

Abstract
Motivation
A single monoclonal broadly neutralizing antibody (bnAb) regimen was recently evaluated in two randomized trials for prevention efficacy against HIV-1 infection. Subsequent trials will evaluate combination bnAb regimens (e.g. cocktails, multi-specific antibodies), which demonstrate higher potency and breadth in vitro compared to single bnAbs. Given the large number of potential regimens, methods for down-selecting these regimens into efficacy trials are of great interest.
Results
We developed Super LeArner Prediction of NAb Panels (SLAPNAP), a software tool for training and evaluating machine learning models that predict in vitro neutralization sensitivity of HIV Envelope (Env) pseudoviruses to a given single or combination bnAb regimen, based on Env amino acid sequence features. SLAPNAP also provides measures of variable importance of sequence features. By predicting bnAb coverage of circulating sequences, SLAPNAP can improve ranking of bnAb regimens by their potential prevention efficacy. In addition, SLAPNAP can improve sieve analysis by defining sequence features that impact bnAb prevention efficacy.
Availabilityand implementation
SLAPNAP is a freely available docker image that can be downloaded from DockerHub (https://hub.docker.com/r/slapnap/slapnap). Source code and documentation are available at GitHub (https://github.com/benkeser/slapnap and https://benkeser.github.io/slapnap/).
1 Introduction
Extensive research has been conducted on passive administration of monoclonal broadly neutralizing antibodies (bnAbs) with the objective of preventing HIV-1 infection (Morris and Mkhize, 2017). BnAbs target epitopes on the HIV-1 envelope (Env) glycoprotein, and recent work has shown that combination bnAb regimens can neutralize most clinical HIV-1 isolates in genetically diverse Env panels (e.g. McCoy and Burton, 2017; Sok and Burton, 2018). BnAb regimens have also been shown to prevent SHIV infection in non-human primate models (Gautam et al., 2016; Hessell et al., 2018; Pegu et al., 2019). These developments position bnAbs as promising tools for the prevention of HIV-1, as well as other infectious diseases, in the near future (Karuna and Corey, 2020).
The two harmonized, randomized, placebo-controlled Antibody Mediated Prevention (AMP) efficacy trials of a single bnAb regimen, VRC01, have been completed (Corey et al., 2021). These trials showed modest overall prevention efficacy (∼20%) and that VRC01 neutralization sensitivity of the acquired virus (IC80) was a strong biomarker of prevention efficacy, with zero estimated efficacy against resistant viruses (IC80 > 3 µg/ml) and 75.4% estimated efficacy (95% confidence interval 45.5–88.9%) against the most sensitive viruses (IC80 < 1 µg/ml). These results highlight the critical need to develop bnAb regimens with broader neutralization coverage of circulating strains, an objective that will require understanding of the neutralization breadth and potency of various candidate bnAb regimens against representative HIV-1 Env panels (Wagh et al., 2016, 2018). Several bnAb combinations targeting distinct Env epitopes have been identified that exhibit greater neutralization breadth and potency than their constituent single bnAbs, with in vitro neutralization coverage rates approaching 100% (Doria-Rose et al., 2012; Kong et al., 2015; Wagh et al., 2016). Thus, ongoing and future clinical trials are focusing on bnAb combination regimens or multi-specific bnAb regimens (e.g. NCT04212091, NCT03928821).
