

Abstract
The metabolic-associated fatty liver disease (MAFLD) is a condition of fat accumulation in the liver in combination with metabolic dysfunction in the form of overweight or obesity and insulin resistance. It is also associated with an increased cardiovascular disease risk, including hypertension and atherosclerosis. Hepatic lipid metabolism is regulated by a combination of the uptake and export of fatty acids, de novo lipogenesis, and fat utilization by β-oxidation. When the balance between these pathways is altered, hepatic lipid accumulation commences, and long-term activation of inflammatory and fibrotic pathways can progress to worsen the liver disease. This review discusses the details of the molecular mechanisms regulating hepatic lipids and the emerging therapies targeting these pathways as potential future treatments for MAFLD.
Keywords: hepatic steatosis, intracellular signaling, non alcoholic fatty liver disease, obesity
Introduction
The levels of obesity are at an all-time high, and this has led to an increase in obesity-associated comorbidities such as insulin-resistant Type 2 diabetes mellitus (T2DM), fatty liver, and cardiovascular diseases. The accumulation of fat in the liver, or simple steatosis, is a benign condition that is a precursor to events that can progress to non-alcoholic fatty liver disease (NAFLD), which is one of the most common causes of chronic liver disease. NAFLD has a global prevalence estimated at 25% [1,2], and its occurrence is increasing in developed and underdeveloped countries [3]. Within the United States, the prevalence is estimated at 24%, with a projected increase of 17.8 million cases from 2015 to 2030 [1,4,5]. In NAFLD, hepatic lipid accumulation is associated with metabolic and cardiovascular diseases like obesity, T2DM, hypertriglyceridemia, and hypertension [6]. Due to this fact, a name change to the metabolic (dysfunction) associated fatty liver disease (MAFLD) has been proposed [2,7]. Patients with MAFLD commonly develop cardiovascular diseases such as hypertension, atherosclerosis, and heart and kidney diseases. For example, 80–90% of obese and 70–80% of T2DM patients typically have MAFLD [8,9]. However, this finding is not universal as others have not revealed an increased risk of cardiovascular disease in MAFLD patients, likely due to the confounding effects of social class [10].
It is currently not understood how MAFLD and T2DM progresses from hepatic insulin resistance to whole-body insulin resistance. One school of thought is that disturbances in lipid metabolism directly influence insulin signaling, leading to impaired secretion of insulin, which contributes to the development and progression of insulin resistance and abnormal glucose metabolism that leads to T2DM [11]. While another concept is that decreased insulin secretion and insulin resistance increase lipolysis, contributing to the development of MAFLD. In addition, increased circulating insulin levels stimulate hepatic de novo lipogenesis in patients with MAFLD [12]. Impaired insulin signaling in adipocytes causes enhanced lipolysis that further worsens the load of lipids routing to the liver. Brown and Goldstein have suggested a selective versus total insulin resistance known as the ‘pathogenic paradox’ [13]. All of these actions might contribute to the development of MAFLD. However, more work is needed to better understand the mechanistic insights into the development of hepatic signaling in MAFLD and whether these are consistent in the latter stages of advanced liver disease, which contain less fat content [14].
The liver controls the metabolism of fatty acids through several molecular mechanisms, including the uptake and release of fatty acids, de novo fatty acid generation, and lipid utilization via fatty acid β-oxidation. In the first part of this review, we discuss the pathophysiology of liver disease and the molecular signaling of pathways in the development of hepatic steatosis in MAFLD. We also discuss the mechanisms that mediate the transition from MAFLD to the worsened state of non-alcoholic steatohepatitis (NASH). The second half of the review focuses on the development of therapies for MAFLD, which arise from our understanding of these pathways and other emerging therapies such as antidiabetic drugs, angiotensin II receptor antagonists, and thyroid receptor β-agonists.
Pathophysiology of liver diseases
Liver disease classification
The onset of MAFLD is characterized by fat accumulation in hepatocytes of at least 5% or more of the liver weight [15]. Hepatic steatosis is classified into various degrees based on lipid percentage in the liver cells (reviewed further in [16]). A healthy liver containing less than 5% of fat accumulation is a grade 0, mild accumulation of 5–33% fat is classified as grade 1, moderate accumulation of 34–66% of fat is grade 2, and severe accumulation with more than 66% of fat is grade 3 steatosis [17] (Figure 1). In obesity with excess calorie intake, the ability of adipose tissue to completely store fats is maximized, which leads to ectopic storage of lipids in tissues like the heart, kidneys, and liver [18,19]. The expansion of abdominal adipose tissue results in the release of cytokines and transcription factors that worsen inflammation associated with obesity [20–23]. Hepatic impairment linked to MAFLD can progress from simple steatosis, NALFD, to NASH [6]. The progression to NASH is the advanced form of MAFLD and is associated with cardiovascular-related mortality that can progress to cirrhosis, liver failure, and hepatocellular carcinoma (HCC) (see later in Figure 5).
Figure 1. MAFLD grade of liver disease and fat content.
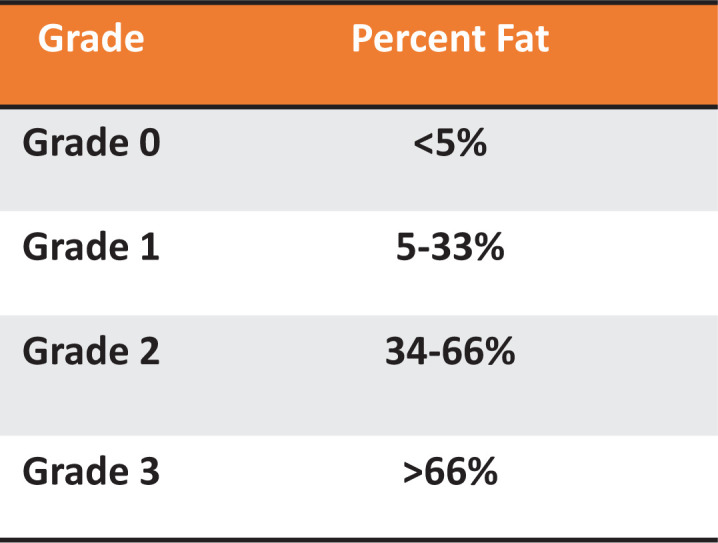
Figure 5. Influence of current and emerging therapies on MAFLD.

Risk factors such as obesity, diet, diabetes mellitus, dyslipidemia, oxidative stress, inflammation, and apoptosis stimulate MAFLD, which can progress to liver fibrosis, cirrhosis, and hepatocellular carcinoma. Lifestyle intervention (diet and exercise) and therapeutic interventions inhibit fatty liver diseases; ARB, angiotensin receptor blocker; DPP-4, dipeptidyl peptidase-4; FGF, fibroblast growth factor; GLP-1RAs, glucagon-like peptide-1 receptor agonists; PPAR, peroxisome proliferator-activated receptor; SGLT-2, sodium-glucose cotransporter-2; THR-β, thyroid hormone receptor-β. Created with Biorender.com
The liver biomarkers typically used in the clinics to measure the level of liver dysfunction are typically plasma levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT), and bilirubin [24] (Figure 2). While AST and ALT are well-established liver biomarkers for hepatocyte injury, the levels of bilirubin have been shown to not be consistent, and there is a discrepancy in the field. This is likely due to the cause of the liver dysfunction, such as fatty liver being induced by diet and excessive calorie intake or by excessive alcohol consumption [2,7,25]. Obesity and MAFLD patients typically have low plasma bilirubin levels unless they are in a stage of decompensated cirrhosis [21,26–28]. Interestingly, preclinical studies demonstrate that increasing plasma bilirubin in the obese reduces adiposity and also lowers the AST and ALT liver enzymes [29–33]. We will discuss the potential of increasing bilirubin levels in patients as a possible therapy for MAFLD later in the section on antioxidants. While AST and ALT have traditionally been utilized as markers of liver function, alterations in plasma ALT do not always accompany the histological changes associated with MAFLD [34]. The search for alternative plasma biomarkers has increased due to the fact that serum ALT levels are not always predictive of MAFLD. Markers like caspase-generated fragmented cytokeratin 18 (fCK18), β-trophin, and adipokines like adiponectin and visfatin are currently being developed as alternatives to ALT and AST for the detection of MAFLD [35–37].
Figure 2. Liver enzymes levels in MAFLD and metabolically fit livers.

The plasma levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) liver biomarker enzymes and bilirubin are typically used in clinics to measure the level of liver dysfunction. The level of de novo lipogenesis and β-oxidation are essential to regulating the liver fat content.
Mechanisms for the transition from MAFLD to NASH
There are several potential mechanisms responsible for the transition from MAFLD to NASH [38,39]. A combination of both extrinsic and intrinsic factors mediates this transition. Adipose tissue inflammation can contribute to insulin resistance and excess release of free fatty acids by lipolysis, which causes hepatic lipid accumulation (Figure 3). The gut microbiota might also play a role in gastrointestinal physiology and the regulation of the intestinal immune system [28]. Changes in the quality and quantity of the gut microbiota, which are associated with deleterious health effects, are referred to as dysbiosis. Studies in mice demonstrate that transferring gut bacteria from humans with MAFLD to lean mice increases insulin resistance and inflammation in response to a high-fat diet, and this results in a greater increase in hepatic steatosis [28,40]. Alterations in the gut microbiota can also result in changes in intestinal permeability to agents like bacterial lipopolysaccharide, which activates Kupffer cells to secrete proinflammatory cytokines, which activates fibrosis pathways by hepatic stellate cells (HSCs) (Figure 4) [41]. Steatosis in hepatocytes can potentially result in the activation of HSCs via several mechanisms [39]. First, lipid accumulation in hepatocytes induces endoplasmic reticulum (ER) stress and increases reactive oxygen species (ROS) production that activates fibrotic pathways, such as the inclusion of fructose with a high-fat diet [39]. Both of these conditions can result in the release of ROS and lipid peroxidation products as well as cytokines, all of which send signals for injury to the liver, such as tumor growth factor-β (TGFβ) that induces HSC activation [39] (Figure 4). Hepatic macrophages produce cytokines and chemokines that directly influence HSC activation, including TGFβ, IL-1β, MCP1, CCL3, and CCL5 [42]. Proinflammatory cytokines such as TGFβ and IL-1β activate HSCs [39]. CCL3 and CCL5 promote hepatic fibrosis through binding with CCR1 and CCR5 on HSCs. Activation of HSCs results in the expression of α-smooth muscle actin (αSMA) [39] and S100 calcium-binding protein A6 (S100A6), the formation of stress fibers, and the deposition of extracellular matrix components. The secretion of profibrotic cytokines promotes the generation of fibrosis, and this interaction with fibrotic tissue further activates HSCs and promotes NASH (Figure 4). Recent data suggest that growth factors such as stem cell growth factor β (SCGF-β), granulocyte-macrophage colony-stimulating factor (GM-CSF), and macrophage colony-stimulating factor (M-CSF) can worsen insulin resistance in males with MAFLD [43].
Figure 3. Pathways governing lipid accumulation in the liver.

Fatty acid uptake and de novo lipogenesis can be up-regulated in MAFLD (green arrows), while fatty acid export and oxidation of fatty acids by the mitochondria and peroxisomes are decreased in MAFLD (red arrows); ACC, acetyl-CoA carboxylase; DAG, diacylglycerol; FAS, fatty acid synthase; SCD1, stearoyl-CoA desaturase-1; VLDL, very-low-density lipoprotein. Created with Biorender.com
Figure 4. Mechanism of hepatic stellate cell activation.

Both adipose tissue inflammation and gut dysbiosis can contribute to hepatocyte steatosis. Hepatocyte steatosis increases endoplasmic reticulum (ER) stress and mitochondrial ROS production, which alters hepatic cytokines resulting in stellate cell activation. Hepatic steatosis and gut dysbiosis also lead to the activation of hepatic macrophages and Kupffer cells that further activates stellate cells to increase collagen synthesis resulting in fibrosis and inflammation and promoting NASH; CCL3/5, chemokine (C-C motif) ligands 3/5; ER, endoplasmic reticulum; IL-1β, interleukin 1β; MCP1, monocyte chemoattractant protein-1; NASH, non-alcoholic steatohepatitis; ROS, reactive oxygen species; TGFβ, transforming growth factor-β. Created with Biorender.com
While MAFLD is widely viewed as a hepatic insulin-resistant disease, most emerging possible therapeutics are to improve hepatic insulin resistance. However, it should be noted that a recent study showed that as the liver disease worsens to NASH and cirrhosis, there could be a change in the insulin signaling in the liver from resistance to sensitivity [39]. An intriguing study by Yen et al. found that patients with liver cirrhosis and insulin-resistant T2DM that use insulin injection therapeutics had significantly elevated risks of death, as well as hypoglycemia, and liver- and cardiovascular complications, compared with those that have the same conditions but did not use insulin injections [44]. Others have also published works indicating that the advanced stages of liver disease might be insulin sensitive for some tissues, such as cirrhotic patients were observed to have greater insulin binding in erythrocytes due to increased insulin receptors on the surface [45]. This work was corroborated by others who showed higher insulin responsiveness in erythrocytes of cirrhotic patients [46–48]. However, not all tissues respond the same. Some studies show reduced insulin receptor activity in immune cells [49] and adipocytes [50]. It is possible that the insulin sensitivity in the liver changes with the progression of the disease and also that other tissues may not be affected in the same manner. This is likely due to hepatocyte death that occurs in the later stages of hepatic disease and HSC activation, which indicates a reorganization of the liver that comprises different cell types in the end stages. Therefore, therapeutics targeting insulin resistance in MAFLD may not be effective in advanced liver diseases with worsened states. Future studies might take a closer analysis of hepatic insulin signaling in the advanced stages of liver disease to determine whether these changes might be occurring, especially since this could impact the life span of the patients. Herein, we will discuss the molecular signaling and mechanistic details that lead to events in MAFLD and the advantages and disadvantages of some of the possible therapeutics.
