

Abstract
The generation of organoids and tissues with programmable cellular complexity, architecture and function would benefit from the simultaneous differentiation of human induced pluripotent stem cells (hiPSCs) into divergent cell types. Yet differentiation protocols for the overexpression of specific transcription factors typically produce a single cell type. Here we show that patterned organoids and bioprinted tissues with controlled composition and organization can be generated by simultaneously co-differentiating hiPSCs into distinct cell types via the forced overexpression of transcription factors, independently of culture-media composition. Specifically, we used such orthogonally induced differentiation to generate endothelial cells and neurons from hiPSCs in a one-pot system containing either neural or endothelial stem-cell-specifying media, and to produce vascularized and patterned cortical organoids within days by aggregating inducible-transcription-factor and wild-type hiPSCs into randomly pooled or multicore-shell embryoid bodies. Moreover, by leveraging multimaterial bioprinting of hiPSC inks without extracellular matrix, we generated patterned neural tissues with layered regions composed of neural stem cells, endothelium and neurons. Orthogonally induced differentiation of stem cells may facilitate the fabrication of engineered tissues for biomedical applications.
Recent innovations in organoid development and three-dimensional (3D) bioprinting offer emerging pathways to create autologous human tissues for drug screening and therapeutic applications1–4. The ability to generate organoids via self-assembly and differentiation of embryoid bodies (EBs), which are aggregated from human induced pluripotent stem cells (hiPSCs), provides a bottom-up approach to creating organ-like microarchitectures5,6. By contrast, multimaterial 3D bioprinting offers a top-down method for fabricating heterogeneous, stem-cell-derived tissues7–12. However, both techniques are limited by the speed, efficiency and scalability of stem cell differentiation. Current protocols for generating cerebral13–15, renal16–18, retinal19 and other organoids often require prolonged culture times ranging from weeks to several months to approach organ-level cellular diversity. Moreover, organoid protocols that generate a wider range of cells and tissues generally result in less reproducible organoids, giving rise to a trade-off between organoid reproducibility and cellular diversity20,21. Similarly, a large number of cell types, each differentiated and rendered into densely cellular bioinks, would be required for multimaterial 3D bioprinting of organ-specific tissues. New approaches that enhance the specificity, efficiency and scalability of stem cell differentiation are therefore needed to generate programmable multicellular organoids and tissues from pluripotent stem cells.
To guide stem cell differentiation, one can either introduce extracellular cues by controlling the media composition or modulate the intracellular state by overexpressing various transcription factors (TFs). Many TF-based protocols synergistically apply external and internal cues to promote rapid and efficient cell differentiation to a single lineage; examples include the (1) derivation of endothelial cells cultured in endothelial cell growth medium, while overexpressing ETS translocation variant 2 (ETV2)22,23, (2) derivation of neurons cultured in neurobasal-A medium with B27 supplement, while overexpressing neurogenin-1 and −2 (NGN1, NGN2)24, and (3) derivation of hepatocytes cultured in hepatocyte medium, while overexpressing combinations of GATA binding protein 4 (GATA4), hepatocyte nuclear factor 1- and 4-alpha (HNF1α, HNF4α), and Forkhead box A1, A2 and A3 (FOXA1, FOXA2, FOXA3)25,26. While these approaches aim to differentiate cells into a singular phenotype, human tissues are composed of multiple cell types organized into hierarchically patterned structures. While the overexpression of certain ‘less-specific’ TFs may create progenitors for multiple cell types27, this strategy provides limited control over the precise composition and distribution of cell types in the resulting tissue. As an alternative strategy, one can overexpress TFs in a subset of cells within a developing organoid to guide their differentiation into specific phenotypes. For example, others recently combined TF-driven differentiation with traditional cortical organoid culture by inducing overexpression of ETV2 in a subset of cells within cortical organoids, giving rise to a vascular endothelium akin to brain microvasculature28. However, the generation of more complex multicellular tissues requires not only a broader range of programmable cell types, but also methods that simultaneously enable control over their spatial patterning.
Recent progress has been made in screening the human genome for specific TFs that, when overexpressed, can drive rapid differentiation from hiPSCs to somatic cell types in a media-independent fashion29. Building on this advance, we posit that an ideal method for generating programmable multicellular organoids and 3D organ-specific tissues would begin by overexpressing TFs in multiple pre-programmed cell populations, each of which is differentiated to a specific cell type of interest with high efficiency. By pooling or printing populations of wild type (WT) and engineered cell lines, multicellular organoids and 3D tissues could then be programmably generated on demand. The successful implementation of this approach requires the ability to simultaneously differentiate multiple hiPSC lines in a one-pot protocol independent of external cues provided by the cell culture media.
Here we report an orthogonally induced differentiation (OID) method for achieving genomically programmed human stem cells, organoids and 3D bioprinted organ-specific tissues. In OID, doxycycline (dox)-induced TF-overexpression in pre-programmed hiPSCs can override media-driven differentiation. We show that forced overexpression of intracellular TFs can operate independently of media-driven differentiation to generate specific cell types, each with near-unity efficiency (Fig. 1a). When applied to randomly pooled or multicore-shell EBs, OID can be used to construct multicellular and spatially patterned organoids (Fig. 1b). Moreover, when this approach is coupled with multimaterial 3D bioprinting of densely cellular, matrix-free WT and inducible-TF hiPSC inks, pluripotent tissues can be patterned and subsequently transformed in situ to multicellular tissue constructs that mimic native tissue architectures (Fig. 1c). While OID can be broadly applied, we demonstrate its utility by creating vascularized cortical organoids in pooled and multicore-shell motifs, as well as 3D cortical tissues composed of multiple cell types patterned in spatially distinct regions.
Fig. 1 |. OiD of stem cells for patterning vascularized organoids and bioprinted tissues.
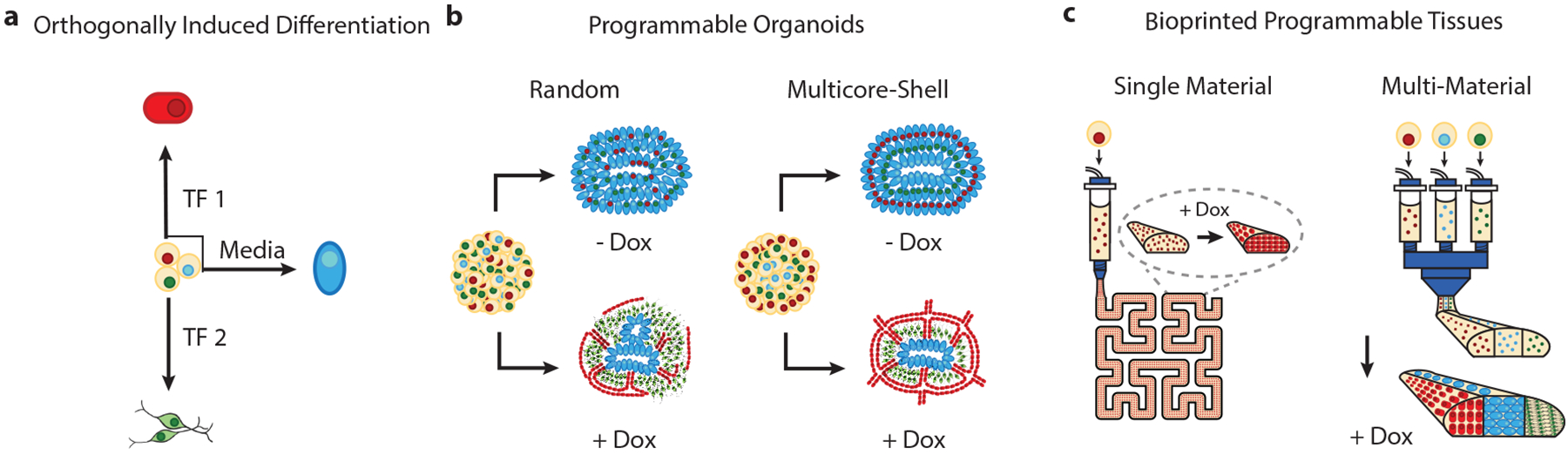
a, Transcription factor overexpression generates distinct cell lineages with near-unity efficiency in identical cell media. b, Programmable and patterned organoids can be generated from randomly pooled and multicore-shell embryoid bodies via OID. c, 3D organ-specific tissues composed of programmable pluripotent cell lines can be bioprinted and transformed in situ via OID.
Results
The derivation of neural tissue from hiPSCs serves as an ideal example to assess the feasibility of OID, given that pluripotent cells are efficiently differentiated into neuroectoderm in a dual-SMAD inhibiting medium30 (Fig. 2). We tested three different Personal Genome Project 1 (PGP1) hiPSC lines, one WT line and two inducible-TF lines, in identical media conditions. WT PGP1 cells form neural stem cells (NSCs) when cultured in neural induction medium (NIM) containing transforming growth factor beta (TGF-β) and bone morphogenetic protein (BMP)-pathway small molecule inhibitors, as previously reported31 (Fig. 2a). For the inducible-TF lines, we utilized a PiggyBac transposon vector to incorporate an all-in-one Tet-On system that enables the rapid and highly efficient doxycycline-induced upregulation of transcription factors. To generate inducible endothelial (iEndo) cells, we overexpressed the transcription factor ETV2, which is known to drive rapid and efficient directed differentiation of pluripotent cells to vascular endothelial cells23 (Fig. 2b). To generate inducible neurons (iNeuron), we upregulated NGN1, as single- or co-expression of neurogenins is known to rapidly generate neurons from hiPSCs24,32 (Fig. 2c).
Fig. 2 |. Programmable differentiation of pluripotent stem cells via OiD under identical media conditions.
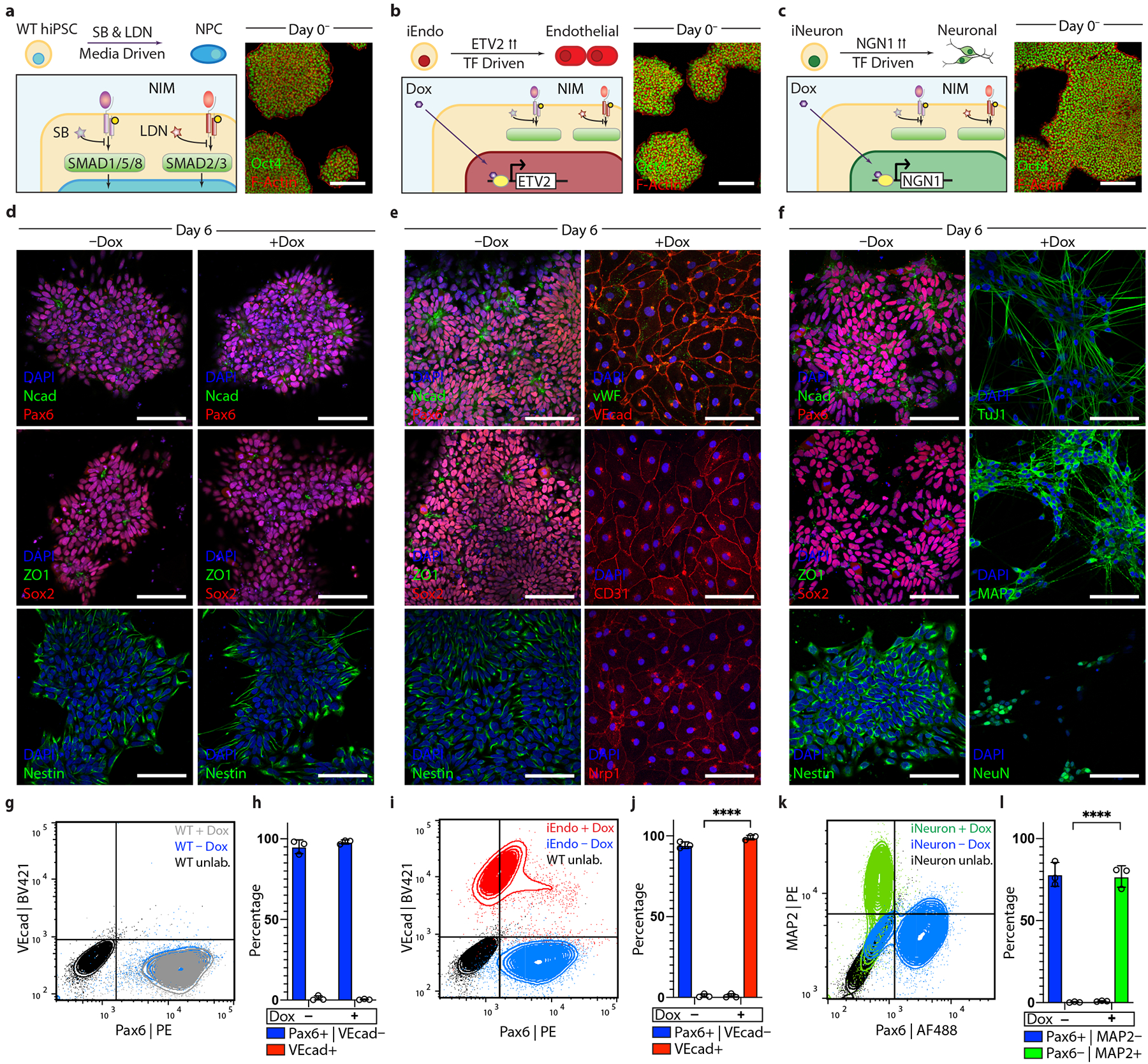
a, Left: schematic detailing WT hiPSC differentiation into neural stem cells in NIM. right: immunostaining of Oct4 and F-actin of WT colonies on day 0. b, Left: schematic detailing iEndo differentiation through doxycycline-induced ETV2 isoform-2 overexpression. right: immunostaining of Oct4 and F-actin of iEndo colonies on day 0. c, Left: schematic detailing iNeuron differentiation through doxycycline-induced NGN1 overexpression in culture. right: immunostaining of Oct4 and F-actin of iNeuron colonies on day 0. d, WT hiPSCs cultured in NIM for 6 d without (left column) or with (right column) doxycycline. Immunostaining of NCAD and PAX6, ZO1 and SOX2, and nestin. e, iEndo cells cultured in NIM for 6 d without (left column) or with (right column) doxycycline. Left: immunostaining of NCAD and PAX6, ZO1 and SOX2, and nestin. right: immunostaining of VWF and VEcad, CD31 and NrP1. f, iNeuron cells cultured in NIM for 6 d without (left column) or with (right column) doxycycline. Left: immunostaining of NCAD and PAX6, ZO1 and SOX2, and nestin. right: immunostaining of Tuj1, MAP2 and NeuN. g, Flow cytometry plots quantifying WT hiPSC differentiation to PAX6+ neural stem cells by 6 d with (grey) and without (blue) doxycycline. BV, Brilliant Violet 421; PE, phycoerythrin. h, Quantification of populations, mean ± s.e.m., n = 3 biological replicates. i, Flow cytometry plots indicating that iEndos differentiate into VEcad+ endothelium in the presence of doxycycline (red) or into PAX6+ neural stem cells in its absence (blue). j, Quantification of populations, mean ± s.e.m., n = 3 biological replicates, **** P = 7.06 × 10−7, unpaired two-tailed t-test. k, Flow cytometry plots indicating that iNeurons differentiate into MAP2+ neurons in the presence of doxycycline (green) or into PAX6+ neural stem cells in its absence (blue). AF488, Alexa Fluor 488. l, quantification of populations, mean ± s.e.m., n = 3 biological replicates, ****P = 3.53 × 10−5, unpaired two-tailed t-test. Scale bars: 100 μm in d–f; 50 μm in a–c.
