

Abstract
The prevalence of homologous recombination–DNA damage response (HR-DDR) genetic alterations is of therapeutic interest in gastroesophageal cancers. This study is a comprehensive assessment of HR-DDR mutation prevalence across gastroesophageal adenocarcinomas and squamous cell carcinomas. Here we investigate the association of HR-DDR mutations with known predictors for immune-checkpoint inhibition [deficiency in mismatch-repair (dMMRP), tumor mutational burden (TMB), and programmed death ligand 1 (PD-L1)]. We confirmed HR-DDR mutations are present in a subset of gastroesophageal adenocarcinomas (23%) and gastroesophageal squamous cell carcinomas (20%). Biomarker expression of dMMRP (18% vs. 1%) and TMB-high with a cutoff of ≥10 mt/MB (27% vs. 9%) was significantly more prevalent in the DDR-mutated cohort compared with the non-DDR-mutated cohort. Mean combined positive score for PD-L1 in the total adenocarcinoma cohort was significantly higher in the DDR-mutated cohort compared with the non–DDR-mutated cohort (10.1 vs. 5.8). We demonstrated that alterations in ARID1A, BRCA2, PTEN, and ATM are correlated with dMMRP, TMB-high, and increased PD-L1 expression in gastroesophageal adenocarcinomas. Our findings show that a subset of gastroesophageal tumors harbor HR-DDR mutations correlated with established immune biomarkers. By better understanding the relationship between HR-DDR mutations and immune biomarkers, we may be able to develop better immunotherapy combination strategies to target these tumors.
Introduction
Despite modest advancements in the treatment of gastroesophageal cancer, patients with metastatic disease continue to have a low five-year survival rate and a median overall survival of only one to two years (1). The treatment of gastroesophageal cancer remains challenging, but novel approaches are currently under investigation with particular interest being paid toward the DNA damage response (DDR) pathway and immune-checkpoint inhibition (ICI). Mutations in the DDR pathway play an important role in the development of cancer (2). DDR genes such as BRCA1 and BRCA2 have been well characterized and are now understood to encode proteins that assist in homology-directed repair. Other DDR genes such as ATM, MRE11, NBN, PALB2, RAD50, and the Fanconi anemia genes encode proteins that interact with BRCA to construct a delicate system for DNA damage repair (Fig. 1; ref. 3). A study examining the prevalence of genetic deficiencies in homologous recombination (HR) across all tumor lineages revealed gastroesophageal cancers have one of the highest frequency of mutations in HR-DDR genes (20.8%; ref. 4).
Figure 1.

Schematic representation of HR in DDR. A DSB is induced on the DNA strand. The MRN complex (MRE11–RAD50–NBS1) recognizes and binds to the area of DNA damage. This leads to the recruitment and activation of the serine/threonine protein kinase ATM (which phosphorylates several key proteins in the HR-DDR pathway such as CHEK2, BRCA1, NBS1, and p53). ATM phosphorylates the histone variant γH2AX on Ser139, which subsequently leads to signal transduction. This leads to BRCA1 and BARD1 recruitment to the site of DNA damage. C-terminal interacting protein (CtIP) is recruited to the 5′ DNA end and begins resecting, whereas replication protein A (RPA) caps the exposed ssDNA. ATR and PALB2 are activated. BRCA1 recruits PALB2, FANCD2, BRCA2, and RAD51. RPA is exchanged for RAD51, which promotes nucleofilament formation and strand invasion to form the D-loop. DNA repair is mediated via the Holliday junction (red and black arrows represent crossover).
The benefit of targeting HR-DDR pathways in gastroesophageal cancers is currently under investigation, and anticancer therapy directed toward HR-DDR is already being used to treat breast, pancreatic, and ovarian cancers (5–7). Recent evidence has demonstrated that DDR mutations are associated with immune biomarkers (e.g., PD-L1 upregulation, increased TMB, and a higher prevalence of tumor infiltrating lymphocytes) that correlate with a higher likelihood of response to immune-checkpoint blockade (ICB; refs. 8–9). Targeting these pathways via DDR inhibitors (e.g., PARP inhibitors) has been shown to modulate the tumor immune environment by potentiating the antitumor effect of PD-L1 blockade and augmenting cytotoxic T-cell infiltration in small cell lung cancer (10). Therefore, targeting the defective DDR pathway has the potential to reset the inflammatory microenvironment of tumors and thus facilitate a proper T-cell immune response. Strategies to target the DDR pathway and immune checkpoints remain a promising treatment option for patients with gastroesophageal cancers (11–13).
The aim of our study was to determine the association of HR-DDR mutations in gastric (GC), esophageal (EC), and gastroesophageal junction (GEJ) cancers with known predictors for immune-checkpoint inhibitors: dMMRP, TMB, and PD-L1. Our study investigated both adenocarcinomas and squamous cell carcinomas. Our first objective was to compare the association of these immune biomarkers in DDR-mutated (DDR-M) versus Non–DDR-mutated gastroesophageal cancers. Our second objective was to correlate specific DDR alterations (e.g., ARID1A, ATRX, BRCA2, PTEN, RAD50, and WRN) in GC, EC, and GEJ with dMMRP, TMB, and PD-L1. By identifying these associations we hoped to better define the subset of patients with gastroesophageal cancer who may benefit from combined drug combinations targeting the HR-DDR and immune-checkpoint pathways.
Materials and Methods
From March 2007 to July 2018, 1,724 gastroesophageal cancer tumors (including both adenocarcinomas and squamous cell carcinomas) were submitted to Caris Life Sciences for genomic analysis. This study was conducted in accordance with guidelines of the Declaration of Helsinki, Belmont report, and U.S. Common rule. In keeping with 45 CFR 46.101(b)(4), this study was performed utilizing retrospective, deidentified clinical data. Therefore, this study was considered IRB exempt and no patient consent was deemed to be necessary.