Analysis of bnAb prevention efficacy trials such as AMP can elucidate the extent to which in vitro markers of neutralization breadth and sensitivity predict in vivo prevention efficacy. Such analyses may support validation of a bnAb-based surrogate endpoint for HIV-1 infection, which could accelerate the development of new prevention modalities, such as new bnAb regimens or novel vaccines that induce bnAbs (Liao et al., 2013; Moody et al., 2016; Williams et al., 2017; Zhang et al., 2016). To realize this potential, analyses of randomized trials will need to be informed by in vitro analyses of bnAb breadth and potency. For example, in preparation for the viral sequence sieve analysis of the AMP trials, Magaret et al. developed models predicting VRC01 neutralization sensitivity using Env amino acid (AA) sequence features (Magaret et al., 2019), based on HIV-1 gp160 pseudoviruses from the Compile, Analyze and Tally NAb Panels (CATNAP) database (Yoon et al., 2015). The super learning ensemble machine learning approach (van der Laan et al., 2007) used to predict right-censored 50% inhibitory concentration titer (IC50) for each pseudovirus yielded a cross-validated area under the receiver operating characteristic (ROC) curve (AUC) of 0.881 (95% confidence interval: 0.813–0.948). Magaret et al. also identified important sequence features for predicting VRC01 sensitivity with the goal to enable the AMP sieve analysis to focus on top-ranked features, thereby improving statistical power. Bricault et al. (2019) conducted similar variable importance signature analyses that generalized to all bnAbs across four antibody classes.
Given the movement toward combination or multi-specific bnAb regimens, we developed Super LeArner Prediction of NAb Panels (SLAPNAP), a publicly available, containerized pipeline that can perform an end-to-end analysis of in vitro neutralization data for bnAb combinations. SLAPNAP analyses of in vitro neutralization data can improve the down-selection of combination or multi-specific bnAb regimens for future efficacy trials by enabling more accurate and precise estimates of potential prevention efficacy. While the normative approach to estimate potential prevention efficacy uses direct in vitro neutralization data against circulating HIV strains as key input data, SLAPNAP can be used to augment these data with predicted in vitro neutralization levels of HIV Env sequences for which neutralization phenotype data are not available. SLAPNAP leverages all data available in the CATNAP database, and given a user-selected bnAb combination and neutralization endpoint, performs a suite of machine learning-based analyses and provides a report that summarizes the predictive results and highlights important pseudovirus sequence features. The goal of the present work is to introduce this tool, compare its performance in predicting sensitivity to a single bnAb to previous approaches, and to validate its performance in predicting sensitivity to combination bnAb regimens.
2 Materials and methods
2.1 Training data and measures of neutralization sensitivity
SLAPNAP is based on in vitro neutralization data available in the CATNAP database. Predictive analysis of neutralization sensitivity can be done for any single bnAb available in this database, or for combinations of any number of available single bnAbs. In the latter case, the neutralization sensitivity of a pseudovirus to the combination bnAb regimen can be estimated based on either an additive or Bliss-Hill model (Wagh et al., 2016).
The models created by SLAPNAP will predict one of three continuous measures of sensitivity [estimated IC50, estimated IC80 and instantaneous inhibitory potential (IIP) (Shen et al., 2008)] and/or two binary measures of sensitivity [whether the estimated ICx < a user-specified cut point (estimated sensitivity), and whether the individual-bnAb ICx < the cut point for a user-specified number of bnAbs in the specified combination (multiple sensitivity), where x can be either 50 or 80].
The features used to make predictions are generated from descriptor variables, viral geometry variables and the amino acid sequence available for each pseudovirus in CATNAP. The descriptor variables include geographic information (binary indicator variables describing the region of origin of each pseudovirus), which is adjusted for as potential confounding variables in all analyses; and the HIV-1 subtype of the virus. Viral geometry variables describe the length, the number of sequons and the number of cysteines in each of the Env, gp120, V2, V3 and V5 regions. Amino acid sequence variables consist of binary indicators of residues containing amino acids, frameshifts, gaps, stops or sequons at each HXB2-referenced site in gp160. In total, there are ∼6000 individual features included in a given SLAPNAP analysis—the exact number of features depends on the pseudoviruses available in CATNAP for the chosen bnAbs.
