

SUMMARY
Caloric restriction and acute fasting are known to reduce seizures but through unclear mechanisms. mTOR signaling has been suggested as a potential mechanism for seizure protection from fasting. We demonstrate that brain mTORC1 signaling is reduced after acute fasting of mice and that neuronal mTORC1 integrates GATOR1 complex-mediated amino acid and tuberous sclerosis complex (TSC)-mediated growth factor signaling. Neuronal mTORC1 is most sensitive to withdrawal of leucine, arginine, and glutamine, which are dependent on DEPDC5, a component of the GATOR1 complex. Metabolomic analysis reveals that Depdc5 neuronal-specific knockout mice are resistant to sensing significant fluctuations in brain amino acid levels after fasting. Depdc5 neuronal-specific knockout mice are resistant to the protective effects of fasting on seizures or seizure-induced death. These results establish that acute fasting reduces seizure susceptibility in a DEPDC5-dependent manner. Modulation of nutrients upstream of GATOR1 and mTORC1 could offer a rational therapeutic strategy for epilepsy treatment.
Graphical abstract
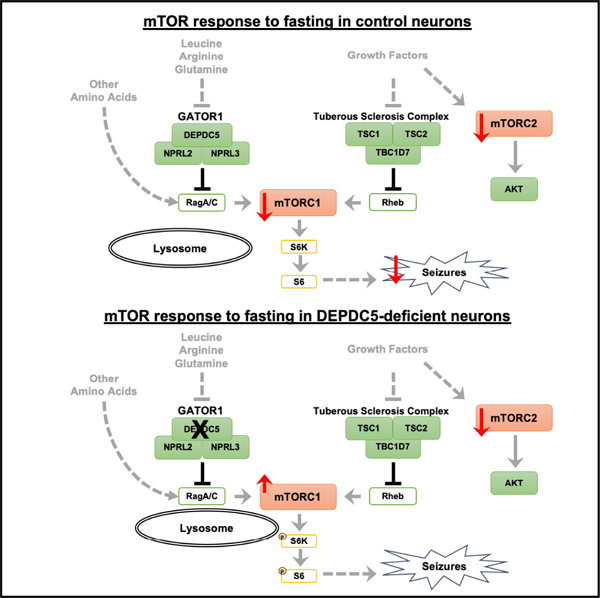
In brief
Yuskaitis et al. find that amino acid sensing through GATOR1 and mTORC1 mediates the protective effects of dietary fasting on seizures. Through their thorough investigation of the upstream regulation of neuronal mTORC1 signaling, they provide insight into nutrient-signaling mechanisms in the brain and how these mechanisms mediate seizures.
INTRODUCTION
Dietary restriction has proven benefits for the health of organisms, including the reduction of seizures in animal models (Bough and Eagles, 1999; DeVivo et al., 1975; Eagles et al., 2003), and is a proposed intervention for drug-resistant epilepsy (Kossoff, 2004; Yuen and Sander, 2014). Acute fasting provides an opportunity to identify the specific molecular changes responsible for the beneficial effects without the confounding effects of the animal adaptation response to intermittent fasting (Mattson et al., 2018). A recent study demonstrated that diet influences levels of circulating extracellular factors that in turn alter neuronal activation (Padamsey et al., 2022). Studies have postulated several intracellular mechanisms to link the dietary changes to neuronal function in the context of epilepsy (Cervenka et al., 2021; deCampo and Kossoff, 2019). An attractive candidate for linking diet to seizures is the mechanistic target of rapamycin (mTOR) pathway. The response of mTOR signaling to fasting and nutrient withdrawal in neurons has yet to be elucidated.
mTORC1 functions as a nutrient/energy sensor integrating upstream amino acid and growth factor signals (Hara et al., 1998; Liu and Sabatini, 2020). Intracellular amino acids are sensed by mTORC1 via Rag GTPases and the Ragulator complex, which recruit mTORC1 to the lysosomal surface (Bar-Peled et al., 2013; Sancak et al., 2010). Once at the lysosomal surface, growth factors signal through the Rheb GTPase to activate mTOR kinase activity (Menon et al., 2014). mTORC1 activation results in phosphorylation of downstream proteins, such as the 70-kDa ribosomal protein S6 kinase (S6K1) and its substrate (S6), to stimulate protein synthesis and cellular growth. A second mTOR-containing complex, mTORC2, is distinct in composition and function. mTORC2 is less well understood, but recent studies suggest that mTORC2 dysfunction alters neuronal synaptic transmission (Chen et al., 2019; McCabe et al., 2020). A readout of mTORC2 activity is phosphorylation of Ser 473 of Akt/protein kinase B. The upstream regulators of these two mTOR complexes to withdrawal of amino acid and growth factors in neurons needs to be understood.