Molecular mechanisms of lipid accumulation in mafld
Excessive lipid uptake
FABP1
Fatty acid-binding protein 1 (FABP1), originally referred to as liver-FABP or L-FABP, is almost exclusively expressed in the liver. FABP1 transports fatty acids between organelles and can also bind cytotoxic free fatty acids and facilitate their oxidation or incorporation into triglycerides [51]. Genetic deletion of FABP1 significantly attenuates fasting-induced increases in hepatic triglyceride uptake and oxidation [52]. FABP1 knockout mice are protected against dietary-induced hepatic steatosis; however, altered energy utilization exhibited by the global loss of FABP1 is a confounding factor in these experiments [53]. More specific hepatocyte targeting using adenoviral vectors to knock down FABP1 demonstrated protection from dietary-induced hepatic steatosis and inflammation [54]. A recent study in human MAFLD patients found higher levels of plasma FABP1 correlated with the degree of hepatic steatosis [55]. However, whether serum FABP1 levels correlate to expression in the liver remains controversial as FABP1 is also expressed in the proximal tubule of the kidney, which may contribute to the plasma levels [56]. Currently, there are no therapeutics specifically targeting FABP1 that are used for MAFLD. More studies on the impact of antagonizing FABP1 in humans and how they affect metabolic dysfunction are needed.
FATP and CD36
Lipid levels in the liver are regulated by the intricate interaction between hepatic lipid uptake and de novo lipogenesis versus lipid β-oxidation and fatty acid export (Figure 3). Hepatic lipid uptake is controlled via the actions of fatty acid transport proteins (FATPs) and the cluster of differentiation 36 (CD36). FATP isoforms 2 and 5 are the major isoforms present in the liver [51]. Genetic deletion of FATP5 resulted in lower hepatic triglyceride and free fatty acid content in mice on a normal fat diet [57]. Adenoviral knockdown of FATP5 in mice with established high-fat diet-induced MAFLD restored insulin sensitivity; however, this improvement in hepatic function was associated with decreased caloric intake and a reduction in body weight of mice administered an adenovirus containing a short hairpin ribonucleic acid (shRNA) targeting FATP5 [58]. In humans, a polymorphism in the FATP5 promoter results in increased FATP5 expression, which correlates with higher hepatic steatosis in male MAFLD patients [59]. The levels of CD36 protein are increased in the liver in response to high-fat diet feeding [60,61]. Human MAFLD patients have increased hepatic CD36 expression, which was associated with high levels of hepatic fat content [62]. Adenoviral overexpression of CD36 increased hepatic lipid uptake and steatosis in mice fed a normal fat diet [60]. A relationship between hepatic lipotoxicity, FATP5, and CD36 in the liver exists; however, the specific mechanisms by which these proteins are altered in MAFLD, as well as their impact on the progression to NASH, is undefined at the present time [63]. Future studies to elucidate if targeting these mechanisms is a useful therapeutic are needed.
De novo lipogenesis
Three essential enzymes, acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), and stearoyl-CoA desaturase-1 (SCD1), regulate de novo lipogenesis in the liver (Figure 3). ACC governs the conversion of acetyl-CoA to malonyl-CoA, which is converted to palmitate by FAS [64]. SCD1 catalyzes the formation of oleate and palmitoleate from stearoyl-CoA and palmitoyl-CoA [64]. These newly generated fatty acids can undergo a wide variety of biological modifications, including desaturation, elongation, and esterification, before either being stored as triglycerides or exported as VLDL particles [42,65]. The levels of these two proteins can increase hepatic steatosis and hypertriglyceridemia [64]. Moreover, increased production of palmitate may lead to steatohepatitis through increases in inflammation and apoptosis [66]. Two transcription factors that regulate FAS and SCD1 to control de novo lipogenesis are the sterol regulatory element-binding protein 1c (SREBP1c) and carbohydrate regulatory element-binding protein (ChREBP). Hepatocyte-specific overexpression of SREBP-1c results in the upregulation of key enzymes in de novo lipogenesis resulting in hepatic lipid accumulation [67]. ChREBP is required for normal lipogenic response following the ingestion of carbohydrates such as sucrose and fructose [68]. ChREBP knockout mice exhibit decreased levels of lipogenic genes such as ACC and FAS in response to a high starch diet suggesting that ChREBP is the transcriptional mediator of carbohydrate induction of lipogenesis in the liver [69]. Liver-specific inhibition of ChREBP in ob/ob mice via adenovirus shRNA suppression improves hepatic steatosis by decreasing lipogenesis resulting in lower levels of plasma triglycerides and nonesterified fatty acids [70]. Enhanced de novo lipogenesis is responsible for a portion of the lipid accumulation in MAFLD and the buildup of toxic lipid species such as ceramides that may promote the transition to NASH. However, care must be taken when proposing blocking specific enzymes in the de novo lipogenesis pathway, as this could promote the accumulation of more toxic lipids [64]. Further studies are needed before this pathway should be considered as a potential therapeutic target for the treatment of MAFLD as there may be major side effects from blockade of normal lipid processing.
β-Oxidation of fatty acids
Oxidation of fatty acids primarily occurs in the mitochondria; however, oxidation of very long-chain fatty acids commences in the peroxisomes and then is finally processed in the mitochondria (Figure 3) [64]. In cases of lipid overload, as seen in obesity, ω-oxidation by cytochrome P450 enzymes also contributes to fatty acid oxidation. However, this pathway’s oxidation of fatty acids generates large amounts of ROS, which promote inflammation and NASH progression [71]. The entry of fatty acids into mitochondria depends on carnitine palmitoyltransferase 1 (CPT1), situated in the outer mitochondrial membrane. One of the major regulators of CPT1 is the peroxisome proliferator-activated receptor-α (PPARα) [31,32,72,73]. Activation of PPARα induces transcription of genes related to fatty acid oxidation in the mitochondria (CPT1), peroxisomes (acyl-CoA oxidase, ACOX), and cytochromes (Cyp4A family) [31,74,75]. Hepatocyte-specific PPARα knockout mice exhibit marked steatosis, inflammation, and suppressed levels of CPT1 as compared with wild-type mice in response to dietary-induced obesity [42]. Glycogen synthase kinase 3β (GSK3β) regulates levels of PPARα in the liver through phosphorylation at serine-73 (Ser73) [73]. Ser73 phosphorylation enhances the ubiquitination of PPARα, resulting in increased protein turnover and decreased activity [73]. In humans, the expression of PPARα decreases with increasing MAFLD activity score and fibrosis score [74]. Targeting this pathway alone or in combination with the PPARγ isoform for the potential treatment of MAFLD will be discussed further below.
Export of hepatic lipids
The export of triglycerides is another important pathway in the regulation of hepatic lipid content. The key components of this process are apolipoprotein B100 (apoB100) and microsomal triglyceride transfer protein (MTTP) (Figure 3). Very low-density lipoprotein (VLDL) particles are formed in the endoplasmic reticulum (ER), where MTTP catalyzes the lipidation of apoB100 [29,31]. The VLDL particles are then transferred to the Golgi apparatus, where they mature and are subsequently secreted into the bloodstream via ApoB100-mediated mechanisms. Moderate exposure to fatty acids increases apoB100 secretion; however, increased levels of fatty acids promote ER stress which subsequently inhibits apoB100 secretion, promoting steatosis [76]. Genetic defects in MTTP affect hepatic triglyceride export and cause MAFLD. In a study of MAFLD susceptibility in the Han Chinese population, genetic polymorphisms in the MTTP gene both increased and decreased the risk of MAFLD [77]. Overexpression of MTTP normalized VLDL secretion and reduced hepatic triglyceride levels in a mouse genetic model of MAFLD [78]. Diminished levels of apoB100 result in decreased secretion of VLDL. NASH patients have lower apoB100 synthesis rates indicating that lower lipid transport could contribute to advanced steatosis in these patients [79]. Taken as a whole, alterations in MTTP and apoB100, especially in conditions of lipid overload, may result in steatosis and lipotoxicity that promotes the progression of MAFLD.
A mechanistic view of therapies for MAFLD
First-line therapies
The most commonly recommended treatment for MAFLD is exercise and dietary restriction (Figure 2) [80]. While there are no FDA-approved therapies specifically for MAFLD, we describe in Figure 5 and below some of the current therapies being used and developments in research that may lead to the first approved treatment regimens.
Antidiabetic drugs
In treating MAFLD, medications targeting diabetes, a disease often associated with MAFLD development, are longstanding (Figure 5). Metformin, a biguanide, is the first-line oral antidiabetic therapy prescribed to T2DM patients functions to lower blood glucose, and this may be beneficial to MAFLD patients with T2DM [5]. Bugianesi et al. determined that metformin had a larger effect on improving aminotransferase levels in patients with MAFLD than in another treatment group with vitamin E [5,81]. The mechanism of action of metformin in the liver involves the activation of AMP-activated protein kinase (AMPK), a mechanism that may be responsible for its wide range of metabolic benefits [82]. Studies have shown that AMPK inhibits ACC, thereby reducing malonyl CoA synthesis, which in turn increases fatty acid oxidation in the mitochondria [73,83]. In MAFLD induced by a high-fat diet, metformin directly decreased fat deposition and inhibited inflammation in the liver by increasing phosphorylation of liver AMPK and ACC but decreased lipogenic enzymes and proinflammatory cytokines [84]. Furthermore, the reversal effect of metformin on insulin resistance and hepatic steatosis is autophagy-dependent via the transcriptional factor EB, a potent regulator of autophagy [85]. Although metformin has been documented in some studies to be beneficial in MAFLD and chronic hepatitis C patients [86,87], its use has been inadvisable in patients with cirrhosis due to increased risk of lactic acidosis [88,89]. Also, its long-term use has been linked with vitamin B12 deficiency [15,90]. Metformin use can also be associated with abdominal discomfort, anxiety, and interaction with other classes of drugs. While metformin reduces serum aminotransferases and insulin resistance in several preclinical studies, several meta-analyses determine no significant improvement in liver histology in MAFLD and NASH patients when treated with metformin therapy [91–93].
Sodium-glucose cotransporter-2 (SGLT2) inhibitors are another class of drugs approved for the treatment of T2DM. SGLT2 inhibitors decrease renal reabsorption of glucose, helping to improve glycemic control. Several studies have evaluated the impact of SGLT2 inhibitors on MAFLD and have found improvements in AST and ALT liver enzymes, as well as in hepatic fat accumulation [5]. SGLT2 inhibitors have been reported to reduce body fluid and body weight; hence, they can be recommended in obese patients with MAFLD [94]. Shiba et al. reported that canagliflozin treatment for 20 weeks delayed the onset of NASH and its progression to hepatocellular carcinoma in a mouse model of human NASH [95]. Likewise, in a human study, treatment with canagliflozin resulted in reduced liver enzymes [ALT, AST, alkaline phosphatase (ALP), and gamma-glutamyl transferase (GGT)], body weight, and an increase in bilirubin [96]. Empagliflozin, another SGLT2-inhibitor, reduces hepatic fat content and significantly improves steatosis and liver fibrosis, as well as reduces AST and ALT in MAFLD patients with or without the condition of T2DM [97–99]. Empagliflozin decreased the expression of hepatic inflammatory genes such as TNF-α, interleukin-6, and monocyte chemoattractant protein-1 (MCP-1) in a mice model of NASH, and when used in combination with linagliptin, a DPP-4 inhibitor, both treatments reduced expression of mRNAs for genes related to fatty acid synthesis, reduced collagen deposition and decreased the expression of αSMA, which is an indicator of fibrosis [39,100]. Empagliflozin also reduced insulin resistance and attenuated diet-induced activation of NLRP-3 inflammasome and triglyceride in the liver [101]. Ipragliflozin reduced plasma and liver biomarkers of oxidative stress such as thiobarbituric acid reactive substances and protein carbonyl and inflammatory markers in T2DM mice with NASH [102]. SGLT2 inhibitors have several side-effects that may limit their use including risk of dehydration, potential for the development of urinary tract infections, and development of ketoacidosis. SGLT2 inhibitors are promising new therapies for MAFLD and other cardiovascular diseases such as congestive heart failure; however, it is unclear if they have a direct effect on hepatic steatosis or work through effects on body weight and insulin sensitivity.
