

Abstract
Sub/supercritical fluid chromatography (SFC) is a green separation technique that has been used to separate a wide variety of compounds and is proven to be immensely useful for chiral separations. However, SFC is currently not thought to be applicable for ionic compounds due to their low solubility in CO2, even with additives and organic modifiers. Recently, a large amount of research has been centered on octahedral complexes of Ru(II) and Os(II) with bidentate polypyridyl ligands due to their ability to serve in cancer treatment and other biological activities. These compounds exist as the delta (Δ) and lambda (Λ) enantiomers. Previously, similar compounds have been enantiomerically separated using HPLC and capillary electrophoresis, but never with SFC. Cyclofructan-6 (CF6) derivatized with (R)-naphthyl ethyl (RN) groups has been proven to be an effective chiral stationary phase for these separations in HPLC. This column chemistry was expanded to SFC to provide the first chiral separation of a wide variety (23 complexes in total) of ionic octahedral polypyridyl complexes. Unexpected behavior for mixing methanol and acetonitrile as the organic modifier will be discussed, along with the effects of additives. Enantioselectivity on CF6-RN chemistry is shown to be dependent on the conjugation level and rigidity of the metal complexes. Mass transfer kinetic behavior is also shown, and high-efficiency baseline resolved rapid separations are shown for fast screening or quantitation of representative coordination complexes.
Keywords: Metal complexes, coordination complexes, sub/supercritical fluid chromatography, cyclofructan derivatives, superficially porous particles, fast chiral separation
Introduction
Modern sub/supercritical fluid chromatography (SFC) systems with CO2 rich eluents have evolved to become an excellent green and reliable replacement for analytical and preparative chiral normal phase liquid chromatography (NPLC) [1–9]. The latter technique often employs highly toxic/carcinogenic solvents such as hexanes and chlorinated alkanes. Solvatochromic studies show that subcritical CO2 (24 °C, 8.75 MPa) has a Nile red transition energy (E(NR)) of 59.54 kcal/mol, which most closely matches liquid pentane (E(NR) =59.12 kcal/mol) in terms of solvent strength [10]. Therefore, a neat pressurized CO2 mobile phase is useful for a small window of nonpolar analytes, including hydrocarbons, fatty acids, waxes, ethers, and esters [11]. In modern SFC, pressurized CO2 is typically modified with small alcohols, and the mobile phase is often held at a back pressure above the critical pressure of CO2, but the modifier brings the fluid into the subcritical state under ambient temperatures [12]. For chromatographic purposes, the physical state of the SFC mobile phase is not of concern since the properties of a supercritical fluid and a liquid are a continuum [13], and the low viscosity advantage of pressurized CO2 can still be maintained. This property, in turn, promotes more efficient mass transfer kinetics as compared to HPLC, making it highly desirable for chiral separations where mass transfer kinetics are often slow [14,15].
Various researchers have attempted to increase the scope of SFC to encompass other classes of compounds that can be separated by traditional reverse phase, ion-pairing, and hydrophilic interaction liquid chromatography [11,16,17]. These classes include hydroxy acids, primary amines, zwitterions, and peptides. Studies have been done using ion-pairing agents to separate permanently ionized compounds in SFC, but early examples are plagued with misunderstandings [18]. The first claimed separation of charged species with a CO2 mobile phase was in 1988, when quaternary ammonium salts were separated. Later that year, it was determined by other authors that the compounds were pre-converted off-line to tertiary amines and then analyzed in their neutral form [19]. In 1990, the first SFC separation of a pharmaceutically relevant ionizable compound was published when propranolol hydrochloride was separated using a cyanopropyl silica packed column with methanol modified CO2 mobile phase containing sodium heptane sulfonate additive [20]. Despite the possibility of over 40 co-solvents that have been used in SFC literature with a variety of additives and applications, SFC is still not routinely applied for the analysis of completely ionized species like metal complexes with large molecular weights [21,22]. The separation of inorganic ions or ionic compounds has been missing from the literature until a recent paper published a method to simultaneously separate cationic and anionic metal ions [23]. We sought to apply the benefits of SFC to enantiomerically separate charged ruthenium (Ru(II)) and osmium (Os(II)) polypyridyl octahedral complexes of clinical importance.
The complexes investigated in this study (Figure 1) have a wide variety of ligand structures. Some of these compounds are potent inhibitors of microtubules (chemotherapy targets) in human cells [24,25], can serve as DNA intercalating agents [26], and can serve as photosensitizing agents for photodynamic (PDT) therapy and photochemotherapy (PCT) [27–29]. A racemate of compound 4 (TLD-1433) is currently in Phase II clinical trials for treating non-muscle invasive bladder cancer (NMIBC) with PDT [30]. Enantiomers of a few similar compounds have been separated by HPLC and capillary electrophoresis [31–33]. These complexes all contain helical chirality due to the bidentate nature of the ligands and the octahedral coordination sites of the metal, creating delta (Δ) and lambda (Λ) enantiomers as shown for TLD-1433 in Figure 2. To the best of our knowledge, TLD-1433 would be the first chiral octahedral drug ever approved by the FDA, making the optimization of these enantioseparations increasingly relevant for drug development. Herein, we report the first chiral SFC separation of ionic octahedral transition metal complexes on a derivatized cyclofructan (CF6) chiral stationary phase bonded to 2.7 μm superficially porous particles (SPP). The significant beneficial effect of blending acetonitrile and methanol is demonstrated, expanding the typical single solvent modifiers found in the literature. The addition of water into the modifier to make a highly efficient quaternary mobile phase is discussed in detail. Enantioselectivity is investigated for the compounds as a function of conjugation level and the resultant molecular rigidity. Kinetic plots are shown for various complexes. Representative enantioseparations are shown at high speeds and efficiencies, demonstrating the true advantage of SFC over HPLC for metal complexes as compared to previously reported works [31,33].
Figure 1:
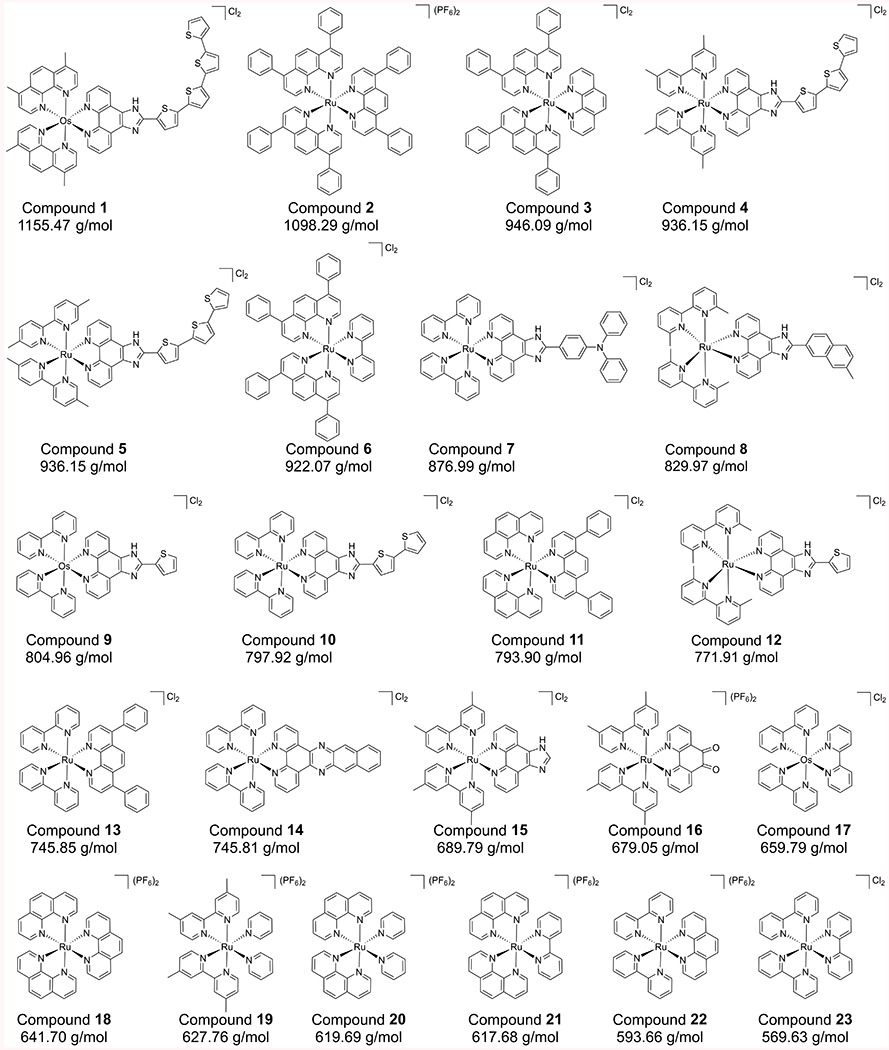
Molecular structures of the Ru(II) and Os(II) polypyridyl octahedral coordination complexes included in this study. Formula weight below the structures corresponds to the mass of the complex without the counter anion.
Figure 2:

Delta (Δ) and lambda (Λ) designations of stereoisomers containing tris(bidentate) metal complexes and other octahedral complexes. Bidentate octahedral complexes contain helical chirality, and the left-handed enantiomer is labeled Λ as the right-handed enantiomer is labeled Δ. Racemate of compound 4 shown here is under Phase-II clinical trial for bladder cancer.
