1 in 10,000 in the UK. 129 subjects were hemizygous or compound heterozygous for 28 Factor 8 (F8 [MIM: 300841]) variants associated with HEMA (Table S7). Twelve subjects were affected (UK Biobank ICD10 code D66; Table S6). Two F8 variants, affecting one subject each, were synonymous and absent from the Centers for Disease Control Hemophilia A Mutation Project Database (Table S7), were removed as likely benign (Figure 3A.vi).55 Sixty-one positive subjects were hemizygous or compound heterozygous for F8 c.396A>C (p.Glu132Asp [GenBank: NM_000132.4], [ClinVar: 10171]), which has a single HEMA ClinVar submission and whose description was limited to one 1995 manuscript.56 Variant pathogenicity assertions from that era are known to be frequently inflated.57 This variant was added to the blocked list. All but five remaining subjects had variants associated with mild HEMA (Tables S6 and S7). Absent trauma or major surgery, such subjects are asymptomatic and may go undiagnosed. Thus, the PPV of genomic NBS for moderate or severe HEMA in UKBB participants was 71% (12 HEMA subjects among 17 positive genotypes).
Third, we removed 545 subjects with ClinVar variant diplotypes for an NBS-rWGS gene and appropriate inheritance but associated with genetic disorders that were not retained (Figure 3A.iv, Table S6). An example is ryanodine receptor 1 (RYR1 [MIM: 180901]), for which 57 of 71 variants were not associated with malignant hyperthermia (MIM: 145600, Table S6). Next, 466 subjects were excluded upon removal of variants that did not fit the pattern of inheritance of the NBS-rWGS disorder (Figure 3A.v). Fifth, 672 false positive diplotypes in recessive disorders occurred as two or more adjacent deleterious variants as haplotypes (in cis) rather than as compound heterozygous (in trans, Table S5): 245 of 248 biotinidase deficiency (MIM: 253260) positive subjects had a haplotype composed of Clinvar variants 373906 (BTD [MIM: 609019], c.40_41del [p.Gly14LeufsTer17] [GenBank: NM_001370658.1]) and 801942 (BTD, c.44_45del [p.Cys15LeufsTer16]). Likewise, all 63 subjects who were positive for pyruvate kinase deficiency (MIM: 266200) had a ClinVar variant 280113 (PKLR [MIM: 609712], c.721G>T [p.Glu241Ter] [GenBank: NM_000298.6]) and 1163645 (PKLR, c.826del [p.Val276TrpfsTer45]) haplotype. In addition, 28 of 32 glycogen storage disease II (Pompe disease [MIM: 232300]) positive subjects had either a ClinVar variant 188484 (GAA [MIM: 606800], c.2237G>C [p.Trp746Ser] [GenBank: NM_000152.5]) and 561162 (GAA, c.2228A>G [p.Gln743Arg]) haplotype or 497032 (GAA, c.1130del [p.Gly377AlafsTer15]) and 497033 (GAA, c.1129G>C [p.Gly377Arg]) haplotype. These haplotypes had not previously been recognized. They were confirmed by examining phased reads (Figure S2). We added one of each variant pair to the blocked list (Table S6): The 5′ variants in BTD and PKLR, a frame-shift and termination codon variant, respectively, were retained and the 3ʹ “silent” variant removed. The better supported GAA variants (188484 and 497032) were retained. This removed 336 positive individuals (Figure 3A.vii). Lastly, we removed 208 subjects associated with variants with poor pathogenicity support (Figure 3A.viii). For example, ClinVar variant 12159 (CYP21A2 [MIM: 613815], c.1360C>T [p.Pro454Ser] [GenBank: NM_000500.9]) is associated with very mild steroid 21-hydroxylase deficiency (MIM: 201910) and has modest effects on enzyme activity.58,59 In toto, feedback loop learning, implemented as root cause analysis, removed 94 (0.3%) of 29,865 variants, reducing likely false positives by 59% to 1,214 (0.27%, 99.7% specificity; Figures 3A and S1B, Table S6). It should be noted that prior medical history information in UKBB participants is self-reported, may be incomplete, and lacks ICD codes for most genetic disorders. Therefore, the nominal PPV for the 388 disorders in middle-aged individuals (12.4%) is a lower limit.
Pathogenicity assessments in NBS-rWGS require knowledge of frequency for each variant genotype (heterozygous, homozygous, hemizygous, or heteroplasmy fraction and frequency).60 Because the number of disorders featured in NBS-rWGS will increase with time, it is important for NBS-rWGS to remain an open system. In practice, both this and the feedback mechanism demonstrated in the UK Biobank data required NBS-rWGS to dynamically calculate the frequency of all possible genotypes at all loci. To accomplish this, the underpinning data management system needed to solve the computational n + 1 problem: That is, the cost to merge the gVCF of 1 newborn (∼5 million genotypes) with a large set (n, ultimately tens of millions) of prior VCFs, and recalculate all genotype frequencies grows super-linearly with number of genomes.61,62 Because time to result is critical for NBS-rWGS, the n + 1 problem cannot be resolved by sample accrual and periodic performance in large batches, the typical informatic solution. Human genomes, however, are 99.8% sparse—only ∼5 million of ∼3 billion positions are non-reference. Therefore, we developed a sparse, cloud-based data management system for NBS-rWGS that employed multi-dimensional arrays (TileDB).39 To benchmark the n + 1 cost of NBS-rWGS, we added one reference gVCF (HG002) to a TileDB array containing 3,202 high coverage VCFs (1KGP)37,38 and calculated frequencies for all genotype possibilities at all 125 million variant positions. With a c6 g.xlarge Amazon Web Services EC2 instance, the n + 1 ingestion and variant frequency refresh took ∼22 min and cost $0.06. Alternatively, by batching the 3,202 gVCFs in thousands, adding HG002 to one batch, and recalculating genotype frequencies in that batch cost $2.18 (a 33-fold increase) with a non-optimized, hierarchical, file-based system and the same cloud provider. Without batching, we anticipate that the n + 1 cost would have been considerably higher.61,62
Newborns with genetic diseases often become critically ill before diagnosis and are admitted to intensive care units, where increasingly they receive singleton or parent-child trio Dx-rWGS.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 We evaluated the analytic performance of NBS-rWGS retrospectively in 2,208 critically ill children with any suspected genetic disorder and 2,168 of their parents, who had received Dx-rWGS.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 We queried their genomes in TileDB with the feedback loop-informed subset of ClinVar P and LP variants that mapped to the 388 NBS-rWGS disease-gene dyads (Figure 3B). Dx-rWGS reported 119 diagnoses, of which 20 where RUSP NBS disorders. 65 (55%) of 119 were positive by NBS-rWGS (Table S8, Figures 3B.i and S2C). The 54 NBS-rWGS false negatives were due to ClinVar absence or conflicting pathogenicity assertions. We supplemented the variant lookup by querying these genomes with the GEM automated interpretation system with a Bayes factor-based cutoff of >0.1 and a generic phenotype (phenotypic abnormality, HP: 0000118, Figure 3B.ii).15 GEM identified an additional 23 diagnoses reported by Dx-rWGS. Of the remaining 32 NBS-rWGS false negatives, 16 were homozygous or hemizygous for a variant in glucose 6-phosphate dehydrogenase (G6PD [MIM:305900], c.292G>A [p.Val98Met] [GenBank: NM_000402.4], ClinVar 37123), which had been removed because the allele frequency was >3%. Adding this variant to the white-list resulted in a total of 104 of 119 (87%) positive by NBS-rWGS and Dx-rWGS (Figures 3B.iii and S1C). In addition, NBS-rWGS identified 15 findings (four probands, 11 parents) that were not reported by Dx-rWGS (Table S7). In toto, the negative predictive value and sensitivity of NBS-rWGS and Dx-rWGS were the same (99.6 and 88.8%). Seventeen of the diagnoses by NBS-rWGS were RUSP core conditions. Fifteen of these had been missed by conventional NBS, including five children with ornithine transcarbamoylase deficiency (OTC [MIM: 311250]) and two with cystic fibrosis (CF [MIM: 219700], Table S9). However, NBS-rWGS did not identify four individuals with RUSP NBS disorders that had been diagnosed by Dx-rWGS (Table S9).