Another class of drugs benefiting MAFLD patients is glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1RAs). Examples of these drugs are exenatide, liraglutide, dulaglutide, and semaglutide. These drugs are commonly used to treat diabetes, and they mimic the action of an incretin hormone called GLP-1, which is released from the L cells of the small intestine after meals to stimulate insulin secretion, reduce glucagon production, and retard gastric emptying [103]. Hence, this antidiabetic drug class induces glucose-mediated insulin secretion and reduces glucagon secretion, promoting weight loss [5]. Interestingly, there is down-regulation of GLP-1 receptors in MAFLD [104]. In preclinical studies, GLP-1 analogs have been demonstrated to inhibit MAFLD by inhibiting NLRP3 inflammasome via enhancement of autophagy/mitophagy pathways, inhibiting macrophage recruitment and activation, and increasing antioxidant defense in the liver [105]. These GLP-1RAs improve insulin sensitivity by reducing JNK phosphorylation and increasing the expression and activity of PPARγ [106]. Several randomized controlled trials (RCTs) have documented its efficacy in reducing liver enzymes, body weight, and hepatic content. In a multicenter clinical trial of 76 MAFLD patients, 24-week treatment with exenatide reduced hepatic steatosis; however, these changes were associated with reductions in body weight, visceral fat, and fasting glucose levels [107]. Taken together, GLP-1RAs have the potential to be developed into therapies for MAFLD as they act on multiple pathways (hepatic lipogenesis and β-oxidation, ER stress, and oxidative stress) and also have beneficial effects to lower body weight and improve insulin sensitivity. GLP-1RA use is associated with minor side-effects such as nausea. All these taken together, GLP-1RAs delay the progression of MAFLD by inhibiting inflammation, oxidative stress, insulin resistance, enzymes involved in hepatic lipogenesis, and enhancing the autophagy/mitophagy pathway, as well as enzymes involved in β-oxidation and offer the potential to be useful therapies.
DPP-4 inhibitors, also called gliptins, are potential medications for the treatment of MAFLD (Figure 5) that work by obstructing the activities of dipeptidyl peptidase-4 (DPP-4) to elevate incretin levels and reduce glucagon release. This action consequently decreases gastric emptying, lowering hepatic glucose production, increasing insulin exocytosis, and fatty acid oxidation in the liver [108]. Sitagliptin, a gliptin drug, was reported to attenuate the expression of SREBP-1c, SCD-1, and FAS which are involved in de novo lipogenesis, and it up-regulated the expression of PPARα in the liver [109]. The SIRT1/AMPK pathway also mediates the anti-steatotic actions of sitagliptin in the liver [110]. Although several studies show that sitagliptin reduces hepatic lipid accumulation in animal models, there are conflicting results in human MAFLD patients. Sitagliptin therapy was initially reported to suppress liver enzymes, body weight, and hepatocyte ballooning in diabetic patients with NASH [111]. However, a follow-up study by Joy et al. showed no significant improvement in the liver fibrosis score, MAFLD activity score, lipid profile, or liver enzymes in patients with biopsy-proven NASH that received sitagliptin for 24 weeks [112]. There are othergliptin drugs that act by improving insulin resistance and hepatic inflammation that could be beneficial in MAFLD patients [113]. Randomized clinical trials of this class of DPP-4 inhibitors are needed in MAFLD patients. In addition, use of DPP-4 inhibitors is associated with upper respiratory tract infection, headaches, and a potential for hypoglycemia especially if used in concert with insulin.
Thiazolidinediones, PPAR- γ agonists, are insulin sensitizers used in the management of diabetes and for the treatment of MAFLD. The two major thiazolidinediones that have been used to treat diabetes are pioglitazone and rosiglitazone. In human studies, pioglitazone improved hepatic steatosis, ballooning necrosis, and inflammation; however, no changes in fibrosis were reported [114,115]. Small clinical trials of rosiglitazone demonstrated the overall tolerance as well as reductions of serum markers ALT and AST and lowering of plasma glucose, insulin, and HbA1c [116,117]. While rosiglitazone treatment was generally well-tolerated in these small clinical trials, effects such as weight gain, fluid retention, osteopenia, prostate and pancreatic cancer, and cardiovascular events have been ascribed to thiazolidinedione use [118].
Inhibiting de novo lipogenesis
ACC is an important enzyme in de novo lipogenesis and an emerging target for the treatment of MAFLD (Figure 5). ACC is present in two isoforms in the liver: ACC1 and ACC2. The use of dual inhibitors of ACC1 and ACC2 was effective in preventing hepatic steatosis in mouse models of dietary-induced MAFLD. However, Goedeke et al. reported that dual inhibition of ACC1 and ACC2 also resulted in plasma hypertriglyceridemia from a combination of increased hepatic very-low-density lipoprotein production and a decrease in triglyceride clearance by lipoprotein lipase (LPL) [119]. Selective ACC1 inhibitors have also been utilized to treat MAFLD in a mouse model of genetic fatty liver [120]. Clinical trials utilizing dual ACC inhibitors alone or in combination with diacylglycerol O-acetyltransferase 2 (DGAT2) are currently underway in adults with MAFLD [121]. In these studies, ACC inhibition resulted in decreases in hepatic steatosis as measured by magnetic resonance imaging (MRI); however, like studies in animals, ACC inhibition resulted in a significant increase in plasma triglyceride levels [121]. Treatment with both ACC and DGAT2 inhibitors also lowered hepatic steatosis but did not result in any significant changes in plasma triglyceride levels [121]. These results suggest that the use of ACC inhibitors, when coupled with drugs that inhibit triglyceride synthesis, may be effective therapies to reduce hepatic steatosis in MAFLD patients.
Targeting excess lipid uptake
The excess lipid uptake mediated through FATP and CD36 contributes to hepatic steatosis in NAFLD (Figure 3). The FATP2 inhibitor, Grassofermata/CB5, was shown to inhibit palmitate-mediated lipid accumulation in HepG2 cells; however, its efficacy has not been tested in animal models [122]. While drugs specifically targeting these pathways are still in development, there are several clinically utilized drugs affecting this pathway that could be beneficial in NAFLD. For example, the PDE4 inhibitor, roflumilast, was clinically approved for the treatment of chronic obstructive pulmonary disease and may be beneficial in treating MAFLD. Roflumilast treatment of dietary-induced obese mice reversed hepatic lipid accumulation [123]. The antitumor and immunosuppressive drug rapamycin [124] was shown to reduce lipid accumulation in 3T3-L1 cells [125] and improve hepatic steatosis by reducing CD36 expression in mice fed a high-fat diet [126]. Rapamycin acts through inhibition of the mammalian target of rapamycin (mTOR) pathway. It also has been shown to suppress hepatic CD36 translational efficiency, decreasing CD36 levels and alleviating hepatic steatosis [126]. However, rapamycin also binds to the FKBP12 [125,127] and has immunosuppressive effects that have been linked to glucose intolerance in mice [128]. There are more studies needed for a better understanding of how targeting these mechanisms might be beneficial, and the efficacy of the compounds should be considered.
Targeting hepatic β-oxidation
PPARα agonists
PPARα is highly expressed in the liver [23,129], where it is vital in triglyceride hydrolysis, fatty acid catabolism, glycerol metabolism, and ketogenesis [130,131]. Studies in mice show that the loss of PPARα in the liver results in simple steatosis on a normal chow diet [129], which is worsened on a high-fat diet [42,132]. Likewise, systemic loss of PPARα coupled with a trans fatty acid-rich diet leads to hepatic steatosis [133]. Fibrates, which target PPARα, are prescribed clinically to lower serum lipids and also have beneficial effects on hepatic steatosis in animal models. In addition, existing data show that fibrate treatment prevents stroke, myocardial infarction, and cardiovascular death [134–136]. The PPARα agonist fenofibrate increases both uptake and β-oxidation of FAs which is for lipid utilization [64]. Fenofibrate protects the liver against MAFLD (Figure 5) by improving insulin sensitivity and by exerting antioxidant, anti-inflammatory, and anti-atherosclerotic effects [137–143]. In mice lacking phosphatidylethanolamine N-methyltransferase (PEMT), fenofibrate treatment completely prevented MAFLD development and partially reversed steatosis and fibrosis in already established MAFLD in this model [144]. Similarly, administration of fenofibrate for 5 days decreased hepatic lipid and glycogen storage in neonatal glucose-6-phosphatase α knockout mice via promotion of β-oxidation of fatty acids and stimulation of hepatic expression of acyl-CoA dehydrogenases [145]. Pemafibrate (K-877), another selective PPARα agonist, improved methionine–choline-deficient diet-induced MAFLD by increasing the expression of hepatic fatty acid oxidation genes in mice [146]. In a phase 3 clinical study, pemafibrate treatment significantly reduced plasma triglycerides in at-risk patients and had a superior safety profile as compared with fenofibrate [147]. It should be noted that fibrates have minimal clinical use as they have varied effects and, at times, show very little response in some patients. Fibrate use is also associated with side effects such as abdominal pain, constipation, headaches and muscle cramps. Preclinical animal studies show that this might be related to estrogen suppression of PPARα, reducing the effectiveness of fibrates in females [148–151]. Hinds et al. showed that female mice have significantly less PPARα expression in the adipose compared with males [23]. Only the males in this study were shown to be affected by the adipose-specific loss of PPARα [23]. This suggests that more studies are needed to better understand the varied effects by which estrogen status affects PPARα expression.
Dual/Pan PPAR-alpha-gamma agonists
The development of MAFLD involves numerous processes, and some novel PPAR-agonists having effects on two or more PPAR isoforms have been shown to be effective in the treatment of MAFLD (Figure 5). Such drugs are classified as dual/pan-PPAR agonists. Saroglitazar is a dual agonist of PPARα and PPAR-γ that is in multiple phase 3 clinical trials. Saroglitazar use is not associated with the side effects (weight gain, edema) linked with activation of PPARγ agonists like pioglitazone [152,153]. In a mouse model of dietary-induced MAFLD, saroglitazar treatment lowered body weight, insulin resistance, triglycerides, total cholesterol, ALT, and improved hepatic steatosis and fibrosis [154]. Compared with pioglitazone and fenofibrate, saroglitazar improved steatosis, fibrosis, and NASH to a greater extent by influencing serum inflammatory cytokines and adiponectin levels in rats fed a high-fat diet [155]. In human clinical trials, saroglitazar reduced ALT levels and improved fatty liver in MAFLD patients with diabetic dyslipidemia [156]. Goyal et al. documented that hepatic enzymes and liver stiffness were improved by saroglitazar in patients with diabetic dyslipidemia and MAFLD [9]. The results from these clinical trials demonstrate that saroglitazar is a promising therapeutic option for MAFLD patients. Elafibranor (GFT-505) is another dual agonist of PPARα and PPARδ [130]. Several studies have demonstrated the role of elafibranor in different diet-induced obese mouse models of NASH. Elafibranor reduced body weight, liver enzymes, histopathological scores of hepatic steatosis and inflammation, and attenuated fibrosis severity in NASH models of C57BL/6J mice and leptin-lacking Lepob/ob fed on high fat, fructose, and cholesterol diet [157]. In another study, elafibranor reduced hepatic steatosis and inflammation and prohibited the progression of fibrosis in a mouse genetic model that exhibited histopathological characteristics of NASH with similar molecular pathways observed in NASH patients [158]. In a human study, abdominally obese insulin-resistant male patients treated with elafibranor for 2 weeks showed reduced serum lipids, improved liver enzymes, and enhanced hepatic insulin sensitivity [159]. In a study carried out for 52 weeks in Europe and United States, patients with NASH without cirrhosis treated with two doses of elafibranor showed resolution of NASH without worsening of fibrosis. In addition, the higher dose of elafibranor reduced liver enzymes, lipids, glucose profiles, and markers of systemic inflammation compared with placebo-treated patients [160]. However, a recent phase 3 clinical trial (NCT02704403, RESOLVE-IT) evaluating the use of elafibranor versus placebo in the treatment of NASH was terminated due to lack of efficacy.
Lanifibranor (IVA337) is a pan-PPAR agonist that activates the three PPAR isoforms (α, γ, δ) proportionately [130]. Lanifibranor controlled body weight gain, improved insulin sensitivity, attenuated hepatic steatosis, inflammation, ballooning, and fibrosis, as well as inhibited the expression of pro-fibrotic and inflammatory genes, and increased the expression of β-oxidation genes in studies with various preclinical models of NASH [161]. Lanifibranor improves insulin sensitivity, lipid profile, inflammation, and fibrosis, and it has been reported to have a good safety profile [162]. In a recently completed phase 2 clinical trial, lanifibrnor decreased the activity part of the Steatosis, Activity, Fibrosis (SAF) score, SAF-A in patients with noncirrhotic NASH [163].