2. Materials and Methods
2.1. Chemicals
All solvents were purchased from Fisher Scientific (Waltham, MA, USA) corporation and were HPLC grade. Additives (acetic acid (AA), trifluoracetic acid (TFA), formic acid (FA), triethylamine (TEA), aqueous ammonia (NH3) (nominal 28-30% NH3 basis, d= 0.9 g/mL), and ammonia formate (AF)) were purchased from Sigma Aldrich (St. Louis, MO, USA) corporation. Analytes were dissolved into HPLC-grade acetonitrile at a ~ 1 mg/mL concentration. Water was purified to 18 MΩ using a Barnstead GenPure Pro UV water purification system from Thermo Fisher Scientific (Waltham, MA, USA). Carbon dioxide was purchased from Airgas (UN1013, Radnor, PA, USA) in a full-length siphon cylinder to deliver liquefied CO2 to the pump heads. Native cyclofructan CF6 was provided by AZYP LLC, and R-(−)-1-(1-Napthyl)ethyl isocyanate to modify CF6 was purchased from TCI (Portland, OR, USA). The bonding chemistry is discussed elsewhere [34]. The functionalized CF6 was bonded to 2.7 micron superficially porous particles (LarihcShell-RN). Chiral columns were synthesized and provided by AZYP, LLC (Arlington, TX, USA). The columns were provided in two lengths (150 mm x 4.6 i.d. mm and 100 mm x 4.6 i.d. mm).
Organic modifiers were prepared using graduated cylinders and volume ratios and mixed prior to incorporating additives. All the quoted percentages for eluents are volume by volume. All reported additive percentages are representative of the percentage of additive added, via micropipette, to a given volume of modifier, e.g., a nominal 2% TFA v/v was prepared by adding 2 mL of TFA into 100 mL of the modifier. Water was added to the modifier in a similar fashion; however, a class-A burette was used to dispense the water into the modifier.
2.2. Synthesis and Characterization of the Ruthenium and Osmium Polypyridyl Complexes
Complexes 4, 8-10, 12, 14-15, and 17 have been previously reported by McFarland and coworkers [28,30,35–41]. Complexes 2, 3, 6, 11, 13, 18, and 21–23 were prepared with the same procedures used in HPLC studies [31]. Complex 7 was prepared following the procedure described by Fan and coworkers [42]. Complexes 16, 19, and 20 were prepared following the procedures described by Chao and coworkers [43]. The synthetic details and characterization data for previously unreported compounds 1 and 5 prepared by McFarland and coworkers are included below. The details of the instrumentation used during synthesis and characterization are provided in the Supporting Information (S1).
[Os(5,6-dmp)2(IP-4T)](Cl)2 (1)
Os(5,6-dmp)2Cl2 (92 mg, 0.2 mmol) and IP-4T (92 mg, 0.168 mmol) were added to a microwave vessel containing argon-purged ethylene glycol (4 mL) and subjected to microwave irradiation at 180 °C for 15 min. The resulting black mixture was then transferred to a separatory funnel with deionized water (25 mL) and CH2Cl2 (25 mL). After gentle agitation, the CH2Cl2 was drained and the remaining aqueous layer was washed with CH2Cl2 (25 mL) until the CH2Cl2 layer was colorless. At that point, another 30 mL of CH2Cl2 was added and allowed to settle to the bottom of the separatory funnel. Then, saturated aqueous KPF6 (5 mL) was added, and the mixture was shaken gently. The CH2Cl2 layer was drained and the PF6 salt of the complex was further extracted from the aqueous layer with CH2Cl2 (25 mL) until the aqueous layer was colorless. The CH2Cl2 extracts were then combined and concentrated under reduced pressure to yield the crude PF6 salt of the product that was subsequently purified using silica gel flash column chromatography with a gradient of ACN, 10% water in ACN, followed by 7.5% water in ACN with 0.5% KNO3. The fractions containing the desired complex were then combined and concentrated under vacuum, then transferred to a separatory funnel with CH2Cl2 (25 mL), deionized water (25 mL), and saturated aqueous KPF6 (1 mL). The resulting mixture was gently mixed and the CH2Cl2 layer was drained. Additional CH2Cl2 (25 mL) was used to extract the remaining product until the aqueous layer was colorless. The CH2Cl2 layers were then combined and dried under vacuum, yielding the PF6 salt of the product as a black solid (44 mg, 18%), which was then converted to its corresponding Cl− salt in quantitative yield using Amberlite IRA-410 with MeOH as the eluent. The Cl− salt was then further purified by dissolving in ~2 mL MeOH and loading onto a Sephadex LH-20 column. The final product was eluted using MeOH, and the purest fractions (determined by TLC) were combined and concentrated under vacuum, yielding complex 1 as a black powder (33 mg, 16%). Full characterization results for this complex are provided in the Supporting Information (S2).
[Ru(5,5-dmb)2(IP-3T)](Cl)2 (5)
Ru(5,5’-dmb)2Cl2 (115 mg, 0.2 mmol) and IP-3T (78 mg, 0.168 mmol) were added to a microwave vessel containing argon-purged ethylene glycol (3 mL) and subjected to microwave irradiation at 180°C for 15 min. The resulting reddish-brown mixture was then transferred to a separatory funnel with deionized water (25 mL) and CH2Cl2 (25 mL). After gentle agitation, the CH2Cl2 was drained, and the remaining aqueous layer was washed with CH2Cl2 (25 mL) until the CH2Cl2 layer was colorless. At that point, another 30 mL of CH2Cl2 was added and allowed to settle to the bottom of the separatory funnel. Then, saturated aqueous KPF6 (5 mL) was added, and the mixture was shaken gently. The CH2Cl2 layer was drained and the PF6 salt of the product was further extracted from the aqueous layer with CH2Cl2 (25 mL) until the aqueous layer was colorless. The CH2Cl2 extracts were then combined and concentrated under reduced pressure. The crude PF6 complex was then purified using silica gel flash column chromatography with a gradient of ACN, 10% water in ACN, followed by 7.5% water in ACN with 0.5% KNO3. The fractions containing the metal complex were then combined and concentrated under vacuum, then transferred to a separatory funnel with CH2Cl2 (25 mL), deionized water (25 mL), and saturated aqueous KPF6 (1 mL). The resulting mixture was gently mixed and the CH2Cl2 layer was drained. Additional CH2Cl2 (25 mL) was used to extract the remaining product until the aqueous layer was colorless. The CH2Cl2 layers were then combined and dried under vacuum, yielding the metal complex PF6 salt that was then converted to its corresponding Cl− salt in quantitative yield using Amberlite IRA-410 with MeOH as the eluent. The Cl− salt was then further purified by dissolving it in ~2 mL MeOH and loading it onto a Sephadex LH-20 column. The product was eluted using MeOH, and the purest fractions (determined by TLC) were combined and concentrated under vacuum, yielding 5 as a red powder (44 mg, 26%). Full characterization results for this complex are provided in the Supporting Information (S2).
2.3. Chromatography
All separations were performed on a Jasco (Tokyo, Japan) semi-prep SFC system (SFC-2000-7), allowing flow rates up to 20 mL/min with a 50 MPa maximum limit. The carbon dioxide and organic modifier were pumped by identical HPLC pumps (PU-2086) with the CO2 pump head kept at a constant-10 °C by a Julabo (Seelbach, Germany) chiller. Following the pumps, the modifier and the CO2 were mixed in a stainless steel mixing chamber and then plumped through an autosampler (AS-2059-SFC) with a 5 μL loop, a thermostatic column oven (CO-2060), a UV detector (UV-2075), and an automatic back pressure regulator (BP-2080). The UV detector contained a high-pressure flow cell and was set to detect at a wavelength of 260 nm. The backpressure regulator was set to a constant 8 MPa and was heated by a Jasco heating controller (HC-2068-01) to 60 °C. PEEK tubings (254 μm i.d.) were used to make connections from the autosampler to the detector since this diameter was found to be optimal for fast SFC [44].
The SFC system was controlled by ChromNav (1.17.01 Build 8) software through an LC-Net II/ADS system. The detector was set to 100 Hz with a response time of 0.05 s (the fastest available for the instrument). The ChromNav software automatically employs a moving filter to reduce noise, and all chromatograms shown were subsequently treated with a 3-point median filter with symmetric zero padding at data ends in MATLAB (R2021b) to eliminate spikes. Efficiency (N) values were calculated by ChromNav using the half-width method.
3. Results and Discussion
The LarihcShell RN columns used in this study consisted of a carbamate coupling of a π-base, R-(−)-1-(1-Napthyl)ethyl isocyanate (RN) to cyclofructan 6 (CF6). This stationary phase shows good enantiomeric selectivity with non-primary amines. Previously, it has been used in polar organic mode and reverse-phase HPLC to separate polypyridyl metal complexes [31,33] on 5 μm fully porous particles, but has never been used with superficially porous particles or with SFC until this study. Interactions of this stationary phase include chiral complexation from the hydrogen bonding donor/acceptor sights of the cyclofructan as well as π-π interactions from the RN group. These interactions are investigated below, and mass transfer kinetics are provided to show the efficiency of this column chemistry on SPP particles.