IHC was performed on full formalin-fixed paraffin-embedded (FFPE) sections of glass slides. Slides were stained using automated staining techniques, per the manufacturer’s instructions, and were optimized and validated per CLIA/CAO and ISO requirements.
The primary antibodies used against PD-L1 were 22c3 (pharmDx, Dako), and CPS was calculated as the number of PD-L1 staining cells (tumor cells, lymphocytes, and macrophages) divided by the total viable tumors cells, multiplied by 100.
Next-generation sequencing (NGS) was performed on genomic DNA isolated from FFPE tumor samples using the NextSeq platform (Illumina, Inc). Matched normal tissue was not sequenced. A custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies). All variants were detected with >99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of >500 and an analytic sensitivity of 5%. Prior to molecular testing, tumor enrichment was achieved by harvesting targeted tissue using manual microdissection techniques. Genetic variants identified were interpreted by board-certified molecular geneticists and categorized as “pathogenic,” “presumed pathogenic,” “variant of unknown significance,” “presumed benign,” or “benign,” according to the American College of Medical Genetics and Genomics standards. When assessing mutation frequencies of individual genes, “pathogenic” and “presumed pathogenic” were counted as mutations while “benign,” “presumed benign” variants, and “variants of unknown significance” were excluded.
TMB was measured [592 genes and 1.4 megabases (MB) sequenced per tumor] by counting all nonsynonymous missense mutations found per tumor that had not been previously described as germline alterations according to dbSNP and 1KG databases. TMB was adjusted by dividing by a factor of 1.2 to ensure that the fraction of TMB-H matched the observed published clinical data (14). A cutoff point of ≥10 mutations per MB was used based on the KEYNOTE-158 pembrolizumab trial, which showed that patients with a TMB of ≥10 mt/MB across several tumor types had higher response rates than patients with a TMB of <10 mt/MB. Median TMB values were used for our cohorts as they were not normally distributed (15). This was done to eliminate biases caused by outliers in this situation.
A combination of multiple test platforms was used to determine dMMRP status of the tumors profiled, including fragment analysis (FA, Promega), IHC (MLH1, M1 antibody: MSH2, G2191129 antibody: MSH6, 44 antibody; and PMS2, EPR3947 antibody; Ventana Medical Systems, Inc.) and NGS (for tumors tested with NextSeq platform, 7,000 target microsatellite loci were examined and compared with the reference genome hg19 from the University of California). FA was determined as dMMRP if two or more mononucleotides out of the five markers included in the assay were abnormal; IHC was considered mismatch-repair deficient if complete absence of protein expression of any of the four proteins was observed; the NGS threshold used to determine dMMRP was ≥46 altered loci per tumor. The three platforms generated highly concordant results and in the rare cases of discordant results, the dMMRP status of the tumor was designated based upon prioritizing the findings from FA, IHC, and NGS in that order.
Results
Patient demographics and DDR selection
Twenty DDR mutations were tested by NGS with a 592-gene panel on a total of 1,724 cancers (657 esophageal; 746 gastric; 321 gastroesophageal junction). Information on the anatomical location of each tumor was obtained from the pathology reports. There were 1,199 tumors classified as primary and 513 tumors classified as metastatic. There were a total of 1,230 males and 494 females across our patient cohort with a median age of 64 (Table 1). Information on race was not available to investigators. The specific DDR genes under investigation in this study were the following: ARID1A, ATM, ATRX, BAP1, BARD1, BLM, BRCA1, BRCA2, BRIP1, CHEK2, FANCA, FANCC, FANCD2, FANCG, MRE11, NBN, PALB2, PTEN, RAD50, and WRN. We aimed for the study to be as inclusive as possible with regard to DDR gene selection. The DDR genes under investigation have been implicated in the HR-DDR pathway and included in biomarker clinical trials (4, 16–26).
Table 1.
Characteristics of tumor and patient cohort.
| Characteristics | Esophageal | Gastric | Gastroesophageal | Total |
|---|---|---|---|---|
|
| ||||
| Tumor count | 657 | 746 | 321 | 1,724 |
| Location | ||||
| Primary | 438 | 521 | 240 | 1,199 |
| Metastatic | 219 | 221 | 73 | 513 |
| Unclear | 0 | 4 | 8 | 12 |
| Gender | ||||
| Male | 536 | 450 | 244 | 1,230 |
| Female | 121 | 296 | 77 | 494 |
| Median age | 65 | 63 | 63 | 64 |
Gastroesophageal DDR mutation rates
Overall, the DDR mutation rate was 23% for all gastroesophageal adenocarcinomas and 20% for all gastroesophageal squamous cell carcinomas (Table 2). These percentages are within the range of findings from prior studies analyzing HR-DDR mutation rates in upper GI malignancies (4). To further define HR-DDR mutation rates among upper GI adenocarcinomas, we observed GC had the highest DDR mutation rate compared with EC and GEJ (27.6% vs. 19.9%, P = 0.0005 and 15.4%, P = 0.0002, respectively; Table 2). We observed esophageal adenocarcinoma had a comparable DDR mutation rate compared with esophageal squamous cell carcinoma (19.9% vs. 19.1%; Table 2).
Table 2.
