

Abstract
Functional studies on echinoderms have been reduced to the use of pharmacological treatments. The ability to modulate the genetic expression of regenerating tissues can elucidate potential effectors during this process. Here we describe an effective transfection protocol that allows the introduction of Dicer-substrate interference RNAs (DsiRNAs) for the modulation of gene expression and its characterization during regeneration.
Keywords: Echinoderm, Sea Cucumber, DsiRNA, RNAi, Regeneration, Electroporation, Gut rudiment
1. Introduction
RNA interference (RNAi) is a well-conserved silencing mechanism found in most eukaryotes [1]. Originally, this mechanism was recognized as an endogenous antiviral protector and gene regulator, but now it has been harnessed as an experimental tool for gene knockdowns [2]. The formal effectors of this silencing mechanism are the double-stranded RNAs (dsRNAs), which are processed into short-interfering RNAs (siRNAs), ~21 – 22 nucleotides in length, by the protein Dicer. Then, these siRNAs are incorporated into the RNA-induced silencing complex (RISC), which recognizes, cleaves and/or represses complementary RNA [3 – 4].
Previously, many researchers synthesize 21-mer siRNAs, which mimic the natural siRNAs obtained from Dicer processing. IDT researchers have advanced this technology by designing a more effective RNAi technology called Dicer-substrate interference RNA (DsiRNA). DsiRNAs are 27-mers RNA duplexes processed by Dicer into 21-mer siRNAs [5]. With this technology scientists provide Dicer with a substrate instead of a product, thus increasing RNAi potency up to 100-fold when compared with usual 21-mers [5].
There are few techniques available to knockdown specific gene expression in members of the phylum Echinodermata, thus limiting the type of studies that can be performed on these animals. Echinoderms, with their key phylogenetic position and their impressive regeneration potential have much to offer to studies of evolution and developmental interest. At present, most studies can only be performed in embryos but not in adult animals [6]. Gene knockdown techniques have been performed recently in certain tissues/organs of adult echinoderms, but still remain highly limited [7].
We have now implemented DsiRNA technology in tissue explants of the sea cucumber Holothuria glaberrima. For this, gut rudiments were electroporated with tetramethylrhodamine-conjugated anionic dextran 3,000 MW lysine fixable using different parameters to elucidate the efficiency of the transfection method. After selecting the optimal parameter, DsiRNA-targeting β-catenin was introduced into the intestinal tissue explants to reduce its abundance and characterize its role on different cellular events during intestinal regeneration.
2. Materials
Prepare all solutions using ultrapure water and analytical-grade reagents. Diligently follow waste disposal regulations when disposing of waste material. All cell culture procedures should be carried out in a laminar flow hood.
2.1. Tissue selection
In our case, we selected the gut rudiments to perform our experiments and followed the procedure of the sea cucumber evisceration, disinfection and gut dissection found in Bello et al. [8], but this protocol can be applied to any tissue of interest.
2.2. Gut Rudiments Culture
L-15 Medium (Leibovitz, Sigma): conditioned for marine species by adding salts to the original composition [9]. Weigh 6.9 g L-15 powder, 6.25 g NaCl, 3.12 g glucose, 1.58 g magnesium sulfate (MgSO4), 172 mg KCl, 96 mg sodium bicarbonate (NaHCO3), 1.33 g magnesium chloride (MgCl2), 150 mg L-glutamine, 745 mg calcium chloride dihydrate (CaCl2·2H2O) and transfer to a 500 mL beaker. Complete to 500 mL with ultrapure water. Mix with a magnetic stirrer and sterilize by filtration (0.22 μm filters). Store in a bottle wrapped in aluminum foil at 4 °C.