2.2 Predictive modeling
2.2.1 Choice of models
The SLAPNAP tool allows predictive models to be built using random forests (Breiman, 2001), boosted regression trees (Friedman, 2001) and the elastic net (Zou and Hastie, 2005). Cross-validation may also be used to select tuning parameters for each algorithm. The algorithms can also be combined using the super learner ensemble framework (van der Laan et al., 2007). A super learner is an ensemble of individual learners with ensemble weights chosen to minimize a cross-validated measure of risk (mean squared error for continuous outcomes and negative log likelihood for binary outcomes). Because SLAPNAP can be used to predict neutralization sensitivity to a vast array of single, multiple-specific and combination bnAbs, and because we often lack a priori knowledge as to which algorithm might provide the best predictions for a given regimen, we generally recommend using the super learner in SLAPNAP. The user may also specify a threshold x for variable screening based on the variability of the binary amino acid sequence features that eliminates residues with fewer than x minority variants.
2.2.2 Measuring predictive performance
SLAPNAP uses V-fold cross-validation to evaluate model performance, where a user can choose the value of V. All tuning parameter selection occurs in a cross-validated way, which ensures honest evaluation of model performance. Because SLAPNAP allows for building and evaluating predictors of bnAb regimens with potentially few observed sequences in the CATNAP database, cross-validated performance measures are reported, rather than measures based on train/test sample splitting. Prior work has demonstrated that this is a propitious approach in small samples (Benkeser et al., 2020).
2.3 Intrinsic and predictive feature importance
In SLAPNAP, we divide the analyses that quantify feature importance into two goals: learning about the underlying biology and understanding how a given prediction algorithm makes use of the features.
We use the population prediction potential of features (Williamson et al., 2020a,b) to address the first goal; this quantifies the intrinsic importance of the features. We measure population prediction potential using non-parametric R2 for continuous outcomes and using the non-parametric AUC for binary outcomes. Two types of intrinsic feature importance may be measured: conditional importance, which describes the increase in population prediction potential when the feature(s) of interest are added to all other remaining features; and marginal importance, which describes the increase in population prediction potential when the feature(s) of interest are added to the geographic confounding variables. Both types of intrinsic importance may be estimated for groups of features or individual features. The feature groups that we consider are: geographic confounders, viral geometry variables (length), gp120 CD4 binding sites, gp120 V2 sites, gp120 V3 sites, gp41 MPER sites, region-specific counts of PNG sites (number of sequons) and cysteine counts. Point estimates and confidence intervals for the difference in R2 or AUC, and P-values for a test of the null hypothesis that the difference in population prediction potential is equal to zero, are computed and displayed for each feature group or individual feature of interest.
For the second goal, algorithm-specific predictive importance is defined in the default manner for each type of learner. For random forests and boosted trees, this is the normalized sum of the decrease in impurity (i.e. the Gini index for binary outcomes or mean squared error for continuous outcomes) over all nodes at which a split on that feature has been conducted; for the elastic net, this is the absolute value of the estimated regression coefficient at the cross-validation-selected tuning parameter value. These commonly reported measures of importance can provide some insight into how a given algorithm makes predictions. If an ensemble is used, the importance measures for the individual learner with the highest weight in the ensemble are reported.
3 Usage
3.1 Running SLAPNAP
SLAPNAP is a Docker container hosted on DockerHub (Docker Inc., 2019). With Docker installed, SLAPNAP can be downloaded by executing the following at the command line:
docker pull slapnap/slapnap: latest
SLAPNAP is executed using the docker run command. For example, the following code will instruct SLAPNAP to create and evaluate a neutralization predictor for the bnAb combination VRC07-523-LS and PGT121:
docker run\
-v path/to/local/save/directory:/home/output/\
-e nab=“VRC07-523-LS; PGT121”\
-e outcomes=“ic50; estsens”\
-e learners=“rf; lasso”\
-e importance_grp=“marg”\
-e importance_ind=“pred”\
slapnap/slapnap: latest
The –v tag specifies the directory on the user’s computer where the report will be saved, and path/to/local/save/directory should be replaced with the desired target directory. Options for the analysis are passed to the container via the -e tag; these options include the bnAbs to include in the analysis (nab), the neutralization outcomes of interest (outcomes), the learners to use in the analysis (learners) and the types of variable importance to compute (importance_grp, for groups of variables; importance_ind, for individual variables). Other output (e.g. the formatted analysis dataset and the fitted learners) can be requested via the return option. Cross-validation is used by default both for parameter tuning (cvtune) and model estimation (cvperf). We recommend the use of both layers of cross-validation to reduce the potential for biased estimation of performance (see, e.g. Varma and Simon, 2006). A full list of options and their syntax are available in the SLAPNAP documentation (https://benkeser.github.io/slapnap/).