DDR-M rates and TMB association with DDR-M stratified by tumor histology and location.
| Median TMB score |
||||
|---|---|---|---|---|
| Type | DDR-M rate | DDR-M cohort | Non–DDR-M cohort | P |
|
| ||||
| Histology | ||||
| Adenocarcinoma (AC) | 23% | 7.0 mt/MB | 6.0 mt/MB | P < 0.001a |
| Squamous cell carcinoma (SCC) | 20% | 8.0 mt/MB | 7.0 mt/MB | P = 0.1601 |
| Location in AC | ||||
| Esophageal (EC) | 19.9% | 7.0 mt/MB | 7.0 mt/MB | P = 0.08460 |
| Gastric (GC) | 27.6% | 8.0 mt/MB | 6.0 mt/MB | P < 0.0001a |
| Gastroesophageal junction (GEJ) | 15.4% | 7.0 mt/MB | 6.0 mt/MB | P = 0.09610 |
| Total gastroesophageal | 23% | 7.0 mt/MB | 6.0 mt/MB | P < 0.0001a |
| Location in SCC | ||||
| Esophageal (EC) | 19.1% | 8.0 mt/MB | 7.0 mt/MB | P = 0.27360 |
We also examined the prevalence of tumors that had multiple DDR mutations based on the assumption that tumors with greater than one mutation are more likely to have a severe DDR phenotype. The total number of cases with greater than one DDR gene mutation was 83 out of 388 (21%). When stratified by tumor histology, we observed the total adenocarcinoma cohort had a higher percentage of cases with more than one mutation compared with the total squamous cell carcinoma cohort (5.1% vs. 2.5%, respectively; Fig. 2A). We observed esophageal adenocarcinoma had a slightly higher percentage of cases with more than one mutation compared with esophageal squamous cell carcinoma (3% vs. 2%, respectively; Fig. 2B). When stratified by location, we observed GC had the highest percentage of cases with more than one mutation compared with GEJ (7.2% vs. 3.4%, respectively; Fig. 2C).
Figure 2.

Prevalence of patients with multiple DDR mutations stratified by (A) adenocarcinoma (AC) vs. squamous cell carcinoma (SCC; B) esophageal AC vs. esophageal SCC, and (C) gastric (combined AC and SCC) vs. gastroesophageal (combined AC and SCC).
Association of immune biomarkers with DDR-M and non–DDR-M cohorts
Next, we examined immune biomarker prevalence among our DDR-M cohort versus our non–DDR-mutated cohort. Here we found dMMRP was significantly more common in the DDR-M cohort compared with the non–DDR-M cohort (18% vs. 1%; P < 0.0001). Similarly, we found TMB-high [defined as ≥10 mutations/megabase (mt/MB)] was more common in the DDR-M cohort compared with the non–DDR-M cohort (27% vs. 9%; P < 0.0001; Fig. 3A). A Kruskal–Wallis nonparametric analysis was done to evaluate the correlation between dMMRP and TMB-high. When only the proficient mismatch-repair protein (pMMRP) cohort was considered, we found the DDR-TMB association was no longer significant across DDR-M tumors compared with non–DDR-M tumors (10.1% vs. 7.8%; P = 0.221; Supplementary Fig. S1A). Overall, our data show both dMMRP and TMB-high had a statistically significant higher prevalence in the DDR-M cohort compared with the non–DDR-M cohort.
Figure 3.
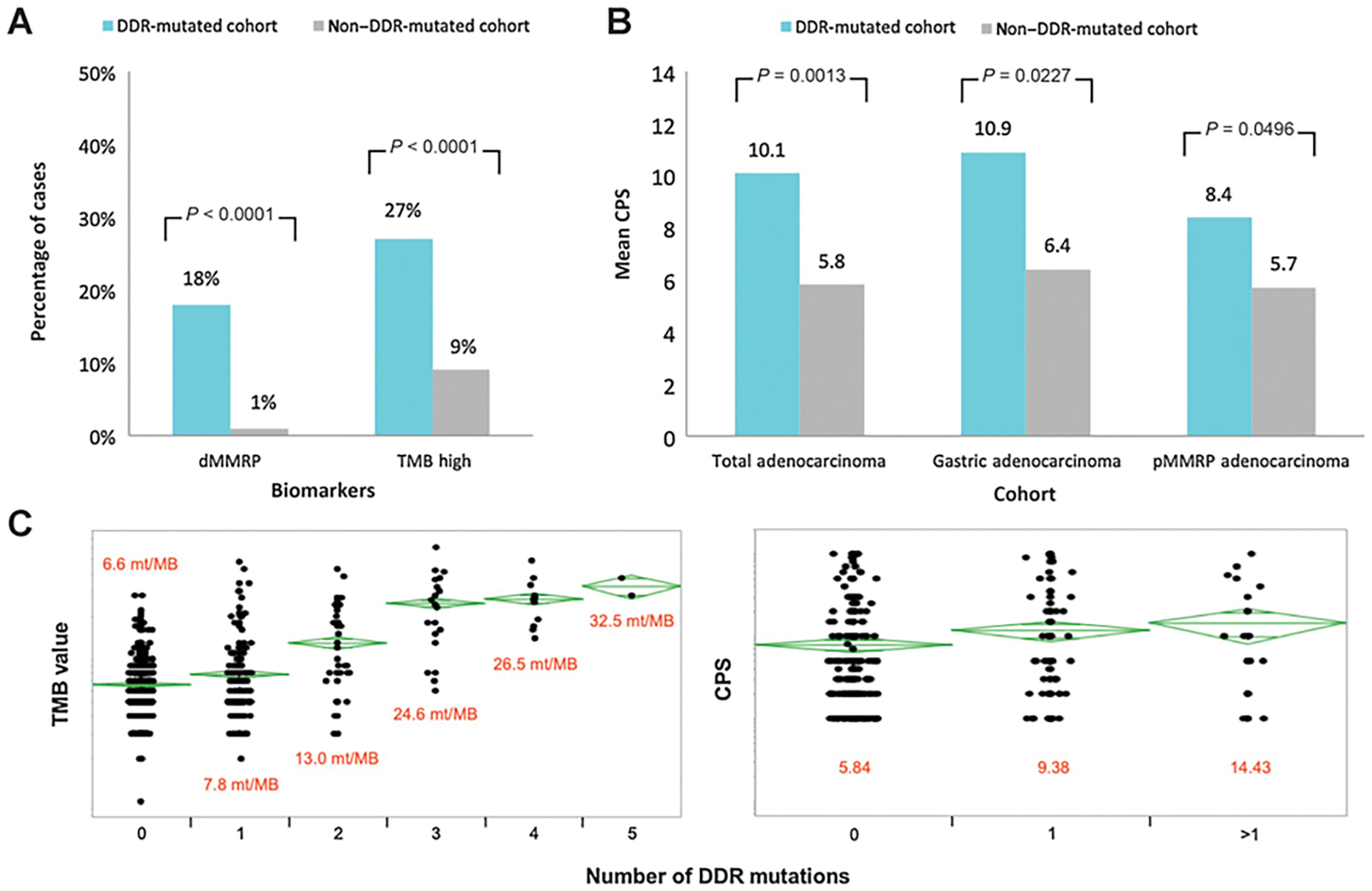
A, dMMRP/TMB biomarker prevalence across DDR cohorts, (B) mean CPSs across total adenocarcinoma, gastric adenocarcinoma, and pMMRP adenocarcinoma cohorts, and (C) TMB and PD-L1 CPS distribution across multiple DDR mutations.