Gut rudiment medium: L-15 medium conditioned for marine species supplemented with antibiotics (100 U/mL Penicillin, 100 μg/mL Streptomycin, 50 μg/mL Gentamicin), antifungal (2.5 μg/mL amphotericin B), 1 × MEM nonessential amino acids, 1 mM sodium pyruvate, 1.75 mg/mL α-tocopherol acetate. Add 500 μL of Penicillin/Streptomycin stock solution (10,000 U Penicillin and 10 mg/mL Streptomycin, respectively), 250 μL of Gentamicin stock solution (10 mg/mL), 50 μL of amphotericin B stock solution (2.5 mg/mL), 500 μL of MEM nonessential amino acid stock solution (100×), 500 μL of sodium pyruvate stock solution (100 mM), and 50 μL of α-tocopherol acetate stock solution (17.5 mg/mL) in a 50 mL tube. Complete 45 mL of the 50 mL with L-15 medium conditioned for marine species. Mix by inversion of the tube and adjust to pH 7.7 – 7.8. Complete to 50 mL adding L-15 medium conditioned for marine species and sterilize by filtration (0.22 μm filters). Store in a bottle wrapped in aluminum foil at 4 °C (see Note 1).
24-well plate: tissue culture-treated polystyrene, flat bottom with lid.
2.3. Gut rudiment Electroporation
Phosphate Buffered Saline (PBS) 0.1 M: Weigh 8 g NaCl and 13.4 g phosphate dibasic heptahydrate (NaH2PO4·7H2O) and transfer to a 1 L beaker. Add 800 mL while of ultrapure water while stirring. Adjust to pH 7.4 and complete to 1 L with ultrapure water.
Tetramethylrhodamine-conjugated anionic dextran (TMRAD) 3,000 MW, lysine fixable: TMRAD 50 mg/mL stock solution. Dilute to 5 μg/μL TMRAD in 0.01 M PBS for every sample (see Note 2).
BTX Harvard Apparatus ECM 830 Square Wave Electroporation System and electroporation cuvettes: 4 mm electroporation cuvettes.
Safety Stands for BTX Cuvettes & Chambers.
2.4. Determining Electroporation Efficiency Using Histological Approaches
4% paraformaldehyde (PFA, Sigma; see Note 3): Weigh 4 g of PFA and dissolve it on 50 mL of ddH2O. Heat the solution up to 60 °C in a chemical hood. Add sodium hydroxide (NaOH), pellet by pellet, until the solution becomes transparent. Add 50 mL of 0.2 M PB. Put the solution on ice and adjust to pH 7.4.
Sucrose: 40% sucrose in 0.1 M PBS.
Mounting media: buffered glycerol solution containing 2 μg/μL 4′,6-diamidino-2-phenylindole (DAPI). To prepare 5 mg/mL DAPI stock solution (14.3 mM), dissolve one vial (10 mg) in 2 mL of ddH2O. Then prepare a 1 mg/mL DAPI working solution adding 200 μL of the stock solution to 800 μL ddH2O. To prepare the mounting media add 50 μL of the working solution to 24.95 mL of 0.1 M PBS in a 50 mL. Then, add 50 mL of glycerol to the 50 mL tube and mix. Wrap the tube with aluminum paper and store at 4 °C.
2.5. DsiRNAs
Nuclease-Free Duplex Buffer from Integrated DNA Technologies (IDT).
DsiRNAs 2 and/or 10 nmol (see Note 4): For 2 nmol and 10 nmol of DsiRNAs, add 20 μL and 100 μL of Nuclease-Free Duplex Buffer, respectively to obtain a stock solution of 100 μM. Heat at 94 °C for 2 min. Dilute the oligonucleotides to 10 μM using Nuclease-Free Duplex Buffer. Store at −20 °C.
2.6. Determination of RNA abundance using qPCR
iQ SYBR Green Supermix (Bio Rad): Add 12.5 μL of iQ SYBR Green Supermix.
Nuclease-Free Water.
qPCR primers 10 μM: For 100 μM stock solution, add Nuclease-Free water to the lyophilized primers (see Note 5). Heat the oligonucleotide at 55 °C for 5 min, then vortex thoroughly. Prepare a 10 μM working solution and store at −20 °C.
3. Methods
3.1. Sea Cucumber Collection and Evisceration
Collect the sea cucumbers from the species Holothuria glaberrima in the Northeast coast of Puerto Rico and keep in indoor saltwater aquaria at 22 – 24 °C.
Eviscerate the sea cucumbers by injecting 0.35 M KCl (3 – 5 mL) into the coelomic cavity. During the process of evisceration, H. glaberrima expels out most of its internal organs, including its intestine, through the cloaca. After evisceration, sea cucumbers are placed in an artificial aquarium for 4 days.