3.1. Requirement of a Blended Methanol and Acetonitrile Modified CO2 Mobile Phase
One of the main concerns in SFC while analyzing completely new charged compounds is the lack of solubility data in pressurized carbon dioxide/organic solvent mixtures. If any compound precipitates to form a solid in the injector or column after encountering the CO2 based mobile phase, it can significantly damage the columns and the chromatograph. To avoid precipitation, the solubility of these inorganic coordination complexes with chloride and hexafluorophosphate (PF6) counter anions was tested in pure methanol and acetonitrile. Most compounds (Figure 1) did not show good solubility in methanol, but they were much more soluble in acetonitrile. However, when acetonitrile (45%) modified CO2 (55%) mobile phase was tested on compound 18 with the strongest additives (0.1% TFA/ 0.1% TEA) determined by polar organic mode HPLC [33], inordinately long retention was observed (k1>100). A methanol (45%) modified CO2 (55%) mobile phase was then tested on compound 18 with the same additives (0.1% TFA/ 0.1% TEA), where retention was observed to be very high (k1 = 60.0), and efficiency was low due to considerable tailing, but elution and full separation was observed (α = 1.50). This is an interesting aspect where a solvent, like acetonitrile, shows high solubility for an analyte in the liquid phase, but when mixed with pressurized CO2, it loses its ability to elute the compound within an acceptable timeframe. In polar organic mode HPLC, blending acetonitrile with methanol created an effective mobile phase for similar complexes [33], but this is not a common technique in the literature for SFC organic modifiers. The use of several blends (ACN/MeOH, ACN/EtOH, and MeOH/EtOH) of organic solvents as the modifier in ultra-high performance SFC has been reported, however, only one to on ratios were tested [45]. Therefore, it was desired to explore the behavior of the ACN/MeOH blended modifier to observe the retention and separation characteristics of these complex ion analytes on the CF6-RN column.
To further investigate how blending methanol and acetonitrile controlled retention for these complexes, compound 18 was tested at six different methanol acetonitrile blends (100/0, 90/10, 80/20, 60/40, 40/60, 30/70), all with 0.1% TFA and 0.1% TEA in Figure 3. Only the composition of the modifier is changed in Figure 3, maintaining the same ratio of CO2 (55%) to modifier (45%). The results are similar U-shaped retention curves analogous to that observed in the polar organic mode for both enantiomers [33], where blending acetonitrile with methanol greatly reduces retention. To find the minima of these curves and the strongest composition of methanol and acetonitrile, a third-order polynomial was fit through the six points (R2 ≥ 0.995). The polynomial equation was then solved to minimize retention, where approximately 65% methanol and 35% acetonitrile were found to be the minima for both curves. The retention study was then repeated for compound 23, where a similar pattern was observed having the same minima position, confirming this is a general trend for these complexes. The optimized methanol to acetonitrile blend (65/35) provided retention (k1) 3.4 times faster than the popular methanol-only modified CO2 mobile phase. These results may support the use of methanol to interact with active cyclofructan sites that lead to strong retention in acetonitrile via hydrogen bonds. Interestingly, enantioselectivity (α) is not significantly affected by the drastic change in retention from the different blends ranging from 1.50 for 100% methanol modifier to 1.42 for 30% methanol 70% acetonitrile for compound 18. This shows that methanol increases selectivity compared to acetonitrile and is necessary in some proportion for any separations on this stationary phase in the SFC mode. For all further studies, this optimized methanol acetonitrile blend was used. This optimized modifier composition lowered retention, allowing for the quantity of CO2 in the mobile phase to be raised to 60% for all additional separations (apart from the Fast Separations section).
Figure 3:
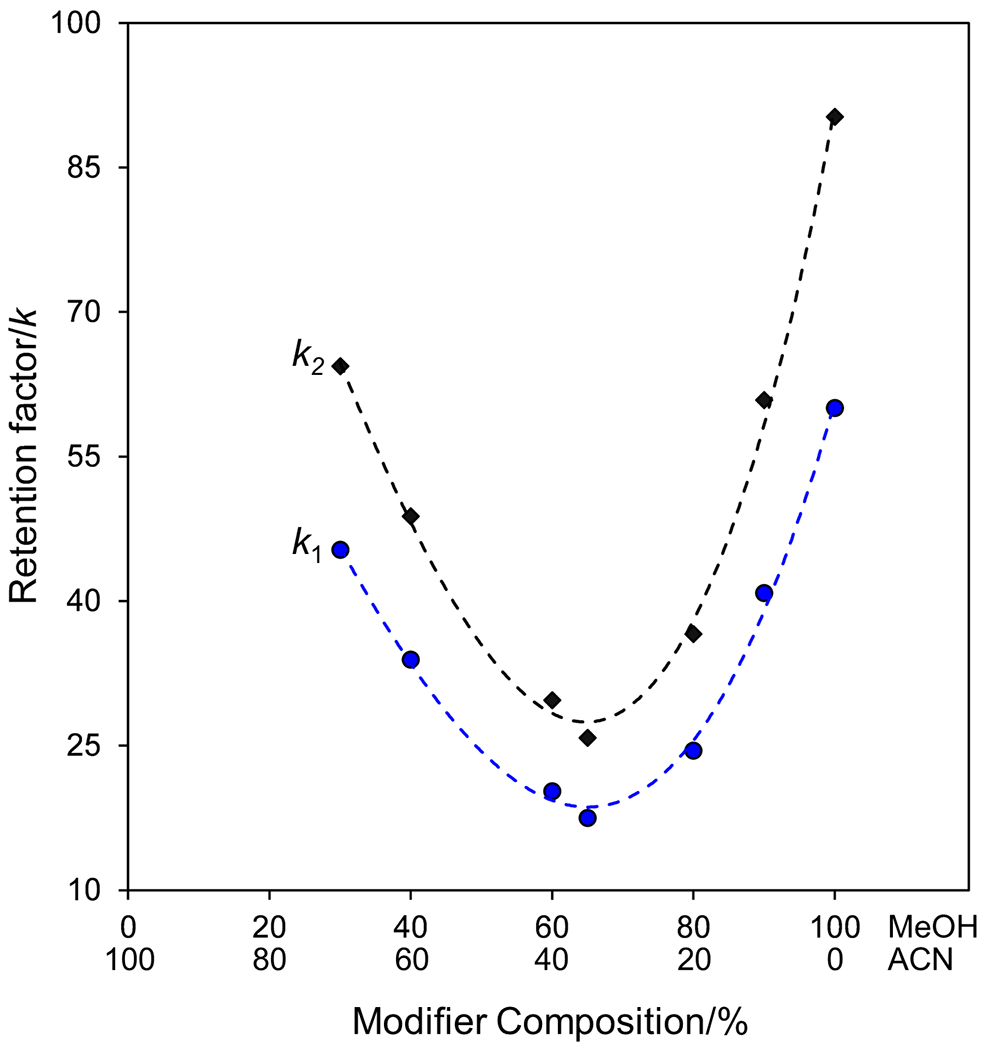
Representative retention curves showing the effect of the composition (MeOH/ACN) of the organic modifier for compound 18 on a 100 mm x 4.6 i.d. mm 2.7μm SPP CF6-RN column. Conditions: 55% CO2, 45% modifier (X% MeOH as X is indicated in the Figure, Y% ACN as Y is indicated in the Figure, 0.1% TFA, 0.1% TEA), 25 °C, 4 mL/min, 8 MPa backpressure, 5 μL injection. The fit shown with dashed lines is a third order polynomial fit.
3.2. Effect of Water Addition to the Acetonitrile Methanol Organic Modifier
After raising the CO2 composition in the mobile phase from 55% to 60%, retention increased substantially. For example, the retention of the first enantiomer of compound 18 raised to 99.5, and the observed efficiency was also low (N1=1550). Previous studies by other authors [2,46,47] and our group [21,48,49] have shown that wetting the modifier in chiral/achiral SFC can greatly increase the efficiency and speed of analysis. The benefits of wetting the mobile phase have been shown in chiral normal phase separations as well [50]. Although pure water is sparingly soluble with pressurized CO2, the presence of a hydrogen bonding solvent in the mobile phase, such as small alcohols (C1-C3), greatly helps in homogenizing H2O with pressurized CO2 [46]. This effect is particularly important for relatively hydrophilic stationary phases, like the one used in this study, where it has been shown that there can be nearly an order of magnitude increase in efficiency by adding small amounts of water to the modifier [21]. This great gain in efficiency is due to water absorbing onto the hydrophilic stationary phase changing the surface properties and altering analyte-stationary interactions improving mass transfer kinetics [51]. To investigate the addition of water into the optimized methanol acetonitrile (65/35) modifier, four mobile phases were made containing different amounts of “added” water (0%, 1%, 5%, 10%) for compounds 9 and 18, which contain different metal centers and different ligand structures. The results of this study are shown in Figure 4.
Figure 4:

The effect of the addition of water into the modifier for compound 18 (A) and compound 9 (B) on a 100 mm x 4.6 i.d. mm 2.7μm SPP CF6-RN column. Conditions: 60% CO2, 40% Modifier (65% MeOH, 35% ACN, 0.1% TFA, 0.1% TEA with X% water as X is indicated in the figure), 25 °C, 4 mL/min, 8 MPa backpressure, 5 μL injection.