We also investigated PD-L1 expression among the DDR-M cohort versus our non–DDR-M cohort. PD-L1 was measured using combined positive score (CPS), which is defined as the percentage of all PD-L1–expressing cells (tumor cells and immune cells combined) among all tumor cells (27). ANOVA statistical analysis was used to compare the mean CPS between the DDR-M and non-DDR-M cohorts and significance was determined by P < 0.05. When considering the total adenocarcinoma cohort, the mean CPS score was significantly higher in the DDR-M cohort compared with the non–DDR-M cohort (10.1 vs. 5.8; P = 0.0013; Fig. 3B). When considering the gastric adenocarcinoma DDR-M cohort, the mean CPS score was significantly higher in the DDR-M cohort compared with the non–DDR-M cohort (10.9 vs. 6.4; P = 0.0277; Fig. 3B). When considering the esophageal adenocarcinoma DDR-M cohort, our data revealed no significant difference across CPS groups. When considering the adenocarcinoma pMMRP cohort, the mean CPS score was higher in the DDR-M cohort compared with the non–DDR-M cohort (8.4 vs. 5.7; P = 0.0496; Fig. 3B).
We further examined the effect a patient’s multiple DDR mutations had on immune biomarker prevalence for TMB and PD-L1. It was previously noted by Heeke and colleagues that in their investigation of HR-DDR mutations, 362 out of 17,566 sequenced tumors harbored at least two HR-DDR pathway mutations (4). At the time, the clinical significance of multiple mutations was unknown. The multiple DDR mutation analysis here was based on the assumption that two or more mutations could increase the severity of DDR phenotype and may be associated with higher TMB. Our data revealed that more DDR mutations were associated with a higher TMB score (13.0 mt/MB associated with 2 mutations vs. 24.6 mt/MB associated with 3 mutations). However, this effect plateaued after 3 DDR mutations (26.5 mt/MB associated with 4 mutations vs. 32.5 mt/MB associated with 5 mutations). Our data also revealed that more DDR mutations were associated with higher PD-L1 expression (mean CPS score of 14.43 associated with more than one DDR mutation compared with mean CPS score of 9.38 and 5.84 associated with one and zero DDR mutations, respectively; Fig. 3C).
We further investigated TMB association with DDR-M across tumor histology and location. A Wilcoxon nonparametric test was used to compare median TMB values. Here we found gastroesophageal adenocarcinoma was associated with a higher median TMB score in the DDR-M cohort compared with the non–DDR-M cohort (median: 7 vs. 6 mt/MB, respectively; P < 0.001). We did not find a significant difference in TMB for gastroesophageal squamous cell carcinoma between the DDR-M cohort and non-DDR-M cohort (median: 8 vs. 7 mt/MB, respectively; P = 0.16; Table 2). When accounting for the DDR-M cohort in adenocarcinoma, GC was associated with the highest TMB score compared with DDR-M EC and GEJ (median: 8 vs. 7 vs. 7 mt/MB, respectively; P < 0.0001). We did not find a significant difference in TMB for esophageal squamous cell carcinoma between the DDR-M cohort and non–DDR-M cohort (median: 8 vs. 7 mt/MB, respectively; P = 0.27; Table 2).
Association of HER2 and DDR mutations
We also investigated HER2 prevalence among the DDR-M cohort versus the non–DDR-M cohort. HER2 was measured using IHC. When considering the total adenocarcinoma DDR-M cohort, our data revealed DDR mutations were not statistically different between the two groups for HER2 (IHC 1+; 30.8% vs. 36.2%) and HER2 (IHC 3+; 8% vs. 11.6%; Supplementary Fig. S1B).