3.2. Sea Cucumber Disinfection and Explants Preparation
Immerse the sea cucumbers (see Note 6) that have regenerated their intestinal tract for 4 days in a dish preparation containing 250 mL of anesthetic solution (RT) for 35 min.
Decontaminate the exterior of the sea cucumbers by immersing the animals one at a time in a 10% sodium hypochlorite solution for 1 min, 95% ethanol for 1 min, and a quick rinse in sterile ultrapure water.
Spray a vinyl pad in a dissecting pad with 70% ethanol and place the sea cucumbers ventral side down. The ventral side of the sea cucumbers can be identified by the presence of the ambulacral tubes. Cut the anterior region (head) near the calcareous ring and then cut with scissors along the longitudinal line that separates the dorsal and ventral axis. Once the body wall is opened, hold down the sea cucumber using pushpins. Dissect the gut rudiments using fine tweezers and microdissection scissors. On day 4 of regeneration, the gut rudiment appears as a pink thickening at the free end of the mesentery.
Collect the gut rudiment (see Note 7) and place each, individually, in 1 mL of the antibiotic/antifungal solution in a 2 mL tube.
Wash the gut rudiments with fresh antibiotic/antifungal solution by placing the tubes in a shaker (moving slowly) for 1 h twice.
3.3. Optimization of the electroporation protocol
Prepare the 24-well plate with 1 mL of the L-15 supplemented with antibiotics and antifungal.
Add 25 μL of 5 μg/μL TMRAD on each electroporation cuvette.
Remove the gut rudiment from the antibiotic/antifungal solution with a tweezer or a pipette tip (see Note 8) and place it inside the electroporation cuvette.
Place the electroporation cuvette in the Safety Stands for Cuvettes and apply the square electric pulses ten times using the following parameters (see Note 9): 15 V, time pulse: 45 ms, interval pulse: 955 ms; 20 V, time pulse: 40 ms, interval pulse: 960 ms; 27 V, time pulse: 30 ms, interval pulse: 970 ms; 35 V, time pulse: 25 ms, 975 ms.
Remove the gut rudiment from the electroporation cuvette and place it in a 24-well plate prepared on step 1.
Incubate the gut rudiment for 2 days in a modular incubator chamber at RT. Check your culture daily to evaluate its morphology and possible contamination.
3.4. Histological studies for electroporation efficiency.
After culturing the gut rudiments for 2 days, use a tweezer or a pipette tip to remove the gut rudiment from the 24-well plate. Place the gut rudiments into the 2 mL tubes with 1 mL of 4% PFA for 24 h and store at 4 °C.
After fixation, remove the 4% PFA from the 2 mL tubes and wash the gut rudiments with 0.1 M PBS three times for 10 min/each.
Add 1 mL of 30 – 40% sucrose to each 2 mL tube containing the gut rudiments for cryoprotection and store at 4 °C for 24 h (see Note 10).
Add Tissue-Tek O. C. T in a metal base mold for the gut rudiment embedding (see Note 11).
Use a tweezer to remove the gut rudiments from the 30 – 40% sucrose and place it in a petri dish to cut ~2.5 cm of the gut rudiment. Place the remaining of the gut rudiments back to the 30 – 40% sucrose and store at 4 °C as backup.
Use a tweezer to put the gut rudiment that has been cut in the metal base mold with the Tissue-Tek O. C. T. (see Note 12).
Place the metal base mold with the gut rudiments into the cryostat (see Note 13) until the Tissue-Tek O. C. T. solidifies.
Place the block of Tissue-Tek O. C. T. in the microtome and perform 20 μm cryosections. Place the cryosections on poly-L-lysine treated slides; 5 or 6 sections per slide.
After cryosectioning, dry the slides for 1 h before doing any histological study.
Mount each slide in buffered glycerol containing DAPI and cover with a 24 × 55 mm coverslip. Seal the slides with nail polish and dry under a blower. DAPI stains the cell nuclei.
Evaluate the slides using a fluorescent microscope or a microscope equipped with the appropriate filters (see Note 14) [Figure 1 near here] [10].
Figure 1.