The national panel of six pediatric geneticists used the Delphi method to evaluate the counterfactual clinical utility of NBS-rWGS, compared with the actual utility at time of diagnosis by Dx-rWGS in 60 of the 104 children with diseases detected by both (Tables 1 and S9). Assuming return of results on day of life 5, NBS-rWGS could have shortened the time to diagnosis by a median of 73 days (average 623 days, range 0–7,912 days). The panel examined which of the observed clinical features were attributable to the molecular diagnosis and the counterfactual clinical utility (the extent to which attributable phenotypes could have been lessened or prevented by implementation on day of life 5 of the GTRx-indicated interventions that had been adjudged to have efficacy).20 In 43 of the 60 newborns, there was sufficient knowledge of the natural history of the disorder with and without early treatment to make an assessment. In 41 of the newborns, the panel consensus was that NBS-rWGS with institution of treatment on day of life 5 could have prevented symptoms to some extent. The consensus was that institution of treatment on day of life 5 would have prevented symptoms completely in seven infants, mostly in 21 infants, partially in 13 infants, and to an unknown extent in two (Table 1).
Table 1.
Delphi method counterfactual analysis of the potential clinical utility of diagnosis by NBS-rWGS on day of life five compared with actual age at diagnosis by diagnostic rWGS in 41 critically ill children
| ID | Diagnosis | Key phenotypes avoided by DOL5 Rx | Days earlier Rx by NBS-rWGS | Consensus phenotype avoidance by NBS-rWGS-based Rx |
|---|---|---|---|---|
| 1 | hyperinsulinemic hypoglycemia 1 | hypoglycemia; encephalopathy; acute kidney injury; seizures; respiratory distress | 93 | mostly |
| 2 | hypoglycemia; hypotonia | 109 | mostly | |
| 5 | pyridoxine-dependent epilepsy | seizures; encephalopathy; respiratory distress | 63 | mostly |
| 6 | hereditary fructosuria | FTT; liver dysfunction; vomiting; diarrhea; hypothyroidism; nephrotic syndrome; electrolyte abnormalities; metabolic acidosis | 79 | completely |
| 8 | seizures; encephalopathy; FTT; liver dysfunction; vomiting; hypoglycemia | 392 | completely | |
| 10 | infantile hypophosphatasia | hypotonia; hypercalcemia | 125 | partially |
| 13 | primary aldosteronism, seizures, and neurologic abnormalities | hypoglycemia; heart block | 6 | mostly |
| 15 | cystic fibrosisa | acute liver failure; hypoglycemia | 210 | partially |
| 16 | cardiorespiratory failure | 58 | partially | |
| 18 | cong. myasthenic syn. 1B, fast-channel | respiratory failure; hypotonia | 22 | partially |
| 21 | dihydrolipoamide dehydrogenase def. | lactic acidosis; metabolic anomalies; bleeding tendency | 7 | partially |
| 23 | ethylmalonic encephalopathy | encephalopathy | 123 | partially |
| 24 | factor XIIIA def. | hemiparesis; cephalohematoma; intracranial hemorrhage | 37 | completely |
| 26 | XL immunodysregulation, poly-endocrinopathy, and enteropathy | diarrhea | 42 | partially |
| 27 | FTT; enteropathy; skin inflammation; diabetes mellitus; hypothyroidism | 134 | mostly | |
| 28 | FTT; hyperinflammation; diabetes mellitus; pancreatitis; pruritis | 406 | mostly | |
| 57 | glycogen storage disease IIa | hypotonia; acute respiratory distress | 187 | partially |
| 58 | mitochondrial trifunctional protein def.a | cardiomyopathy; hypotonia; lactic acidosis | 5 | partially |
| 87 | methylmalonic aciduria and homocystinuria, cblC typea | neutropenia; feeding difficulties; metabolic abnormalities; hypotonia | 8 | mostly |
| 88 | methylmalonic aciduriaa | metabolic abnormalities | 31 | mostly |
| 91 | AD pseudohypoaldosteronism I | cardiac arrest; arrhythmia; electrolyte abnormalities | 17 | completely |
| 92 | respiratory failure; electrolyte abnormalities; feeding difficulties | 26 | completely | |
| 93 | ornithine transcarbamylase def.a | hyperornithinemia; electrolyte abnormalities; glucosuria; pancytopenia | 1,500 | mostly |
| 94 | hyperammonemia; decreased liver function; developmental regression | 835 | mostly | |
| 95 | metabolic acidosis; hypoglycemia; respiratory distress; hyperammonemia; oroticaciduria; uraciluria | 7 | mostly | |
| 97 | hyperammonemia; vomiting; seizures | 161 | mostly | |
| 99 | hypertonia; seizures; respiratory failure; anuria; coagulopathy; hypocalcemia; hyperammonemia; hypotension; | 2 | mostly | |
| 100 | propionic acidemiaa | propionic acidemia | 11 | mostly |
| 101 | cong. dis. of glycosylation 1t | hypoglycemia; FTT | 112 | mostly |
| 102 | pyruvate kinase def. | anemia; hyperbilirubinemia | 135 | partially |
| 103 | early-onset, vitamin B6-dependent epilepsy | seizures | 1,393 | mostly |
| 104 | pyridoxal phosphate-responsive seizures | status epilepticus | 1 | mostly |
| 105 | seizures; metabolic acidosis; respiratory distress; pancytopenia; hypertension | 3,484 | mostly | |
| 106 | familial hemophagocytic lymphohistiocytosis 2 | pancytopenia; fever; increased serum ferritin; increased inflammatory response | 92 | mostly |
| 107 | cong. myasthenic syn. 11 (acetylcholine receptor def.) | hypotonia; respiratory distress | 12 | partially |
| 120 | susc. to malignant hyperthermia 1 | malignant hyperthermia | 136 | completely |
| 123 | Shwachman-Diamond syn. 1 | pancytopenia | 2,290 | completely |
| 139 | long QT syn. 3 | aborted sudden cardiac death | 2,727 | mostly |
| 144 | sucrase-isomaltase def. | FTT; diarrhea | 183 | mostly |
| 145 | biotin-responsive basal ganglia dis. | seizures; encephalopathy; FTT | 37 | partially |
| 147 | cong. myasthenic syn. 18 | feeding difficulties; respiratory distress | 67 | partially |
Table S9 shows the genetic findings, full clinical features, GTRx-indicated interventions, and individual reviewer assessments in these individuals and in two in whom the consensus clinical utility was unknown. Reversible phenotypes attributable to the molecular diagnosis were identified from MIM, Genetic and Rare Diseases Information Center, and MEDLINE searches. Newborn treatments and their efficacy are from GTRx.20
ID, subject ID; Rx, treatment; DOL, day of life; FTT, failure to thrive; susc., susceptibility; syn., syndrome; dis., disease; def., deficiency; cong., congenital.