Angiotensin receptor blockers (ARBS)
Angiotensin receptor blockers (ARBs), also known as angiotensin II receptor antagonists, are traditionally used to treat hypertension, heart failure, and chronic kidney disease but have potential in the treatment of MAFLD (Figure 5). In animal models, ARB treatment lessened hepatic steatosis and fibrosis, reduced markers of inflammation, and improved hepatic mitochondrial function [164,165]. ARB treatment attenuated transhepatic pressure gradients and intrahepatic vascular resistance in the high-fat, high-fructose diet-induced NASH model [166]. ARB treatment was also found to reverse mitochondrial dysfunction and reduce the proinflammatory M1 macrophage population [165]. Studies have shown that increasing the M1 macrophage population in the liver of mice induces fat accumulation [167]. However, a confounding study reported that dietary intervention but not losartan reversed obesity, insulin resistance, autophagic flux, and histological features of NASH [168]. ARBs also elevate circulating adiponectin levels which can exert an antifibrotic effect, improving both MAFLD and NASH [169,170].
Several phase 2 clinical trials have examined the efficacy of ARBs in MAFLD. In a phase 2 clinical trial study, MAFLD pediatric patients treated with losartan for 8 weeks showed improved ALT, AST, and insulin resistance compared with placebo [171]. In the fatty liver protection trial by telmisartan or losartan study (FANTASY), telmisartan showed more beneficial effects in improving fatty liver than losartan in hypertensive MAFLD patients with T2DM [172]. This is likely due to the fact that telmisartan activates PPARγ [173]. ARBs not only are effective anti-hypertensive drugs, but they may also prove beneficial in the treatment of patients with MAFLD. More studies are needed to better understand the potential uses of the ARBs in treating MAFLD.
Thyroid hormone receptor-β agonist
Resmetirom (MGL-3196) is an orally administered selective thyroid hormone receptor-β (THR-β) agonist. The thyroid hormone acts via THR-β in the liver to increase lipid metabolism and prevent pathological lipid accumulation in the liver. Several studies have shown the involvement of THR-β in MAFLD/NASH metabolic pathways [174–176]. In a mouse model of NASH and fibrosis, resmetirom improved MAFLD activity score and decreased hepatic fibrosis by lowering the content of αSMA and down-regulating genes involved in fibrogenesis such as collagen 1α1, lysyl oxidase-like 2, and hydroxysteroid 17-β dehydrogenase 13 [176]. THR-β agonists improve mitochondrial β-oxidation and reduce hepatic lipid accumulation in NASH patients [177]. In a phase 2 trial, resmetirom reduced hepatic fat accumulation after 12 weeks and 36 weeks of treatment in patients with NASH [178]. Presently, there are two ongoing phase 3 clinical trials; one is to evaluate the efficacy and safety of MGL-3196 in NASH patients with advanced fibrosis (MAESTRO-NASH), and the other is to evaluate the safety of MGL-3196 in MAFLD patients (MAESTRO-MAFLD-1) [175]. These clinical studies should reveal the usefulness of these compounds and if targeting THR-β is useful for treating MALFD. Resmetirom use is associated with mild side effects such as transient nausea.
Antioxidants
Antioxidants such as Vitamin E has been used for MAFLD treatment (Figure 5) in hopes of combating the ROS and oxidative stress that is known to occur [179]. A meta-analysis by Abdel-Maboud et al. analyzing 1317 patients from 15 randomized controlled trials showed that vitamin E was efficacious at reducing MAFLD activity score and AST and ALT liver enzymes [180]. There was a clinical trial (NCT01792115) to determine the dosage of vitamin E combined with exercise that provides the best protection. The study only found modestly reduced liver fat content with the highest dose. Another antioxidant, bilirubin [181], has gained attention for its recently discovered role that it is also a hormone that activates PPARα [21,38,182,183]. Bilirubin strongly reduces oxidative stress in hepatocytes [184] and in other cell types [185–187]. Preclinical studies show that bilirubin suppresses hepatic fat content in obese mice and lowers hepatocyte injury markers AST and ALT [29,31,32]. Plasma bilirubin levels are lower in patients with MAFLD [188–190]. However, other studies have not found that bilirubin levels are changed in MAFLD [191]. It likely depends on the metabolic state of the patients, as patients exhibiting mildly elevated (>12 μM) bilirubin levels were shown to have significantly fewer metabolic disorders such as NAFLD, obesity, or T2DM [192–200]. This is supported by a study by Lin et al. that found that obese children with the UGT1A1*6 variant with slightly elevated plasma bilirubin levels were less likely to be diagnosed with MAFLD [201]. One of the current therapeutics used for MAFLD is dietary restriction and exercise. Interestingly, plasma bilirubin levels are increased during exercise [181,202], which may be one of the mechanistic actions that exercise improves liver fat content. However, more studies are needed to better understand this aspect. Bilirubin can also impact hepatic steatosis by acting as an activator of the nuclear hormone receptor PPARα [32,203]. Bilirubin is able to increase the levels of CPT1 and Cya4A family isoforms to increase the oxidation of fatty acids [32]. Treatment with bilirubin nanoparticles also attenuates dietary obesity-induced hepatic steatosis and increases plasma ketone levels [29]. Other options of possibly increasing bilirubin levels in the blood are to target the enzyme that conjugates it, UGT1A1 [31,204]. More work is needed to reveal if antioxidants might be useful for treating MAFLD. It could be that they might be better in combination therapy in which one reduces ROS and the other burns fat, and they may be more efficacious together at reducing MAFLD.
Conclusions
MAFLD is becoming a common cause of chronic liver disease and significantly impacts our healthcare system. With very few FDA-approved drugs explicitly for the treatment of MAFLD, patients must rely on weight loss from lifestyle changes or medications to treat comorbidities such as diabetes and hypertension. It is clear from the scope of this review and others that advances in our understanding of the molecular pathways regulating hepatic lipid accumulation can lead to promising therapies for MAFLD [205]. In addition, several drugs used for conditions like diabetes, obesity, and hypertension are being repurposed for use in the treatment of MAFLD (Figure 5). By systematic dissection of the pathways responsible for hepatic lipid accumulation and the progression of MAFLD, new therapeutic targets for MAFLD should be identified. It is hopeful that the emerging therapies will be successful in attenuating the emerging epidemic of MAFLD and its associated pathologies.
Acknowledgements
Figure 2 was created by Matthew Hazzard at the University of Kentucky. Figures 3–5 were made using BioRender.com.
Abbreviations
- ACC
acetyl-CoA carboxylase
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AMPK
AMP-activated protein kinase
- apoB100
apolipoprotein B100
- ARB
angiotensin receptor blocker
- AST
aspartate aminotransferase
- CCL3
chemokine (C-C motif) ligand 3
- CCL5
chemokine (C-C motif) ligand 5
- CCR1
C-C chemokine receptor type 1
- CCR5
C-C chemokine receptor type 5
- CD36
cluster of differentiation 36
- ChREBP
carbohydrate regulatory element-binding protein
- CPT1
carnitine palmitoyltransferase 1
- DGAT2
diacylglycerol O-acetyltransferase 2
- DPP-4
dipeptidyl peptidase-4
- ER
endoplasmic reticulum
- FABP1
fatty acid-binding protein 1
- FAS
fatty acid synthase
- FATP
fatty acid transport proteins
- fCK18
caspase-generated fragmented cytokeratin 18
- FDA
Food and Drug Administration
- FGF
fibroblast growth factor
- GGT
γ-glutamyl tranferase
- GLP-1
glucagon-like peptide-1
- GSK3β
glycogen synthase kinase 3β
- HCC
hepatocellular carcinoma
- HSC
hepatic stellate cell
- HTN
hypertension
- IL-1β
interleukin 1β
- IR
insulin resistance
- MAFLD
metabolic-associated fatty liver disease
- MCP1
monocyte chemoattractant protein-1
- mTOR
mammalian target of rapamycin
- MTTP
microsomal triglyceride transfer protein
- NASH
non-alcoholic steatohepatitis
- NLRP3
NLR family pyrin domain containing 3
- PDE4
phosphodiesterase-4
- PEMT
phosphatidylethanolamine N-methyltransferase
- PPAR
peroxisome proliferator-activated receptor
- RCTs
randomized controlled trial
- SCD1
stearoyl-CoA desaturase-1
- SGLT2
sodium-glucose cotransporter-2
- shRNA
short hairpin RNA
- SIRT1
Sirtuin 1
- SREBP1c
sterol regulatory element-binding protein 1c
- TGFβ
transforming growth factor-β
- THR-β
thyroid hormone receptor-β
- T2DM
Type 2 diabetes mellitus
- VLDL
very low-density lipoprotein
Contributor Information
Terry D. Hinds, Jr, Email: Terry.Hinds@uky.edu.
David E. Stec, Email: dstec@umc.edu.
Data Availability
All data are included within the main manuscript file.
Competing Interests
T.D.H.J. and D.E.S. have submitted patents on bilirubin and obesity-related disorders.
Funding
This work was supported by the National Institutes of Health [grant numbers R01DK126884 (to D.E.S.) and R01DK121797 (to T.D.H.J.)]; the National Heart, Lung and Blood Institute [grant number P01HL05197-11 (to D.E.S.)]; and the National Institute of General Medical Sciences [grant number P20GM104357-02 (to D.E.S.)].
Open Access
Open access for this article was enabled by the participation of University of Mississippi in an all-inclusive Read & Publish agreement with Portland Press and the Biochemical Society.
CRediT Author Contribution
Olufunto O. Badmus: Conceptualization, Visualization, Writing—original draft, Writing—review & editing. Sarah A. Hillhouse: Conceptualization, Writing—original draft, Writing—review & editing. Christopher D. Anderson: Conceptualization, Writing—original draft, Writing—review & editing. Terry D. Hinds, Jr: Conceptualization, Funding acquisition, Writing—original draft, Writing—review & editing. David E. Stec: Conceptualization, Supervision, Funding acquisition, Validation, Visualization, Writing—original draft, Writing—review & editing.