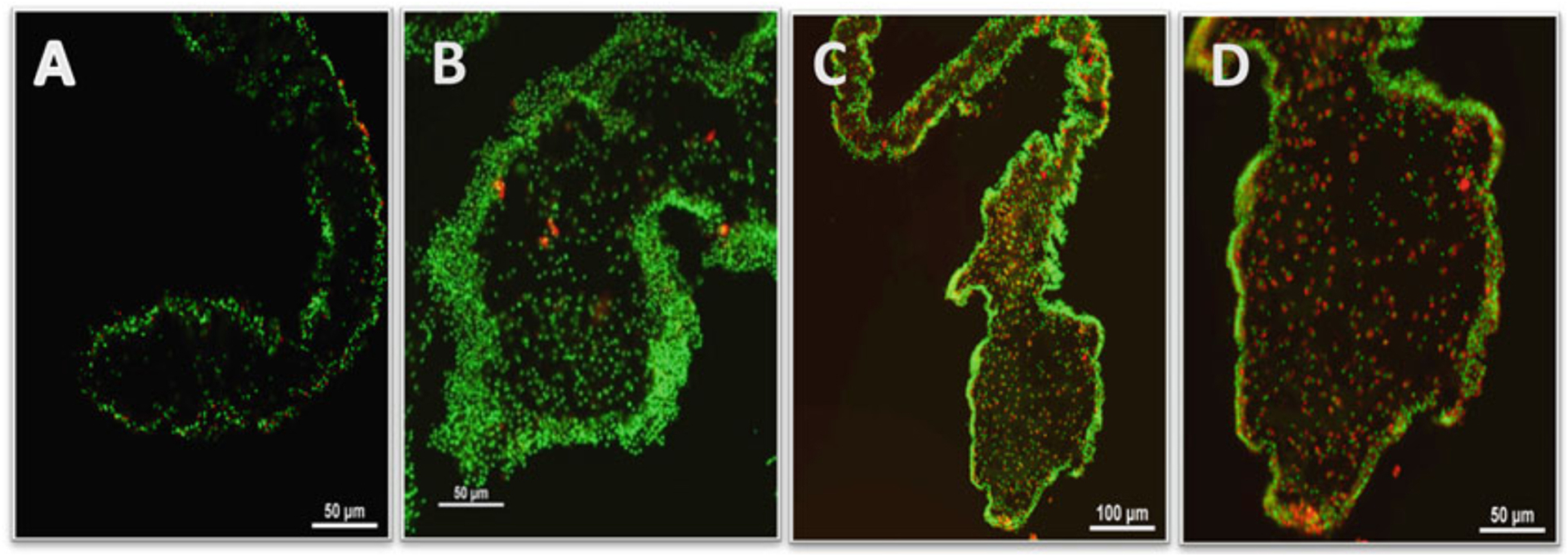
Electroporation of tetramethylrhodamine-conjugated anionic dextran in explants of the sea cucumber H. glaberrima. Nuclei are stained with DAPI (green) and tetramethylrhodamine-conjugated anionic dextran stained the cytosol (red). (A) Electroporation of the explant with 0.1 M PBS. (B) Tetramethylrhodamine-conjugated anionic dextran alone (without electroporation). (C) Electroporation of the explant with tetramethylrhodamine-conjugated anionic dextran. (D) Higher magnification of (C).
3.5. siRNA Design
Access siDirect version 2.0 (for example, at http:sidirect2.rnai.jp) and paste the nucleotide sequence of interest to choose the target sequence and design the siRNA. This program will retrieve different effective siRNA candidates and a graphical view of these candidates [10].
Select the candidates with a seed-duplex stability Tm < 21.5 °C. These are off-target reduced siRNAs, which are siRNAs that interfere specifically the sequence of interest and have a less probability to interfere other sequences.
- After selecting the siRNA duplex, use the guidelines of Integrated DNA Technologies (IDT), Inc to design the DsiRNA-targeting β-catenin:
- Target sequence and siRNA duplexes:
- 5′ – GGGTGTAACAACTTGACAATTAC – 3′
- 5′ – GUGUAACAACUUGACAAUUAC – 3′ Passenger (sense) strand
- 3′ – CCCACAUUGUUGAACUGUUAA – 5′ Guide (antisense) strand
- To create a Dicer substrate RNAi, add 4 bases to the 3′-end of the sense strand and 6 bases to the 5′-end of the antisense strand to create an asymmetric blunt ended and 3′ overhang molecule.