NBS RUSP disorders.
While not part of the prototypic NBS-rWGS system described herein, several optional future uses were modeled in detail for the data generated by NBS-rWGS. A desirable, optional secondary use was phenotype-informed diagnostic interpretation of the entire set of ∼5 million genomic variants upon physician order when symptoms arise later in life that suggest a genetic disorder (Figure 2C). This use case would require the WGS-derived variant call file (gVCF) to reside in a secure, parent-controlled data platform with transparent rights that convey to the child, persist across the lifetime, and that can be integrated into future medical care in a manner consistent with informed consent. Such a system has been developed by Luna PBC for natural history studies in several childhood genetic diseases. Data security consistent with the General Data Protection Regulation is implemented in overlapping envelopes, such as multi-factor authentication at account creation and login, and data encryption and data fragmentation between secure, isolated trusted environments. For example, each type of each person’s data is uniquely tagged with a character sequence determined by a one-way hash function that is designed to prevent reverse-engineering the given value. Data security controls are documented, audited, and tested regularly and evolve with time. In contrast, data privacy policies are codified through the platform design, with a set of transparent rights guaranteed to individual parents to access, correct, share, un-share, restrict, transport, and delete their newborn’s data. Integration into future medical care occurs through, for example, a pediatrician order for genome re-interpretation being placed in the EHR, and parental approval by cell phone for their child’s genome and EHR phenotype data to be accessed by the interpreting laboratory (Figure 2C). The resultant diagnostic report is returned to the EHR and genomics data platform, with links to management guidance (Genome-to-Treatment [GTRx]). Another optional secondary use would be genome re-screening at the individual’s request in early adulthood for pathogenic and likely pathogenic variants associated with later onset disorders that can be prevented or ameliorated by early treatment that are part of the ACMG secondary findings list.24,26,27,34
Discussion
Herein, we demonstrate the feasibility of NBS-rWGS for early treatment of several hundred childhood genetic disorders. While the concept of genomic NBS was part of the promise of the genome project, genomic knowledge and biotechnology and informatic tools hitherto lacked sufficient maturity for practical performance.21,23,24 In addition to an exponential decrease in WGS cost and improved time to result,20 three recent developments were instrumental in engendering NBS-rWGS. Firstly, broad diagnostic use of WES and WGS in affected children has allowed establishment of large databases of variant pathogenicity assessments, which provided qualified variants for genomic NBS.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 Secondly, very large sets of genomes and linked phenotypes are now queryable, enabling in silico training for retention of loci and variants with suitable analytic performance for NBS.40,42 Thirdly, a virtual, acute management guidance system for genetic disorders that cause critical illness in children both enabled examination of established NBS criteria in hundreds of disorders and serves as a general mechanism to translate positive results into treatments and, thereby, improved outcomes.20 Importantly, the prototypic NBS-rWGS system described herein has the capacity to accomplish both screening and diagnosis (in contrast to traditional NBS, which performs screening alone). The NBS-rWGS system also features root cause analysis that acts to refine and increase the screened variants, loci, and treatments with time, results of NBS-rWGS, and as variant databases and population datasets expand. While the latter was performed manually herein, each root cause can be codified and performed automatically in the future. In an era of rapid impending growth in cell and gene therapies and orphan drugs, NBS-rWGS will enable conditions with newly approved, highly effective interventions to be screened without delay.3 We anticipate that ∼1,000 genetic disorders may meet criteria for NBS by 2030.3 Unlike panel tests with fixed content, NBS-rWGS conditions can be added or removed dynamically on the basis of individual, regional, or societal preferences.
Feasibility pilots over the last decade found that many of the initial ethical, legal, and social implications (ELSIs) of genomic NBS were not observed in practice.23, 24, 25,46,63, 64, 65 Many ELSIs are solved by adherence to the original criteria for NBS disorder selection and requiring informed parental consent.31,46 Practical concerns, however, will be how to maintain the current 98% participation in NBS despite a requirement for consent and the allowable secondary use of genomic information. For example, the individual benefit of retaining uninterpreted genome information for future diagnostic analysis at onset of a suspected genetic illness upon physician request and individual consent should be weighed against the potential risks to privacy and confidentiality.24,26,27 Similarly, the individual risks versus societal benefits of enriching an anonymized database of NBS-rWGS variant pathogenicity with race, ethnicity, and ancestral group imputations should be considered. This is important because under-representation of many groups in existing variant frequency-by-zygosity sets makes pathogenicity assessment challenging. Broad participation and optional secondary uses will be facilitated through use of a secure, private, parent-controlled data platform with transparent rights that convey to the child at age of consent.66
The analytic performance of this prototype was sufficiently good to start prospective studies in late 2022. In 454,707 UK Biobank participants, NBS for 388 genetic diseases with a combined incidence of ∼0.8% had a 0.27% upper estimate of false positives, making a target PPV of 50% attainable. For HEMA, PPV was 71%. As additional UK Biobank and All of Us datasets are made available, it will be possible to calculate PPV for additional disorders and variants. RYR1, the locus with the second highest number of positive subjects (0.03%), is a risk factor for malignant hyperthermia, rather than highly penetrant. The Delphi panel retained it because the benefits were clear – avoidance of depolarizing muscle relaxants and having dantrolene on hand during general anesthesia – and one infant could have avoided malignant hyperthermia in our retrospective analysis. For 90% of disorders selected for NBS, the upper estimate of false positives was less than 1 in 100,000. This agreed with two prior estimates of the frequency of severe pediatric disease alleles in large genomic datasets.67,68 It should be noted, however, that these are not representative of global genomic diversity and evaluations were limited to nucleotide variants. For NBS disorders, such as type 1 spinal muscular atrophy (MIM: 253300), Duchenne muscular dystrophy (MIM: 310200), HEMA, and alpha thalassemia (MIM: 604131), the most prevalent causes are deletions. A low false positive rate was achieved by retention of only rare, known pathogenic, and likely pathogenic variants and informed by root cause analysis. In 119 affected children who had been diagnosed by rWGS, 87% were positive by NBS-rWGS. The diagnostic sensitivity of NBS-rWGS can be further increased by inclusion of variants identified by artificial intelligence-assisted literature curation or interpretation (Table S10) and imputation of truncating variants in known loss-of-function loci.15 However, increasing the number of variants screened will increase the false positive rate, reinforcing the need for ongoing root cause analysis.