References
- 1.Younossi Z.M., Koenig A.B., Abdelatif D., Fazel Y., Henry L. and Wymer M. (2016) Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 10.1002/hep.28431 [DOI] [PubMed] [Google Scholar]
- 2.Eslam M., Sanyal A.J., George J. and International Consensus P (2020) MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999e1–2014e1 10.1053/j.gastro.2019.11.312 [DOI] [PubMed] [Google Scholar]
- 3.Ge X., Zheng L., Wang M., Du Y. and Jiang J. (2020) Prevalence trends in non-alcoholic fatty liver disease at the global, regional and national levels, 1990-2017: a population-based observational study. BMJ Open 10, e036663 10.1136/bmjopen-2019-036663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman S.L., Neuschwander-Tetri B.A., Rinella M. and Sanyal A.J. (2018) Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922 10.1038/s41591-018-0104-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mundi M.S., Velapati S., Patel J., Kellogg T.A., Abu Dayyeh B.K. and Hurt R.T. (2020) Evolution of NAFLD and its management. Nutr. Clin. Pract. 35, 72–84 10.1002/ncp.10449 [DOI] [PubMed] [Google Scholar]
- 6.Lonardo A., Nascimbeni F., Mantovani A. and Targher G. (2018) Hypertension, diabetes, atherosclerosis and NASH: cause or consequence? J. Hepatol. 68, 335–352 10.1016/j.jhep.2017.09.021 [DOI] [PubMed] [Google Scholar]
- 7.Eslam M., Newsome P.N., Sarin S.K., Anstee Q.M., Targher G., Romero-Gomez M.et al. (2020) A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J. Hepatol. 73, 202–209 10.1016/j.jhep.2020.03.039 [DOI] [PubMed] [Google Scholar]
- 8.Bellentani S., Scaglioni F., Marino M. and Bedogni G. (2010) Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 28, 155–161 10.1159/000282080 [DOI] [PubMed] [Google Scholar]
- 9.Goyal O., Nohria S., Goyal P., Kaur J., Sharma S., Sood A.et al. (2020) Saroglitazar in patients with non-alcoholic fatty liver disease and diabetic dyslipidemia: a prospective, observational, real world study. Sci. Rep. 10, 21117 10.1038/s41598-020-78342-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexander M., Loomis A.K., van der Lei J., Duarte-Salles T., Prieto-Alhambra D., Ansell D.et al. (2019) Non-alcoholic fatty liver disease and risk of incident acute myocardial infarction and stroke: findings from matched cohort study of 18 million European adults. BMJ 367, l5367 10.1136/bmj.l5367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savage D.B., Petersen K.F. and Shulman G.I. (2007) Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 87, 507–520 10.1152/physrev.00024.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith G.I., Shankaran M., Yoshino M., Schweitzer G.G., Chondronikola M., Beals J.W.et al. (2020) Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Invest. 130, 1453–1460 10.1172/JCI134165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown M.S. and Goldstein J.L. (2008) Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7, 95–96 10.1016/j.cmet.2007.12.009 [DOI] [PubMed] [Google Scholar]
- 14.Wanless I.R. and Shiota K. (2004) The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin. Liver Dis. 24, 99–106 10.1055/s-2004-823104 [DOI] [PubMed] [Google Scholar]
- 15.Chalasani N., Younossi Z., Lavine J.E., Charlton M., Cusi K., Rinella M.et al. (2018) The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357 10.1002/hep.29367 [DOI] [PubMed] [Google Scholar]
- 16.Oliveros-Montiel A., Santos-Lopez G. and Sedeno-Monge V. (2020) Proteins involved in lipid metabolism as possible biomarkers or predisposing factors for non-alcoholic fatty liver disease. Acta Gastroenterol. Belg. 83, 622–630 [PubMed] [Google Scholar]
- 17.Qayyum A., Nystrom M., Noworolski S.M., Chu P., Mohanty A. and Merriman R. (2012) MRI steatosis grading: development and initial validation of a color mapping system. AJR. Am. J. Roentgenol. 198, 582–588 10.2214/AJR.11.6729 [DOI] [PubMed] [Google Scholar]
- 18.Nassir F., Rector R.S., Hammoud G.M. and Ibdah J.A. (2015) Pathogenesis and prevention of hepatic steatosis. Gastroenterol Hepatol (N Y) 11, 167–175 [PMC free article] [PubMed] [Google Scholar]
- 19.John K., Marino J.S., Sanchez E.R. and Hinds T.D. Jr (2016) The glucocorticoid receptor: cause of or cure for obesity? Am. J. Physiol. Endocrinol. Metab. 310, E249–E257 10.1152/ajpendo.00478.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Brien L., Hosick P.A., John K., Stec D.E. and Hinds T.D. Jr (2015) Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 26, 212–220 10.1016/j.tem.2015.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creeden J.F., Gordon D.M., Stec D.E. and Hinds T.D. Jr (2021) Bilirubin as a metabolic hormone: the physiological relevance of low levels. Am. J. Physiol. Endocrinol. Metab. 320, E191–E207 10.1152/ajpendo.00405.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stec D.E., Gordon D.M., Nestor-Kalinoski A.L., Donald M.C., Mitchell Z.L., Creeden J.F.et al. (2020) Biliverdin reductase A (BVRA) knockout in adipocytes induces hypertrophy and reduces mitochondria in white fat of obese mice. Biomolecules 10,1–14 10.3390/biom10030387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinds T.D. Jr, Kipp Z.A., Xu M., Yiannikouris F.B., Morris A.J., Stec D.F.et al. (2021) Adipose-specific PPARalpha knockout mice have increased lipogenesis by PASK-SREBP1 signaling and a polarity shift to inflammatory macrophages in white adipose tissue. Cells 11,1-20 10.3390/cells11010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall P. and Cash J. (2012) What is the real function of the liver ‘function’ tests? Ulster Med. J. 81, 30–36 [PMC free article] [PubMed] [Google Scholar]
- 25.Osna N.A., Donohue T.M. Jr and Kharbanda K.K. (2017) Alcoholic liver disease: pathogenesis and current management. Alcohol Res. 38, 147–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinds T.D. Jr and Stec D.E. (2019) Bilirubin safeguards cardiorenal and metabolic diseases: a protective role in health. Curr. Hypertens. Rep. 21, 87 10.1007/s11906-019-0994-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinds T.D. Jr and Stec D.E. (2018) Bilirubin, a cardiometabolic signaling molecule. Hypertension 72, 788–795 10.1161/HYPERTENSIONAHA.118.11130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamoud A.R., Weaver L., Stec D.E. and Hinds T.D. Jr (2018) Bilirubin in the liver-gut signaling axis. Trends Endocrinol. Metab. 29, 140–150 10.1016/j.tem.2018.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinds T.D. Jr, Creeden J.F., Gordon D.M., Stec D.F., Donald M.C. and Stec D.E. (2020) Bilirubin nanoparticles reduce diet-induced hepatic steatosis, improve fat utilization, and increase plasma beta-hydroxybutyrate. Front. Pharmacol. 11, 594574 10.3389/fphar.2020.594574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon D.M., Neifer K.L., Hamoud A.A., Hawk C.F., Nestor-Kalinoski A.L., Miruzzi S.A.et al. (2020) Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 10.1074/jbc.RA120.013700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hinds T.D. Jr, Hosick P.A., Hankins M.W., Nestor-Kalinoski A. and Stec D.E. (2017) Mice with hyperbilirubinemia due to Gilbert's Syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARalpha. Am. J. Physiol. Endocrinol. Metab. 2016, ajpendo 00396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stec D.E., John K., Trabbic C.J., Luniwal A., Hankins M.W., Baum J.et al. (2016) Bilirubin Binding to PPARalpha Inhibits Lipid Accumulation. PloS ONE 11, e0153427 10.1371/journal.pone.0153427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stec D.E. and Hinds T.D. Jr (2020) Natural Product Heme Oxygenase Inducers as Treatment for Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 21, 10.3390/ijms21249493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong V.W., Wong G.L., Tsang S.W., Hui A.Y., Chan A.W., Choi P.C.et al. (2009) Metabolic and histological features of non-alcoholic fatty liver disease patients with different serum alanine aminotransferase levels. Aliment. Pharmacol. Ther. 29, 387–396 10.1111/j.1365-2036.2008.03896.x [DOI] [PubMed] [Google Scholar]
- 35.Eguchi A., Iwasa M., Yamada M., Tamai Y., Shigefuku R., Hasegawa H.et al. (2022) A new detection system for serum fragmented cytokeratin 18 as a biomarker reflecting histologic activities of human nonalcoholic steatohepatitis. Hepatol. Commun. 6, 1987–1999 10.1002/hep4.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cengiz M., Ozenirler S. and Kocabiyik M. (2016) Serum beta-trophin level as a new marker for noninvasive assessment of nonalcoholic fatty liver disease and liver fibrosis. Eur. J. Gastroenterol. Hepatol. 28, 57–63 10.1097/MEG.0000000000000502 [DOI] [PubMed] [Google Scholar]
- 37.Jamali R., Arj A., Razavizade M. and Aarabi M.H. (2016) Prediction of Nonalcoholic Fatty Liver Disease Via a Novel Panel of Serum Adipokines. Medicine (Baltimore). 95, e2630 10.1097/MD.0000000000002630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weaver L., Hamoud A.R., Stec D.E. and Hinds T.D. Jr (2018) Biliverdin reductase and bilirubin in hepatic disease. Am. J. Physiol. Gastrointest. Liver Physiol. 314, G668–G676 10.1152/ajpgi.00026.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Creeden J.F., Kipp Z.A., Xu M., Flight R.M., Moseley H.N.B., Martinez G.J.et al. (2022) Hepatic Kinome Atlas: An In-Depth Identification of Kinase Pathways in Liver Fibrosis of Humans and Rodents. Hepatology 10.1002/hep.32467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burz S.D., Monnoye M., Philippe C., Farin W., Ratziu V., Strozzi F.et al. (2021) Fecal Microbiota Transplant from Human to Mice Gives Insights into the Role of the Gut Microbiota in Non-Alcoholic Fatty Liver Disease (NAFLD). Microorganisms 9, 10.3390/microorganisms9010199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seki E., Tsutsui H., Nakano H., Tsuji N., Hoshino K., Adachi O.et al. (2001) Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J. Immunol. 166, 2651–2657 10.4049/jimmunol.166.4.2651 [DOI] [PubMed] [Google Scholar]
- 42.Stec D.E., Gordon D.M., Hipp J.A., Hong S., Mitchell Z.L., Franco N.R.et al. (2019) Loss of hepatic PPARalpha promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 317, R733–R745 10.1152/ajpregu.00153.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tarantino G., Citro V., Balsano C. and Capone D. (2020) Could SCGF-Beta Levels Be Associated with Inflammation Markers and Insulin Resistance in Male Patients Suffering from Obesity-Related NAFLD? Diagnostics (Basel) 10, 10.3390/diagnostics10060395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yen F.S., Lai J.N., Wei J.C., Chiu L.T., Hsu C.C., Hou M.C.et al. (2021) Is insulin the preferred treatment in persons with type 2 diabetes and liver cirrhosis? BMC Gastroenterol. 21, 263 10.1186/s12876-021-01773-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peterson S.W., Angelico M., Masella R., Foster K., Gandin C. and Cantafora A. (1992) Altered insulin receptor processing and membrane lipid composition in erythrocytes of cirrhotic patients. Ital. J. Gastroenterol. 24, 65–71 [PubMed] [Google Scholar]
- 46.Okada Y. (1981) Increased insulin binding to erythrocytes in chronic liver disease. Acta Med. Okayama 35, 155–164 [DOI] [PubMed] [Google Scholar]
- 47.Petrides A.S., Passlack W., Reinauer H., Stremmel W. and Strohmeyer G. (1987) Insulin binding to erythrocytes in hyperinsulinemic patients with precirrhotic hemochromatosis and cirrhosis. Klin. Wochenschr. 65, 873–878 10.1007/BF01737009 [DOI] [PubMed] [Google Scholar]
- 48.Teng C.S., Ho P.W. and Yeung R.T. (1982) Down-regulation of insulin receptors in postnecrotic cirrhosis of liver. J. Clin. Endocrinol. Metab. 55, 524–530 10.1210/jcem-55-3-524 [DOI] [PubMed] [Google Scholar]
- 49.Greco A.V., Bertoli A., Ghirlanda G., Manna R., Altomonte L. and Rebuzzi A.G. (1980) Insulin resistance in liver cirrhosis: decreased insulin binding to circulating monocytes. Hormone and Metabolic Research = Hormon- Und Stoffwechselforschung = Hormones et Metabolisme 12, 577–581 10.1055/s-2007-999204 [DOI] [PubMed] [Google Scholar]
- 50.Harewood M.S., Proietto J., Dudley F. and Alford F.P. (1982) Insulin action and cirrhosis: insulin binding and lipogenesis in isolated adipocytes. Metabolism 31, 1241–1246 10.1016/0026-0495(82)90011-7 [DOI] [PubMed] [Google Scholar]
- 51.Ipsen D.H., Lykkesfeldt J. and Tveden-Nyborg P. (2018) Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci.:CMLS 75, 3313–3327 10.1007/s00018-018-2860-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newberry E.P., Xie Y., Kennedy S., Han X., Buhman K.K., Luo J.et al. (2003) Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J. Biol. Chem. 278, 51664–51672 10.1074/jbc.M309377200 [DOI] [PubMed] [Google Scholar]
- 53.Newberry E.P., Xie Y., Kennedy S.M., Luo J. and Davidson N.O. (2006) Protection against Western diet-induced obesity and hepatic steatosis in liver fatty acid-binding protein knockout mice. Hepatology 44, 1191–1205 10.1002/hep.21369 [DOI] [PubMed] [Google Scholar]