- 1. 5′ – GGGTGTAACAACTTGACAATTAC – 3′
- 5′ – GUGUAACAACUUGACAAUUACAGGA – 3′
- 3′ – CCCACAUUGUUGAACUGUUAAUGUCCU – 5′
- Replace 2 bases of the 3′-end of the sense strands with DNA bases when necessary.
- 1. 5′ – GGGTGTAACAACTTGACAATTAC – 3′
- 5′ – GUGUAACAACUUGACAAUUACAGGA – 3′
- 3′ – CCCACAUUGUUGAACUGUUAAUGUCCU – 5′
3.7. Electroporation of Gut Rudiments with DsiRNAs
Follow Method 3.2.
Prepare the 24-well plate with 1 mL of the L-15 supplemented with antibiotics and antifungal.
Add 5 μL of three different concentrations of DsiRNA-targeting β-catenin on each electroporation cuvette: 1 μM, 10 μM and 100 μM. Use biological replicates.
Remove the gut rudiment from the antibiotic/antifungal solution with a tweezer or a pipette tip (see Note 8) and place it inside the electroporation cuvette.
Place the electroporation cuvette in the Safety Stands for Cuvettes and apply the square electric pulses ten times using the following parameters: 35 V, time pulse: 25 ms, 975 ms.
Remove the gut rudiment from the electroporation cuvette and place it in a 24-well plate prepared in step 2.
Incubate the gut rudiment for 2 days in a modular incubator chamber at RT. Check your culture daily to evaluate its morphology and possible contamination.
After 2 days in culture, remove the gut rudiments from the 24-well plate and store at −20 °C in 1 mL of RNAlater™ in a 2 mL tube.
3.8. RNA extraction
Place the 2 mL tubes with the gut rudiments on ice.
Prepare five 15 mL tubes: three 15 mL tubes with 10 mL ddH2O, one with 10 mL 70% ethanol and one with 10 mL ZAP. Place the tubes in the following order: 2 ddH2O, 70% ethanol, ZAP and ddH2O.
Take out the RNA later from the 2 mL tubes of Step 8 from section 3.7 and add 1 mL of Trizol (maintain the tubes on ice).
Homogenize the gut rudiments at low/medium level using a homogenizer until the tissue is completely homogenized.
After homogenizing the gut rudiments, place the tubes on ice for 30 min.
After 30 min on ice, add 200 μL of chloroform and mix vigorously.
Incubate the tubes at RT for 10 min.
Centrifuge the tubes for 15 min at 12,000 rpm at 4 °C (see Note 15).
Remove the aqueous phase (RNA is here! – see Note 16) and place it in a 1.5 mL RNase-free tube.
Follow the Qiagen RNeasy Protocol for RNA purification, including the DNase treatment (Cat. Num. 74104).
Place the 1.5 mL tubes with the purified RNA on ice and determine the RNA concentration using the NanoDrop 1000 spectrophotometer. Write down the concentration, the 260/280, and the 260/230 ratios.
3.9. cDNA synthesis
Calculate the volume needed for 1 μg of RNA extracted from the gut rudiments starting from the RNA concentration obtained in Methods 3.8.
In a 0.2 mL tube add 2 μL of Oligo dT, the volume calculated for 1 μg of RNA and complete to 10 μL with Nuclease-free water.
Mix, spin briefly and heat for 5 min at 70 °C.
Add the remaining components to the 0.2 mL tube (see Note 17): 4 μL Improm-2 5× Reaction Buffer, 3 μL MgCl2, 1 μL dNTPs, 1 μL RNasin Ribonuclease inhibitor and 1 μL Improm-2 Reverse transcriptase to obtain a final volume of 20 μL.
Mix by pipetting up and down, incubate at 25 °C (RT) for 5 min, 42 °C for 1 h and 70 °C for 15 min (see Note 18).
3.10. qPCR to determine the efficiency of the interference
Place the cDNA samples, the primers (see Note 19), the iQ SYBR Green Supermix on ice.