Case-based factual-counterfactual analyses have been useful in demonstrating the net clinical utility and cost effectiveness of diagnostic rWGS in newborns compared with standard genetic testing.9, 10, 11, 12, 13, 14, 15, 16 Here, we used such methods to examine the potential of NBS-rWGS to improve outcomes when compared with first tier use of Dx-rWGS. Had NBS-indicated treatments been started on day of life 5, it was likely NBS-rWGS could have completely avoided morbidity and improved outcomes in seven of 2,208 probands, mostly avoided these in 21 infants, and partially in 13 infants. For example, infant 24 (factor XIIIA, F13A1 [MIM: 134570], deficiency [MIM: 613225]) was admitted at 5 weeks of age with hemiparesis following an intracranial hemorrhage. Initiation of factor XIII replacement in the first week of life could have avoided this catastrophic event. Most neonates in ICUs, however, do not receive first tier Dx-rWGS. They experience considerably longer diagnostic odysseys and may die undiagnosed.19,69 Such neonates could have greater morbidity and mortality associated with further delayed treatment and could derive additional benefit from NBS-rWGS. Large prospective studies are now needed to evaluate the clinical utility and cost effectiveness of NBS-rWGS, particularly for disorders in which treatment would not be instituted until symptom onset and loci with considerable phenotypic heterogeneity. Examples are subjects 71–83 and 124–133 with variants in KCNQ2 [MIM: 602235], SCN1A [MIM: 182389], and SCN2A [MIM: 182390], loci that are associated both with epileptic encephalopathies (developmental and epileptic encephalopathy 7, DEE7 [MIM: 613720], DEE6B [MIM: 619317], and DEE11 [MIM: 613721], respectively) and benign seizures (benign neonatal seizures 1 [MIM: 121200], familial febrile seizures 3A [MIM: 604403], and benign familial infantile seizures 3 [MIM: 607745], respectively). While positive results would increase scrutiny for seizures, enable prompt, targeted, anti-seizure medicine therapy, and reduce iatrogenesis,70,71 prospective studies are needed to evaluate the positive predictive value of NBS for channelopathies. It is important to note that the panel of disorders presented herein is the initial version intended to be suitable for evaluation in prospective clinical studies. As evidenced by the group B disorders, many rare genetic diseases currently lack sufficient published data regarding natural history or treatment efficacy to judge their viability for newborn screening. We invite individuals and organizations both to nominate disorders for consideration for inclusion and to communicate concerns regarding the 388 current NBS-rWGS disorders.
Cost effectiveness studies of NBS-rWGS have not yet been performed. While NBS-rWGS is intended to supplement NBS-MS, not replace it, the current cost of NBS-MS for the 35 core disorders on the RUSP provides a reference point for what is likely to be acceptable for public-health-funded NBS-rWGS. Most states publish the fees charged for NBS-MS, which represent part of the total cost. The highest such fee is $220 per newborn. Diagnostic rWGS costs RCIGM ∼$8,500 per newborn. However, the interpretation burden of NBS-rWGS is about one thousandth that of Dx-rWGS and several biotechnology companies have indicated that $100 rWGS will be possible in the relatively near future.20 The prerequisites for inexpensive NBS-rWGS are performance at massive scale and near complete automation.
Because NBS-rWGS and NBS-MS use orthogonal methods, they have considerable potential complementarity.23 The Newborn Sequencing in Genomic Medicine and Public Health (NSIGHT) program found that NBS-MS was more sensitive for RUSP conditions than NBS by whole-exome sequencing (WES): WES had 88% sensitivity for RUSP disorders in 691 positive samples by NBS-MS.43,49,50 Separately, Cho et al. reported 93% sensitivity of WES in 81 children with core NBS metabolic disorders.53 Battacharjee et al. reported 75% sensitivity of a gene panel in 36 children with the same conditions.51 Bodian et al. reported 89% concordance of WGS and NBS in 1,696 newborns.52 However, the NSIGHT projects also found that WES identified three NBS-related disorders in 159 infants and four “actionable” findings in 106 infants that were missed by NBS-MS.43,49 Very recently, Jian et al. reported that NBS-WGS for 251 genes (with 16–24 weeks turnaround time) had superior analytic performance than traditional MS for 51 disorders in 321 newborns.72 Confirmatory testing showed a false positive for 3-methylcrotonyl-CoA carboxylase 1 deficiency [MIM: 210200] identified by traditional MS was correctly identified as an MCCC1 [MIM: 609010] carrier by NBS-WGS, and six newborns with GJB2 [MIM: 121011]-associated autosomal recessive deafness 1A [MIM: 220290] or MT-RNR1 [MIM: 561000]-associated aminoglycoside-induced deafness [MIM: 580000] were identified by NBS-WGS and not by traditional NBS. Herein, NBS-rWGS identified 15 findings that were not reported by Dx-rWGS. Complementarity of NBS-rWGS and NBS-MS was evident in 15 children herein. In two newborns with positive NBS T lymphocyte receptor excision circle assays, Dx-rWGS rapidly identified the specific immunodeficiency loci and variants, knowledge of which is needed for precision therapy. Fifteen children were diagnosed with RUSP disorders by rWGS, which were screened but not detected by NBS-MS. NBS-rWGS for RUSP disorders will be particularly useful in premature and low birthweight newborns, in whom NBS-MS suffers frequent false positives and negatives.23,45
In summary, NBS-rWGS is feasible for hundreds of severe, early childhood-onset genetic disorders that progress rapidly if untreated and have effective therapies. Given the very rapid evolution of genome science and gene therapy, NBS-rWGS requires an open system to remain current.20 Acceptable analytic performance and turnaround time were achieved by combining screening, diagnosis, large genome-phenotype datasets, and learning feedback loops. We invite groups worldwide to join the BeginNGS (Newborn Genomic Sequencing) consortium in implementation studies of NBS-rWGS in diverse populations.