- 54.Mukai T., Egawa M., Takeuchi T., Yamashita H. and Kusudo T. (2017) Silencing of FABP1 ameliorates hepatic steatosis, inflammation, and oxidative stress in mice with nonalcoholic fatty liver disease. FEBS Open Bio. 7, 1009–1016 10.1002/2211-5463.12240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu Y.C., Chang C.C., Wang C.P., Hung W.C., Tsai I.T., Tang W.H.et al. (2020) Circulating fatty acid-binding protein 1 (FABP1) and nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. Int. J. Med. Sci. 17, 182–190 10.7150/ijms.40417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang G., Bonkovsky H.L., de Lemos A. and Burczynski F.J. (2015) Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 56, 2238–2247 10.1194/jlr.R056705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doege H., Baillie R.A., Ortegon A.M., Tsang B., Wu Q., Punreddy S.et al. (2006) Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis. Gastroenterology 130, 1245–1258 10.1053/j.gastro.2006.02.006 [DOI] [PubMed] [Google Scholar]
- 58.Doege H., Grimm D., Falcon A., Tsang B., Storm T.A., Xu H.et al. (2008) Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J. Biol. Chem. 283, 22186–22192 10.1074/jbc.M803510200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Auinger A., Valenti L., Pfeuffer M., Helwig U., Herrmann J., Fracanzani A.L.et al. (2010) A promoter polymorphism in the liver-specific fatty acid transport protein 5 is associated with features of the metabolic syndrome and steatosis. Hormone and Metabolic Res. = Hormon- Und Stoffwechselforschung = Hormones et Metabolisme 42, 854–859 10.1055/s-0030-1267186 [DOI] [PubMed] [Google Scholar]
- 60.Koonen D.P., Jacobs R.L., Febbraio M., Young M.E., Soltys C.L., Ong H.et al. (2007) Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 56, 2863–2871 10.2337/db07-0907 [DOI] [PubMed] [Google Scholar]
- 61.Buttet M., Poirier H., Traynard V., Gaire K., Tran T.T., Sundaresan S.et al. (2016) Deregulated Lipid Sensing by Intestinal CD36 in Diet-Induced Hyperinsulinemic Obese Mouse Model. PloS ONE 11, e0145626 10.1371/journal.pone.0145626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miquilena-Colina M.E., Lima-Cabello E., Sanchez-Campos S., Garcia-Mediavilla M.V., Fernandez-Bermejo M., Lozano-Rodriguez T.et al. (2011) Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 60, 1394–1402 10.1136/gut.2010.222844 [DOI] [PubMed] [Google Scholar]
- 63.Rada P., Gonzalez-Rodriguez A., Garcia-Monzon C. and Valverde A.M. (2020) Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis. 11, 802 10.1038/s41419-020-03003-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hong S., Gordon D., Stec D.E. and Hinds T.D. (2021) Bilirubin: A Ligand of the PPARα Nuclear Receptor. In Nuclear Receptors: The Art and Science of Modulator Design and Discovery(Badr M.Z., ed.), pp. 463–482, Springer International Publishing, Cham: 10.1007/978-3-030-78315-0_17 [DOI] [Google Scholar]
- 65.Sanders F.W. and Griffin J.L. (2016) De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 91, 452–468 10.1111/brv.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joshi-Barve S., Barve S.S., Amancherla K., Gobejishvili L., Hill D., Cave M.et al. (2007) Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 46, 823–830 10.1002/hep.21752 [DOI] [PubMed] [Google Scholar]
- 67.Knebel B., Haas J., Hartwig S., Jacob S., Kollmer C., Nitzgen U.et al. (2012) Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PloS ONE 7, e31812 10.1371/journal.pone.0031812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iizuka K., Takao K. and Yabe D. (2020) ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. (Lausanne) 11, 587189 10.3389/fendo.2020.587189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iizuka K., Bruick R.K., Liang G., Horton J.D. and Uyeda K. (2004) Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. U. S. A. 101, 7281–7286 10.1073/pnas.0401516101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dentin R., Benhamed F., Hainault I., Fauveau V., Foufelle F., Dyck J.R.et al. (2006) Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 55, 2159–2170 10.2337/db06-0200 [DOI] [PubMed] [Google Scholar]
- 71.Moreno-Fernandez M.E., Giles D.A., Stankiewicz T.E., Sheridan R., Karns R., Cappelletti M.et al. (2018) Peroxisomal beta-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI Insight 3, 10.1172/jci.insight.93626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hinds T.D. Jr., Adeosun S.O., Alamodi A.A. and Stec D.E. (2016) Does bilirubin prevent hepatic steatosis through activation of the PPARalpha nuclear receptor? Med. Hypotheses 95, 54–57 10.1016/j.mehy.2016.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hinds T.D. Jr., Burns K.A., Hosick P.A., McBeth L., Nestor-Kalinoski A., Drummond H.A.et al. (2016) Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3beta phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) alpha. J. Biol. Chem. 10.1074/jbc.M116.731703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Francque S., Verrijken A., Caron S., Prawitt J., Paumelle R., Derudas B.et al. (2015) PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 63, 164–173 10.1016/j.jhep.2015.02.019 [DOI] [PubMed] [Google Scholar]
- 75.Wang Y., Nakajima T., Gonzalez F.J. and Tanaka N. (2020) PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 21, 10.3390/ijms21062061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ota T., Gayet C. and Ginsberg H.N. (2008) Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J. Clin. Invest. 118, 316–332 10.1172/JCI32752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peng X.E., Wu Y.L., Lu Q.Q., Hu Z.J. and Lin X. (2014) MTTP polymorphisms and susceptibility to non-alcoholic fatty liver disease in a Han Chinese population. Liver Int. 34, 118–128 10.1111/liv.12220 [DOI] [PubMed] [Google Scholar]
- 78.Shindo N., Fujisawa T., Sugimoto K., Nojima K., Oze-Fukai A., Yoshikawa Y.et al. (2010) Involvement of microsomal triglyceride transfer protein in nonalcoholic steatohepatitis in novel spontaneous mouse model. J. Hepatol. 52, 903–912 10.1016/j.jhep.2009.12.033 [DOI] [PubMed] [Google Scholar]
- 79.Charlton M., Sreekumar R., Rasmussen D., Lindor K. and Nair K.S. (2002) Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology 35, 898–904 10.1053/jhep.2002.32527 [DOI] [PubMed] [Google Scholar]
- 80.Whitsett M. and VanWagner L.B. (2015) Physical activity as a treatment of non-alcoholic fatty liver disease: A systematic review. World J. Hepatol. 7, 2041–2052 10.4254/wjh.v7.i16.2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bugianesi E., Gentilcore E., Manini R., Natale S., Vanni E., Villanova N.et al. (2005) A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am. J. Gastroenterol. 100, 1082–1090 10.1111/j.1572-0241.2005.41583.x [DOI] [PubMed] [Google Scholar]
- 82.Zang M., Zuccollo A., Hou X., Nagata D., Walsh K., Herscovitz H.et al. (2004) AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 279, 47898–47905 10.1074/jbc.M408149200 [DOI] [PubMed] [Google Scholar]
- 83.Steinberg G.R., Macaulay S.L., Febbraio M.A. and Kemp B.E. (2006) AMP-activated protein kinase–the fat controller of the energy railroad. Can. J. Physiol. Pharmacol. 84, 655–665 10.1139/y06-005 [DOI] [PubMed] [Google Scholar]
- 84.Woo S.L., Xu H., Li H., Zhao Y., Hu X., Zhao J.et al. (2014) Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PloS ONE 9, e91111 10.1371/journal.pone.0091111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang D., Ma Y., Liu J., Deng Y., Zhou B., Wen Y.et al. (2021) Metformin Alleviates Hepatic Steatosis and Insulin Resistance in a Mouse Model of High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease by Promoting Transcription Factor EB-Dependent Autophagy. Front. Pharmacol. 12, 689111 10.3389/fphar.2021.689111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Romero-Gomez M., Diago M., Andrade R.J., Calleja J.L., Salmeron J., Fernandez-Rodriguez C.M.et al. (2009) Treatment of insulin resistance with metformin in naive genotype 1 chronic hepatitis C patients receiving peginterferon alfa-2a plus ribavirin. Hepatology 50, 1702–1708 10.1002/hep.23206 [DOI] [PubMed] [Google Scholar]
- 87.Duseja A., Das A., Dhiman R.K., Chawla Y.K., Thumburu K.T., Bhadada S.et al. (2007) Metformin is effective in achieving biochemical response in patients with nonalcoholic fatty liver disease (NAFLD) not responding to lifestyle interventions. Ann. Hepatol. 6, 222–226 10.1016/S1665-2681(19)31902-7 [DOI] [PubMed] [Google Scholar]
- 88.Cone C.J., Bachyrycz A.M. and Murata G.H. (2010) Hepatotoxicity associated with metformin therapy in treatment of type 2 diabetes mellitus with nonalcoholic fatty liver disease. Ann. Pharmacother. 44, 1655–1659 10.1345/aph.1P099 [DOI] [PubMed] [Google Scholar]
- 89.Miralles-Linares F., Puerta-Fernandez S., Bernal-Lopez M.R., Tinahones F.J., Andrade R.J. and Gomez-Huelgas R. (2012) Metformin-induced hepatotoxicity. Diabetes Care. 35, e21 10.2337/dc11-2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.American Diabetes A. (2019) 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2019. Diabetes Care. 42, S90–S102 10.2337/dc19-S009 [DOI] [PubMed] [Google Scholar]
- 91.Rakoski M.O., Singal A.G., Rogers M.A. and Conjeevaram H. (2010) Meta-analysis: insulin sensitizers for the treatment of non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 32, 1211–1221 10.1111/j.1365-2036.2010.04467.x [DOI] [PubMed] [Google Scholar]
- 92.Li Y., Liu L., Wang B., Wang J. and Chen D. (2013) Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 1, 57–64 10.3892/br.2012.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Musso G., Gambino R., Cassader M. and Pagano G. (2010) A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 52, 79–104 10.1002/hep.23623 [DOI] [PubMed] [Google Scholar]
- 94.Pereira M.J. and Eriksson J.W. (2019) Emerging Role of SGLT-2 Inhibitors for the Treatment of Obesity. Drugs 79, 219–230 10.1007/s40265-019-1057-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shiba K., Tsuchiya K., Komiya C., Miyachi Y., Mori K., Shimazu N.et al. (2018) Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 8, 2362 10.1038/s41598-018-19658-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Leiter L.A., Forst T., Polidori D., Balis D.A., Xie J. and Sha S. (2016) Effect of canagliflozin on liver function tests in patients with type 2 diabetes. Diabetes Metab. 42, 25–32 10.1016/j.diabet.2015.10.003 [DOI] [PubMed] [Google Scholar]
- 97.Sattar N., Fitchett D., Hantel S., George J.T. and Zinman B. (2018) Empagliflozin is associated with improvements in liver enzymes potentially consistent with reductions in liver fat: results from randomised trials including the EMPA-REG OUTCOME(R) trial. Diabetologia 61, 2155–2163 10.1007/s00125-018-4702-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Taheri H., Malek M., Ismail-Beigi F., Zamani F., Sohrabi M., Reza Babaei M.et al. (2020) Effect of Empagliflozin on Liver Steatosis and Fibrosis in Patients With Non-Alcoholic Fatty Liver Disease Without Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. Adv. Ther. 37, 4697–4708 10.1007/s12325-020-01498-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lai L.L., Vethakkan S.R., Nik Mustapha N.R., Mahadeva S. and Chan W.K. (2020) Empagliflozin for the Treatment of Nonalcoholic Steatohepatitis in Patients with Type 2 Diabetes Mellitus. Dig. Dis. Sci. 65, 623–631 10.1007/s10620-019-5477-1 [DOI] [PubMed] [Google Scholar]
- 100.Jojima T., Tomotsune T., Iijima T., Akimoto K., Suzuki K. and Aso Y. (2016) Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetol. Metab. Syndr 8, 45 10.1186/s13098-016-0169-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Benetti E., Mastrocola R., Vitarelli G., Cutrin J.C., Nigro D., Chiazza F.et al. (2016) Empagliflozin Protects against Diet-Induced NLRP-3 Inflammasome Activation and Lipid Accumulation. J. Pharmacol. Exp. Ther. 359, 45–53 10.1124/jpet.116.235069 [DOI] [PubMed] [Google Scholar]
- 102.Tahara A., Kurosaki E., Yokono M., Yamajuku D., Kihara R., Hayashizaki Y.et al. (2013) Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur. J. Pharmacol. 715, 246–255 10.1016/j.ejphar.2013.05.014 [DOI] [PubMed] [Google Scholar]
- 103.Kenny P.R., Brady D.E., Torres D.M., Ragozzino L., Chalasani N. and Harrison S.A. (2010) Exenatide in the treatment of diabetic patients with non-alcoholic steatohepatitis: a case series. Am. J. Gastroenterol. 105, 2707–2709 10.1038/ajg.2010.363 [DOI] [PubMed] [Google Scholar]
- 104.Sofogianni A., Filippidis A., Chrysavgis L., Tziomalos K. and Cholongitas E. (2020) Glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: An update. World J. Hepatol. 12, 493–505 10.4254/wjh.v12.i8.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shao N., Yu X.Y., Ma X.F., Lin W.J., Hao M. and Kuang H.Y. (2018) Exenatide Delays the Progression of Nonalcoholic Fatty Liver Disease in C57BL/6 Mice, Which May Involve Inhibition of the NLRP3 Inflammasome through the Mitophagy Pathway. Gastroenterol Res. Pract. 2018, 1864307 10.1155/2018/1864307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Svegliati-Baroni G., Saccomanno S., Rychlicki C., Agostinelli L., De Minicis S., Candelaresi C.et al. (2011) Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 31, 1285–1297 10.1111/j.1478-3231.2011.02462.x [DOI] [PubMed] [Google Scholar]