Add 12.5 μL iQ SYBR Green Supermix, 0.4 μL primer forward, 0.4 μL primer reverse, 5.7 μL Nuclease-free water and 1 μL of the cDNA sample on each well on a 96-well plate (see Note 20). Perform technical replicates of each biological replicates. Use primers for β-catenin and NADH (reference gene).
- Put the 96-well plate in a Mastercycler Ep realplex and use the following parameters:
- Step 1: 95 °C for 3:00 min
- Step 2: 95 °C for 15 s
- Step 3: 57 °C for 30 s
- Step 4: 60 °C for 40 s
- Melting curve
Analyze the data using the Livak Method or the analysis of relative gene expression. Perform a one-way ANOVA for statistical analyses (see Note 21) [Figure 2 near here][11].
Figure 2.

RNA interference promotes β-catenin downregulation. To evaluate the efficiency of the interference of β-catenin transcript, qRT-PCR of intestinal explants was performed after 2 days of DsiRNA electroporation. Expression values were plotted as fold change relative to DsiRNA-targeting GFP (negative control) treatment in log2 scale. N = 4.
4. Notes
Antibiotics and antifungals lose their activity in ~ 3 – 5 days in culture medium. Also, other supplements lose their activity in a few days in culture. Thus, it is recommended to supplement the media one day before the experiment and change the media every 3 – 5 days. The L-15 supplemented medium can be stored at 4 °C up to 2 weeks.
This dilution corresponds to one gut rudiment. For more samples, take into account the total volume needed on each electroporation cuvette for each gut rudiment and prepare a master mix.
For 4% PFA preparation wear gloves, laboratory glasses and a face mask. Prepare the solution inside a chemical hood. For 0.2 M Phosphate Buffer (PB), prepare Solution A dissolving 13.8 g of NaH2PO4·H2O in 500 mL ddH2O and for Solution B dissolve 14.2 g of Na2HPO4 -anhydrous in 500 mL ddH2O. Mix 19 mL of Solution A and 81 mL of Solution B to obtain 0.2 M PB.
Purchase the amount of DsiRNAs according to the number of samples and the concentration needed. Centrifuge the tube with DsiRNA previous to its resuspension to avoid any loss of material.
The amount of Nuclease-free water depends on the nmol of the primers. The formula used is: nmol × 10, according to IDT.
The number of sea cucumbers is not established in this part of the protocol because it depends on the requirements of the experiments.
In this particular experiment, we used the gut rudiment, but this protocol can be applied to the tissue of interest.
Gut rudiments are delicate during this regenerative stage. Thus, use a pipette tip as a hook to transport the gut rudiments from one place to another. If using a tweezer, try not to squeeze the gut rudiment.
In this part of the experiment, we used 18 sea cucumbers, three sea cucumbers per parameter. We were trying to elucidate the optimal parameter to accomplish the transfection of the TMRAD and, eventually the transfection of the DsiRNAs. This part of the protocol is very important for elucidating the optimal parameter of a particular tissue. It is very important to mention that every tissue has a different response to the process of electroporation. So, these parameters can be used as a reference for the electroporation of other tissues.
Gut rudiments are placed in 30 – 40% sucrose for cryoprotection. 24 h is enough for the sucrose to be absorbed by the gut rudiments, but another signal is when the tissues sink to the bottom of the 2 mL tube containing the 30 – 40% sucrose.
Gut rudiments embedding are prepared at RT because Tissue-Tek O. C. T. freezes at lower temperatures.
As gut rudiments are long, but thin, several replicates can be embedded in a metal base mold.
Set the cryostat temperature on −35 °C. Perform the cryosections from −27 to −35 °C to obtain better results. Set the temperature 1 h before starting to cut.
In our laboratory, histological studies are examined and photographed using a Nikon Eclipse Ni fluorescent microscope equipped with DAPI, FITC and R/D2 filters and a DS-Qi2 camera. Images were obtained using the software NIS-Elements.
Set the temperature of the centrifuge to 4 °C before starting the RNA extraction protocol. After this centrifugation, set the temperature to 22 – 24 °C and press the “fast temp” button; RNA purification is performed at room temperature.