Acknowledgments
We thank David Hale for reviewing this manuscript. This manuscript was supported by NIH grants UL1TR002550 from NCATS to E.J. Topol (with sub-award to S.F.K.) and R01HD101540, Rady Children’s Institute for Genomic Medicine, Rady Children’s Hospital, a grant from Alexion Pharmaceuticals, Horizon Therapeutics, Sarepta Therapeutics, and Travere Therapeutics and in-kind support from Alexion Pharmaceuticals, Illumina Inc., TileDB Inc., Genomenon Inc., and Fabric Genomics Inc. A Deo lumen, ab amicis auxilium.
Declaration of interests
K.P.H., C.M.K., S.S.M., and D.T. are employees and shareholders of Illumina, Inc. G.D,A., B.M., S.L., and T.D. are employees and shareholders of Alexion Pharmaceuticals. E.F. and M.G.R. are employees and shareholders of Fabric Genomics, Inc. M.K. and S.S. are employees and shareholders of Genomenon, Inc. C.K., G.P., S.S., S.P., and A.R.W. are employees and shareholders of TileDB, Inc. S.K. is an employee and shareholder of Luna PBC, Inc. S.K. has filed a patent related to this work.
Published: August 24, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.08.003.
Web resources
BeginNGS consortium, https://radygenomics.org/begin-ngs-newborn-sequencing/
BeginNGS consortium (in Greek), https://beginnings.gr/
Genome to Treatment (GTRx), https://gtrx.rbsapp.net/
The Newborn Screening Condition Resource, https://nbstrn.org/tools/nbs-cr
The UK NBS panel, https://www.gov.uk/guidance/newborn-blood-spot-screening-programme-overview#conditions-screened-for
The US Recommended Uni-form Newborn Screening Panel, https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html
Supplemental information
Data and code availability
Consented proband and parent data analyzed in this study and non-human subjects data generated during this study are available at the Longitudinal Pediatric Data Resource (LPDR) under accession code nbs000003.v1.p at https://nbstrn.org/. Qualified researchers can obtain access by registration at https://nbstrn.org/login?token-expired=true&rel=/tools/lpdr. There are restrictions to the availability of raw individual data because of data privacy and confidentiality laws. Anonymized and pseudonymized individual data generated in this study, subject to the terms of informed written consent documents and state and federal laws, are provided in the supplemental information.
GTRx and the GTRx REDCap instance is available at https://gtrx.rbsapp.net/ and code is available from Christian Hansen (chansen@rchsd.org) and at https://github.com/rao-madhavrao-rcigm/gtrx. The DRAGEN Platform and Illumina Connected Analytics are available from Illumina (Shyamal Mehtalia, smehtalia@illumina.com). GEM is available from Fabric Genomics (info@fabricgenomics.com). TileDB v2.8.0 is available at https://github.com/TileDB-Inc/TileDB. TileDB-VCF v0.15.0 is available at https://github.com/tiledb-inc/tiledb-vcf.
References
- 1.Therrell B.L., Padilla C.D., Loeber J.G., Kneisser I., Saadallah A., Borrajo G.J.C., Adams J. Current status of newborn screening worldwide. Semin. Perinatol. 2015;39:171–187. doi: 10.1053/j.semperi.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 2.American Academy of Pediatrics Newborn Screening Task Force Newborn screening: a blueprint for the future. Pediatrics. 2000;106(Suppl. 2):383–427. [Google Scholar]
- 3.Kayki-Mutlu G., Aksoyalp Z.S., Wojnowski L., Michel M.C. A year in pharmacology: new drugs approved by the US Food and Drug Administration in 2021. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2022;395:867–885. doi: 10.1007/s00210-022-02250-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sontag M.K., Yusuf C., Grosse S.D., Edelman S., Miller J.I., McKasson S., Kellar-Guenther Y., Gaffney M., Hinton C.F., Cuthbert C., et al. Infants with Congenital Disorders Identified Through Newborn Screening — United States, 2015–2017. MMWR Morb. Mortal. Wkly. Rep. 2020;69:1265–1268. doi: 10.15585/mmwr.mm6936a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention CDC Impact of expanded newborn screening—United States, 2006. MMWR Morb. Mortal. Wkly. Rep. 2008;57:1012–1015. [PubMed] [Google Scholar]
- 6.Amberger J.S., Bocchini C.A., Scott A.F., Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019;47:D1038–D1043. doi: 10.1093/nar/gky1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willig L.K., Petrikin J.E., Smith L.D., Saunders C.J., Thiffault I., Miller N.A., Soden S.E., Cakici J.A., Herd S.M., Twist G., et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 2015;3:377–387. doi: 10.1016/S2213-2600(15)00139-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrikin J.E., Cakici J.A., Clark M.M., Willig L.K., Sweeney N.M., Farrow E.G., Saunders C.J., Thiffault I., Miller N.A., Zellmer L., et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom. Med. 2018;3:6. doi: 10.1038/s41525-018-0045-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farnaes L., Hildreth A., Sweeney N.M., Clark M.M., Chowdhury S., Nahas S., Cakici J.A., Benson W., Kaplan R.H., Kronick R., et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom. Med. 2018;3:10. doi: 10.1038/s41525-018-0049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark M.M., Stark Z., Farnaes L., Tan T.Y., White S.M., Dimmock D., Kingsmore S.F. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 2018;3:16. doi: 10.1038/s41525-018-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanford E.F., Clark M.M., Farnaes L., Williams M.R., Perry J.C., Ingulli E.G., Sweeney N.M., Doshi A., Gold J.J., Briggs B., et al. Rapid Whole Genome Sequencing Has Clinical Utility in Children in the PICU. Pediatr. Crit. Care Med. 2019;20:1007–1020. doi: 10.1097/PCC.0000000000002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark M.M., Hildreth A., Batalov S., Ding Y., Chowdhury S., Watkins K., Ellsworth K., Camp B., Kint C.I., Yacoubian C., et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 2019;11:eaat6177. doi: 10.1126/scitranslmed.aat6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kingsmore S.F., Cakici J.A., Clark M.M., Gaughran M., Feddock M., Batalov S., Bainbridge M.N., Carroll J., Caylor S.A., Clarke C., et al. A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am. J. Hum. Genet. 2019;105:719–733. doi: 10.1016/j.ajhg.