Figure 4 shows the addition of small amounts of added water to the modifier reduces retention to around a third of the time of the dry system and almost doubles the efficiency for enantiomers of both compounds. This “greens” the SFC method employed in this work by using significantly less organic solvent than a water-free system and peak tailing is reduced as well. In chiral chromatography with polysaccharides, there is often a semi-permanent change in enantioselectivity after the column absorbs trace amounts of water. This effect is known as a “memory effect” [52] or a display of hysteresis. To test this, the column used in this study was wetted using the modifier containing 10% water and then dried using 200 proof ethanol (0.5 mL/min) overnight (>8 hours). It was found that the column could go back to its “dry” state reproducibly during several cycles. However, a small amount of water may be present in the solvent and adsorbed on the column explaining the negligible change in retention from 0% water to 1% water in Figure 4.
In general, the introduction of water into the mobile phase also leads to more reproducible retention times as the effect of small amounts of water in all solvents no longer is a concern and has the additional benefit of eliminating silyl ether formation (SEF) in the long term. In SFC, silyl ethers can form by reacting alcohols in the modifier with those on the surface of the particles of the stationary phase [53] if the SFC experiment is continuously run for a long time. This reaction lowers the hydrophilicity of the column and changes the retention and selectivity of the column for many achiral molecules [53]. Water prevents such reactions and keeps the column reproducible for a longer time [53]. For all further studies, 10% water was added into the modifier.
3.3. Effect of Additives and Additive Concentration
Additives are necessary for efficient separations of polar or ionic analytes, although there is some debate as to the exact mechanism [23,54,55]. For eluting ionic or ionizable compounds in chiral SFC, small amounts (~1-2 % v/v) of additive in the organic modifier are required. For example, the addition of a small amount of trifluoracetic acid (TFA) has been proven by solvatochromic analysis to increase the overall polarity of the mobile phase [54]. This helps increase the solubility of polar and ionic analytes in the mobile phase. TFA alone is a very strong acid, and it can cleave siloxane bonds when in the presence of water. To avoid this concern, TFA was combined with TEA to provide a “buffering” action.
In this study, eight different additive compositions (0.2% TFA/0.2% TEA, 0.1% AA/ 0.1% TEA, 0.1% FA /0.1%TEA, 0.1% FA, 0.1% AA, 0.2% TFA/0.09% NH3, 0.1% AF, and 0.1% TEA). were tested for compound 18 and compound 9 (Figure 1) using 40% of the optimized modifier (65% Methanol, 35% Acetonitrile, 10% water) with 60% CO2. Of these combinations, the only options that provided acceptable elution (~ 1< k1 < 40) was trifluoracetic acid with triethylamine (0.1% TFA/ 0.1% TEA) and trifluoracetic acid with aqueous ammonia (0.2% TFA/ 0.09% NH3). These results confirm observations from HPLC studies of similar complexes with the same stationary phase functionality, where TFA/TEA was consistently the optimal combination in the eluent [31,33]. Since the mobile phase is weakened for these complexes by the addition of CO2, it is advantageous to use the strongest additives possible. The effect of the concentration of TFA/TEA (0.01% to 2%) on retention (A) and efficiency (B) for compound 18 and compound 9 is shown in Figure 5. For both compounds, the retention factor was increased from 0.01% to 0.1% and then decreased from 0.1% to 2%. Efficiency, however, was always increased as the additive concentration was raised. The fastest and most efficient separations were found at 2% TFA/ 2% TEA, so all compounds were tested under these conditions, as shown in Table 1. The same molar equivalence was tested for TFA and aqueous ammonia (Table 1), where it was shown that resolution and efficiency were slightly worse compared to using TEA as a base. Separations were always better when there was a slight molar excess of the acid additive. Note that we did not try a higher concentration than 2% of TFA or a higher molar ratio of TFA since such additives can potentially add a burden to any preparative work-up.
Figure 5:

The effect of TFA and TEA concentration modifier on efficiency (N1) and retention (k1) in the modifier for compound 18 (orange) and compound 9 (blue) on a 100 mm x 4.6 i.d. mm 2.7μm SPP CF6-RN column. Samples were analyzed in triplicate and the relative standard deviation was less than 2.5% for efficiency and retention factor. Conditions: 60% CO2, 40% modifier (65% MeOH, 35% ACN, 10% H2O, X% TFA, and X% TEA as X is indicated in the figure), 25°C, 4 mL/min, 8 MPa backpressure, 5 μL injection.
Table 1:
Chromatographic figures of merit of inorganic complexes using 100 mm x 4.6 mm i.d. CF6-RN column in SFC mode.
| Compound ID | 0.26 M TFA, 0.14 M TEA* | 0.26 M TFA, 0.14 M NH3* | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| k 1 | α | Rs | N 1 | N 2 | k 1 | α | Rs | N 1 | N 2 | |
| 2 | 3.68 | 1.53 | 7.0 | 6700 | 6300 | 3.19 | 1.50 | 6.4 | 6700 | 6400 |
| 3 | 4.57 | 1.49 | 6.7 | 6700 | 6400 | 4.10 | 1.44 | 6.1 | 6600 | 6600 |
| 4 | 9.93 | 1.50 | 6.5 | 5200 | 4600 | 11.01 | 1.43 | 6.0 | 5400 | 5000 |
| 5 | 8.84 | 1.44 | 6.9 | 6400 | 5900 | 9.99 | 1.37 | 5.7 | 6800 | 6100 |
| 6 | 3.99 | 1.32 | 4.6 | 7000 | 6700 | 3.64 | 1.29 | 4.0 | 6700 | 6000 |
| 7 | 16.48 | 1.30 | 4.2 | 5100 | 4700 | 16.31 | 1.26 | 3.4 | 4600 | 4400 |
| 9 | 18.05 | 1.26 | 3.9 | 5000 | 5100 | 18.48 | 1.22 | 3.2 | 4600 | 4800 |
| 10 | 20.64 | 1.32 | 4.9 | 5600 | 5100 | 20.64 | 1.28 | 4.1 | 5100 | 4600 |
| 11 | 8.14 | 1.41 | 5.9 | 6200 | 5900 | 7.70 | 1.34 | 5.1 | 5800 | 5700 |
| 12 | 9.59 | 1.04 | - | - | - | 10.16 | 1.04 | - | - | - |
| 13 | 7.17 | 1.14 | 2.4 | 6600 | 6400 | 7.21 | 1.12 | 2.1 | 6100 | 5900 |
| 14 | 23.30 | 1.53 | 6.6 | 4700 | 4000 | 22.15 | 1.46 | 5.6 | 4000 | 3600 |
| 15 | 6.56 | 1.30 | 3.9 | 4700 | 4500 | 6.66 | 1.25 | 3.3 | 4600 | 4500 |
| 16 | 3.65 | 1.15 | 1.1 | 1400 | 1600 | 3.93 | 1.14 | 1.2 | 1800 | 1900 |
| 17 | 15.82 | 1.07 | 1.2 | 5300 | 5200 | 17.62 | 1.06 | 0.95 | 4800 | 4600 |
| 18 | 16.17 | 1.37 | 5.2 | 4900 | 4700 | 16.90 | 1.31 | 4.1 | 4200 | 4100 |
| 19 | 1.37 | 1.09 | 1.1 | 6700 | 6800 | 1.81 | 1.08 | 0.9 | 4700 | 5300 |
| 20 | 9.23 | 1.14 | 2.4 | 6800 | 6800 | 11.9 | 1.12 | 1.9 | 5100 | 4900 |
| 21 | 15.69 | 1.24 | 3.6 | 5000 | 4900 | 15.85 | 1.20 | 2.8 | 4200 | 4200 |
| 22 | 14.67 | 1.14 | 2.0 | 4600 | 4300 | 14.57 | 1.11 | 1.6 | 3700 | 3500 |
| 23 | 14.07 | 1.07 | - | - | - | 14.52 | 1.06 | - | - | - |
Conditions: 60% CO2:40% Modifier (65% MeOH, 35% ACN with 10% water and additives listed above. *Molarity of TFA and TEA corresponds to 2% v/v, NH3 (aq) was adjusted to 0.9% v/v to keep the molar ratio of acid to base the same), 25 °C, 4 mL/min, 8 MPa back pressure, 5 μL injection, samples ~1.0 mg/mL in ACN
3.4. Investigation of Ru(II)/Os(II) Polypyridyl Complexes
Table 1 provides a summary of the chromatographic figures of merit, including the retention factor for the first enantiomer (k1), enantioselectivity (α), and resolution between enantiomers (Rs) and the number of theoretical plates (N) for both enantiomers. In almost all cases, the TFA/TEA additives provided slightly better efficiency and resolution than TFA/NH3. The representative chromatograms in Figure 6A show a baseline separation (Rs >1.5) for all compounds except compounds 8, 12, 16, 17, 19 and 23. To further resolve these compounds, a 150 mm column was used along with a lower flow rate (1.1 mL/min) and a cooled column from 25 °C to 8 °C (lowest temperature possible on the instrument). Compound 1 showed extreme retention and different additive concentration was used (Figure 6B). The improved resolutions and chromatographic information are provided in Table 2 with representative chromatograms in Figure 6C. This leaves all compounds at or near baseline separation, except for compound 8, which has slight chiral discrimination shown by the shoulder in Figure 6C. Compound 8 does not separate under the initial screening conditions or have a quantifiable separation under the improved conditions, so it is not included in Figure 7, Table 1, or Table 2.