The RNA extraction using trizol/chloroform produce 3 phases after the centrifugation. Proteins are extracted to the organic phase (bottom of the tube), DNA resolves at the interface and RNA remains in the aqueous phase (top of the tube).
In our laboratory, we prepare master mixes for the remaining reagents of the cDNA synthesis. Thus, we multiply the volume of each reagent by the total number of samples including an extra sample.
We set these parameters in a thermocycler. We add our first 10 μL of the protocol (Oligo dT, Nuclease-free water and cDNA), put the tubes in the thermocycler to start the first part of the protocol. Then, we add the remaining components to each tube and put the tubes in the thermocycler to finish the protocol.
The primers were designed to flank the target sequence of the RNAi to assure that the knockdown was efficiently done.
To prepare the 96-well plate, we prepare master mixes for each pair of primers. In this case, we prepare master mixes for β-catenin and NADH. The master mixes are prepared taking into account the total number of samples and technical replicates of the biological replicates. We prepare 3 technical replicates for each sample. Thus, if we are going to calculate the total volume for the forward and/or reverse primers, we multiply 0.4 μL × 3.5 (technical replicates) × 13 (extra sample). This apply also for the iQ SYBR Green Supermix and the Nuclease-free water. After that, we prepare twelve 0.2 mL tubes (one per sample) and add 66.5 μL of the master mix for β-catenin and 3.5 μL of the samples. Thus, we have 12 mini-master mixes with 70 μL corresponding to the technical replicates of each biological sample. We perform the same for the master mix of NADH and then we transfer the reaction to the 96-well plate. Each well will have 20 μL of 3 technical replicates for each biological sample.
Results present three different concentration treatments, 1, 10 and 100 μM. By doing this gradient, we wanted to elucidate the optimal DsiRNA concentration for β-catenin knockdown.
Acknowledgments
This project was supported by NIH (Grants R15GM124595, R21AG057974), and the University of Puerto Rico.
Contributor Information
Miosotis Alicea-Delgado, University of Puerto Rico – Río Piedras Campus, San Juan, P.R..
Samir A. Bello-Melo, University of Puerto Rico – Río Piedras Campus, San Juan, P.R.
José E. García-Arrarás, University of Puerto Rico – Río Piedras Campus, San Juan, P.R.
References
- 1.Hannon GJ (2002) RNA interference. Nature 418:244–251 [DOI] [PubMed] [Google Scholar]
- 2.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811 [DOI] [PubMed] [Google Scholar]
- 3.Elbashir SM, Lendeckel W, Tuschl T (2001) RNA interference is mediated by 21- and 22- nucleotide RNAs Genes Dev. 15:188–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meister G, Tuschi T (2004) Mechanisms of gene silencing by double-stranded RNA. Nature 431:343–349 [DOI] [PubMed] [Google Scholar]
- 5.Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ (2005) Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol 23:222–226 [DOI] [PubMed] [Google Scholar]
- 6.Bogarad L, Arnone MI, Chang C, Davidson EH (1998) Interference with gene regulation in living sea urchin embryos: Transcription factor Knock Out (TKO), a genetically controlled vector for blockade of specific transcription factor. PNAS 25:14827–14832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mashanov VS, Zueva OR, García-Arrarás JE (2015) Myc regulated programmed-cell death and radial glia dedifferentiation after neural injury in an echinoderm. BMC Developmental Biology 15:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bello SA, Abreu-Irizarry RJ, García-Arrarás JE (2015) Primary cell cultures of regenerating holothurian tissues. Methods Mol Biol 1189:283–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schacher S, Proshansky E (1983) Neurite regeneration by Aplysia neurons in dissociated cell culture: modulation by Aplysia hemolymph and the presence of the initial axonal segment. J Neurosci 3:2403–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naito Y, Yoshimura J, Morishita S, Ui-Tei K (2009) siDirect 2.0: updated software for designing functional siRNA with reduced seed-dependent off-target effect. BMC Bioinformatics 10:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alicea-Delgado M (2019) Functional role of β-catenin and Myc as active players in the canonical Wnt signaling pathway during intestinal regeneration of the sea cucumber Holothuria glaberrima. Thesis, University of Puerto Rico – Río Piedras Campus [Google Scholar]