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dimmock D.P., Clark M.M., Gaughran M., Cakici J.A., Caylor S.A., Clarke C., Feddock M., Chowdhury S., Salz L., Cheung C., et al. An RCT of Rapid Genomic Sequencing among Seriously Ill Infants Results in High Clinical Utility, Changes in Management, and Low Perceived Harm. Am. J. Hum. Genet. 2020;107:942–952. doi: 10.1016/j.ajhg.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De La Vega F.M., Chowdhury S., Moore B., Frise E., McCarthy J., Hernandez E.J., Wong T., James K., Guidugli L., Agrawal P.B., et al. Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases. Genome Med. 2021;13:153. doi: 10.1186/s13073-021-00965-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimmock D., Caylor S., Waldman B., Benson W., Ashburner C., Carmichael J.L., Carroll J., Cham E., Chowdhury S., Cleary J., et al. Project Baby Bear: Rapid precision care incorporating rWGS in 5 California children's hospitals demonstrates improved clinical outcomes and reduced costs of care. Am. J. Hum. Genet. 2021;108:1231–1238. doi: 10.1016/j.ajhg.2021.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maron J.L., Kingsmore S.F., Wigby K., Chowdhury S., Dimmock D., Poindexter B., Suhrie K., Vockley J., Diacovo T., Gelb B.D., et al. Novel Variant Findings and Challenges Associated With the Clinical Integration of Genomic Testing: An Interim Report of the Genomic Medicine for Ill Neonates and Infants (GEMINI) Study. JAMA Pediatr. 2021;175:e205906. doi: 10.1001/jamapediatrics.2020.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sweeney N.M., Nahas S.A., Chowdhury S., Batalov S., Clark M., Caylor S., Cakici J., Nigro J.J., Ding Y., Veeraraghavan N., et al. Rapid whole genome sequencing impacts care and resource utilization in infants with congenital heart disease. NPJ Genom. Med. 2021;6:38. doi: 10.1038/s41525-021-00192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kingsmore S.F., Cole F.S. The role of genome sequencing in the NICU. Annu. Rev. Genomics Hum. Genet. 2022;23 doi: 10.1146/annurev-genom-120921-103442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Owen M.J., Lefebvre S., Hansen C., Kunard C.M., Dimmock D.P., Smith L.D., Scharer G., Mardach R., Willis M.J., Feigenbaum A., et al. An automated 13.5 hour system for scalable diagnosis and acute management guidance for genetic diseases. Nat. Commun. 2022;13:4057. doi: 10.1038/s41467-022-31446-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins F.S. Harper Collins; 2010. The Language of Life: DNA and the Revolution in Personalized Medicine; p. 208. [Google Scholar]
- 22.Kingsmore S.F., Lantos J.D., Dinwiddie D.L., Miller N.A., Soden S.E., Farrow E.G., Saunders C.J. Next-generation community genetics for low- and middle-income countries. Genome Med. 2012;4:25. doi: 10.1186/gm324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woerner A.C., Gallagher R.C., Vockley J., Adhikari A.N. The Use of Whole Genome and Exome Sequencing for Newborn Screening: Challenges and Opportunities for Population Health. Front. Pediatr. 2021;9:663752. doi: 10.3389/fped.2021.663752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biesecker L.G., Green E.D., Manolio T., Solomon B.D., Curtis D. Should all babies have their genome sequenced at birth? BMJ. 2021;375:2679. doi: 10.1136/bmj.n2679. [DOI] [PubMed] [Google Scholar]
- 25.Pichini A., Ahmed A., Patch C., Bick D., Leblond M., Kasperaviciute D., Deen D., Wilde S., Garcia Noriega S., Matoko C., et al. Developing a National Newborn Genomes Program: An Approach Driven by Ethics, Engagement and Co-design. Front. Genet. 2022;13:866168. doi: 10.3389/fgene.2022.866168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ceyhan-Birsoy O., Machini K., Lebo M.S., Yu T.W., Agrawal P.B., Parad R.B., Holm I.A., McGuire A., Green R.C., Beggs A.H., Rehm H.L. A curated gene list for reporting results of newborn genomic sequencing. Genet. Med. 2017;19:809–818. doi: 10.1038/gim.2016.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milko L.V., O'Daniel J.M., DeCristo D.M., Crowley S.B., Foreman A.K.M., Wallace K.E., Mollison L.F., Strande N.T., Girnary Z.S., Boshe L.J., et al. An Age-Based Framework for Evaluating Genome-Scale Sequencing Results in Newborn Screening. J. Pediatr. 2019;209:68–76. doi: 10.1016/j.jpeds.2018.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeCristo D.M., Milko L.V., O'Daniel J.M., Foreman A.K.M., Mollison L.F., Powell B.C., Powell C.M., Berg J.S. Actionability of commercial laboratory sequencing panels for newborn screening and the importance of transparency for parental decision-making. Genome Med. 2021;13:50. doi: 10.1186/s13073-021-00867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bick D., Bick S.L., Dimmock D.P., Fowler T.A., Caulfield M.J., Scott R.H. An online compendium of treatable genetic disorders. Am. J. Med. Genet. C Semin. Med. Genet. 2021;187:48–54. doi: 10.1002/ajmg.c.31874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brower A., Chan K., Taylor J., Wiebenga R., Tona G., Unnikumaran Y., Barnes L. Accelerating the Pace of Newborn Screening Research to Advance Disease Understanding and Improve Health Outcomes: Key Efforts of the Newborn Screening Translational Research Network (NBSTRN) Dela. J. Public Health. 2021;7:36–37. doi: 10.32481/djph.2021.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson J.M.G., Jungner G., World Health Organization . World Health Organization; 1968. Principles and Practice of Screening for Disease.https://apps.who.int/iris/handle/10665/37650 [Google Scholar]
- 32.Watson M.S., Mann M.Y., Lloyd-Puryear M.A., Rinaldo P., Howell R.R. Newborn Screening: Towards a Uniform Screening Panel and System. Genet. Med. 2006;8:1S–11S. doi: 10.1097/01.gim.0000223891.82390.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kemper A.R., Green N.S., Calonge N., Lam W.K.K., Comeau A.M., Goldenberg A.J., Ojodu J., Prosser L.A., Tanksley S., Bocchini J.A., Jr. Decision-making process for conditions nominated to the recommended uniform screening panel: statement of the US Department of Health and Human Services Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children. Genet. Med. 2014;16:183–187. doi: 10.1038/gim.2013.98. [DOI] [PubMed] [Google Scholar]