Association of specific DDR mutations across esophageal cancers
We investigated the correlation between mutations in specific DDR genes (e.g., ARID1A, ATRX, BRCA2, PTEN, RAD50, WRN) across esophageal adenocarcinoma and esophageal squamous cell carcinoma. We looked at 20 DDR genes and examined which genetic alterations were associated with each tumor histology type. For esophageal adenocarcinoma, our data revealed ARID1A, ATRX, and RAD50 were the most prevalent DDR mutations (11.2%, 0.6%, 0.2%, respectively). FANCA, FANCG, and MRE11 were among the lowest DDR mutations (0%, 0%, 0%, respectively). For esophageal squamous cell carcinoma, our data revealed ARID1A and PTEN were the most prevalent DDR mutations (5.3% and 7.4%, respectively). BARD1 and RAD50 were among the lowest DDR mutations (0% and 0%, respectively; Fig. 4A).
Figure 4.

Percentage of cases with mutation in designated gene stratified by (A) esophageal adenocarcinoma vs. esophageal squamous cell carcinoma. Percentage of cases with mutation in designated gene stratified by (B) dMMRP, (C) TMB (≥10 mt/MB), and (D) PD-L1 (CPS ≥ 1) across total adenocarcinoma cohort (combined EC, GC, and GEJ).
Association of specific DDR mutations with immune biomarkers in gastroesophageal adenocarcinoma
We then investigated the correlation between mutations in specific DDR genes (e.g., ARID1A, ATRX, BRCA2, PTEN, RAD50, and WRN) with dMMRP, TMB, and PD-L1 in gastroesophageal adenocarcinoma. Here we looked at 20 DDR mutations and examined which genetic alterations were associated with the highest biomarker response. For dMMRP, our data revealed that ARID1A, BRCA2, PTEN, and ATM were the most prevalent DDR mutations (72%, 27.2%, 23.5%, and 16%, respectively). BARD1, FANCA, FANCC, and FANCD2 were among the lowest DDR mutations associated with dMMRP (0%, 0%, 0%, and 0%, respectively; Fig. 4B). Similarly for the TMB-high subgroup, our data revealed that ARID1A, BRCA2, PTEN, and ATM were the most prevalent DDR mutations (38.0%, 13.5%, 10.7%, and 8.3%, respectively). FANCG, BRIP1, BARD1, and FANCD2 were among the lowest DDR mutations associated with TMB-high (1.0%, 0.5%, 0.5%, and 0%, respectively; Fig. 4C). Similarly for the PD-L1 CPS ≥ 1 subgroup, our data revealed that ARID1A, ATM, BRCA2, and PTEN were the most prevalent DDR mutations (13.9% vs. 3.4% vs. 2.7% vs. 1.8%). BARD1, FANCC, MRE11, and BRIP1 were among the lowest DDR mutations associated with PD-L1 (0.2%, 0.2%, 0.2%, and 0%, respectively; Fig. 4D).
We further investigated the association between dMMRP and the DDR-M cohort by analyzing specific mismatch-repair protein changes. An analysis of specific gene mutations in ARID1A, BRCA2, PTEN, and CHEK2 showed that whereas some mutations were seen more commonly in dMMRP cases, there were a few notable gene mutations that were seen equally or more frequently in pMMRP cases. Q583fs and R1276X protein changes in ARID1A were more common in pMMRP compared with dMMRP (2 vs. 1 and 5 vs. 1, respectively). N1544fs and N1784fs protein changes in BRCA2 were seen equally between pMMRP and dMMRP (1 vs. 1 and 1 vs. 1, respectively). I157T and T367fs protein changes in CHEK2 were more common in pMMRP compared with dMMRP (3 vs. 1 and 5 vs. 1, respectively). Conversely, all noted protein changes in PTEN were only seen in the dMMRP cohort (Fig. 5).
Figure 5.

Protein changes across total adenocarcinoma DDR-M cohort in dMMRP vs. pMMRP (dMMRP gene prevalence cutoff 23.5%) involving ARID1A, BRCA2, PTEN, and CHEK2.
We further investigated the prevalence of commonly comutated DDR genes. ARID1A was the most prevalent mutation with 226 cases in the DDR-M cohort. Of these 226 ARID1A mutations, 70 cases had comutations with other DDR genes. When examining the total number of cases with greater than one DDR gene mutation in our cohort, ARID1A accounted for 70 of 83 cases (84%). Of the 83 cases with greater than one DDR mutation, BRCA2 was the most commonly comutated DDR gene with ARID1A followed by ATM and WRN (31% vs. 22% vs. 11%, respectively; Fig. 6). ATM and ATRX mutations were mutually exclusive in our cohort.
Figure 6.

Oncoprint depicting the prevalence of comutated DDR genes with ARID1A.
Discussion
Our study is a comprehensive assessment of HR-DDR prevalence across gastroesophageal cancers. This is a timely analysis given the surge in therapies currently focused on inhibiting the DDR pathway. PARP inhibitors like olaparib have shown considerable efficacy against tumors harboring mutations in BRCA1 and BRCA2 (28). DDR-targeted agents in cancer treatment maximize DNA damage in the G1-phase and S-phase of the cell cycle, thereby preventing DNA repair in G2 (29–30). When cells proceed into mitosis unrepaired DNA double-strand breaks (DSB) result in cell death (31–32). DDR-based therapies can potentially have greater therapeutic efficacy compared with traditional chemotherapy in part because of their ability to increase tumor cell immunogenicity. DDR inhibition facilitates cell-extrinsic immunogenicity by activating stimulator of interferon genes (STING) and NF-kB transcription factors (33–34). This promotes the release of proinflammatory signals (e.g., chemokines such as CXCL10 and CCL5) that induce cytotoxic T lymphocytes (35). DDR inhibition can also facilitate cell-intrinsic immunogenicity through upregulation of PD-L1, thereby making tumor cells visible to T cells via modulation of their cell-surface phenotype (36). Given that inhibition of DDR promotes inflammation and immune priming, DDR inhibitors such as olaparib can serve a role in making tumors more receptive to ICB. Therefore, there is a strong role for pursuing studies combining DDR agents with immunotherapy drugs in select populations.