OID of stem cells.
We characterized the differentiation of WT and inducible-TF hiPSC lines in the absence (control) and presence of doxycycline. When cultured for 6 d in NIM, WT hiPSCs experienced a loss of pluripotency and efficiently differentiated into a neural stem cell fate in both cases, as indicated by the formation of characteristic polarized neural rosettes and the expression of neural cadherin (NCAD), paired-box gene 6 (PAX6), SRY-box transcription factor 2 (SOX2) and nestin (Fig. 2d). Similarly, iEndo cells undergo media-driven differentiation to neural stem cells in the absence of doxycycline (Fig. 2e). However, in the presence of doxycycline, the overexpression of ETV2 drives the rapid differentiation to a vascular endothelial cell phenotype, as evidenced by the formation of a confluent cobblestone morphology, the expression of vascular endothelial cadherin (VEcad), von-Willebrand factor (VWF), cluster of differentiation 31 (CD31) and the vascular endothelial growth factor receptor neuropilin 1 (NRP1). iNeuron cells also differentiated into neural stem cells in the absence of doxycycline (Fig. 2f). In the presence of doxycycline, iNeurons formed neurons with a bipolar morphology and expressed neural markers neuron-specific class III beta-tubulin (Tuj1), microtubule associated protein 2 (MAP2) and neuronal nuclei (NeuN). When induced, both iEndo and iNeuron cells efficiently and rapidly differentiated, confirming the feasibility of OID (Fig. 2g–l and Supplementary Fig. 1). Indeed, the contrast between the differentiation of iEndo in the presence (99% vascular endothelium) and absence (94% neural stem cells) of doxycycline serves as an illustration of the orthogonality of externally driven (media directed) versus internally driven (TF-directed) cell differentiation.
To assess the generalizability of OID, we subjected WT and iNeuron hiPSCs to media conditions that drive endothelial differentiation via an established protocol that uses glycogen synthase kinase 3 beta (GSK-3β) inhibition to direct cells into mesendoderm before applying growth factors to specify endothelium33. After 8 d of culture, WT cells differentiated into endothelium expressing VEcad, VWF, CD31, and NRP1 with a typical cobblestone morphology irrespective of the presence of doxycycline (Supplementary Fig. 2a). Without doxycycline, iNeuron cells similarly differentiated into endothelium. However, in the presence of doxycycline, iNeuron cells differentiated into neurons with a bipolar morphology that express Tuj1, MAP2 and NeuN (Supplementary Fig. 2b). Taken together, these data demonstrate that intracellular TF-driven differentiation can fully override the otherwise strong and specific extracellular differentiation cues.
Next we investigated whether simultaneous OID of a mixed initial population of hiPSCs would give rise to a heterogeneous differentiated cell population with programmed composition. Specifically, we seeded different proportions of WT, iEndo and iNeuron hiPSCs onto a Matrigel surface and cultured them in NIM with doxycycline. We observed that distinct multicellular populations form after 6 d in one-pot culture conditions (Supplementary Fig. 3). Furthermore, under these conditions, iEndo cells self-assembled into a network-like microvasculature that is consistent with endothelial tubulogenesis assays. In contrast, WT cells formed neurospheres that rose above the surface of the gel, while iNeuron cells formed a network of neurites. Notably, when iEndo and WT cells were cultured together, the differentiated cells formed a distinct network pattern in which endothelial cells comprise the edges and neurospheres comprise the nodes. Using iEndo-mKate2- and WT-green fluorescent protein (GFP)-labelled cells, we confirmed that the vascular and neurosphere components are indeed composed of iEndo and WT cells, respectively (Supplementary Video 1). When iEndo and iNeuron cells were cultured together, the resulting endothelial and neuronal cells formed overlapping networks in which neurites extend along the vascular network. To demonstrate simultaneous OID, we pooled all three cell lines together and cultured them in NIM in the absence (control) and presence of doxycycline. Without doxycycline, all three cell lines differentiated into neurospheres, with no visible VEcad or MAP2 staining. However, in the presence of doxycycline, WT, iEndo and iNeuron cells differentiated into SOX2+ neurospheres, VEcad+ vascular endothelium and MAP2+ neurons, respectively. Importantly, our results show that OID offers several distinct advantages over traditional organoid differentiation protocols, which typically aim to derive cells from a single germ layer13–15,20,21.
We then investigated whether OID of pre-programmed cells in co-culture results in changes in gene expression due to the presence of multiple cell types. Using fluorescently labelled mKate2-expressing iEndo cells and super folder GFP (sfGFP)-expressing iNeuron cells, we isolated iEndos and iNeurons from co-culture and performed bulk RNA sequencing (Supplementary Figs. 4a,b and 5a). We found that the phenotype of iNeurons is largely unaffected by co-differentiation over 7 d with either WT neural stem cells or iEndo-derived endothelium (Supplementary Fig. 5b,c). Gene ontology analysis of significantly upregulated genes in iEndo cells co-cultured with WT neural stem cells showed an enrichment in genes related to wound healing and extracellular matrix organization, suggesting increased matrix remodelling activity (Supplementary Fig. 5d). However, iEndo cells cultured with iNeurons remain similar to iEndo cells in monoculture (Supplementary Fig. 5e,f). Overall, our findings suggest that OID-derived cells remain stable when co-differentiated with other cell types. Hence, orthogonally induced differentiation can be used to programmably define cell subpopulations within stem-cell-derived tissues.
Programmable cortical organOIDs.
Having demonstrated that OID is capable of programming stem cell co-differentiation with near-unity efficiency, we next addressed the challenge of organoid vascularization, since traditional cerebral (and cortical) organoid protocols fail to generate an embedded vascular network13–15. One strategy to achieve this goal is to simply mix endothelial cells with hiPSCs to form a multicellular embryoid body that subsequently differentiates into the desired vascularized organoid34. However, we found that human umbilical vein endothelial cells (HUVECs), when mixed with WT hiPSCs in microwells, underwent phase separation within 24 h (Extended Data Fig. 1a). This observation probably arises due to differences in cell adhesion35, as endothelial cells and epithelial hiPSCs express different cell adhesion molecules. Importantly, when iEndo-mKate2 and WT-GFP hiPSCs were mixed and cultured in microwells, single embryoid bodies were formed with interspersed cells (Fig. 3b,c and Extended Data Fig. 1b). By controlling the ratio of WT-to-iEndo cells seeded in each microwell, we can precisely tailor the resulting endothelium after their orthogonally induced differentiation to form organOIDs (Extended Data Fig. 1c,d). To illustrate this, we produced vascularized cortical organOIDs by pooling 67% WT hiPSCs and 33% iEndo cells into embryoid bodies that are cultured in dual-SMAD inhibiting media to direct dorsal forebrain formation. After 3 d in suspension culture, the embryoid bodies were embedded into a Matrigel-collagen gel droplet. After 10 d, the avascular WT-only organoids (control) retained a smooth border, while the organOIDs composed of a 2:1 ratio of WT-to-iEndo cells formed extensive sprouted vascular networks that penetrated into the surrounding gel, as marked by lectin (Ulex Europaeus Agglutinin, UEA1) and visualized using bright-field (Fig. 3d) and fluorescence microscopy (Fig. 3e and Extended Data Fig. 2a–e). The complete absence of microvascular markers in the WT organoids is consistent with previous reports that cortical organoids generated by traditional differentiation protocols lack a vascular network.
Fig. 3 |. Programmable vascularization of cortical organOiDs.
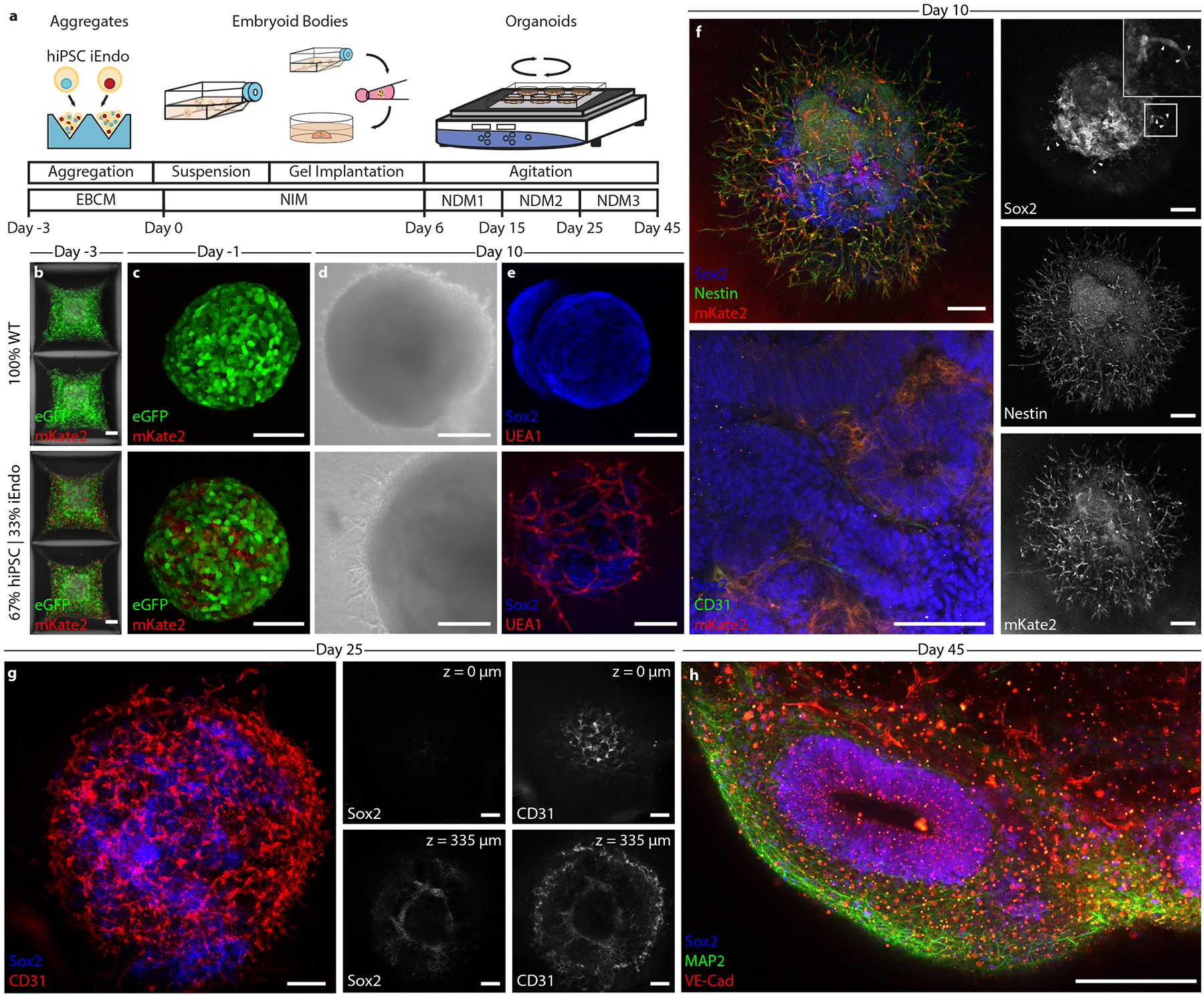
a, Schematic of the vascularized cortical organoid protocol. b, Fluorescent images of hiPSCs in microwells 3 d before suspension culture. Top: 100% WT-eGFP hiPSCs. Bottom: 67% WT-eGFP and 33% iEndo-mKate2 randomly pooled hiPSCs. c, Fluorescent images of resulting EBs. Top: 100% WT-eGFP EBs. Bottom: 67% WT-eGFP and 33% iEndo-mKate2 randomly pooled EBs. d, Brightfield images of organoids derived from EBs cultured for 10 d. Top: 100% WT-eGFP organoids. Bottom: 67% WT-eGFP and 33% iEndo-mKate2 pooled organoids. e, Immunostaining of SOX2 and UEA1 labelling of organoids cultured for 10 d. Top: 100% WT organoids. Bottom: 67% WT and 33% iEndo pooled organOIDs. f, Left, top: confocal maximum intensity z-projections obtained from immunostaining of SOX2 and nestin of 67% WT and 33% iEndo-mKate2 pooled organOIDs cultured for 10 d; right column: individual channels for SOX2, nestin and mKate2. Arrowheads indicate SOX2+ positive cells that co-localize with the vasculature. Left, bottom: immunostaining of SOX2 and CD31 with iEndo-mKate2 of 67% WT and 33% iEndo-mKate2 pooled organOIDs cultured for 10 d. g, Left: maximum intensity z-projection with immunostaining of SOX2 and CD31 of 67% WT and 33% iEndo pooled organOIDs cultured for 25 d. right: optical slices of SOX2 and CD31 channels at depths of z = 0 μm (top row) and z = 335 μm (bottom row). h, Maximum intensity z-projection with immunostaining of SOX2, MAP2 and VEcad of 67% WT and 33% iEndo pooled organOIDs cultured for 45 d. Scale bars: 200 μm in d, f (top), g and h; 100 μm in b, c, f (bottom) and e.