Figure 6:

Representative chromatograms of the compounds shown in Figure 2 on CF6-RN column chemistry. Conditions: A) as stated in Table 1. B) Compound 1 was analyzed at 8 mL/min flow rate on a 100 mm x 4.6 i.d. mm 2.7μm SPP CF6-RN column (Conditions: 60% CO2, 40% modifier (65% MeOH, 35% ACN, 10% H2O, 0.2% TFA, and 0.2 TEA%). C) as stated in Table 2. In the chromatogram, the highest intensity peak is normalized to unity.
Table 2:
Chromatographic figures of merit of inorganic complexes using a 150 x4.6 mm CF6-RN column in SFC mode.
| Compound ID | 0.26 M TFA, 0.14 M TEA |
||||
|---|---|---|---|---|---|
| k 1 | α | Rs | N 1 | N 2 | |
| 12 | 12.87 | 1.07 | 1.1 | 5800 | 5100 |
| 16 | 3.13 | 1.29 | 2.2 | 1900 | 2100 |
| 17 | 14.48 | 1.13 | 1.5 | 2700 | 2800 |
| 19 | 1.91 | 1.16 | 2.0 | 6700 | 6000 |
| 23 | 12.71 | 1.12 | 1.5 | 3000 | 2900 |
Conditions: 60% CO2:40% Modifier (65% MeOH, 35% ACN with 10% water and additives listed above), 8 °C, 1.1 mL/min, 8 MPa back pressure, 5 μL injection, samples ~1.0 mg/mL in ACN
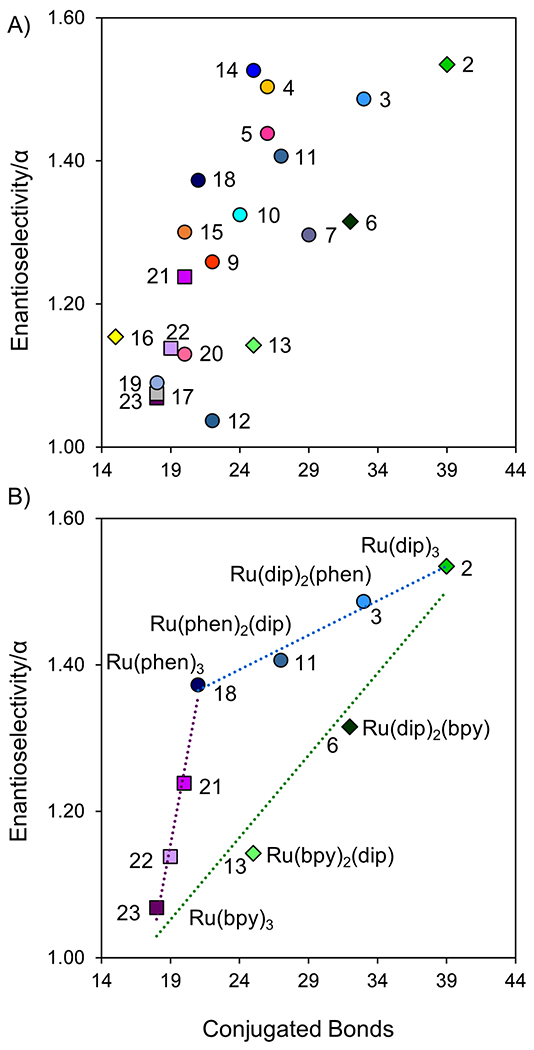
Figure 7:
Selectivity plot for compounds 2-23, excluding compound 8 on a 100 mm x 4.6 i.d. mm 2.7μm SPP CF6-RN column. Conditions: 60% CO2, 40% modifier (65% MeOH, 35% ACN, 2% TFA, 2% TEA, and 10% H2O), 25 °C, 4 mL/min, 8 MPa back pressure, 5 μL injection.
A previous study has shown that there was no chiral recognition of Ru(II) polypyridyl complexes with cyclofructan six (CF6) when derivatized with aliphatic functional groups [31]. This result indicated that π-π interactions between the analyte’s ligands and the (R)-naphthyl ethyl (RN) groups of the stationary phase play a key role in the separation mechanism for these enantiomers. To investigate this, enantioselectivity (α) was viewed in terms of the amount of conjugation provided by the complex’s ligands. All conjugated bonds were counted for each of the complex’s ligands and plotted against α, shown in Figure 7A. Figure 7A shows a clear upward trend, with the ligands providing the best enantioselectivity having the largest number of conjugated double bonds. All compounds that have a low resolution in this study have a smaller number of double bonds and lower enantioselectivity. This general trend supports the findings in HPLC that π-π interactions are imperative for chiral discrimination of these complexes on a cyclofructan-based stationary phase. Also, it should be noted that bidentate ligands that are more highly conjugated are generally much more rigid and have fewer or no rotational degrees of freedom. Likely, this rigidity enhances the analytes’ chiral recognition by the chiral stationary phase.
Within a class of complexes, the enantioselectivity is linear with the number of double bonds, as shown in Figure 7B. The complexes in Figure 7B were selectively synthesized with a range of lipophobicity. To do this, nine complexes (Compounds 2, 3, 6, 11, 13, 18, 21, 22, and 23) were synthesized using combinations of three ligands with increasing lipophobicity: (i) 2,2′-bipyridyl (bpy), (ii) 1,10-phenanthroline (phen), (iii) 4,7-diphenyl-1,10-phenanthroline (dip). The combination of these ligands makes nine complexes in three series (bpy-phen, bpy-dip, phen-dip), shown by the three linear trendlines in Figure 7B. The bpy-dip series and the phen-dip series are annotated, showing how the combination of molecules forms a series. This linearity better proves that there is a relationship between complex conjugation and enantioselectivity provided by naphthyl groups on CF6.
3.5. Mass Transfer Kinetic Plots.
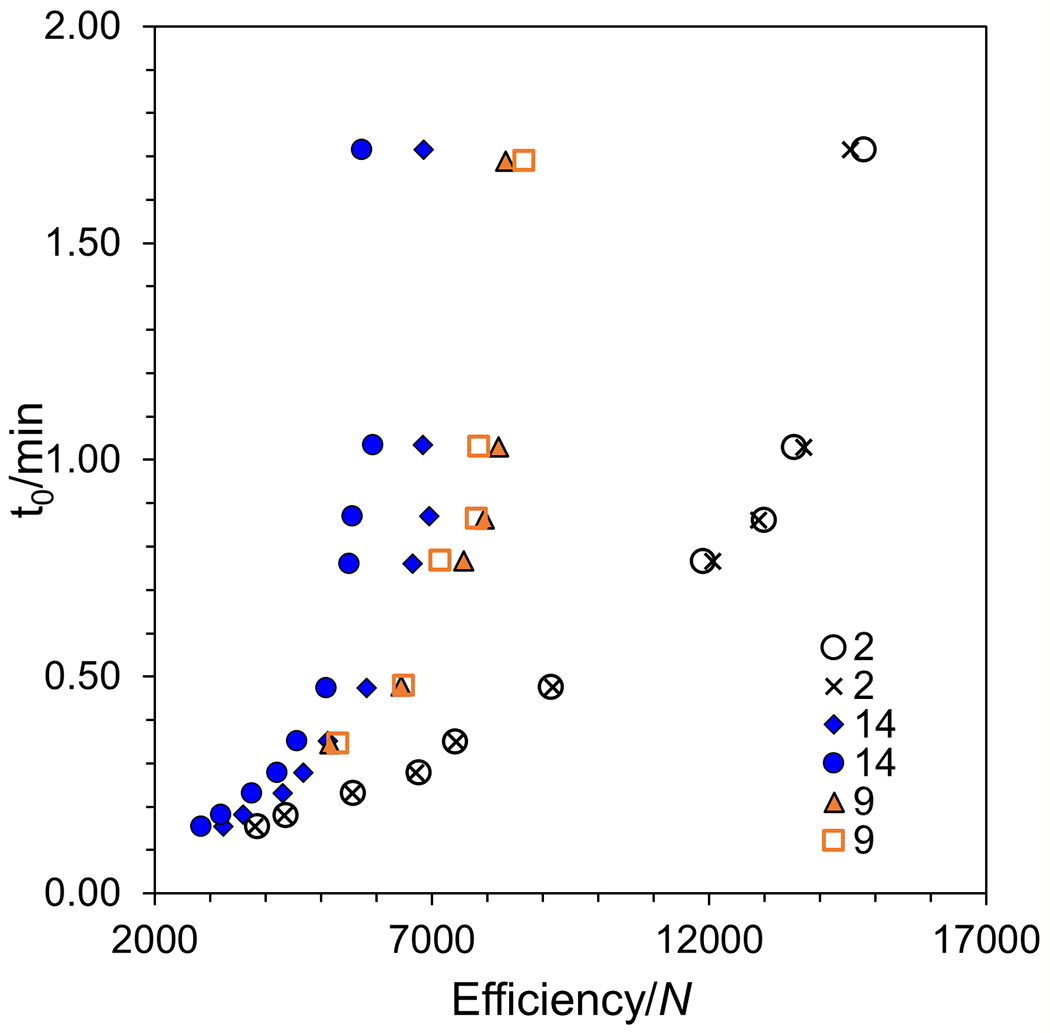
The performance of this column chemistry packed with superficially porous particles was examined in the SFC mode. The SPP particle contains a 1.5 μm solid core with a 0.6 μm porous shell and 120 Å pore size. SPP provides lower axial diffusion due to its solid core and lowers eddy dispersion due to its narrow particle size distribution when compared to fully porous particles (FPP) [9,56]. This becomes even more apparent at higher flow rates (like those seen in SFC) and for larger analytes [9]. The SPP particle provides efficiencies when packed into a 100 mm x 4.6 mm column, similar to a 1.7 μm FPP at a back pressure two to three times lower [56,57]. To study the mass transfer effects of this new column chemistry, kinetic plots for compounds 2, 9, and 14 are shown in Figure 8. The efficiency of the 100 mm column is plotted on the x-axis, and dead time is on the y-axis. These plots show a maximum efficiency of 14,790 plates/10 cm in SFC compared to the maximum efficiency of 1200 plates/5 cm for ruthenium polypyridyl complexes in HPLC [33]. The kinetic plot shows that high plate counts can be obtained in the short time of 1.72 min (as dead time).