- 34.Miller D.T., Lee K., Chung W.K., Gordon A.S., Herman G.E., Klein T.E., Stewart D.R., Amendola L.M., Adelman K., Bale S.J., et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG) Genet. Med. 2021;23:1381–1390. doi: 10.1038/s41436-021-01172-3. [DOI] [PubMed] [Google Scholar]
- 35.Gudmundsson S., Singer-Berk M., Watts N.A., Phu W., Goodrich J.K., Solomonson M., O'Donnell-Luria A., Genome Aggregation Database Consortium. MacArthur D.G. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2022;43:1012–1030. doi: 10.1002/humu.24309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding Y., Owen M., Le J., Batalov S., Chau K., Van Der Kraan L., Bezares-Orin Z., Zhu Z., Veeraraghavan N., Gleeson J., et al. Scalable, high quality, whole genome sequencing from archived, newborn, dried blood spots. medRxiv. 2022 doi: 10.1101/2022.07.27.22278102. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Byrska-Bishop M., Evani U.S., Zhao X., Basile A.O., Abel H.J., Regier A.A., Corvelo A., Clarke W.E., Musunuri R., Nagulapalli K., et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv. 2021 doi: 10.1101/2021.02.06.430068. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zook J.M., McDaniel J., Olson N.D., Wagner J., Parikh H., Heaton H., Irvine S.A., Trigg L., Truty R., McLean C.Y., et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol. 2019;37:561–566. doi: 10.1038/s41587-019-0074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papadopoulos S., Datta K., Madden S., Mattson T. The TileDB Array Data Storage Manager. Proceedings VLDB Endowment. 2016;10:349–360. [Google Scholar]
- 40.Backman J.D., Li A.H., Marcketta A., Sun D., Mbatchou J., Kessler M.D., Benner C., Liu D., Locke A.E., Balasubramanian S., et al. Exome sequencing and analysis of 454, 787 UK Biobank participants. Nature. 2021;599:628–634. doi: 10.1038/s41586-021-04103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fry A., Littlejohns T.J., Sudlow C., Doherty N., Adamska L., Sprosen T., Collins R., Allen N.E. Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants With Those of the General Population. Am. J. Epidemiol. 2017;186:1026–1034. doi: 10.1093/aje/kwx246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szustakowski J.D., Balasubramanian S., Kvikstad E., Khalid S., Bronson P.G., Sasson A., Wong E., Liu D., Wade Davis J., Haefliger C., et al. Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat. Genet. 2021;53:942–948. doi: 10.1038/s41588-021-00885-0. [DOI] [PubMed] [Google Scholar]
- 43.Wojcik M.H., Zhang T., Ceyhan-Birsoy O., Genetti C.A., Lebo M.S., Yu T.W., Parad R.B., Holm I.A., Rehm H.L., Beggs A.H., et al. Discordant results between conventional newborn screening and genomic sequencing in the BabySeq Project. Genet. Med. 2021;23:1372–1375. doi: 10.1038/s41436-021-01146-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braun D., Braun E., Chiu V., Burgos A.E., Gupta M., Volodarskiy M., Getahun D. Trends in Neonatal Intensive Care Unit Utilization in a Large Integrated Health Care System. JAMA Netw. Open. 2020;3:e205239. doi: 10.1001/jamanetworkopen.2020.5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwon C., Farrell P.M. The magnitude and challenge of false-positive newborn screening test results. Arch. Pediatr. Adolesc. Med. 2000;154:714–718. doi: 10.1001/archpedi.154.7.714. [DOI] [PubMed] [Google Scholar]
- 46.Clayton E.W. Currents in contemporary ethics. State run newborn screening in the genomic era, or how to avoid drowning when drinking from a fire hose. J. Law Med. Ethics. 2010;38:697–700. doi: 10.1111/j.1748-720X.2010.00522.x. [DOI] [PubMed] [Google Scholar]
- 47.Hall P.L., Marquardt G., McHugh D.M.S., Currier R.J., Tang H., Stoway S.D., Rinaldo P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014;16:889–895. doi: 10.1038/gim.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.ACMG ACT Sheets and Algorithms. American College of Medical Genetics and Genomics; 2001. https://www.ncbi.nlm.nih.gov/books/NBK55832/ [PubMed] [Google Scholar]
- 49.Roman T.S., Crowley S.B., Roche M.I., Foreman A.K.M., O'Daniel J.M., Seifert B.A., Lee K., Brandt A., Gustafson C., DeCristo D.M., et al. Genomic Sequencing for Newborn Screening: Results of the NC NEXUS Project. Am. J. Hum. Genet. 2020;107:596–611. doi: 10.1016/j.ajhg.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adhikari A.N., Gallagher R.C., Wang Y., Currier R.J., Amatuni G., Bassaganyas L., Chen F., Kundu K., Kvale M., Mooney S.D., et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat. Med. 2020;26:1392–1397. doi: 10.1038/s41591-020-0966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhattacharjee A., Sokolsky T., Wyman S.K., Reese M.G., Puffenberger E., Strauss K., Morton H., Parad R.B., Naylor E.W. Development of DNA confirmatory and high-risk diagnostic testing for newborns using targeted next-generation DNA sequencing. Genet. Med. 2015;17:337–347. doi: 10.1038/gim.2014.117. [DOI] [PubMed] [Google Scholar]
- 52.Bodian D.L., Klein E., Iyer R.K., Wong W.S.W., Kothiyal P., Stauffer D., Huddleston K.C., Gaither A.D., Remsburg I., Khromykh A., et al. Utility of whole-genome sequencing for detection of newborn screening disorders in a population cohort of 1, 696 neonates. Genet. Med. 2016;18:221–230. doi: 10.1038/gim.2015.111. [DOI] [PubMed] [Google Scholar]
- 53.Cho Y., Lee C.H., Jeong E.G., Kim M.H., Hong J.H., Ko Y., Lee B., Yun G., Kim B.J., Jung J., et al. Prevalence of rare genetic variations and their implications in NGS-data interpretation. Sci. Rep. 2017;7:9810. doi: 10.1038/s41598-017-09247-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wagner J., Olson N.D., Harris L., Khan Z., Farek J., Mahmoud M., Stankovic A., Kovacevic V., Yoo B., Miller N., et al. Benchmarking challenging small variants with linked and long reads. Cell Genomics. 2022;2:100128. doi: 10.1016/j.xgen.2022.100128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Payne A.B., Miller C.H., Kelly F.M., Michael Soucie J., Craig Hooper W. The CDC Hemophilia A Mutation Project (CHAMP) mutation list: a new online resource. Hum. Mutat. 2013;34:E2382–E2391. doi: 10.1002/humu.22247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Antonarakis S.E., Kazazian H.H., Tuddenham E.G. Molecular etiology of factor VIII deficiency in hemophilia A. Hum. Mutat. 1995;5:1–22. doi: 10.1002/humu.1380050102. [DOI] [PubMed] [Google Scholar]