By pooling fluorescent mKate2-expressing iEndo cells, we confirmed that the embedded and sprouting vascular network observed within these organOIDs arises solely from those cells, as evidenced by the co-localization of CD31 and mKate2 (Fig. 3f). Notably, we see evidence of SOX2+ neural stem cells co-localizing along the sprouting vasculature (arrowheads, Fig. 3f). This interaction is consistent with our observations in co-culture of neural stem cells and iEndo endothelium on Matrigel (Supplementary Fig. 3), as well as known in vivo interactions of neural stem cells and endothelium in the neural stem cell niche in the subventricular zone36. Notably, endothelial cells reside in between the rosette or ventricle-like structures, whereas the SOX2+ ventricular zone-like structures are devoid of these cells, consistent with observed vascular patterns in neurodevelopment37. After 25 d in culture, the vascularized cortical organ-OIDs developed NeuN positive neural populations (Extended Data Fig. 2f,g and Supplementary Fig. 6a) and only organOIDs formed using 33% iEndo cells possessed an embedded vascular network (Supplementary Fig. 6b). Using whole-mount immunofluorescence of optically cleared organoids, we confirmed that this vascular network exists both at the organOID surface and within their core (Fig. 3g and Supplementary Video 2). Embedded vascular networks were imaged in multiple slices within cleared organoids, revealing the difference between the WT-only and the WT + iEndo cortical organoids (Fig. 3g, and Supplementary Figs. 7 and 8). As expected, there was no detectable vasculature within the WT-only organoids. In contrast, by day 45 of culture, WT + iEndo cortical organOIDs developed ventricle-like architectures that are surrounded by neural cell bodies, neurites and a pervasive vascular network (Fig. 3h and Extended Data Fig. 2f,g). Using quantitative reverse transcription polymerase chain reaction (RT-qPCR), we found that these vascularized cortical organOIDs have significantly enhanced expression of vasculature-related genes (CDH5, CD31), while neural stem cell, neural and pluripotent gene expression remain similar to the WT-only (control) organoids (Supplementary Fig. 9). This result demonstrates that OID promotes vascular network formation without adversely affecting the neural phenotype within these organOIDs.
Moving beyond the random incorporation of iEndo cells into pooled embryoid bodies to form vascularized organoids, we then sought to create spatially patterned cortical organOIDs. We combined WT, iEndo and iNeuron cells in a multicore-shell organOID architecture that is reminiscent of a developing brain, that is, having a germinal zone (GZ) (inner core), surrounding neurons (middle layer) and an encapsulating perineural vascular plexus (outer shell) (Fig. 4a). Additionally, the incorporation of iNeuron cells within these organOIDs enables faster neurogenesis when compared with cortical organoids produced by traditional protocols, which typically require ~30 d of culture to form a NeuN+ neuronal population38. Stepwise addition of hiPSCs in a U-well plate has previously been shown to generate a Janus embryoid body that can be used in conjunction with an inducible Sonic Hedgehog-expressing hiPSC line to encourage dorsoventral axis formation39. By using V-shaped wells, we created organoids with radial-symmetric multicore-shell architectures, rather than the two hemispherical compartments seen for Janus organoids assembled in U-wells (Fig. 4b). Embryoid bodies formed from WT-only, randomly pooled (WT, iNeuron, iEndo) triple populations and stepwise multicore-shell organoids all formed cohesive Oct4+ embryoid bodies after 1 d in culture. Consistent with other cortical organoid protocols38,40, those containing WT-only cells contained few neurons at day 10, as evidenced by a minimal number of NeuN expressing cells (Fig. 4c). In contrast, the incorporation of iNeuron cells in both random and multicore-shell organOIDs resulted in a large NeuN+ neuronal population by day 10 (Fig. 4d,e). These neuronal populations surround the ventricle-like structures within the organOIDs, forming a neuron-rich layer that resides more superficially than the deeper GZs. We also found clusters of NeuN+ neurons deep in the centre of randomly incorporated organOIDs that are absent in multicore-shell patterned organOIDs (Fig. 4d,e), suggesting that the initial spatial patterning imposed by stepwise aggregation informs later layered organization within cortical organoids. Furthermore, a pervasive CD31+ vascular network is present throughout both randomly pooled and multicore-shell patterned organoids. Notably, orthogonally induced differentiation of patterned multicore-shell embryoid bodies gives rise to cortical organOIDs with larger ventricle-like structures, as measured by the length of apical NCAD+ neuroepithelium, when compared with those that form in randomly pooled embryoid bodies and are similar in size to those found in WT-only organoids (Extended Data Fig. 3). This suggests that neural self-assembly processes can be preserved in organOIDs by imposing spatial patterning.
Fig. 4 |. Multicore-shell cortical organOiDs.
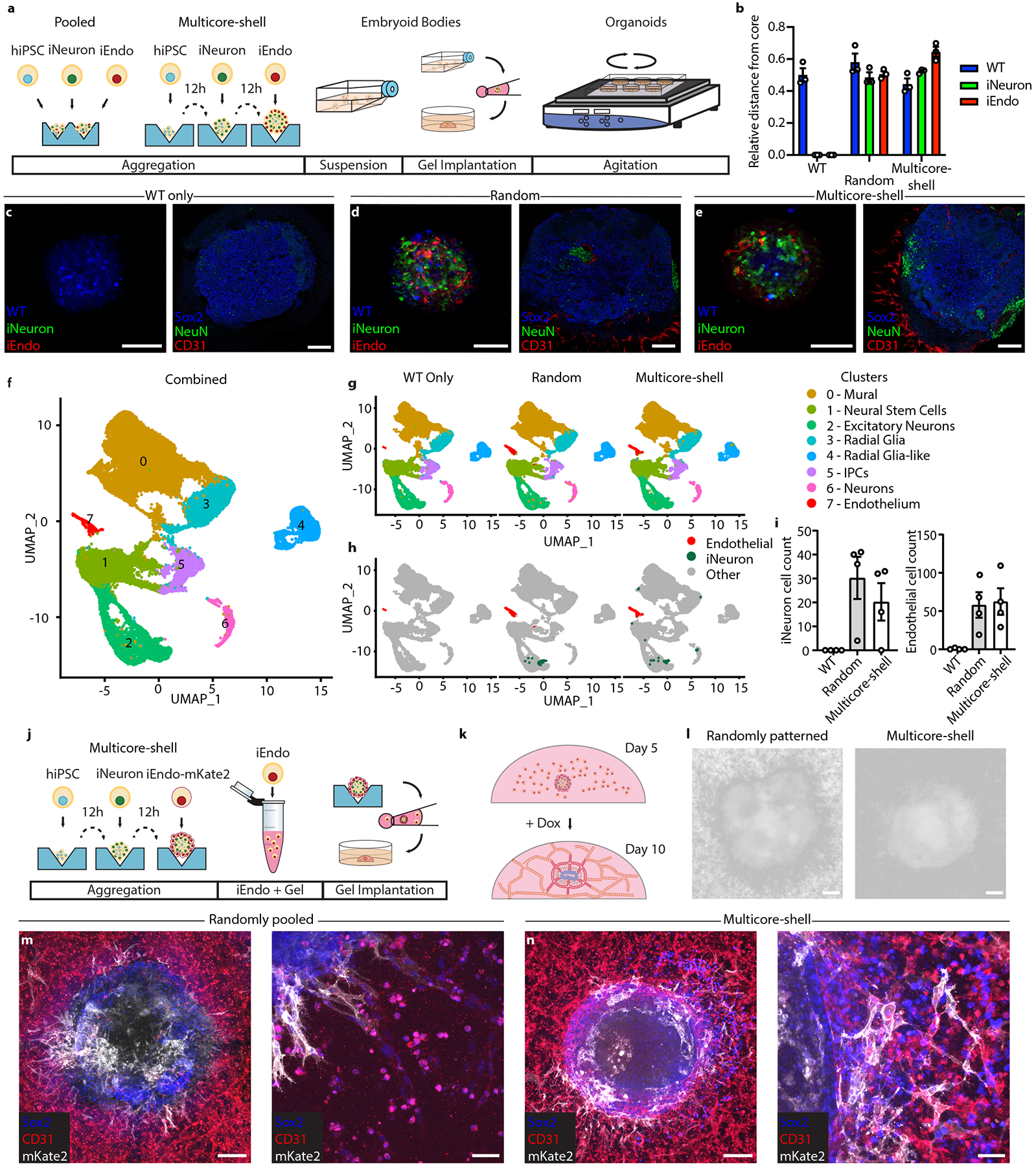
a, Schematic of the pooled and multicore-shell organoid formation protocols. b, Quantification of hiPSCs seeding distribution of WT, iEndo and iNeurons in WT-only, pooled and multicore-shell approaches. Data represent the mean ± s.e.m. (n = 3, from 3 independent batches). c, WT-only organoids. Left: fluorescence image of CellTracker-labelled WT-only organoids cultured for 1 d; right: immunostaining of SOX2, NeuN and CD31 of WT-only organoids cultured for 10 d. d, randomly pooled organOIDs. Left: fluorescence image of CellTracker-labelled randomly pooled organoids for 1 d. right: immunostaining of SOX2, NeuN and CD31 of randomly pooled organOIDs cultured for 10 d. e, Multicore-shell organOIDs. Left: fluorescence image of CellTracker-labelled multicore-shell organOID cultured for 1 d. right: immunostaining of SOX2, NeuN and CD31of multicore-shell organOIDs cultured for 10 d. f, uniform manifold approximation and projection (UMAP) plot of combined cellular composition of organoids for 25 d. g, UMAP plots of WT, random and multicore-shell organoids. h, UMAP plots highlighting endothelial and barcoded iNeuron populations. i, Left: total number of barcoded iNeurons for WT, random and multicore-shell organoids. right: total number of cells in cluster 7 for WT, random and multicore-shell organoids. Data represent mean ± s.e.m. (n = 4, from 2 independent batches). j, Schematic of embedding organoids in vascularized gels. k, Schematic of the organoid gel vascularization process. l, Brightfield image of randomly pooled (left) and multicore-shell (right) organoid embedded in a vascularized gel. m, Immunofluorescent images of SOX2, CD31 and mKate2 in randomly pooled organoids. Left: whole organoid view. right: high-magnification image of gel–organoid vascular anastomosis. n, Immunofluorescent images of SOX2, CD31 and mKate2 in multicore-shell organoids. Left: whole organoid view. right: high-magnification image of gel–organoid vascular anastomosis. Scale bars: 200 μm in l, m (left), and n (left); 100 μm in c–e; 50 μm in m (right) and n (right).
We next employed single-cell RNA sequencing (scRNA-seq) to investigate the cellular composition of day 25 organoids using the 10x Genomics pipeline (Fig. 4f,g). To annotate the clusters, we employed the quantitative classification tool SingleCellNet41 (Supplementary Fig. 10) to compare primary brain tissue42 to our single-cell datasets and plotted the top 10 most significantly differentially expressed genes in each cluster for validation (Supplementary Fig. 11). We found that hiPSCs without a dox-inducible transgene (WT cells) differentiated into clusters of neural stem cells (cluster 1), excitatory/inhibitory neurons (cluster 2), radial glia (clusters 3 and 4), intermediate progenitor cells (cluster 5) and neurons (cluster 6) (Fig. 4g). Cluster 0 is classified as cells of a non-neural lineage, with significant upregulation of mural cell markers such as collagen type III alpha 1 chain (COL3A1) and caldesmon 1 (CALD1)43, which is consistent with previous observations of migratory stromal cells emerging from cerebral organoids that are embedded in Matrigel15. Endothelial cells (cluster 7) were nearly absent from WT-only organoids, but notably increased in both random and multicore-shell organOIDs as measured by total cell number (Fig. 4h,i and Supplementary Fig. 12a,b). Furthermore, the cells in cluster 7 exhibited significant upregulation of endothelial markers compared with the clusters that exhibited some mesodermal lineage markers, clusters 0 and 3, including CD31 and CDH5 (encoding VEcad) that are significantly enriched with respect to other clusters, strongly suggesting that cluster 7 is endothelium derived from iEndo cells (Supplementary Fig. 13a). Finally, we investigated the identity of iNeurons in organOID culture. To ensure accurate tracking of neuronal-like cells, we placed a barcode on the 3’ end of the transgene cassette. We found that iNeurons are closest transcriptionally to mature neurons (Supplementary Fig. 13b), indicating that they can be used to increase neuronal populations within early developing organoids. While the majority of barcoded iNeuron cells exhibited a neuronal phenotype, a minority of these cells differentiated into non-neuronal phenotypes. This observation is consistent with the lower efficiency (~75%) of iNeuron cell differentiation into neurons in 2D culture, and whether this minority of cells are similarly differentiating to WT cells warrants future study (Fig. 2l).
We then used our scRNA-seq data to compare the cell populations found in WT-only, randomly pooled and multicore-shell organoids. Differential gene analysis on a cluster-by-cluster basis among all three organoids reveals that gene expression remains consistent for all clusters across all conditions and replicates (Supplementary Figs. 12b and 14–16). We found 33 genes with statistically significant differences in expression between randomly pooled and multicore-shell organOIDs across all genes. Specifically, only 7 genes were significantly differentially expressed in random and multicore-shell organOIDs among clusters 0–5. Gene ontology analysis of the remaining 26 differentially expressed genes in clusters 6 and 7 of randomly pooled and multicore-shell organoids shows an enrichment of genes associated with cell division in the neuronal cluster 6 and an enrichment of genes associated with cell migration in the endothelial cluster 7 (Supplementary Fig. 17). These findings suggest that these inducible cell lines can be added into existing cortical organoid protocols to generate additional specific cell types without major alterations to existing protocols. Hence, OID can be used to enhance those protocols by programming cell fate via deterministic design principles.
To demonstrate applications towards in vitro vascularization of organoids, we embedded randomly pooled and multicore-shell organoids in endothelial cell-laden gels (Fig. 4j,k). After 4 d of culture, an extensive microvascular network formed between the nascent vasculature within the gel and organoids (Fig. 4l). When embedded in gel, randomly pooled organoids became disorganized, losing their originally well-defined (organoid–gel) boundary. By contrast, multicore-shell organoids retained a clear (organoid–gel) boundary, indicating that their patterned architecture leads to improved integrity. Immunostaining of both randomly pooled and multicore-shell organoids reveals that the iEndo-mKate2+ vasculature within these organoids undergoes anastomosis with the microvessels in the gel (Fig. 4m,n). Looking ahead, we plan to build upon these observations to create vascularized cerebral organoids that are perfusable.
Programmable 3D neural tissues via bioprinting.