The value of investigating the association between HR-DDR and known immune biomarkers of ICB is not unique to gastric and GEJ cancers. Studies have shown colorectal cancers with tumors harboring mutations in DDR-related genes more commonly exhibit dMMRP (37). Pancreatic ductal adenocarcinomas with BRCA1/2 mutations exhibit increased PD-L1 expression (38). Similar findings were observed in patients with high-grade serous ovarian cancers (39). Another study found significantly higher TMB in breast carcinomas with DDR mutations (40).
The potential role for HR-DDR–targeted therapy can be seen in previous studies such as the GOLD trial. This study evaluated the efficacy of olaparib plus paclitaxel in second-line metastatic gastric/GEJ cancer. Phase II of this study showed an improved overall survival (OS) benefit in ATM-deficient gastric cancers when treated with olaparib plus paclitaxel versus paclitaxel alone (41). However, the phase III study did not show a difference in OS between the treatment groups (42). Although it remains unclear why the phase III study did not meet its primary objective by showing PARP inhibitor efficacy, there could still be a role in targeting the HR pathway in these cancers predicated on a patient’s genetic profile of HR-DDR mutations (as suggested in phase II). The GOLD study shows paclitaxel may not be the best partner for olaparib. In addition, tumors may need to be enriched for DDR alterations in order to respond to therapy or be used in combination with ICB. A vital step toward improving the use of immunotherapy in GC is identifying predictive biomarkers that can help delineate which patients might benefit in the clinical setting.
A number of clinical trials are currently ongoing investigating the DDR–ICB combination. Preliminary data from NCT02484404, a clinical study investigating the efficacy of olaparib and durvalumab (a PD-L1 inhibitor) in patients with metastatic castration-resistant prostate cancer, demonstrated that DDR mutation was a favorable biomarker indicating an improved likelihood response to combination therapy (12-month progression-free survival probability of DDR-M vs. non–DDR-M, 83.3% vs. 36.4%, P = 0.03; refs. 43–44). A phase I/II study evaluating the efficacy of niraparib (a PARP inhibitor) and pembrolizumab in metastatic triple-negative breast cancer or recurrent ovarian cancer demonstrated that the therapeutic response rate was higher in the cohorts with BRCA1/2 mutated breast and ovarian tumors (NCT02657889; refs. 45–46). These DDR-immunotherapy drug regimens show promising results and highlight the potential to use this combination to improve patient outcomes.
The data in our study support a significant prevalence of HR-DDR mutations across upper gastrointestinal (GI) malignancies. We confirmed DDR mutations are prevalent across all gastroesophageal adenocarcinomas (23%) and gastroesophageal squamous cell carcinomas (20%). Among the esophageal, gastric, and gastroesophageal junction cancers tested, DDR mutations were most pronounced in gastric adenocarcinomas (27.6%). In comparing adenocarcinoma to squamous histology, there was a similar rate of DDR mutations between the histologic subtypes, making both groups amenable to trials investigating HR-DDR therapy.
When analyzing the association of DDR-M with known immune biomarkers to ICB, we confirmed biomarker expression of dMMRP (18% vs. 1%) and TMB-high with a cutoff of ≥10 mt/MB (27% vs. 9%) was significantly more prevalent in the DDR-M cohort compared with the non–DDR-M cohort. A previous study investigating the correlation between HR gene mutations in gastric adenocarcinoma also showed that HR genes were associated with elevated TMB (13). Importantly, tumors that harbored HR gene mutations had improved survival when treated with ICI (13). It should be noted that the DDR–TMB association was not statistically significant when focusing only on the pMMRP cohort.
PD-L1 expression in the total adenocarcinoma cohort was significantly higher in the DDR-M cohort compared with the non–DDR-M cohort (CPS 10.1 vs. 5.8; P = 0.0013). We consider this to be a relevant finding as higher PD-L1 CPS is now an established biomarker for immune-checkpoint inhibitor response in gastroesophageal cancers. These data lend support to investigating therapeutics focusing on immune therapies combinations in tumors that harbor DDR mutations.
Multiple DDR mutations were associated with higher TMB and PD-L1 CPS. We concluded that malignancies with multiple DDR mutations could result in stronger immunogenicity of the tumor. However, whether tumors with greater than one mutation are likely to have more severe DDR phenotype deserves further investigation.
The data in our study further delineate the similarities and differences between esophageal adenocarcinoma and esophageal squamous cell carcinoma. The adenocarcinoma cohort had comparable DDR mutation rates compared with the squamous cell carcinoma cohort (19.9% vs. 19.1%). Similarly, the adenocarcinoma cohort had a comparable rate of more than one mutation compared with the squamous cell carcinoma cohort (3% vs. 2%), demonstrating that having more than one DDR mutation was rare across both cohorts. Among the 20 DDR genes examined, our data revealed ARID1A, ATRX, and RAD50 were the most prevalent DDR mutations in the adenocarcinoma cohort. ARID1A and PTEN were the most prevalent DDR mutations in the squamous cell carcinoma cohort.
Among the 20 DDR mutations examined across total gastroesophageal adenocarcinomas, alterations in ARID1A, BRCA2, PTEN, and ATM were the most commonly mutated HR-DDR genes among tumors that were classified as dMMRP and TMB-high. Similarly, ARID1A, BRCA2, PTEN, and ATM were also the most commonly mutated HR-DDR genes correlated with increased PD-L1 expression. A further analysis of specific gene mutations in ARID1A, BRCA2, PTEN, and CHEK2 showed that whereas some mutations were seen more commonly in dMMRP tumors, there were a few notable gene mutations that were seen equally or more frequently in pMMRP tumors. These specific mutations warrant further investigation in future studies. Notably, PTEN protein changes were seen only in dMMRP tumors, and none were seen in pMMRP tumors.