3.2 HTML report and requested output
Following the completion of a SLAPNAP run, an HTML report—along with any other requested output—will be saved to the output directory and the SLAPNAP container will shut down. Excerpts from the report are shown in Figure 1. Each report begins with a section summarizing data extraction and key results (Panel A). Descriptive statistics and plots are provided for each requested bnAb and for the estimated sensitivity measures for the combined bnAbs (Panel B). The performance of the learners for predicting binary sensitivity endpoints is estimated using cross-validated ROC curve analysis (Panel C). Feature importance is determined by a variety of methods and summarized in tables and figures (Panel D). Full reports are available in the SLAPNAP documentation (https://benkeser.github.io/slapnap/6-sec-report.html#example-reports).
Fig. 1.
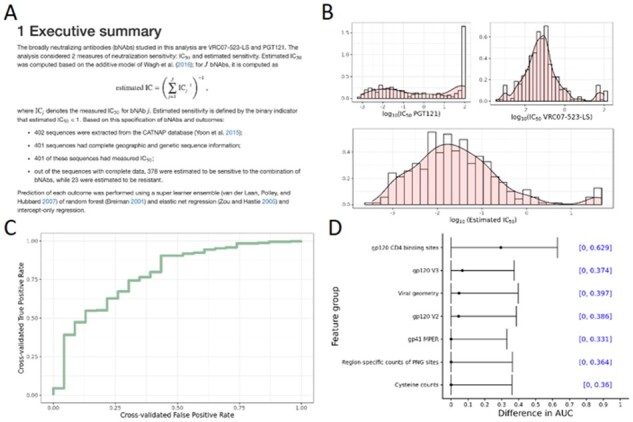
Example text and plots from a SLAPNAP report. This report analyzed the combination of bnAbs VRC07-523-LS and PGT121. (A) Executive summary, including outcome definitions and the number of sequences analyzed; (B) plots of individual IC50 for each bnAb and estimated combination IC50; (C) cross-validated receiver operating characteristic curve for predicting the binary endpoint IC50 < 1 µg/mL; and (D) estimated intrinsic feature importance for the groups of amino acid features, measured via the difference in population AUC comparing a feature group to geographic confounders
4 Results
4.1 SLAPNAP produces accurate predictions of sensitivity to single bnAbs across epitopes
We compared the performance of SLAPNAP for predicting neutralization sensitivity of HIV-1 viruses to single bnAbs to two other methods: the support vector machine (SVM)-based method of Hake and Pfeifer (2017) and the gradient boosted machine-based method of Rawi et al. (2019). We considered all bnAbs in CATNAP analyzed by either Hake and Pfeifer (2017) or Rawi et al. (2019) including bnAbs targeting the V1/V2 region, the V3 loop, the CD4 binding site, the fusion peptide, the subunit interface and the membrane proximal external region.