Most bioprinting methods produce multicellular tissues by printing inks composed of different primary human cells suspended in hydrogel matrices. Recently, human mesenchymal stem cell (hMSC)44 and hiPSC12,45–48-laden inks have been printed and differentiated in situ to form 3D tissue constructs. However, these stem cell-derived tissues contained cell types defined solely by media cues. By leveraging the proliferative capacity of hiPSCs along with the programmability afforded by our OID method, single and patterned multicellular 3D human tissues can be created with high cell densities. As a simple demonstration, we first created concentrated, matrix-free hiPSC bioinks by pelleting single-cell suspensions of hiPSCs and then directly printing these pluripotent inks onto transwell membranes (Fig. 5a–c and Supplementary Video 3). The printed filamentary features, which contained a remarkably high cellular density exceeding 5 × 108 Oct4+ cells per ml (Fig. 5d), were subsequently encased in a gelatin-fibrinogen extracellular matrix to support the tissue and enable 3D cell migration. Through calcein-AM/ethidium homodimer-2 live/dead assays of matrix-free hiPSC inks printed through 50 μm and 100 μm tapered nozzles at speeds of up to 20 mm s−1, we confirmed high cell viability akin to that observed for casted (control) tissues (Fig. 5e). We observed improved cell viability at higher print speeds, suggesting that shear forces have little effect over the range of printing conditions explored. We attribute the reduced cell viability observed at lower printing speeds to water evaporation from the printed cell-laden inks before encasing them within the gelatin-fibrinogen (gelbrin) extracellular matrix. The pluripotent filaments were printed with a high resolution, with widths measuring 132 μm and 182 μm (full width, half-maximum (FWHM)) for bioinks printed using 50 μm and 100 μm nozzles, respectively (Fig. 5f). Next, we printed planar patterns in the form of third-order pseudo-Hilbert curves composed of filamentary features produced from WT, iEndo or iNeuron inks (Fig. 5g,h). Printed WT filaments exhibited a slight compaction when differentiated in NIM containing doxycycline, forming a neuroectoderm filament with small NCAD+ ventricle-like structures or rosettes punctuating its length. By contrast, printed and differentiated iEndo filaments exhibited vasculo-genesis, resulting in the formation of a microvascular network over time. Finally, printed iNeuron filaments differentiated into densely packed neurons (NeuN+ cells) that formed a pervasive network of protruding Tuj1+ neurites (Supplementary Video 4).
Fig. 5 |. Multicellular neural tissues via 3D bioprinting coupled with OiD.
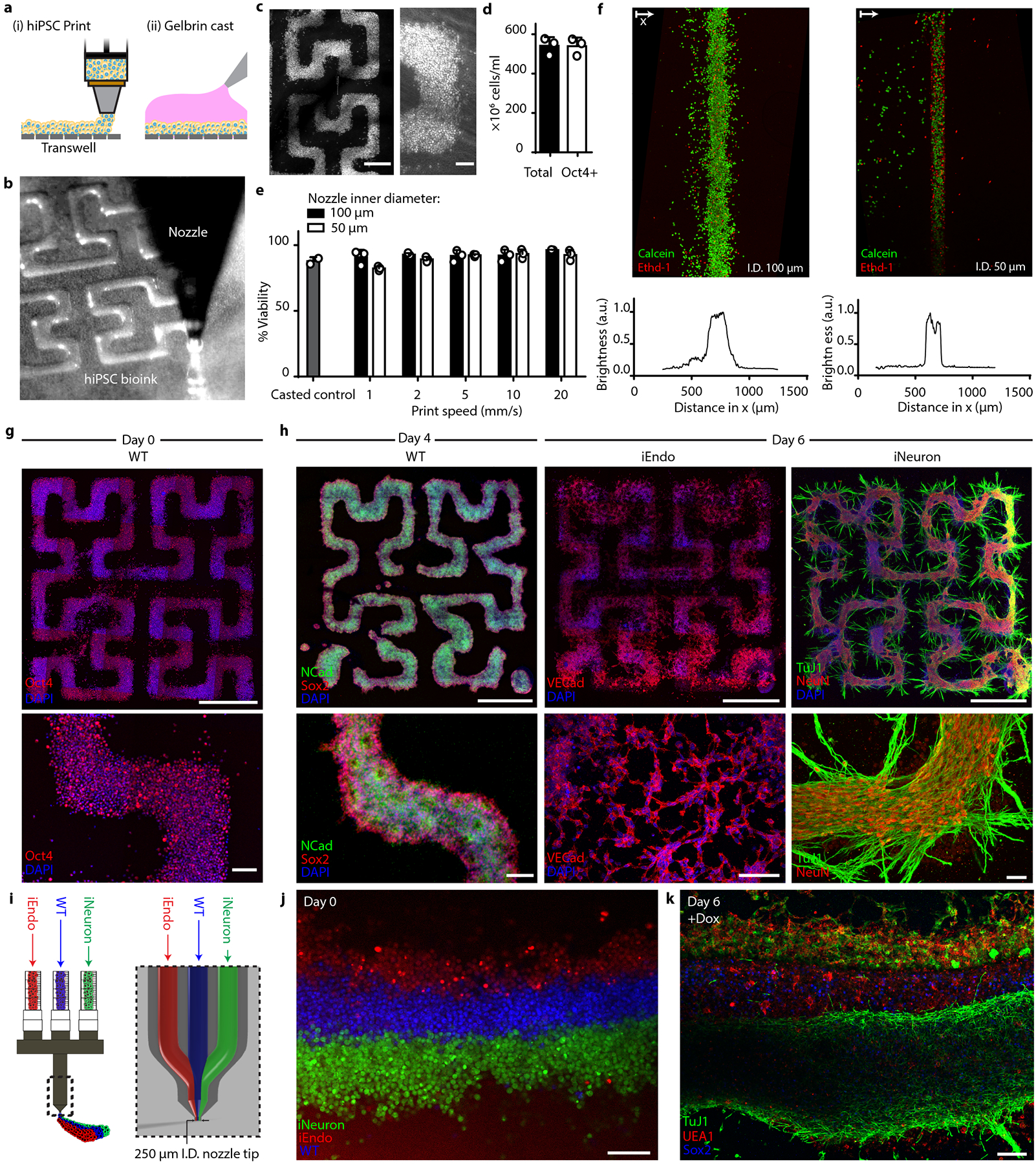
a, Schematic of the bioprinting and gel casting process. b, Image of bioink extrusion through the 50 μm nozzle during the bioprinting process. c, Brightfield images of bioprinted filamentary features. Left: overview image of the printed pattern. right: closeup image of the cell-dense printed pattern. d, Total cell number and flow cytometry quantification of Oct4+ cells within bioprinted filaments immediately after printing. Data represent mean ± s.d. (n = 3, from 3 independent cell ink batches). e, hiPSC viability within bioprinted filaments at different speeds and casted (control) samples, as measured using calcein-AM/ethidium homodimer live/dead assays. Data represent mean ± s.e.m. (n = 3, from 3 independent batches). f, Live/dead staining of bioprinted filaments produced using nozzle diameters of 100 μm (top, left) and 50 μm (top, right) and the corresponding distributions (bottom) of their fluorescence intensity. g, Immunostaining of Oct4 in patterns fixed on day 0, immediately after printing. h, Bioprinted tissue architectures cultured in NIM. Top: overview images of printed patterns. Bottom: close-up images of hiPSC differentiation. Left: immunostaining of NCAD and SOX2 for WT printed patterns at day 4. Middle: immunostaining of VEcad for iEndo printed patterns at day 6. right: immunostaining of Tuj1 and NeuN of iNeuron printed patterns at day 6. i, Design of tri-material nozzle for multimaterial bioprinting. j, Fluorescence image of bioprinted CellTracker-labelled WT, iEndo and iNeuron inks. k, Immunostaining of Tuj1, UEA1 and SOX2 of 3D-printed multicellular tissues at day 6. Scale bars: 1000 μm in g (top) and h (top); 500 μm in c (left); 100 μm in c (right), g (bottom) and h (bottom), j and k.
To understand tissue evolution during OID, we printed multicellular tissues composed of fluorescently labelled WT and iEndo cells that initially contained a well-defined boundary between the two cell types (Supplementary Fig. 18a). Time-lapse imaging reveals that this boundary evolved over time, with a notable bulk expansion of the WT neural stem cells that grew into the iEndo cell-rich region. Concomitantly, the iEndo cells protruded into the WT cell-rich region (Supplementary Fig. 18b–e). Consequently, the border between the two cell regions widened by nearly 3-fold over the course of 6 d (Supplementary Fig. 18f). The observed changes in printed tissue morphology are akin to those found in patterned organoids, where WT hiPSCs develop into neural stem cells and expand in bulk, while iEndo-derived vasculature forms protrusions throughout the tissue and surrounding environment. This seemingly contrasts with our scRNA-seq data that indicate a down-regulation of migration-associated genes when iEndo cells are programmably patterned versus randomly distributed among WT cells (Supplementary Fig. 17b). Future studies, such as single-cell tracking, could elucidate the distinct roles that cellular migration and tissue compaction have on the evolution of printed organOID architectures.
As a final demonstration, we created multicellular tissue architectures by co-printing three bioinks composed of WT, iEndo and iNeuron hiPSCs (Fig. 5i and Supplementary Fig. 19). The printhead contained three independent ink delivery channels that merged shortly upstream of the 250-μm-wide nozzle. Under laminar flow, these three inks converged to form tri-layer filaments that exit the nozzle. Using fluorescent dye-labelled WT, iEndo and iNeuron hiPSCs, we showed that this multimaterial printhead can generate multicellular pluripotent filaments in layered architectures with high fidelity, with a filament width of 269 μm (FWHM) (Fig. 5j). After printing, we added doxycycline to induce the simultaneous differentiation of neural stem cells, vascular endothelium and neurons, and observed that the layered architecture was preserved over 6 d of culture. (Fig. 5k). Finally, using this integrated OID-based bioprinting method, we recapitulated the geometry of a developing human dorsal forebrain coronal section by co-printing densely cellular WT and iNeuron hiPSC inks (Supplementary Fig. 20a–c and Video 5). After printing, the printed inks underwent OID to form a neuroectoderm and overlying neuron-dense layer (Supplementary Fig. 20d). As new protocols for deriving specific neuronal cell types emerge, OID will enable patterning of multilayer cerebral architectures from an even broader array of inducible-TF hiPSCs.
Discussion
We have shown an orthogonally induced differentiation method for rapidly programming and patterning human stem cells, organOIDs, and bioprinted organ-specific tissues within days. We found that the overexpression of ETV2 and NGN1 efficiently overrides a broad range of media cues, enabling the simultaneous generation of vascular endothelium and neurons in a one-pot system. Importantly, in co-culture, OID-derived cells retain their pre-programmed identity in the presence of other differentiating cell types. Leveraging this capability, we created both vascularized and multicore-shell cortical organOIDs from pooled and patterned embryoid bodies, respectively, and showed that neural self-assembly processes can be preserved by spatially patterning orthogonal cell types. Through multimaterial 3D bioprinting of matrix-free wild type and inducible-TF hiPSC inks, we generated programmable, multicellular neural tissues in layered architectures that can be differentiated on demand.
The discovery of additional TFs that can efficiently reprogramme cells to specific cell types, such as NK3 Homeobox 1 (NKX3–1) for fibroblasts and SOX9 for oligodendrocytes29, will expand the library of cells that can be generated via OID. With further advancement of stem cell differentiation protocols, it may soon be possible to efficiently and scalably produce the multitude of cells present in the human body. Looking ahead, this strategy offers a facile route for creating many types of programmable organOIDs and organ-specific tissues of interest for drug screening, disease modelling and therapeutic applications.
Methods
Cell culture.
PGP1 hiPSCs were utilized for this study (Coriell, GM23338). Cells were verified for pluripotency by flow cytometry and cultured between passages 20 and 50. PGP1s were cultured and passaged without antibiotics in mTeSR1 medium (STEMCELL Technologies, 05850) on tissue-culture plates coated with human embryonic stem cell-qualified Matrigel (Corning, 354277) during inducible cell line generation, or with growth factor-reduced Matrigel (Corning 354230), for growing hiPSCs before differentiation, organoid formation or bioprinting. For passaging, hiPSCs were washed with phosphate buffered saline (PBS) without calcium and magnesium (Gibco, 14190250), dissociated using TrypLE Express (Life Technologies, 12604013), and then seeded at 300,000 cells per well in a 6-well plate supplemented with 10 μM Y-27632 (Selleck Chemicals, S1049) for 1 d, and subsequently maintained in mTeSR1 with daily media changes. For cryo-storage, cells were dissociated with TrypLE Express, counted and resuspended in mFreSR (STEMCELL Technologies, 05854) at a concentration of 106 cells per ml (STEMCELL Technologies, 5854) using a CoolCell LX freezing block (Biocision, BCS-405) overnight at −80 °C, then stored in liquid nitrogen for long-term storage.
TF-induced clonal hiPSC cell lines.
A Gateway compatible, doxycycline-inducible (Tet-On), PiggyBac vector with a puromycin resistance gene was provided by the Church Lab. pDONR221-NEUROG1 (HsCD00040492) from the DNASU49,50 repository was cloned into a barcoded version of PBAN001 using a Gateway reaction with Gateway LR Clonase II Enzyme mix (Invitrogen, 12538200) with a 20 bp barcode:5’ CAAAGTGAAACCAGAGTCGC 3’. The construct was delivered into PGP1 cells via Super PiggyBac Transposase Expression vector (SBI, PB210PA-1) by nucleofection at a mass ratio of 1:4 (PBAN-NGN1):(SPB) in 2 μl of total reaction volume, utilizing the Lonza 4D Nucleofector X-Unit (Lonza, AAF-1002X) with a 20 μl P3 solution kit (Lonza, V4XP-3032) with 600,000 WT cells in each 20 μl reaction well as per manufacturer’s instructions. After nucleofection, cells in solution were plated on a 6-well tissue-culture treated plate coated with Matrigel, and subsequently cultured in mTeSR1 supplemented with 10 μM Y-27632 for 1 d. The media was changed daily to mTeSR1 until cells reached 70% confluency. Then, nucleofected cells were selected for by addition of 1 μg ml−1 puromycin (Gibco, A1113803) cells with 5 μM Y-27632 for 1 d. Cells were washed once using PBS without calcium or magnesium and maintained in mTeSR1 supplemented with 10 μM Y-27632 until colonies were visible.
Confluent polyclonal iNeuron colonies were then dissociated with TrypLE for 7 min, centrifuged at 300 g for 5 min, resuspended in 500 μl mTeSR1 supplemented with 10 μM Y-27632 and penicillin–streptomycin (MilliporeSigma, P4333), strained through a 35 μm cell strainer (Falcon, 352235) and immediately placed on ice. We used the SH800S Sony cell sorter (100 μm chip nozzle) to sort single cells into Matrigel-coated 96-well plates, with each well containing 150 μl mTeSR1 supplemented with CloneR (STEMCELL Technologies, 05888). Following the manufacturer’s instructions, we maintained the cells until confluent and seeded candidate colonies into two 48-well plate wells, one for validation through induction via addition of 500 ng ml−1 doxycycline hyclate (MilliporeSigma, D9891) and the other for further expansion/passaging. iEndo cells, a clonal PGP1 ETV2 isoform-2 line, was constructed in a manner similar to the iNeurons using the TFome transcription factor library29.