- 57.Bell C.J., Dinwiddie D.L., Miller N.A., Hateley S.L., Ganusova E.E., Mudge J., Langley R.J., Zhang L., Lee C.C., Schilkey F.D., et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 2011;3:65ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barbaro M., Soardi F.C., Östberg L.J., Persson B., de Mello M.P., Wedell A., Lajic S. In vitro functional studies of rare CYP21A2 mutations and establishment of an activity gradient for nonclassic mutations improve phenotype predictions in congenital adrenal hyperplasia. Clin. Endocrinol. 2015;82:37–44. doi: 10.1111/cen.12526. [DOI] [PubMed] [Google Scholar]
- 59.de Paula Michelatto D., Karlsson L., Lusa A.L.G., Silva C.D.M., Östberg L.J., Persson B., Guerra-Júnior G., de Lemos-Marini S.H.V., Barbaro M., de Mello M.P., Lajic S. Functional and structural consequences of nine CYP21A2 mutations ranging from very mild to severe effects. Int. J. Endocrinol. 2016;2016:4209670. doi: 10.1155/2016/4209670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whiffin N., Minikel E., Walsh R., O'Donnell-Luria A.H., Karczewski K., Ing A.Y., Barton P.J.R., Funke B., Cook S.A., MacArthur D., Ware J.S. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017;19:1151–1158. doi: 10.1038/gim.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li R., Chang C., Tanigawa Y., Narasimhan B., Hastie T., Tibshirani R., Rivas M.A. Fast Numerical Optimization for Genome Sequencing Data in Population Biobanks. Bioinformatics. 2021;37:4148–4155. doi: 10.1093/bioinformatics/btab452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang L., Zheng Z., Fang H., Yang J. A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 2021;53:1616–1621. doi: 10.1038/s41588-021-00954-4. [DOI] [PubMed] [Google Scholar]
- 63.Genetti C.A., Schwartz T.S., Robinson J.O., VanNoy G.E., Petersen D., Pereira S., Fayer S., Peoples H.A., Agrawal P.B., Betting W.N., et al. Parental interest in genomic sequencing of newborns: enrollment experience from the BabySeq Project. Genet. Med. 2019;21:622–630. doi: 10.1038/s41436-018-0105-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pereira S., Robinson J.O., Gutierrez A.M., Petersen D.K., Hsu R.L., Lee C.H., Schwartz T.S., Holm I.A., Beggs A.H., Green R.C., et al. Perceived Benefits, Risks, and Utility of Newborn Genomic Sequencing in the BabySeq Project. Pediatrics. 2019;143:S6–S13. doi: 10.1542/peds.2018-1099C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moultrie R.R., Paquin R., Rini C., Roche M.I., Berg J.S., Powell C.M., Lewis M.A. Parental Views on Newborn Next Generation Sequencing: Implications for Decision Support. Matern. Child Health J. 2020;24:856–864. doi: 10.1007/s10995-020-02953-z. [DOI] [PubMed] [Google Scholar]
- 66.Miyachi K., Mackey T.K. hOCBS: a privacy-preserving blockchain framework for healthcare data leveraging an on-chain and offchain system design. Inf. Process. Manag. 2021;58:102535. [Google Scholar]
- 67.Gold N.B., Harrison S.M., Rowe J.H., Gold J., Furutani E., Biffi A., Duncan C.N., Shimamura A., Lehmann L.E., Green R.C. Low frequency of treatable pediatric disease alleles in gnomAD: An opportunity for future genomic screening of newborns. HGG Adv. 2022;3:100059. doi: 10.1016/j.xhgg.2021.100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen R., Shi L., Hakenberg J., Naughton B., Sklar P., Zhang J., Zhou H., Tian L., Prakash O., Lemire M., et al. Analysis of 589, 306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotechnol. 2016;34:531–538. doi: 10.1038/nbt.3514. [DOI] [PubMed] [Google Scholar]
- 69.Diaby V., Babcock A., Huang Y., Moussa R.K., Espinal P.S., Janvier M., Soler D., Gupta A., Jayakar P., Diaz-Barbosa M., et al. Real-world economic evaluation of prospective rapid whole-genome sequencing compared to a matched retrospective cohort of critically ill pediatric patients in the United States. Pharmacogenomics J. 2022;18:1–7. doi: 10.1038/s41397-022-00277-5. [DOI] [PubMed] [Google Scholar]
- 70.de Lange I.M., Gunning B., Sonsma A.C.M., van Gemert L., van Kempen M., Verbeek N.E., Nicolai J., Knoers N.V.A.M., Koeleman B.P.C., Brilstra E.H. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A-related seizure phenotypes. Epilepsia. 2018;59:1154–1165. doi: 10.1111/epi.14191. [DOI] [PubMed] [Google Scholar]
- 71.Beltrán-Corbellini Á., Aledo-Serrano Á., Møller R.S., Pérez-Palma E., García-Morales I., Toledano R., Gil-Nagel A. Epilepsy genetics and precision medicine in adults: a new landscape for developmental and epileptic encephalopathies. Front. Neurol. 2022;13:777115. doi: 10.3389/fneur.2022.777115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jian M., Wang X., Sui Y., Fang M., Feng C., Huang Y., Liu C., Guo R., Guan Y., Gao Y., et al. A pilot study of assessing whole genome sequencing in newborn screening in unselected children in China. Clin. Transl. Med. 2022;12:e843. doi: 10.1002/ctm2.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Consented proband and parent data analyzed in this study and non-human subjects data generated during this study are available at the Longitudinal Pediatric Data Resource (LPDR) under accession code nbs000003.v1.p at https://nbstrn.org/. Qualified researchers can obtain access by registration at https://nbstrn.org/login?token-expired=true&rel=/tools/lpdr. There are restrictions to the availability of raw individual data because of data privacy and confidentiality laws. Anonymized and pseudonymized individual data generated in this study, subject to the terms of informed written consent documents and state and federal laws, are provided in the supplemental information.
GTRx and the GTRx REDCap instance is available at https://gtrx.rbsapp.net/ and code is available from Christian Hansen (chansen@rchsd.org) and at https://github.com/rao-madhavrao-rcigm/gtrx. The DRAGEN Platform and Illumina Connected Analytics are available from Illumina (Shyamal Mehtalia, smehtalia@illumina.com). GEM is available from Fabric Genomics (info@fabricgenomics.com). TileDB v2.8.0 is available at https://github.com/TileDB-Inc/TileDB. TileDB-VCF v0.15.0 is available at https://github.com/tiledb-inc/tiledb-vcf.