Figure 8:
Kinetic plots for the enantiomers of compounds 2, 9, and 14 on a 100 mm x 4.6 i.d. mm 2.7μm SPP CF6-RN column. The deadtime (t0) was estimated from the change in refractive index. Conditions: 60% CO2, 40% modifier (65% MeOH, 35% ACN, 2% TFA, 2% TEA, and 10% H2O), 25 °C, 0.7 mL/min to 9 mL/min, 8 MPa back pressure, 5 μL injection.
To achieve an enantiomeric separation, there must be a strong interaction between the stationary phase and at least one of the analyte’s enantiomers. Such strong interactions slow mass transfer for the more retained enantiomer leading to tailing [14] and an expected lower efficiency (N2) for the second peak. Kinetic plots of three different racemic analytes were analyzed with a variety of retentions. Compound 2 has low retention (k1 = 3.68) but has high efficiency (N1 = 6700) and high enantioselectivity (α = 1.53). In this case, through the range of flow rates (0.7 to 9 mL/min), the enantiomers remain almost identical in efficiency. Since the efficiencies are calculated using the half width method, the second enantiomer’s peak must have broadened at the same rate it gained retention time. Therefore, the ratio between the peak’s width and its retention time remains the same compared to the first enantiomer’s resulting in no loss of efficiency. Compound 9 is moderately retained (k1 = 18.05) with moderate efficiency (N1 = 5000) and shows a slight reduction in efficiency for the second enantiomer. Compound 14 is well retained (k1 = 23.3) with low efficiency (N1 = 4700) and shows the typical lower efficiency for the second enantiomer, where the second enantiomer’s broadening exceeds its gain in retention time
3.6. Fast Separations with Enhanced Fluidity Liquid Chromatography
Fast separations is an emerging trend in analytical separations, enhancing throughput as never before [58]. Mobile phases containing a compressed gas are very well suited for fast, high efficiency separations because their low viscosity allows for high flow rates at moderate pressures, and faster mass transfer kinetics compared to pure liquid mobile phases. To enter into this chromatographic mode, the amount of optimized modifier was raised to 60%, and CO2 was lowered to40%, providing conditions closer to enhanced fluidity chromatography (EFLC) than SFC [59,60]. The flow rate was then raised to 6 mL/min, and the pressure still was only 410 bar on a 10 cm column with 2.7 μm SPP. The fast results for compounds 2, 3, 4, and 5 are shown in Figure 9. For these separations, baseline resolutions were maintained with run times under 60 seconds. This method is useful for high throughput chiral analysis of these complex’s enantiomers and is extremely green, using less than 3.6 mL of organic solvent to separate each compound.
Figure 9:
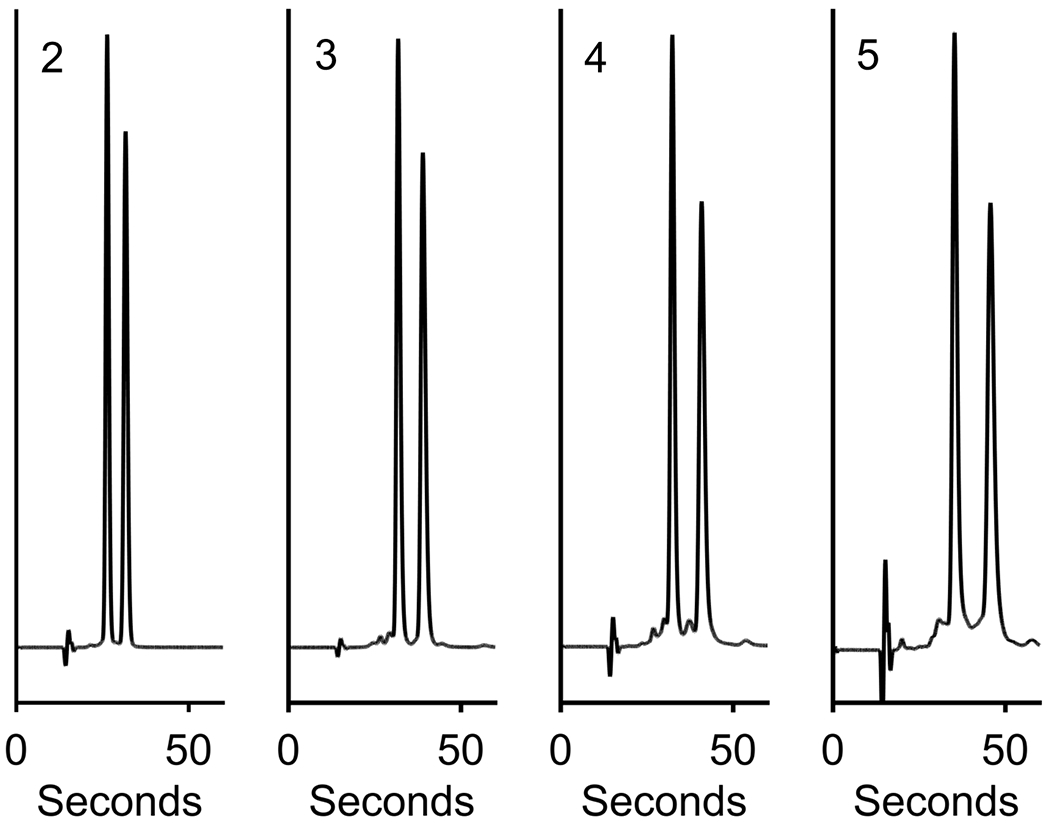
Fast separations (tr2 < 60 s) of compounds 2, 3, 4, and 5 on a 100 mm x 4.6 i.d. mm 2.7μm SPP CF6-RN column. Conditions: 40% CO2, 60% modifier (65% MeOH, 35% ACN, 2% TFA, 2% TEA, and 10% H2O), 25 °C, 6 mL/min, 8 MPa back pressure, 5 μL injection, samples ~1.0 mg/mL in ACN. The y-axes are scaled for enhanced visualization.
4. Conclusions
This work shows the first successful separation of twenty-three cationic chiral octahedral Ru(II) and Os(II) complexes in the SFC mode. The results show promising applications of the LarihcShell-RN chiral stationary phase for separating chiral coordination complexes. The optimization of the quaternary mobile phase was conducted, and the changing the ratio of the modifier to CO2 was shown to tailor the speed as well as peak-to-peak separation of the analyte’s enantiomers. No solubility or precipitation issues were observed. It was shown that the greater the conjugation, the better the chiral selectivity for these metallic complexes. Kinetic plots showed the efficiency advantage of SPPs in SFC. Future work will involve developing a preparative method to isolate enantiomers of similar complexes using SFC. The pure enantiomers can then be tested to see if their pharmacological activity differs. The application of this stationary phase on SPP particles to conduct highly efficient ultrafast separations in both EFLC and UHPLC polar organic mode is of interest. Further development of these methods will provide a high throughput method to screen for optical purity and enantiomeric excess.
Supplementary Material
Acknowledgments
The authors would like to thank the Robert A. Welch Foundation (Y-0026) for financially supporting this work. The authors would also like to acknowledge AZYP, LLC for synthesizing and providing chiral columns. S.A.M. thanks the National Cancer Institute (NCI) of the National Institutes of Health (NIH) (Award R01CA222227) for partial support. The content in this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.A.M. and Collin G. Cameron also thank the National Science Foundation (award 2102459) for partial support of this work.