To engineer fluorescently labelled hiPSC lines iEndo-mKate2 and eGFP-WT hiPSCs, the FUGW plasmid (Addgene, 14883), which encodes constitutively expressed GFP, was modified to express membrane-bound mKate2 (FUmemKW). These two plasmids were packaged into lentiviruses as described previously29, and FUGW and FUmemKW lentiviruses were transduced into WT PGP1 and iEndo hiPSCs to generate green and red cell lines, respectively. hiPSCs were sorted on a FACS Aria to obtain a pure population of cells that express the fluorescent proteins uniformly.
iNeuron-sfGFP line generation.
sfGFP plasmid51 gifted by David Thompson from the Church Lab was cloned into a pLenti CMV Puro DEST (Addgene, 17452) vector using LR Clonase II and transformed into DH5alpha cells (NEB, C2987H). Purified pLenti-sfGFP plasmid DNA (37.5 ug) was diluted into 1 ml Opti-MEM I (ThermoFisher, 319850672) with lentivirus pCMV-VSV-G envelope (Addgene, 8454) and psPAX2 packaging plasmid (Addgene, 12260) in a 3:3:1 mass ratio. FuGene6 (56.25 μl; Promega, E2691) was added to the mixture and incubated for 20 min at r.t. The transfection mix was then diluted in 15 ml antibiotic-free media consisting of Dulbecco’s modified Eagle medium (DMEM), high glucose (Gibco, 11965118), 10% fetal bovine serum, 0.1 mM minimal essential media non-essential amino acids (MEM-NEAA) (Gibco, 11140050), 6 mM l-glutamine, and 1 mM sodium pyruvate, and added to a T75 flask that was plated 1 d before transfection with 5.5 million human embryonic kidney (HEK) 293T cells. After 3 d, media with packaged lentivirus were collected and concentrated 50× using Lenti-X concentrator (Takara Bio, 631231), aliquoted and frozen at −80 °C. Single-cell clones of PGP1-SV NGN1 were subjected to transduction by packaged lentivirus for 1 d. Cells were grown until confluent, flow-sorted in a Sony SH800S cell sorter for the top 10% of sfGFP+ cells, expanded and frozen.
Two-dimensional OID assays.
To study OID in neural-specifying conditions, 300,000 WT PGP1, iEndo-PGP1 or iNeuron-PGP1 cells were seeded in each well of Matrigel-coated 6-well plates in NIM, consisting of 1:1 DMEM/Ham’s F-12 (DMEM/F12) with GlutaMAX (Gibco, 10565018) supplemented with 1:100 N2 supplement (Gibco, 17502048), 1:100 MEM-NEAA, 1 μg ml−1 heparin (Sigma, H3149), 5 μM SB431542 (BioGems, 3014193) and 100 nM LDN193189 (BioGems, 1066208), with or without 500 ng ml−1 doxycycline hyclate. NIM was further supplemented with 10 μM Y-27632 at the time of seeding. At 24 h after plating, the medium was changed to NIM with or without doxycycline. The medium was changed every 48 h for 6 d of culture.
To study OID in endothelial specifying conditions, WT or iNeuron-PGP1 cells were grown to 60–70% confluency in mTeSR1, at which point cells were rinsed once in PBS and the medium was changed to RPMI 1640 (Gibco, 11875093) supplemented with 1:50 B27 minus insulin (Gibco, A1895601) and 6 μM CHIR99021 (BioGems, 2520691), with or without 500 ng ml−1 doxycycline, for 2 d. On day 2, the medium was changed to RPMI 1640 with 1:50 B27 minus insulin and 3 μM CHIR99021. On day 4, the medium was changed to endothelial growth media-2 (EGM-2) (PromoCell, C-22111) with 10 μM SB431542, 50 ng ml−1 vascular endothelial growth factor (VEGF) (PeproTech, 100–20) and 25 ng ml−1 fibroblast growth factor 2 (FGF2) (PeproTech, 100–18B), with or without 500 ng ml−1 doxycycline. The medium was changed again on day 6 and cells were kept in culture until day 8.
Fluorescent time-lapse imaging during stem cell differentiation.
Cold Matrigel (500 μl) was added to a single well in a 6-well tissue-culture treated plate and allowed to gel by warming to 37 °C for 30 min. WT-eGFP PGP1 cells (150,000) and iEndo-mKate2 PGP1 cells (150,000) were seeded in mTeSR1 supplemented with Y-27632 and 500 ng ml−1 doxycycline and cultured for 1 d. On day 2 of culture, the mTeSR1 medium was removed and the plate was gently rinsed with PBS before replacing with NIM, with 500 ng ml−1 doxycycline hyclate added. The plate was transferred to an inverted fluorescence microscope (Zeiss Axio Observer Z1) with an environmental chamber (CO2 and temperature controlled) and imaged through a ×10 objective onto a Photometrics Evolve electron-multiplying charge-coupled device (EMCCD), tiling over a 4 × 4 grid every 30 min. The medium was changed daily and images were captured over a 4 d period. Tiled images were stitched automatically in Zeiss’ Zen Blue software with a 5% tile overlap and processed using ImageJ.
Flow cytometry.
For intracellular and surface marker staining, cells were dissociated using 0.05% trypsin-ethylenediaminetetraacetic acid (EDTA) (Gibco, 25300054), fixed for 15 min in BD Cytofix (BD, 554714) and washed 3× in PBS with 3% w/v bovine serum albumin (BSA) (MilliporeSigma, A305). Samples were then aliquoted in a 96-well plate at 5 × 105 cells per well, permeabilized in BD Perm/Wash (BD, 554723) solution for 15 min, pelleted via centrifugation at 300 g, and the supernatant was aspirated. Samples were incubated for 45 min in antibodies diluted in BD Perm/Wash at a concentration of 107 cells per ml per dilution in the dark and washed 3× in BD Perm/Wash before flow cytometry experiment was performed. Colour compensation was performed on each run utilizing BD anti-mouse immunoglobulin (BD, 552843) and fluorescently conjugated antibodies. All flow cytometry measurements were performed on the BD LSRFortessa cell analyser. Flow files (.fcs) were processed in FlowJo 10.6.1 (gating strategy included in Supplementary Fig. 1). Plots were generated with Prism 8.4.0. Fluorescently conjugated antibodies utilized in flow cytometry assays are listed in Supplementary Table 1.
RNA sequencing of 2D OID co-culture assays.
To study the effect of co-differentiation on OID-derived endothelium, a mixture of 150,000 iEndo-mKate2 PGP1 cells with 150,000 cells of either iEndo-mKate2 PGP1, WT PGP1 or iNeuron PGP1 was seeded onto Matrigel-coated plates in NIM supplemented with 10 μM Y-27632 at the time of seeding. At 24 h after seeding and every 48 h thereafter, the medium was changed to NIM without additional supplements. To study the effect of co-differentiation on OID-derived endothelium, a mixture of 150,000 iNeuron-sfGFP PGP1 cells with 150,000 cells of either iNeuron-sfGFP PGP1, WT PGP1 or iEndo PGP1 was seeded onto Matrigel-coated plates in NIM supplemented with 10 μM Y-27632 at the time of seeding. At 24 h after seeding and every 48 h thereafter, the medium was changed to NIM without additional supplements.
At 5 d after seeding, cells were dissociated in 0.05% trypsin-EDTA and single-cell-filtered. Flow cytometry sorting of mKate2+ ETV2 cells and, separately, SFGFP+ iNeuron cells was performed on BD FACSAria II. Gating strategies are noted in Supplementary Fig. 4. RNA extraction was performed utilizing the Ambion PureLink RNA mini kit. Extracted RNA samples were validated utilizing an Agilent 2200 TapeStation for RNA integrity number (RIN) score quantification, with all samples utilized validated to be above 8.7. Library preparation was performed utilizing low-input RNA kit (Takara, 634894) and samples were sequenced on a Novaseq 6000 SP flow cell with 2 × 50 bp read length, with a target total read count of ~20 million reads per sample. Reads from the two lanes were merged and pseudocount alignment was performed in Salmon 1.4.0 with the GRCh38 GENCODE v38 reference genome. Differential gene expression analysis was performed utilizing the DESeq2 package in R version 4.1.0. To account for sorting error, documented neuron-elevated genes from the Human Protein Atlas were removed from all endothelial co-culture analysis. Similarly, endothelial-elevated genes from the Human Protein Atlas were removed from all iNeuron related analyses. Gene ontology analysis was performed utilizing the ClusterProfiler R package.
HUVEC-hiPSC aggregate cohesion assay.
HUVEC-RFP cells (Angio-Proteomie, cAP-0001RFP), WT-eGFP PGP1 cells and iEndo-mKate2 PGP1 cells were dissociated from 70–80% confluency by incubating cells in Gentle Cell Dissociation Reagent (STEMCELL Technologies, 07174) for 12 min at 37 °C, then resuspended in DMEM/F12 with 2-(4-(2-Hydroxyethyl)piperazin-1-yl) ethane-1-sulfonic acid (HEPES) (Gibco, 11330032). Cells were then centrifuged at 250 g for 5 min before resuspending and single-cell filtering in either EB culture medium (EBCM) consisting of mTeSR1 supplemented with 4 mg ml−1 polyvinyl alcohol (PVA, MilliporeSigma, P8136) for 1:1 co-cultures of iEndo-mKate2 cells and WT-eGFP cells, or a 1:1 mix of EBCM and EGM-2 for 1:1 co-cultures of HUVEC-RFPs:WT-eGFPs. The PVA stock solution was prepared by fully dissolving PVA in stirred deionized water at 90 °C to a stock concentration of 200 mg ml−1. Wells of a 24-well AggreWell 400 plate (STEMCELL Technologies, 34411) were seeded with either 1:1 iEndo-mKate2:WT-eGFP cells in EBCM with 10 μM Y-27632, or 1:1 HUVEC-RFP cells:WT-eGFP cells in a 1:1 mix of EBCM and EGM-2, with 10 μM Y-27632. At 24 h after plating, media in both conditions were changed to EBCM without Y-27632. At 48 h after plating, aggregates were transferred to a suspension culture in non-adherent T25 flasks in EBCM on an orbital shaker rotating at 53 r.p.m. Aggregates were imaged 24 h and 48 h after plating on a Zeiss LSM710 confocal microscope.
Vascularized cortical organoid culture.
Cortical organoid culture was adapted from previously established protocols13,52,53. EBs were formed on day –3 by dissociating hiPSC monolayers at 70–80% confluency with EDTA dissociation reagent (EDR) for 12 min at 37 °C and 5% CO2, EDR consisting of 0.5 mM EDTA in PBS supplemented with an additional 0.03 M NaCl. Cells were then resuspended in DMEM/F12 with HEPES and centrifuged at 250 g for 5 min. Wells of a 24-well AggreWell 800 plate (STEMCELL Technologies, 34815) were treated with an anti-adhesive coating of 0.2% Pluronic F-127 in deionized water and seeded with 1.5 × 106 cells per well using either WT PGP1 hiPSCs or a 1:2 ratio of iEndo:WT PGP1 hiPSCs in EBCM supplemented with 10 μM Y-27632. The day after aggregation (day –2), the EBCM medium was replaced with fresh EBCM to remove Y-27632 within the AggreWell 800 plates. On day –1, EBs were transferred into suspension culture in fresh EBCM in non-adherent T25 flasks on an orbital shaker rotating at 53 r.p.m.
Neural differentiation began on day 0 (after 1 d of suspension culture). EBs were transferred to NIM with doxycycline. After 3 d in NIM, organoids were transferred into 80 μl cold collagen/Matrigel gel droplets formed on dimpled parafilm, which was prepared similarly to a previously established protocol44. Final collagen and Matrigel concentrations were 4 mg ml−1 rat tail collagen type I (Corning, 354249) and 25% growth factor-reduced Matrigel, respectively. Gel droplets containing organoids were gelled at 37 °C for 15 min before being transferred back into NIM in ultra-low-adherence 6-well plates (Corning, 3471), which were held stationary. At 3 d after implanting organoids into gels (6 d after the start of neural induction), organoids were transferred onto an orbital shaker rotating at 90 r.p.m. and the medium was changed to neural differentiation media 1 (NDM1), consisting of a 1:1 mix of DMEM/F12 with GlutaMAX and Neurobasal media (Gibco, 21103049) supplemented with 1:200 GlutaMAX (Gibco, 35050061), 1:200 MEM-NEAA, 1:200 N2 supplement, 1:100 B27 supplement without vitamin A (Gibco, 12587010), 1:4,000 insulin (MilliporeSigma, I9278), 10 ng ml−1 VEGF, 20 ng ml−1 epidermal growth factor (EGF) (PeproTech, AF-100–15), 20 ng ml−1 FGF2, 50 μM β-mercaptoethanol (MilliporeSigma, M6250) and 500 ng ml−1 doxycycline. After 4 d, half the medium was replaced with fresh NDM1. At 13 d after the start of neural induction, a full medium change was performed to replace all media with neural differentiation media 2 (NDM2), consisting of a 1:1 mix of DMEM/F12 with GlutaMAX and Neurobasal media supplemented with 1:200 GlutaMAX, 1:200 MEM-NEAA, 1:200 N2 supplement, 1:100 B27 supplement (Gibco, 17504044), 1:4,000 insulin, 10 ng ml−1 VEGF, 20 ng ml−1 EGF, 20 ng ml−1 FGF2, 50 μM β-mercaptoethanol and 500 ng ml−1 doxycycline. Half media changes of NDM2 were performed every 4 d until 25 d after neural induction. At 25 d after the start of neural induction, a full medium change was performed to replace all media with NDM3, consisting of a 1:1 mix of DMEM/F12 with GlutaMAX and Neurobasal media supplemented with 1:200 GlutaMAX, 1:200 MEM-NEAA, 1:200 N2 supplement, 1:100 B27 supplement with vitamin A (Gibco, 12587010), 1:4,000 insulin, 10 ng ml−1 VEGF, 20 ng ml−1 brain derived neurotrophic factor (BDNF) (PeproTech, 450–02), 50 μM β-mercaptoethanol and 500 ng ml−1 doxycycline. Half media changes of NDM3 were performed as needed every 1–4 d for the duration of organoid culture. Vascularized cortical organoids were cultured for up to 45 d.
Multicore-shell cortical organoid culture.