The majority of the tumors evaluated were from the primary site versus metastatic sites (1,199 vs. 513). Whether there are differential DDR mutation patterns such as number and type of DDR mutations in the primary versus metastatic site deserve further investigation.
Looking forward, investigations should focus on characterizing which of these HR-DDR gene alterations function as driver mutations in gastroesophageal cancers. A limitation of our study is that of the 20 DDR mutations examined, while the roles of BRCA1 and BRCA2 are well characterized in cancer pathogenesis (47–49), the roles that DDR genes such as ARID1A, ATM, and PTEN play in gastrointestinal cancer progression currently needs further review (50–53). By better elucidating the HR-DDR genes relevant in gastroesophageal cancers, it would be possible to develop a validated HR-DDR assay to help investigators better identify gastroesophageal cancers that would benefit from therapies aimed at the HR-DDR pathway.
In conclusion, we confirmed HR-DDR mutations are a clinically significant finding across gastroesophageal adenocarcinomas and squamous cell carcinomas. We confirmed immune biomarker expression is more prevalent in DDR-mutated tumors, and we examined the association between these biomarkers and specific DDR alterations. Our findings could lead to validation of clinical criteria that would help identify patients who would benefit from novel combination therapy involving both HRD-directed therapy and immunotherapy in future clinical trials. By further identifying associations of DDR alterations and immunotherapy-predictive biomarkers, rational drug combinations have strong promise in the treatment of gastroesophageal cancers.
Supplementary Material
{kind=link}
Footnotes
Authors’ Disclosures
J. Xiu reports other support from Caris Life Sciences during the conduct of the study. A. Grothey reports grants, personal fees, and nonfinancial support from Bayer and Roche/Genentech, grants and personal fees from Merck, grants from OBI Pharmaceuticals, Boston Biomedicals, Natera, Amgen, and BMS outside the submitted work. M.J. Pishvaian reports personal fees from Merck, Astra Zeneca, Perthera, and Foundation Medicine, other support from Seattle Genetics, Pfizer, Genentech, AbbVie, Tesaro, Novartis, Celldex, Curegenix, BMS, Regeneron, Fibrogen, ARMO, GSK, RenovoRx, Bayer, and Calithera and personal fees and other support from Halozyme outside the submitted work; in addition, M.J. Pishvaian has a patent for Perthera issued to Myself and a patent for AbbVie pending to Myself. J.J. Hwang reports personal fees from Bristol Myers Squibb, Boehringer Ingelheim, Pfizer, Incyte, QED, Eisai, Deciphera, Genentech/Roche, Amgen, and Ipsen outside the submitted work. J.L. Marshall reports personal fees from Caris Life Science outside the submitted work; and Indivumed- Oncology CMO. A.M. VanderWalde reports nonfinancial support from Caris Life Sciences during the conduct of the study; grants from Amgen, personal fees and nonfinancial support from AstraZeneca, personal fees from Bristol Myers Squibb, Roche/Genentech, Caris Life Sciences, Elsevier, Mirati, ConcertAI, and Bayer outside the submitted work. A.F. Shields reports personal fees and other support from Caris Life Sciences outside the submitted work. W. Korn reports personal fees from Merck outside the submitted work; and Caris Life Sciences, employment, stock options, stock ownership. M. Salem reports other support from Taiho, BMS, Merck, AZ/Daiichi Sankyo, and Exelixis during the conduct of the study. R.M. Goldberg reports personal fees from Amgen, Bayer, Astra Zeneca, Merck, Novartis, Genentech, adaptimmune, GlaxoSmithKline, Compass Therapeutics, and GI Therapeutics outside the submitted work. J. Zeng reports personal fees from Caris Life Sciences during the conduct of the study. S.S. Kim reports Astellas, Merck, and Daiichi outside the submitted work. No disclosures were reported by the other authors.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
References
- 1.Yang D, Hendifar A, Lenz C, Togawa K, Lenz F, Lurje G, et al. Survival of metastatic gastric cancer: Significance of age, sex and race/ethnicity. J Gastrointest Oncol 2011;2:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin PH, Kuo WH, Huang AC, Lu YS, Lin CH, Kuo SH, et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016;7:8310–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol 2016;27:1449–55. [DOI] [PubMed] [Google Scholar]
- 4.Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen WJ, et al. Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol 2018;2018:PO.17.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for germline-mutated metastatic pancreatic cancer. N Engl J Med 2019;381:317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 2017;377:523–33. [DOI] [PubMed] [Google Scholar]
- 8.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res 2017;23:3711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mouw KW, Goldberg MS, Konstantinopoulos PA, D’andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov 2017;7:675–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 2019;9:646–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol 2018;36:1685–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mouw KW, D’andrea AD. DNA repair deficiency and immunotherapy response. J Clin Oncol 2018;36:1710–3. [DOI] [PubMed] [Google Scholar]
- 13.Fan Y, Ying H, Wu X, Chen H, Hu Y, Zhang H, et al. The mutational pattern of homologous recombination (HR)-associated genes and its relevance to the immunotherapeutic response in gastric cancer. Cancer Biol Med 2020;17:1002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 2019;30:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncology 2020;21:1353–65. [DOI] [PubMed] [Google Scholar]