References
- [1].Miller L, Potter M, Preparative chromatographic resolution of racemates using HPLC and SFC in a pharmaceutical discovery environment, J. Chromatogr. B 875 (2008) 230–236. 10.1016/j.jchromb.2008.06.044. [DOI] [PubMed] [Google Scholar]
- [2].Liu J, Regalado EL, Mergelsberg I, Welch CJ, Extending the range of supercritical fluid chromatography by use of water-rich modifiers, Org. Biomol. Chem 11 (2013) 4925–4929. 10.1039/c3ob41121d. [DOI] [PubMed] [Google Scholar]
- [3].Tarafder A, Metamorphosis of supercritical fluid chromatography to SFC: An Overview, TrAC - Trends Anal. Chem 81 (2016) 3–10. 10.1016/j.trac.2016.01.002. [DOI] [Google Scholar]
- [4].Miller L, Preparative enantioseparations using supercritical fluid chromatography, J. Chromatogr. A 1250 (2012) 250–255. 10.1016/j.chroma.2012.05.025. [DOI] [PubMed] [Google Scholar]
- [5].Armstrong DW, Tang Y, Ward T, Nichols M, Devitalized Cyclodextrins Immobilized on Fused-Silica Capillaries for Enantiomeric Separations via Capillary Electrophoresis, Gas Chromatography, or Supercritical Fluid Chromatography, Anal. Chem 65 (1993) 3543–3543. 10.1021/ac00071a600. [DOI] [Google Scholar]
- [6].Vozka J, Kalíková K, Roussel C, Armstrong DW, Tesařová E, An insight into the use of dimethylphenyl carbamate cyclofructan-7 chiral stationary phase in supercritical fluid chromatography: The basic comparison with HPLC, J. Sep. Sci 36 (2013) 1711–1719. 10.1002/jssc.201201174. [DOI] [PubMed] [Google Scholar]
- [7].Breitbach AS, Lim Y, Xu QL, Kürti L, Armstrong DW, Breitbach ZS, Enantiomeric separations of α-aryl ketones with cyclofructan chiral stationary phases via high performance liquid chromatography and supercritical fluid chromatography, J. Chromatogr. A 1427 (2016) 45–54. 10.1016/j.chroma.2015.11.069. [DOI] [PubMed] [Google Scholar]
- [8].Liu Y, Berthod A, Mitchell CR, Xiao TL, Zhang B, Armstrong DW, Super/subcritical fluid chromatography chiral separations with macrocyclic glycopeptide stationary phases, J. Chromatogr. A 978 (2002) 185–204. 10.1016/S0021-9673(02)01356-0. [DOI] [PubMed] [Google Scholar]
- [9].Folprechtová D, Kozlov O, Armstrong DW, Schmid MG, Kalíková K, Tesařová E, Enantioselective potential of teicoplanin- and vancomycin-based superficially porous particles-packed columns for supercritical fluid chromatography, J. Chromatogr. A 1612 (2020). 10.1016/j.chroma.2019.460687. [DOI] [PubMed] [Google Scholar]
- [10].Deye JF, Berger TA, Anderson AG, Nile Red as a Solvatochromic Dye for Measuring Solvent Strength in Normal Liquids and Mixtures of Normal Liquids with Supercritical and Near Critical Fluids, Anal. Chem 62 (1990) 1552. 10.1021/ac00213a044. [DOI] [Google Scholar]
- [11].Berger TA, Supercritical Fluid Chromatography, Agilent Technolgies, Inc., Santa Clara, CA, 2015. [Google Scholar]
- [12].Leu AD, Chung SYK, Robinson DB, The equilibrium phase properties of (carbon dioxide + methanol), J. Chem. Thermodyn 23 (1991) 979–985. 10.1016/S0021-9614(05)80178-8. [DOI] [Google Scholar]
- [13].West C, How Good is SFC for Polar Analytes ?, Chromatogr. Today (2013) 22–27. [Google Scholar]
- [14].Fornstedt T, Zhong G, Guiochon G, Peak tailing and mass transfer kinetics in linear chromatography, J. Chromatogr. A 741 (1996) 1–12. 10.1016/0021-9673(96)00152-5. [DOI] [Google Scholar]
- [15].Płotka JM, Biziuk M, Morrison C, Namieśnik J, Pharmaceutical and forensic drug applications of chiral supercritical fluid chromatography, TrAC - Trends Anal. Chem 56 (2014) 74–89. 10.1016/j.trac.2013.12.012. [DOI] [Google Scholar]
- [16].Berger TA, Deye JF, Separation of benzene polycarboxylic acids by packed column supercritical fluid chromatography using methanol-carbon dioxide mixtures with very polar additives, J. Chromatogr. Sci 29 (1991) 141–146. 10.1093/chromsci/29.4.141. [DOI] [Google Scholar]
- [17].Firooz SK, Wahab MF, Yu J, Armstrong DW, High efficiency functionalized hydrophilic cyclofructans as stationary phases in sub/supercritical fluid chromatography, Talanta. 232 (2021) 122308. 10.1016/j.talanta.2021.122308. [DOI] [PubMed] [Google Scholar]
- [18].Taylor L, Separation of ionic analytes using supercritical fluid chromatography, LC GC Eur 22 (2009). [Google Scholar]
- [19].David F, Sandra P, Analysis of aliphatic amines and quaternary ammonium salts by capillary supercritical fluid chromatography, J. High Resolut. Chromatogr 11 (1988) 897–898. 10.1002/jhrc.1240111211. [DOI] [Google Scholar]
- [20].Steuer W, Baumann J, Erni F, Separation of ionic drug substances by supercritical fluid chromatography, J. Chromatogr. A 500 (1990) 469–479. 10.1016/S0021-9673(00)96086-2. [DOI] [PubMed] [Google Scholar]
- [21].Roy D, Wahab MF, Berger TA, Armstrong DW, Ramifications and Insights on the Role of Water in Chiral Sub/Supercritical Fluid Chromatography, Anal. Chem 91 (2019) 14672–14680. 10.1021/acs.analchem.9b03908. [DOI] [PubMed] [Google Scholar]
- [22].Page SH, Sumpter SR, Lee ML, Fluid phase equilibria in supercritical fluid chromatography with CO2-based mixed mobile phases: A review, J. Microcolumn Sep. 4 (1992) 91–122. 10.1002/mcs.1220040202. [DOI] [Google Scholar]
- [23].Foulon C, Di Giulio P, Lecoeur M, Simultaneous determination of inorganic anions and cations by supercritical fluid chromatography using evaporative light scattering detection, J. Chromatogr. A 1534 (2018) 139–149. 10.1016/j.chroma.2017.12.047. [DOI] [PubMed] [Google Scholar]
- [24].Alatrash N, Issa FH, Bawazir NS, West SJ, Van Manen-Brush KE, Shelor CP, Dayoub AS, Myers KA, Janetopoulos C, Lewis EA, MacDonnell FM, Disruption of microtubule function in cultured human cells by a cytotoxic ruthenium(ii) polypyridyl complex, Chem. Sci 11 (2020) 264–275. 10.1039/c9sc05671h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Alatrash N, Narh ES, Yadav A, Kim MJ, Janaratne T, Gabriel J, MacDonnell FM, Synthesis DNA Cleavage Activity, Cytotoxicity, Acetylcholinesterase Inhibition, and Acute Murine Toxicity of Redox-Active Ruthenium(II) Polypyridyl Complexes, ChemMedChem 12 (2017) 1055–1069. 10.1002/cmdc.201700240. [DOI] [PubMed] [Google Scholar]
- [26].Shi S, Liu J, Li J, Zheng KC, Tan CP, Chen LM, Ji LN, Electronic effect of different positions of the –NO2 group on the DNA-intercalator of chiral complexes [Ru(bpy)2L]2+(L =o-npip, m-npip and p-npip), Dalt. Trans (2005) 2038. 10.1039/b501112d. [DOI] [PubMed] [Google Scholar]
- [27].Roque JA, Barrett PC, Cole HD, Lifshits LM, Shi G, Monro S, Von Dohlen D, Kim S, Russo N, Deep G, Cameron CG, Alberto ME, McFarland SA, Breaking the barrier: An osmium photosensitizer with unprecedented hypoxic phototoxicity for real world photodynamic therapy, Chem. Sci 11 (2020) 9784–9806. 10.1039/d0sc03008b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shi G, Monro S, Hennigar R, Colpitts J, Fong J, Kasimova K, Yin H, DeCoste R, Spencer C, Chamberlain L, Mandel A, Lilge L, McFarland SA, Ru(II) dyads derived from α-oligothiophenes: A new class of potent and versatile photosensitizers for PDT, Coord. Chem. Rev 282–283 (2015) 127–138. 10.1016/j.ccr.2014.04.012. [DOI] [Google Scholar]
- [29].Cole HD, Roque JA, Shi G, Lifshits LM, Ramasamy E, Barrett PC, Hodges RO, Cameron CG, Mcfarland SA, Anticancer Agent with Inexplicable Potency in Extreme Hypoxia: Characterizing a Light-Triggered Ruthenium Ubertoxin, J. Am. Chem. Soc (2021). 10.1021/jacs.1c09010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McFarland SA, Mandel A, Dumoulin-White R, Gasser G, Metal-based photosensitizers for photodynamic therapy: the future of multimodal oncology? Curr. Opin. Chem. Biol 56 (2020) 23–27. 10.1016/j.cbpa.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shu Y, Breitbach ZS, Dissanayake MK, Perera S, Aslan JM, Alatrash N, MacDonnell FM, Armstrong DW, Enantiomeric Separations of Ruthenium (II) Polypyridyl Complexes Using HPLC With Cyclofructan Chiral Stationary Phases, Chirality. 27 (2015) 64–70. 10.1002/chir.22389. [DOI] [PubMed] [Google Scholar]
- [32].Jiang C, Tong M-Y, Armstrong DW, Perera S, Bao Y, MacDonnell FM, Enantiomeric separation of chiral ruthenium(II) complexes using capillary electrophoresis, Chirality. 21 (2009) 208–217. 10.1002/chir.20641. [DOI] [PubMed] [Google Scholar]
- [33].Armstrong DW, Yu J, Cole HD, McFarland SA, Nafie J, Chiral resolution and absolute configuration determination of new metal-based photodynamic therapy antitumor agents, J. Pharm. Biomed. Anal 204 (2021) 114233. 10.1016/j.jpba.2021.114233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sun P, Wang C, Breitbach ZS, Zhang Y, Armstrong DW, Development of new HPLC chiral stationary phases based on native and derivatized cyclofructans, Anal. Chem 81 (2009) 10215–10226. 10.1021/ac902257a. [DOI] [PubMed] [Google Scholar]
- [35].Cole HD, Roque JA, Lifshits LM, Hodges R, Barrett PC, Havrylyuk D, Heidary D, Ramasamy E, Cameron CG, Glazer EC, McFarland SA, Fine-Feature Modifications to Strained Ruthenium Complexes Radically Alter Their Hypoxic Anticancer Activity†, Photochem. Photobiol 98 (2022) 73–84. 10.1111/php.13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].McFarland SA, Metal-based Thiophene Photodynamic Compounds and Their Use, US Patent 9,676,806 B2, 2017.