We ran SLAPNAP for each of these bnAbs with neutralization sensitivity defined by the binary indicator that IC50 was less than 50 µg/mL so that our analysis was harmonized with those of Hake and Pfeifer (2017) and Rawi et al. (2019). In each case, we used an ensemble with a library consisting of random forests, lasso regression and gradient boosted trees implemented in H2O (H20.ai, 2016) and minimum variability thresholds of zero, four and eight for the binary amino acid sequence features. To construct our super learner, we used five-fold cross-validation to determine the optimal convex combination of these learners and screens that minimized the cross-validated negative log likelihood. We additionally used an outer layer of five-fold cross-validation to assess prediction performance. We compare SLAPNAP to previous results by comparing the SLAPNAP-estimated cross-validated AUC (CV-AUC) to the reported AUCs of those works. SLAPNAP is at a disadvantage in these comparisons, since the tool adaptively chooses tuning parameters, whereas both sets of previously reported results optimized tuning parameters on the CATNAP data before computing their performance measures. We further analyzed the performance of SLAPNAP in predicting continuous IC50 and IC80, and for these outcomes report cross-validated R2 (CV-R2) along with 95% confidence intervals; these outcomes were not analyzed by either Hake and Pfeifer (2017) or Rawi et al. (2019). The full SLAPNAP specification for each of these analyses is available on GitHub (https://github.com/benkeser/slapnap_supplemental).
All methods were found to have good classification performance (all CV-AUC > 0.6) across epitopes (Fig. 2). Additionally, while there are differences in performance on individual bnAbs, no one method dominates the others across epitopes (Table 1). The median estimated CV-AUCs across all bnAbs were 0.71, 0.84 and 0.81 for Hake and Pfeifer (2017), Rawi et al. (2019) and SLAPNAP, respectively.
Fig. 2.

Cross-validated performance for predicting neutralization sensitivity across bnAb epitopes for single monoclonal bnAbs. The figure compares SLAPNAP (squares); the SVM-based method of Hake and Pfeifer (2017; circles); and the gradient boosted machine-based method of Rawi et al. (2019; triangles)
Table 1.
MedianCV-AUC for predicting neutralization sensitivity for single monoclonal bnAbs across epitopes
| Epitope | Hake and Pfeifer (2017) | Rawi et al (2019 ) | SLAPNAP |
|---|---|---|---|
| CD4bs | 0.71 | 0.79 | 0.79 |
| Fusion peptide | NA | 0.78 | 0.81 |
| MPER | NA | 0.90 | 0.79 |
| Subunit interface | 0.65 | 0.77 | 0.80 |
| V1V2 | 0.69 | 0.86 | 0.84 |
| V3 | 0.80 | 0.92 | 0.90 |
|
| |||
| Overall | 0.71 | 0.84 | 0.81 |
The prediction performance of SLAPNAP for the continuous outcomes was mixed (Fig. 3). For some bnAbs (e.g. 10-1074 and 8ANC195), SLAPNAP achieves a high CV-R2; for other bnAbs (e.g. HJ16 and 35O22), the estimated CV-R2 is ∼0. This variability reflects both the increased difficulty of predicting continuous outcomes compared to predicting binary outcomes and the differing number of sequences with observed IC50 and IC80 values in CATNAP. In particular, IC80 values are missing for many sequences, leading to a small sample (fewer than 200 sequences) on which to train and evaluate SLAPNAP. The cross-validated predicted outcomes (i.e. each predicted outcome is based on a learner trained on data excluding the corresponding observed outcome) are moderately correlated with the observed outcomes: the median correlation over all bnAbs is 0.65 and 0.52 for IC50 and IC80, respectively. Taken together, these results suggest modest capability to predict the continuous outcomes.
Fig. 3.