WT PGP1 hiPSCs were dissociated using EDR and seeded into 0.2% Pluronic F-127 treated V-bottom 96-well plates (Corning, 3894) at a density of 1,000 cells per 96-well well in EBCM + 10 μM Y-27632 for 12 h to generate organoid cores. After 12 h, iNeurons were dissociated using EDR, added to each EB at an additional 2,000 cells per 96-well well and allowed to aggregate for 12 h in EBCM with 10 μM Y-27632. Then, iEndo cells were dissociated using EDR and an additional 2,000 cells were added to each EB and allowed to aggregate over 12 h in EBCM + 10 μM Y-27632. At 12 h after the final aggregation, the medium was fully replaced with EBCM. Neural induction was initiated 48 h after the start of aggregation by changing the medium to NIM within the 96-well plate. At 24 h after initiating neural induction, organoids were collected from the 96-well plate and implanted into 80 μl collagen/Matrigel gel droplets. Multicore-shell organoids were then cultured using the vascularized cortical organoid protocol.
Cryo-sectioning and immunostaining.
Organoids were fixed in 4% paraformaldehyde for 30 min and rinsed 3× in PBS. For cryo-sections, fixed organoids were incubated for 2 d at 4 °C in PBS containing 30% w/v sucrose and then transferred into a 1:1 solution of Optimal Cutting Temperature compound (OCT) (Tissue-Tek, 4583) and PBS containing 30% w/v sucrose for 90 min. Next, the tissue was placed into a cryostat tissue mould, which was subsequently filled with 100% OCT solution and frozen at −20 °C on a cryostat Peltier cooler. The tissue was sectioned using 40–60 μm slices and transferred onto a Superfrost Plus glass slide (VWR, 48311–703). Sections were stored at −20 °C before immunostaining.
For immunostaining of cryo-sections and whole cortical organoids, tissues were permeabilized for 30 min in PBS containing 0.1% Triton-X (MilliporeSigma, T8787), then blocked for >1 h in PBS containing 2% donkey serum. Next, tissue sections were incubated overnight and whole-mount organoids were incubated for 1–3 d in primary antibodies in PBS containing 2% donkey serum (MilliporeSigma, D9663). Tissues were rinsed 3× in PBS containing 0.05% Tween-20 (PBST) (MilliporeSigma, P9416), then incubated in secondary antibodies in PBS with 2% donkey serum for an equal amount of time as in primary antibodies. Cell nuclei were labelled with 300 nM of 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 10 min, followed by 3 rinses in PBST. Tissue sections and whole-mount organoids were imaged on a Zeiss LSM710 confocal microscope. All primary and secondary antibodies used in immunostaining are listed in Supplementary Table 2.
iDISCO+ tissue clearing.
Cortical organoids were cleared and immunolabelled using an adapted version of the immunolabelling-enabled three-dimensional imaging of solvent cleared organs (iDISCO+) protocol54. Briefly, the organoids were dehydrated using a methanol/water gradient over the course of 6 h, then delipidated using a 67% dichloromethane (DCM)/33% methanol solution for 3 h before bleaching in 5% hydrogen peroxide in methanol overnight. Next, they were rehydrated in a reverse methanol/water gradient over the course of 6 h before being incubated in PBS overnight. They were then immunolabelled using the same immunolabelling protocol described above and dehydrated a second time using a methanol/water gradient over 6 h before further delipidation in 67% DCM/33% methanol for 3 h. The organoids were then rinsed twice in 100% DCM before rehydration over 6 h with a reverse methanol/water gradient. Finally, they were index matched using EasyIndex (LifeCanvas Technologies EI-Z1001) and imaged using a Zeiss LSM710 confocal microscope. Three-dimensionsl renderings of cleared cortical organoids were made using the 3Dscript ImageJ plugin55.
Angiotool analysis of vascularized cortical organoids.
Confocal z-stacks were taken of iDISCO+-cleared WT-only and WT + iEndo organoids. Individual optical sections at z = 10.8, 119, 227, 335 and 442 μm within a single organoid were used for Angiotool analysis56. Optical sections were preprocessed by raising the lookup table threshold to 20 to eliminate background noise. Angiotool analysis was conducted using a vessel diameter of 5 and a small-particles filter of 40.
RNA extraction and RT-qPCR of cortical organoids.
Cortical organoids were cultured using protocols described above. RNA extraction was performed utilizing the Ambion PureLink RNA mini kit (Invitrogen, 12183025). Homogenization of single organoids suspended in Matrigel/collagen hydrogel droplets was performed utilizing a hand-held homogenizer (Bel-Art, F65000–0000) with accompanying RNAse-free, DNAse-free single-use pestles (Bel-Art, F65000–0006), with organoids suspended in 600 μl lysis buffer provided in the Ambion PureLink kit. On-column DNAse I (Invitrogen, AM2222) digestion was also performed during the RNA extraction process. Extracted RNA samples were validated utilizing an Agilent 2200 TapeStation for RIN score quantification, with all samples utilized in this study validated to be above 8.8. Complementary DNA (cDNA) synthesis was performed using the SuperScript IV First-Strand Synthesis System (Invitrogen, 18091050) with Oligo d(T) primer and with the input cDNA for each sample normalized to 10 ng μl−1 for a total of 110 ng per reaction. cDNA was validated with included kit controls by gel electrophoresis. RT-qPCR experiments were performed with IDT’s PrimeTime qPCR Probe Assays (ZEN/FAM), utilizing IDT’s PrimeTime Gene Expression Master Mix (IDT, 1055771). Curves were obtained on a BioRad CFX96 qPCR machine. Scripts for data processing of quantification cycle (Cq) values were created in Python and graphs were made utilizing Prism 8.4.0. A list of all primers and probes used is provided in Supplementary Table 3.
Single-cell sequencing of day 25 organoids.
WT hiPSC, randomly pooled and multicore-shell cortical organoids were cultured using the previously described methods until day 25. Dissociation was performed using a previously described protocol14. Cell encapsulation (gel bead-in emulsions (GEM) generation) was performed using the Chromium Next GEM Chip G Single Cell Kit (10x Genomics, PN-1000127) and accompanying Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.0 (10x Genomics, PN-1000128) on the 10x Chromium Controller (10x Genomics, PN-1000202), with a targeted recovery of 5,000 single cells per sample. Post GEM-reverse transcription cleanup and cDNA amplification were performed using the recommended protocols. cDNA quality control was performed on the Qiagen Qubit 3.0 platform using the Qubit double stranded DNA HS assay kit (ThermoFisher Scientific, Q32854). Quantification was performed using the Agilent High Sensitivity DNA Analysis Kit (Agilent, 5067–4626) on the Agilent 2100 Bioanalyzer Instrument (Agilent, G2939BA). A 3’ gene expression library was constructed, and samples were indexed using the Single Index Kit T Set A (10x Genomics, PN-1000213). Indexed samples were further validated for quality using Qubit and Bioanalyzer kits and normalization of concentration was performed using the manufacturer’s suggested methodology before pooling for submission for sequencing. The pooled library was quality controlled using qPCR and ran on a single lane of a NovaSeq S4 flow cell with v1.5 reagent kits and 2 × 150 bp, with a targeted depth of 50,000 reads per cell. Raw reads were processed using 10x Genomics’ CellRanger 5.0 software suite against the GRCh38 human reference genome.
In Seurat, cells with more than 3,000 unique molecular identifiers, 1,300 genes and less than 20% counts from mitochondria genes were used for further processing. Counts were normalized using SCTransform within each batch, followed by integration of two batches using canonical correlation analysis (CCA) and mutual nearest neighbours (MNNs) in Seurat. Cluster identification was performed using the R Package Seurat 4.0 ‘FindClusters’, UMAP analysis was performed by the ‘RunUMAP’ function, and UMAP plotting was performed by running the ‘Featureplot’ function. Differential expression analysis was performed on the basis of the Wilcoxon rank-sum test and the ‘FindMarkers’ function. The P values were adjusted for multiple testing using the Bonferroni correction. Gene ontology analysis was performed using the clusterProfiler 3.8.1 R package using the ‘enrichGO’ function and plotted using its ‘dotplot’ function.
Automated annotations using SingleCellNet.
Cluster generation was annotated using the SingleCellNet41 R package, with the training reference dataset being primary human brain samples GSE132672. The classifier was trained against the ‘subtype’ class detailed in their metadata, with the cells implied in subtype ‘outlier’ being removed for clarity. The classifier was utilized to suggest identities for clusters in 0.1 resolution.
CellTracker labelling of hiPSCs.
For CellTracker studies of aggregated organoids and printed tissue filaments, CellTracker fluorescent dye was reconstituted in DMSO, then diluted to the working concentration in mTeSR1. The hiPSCs were then incubated with dye for 30 min at 37 °C. CellTracker Green 5-chloromethylfluorescein diacetate CMFDA (2.5 μM; Molecular Probes, C7025) was used to label WT-PGP1 cells, 2.5 μM of CellTracker Orange 5-(and-6)-(((4-chloromethyl)benzoyl)amino)tetramethyl-rhodamine CMTMR (Molecular Probes, C2927) was used to label iNeuron cells, and 250 nM of CellTracker Deep Red (Molecular Probes, C34565) was used to label iEndo cells. In triple prints, WT-PGP1 cells were labelled with 5 μM 7-amino-4-chloromethylcoumarin (CellTracker Blue CMAC) (Molecular Probes, C2110), iNeuron-PGP1 cells were labelled with 2.5 μM 5-chloromethylfluorescein diacetate (CellTracker Green CMFDA) (Molecular Probes, C7025), and iEndo-PGP1 cells were labelled with 2.5 μM Chloromethyl 6-(4(5)-amino-2-carboxyphenyl)-1,2,2,4,8,10,10,11-octamethyl-1,2,10,11-tetrahydrodipyrido(3,2-b: 2,3-i) xanthylium (CellTracker Red CMTPX) (Molecular Probes, C34552). Cells were then washed with PBS without calcium or magnesium and dissociated for printing or organoid aggregation. Organoids were fixed 24 h after the final aggregation and bioprinted tissues were fixed 4 h after printing in 4% paraformaldehyde for 30 min.
Quantification of organoid patterning.
Organoids were mounted in EasyIndex solution and imaged on a Zeiss LSM710 confocal microscope. A custom MATLAB script was written to analyse CellTracker-labelled organoids. Each pixel was assigned to one of the three colours used in the CellTracker study on the basis of the fluorescence intensity and pictures were binarized using a maximum entropy thresholding function. Ref.57 provided the following formula to account for the focus shift that results from imaging into high refractive index media:
where AFP is the actual focus position (z position of the voxel), NFP is the nominal focus position (imaged z position of the voxel), and n1 and n2 are the refractive indices of air (n1 = 1) and EasyIndex (n2 = 1.47), respectively. EasyIndex is assumed to have minimal dispersion across visible wavelengths of light. The centre position of each organoid was identified, and the distance between each voxel and the centre position for all voxels was calculated. Distances were normalized to the radius of each organoid and were summed up to a histogram plot normalized to histogram surface area.
To measure the length of apical neuroepithelium in ventricles, organoid sections were stained with NCAD and SOX2, and imaged on a Zeiss LSM710 confocal microscope. NCAD positive regions surrounded by radially organized SOX2+ cells were considered to be ventricles and traced in ImageJ.
Vascular anastomosis in gels.
To make endothelial cell-laden gels, iEndo hiPSCs were dissociated from confluency with TrypLE Express and centrifuged at 250 g for 5 min. iEndo hiPSCs were then plated onto Matrigel-coated T75 flasks containing 15 ml EGM-2 supplemented with 10 μM Y-27632 and 500 ng ml−1 doxycycline to begin differentiation into endothelium. The next day, the medium was changed to 15 ml EGM-2 supplemented only with 500 ng ml−1 doxycycline. The medium was changed every 2 d thereafter until 8 d of culture. In parallel, randomly pooled and multicore-shell organoids were made as described above. At 8 d of culture, iEndo-derived endothelium was dissociated with TrypLE, diluted in DMEM/F12 and centrifuged at 250 g for 5 min. Endothelial cells were then resuspended in collagen/Matrigel gel at a concentration of 4 × 106 cells per ml at 4 °C. Simultaneously, 5-day-old randomly pooled and multicore-shell patterned organoids were collected and resuspended in endothelial cell-laden gel at a density of 24 organoids per ml. The organoid/endothelial cell-laden gel mixture (250 μl) was then plated onto a glass-bottom Petri dish and allowed to gel at 37 °C for 30 min. Afterwards, organoids were maintained in 2.5 ml NIM for 24 h before changing the medium to 2.5 ml NDM1. Organoids were maintained in NDM1 for 4 d, with media changes every 2 d, before fixation in 4% paraformaldehyde. Organoids were then stained and imaged on a Zeiss LSM980 confocal microscope.
Three-dimensional bioprinting.
A triple-material co-flow nozzle (printhead) was designed using Fusion360 (Autodesk) and exported as a stereolithography (.stl) file. The nozzles were printed using a stereolithography 3D printer (Perfactory Aureus, EnvisionTEC), using HTM140v2 resin (EnvisionTEC) with a layer height of 50 μm and a calibrated power of 700 mW. Printed nozzles were first rinsed and their internal channels were flushed using isopropyl alcohol. They were then dried under a stream of air and further cured under ultraviolet (UV) illumination using an Omnicure lamp (EXFO).
Densely cellular bioinks were created from one 90% confluent T225 flask of hiPSCs, which were loaded into a syringe before printing. These hiPSCs were first rinsed in PBS without calcium or magnesium, incubated in TrypLE for 7 min at 37 °C and 5% CO2, lifted off and added into 37 °C DMEM/F12 with HEPES, and centrifuged at 250 g for 5 min. After the supernatant was removed, the cells were resuspended in DMEM/F12 with HEPES, and the suspension was filtered through a 40 μm cell strainer (BD Falcon, 352340) to generate a single-cell suspension, which was subsequently pelleted via centrifugation at 250 g for 5 min. The supernatant was aspirated, the remaining cell pellet was resuspended in 250 μl mTeSR1 containing 10 μM Y-27632, transferred to a 1.6 ml Eppendorf tube and front loaded into a 1 ml syringe (Covidien Kendall, 8881901014). The 1 ml syringe was centrifuged with the tip facing upwards, at 750 g for 5 min to form a cell pellet that rests upon the syringe plunger. The supernatant was aspirated inside the syringe by inserting a 1.5-inch-long aspiration nozzle via the tip of the syringe, leaving the pellet intact. Next, the pellet was brought to the tip of the 1 ml syringe by gently manipulating the syringe plunger. The tip of the syringe was then pressed against the back-end of a 250 μl gas-tight glass syringe (Hamilton 81120) without its plunger in place, and the pellet was back-loaded into the glass syringe by gently pressing the 1 ml syringe plunger. Next, the gas-tight plunger was inserted into the glass syringe to bring the rear-loaded pellet up to the tip of the syringe, taking care to avoid introducing air at the plunger–pellet interface. The custom-built printhead was attached to the syringe, which was then mounted on a 6-axis motion control stage9 fitted with a custom-built syringe pump10, for 3D bioprinting.