- 16.Mansour WY, Tennstedt P, Volquardsen J, Oing C, Kluth M, Hube-Magg C, et al. Loss of PTEN-assisted G2–M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci Rep 2018;8:3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niedermaier B, Sak A, Zernickel E, Xu S, Groneberg M, Stuschke M. Targeting ARID1A-mutant colorectal cancer: depletion of ARID1B increases radiosensitivity and modulates DNA damage response. Sci Rep 2019;9:18207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006;66:8109–15. [DOI] [PubMed] [Google Scholar]
- 19.Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov 2015;5:752–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A 2014;111:285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sung P, Klein H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 2006;7:739–50. [DOI] [PubMed] [Google Scholar]
- 22.Zhao W, Steinfeld JB, Liang F, Chen X, Maranon DG, Jian Ma C, et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017;550:360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aarts M, Bajrami I, Herrera-Abreu MT, Elliott R, Brough R, Ashworth A, et al. Functional genetic screen identifies increased sensitivity to WEE1 inhibition in cells with defects in Fanconi anemia and HR pathways. Mol Cancer Ther 2015;14:865–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res 2014;20:764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res 2010;70:5389–98. [DOI] [PubMed] [Google Scholar]
- 26.Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 2007;21:3073–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulangara K, Zhang N, Corigliano E, Guerrero L, Waldroup S, Jaiswal D, et al. Clinical utility of the combined positive score for programmed death ligand-1 expression and the approval of pembrolizumab for treatment of gastric cancer. Arch Pathol Lab Med 2019;143:330–7. [DOI] [PubMed] [Google Scholar]
- 28.MJ O’. Targeting the DNA damage response in cancer. Mol Cell 2015;60:547–60. [DOI] [PubMed] [Google Scholar]
- 29.Ceccaldi R, Rondinelli B, D’andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 2016;26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009;461:1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004;23:2825–37. [DOI] [PubMed] [Google Scholar]
- 32.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013;155:1088–103. [DOI] [PubMed] [Google Scholar]
- 33.Stewart RA, Pilié PG, Yap TA. Development of PARP and immune-checkpoint inhibitor combinations. Cancer Res 2018;78:6717–25. [DOI] [PubMed] [Google Scholar]
- 34.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 2016;17:1142–9. [DOI] [PubMed] [Google Scholar]
- 35.Talens F, Van Vugt M. Inflammatory signaling in genomically instable cancers. Cell Cycle 2019;18:1830–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li A, Yi M, Qin S, Chu Q, Luo S, Wu K. Prospects for combining immune checkpoint blockade with PARP inhibition. J Hematol Oncol 2019;12:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arai H, Elliott A, Xiu J, Wang J, Battaglin F, Kawanishi N, et al. The landscape of alterations in DNA damage response pathways in colorectal cancer. Clin Cancer Res 2021;27:3234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, et al. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol 2017;3:774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016;7:13587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mei P, Freitag CE, Wei L, Zhang Y, Parwani AV, Li Z. High tumor mutation burden is associated with DNA damage repair gene mutation in breast carcinomas. Diagn Pathol 2020;15:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bang YJ, Im SA, Lee KW, Cho JY, Song EK, Lee KH, et al. Randomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol 2015;33:3858–65. [DOI] [PubMed] [Google Scholar]
- 42.Bang YJ, Xu RH, Chin K, Lee KW, Park SH, Rha SY, et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1637–51. [DOI] [PubMed] [Google Scholar]
- 43.Karzai F, Madan RA, Owens H, Couvillon A, Hankin A, Williams M, et al. A phase 2 study of olaparib and durvalumab in metastatic castrate-resistant prostate cancer (mCRPC) in an unselected population. J Clin Oncol 2018;36:163. [Google Scholar]
- 44.Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer 2018;6:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vinayak S, Tolaney SM, Schwartzberg LS, Mita MM, McCann GA-L, Tan AR, et al. TOPACIO/Keynote-162: Niraparib + pembrolizumab in patients (pts) with metastatic triple-negativebreast cancer (TNBC), a phase 2 trial. JClin Oncol 2018;36:1011. [Google Scholar]
- 46.Konstantinopoulos PA, Waggoner SE, Vidal GA, Mita MM, Fleming GF, Holloway RW, et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—results from ROC cohort. J Clin Oncol 2018;36:106. [Google Scholar]
- 47.Mehrgou A, Akouchekian M. The importance of BRCA1 and BRCA2 genes mutations in breast cancer development. Med J Islam Repub Iran 2016;30:369. [PMC free article] [PubMed] [Google Scholar]
- 48.Friebel TM, Domchek SM, Rebbeck TR. Modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: systematic review and meta-analysis. J Natl Cancer Inst 2014;106:dju091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Godet I, Gilkes DM. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr Cancer Sci Ther 2017;4: 10.15761/ICST.1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou H, Tan S, Li H, Lin X. Expression and significance of EBV, ARID1A and PIK3CA in gastric carcinoma. Mol Med Rep 2019;19:2125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bi C, Liu M, Rong W, Wu F, Zhang Y, Lin S, et al. High Beclin-1 and ARID1A expression corelates with poor survival and high recurrence in intrahepatic cholangiocarcinoma: a histopathological retrospective study. BMC Cancer 2019;19:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klempner SJ, Bhangoo MS, Luu HY, Kim ST, Chao J, Kim KM, et al. Low ATM expression and progression-free and overall survival in advanced gastric cancer patients treated with first-line XELOX chemotherapy. J Gastrointest Oncol 2018;9:1198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoppe MM, Sundar R, Tan DSP, Jeyasekharan AD. Biomarkers for homologous recombination deficiency in cancer. J Natl Cancer Inst 2018;110:704–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.