- [37].Monro S, Colón KL, Yin H, Roque J, Konda P, Gujar S, Thummel RP, Lilge L, Cameron CG, McFarland SA, Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433, Chem. Rev 119 (2019) 797–828. 10.1021/acs.chemrev.8b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Reichardt C, Monro S, Sobotta FH, Colón KL, Sainuddin T, Stephenson M, Sampson E, Roque J, Yin H, Brendel JC, Cameron CG, McFarland S, Dietzek B, Predictive Strength of Photophysical Measurements for in Vitro Photobiological Activity in a Series of Ru(II) Polypyridyl Complexes Derived from π-Extended Ligands, Inorg. Chem 58 (2019) 3156–3166. 10.1021/acs.inorgchem.8b03223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roque JA, Barrett PC, Cole HD, Lifshits LM, Bradner E, Shi G, von Dohlen D, Kim S, Russo N, Deep G, Cameron CG, Alberto ME, McFarland SA, Os(II) Oligothienyl Complexes as a Hypoxia-Active Photosensitizer Class for Photodynamic Therapy, Inorg. Chem 59 (2020) 16341–16360. 10.1021/acs.inorgchem.0c02137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sainuddin T, Pinto M, Yin H, Hetu M, Colpitts J, McFarland SA, Strained ruthenium metal–organic dyads as photocisplatin agents with dual action, J. Inorg. Biochem 158 (2016) 45–54. 10.1016/j.jinorgbio.2016.01.009. [DOI] [PubMed] [Google Scholar]
- [41].Yin H, Stephenson M, Gibson J, Sampson E, Shi G, Sainuddin T, Monro S, McFarland SA, In vitro multiwavelength PDT with 3IL States: Teaching old molecules new tricks, Inorg. Chem 53 (2014) 4548–4559. 10.1021/ic5002368. [DOI] [PubMed] [Google Scholar]
- [42].Zheng HQ, Guo YP, Yin MC, Fan YT, Synthesis, characterization of a new photosensitive compound [Ru(bpy)2 (TPAD)](PF6)2 and its application for photocatalytic hydrogen production, Chem. Phys. Lett 653 (2016) 17–23. 10.1016/j.cplett.2016.04.064. [DOI] [Google Scholar]
- [43].Chao H, Liu J-G, Jiang C-W, Ji L-N, Li X-Y, Feng C-L, Stereoisomerically controlled supramolecular architectures: a new strategy for the construction of enantio- and diastereomerically pure multinuclear Ru(II) complexes, Inorg. Chem. Commun 4 (2001) 45–48. 10.1016/S1387-7003(00)00186-6. [DOI] [Google Scholar]
- [44].Barhate CL, Wahab MF, Tognarelli DJ, Berger TA, Armstrong DW, Instrumental Idiosyncrasies Affecting the Performance of Ultrafast Chiral and Achiral Sub/Supercritical Fluid Chromatography, Anal. Chem 88 (2016) 8664–8672. 10.1021/acs.analchem.6b01898. [DOI] [PubMed] [Google Scholar]
- [45].Plachká K, Švec F, Nováková L, Ultra-high performance supercritical fluid chromatography in impurity control: Searching for generic screening approach, Anal. Chim. Acta 1039 (2018) 149–161. 10.1016/j.aca.2018.07.008. [DOI] [PubMed] [Google Scholar]
- [46].Li J, Thurbide KB, A comparison of methanol and isopropanol in alcohol/water/CO2 mobile phases for packed column supercritical fluid chromatography, Can. J. Anal. Sci. Spectrosc 53 (2008) 59–65. [Google Scholar]
- [47].Liu J, Makarov AA, Bennett R, Haidar Ahmad IA, Dasilva J, Reibarkh M, Mangion I, Mann BF, Regalado EL, Chaotropic Effects in Sub/Supercritical Fluid Chromatography via Ammonium Hydroxide in Water-Rich Modifiers: Enabling Separation of Peptides and Highly Polar Pharmaceuticals at the Preparative Scale, Anal. Chem 91 (2019) 13907–13915. 10.1021/acs.analchem.9b03408. [DOI] [PubMed] [Google Scholar]
- [48].Khvalbota L, Roy D, Wahab MF, Firooz SK, Machyňáková A, Špánik I, Armstrong DW, Enhancing supercritical fluid chromatographic efficiency: Predicting effects of small aqueous additives, Anal. Chim. Acta 1120 (2020) 75–84. 10.1016/j.aca.2020.04.065. [DOI] [PubMed] [Google Scholar]
- [49].Roy D, Wahab MF, Talebi M, Armstrong DW, Replacing methanol with azeotropic ethanol as the co-solvent for improved chiral separations with supercritical fluid chromatography (SFC), Green Chem 22 (2020) 1249–1257. 10.1039/c9gc04207e. [DOI] [Google Scholar]
- [50].Aslani S, Wahab MF, Kenari ME, Berthod A, Armstrong DW, An examination of the effects of water on normal phase enantioseparations, Anal. Chim. Acta 1200 (2022) 339608. 10.1016/j.aca.2022.339608. [DOI] [PubMed] [Google Scholar]
- [51].Roy D, Tarafder A, Miller L, Effect of water addition to super/sub-critical fluid mobile-phases for achiral and chiral separations, TrAC - Trends Anal. Chem 145 (2021) 116464. 10.1016/j.trac.2021.116464. [DOI] [Google Scholar]
- [52].Putnam J, Guiochon G, The influence of water on the memory effect of the amylose tris(3,5-dimethylphenyl carbamate) stationary phase, J. Chromatogr. A 1217 (2010) 8146–8153. 10.1016/j.chroma.2010.10.054. [DOI] [PubMed] [Google Scholar]
- [53].Fairchild JN, Brousmiche DW, Hill JF, Morris MF, Boissel CA, Wyndham KD, Chromatographic evidence of silyl ether formation (SEF) in supercritical fluid chromatography, Anal. Chem 87 (2015) 1735–1742. 10.1021/ac5035709. [DOI] [PubMed] [Google Scholar]
- [54].Berger TA, Deye JF, Role of additives in packed column supercritical fluid chromatography: suppression of solute ionization, J. Chromatogr. A 547 (1991) 377–392. 10.1016/S0021-9673(01)88661-1. [DOI] [Google Scholar]
- [55].Zheng J, Taylor LT, Pinkston JD, Mangels ML, Effect of ionic additives on the elution of sodium aryl sulfonates in supercritical fluid chromatography, J. Chromatogr. A 1082 (2005) 220–229. 10.1016/j.chroma.2005.04.086. [DOI] [PubMed] [Google Scholar]
- [56].Gritti F, Cavazzini A, Marchetti N, Guiochon G, Comparison between the efficiencies of columns packed with fully and partially porous C18-bonded silica materials, J. Chromatogr. A 1157 (2007) 289–303. 10.1016/j.chroma.2007.05.030. [DOI] [PubMed] [Google Scholar]
- [57].Gritti F, Guiochon G, Facts and Legends on Columns Packed with Sub-3-μm Core-Shell Particles, LCGC North Am. 30 (2012) 586–595. [Google Scholar]
- [58].Wahab MF, Roy D, Armstrong DW, The theory and practice of ultrafast liquid chromatography: A tutorial, Anal. Chim. Acta 1151 (2021) 238170. 10.1016/j.aca.2020.12.045. [DOI] [PubMed] [Google Scholar]
- [59].Sun Q, Olesik SV, Chiral separations performed by enhanced-fluidity liquid chromatography on a macrocyclic antibiotic chiral stationary phase, Anal. Chem 71 (1999) 2139–2145. 10.1021/ac981134m. [DOI] [PubMed] [Google Scholar]
- [60].Cui Y, Olesik SV, High-Performance Liquid Chromatography Using Mobile Phases with Enhanced Fluidity, Anal. Chem 63 (1991) 1812–1819. 10.1021/ac00017a028. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.