- 107.Liu L., Yan H., Xia M., Zhao L., Lv M., Zhao N.et al. (2020) Efficacy of exenatide and insulin glargine on nonalcoholic fatty liver disease in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 36, e3292 10.1002/dmrr.3292 [DOI] [PubMed] [Google Scholar]
- 108.Sayari S., Neishaboori H. and Jameshorani M. (2018) Combined effects of synbiotic and sitagliptin versus sitagliptin alone in patients with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 24, 331–338 10.3350/cmh.2018.0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shirakawa J., Fujii H., Ohnuma K., Sato K., Ito Y., Kaji M.et al. (2011) Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes 60, 1246–1257 10.2337/db10-1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shen T., Xu B., Lei T., Chen L., Zhang C. and Ni Z. (2018) Sitagliptin reduces insulin resistance and improves rat liver steatosis via the SIRT1/AMPKalpha pathway. Exp. Ther. Med. 16, 3121–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yilmaz Y., Yonal O., Deyneli O., Celikel C.A., Kalayci C. and Duman D.G. (2012) Effects of sitagliptin in diabetic patients with nonalcoholic steatohepatitis. Acta Gastroenterol. Belg. 75, 240–244 [PubMed] [Google Scholar]
- 112.Joy T.R., McKenzie C.A., Tirona R.G., Summers K., Seney S., Chakrabarti S.et al. (2017) Sitagliptin in patients with non-alcoholic steatohepatitis: A randomized, placebo-controlled trial. World J. Gastroenterol. 23, 141–150 10.3748/wjg.v23.i1.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen L., Zhang X., Zhang L. and Zheng D. (2020) Effect of Saxagliptin, a Dipeptidyl Peptidase 4 Inhibitor, on Non-Alcoholic Fatty Liver Disease. Diab. Metab. Syndrome Obesity:Targets Therapy 13, 3507–3518 10.2147/DMSO.S262284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Belfort R., Harrison S.A., Brown K., Darland C., Finch J., Hardies J.et al. (2006) A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 355, 2297–2307 10.1056/NEJMoa060326 [DOI] [PubMed] [Google Scholar]
- 115.Bril F., Kalavalapalli S., Clark V.C., Lomonaco R., Soldevila-Pico C., Liu I.C.et al. (2018) Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 16, 558e2–566e2 10.1016/j.cgh.2017.12.001 [DOI] [PubMed] [Google Scholar]
- 116.Saryusz-Wolska M., Szymanska-Garbacz E., Jablkowski M., Bialkowska J., Pawlowski M., Kwiecinska E.et al. (2011) Rosiglitazone treatment in nondiabetic subjects with nonalcoholic fatty liver disease. Pol. Arch. Med. Wewn. 121, 61–66 10.20452/pamw.1023 [DOI] [PubMed] [Google Scholar]
- 117.Wang C.H., Leung C.H., Liu S.C. and Chung C.H. (2006) Safety and effectiveness of rosiglitazone in type 2 diabetes patients with nonalcoholic Fatty liver disease. J. Formos. Med. Assoc. 105, 743–752 10.1016/S0929-6646(09)60202-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sumida Y. and Yoneda M. (2018) Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 53, 362–376 10.1007/s00535-017-1415-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goedeke L., Bates J., Vatner D.F., Perry R.J., Wang T., Ramirez R.et al. (2018) Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 68, 2197–2211 10.1002/hep.30097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tamura Y.O., Sugama J., Iwasaki S., Sasaki M., Yasuno H., Aoyama K.et al. (2021) Selective Acetyl-CoA Carboxylase 1 Inhibitor Improves Hepatic Steatosis and Hepatic Fibrosis in a Preclinical Nonalcoholic Steatohepatitis Model. J. Pharmacol. Exp. Ther. 379, 280–289 10.1124/jpet.121.000786 [DOI] [PubMed] [Google Scholar]
- 121.Calle R.A., Amin N.B., Carvajal-Gonzalez S., Ross T.T., Bergman A., Aggarwal S.et al. (2021) ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 27, 1836–1848 10.1038/s41591-021-01489-1 [DOI] [PubMed] [Google Scholar]
- 122.Saini N., Black P.N., Montefusco D. and DiRusso C.C. (2015) Fatty acid transport protein-2 inhibitor Grassofermata/CB5 protects cells against lipid accumulation and toxicity. Biochem. Biophys. Res. Commun. 465, 534–541 10.1016/j.bbrc.2015.08.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tao X., He H., Peng J., Xu R., Fu J., Hu Y.et al. (2022) Overexpression of PDE4D in mouse liver is sufficient to trigger NAFLD and hypertension in a CD36-TGF-beta1 pathway: therapeutic role of roflumilast. Pharmacol. Res. 175, 106004 10.1016/j.phrs.2021.106004 [DOI] [PubMed] [Google Scholar]
- 124.Smedlund K.B., Sanchez E.R. and Hinds T.D. Jr (2021) FKBP51 and the molecular chaperoning of metabolism. Trends Endocrinol. Metab. 32, 862–874 10.1016/j.tem.2021.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hinds T.D., John K., McBeth L., Trabbic C.J. and Sanchez E.R. (2016) Timcodar (VX-853) Is a Non-FKBP12 Binding Macrolide Derivative That Inhibits PPARγand Suppresses Adipogenesis. PPAR Res. 2016, 1–10 10.1155/2016/6218637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang C., Yan Y., Hu L., Zhao L., Yang P., Moorhead J.F.et al. (2014) Rapamycin-mediated CD36 translational suppression contributes to alleviation of hepatic steatosis. Biochem. Biophys. Res. Commun. 447, 57–63 10.1016/j.bbrc.2014.03.103 [DOI] [PubMed] [Google Scholar]
- 127.Hinds T.D., Stechschulte L.A., Elkhairi F. and Sanchez E.R. (2014) Analysis of FK506, timcodar (VX-853) and FKBP51 and FKBP52 chaperones in control of glucocorticoid receptor activity and phosphorylation. Pharmacol. Res. Perspect. 2, e00076 10.1002/prp2.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chang G.R., Wu Y.Y., Chiu Y.S., Chen W.Y., Liao J.W., Hsu H.M.et al. (2009) Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin. Pharmacol. Toxicol. 105, 188–198 10.1111/j.1742-7843.2009.00427.x [DOI] [PubMed] [Google Scholar]
- 129.Stec D.E., Gordon D.M., Hipp J.A., Hong S., Mitchell Z.L., Franco N.R.et al. (2019) The loss of hepatic PPARalpha promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 10.1152/ajpregu.00153.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fougerat A., Montagner A., Loiseau N., Guillou H. and Wahli W. (2020) Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 9, 10.3390/cells9071638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liss K.H. and Finck B.N. (2017) PPARs and nonalcoholic fatty liver disease. Biochimie 136, 65–74 10.1016/j.biochi.2016.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Montagner A., Polizzi A., Fouche E., Ducheix S., Lippi Y., Lasserre F.et al. (2016) Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 65, 1202–1214 10.1136/gutjnl-2015-310798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hu X., Tanaka N., Guo R., Lu Y., Nakajima T., Gonzalez F.J.et al. (2017) PPARalpha protects against trans-fatty-acid-containing diet-induced steatohepatitis. J. Nutr. Biochem. 39, 77–85 10.1016/j.jnutbio.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang D., Liu B., Tao W., Hao Z. and Liu M. (2015) Fibrates for secondary prevention of cardiovascular disease and stroke. Cochrane Database Syst. Rev. CD009580 10.1002/14651858.CD009580.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Scott R., O'Brien R., Fulcher G., Pardy C., D'Emden M., Tse D.et al. (2009) Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care. 32, 493–498 10.2337/dc08-1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim N.H., Han K.H., Choi J., Lee J. and Kim S.G. (2019) Use of fenofibrate on cardiovascular outcomes in statin users with metabolic syndrome: propensity matched cohort study. BMJ 366, l5125 10.1136/bmj.l5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fatani S., Itua I., Clark P., Wong C. and Naderali E.K. (2011) The effects of diet-induced obesity on hepatocyte insulin signaling pathways and induction of non-alcoholic liver damage. Int. J. Gen. Med. 4, 211–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang D., Niu S., Ma Y., Chen H., Wen Y., Li M.et al. (2021) Fenofibrate Improves Insulin Resistance and Hepatic Steatosis and Regulates the Let-7/SERCA2b Axis in High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease Mice. Front. Pharmacol. 12, 770652 10.3389/fphar.2021.770652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Harano Y., Yasui K., Toyama T., Nakajima T., Mitsuyoshi H., Mimani M.et al. (2006) Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int. 26, 613–620 10.1111/j.1478-3231.2006.01265.x [DOI] [PubMed] [Google Scholar]
- 140.Kostapanos M.S., Kei A. and Elisaf M.S. (2013) Current role of fenofibrate in the prevention and management of non-alcoholic fatty liver disease. World J. Hepatol. 5, 470–478 10.4254/wjh.v5.i9.470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Abdelmoneim D., El-Adl M., El-Sayed G. and El-Sherbini E.S. (2021) Protective effect of fenofibrate against high-fat-high-fructose diet induced non-obese NAFLD in rats. Fundam. Clin. Pharmacol. 35, 379–388 10.1111/fcp.12597 [DOI] [PubMed] [Google Scholar]
- 142.Shiri-Sverdlov R., Wouters K., van Gorp P.J., Gijbels M.J., Noel B., Buffat L.et al. (2006) Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J. Hepatol. 44, 732–741 10.1016/j.jhep.2005.10.033 [DOI] [PubMed] [Google Scholar]
- 143.Baron M., Leroyer A.S., Majd Z., Lalloyer F., Vallez E., Bantubungi K.et al. (2011) PPARalpha activation differently affects microparticle content in atherosclerotic lesions and liver of a mouse model of atherosclerosis and NASH. Atherosclerosis 218, 69–76 10.1016/j.atherosclerosis.2011.03.009 [DOI] [PubMed] [Google Scholar]
- 144.van der Veen J.N., Lingrell S., Gao X., Takawale A., Kassiri Z., Vance D.E.et al. (2017) Fenofibrate, but not ezetimibe, prevents fatty liver disease in mice lacking phosphatidylethanolamine N-methyltransferase. J. Lipid Res. 58, 656–667 10.1194/jlr.M070631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yavarow Z.A., Kang H.R., Waskowicz L.R., Bay B.H., Young S.P., Yen P.M.et al. (2020) Fenofibrate rapidly decreases hepatic lipid and glycogen storage in neonatal mice with glycogen storage disease type Ia. Hum. Mol. Genet. 29, 286–294 10.1093/hmg/ddz290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Takei K., Han S.I., Murayama Y., Satoh A., Oikawa F., Ohno H.et al. (2017) Selective peroxisome proliferator-activated receptor-alpha modulator K-877 efficiently activates the peroxisome proliferator-activated receptor-alpha pathway and improves lipid metabolism in mice. J. Diabetes Investig. 8, 446–452 10.1111/jdi.12621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Arai H., Yamashita S., Yokote K., Araki E., Suganami H., Ishibashi S.et al. (2018) Efficacy and Safety of Pemafibrate Versus Fenofibrate in Patients with High Triglyceride and Low HDL Cholesterol Levels: A Multicenter, Placebo-Controlled, Double-Blind, Randomized Trial. J. Atheroscler. Thromb. 25, 521–538 10.5551/jat.44412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yoon M. (2010) PPARalpha in Obesity: Sex Difference and Estrogen Involvement. PPAR Res. 2010, 10.1155/2010/584296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jeong S. and Yoon M. (2007) Inhibition of the actions of peroxisome proliferator-activated receptor alpha on obesity by estrogen. Obesity (Silver Spring) 15, 1430–1440 10.1038/oby.2007.171 [DOI] [PubMed] [Google Scholar]
- 150.Yoon M. (2009) The role of PPARalpha in lipid metabolism and obesity: focusing on the effects of estrogen on PPARalpha actions. Pharmacol. Res. 60, 151–159 10.1016/j.phrs.2009.02.004 [DOI] [PubMed] [Google Scholar]
- 151.Lee H. and Yoon M. (2013) 17β-estradiol inhibits PPARα of skeletal muscle. Animal Cells and Systems 17, 331–340 10.1080/19768354.2013.831772 [DOI] [Google Scholar]
- 152.Jain M.R., Giri S.R., Trivedi C., Bhoi B., Rath A., Vanage G.et al. (2015) Saroglitazar, a novel PPARalpha/gamma agonist with predominant PPARalpha activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharmacol. Res. Perspect. 3, e00136 10.1002/prp2.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chiu M., McBeth L., Sindhwani P. and Hinds T.D. (2017) Deciphering the Roles of Thiazolidinediones and PPARgamma in Bladder Cancer. PPAR Res. 2017, 4810672 10.1155/2017/4810672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kumar D.P., Caffrey R., Marioneaux J., Santhekadur P.K., Bhat M., Alonso C.et al. (2020) The PPAR alpha/gamma Agonist Saroglitazar Improves Insulin Resistance and Steatohepatitis in a Diet Induced Animal Model of Nonalcoholic Fatty Liver Disease. Sci. Rep. 10, 9330 10.1038/s41598-020-66458-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Akbari R., Behdarvand T., Afarin R., Yaghooti H., Jalali M.T. and Mohammadtaghvaei N. (2021) Saroglitazar improved hepatic steatosis and fibrosis by modulating inflammatory cytokines and adiponectin in an animal model of non-alcoholic steatohepatitis. BMC Pharmacol. Toxicol. 22, 53 10.1186/s40360-021-00524-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kaul U., Parmar D., Manjunath K., Shah M., Parmar K., Patil K.P.et al. (2019) New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: integrated analysis of the real world evidence. Cardiovasc. Diabetol. 18, 80 10.1186/s12933-019-0884-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tolbol K.S., Kristiansen M.N., Hansen H.H., Veidal S.S., Rigbolt K.T., Gillum M.P.et al. (2018) Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J. Gastroenterol. 24, 179–194 10.3748/wjg.v24.i2.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.van den Hoek A.M., Verschuren L., Caspers M.P.M., Worms N., Menke A.L. and Princen H.M.G. (2021) Beneficial effects of elafibranor on NASH in E3L.CETP mice and differences between mice and men. Sci. Rep. 11, 5050 10.1038/s41598-021-83974-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cariou B., Hanf R., Lambert-Porcheron S., Zair Y., Sauvinet V., Noel B.et al. (2013) Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care. 36, 2923–2930 10.2337/dc12-2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ratziu V., Harrison S.A., Francque S., Bedossa P., Lehert P., Serfaty L.et al. (2016) Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 150, 1147–1159, e5 10.1053/j.gastro.2016.01.038 [DOI] [PubMed] [Google Scholar]