Cross-validated performance of SLAPNAP for predicting continuous neutralization outcomes (IC50 and IC80) across bnAb epitopes for single monoclonal bnAbs. The outcomes are differentiated by color (IC50 is red, while IC80 is teal); the size of the point corresponds to the number of available outcomes, with the largest size denoting a large number of outcomes. Bars represent 95% confidence intervals for CV-R2
4.2 SLAPNAP produces accurate predictions of sensitivity to combinations of bnAbs
To validate the performance of the tool for the purpose of predicting combination sensitivity, we evaluated all bnAb combinations that are available in the CATNAP database that satisfied the following criteria: each bnAb in the combination had individual sensitivity measurements available, combination sensitivity was measured on at least 100 pseudoviruses (to provide sufficient data to evaluate the learners), at least five pseudoviruses were resistant (defined for this analysis as having IC50 > 1 μg/mL) to each of the individual bnAbs (to provide sufficient data to train the learners), and at least one pseudovirus was resistant to the combination bnAb (to provide sufficient data to evaluate performance). For the 16 qualifying bnAbs, we used SLAPNAP to train models using the single bnAb sensitivity data available in CATNAP, using both proposed measures of estimated combination sensitivity based on an additive model (described in Section 2.1). The Super Learner ensembles used in this section were based on a different implementation of boosting (xgboost; Chen and Guestrin, 2016) than in the previous section, but otherwise the ensemble was the same; this illustrates the ease of using different learning algorithms within SLAPNAP, a point that we elaborate on further in the Discussion. We then used the trained models to obtain out-of-sample predictions of sensitivity on the pseudoviruses with measured sensitivity to the combination bnAb regimen and computed AUC.
We found that SLAPNAP predictions of combination sensitivity lead to an AUC > 0.95 for 14 of 16 combinations and an AUC > 0.66 for the remaining two combinations (Fig. 4). We did not observe a consistent trend in terms of predictive performance across the two definitions of estimated sensitivity available in SLAPNAP. The average difference in AUC between the two approaches was -0.004 with the largest observed difference only 0.038.
Fig. 4.

Out-of-sample AUC of SLAPNAP models trained used single-bnAb neutralization data for predicting measured sensitivity (IC50 < 1 µg/mL) to the combination bnAb regimen. The out-of-sample AUC is computed based on 125 pseudoviruses for all combinations except BG1 + BG18 + NC37, which is computed based on 119 pseudoviruses. The number of pseudoviruses measured to be resistant (IC50 ≥ 1 µg/mL) to each combination is shown across the bottom
We further evaluated the performance of SLAPNAP by estimating prediction performance based on viruses from two studies of the combination regimen 10-1074 + 3BNC117 that are not available in CATNAP (Bar-On et al., 2018; Mendoza et al., 2018). We matched the neutralization values from these two studies (provided in Supplementary Table S4 of Mendoza et al., 2018 and Supplementary Table S6 of Bar-On et al., 2018) with the viral sequence data from these studies that are publicly available on GenBank (details for accessing this information are available in Mendoza et al., 2018; Bar-On et al., 2018). After restricting to a single virus per participant, 21 viruses with complete outcome and sequence information remained, of which 19 were estimated to be sensitive to the combination (based on the 2 µg/mL sensitivity threshold used in Mendoza et al., 2018 and Bar-On et al., 2018) and 14 were sensitive to at least one of the bnAbs in the combination. The test-set AUCs for predicting these outcomes were 0.79 [0.55, 0.92] and 0.86 [0.58, 0.96] for estimated and multiple sensitivity, respectively; the test-set R2 for predicting continuous IC50 was 0.13 [0, 0.34].
5 Discussion
Our results indicate that SLAPNAP achieves comparable performance to existing approaches for predicting single-bnAb sensitivity based on Env amino acid sequences without pre-optimizing any tuning parameters. Moreover, the tool offers the ability to extend prediction of sensitivity to regimens containing multiple bnAbs, thereby validating its utility as a tool for various applications including improving down-selection of multiple-bnAb regimens into future HIV-1 prevention efficacy trials and improving analytic plans for sieve analysis.