For bioprinting single-cell filaments, tapered milled metal nozzles with an inner diameter (I.D.) of 50 μm (GPD Global, 10/4794) or 100 μm (GPD Global, 10/4793) were attached to the tip of a glass syringe loaded with the cell pellet. For printing multicellular filaments, a 3D-printed triple nozzle was connected to three loaded syringes that were mounted to a single syringe pump that simultaneously drives extrusion of all three inks. Ink extrusion was controlled using an Arduino microcontroller and custom-built stepper motor driver. Printer motion was controlled using a manually written G-code. The G-code for the cortical tissue architecture was created from an image taken of a GW11 human brain section58. PGP1 hiPSC filaments were printed onto ThinCert transparent 0.4-μm-pore-sized transwells in a 6-well plate (Greiner Bio-One, 657641). Immediately after printing, the two parts of the gelatin–fibrin pre-gel solution, as described below, were mixed at a 4:1 ratio and cast overtop the printed filament(s). A combined total of 500 μl of mixed gelatin–fibrin gel was used to encapsulate the printed tissue. EBCM (1 ml) supplemented with 10 μM Y-27632 and 1 U ml−1 thrombin (MilliporeSigma, T4648) was added beneath the transwell to keep the cells hydrated. Prints were first incubated for 10 min at room temperature to cross-link the fibrin gel, then transferred to an incubator at 37 °C with 5% CO2 for 30 min. The medium was then removed and replaced with 4 ml per well EBCM supplemented with 10 μM Y-27632, 100 U ml−1 penicillin–streptomycin, 11.5 KiU ml−1 aprotinin and 500 ng ml−1 doxycycline. The next day, the medium was changed to NIM supplemented with 100 U ml−1 penicillin–streptomycin and 11.5 KiU ml−1 aprotinin. Prints were maintained at 37 °C and 5% CO2, and a full medium change was performed every other day until day 6.
After printing, the densely cellular filamentary features were encapsulated in a gelatin–fibrin gel, which was prepared using a modified version of a previous protocol44. Briefly, a 15% w/v gelatin solution was produced by adding gelatin powder (MilliporeSigma, G2500) to PBS without calcium or magnesium, stirring for 12 h at 70 °C and adjusting the pH to 7.5 using 1 M NaOH. Part 1 of the gel solution was made by diluting the 15% w/v gelatin 1:1 with mTeSR1 and adding 2.5 mM CaCl2, 10 μM Y-27632 and 1 U ml−1 thrombin for a final 7.5% w/v gelatin mix. Part 2 of the gel solution was produced by dissolving lyophilised bovine blood plasma fibrinogen (MilliporeSigma, 341576) at 37 °C in sterile PBS at 50 mg ml−1. Both parts of the pre-gel solution were maintained in separate tubes at 37 °C before use.
The density of hiPSCs in bioinks was estimated by dispensing 100 μl of bioink from the glass syringe, resuspending the cells in 4 ml mTeSR1 and counting the number of cells using a cell counter (Invitrogen, AMQAX1000). The density of Oct4+ cells was calculated via multiplying the total number of cells per ml in the bioink by the percentage of cells that were Oct4+, as measured by flow cytometry using cells obtained from bioink samples.
Live/dead quantification of printed filaments.
Before printing, a solution containing LIVE/DEAD viability/cytotoxicity kit (Invitrogen, L3224) reagents was prepared in EBCM containing 20 μM Y-27632, 4 μM calcein-AM and 8 μM ethidium homodimer-1 (EthD-1). Then single-cell filaments were printed as described above. After printing, the LIVE/DEAD solution was used to resuspend the printed cellular filament into a single-cell filament. The cell suspension was then mixed 1:1 with 15% w/v gelatin for a final gel concentration of 7.5% w/v gelatin, 10 μM Y-27632, 2 μM calcein-AM and 4 μM EthD-1. The gel was then set at r.t. and the cells were imaged on a Zeiss LSM700 confocal microscope. Image processing was done in ImageJ to count the number of cells staining for calcein-AM and EthD-1.
Measurement of printed filament width.
Single-cell filaments were stained with LIVE/DEAD reagents and multicellular filaments were stained with CellTracker Blue CMAC, Green CMFDA and Red CMTPX, and imaged under a Zeiss LSM700 confocal microscope. The total signal intensity from all channels was then converted to greyscale in ImageJ and averaged parallel to the filament to create a longitudinal average intensity profile. Filament widths were calculated as the FWHM of the averaged intensity profile.
Cell migration assay.
iEndo-mKate2 PGP1 cells and WT-eGFP PGP1 hiPSCs were rinsed in PBS without calcium and magnesium, dissociated in TrypLE for 7 min at 37 °C and 5% CO2, lifted off and added into 37 °C DMEM/F12 with HEPES and centrifuged at 250 g for 5 min. After the supernatant was removed, both cells were resuspended in DMEM/F12 with HEPES and transferred into a 1.6 ml Eppendorf tube and centrifuged at 300 g for 3 min. The supernatant was then aspirated and 5 μl of each concentrated cell pellet was pipetted onto a glass-bottom Petri dish, taking care to ensure that the two pellets were in contact. The cell pellets were then encapsulated in a gelatin–fibrin gel and incubated at 37 °C and 5% CO2 for 30 min. Encapsulated cells were then fed with 2 ml per dish of mTeSR supplemented with 10 μM Y-27632, 11.7 KiU ml−1 aprotinin, 100 U ml−1 antibiotic-antimycotic (Gibco, 15240062) and 500 ng ml−1 doxycycline. The following day, the medium was changed to 2 ml per dish NIM supplemented with 11.7 KiU ml−1 aprotinin, 100 U ml−1 antibiotic-antimycotic and 500 ng ml−1 doxycycline. The medium was refreshed every 2 d until day 6 of culture. Images were taken daily on a Zeiss LSM980 inverted confocal microscope. Border lengths were calculated using a custom MATLAB script.
Statistics and reproducibility.
For Fig. 2g–i: n = 3 independent biological replicates from cells differentiated from different passages; unpaired two-tailed t-test; g, not significant; h, P = 7.06 × 10−7; i, P = 3.53 × 10−5. For Supplemental Fig. 8: n = 5 optical sections from single whole-mount WT and WT + iEndo organoids; unpaired two-tailed t-test, P = 4.23 × 10−5. For Supplemental Fig. 9: n = 6 organoids from 3 independent batches. For Fig. 4b: n = 3 organoids from 3 independent batches. For Fig. 5d: n = 3 independent bioink batches. For Fig. 5e: n = 2 (casted control), n = 3 (otherwise) independent batches of bioinks. For Extended Data Fig. 3g: P = 0.0169 between WT-only and randomly pooled organoids, P = 0.0286 between randomly pooled and multicore-shell organoids (n = 72 WT ventricles, n = 10 randomly pooled ventricles, n = 30 multicore-shell ventricles, unpaired two-tailed t-test).
Extended Data
Extended Data Fig. 1 |. Pooled hiPSCs enable the formation of cohesive embryoid bodies with tailorable cellular composition.
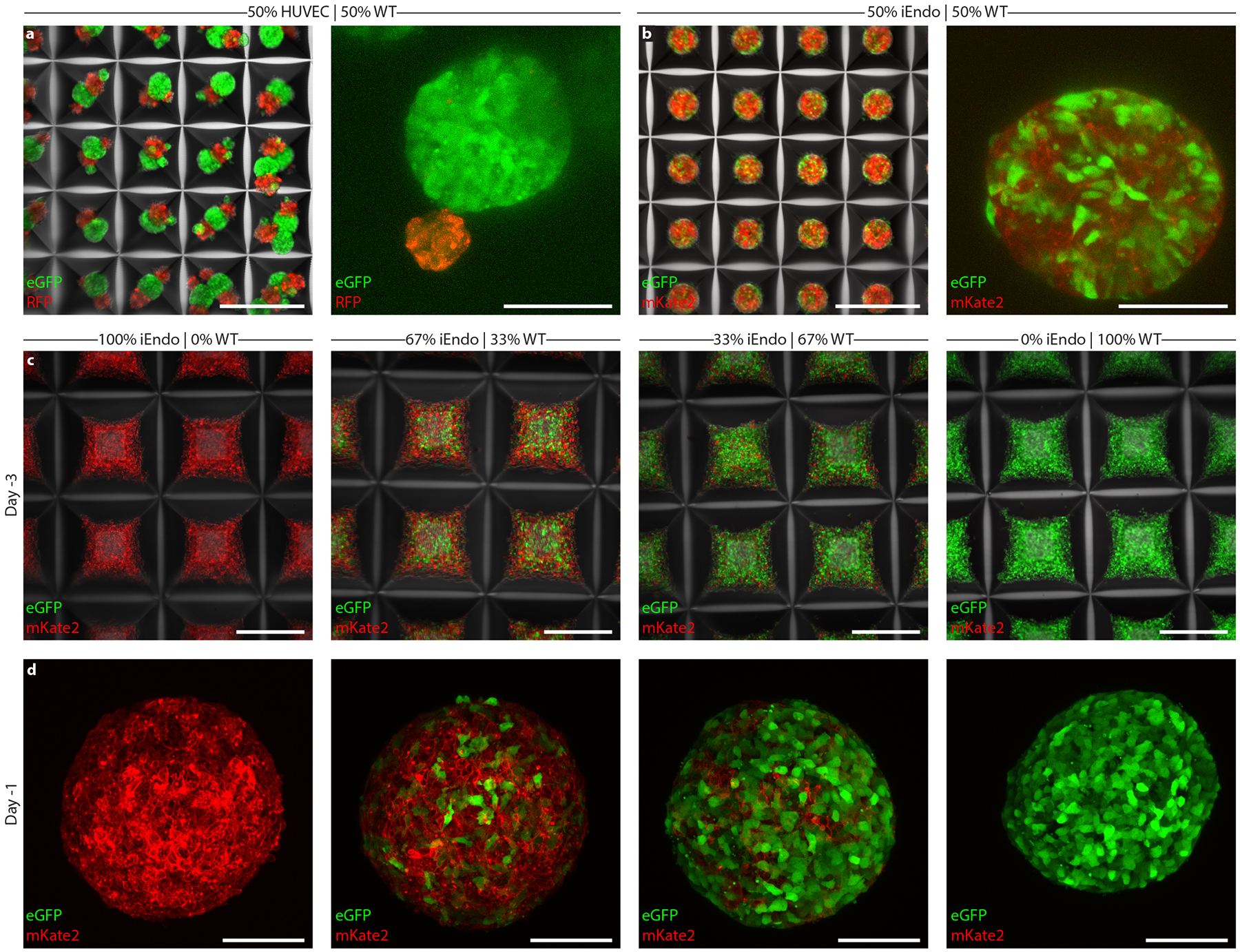
a, Left, 50% WT-eGFP and 50% rFP HUVEC EBs in microwell arrays cultured for 1 day. right, 50% WT-eGFP and 50% rFP HUVEC EBs cultured for 3 days. b, Left, 50% WT-eGFP and 50% iEndo-mKate2 EBs in microwell arrays cultured for 1 day. right, WT-eGFP and 50% iEndo-mKate2 EBs cultured for 3 days. c, Different seeded proportions of WT-eGFP and iEndo-mKate2 hiPSC aggregates in microwell arrays 3 days before suspension culture. Left, 100% iEndo-mKate2. Middle-left, 67% iEndo-mKate2 and 33% WT-eGFP. Middle-right 33% iEndo-mKate2 and 67% WT-eGFP. right, 100% WT-eGFP. d, WT-eGFP and iEndo-mKate2 EBs 1 day before suspension culture. Left, 100% iEndo-mKate2. Middle-left, 67% iEndo-mKate2 and 33% WT-eGFP. Middle-right 33% iEndo-mKate2 and 67% WT-eGFP. right, 100% WT-eGFP. Scale bars: 500 μm in a (left), b (left), and c; 100 μm in a (right), b (right), and d.
Extended Data Fig. 2 |. Programmable vascularized cortical organOiDs.
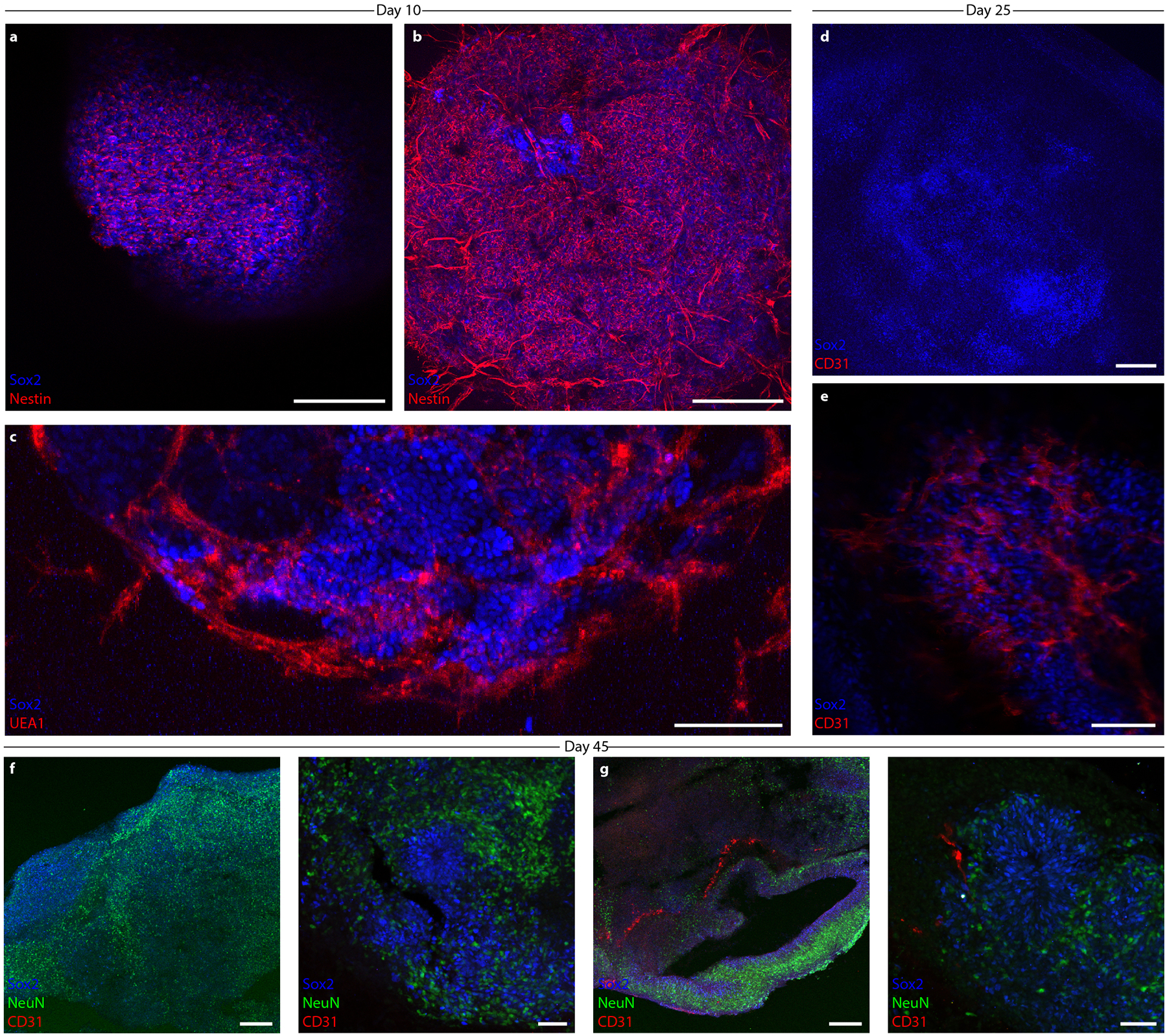
a, Immunostaining of SOX2 and nestin in 100% WT organoids cultured for 10 days. b, Immunostaining of SOX2 and nestin in 67% WT and 33% iEndo organOIDs cultured for 10 days. c, Immunostaining for SOX2 and UEA1 for 67% WT and 33% iEndo organoid cultured for 10 days. d, Immunostaining of SOX2 and CD31 in 100% WT organoids cultured for 25 days. e, Immunostaining of SOX2 and CD31 in 67% WT and 33% iEndo organOIDs cultured for 10 days. f, Immunostaining of SOX2, NeuN, and CD31 for 100% WT organoids cultured for 45 days. g, Immunostaining for SOX2, NeuN, and CD31 in a 67% WT and 33% iEndo organOIDs cultured for 45 days. Scale bars: 200 μm in a, b, d, f (left), and g (left), 100 μm in c and e, and 50 μm in f (right) and g (right).