- 161.Wettstein G., Luccarini J.M., Poekes L., Faye P., Kupkowski F., Adarbes V.et al. (2017) The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 1, 524–537 10.1002/hep4.1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dewidar B., Kahl S., Pafili K. and Roden M. (2020) Metabolic liver disease in diabetes - From mechanisms to clinical trials. Metabolism 111S, 154299 10.1016/j.metabol.2020.154299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Francque S.M., Bedossa P., Ratziu V., Anstee Q.M., Bugianesi E., Sanyal A.J.et al. (2021) A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 385, 1547–1558 10.1056/NEJMoa2036205 [DOI] [PubMed] [Google Scholar]
- 164.Qiang G., Zhang L., Yang X., Xuan Q., Shi L., Zhang H.et al. (2012) Effect of valsartan on the pathological progression of hepatic fibrosis in rats with type 2 diabetes. Eur. J. Pharmacol. 685, 156–164 10.1016/j.ejphar.2012.04.028 [DOI] [PubMed] [Google Scholar]
- 165.Wang C.H., Liu H.M., Chang Z.Y., Huang T.H. and Lee T.Y. (2021) Losartan Prevents Hepatic Steatosis and Macrophage Polarization by Inhibiting HIF-1alpha in a Murine Model of NAFLD. Int. J. Mol. Sci. 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.van der Graaff D., Chotkoe S., De Winter B., De Man J., Casteleyn C., Timmermans J.P.et al. (2022) Vasoconstrictor antagonism improves functional and structural vascular alterations and liver damage in rats with early NAFLD. JHEP Rep. 4, 100412 10.1016/j.jhepr.2021.100412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Marino J.S., Stechschulte L.A., Stec D.E., Nestor-Kalinoski A., Coleman S. and Hinds T.D. Jr (2016) Glucocorticoid Receptor beta Induces Hepatic Steatosis by Augmenting Inflammation and Inhibition of the Peroxisome Proliferator-activated Receptor (PPAR) alpha. J. Biol. Chem. 291, 25776–25788 10.1074/jbc.M116.752311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Verbeek J., Spincemaille P., Vanhorebeek I., Van den Berghe G., Vander Elst I., Windmolders P.et al. (2017) Dietary intervention, but not losartan, completely reverses non-alcoholic steatohepatitis in obese and insulin resistant mice. Lipids in Health Dis. 16, 46 10.1186/s12944-017-0432-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yu F., Takahashi T., Moriya J., Kawaura K., Yamakawa J., Kusaka K.et al. (2006) Angiotensin-II receptor antagonist alleviates non-alcoholic fatty liver in KKAy obese mice with type 2 diabetes. J. Int. Med. Res. 34, 297–302 10.1177/147323000603400309 [DOI] [PubMed] [Google Scholar]
- 170.Kim M.Y., Cho M.Y., Baik S.K., Jeong P.H., Suk K.T., Jang Y.O.et al. (2012) Beneficial effects of candesartan, an angiotensin-blocking agent, on compensated alcoholic liver fibrosis - a randomized open-label controlled study. Liver Int. 32, 977–987 10.1111/j.1478-3231.2012.02774.x [DOI] [PubMed] [Google Scholar]
- 171.Vos M.B., Jin R., Konomi J.V., Cleeton R., Cruz J., Karpen S.et al. (2018) A randomized, controlled, crossover pilot study of losartan for pediatric nonalcoholic fatty liver disease. Pilot Feasibility Stud. 4, 109 10.1186/s40814-018-0306-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hirata T., Tomita K., Kawai T., Yokoyama H., Shimada A., Kikuchi M.et al. (2013) Effect of Telmisartan or Losartan for Treatment of Nonalcoholic Fatty Liver Disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY). Int. J. Endocrinol. 2013, 587140 10.1155/2013/587140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Benson S.C., Pershadsingh H.A., Ho C.I., Chittiboyina A., Desai P., Pravenec M.et al. (2004) Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension 43, 993–1002 10.1161/01.HYP.0000123072.34629.57 [DOI] [PubMed] [Google Scholar]
- 174.Luong X.G., Stevens S.K., Jekle A., Lin T.I., Gupta K., Misner D.et al. (2020) Regulation of gene transcription by thyroid hormone receptor beta agonists in clinical development for the treatment of non-alcoholic steatohepatitis (NASH). PloS ONE 15, e0240338 10.1371/journal.pone.0240338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fraile J.M., Palliyil S., Barelle C., Porter A.J. and Kovaleva M. (2021) Non-Alcoholic Steatohepatitis (NASH) - A Review of a Crowded Clinical Landscape, Driven by a Complex Disease. Drug Des. Devel. Ther. 15, 3997–4009 10.2147/DDDT.S315724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kannt A., Wohlfart P., Madsen A.N., Veidal S.S., Feigh M. and Schmoll D. (2021) Activation of thyroid hormone receptor-beta improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br. J. Pharmacol. 178, 2412–2423 10.1111/bph.15427 [DOI] [PubMed] [Google Scholar]
- 177.Sinha R.A. and Yen P.M. (2016) Thyroid hormone-mediated autophagy and mitochondrial turnover in NAFLD. Cell Biosci. 6, 46 10.1186/s13578-016-0113-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Harrison S.A., Bashir M.R., Guy C.D., Zhou R., Moylan C.A., Frias J.P.et al. (2019) Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 394, 2012–2024 10.1016/S0140-6736(19)32517-6 [DOI] [PubMed] [Google Scholar]
- 179.Pacana T. and Sanyal A.J. (2012) Vitamin E and nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 15, 641–648 10.1097/MCO.0b013e328357f747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Abdel-Maboud M., Menshawy A., Menshawy E., Emara A., Alshandidy M. and Eid M. (2020) The efficacy of vitamin E in reducing non-alcoholic fatty liver disease: a systematic review, meta-analysis, and meta-regression. Therap. Adv. Gastroenterol. 13, 1756284820974917 10.1177/1756284820974917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Thomas D.T., DelCimmuto N.R., Flack K.D., Stec D.E. and Hinds T.D. Jr (2022) Reactive Oxygen Species (ROS) and Antioxidants as Immunomodulators in Exercise: Implications for Heme Oxygenase and Bilirubin. Antioxidants (Basel) 11, 10.3390/antiox11020179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gordon D.M., Hong S.H., Kipp Z.A. and Hinds T.D. Jr (2021) Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha). Molecules 26, 10.3390/molecules26102975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gordon D.M., Blomquist T.M., Miruzzi S.A., McCullumsmith R., Stec D.E. and Hinds T.D. Jr (2019) RNA sequencing in human HepG2 hepatocytes reveals PPAR-alpha mediates transcriptome responsiveness of bilirubin. Physiol. Genomics 51, 234–240 10.1152/physiolgenomics.00028.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gordon D.M., Adeosun S.O., Ngwudike S.I., Anderson C.D., Hall J.E., Hinds T.D. Jret al. (2019) CRISPR Cas9-mediated deletion of biliverdin reductase A (BVRA) in mouse liver cells induces oxidative stress and lipid accumulation. Arch. Biochem. Biophys. 672, 108072 10.1016/j.abb.2019.108072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Adeosun S.O., Moore K.H., Lang D.M., Nwaneri A.C., Hinds T.D. Jr and Stec D.E. (2018) A Novel Fluorescence-Based Assay for the Measurement of Biliverdin Reductase Activity. React. Oxyg. Species (Apex) 5, 35–45 10.20455/ros.2018.809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Adeosun S.O., Gordon D.M., Weeks M.F., Moore K.H., Hall J.E., Hinds T.D. Jret al. (2018) Loss of biliverdin reductase-A promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Renal. Physiol. 315, F323–F331 10.1152/ajprenal.00495.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sundararaghavan V.L., Binepal S., Stec D.E., Sindhwani P. and Hinds T.D. Jr (2018) Bilirubin, a new therapeutic for kidney transplant? Transplant. Rev. (Orlando) 10.1016/j.trre.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Puri K., Nobili V., Melville K., Corte C.D., Sartorelli M.R., Lopez R.et al. (2013) Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J. Pediatr. Gastroenterol. Nutr. 57, 114–118 10.1097/MPG.0b013e318291fefe [DOI] [PubMed] [Google Scholar]
- 189.Salomone F., Li Volti G., Rosso C., Grosso G. and Bugianesi E. (2013) Unconjugated bilirubin, a potent endogenous antioxidant, is decreased in patients with non-alcoholic steatohepatitis and advanced fibrosis. J. Gastroenterol. Hepatol. 28, 1202–1208 10.1111/jgh.12155 [DOI] [PubMed] [Google Scholar]
- 190.Hjelkrem M., Morales A., Williams C.D. and Harrison S.A. (2012) Unconjugated hyperbilirubinemia is inversely associated with non-alcoholic steatohepatitis (NASH). Aliment. Pharmacol. Ther. 35, 1416–1423 10.1111/j.1365-2036.2012.05114.x [DOI] [PubMed] [Google Scholar]
- 191.Kunutsor S.K., Frysz M., Verweij N., Kieneker L.M., Bakker S.J.L. and Dullaart R.P.F. (2020) Circulating total bilirubin and risk of non-alcoholic fatty liver disease in the PREVEND study: observational findings and a Mendelian randomization study. Eur. J. Epidemiol. 35, 123–137 10.1007/s10654-019-00589-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Jang B.K. (2012) Elevated serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 18, 357–359 10.3350/cmh.2012.18.4.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kwak M.S., Kim D., Chung G.E., Kang S.J., Park M.J., Kim Y.J.et al. (2012) Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 18, 383–390 10.3350/cmh.2012.18.4.383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Choi S.H., Yun K.E. and Choi H.J. (2011) Relationships between serum total bilirubin levels and metabolic syndrome in Korean adults. Nutr. Metab. Cardiovasc. Dis. [DOI] [PubMed] [Google Scholar]
- 195.Wu Y., Li M., Xu M., Bi Y., Li X., Chen Y.et al. (2011) Low serum total bilirubin concentrations are associated with increased prevalence of metabolic syndrome in Chinese. J. Diab. 3, 217–224 10.1111/j.1753-0407.2011.00138.x [DOI] [PubMed] [Google Scholar]
- 196.Cheriyath P., Gorrepati V.S., Peters I., Nookala V., Murphy M.E., Srouji N.et al. (2010) High Total Bilirubin as a Protective Factor for Diabetes Mellitus: An Analysis of NHANES Data From 1999 - 2006. J. Clin. Med. Res. 2, 201–206 10.4021/jocmr425w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Guzek M., Jakubowski Z., Bandosz P., Wyrzykowski B., Smoczynski M., Jabloiska A.et al. (2012) Inverse association of serum bilirubin with metabolic syndrome and insulin resistance in Polish population. Przegl. Epidemiol. 66, 495–501 [PubMed] [Google Scholar]
- 198.Kwon K.M., Kam J.H., Kim M.Y., Kim M.Y., Chung C.H., Kim J.K.et al. (2011) Inverse association between total bilirubin and metabolic syndrome in rural korean women. J. Womens Health 20, 963–969 10.1089/jwh.2010.2453 [DOI] [PubMed] [Google Scholar]
- 199.Jo J., Yun J.E., Lee H., Kimm H. and Jee S.H. (2011) Total, direct, and indirect serum bilirubin concentrations and metabolic syndrome among the Korean population. Endocrine 39, 182–189 10.1007/s12020-010-9417-2 [DOI] [PubMed] [Google Scholar]
- 200.Han S.S., Na K.Y., Chae D.W., Kim Y.S., Kim S. and Chin H.J. (2010) High serum bilirubin is associated with the reduced risk of diabetes mellitus and diabetic nephropathy. Tohoku J. Exp. Med. 221, 133–140 10.1620/tjem.221.133 [DOI] [PubMed] [Google Scholar]
- 201.Lin Y.C., Chang P.F., Hu F.C., Chang M.H. and Ni Y.H. (2009) Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics 124, e1221–e1227 10.1542/peds.2008-3087 [DOI] [PubMed] [Google Scholar]
- 202.Hinds T.D. Jr, Creeden J.F., Gordon D.M., Spegele A.C., Britton S.L., Koch L.G.et al. (2020) Rats Genetically Selected for High Aerobic Exercise Capacity Have Elevated Plasma Bilirubin by Upregulation of Hepatic Biliverdin Reductase-A (BVRA) and Suppression of UGT1A1. Antioxidants (Basel) 9, 10.3390/antiox9090889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Molzer C., Wallner M., Kern C., Tosevska A., Schwarz U., Zadnikar R.et al. (2016) Features of an altered AMPK metabolic pathway in Gilbert's Syndrome, and its role in metabolic health. Sci. Rep. 6, 30051 10.1038/srep30051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sundararaghavan V.L., Sindhwani P. and Hinds T.D. Jr (2017) Glucuronidation and UGT isozymes in bladder: new targets for the treatment of uroepithelial carcinomas? Oncotarget 8, 3640–3648 10.18632/oncotarget.12277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Negi C.K., Babica P., Bajard L., Bienertova-Vasku J. and Tarantino G. (2022) Insights into the molecular targets and emerging pharmacotherapeutic interventions for nonalcoholic fatty liver disease. Metabolism 126, 154925 10.1016/j.metabol.2021.154925 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included within the main manuscript file.