In our test-set data analysis of the combination regimen 3BNC117 + 10-1074, we observed reduced AUC relative to the cross-validated in-sample performance for this combination (estimated to be greater than 0.95 for both binary outcomes). There were also several limitations to this analysis. First, there were only 30 participants enrolled in one of the two trials that gave rise to these data; it is possible that with a larger sample size we could see improved performance. Second, these participants were chosen in part due to sensitivity to either bnAb, leading to an imbalance between the number of sensitive and resistant viruses. Finally, SLAPNAP currently does not have an option to restrict training to a specified HIV-1 subtype; the viruses in this test-set analysis were all subtype B, while the training data were only 30% subtype B viruses. We will implement such a filter in a future iteration of SLAPNAP.
SLAPNAP provides information beyond point estimates of neutralization potential, and additional flexibility that make it a general-purpose tool. Users can select different combinations of neutralization outcomes, thresholds for neutralization sensitivity, learners and variable screens, among other control arguments making SLAPNAP useful in a wide variety of contexts. The confidence interval estimates that are reported provide important context for how to interpret the neutralization results and can aid in the down-selection of bnAb regimens into future trials. Variable importance estimates can be useful in interpreting the results as well.
There are several important limitations of SLAPNAP that we hope to address in future iterations. First, as the SVM approach of Hake and Pfeifer (2017) was found to perform nearly as well as our super learner approach and the gradient boosting approach of Rawi et al. (2019) for some bnAbs, we will in future releases build in support for an implementation of SVM in SLAPNAP. A strength of the super learner framework is that it is agnostic to its constituent learners, so addition of an SVM or switching between the two available boosted machine implementations (Chen and Guestrin, 2016; H2O.ai, 2016) is quite natural in this context. Second, obtaining SLAPNAP-based predictions of sensitivity for (pseudo-)viruses not derived from the CATNAP database requires considerable effort from the SLAPNAP user. In future iterations, we plan to include native sequence alignment tools in SLAPNAP, so that the proper formatting is done internally and no additional effort is required. Finally, Docker requires elevated privileges to run and so SLAPNAP may be inaccessible to some users. In the future, we hope to design a web-based user interface to ease the use of SLAPNAP.
Acknowledgements
The authors thank Adam Dingens for assistance in defining the relevant HXB2 sites so that SLAPNAP works well across epitopes, and thank Bhavesh Borate for insightful comments that helped improve the report returned by SLAPNAP.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institute of Health [UM1AI068635 (HVTN SDMC) to P.B.G.] and the National Institutes of Health [S10OD028685].
Conflict of Interest: none declared.
Contributor Information
Brian D Williamson, Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Craig A Magaret, Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Peter B Gilbert, Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA; Department of Biostatistics, University of Washington, Seattle, WA 98195, USA.
Sohail Nizam, Department of Biostatistics and Bioinformatics, Rollins School of Public Health, Emory University, Atlanta, GA 30322, USA.
Courtney Simmons, Department of Biostatistics and Bioinformatics, Rollins School of Public Health, Emory University, Atlanta, GA 30322, USA.
David Benkeser, Department of Biostatistics and Bioinformatics, Rollins School of Public Health, Emory University, Atlanta, GA 30322, USA.
References
- Bar-On Y. et al. (2018) Safety and antiviral activity of combination HIV-1 broadly neutralizing antibodies in viremic individuals. Nat. Med., 24, 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkeser D. et al. (2020) Improved small-sample estimation of nonlinear cross-validated prediction metrics. J. Am. Stat. Assoc., 115, 1917–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- Bricault C.A. et al. (2019) HIV-1 neutralizing antibody signatures and application to epitope-targeted vaccine design. Cell Host Microbe, 25, 59–72.e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docker Inc. (2019) Docker Hub Quickstart. Docker Docs. https://docs.docker.com/docker-hub/ (30 March 2020, date last accessed).
- Chen T., Guestrin C. (2016) XGBoost: a scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Discovery and Data Mining, San Francisco, CA, USA, pp. 785–794.