Extended Data Fig. 3 |. Large ventricular architectures in programmable multicore–shell cortical organOiDs.
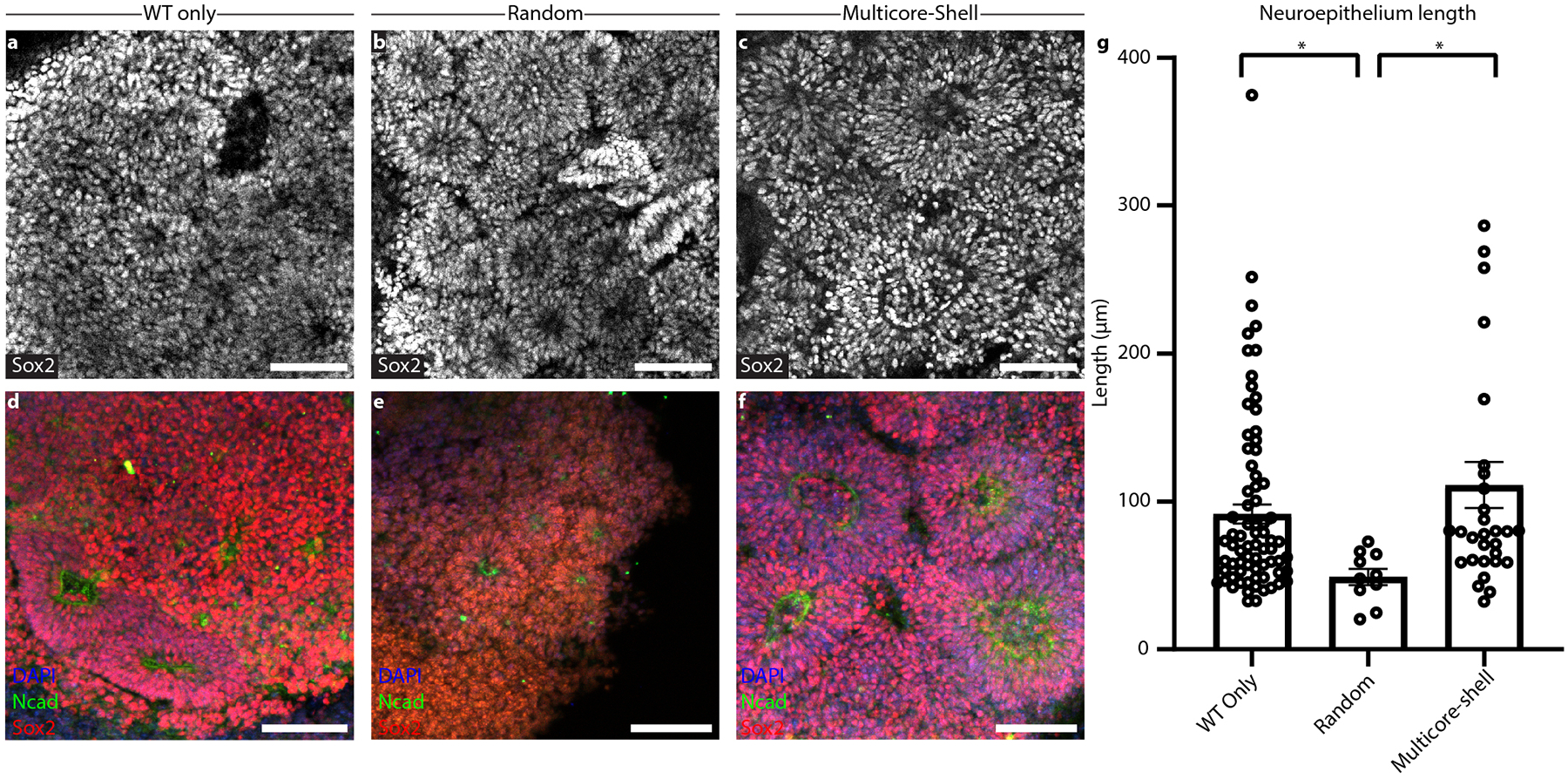
a, Immunostaining of SOX2 in 10-day-old WT-only organoids. b, Immunostaining of SOX2 in 10 day-old randomly pooled organOIDs c, Immunostaining of SOX2 in 10-day-old multicore-shell organOIDs. d, Immunostaining of SOX2 and NCAD in 10-day old WT-only organoids. e, Immunostaining of SOX2 and NCAD in 10-day old randomly pooled organOIDs. f, Immunostaining of SOX2 and NCAD in 10-day old multicore-shell organOIDs. g, Lengths of neuroepithelium within ventricles as measured by the length of NCAD expression within ventricles. Data represents mean ± s.e.m. * P = 0.0169 between WT-only organoids and randomly pooled organOIDs, * P = 0.0286 between randomly pooled and multicore-shell organOIDs (n = 72 WT ventricles, n = 10 randomly pooled ventricles, n = 30 multicore-shell ventricles, unpaired two-tailed t-test). Scale bars: 100 μm in a-f.
Supplementary Material
Acknowledgements
This research was sponsored by the National Human Genome Research Institute of the NIH under award number RM1HG008525, NIH Brain Initiative under award number R01MH123977-01, and by the Vannevar Bush Faculty Fellowship Program, sponsored by the Basic Research Office of the Assistant Secretary for Defense for Research and Engineering through the Office of Naval Research Grants N00014-16-1-2823 and N00014-21-1-2958. A.L. received fellowship support from the Charles Stark Draper Laboratory. We thank T. Ferrante and the Harvard Center for Biological Imaging for microscopy infrastructure and support; the Bauer Core Facility at Harvard University for Illumina sequencing of organoid single-cell libraries and flow cytometry; the Harvard Biopolymers Facility for RNA sequencing support; S. Uzel, S. Han, M. Mata and Z. Niziolek for experimental assistance; and J. Coppeta and J. Aach for useful discussions.
Footnotes
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
The custom code for measuring multicore-shell organoid cell distributions and the code for measuring cell migration in printed tissues is available at https://doi.org/10.5281/zenodo.5823604. The scripts for the analysis of processed single-cell and bulk RNA-sequencing data are available at https://github.com/churchlab/Vascularized-Organoids.
Competing interests
The authors have submitted a patent application associated with this work (U.S Provisional Application Serial No. 63/048,502, filed July 6, 2020).
Extended data is available for this paper at https://doi.org/10.1038/s41551-022-00856-8.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-022-00856-8.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. Source data for the figures are provided with this paper. Raw and processed sequence data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through the GEO series accession number GSE193149. Flow cytometry data, RT-qPCR data and processed bulk RNA-sequencing data are available at https://github.com/churchlab/Vascularized-Organoids. Source data are provided with this paper.
References
- 1.Yin X et al. Engineering stem cell organoids. Cell Stem Cell 18, 25–38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takebe T & Wells JM Organoids by design. Science 364, 956–959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laurent J et al. Convergence of microengineering and cellular self-organization towards functional tissue manufacturing. Nat. Biomed. Eng 1, 939–956 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Brassard JA & Lutolf MP Engineering stem cell self-organization to build better organoids. Cell Stem Cell 24, 860–876 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Lancaster MA & Knoblich JA Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Clevers H Modeling development and disease with organoids. Cell 165, 1586–1597 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Lee A et al. 3D bioprinting of collagen to rebuild components of the human heart. Science 365, 482–487 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Lee W et al. Multi-layered culture of human skin fibroblasts and keratinocytes through three-dimensional freeform fabrication. Biomaterials 30, 1587–1595 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Kolesky DB et al. 3D bioprinting of vascularized, heterogeneous cell-laden tissue constructs. Adv. Mater 26, 3124–3130 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Skylar-Scott MA et al. Biomanufacturing of organ-specific tissues with high cellular density and embedded vascular channels. Sci. Adv 5, eaaw2459 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang H-W et al. A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat. Biotechnol 34, 312–319 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Gu Q, Tomaskovic-Crook E, Wallace GG & Crook JM 3D bioprinting human induced pluripotent stem cell constructs for in situ cell proliferation and successive multilineage differentiation. Adv. Healthc. Mater 6, 1–11 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Yoon S-J et al. Reliability of human cortical organoid generation. Nat. Methods 16, 75–78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Velasco S et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lancaster MA et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takasato M et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–568 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Morizane R et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol 33, 1193–1200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homan KA et al. Flow-enhanced vascularization and maturation of kidney organoids in vitro. Nat. Methods 16, 255–262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eiraku M et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51–56 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Kelava I & Lancaster MA Stem cell models of human brain development. Cell Stem Cell 18, 736–748 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Phipson B et al. Evaluation of variability in human kidney organoids. Nat. Methods 16, 79–87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morita R et al. ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc. Natl Acad. Sci. USA 112, 160–165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elcheva I et al. Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat. Commun 5, 4372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busskamp V et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol 10, 760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sekiya S & Suzuki A Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 475, 390–393 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Huang P et al. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 475, 386–389 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Guye P et al. Genetically engineering self-organization of human pluripotent stem cells into a liver bud-like tissue using Gata6. Nat. Commun 7, 10243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cakir B et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 16, 1169–1175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng AHM et al. A comprehensive library of human transcription factors for cell fate engineering. Nat. Biotechnol 39, 510–519 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D-S et al. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. Rep 6, 270–281 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Chambers SM et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27, 275–280 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsushita M et al. Neural differentiation of human embryonic stem cells induced by the transgene-mediated overexpression of single transcription factors. Biochem. Biophys. Res. Commun 490, 296–301 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Gu M Efficient differentiation of human pluripotent stem cells to endothelial cells. Curr. Protoc. Hum. Genet 98, e64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Y et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 18, 1–29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinberg MS On the mechanism of tissue reconstruction by dissociated cells. I. Population kinetics, differential adhesiveness, and the absence of directed migration. Proc. Natl Acad. Sci. USA 48, 1577–1582 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldberg JS & Hirschi KK Diverse roles of the vasculature within the neural stem cell niche. Regen. Med 4, 879–897 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi T et al. Angiogenesis in the developing spinal cord: blood vessel exclusion from neural progenitor region is mediated by VEGF and its antagonists. PLoS ONE 10, e0116119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian X et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cederquist GY et al. Specification of positional identity in forebrain organoids. Nat. Biotechnol 37, 436–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pasca AM et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan Y & Cahan P SingleCellNet: a computational tool to classify single cell RNA-seq data across platforms and across species. Cell Syst. 9, 207–213.e2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhaduri A et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He L et al. Analysis of the brain mural cell transcriptome. Sci. Rep 6, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kolesky DB, Homan KA, Skylar-Scott MA & Lewis JA Three-dimensional bioprinting of thick vascularized tissues. Proc. Natl Acad. Sci. USA 113, 3179–3184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kupfer ME et al. In situ expansion, differentiation and electromechanical coupling of human cardiac muscle in a 3D bioprinted, chambered organoid. Circ. Res 127, 207–224 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu Q et al. Functional 3D neural mini-tissues from printed gel-based bioink and human neural stem cells. Adv. Healthc. Mater 5, 1429–1438 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Koch L et al. Laser bioprinting of human induced pluripotent stem cells - the effect of printing and biomaterials on cell survival, pluripotency, and differentiation. Biofabrication 10, 035005 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Li Y et al. 3D printing human induced pluripotent stem cells with novel hydroxypropyl chitin bioink: scalable expansion and uniform aggregation. Biofabrication 10, 044101 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Seiler CY et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 42, D1253–D1260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiemann S et al. The ORFeome Collaboration: a genome-scale human ORF-clone resource. Nat. Methods 13, 191–192 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Thompson DB, Villaseñor R, Dorr BM, Zerial M & Liu DR Cellular uptake mechanisms and endosomal trafficking of supercharged proteins. Chem. Biol 19, 831–843 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lancaster MA & Knoblich JA Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc 9, 2329–2340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Birey F et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Renier N et al. Mapping of brain activity by automated volume analysis of immediate early genes. Cell 165, 1789–1802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmid B et al. 3Dscript: animating 3D/4D microscopy data using a natural-language-based syntax. Nat. Methods 16, 278–280 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Zudaire E, Gambardella L, Kurcz C & Vermeren S A computational tool for quantitative analysis of vascular networks. PLoS ONE 6, e27385 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hell S, Reiner G, Cremer C & Stelzer EHK Aberrations in confocal fluorescence microscopy induced by mismatches in refractive index. J. Microsc 169, 391–405 (1993). [Google Scholar]
- 58.Bayer SA & Altman J The Human Brain During the Third Trimester (CRC Press, 2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. Source data for the figures are provided with this paper. Raw and processed sequence data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through the GEO series accession number GSE193149. Flow cytometry data, RT-qPCR data and processed bulk RNA-sequencing data are available at https://github.com/churchlab/Vascularized-Organoids. Source data are provided with this paper.