- Corey L. et al. (2021) Two randomized trials of neutralizing antibodies to prevent HIV-1 acquisition. NEJM, 384, 1003–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria-Rose N.A. et al. (2012) HIV-1 neutralization coverage is improved by combining monoclonal antibodies that target independent epitopes. J. Virol., 86, 3393–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J.H. (2001) Greedy function approximation: a gradient boosting machine. Ann. Stat., 29, 1189–1232. [Google Scholar]
- Gautam R. et al. (2016) A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature, 533, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- H2O.ai. (2016) R Interface for H2O, R package version 3. https://github.com/h2oai/h2o-3. (1 February 2021, date last accessed).
- Hake A., Pfeifer N. (2017) Prediction of HIV-1 sensitivity to broadly neutralizing antibodies shows a trend towards resistance over time. PLoS Comput. Biol., 13, e1005789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessell A.J. et al. (2018) Passive and active antibody studies in primates to inform HIV vaccines. Expert Rev. Vaccines, 17, 127–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuna S.T., Corey L. (2020) Broadly neutralizing antibodies for HIV prevention. Annu. Rev. Med., 71, 329–346. [DOI] [PubMed] [Google Scholar]
- Kong R. et al. (2015) Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. J. Virol., 89, 2659–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H.X. et al. ; NISC Comparative Sequencing Program. (2013) Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature, 496, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magaret C.A. et al. (2019) Prediction of VRC01 neutralization sensitivity by HIV-1 gp160 sequence features. PLoS Comput. Biol., 15, e1006952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy L.E., Burton D.R. (2017) Identification and specificity of broadly neutralizing antibodies against HIV. Immunol. Rev., 275, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza P. et al. (2018) Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature, 561, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody M.A. et al. (2016) Immune perturbations in HIV-1-infected individuals who make broadly neutralizing antibodies. Sci. Immunol., 1, aag0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris L., Mkhize N.N. (2017) Prospects for passive immunity to prevent HIV infection. PLoS Med., 14, e1002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegu A. et al. (2019) A meta-analysis of passive immunization studies shows that serum-neutralizing antibody titer associates with protection against SHIV challenge. Cell Host Microbe, 26, 336–346.e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawi R. et al. (2019) Accurate prediction for antibody resistance of clinical HIV-1 isolates. Sci. Rep., 9, 14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L. et al. (2008) Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med., 14, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sok D., Burton D.R. (2018) Recent progress in broadly neutralizing antibodies to HIV. Nat. Immunol., 19, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan M.J. et al. (2007) Super learner. Stat. Appl. Genet. Mol. Biol., 6, Article25. [DOI] [PubMed] [Google Scholar]
- Varma S., Simon R. (2006) Bias in error estimation when using cross-validation for model selection. BMC Bioinformatics, 7, 91.( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh K. et al. (2016) Optimal combinations of broadly neutralizing antibodies for prevention and treatment of HIV-1 clade C infection. PLoS Pathog., 12, e1005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh K. et al. (2018) Potential of conventional & bispecific broadly neutralizing antibodies for prevention of HIV-1 subtype A, C & D infections. PLoS Pathog., 14, e1006860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams W.B. et al. (2017) Initiation of HIV neutralizing B cell lineages with sequential envelope immunizations. Nat. Commun., 8, 1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson B.D. et al. (2020a) Nonparametric variable importance assessment using machine learning techniques (with discussion). Biometrics, 77, 9–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson B.D. et al. (2020b) A unified approach for inference on algorithm-agnostic variable importance. arXiv, 03683. [stat.ME]. [DOI] [PMC free article] [PubMed]
- Yoon H. et al. (2015) CATNAP: a tool to compile, analyze and tally neutralizing antibody panels. Nucleic Acids Res., 43, W213–W219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R. et al. (2016) Initiation of immune tolerance-controlled HIV gp41 neutralizing B cell lineages. Sci. Transl. Med., 8, 336ra362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H., Hastie T. (2005) Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B (Stat. Methodol.), 67, 301–320. [Google Scholar]