Negative regulators of the amino acid- and growth factor-mediated mTORC1 signaling pathways are essential for maintaining metabolic homeostasis. A main negative regulator of the amino acid sensing machinery is GATOR1, a protein complex composed of DEP domain-containing protein 5 (DEPDC5), NPR2 like (NPRL2), and NPR3 like (NPRL3), which serves as a GTPase-activating protein (GAP) for RagA and RagB (Bar-Peled et al., 2013). The main regulator of the growth factor sensing machinery is the tuberous sclerosis complex (TSC) protein complex, composed of TSC1 (hamartin), TSC2 (tuberin), and TBC1D7, which serves as a GAP for Rheb (Dibble et al., 2012; Menon et al., 2014). In nutrient-limited states such as fasting, mTORC1 inhibition decreases anabolic processes leading to energy and nutrient conservation. Sufficient amino acids and growth factors are required for maximal activation of mTORC1 in non-neuronal cells (Valvezan and Manning, 2019); however, the signaling mechanisms upstream of mTORC1 have yet to be determined in post-mitotic neurons. Studies of DEPDC5-deficient neurons have not evaluated the response to amino acid and growth factors independently (Iffland et al., 2020; Klofas et al., 2020a). In non-neuronal cells, leucine, arginine, and methionine sensors have been identified upstream of GATOR1 and Rag-mediated regulation of mTORC1 (Chantranupong et al., 2016; Gu et al., 2017; Wolfson et al., 2016, 2017); however, the mTORC1 response to different amino acids appears to be cell-type dependent and not fully characterized in neurons (Carroll et al., 2016; Meng et al., 2020).
We and others have demonstrated a role for mTOR complex 1 (mTORC1) signaling in epilepsy. Inhibition of mTORC1 reduces seizures in hyperactive mTORC1 models (Switon et al., 2017) with mTOR inhibitors showing promise as anticonvulsants for epilepsy in patients with TSC (French et al., 2016; Krueger et al., 2016). Animal models and patients with loss of function variants of DEPDC5 exhibit epilepsy and hyperactivation of mTORC1 in neurons as measured by pS6 (Yuskaitis et al., 2018). mTOR inhibitors reduce seizures and seizure-induced death in Depdc5 knockout models (Klofas et al., 2020b; Yuskaitis et al., 2019). mTOR mediates epileptogenesis in models independent of TSC or other mTOR pathologies (Buckmaster et al., 2009; Zeng et al., 2009). Time-dependent changes in mTOR occur after induction of seizures in pentylenetetrazol (PTZ)-induced seizures (Zhang and Wong, 2012) and mouse models of temporal lobe epilepsy (Ahmed et al., 2021). Thus, reduction of mTOR may be a rational approach in epilepsy management regardless of the etiology (Griffith and Wong, 2018).
mTORC1 activity is inhibited by acute fasting in a tissue-specific manner outside of the nervous system (Cummings and Lamming, 2017). Despite the brain being largely protected from acute nutritional deficits, fasting results in fluctuations in the concentration of amino acids between the blood and brain (Yudkoff et al., 2005, 2006). A mechanistic understanding of neuronal mTOR regulation by amino acids is a key step in the development of targeted dietary therapies for epilepsy. Here, we demonstrate that anticonvulsant effects of fasting are associated with reduced neuronal mTOR activity and are abolished with DEPDC5 loss. In neurons, leucine, arginine, and glutamine sensing by mTORC1 is dependent on DEPDC5, whereas growth factor sensing is dependent on TSC2. Furthermore, the mTORC2 response is selective for growth factors and independent of DEPDC5 and TSC2. In mouse cortical samples, metabolomic analysis demonstrates decreased amino acid levels after fasting. DEPDC5-deficient mice failed to respond to these changes, and fasting did not protect them from seizures. Collectively, we highlight the critical role of amino acid-mediated mTOR signaling in neurons and provide evidence that intact mTOR signaling is required for the protective effect of fasting on seizures.
RESULTS
Acute fasting protects from seizures and reduces mTOR activity in the brain
We first tested if 24-h fasting protects mice from seizures using an acute PTZ-induced seizure paradigm (Figure 1A) (Yuskaitis et al., 2021). Fasting for 24 h significantly reduced PTZ-induced seizures from 56% having seizures in the fed state to 22% in fasted mice (Figure 1B). There was no effect on seizure latency or seizure duration (Figures S1A and S1B). Weight and glucose were significantly reduced in the fasted mice compared with fed mice (Figures S1C and S1D). Next, we tested if mTOR activity is decreased in the cerebral cortex after fasting in a separate cohort of mice to avoid the known confound of seizure-induced changes in mTOR activity. By western blot analysis of cortical lysates, fasting significantly reduced phosphorylation of Ser240/244 on S6 (pS6) and Ser473 on AKT (pAKT), as markers of mTORC1 and mTORC2 activity, respectively (Figures 1C and 1D). We demonstrate that mTOR activity in the brain is modified by acute dietary fasting.
Figure 1. Brain mTORC1 activity and PTZ-induced seizures were insensitive to protective effects of fasting in Depdc5 neuronal knockout mice.
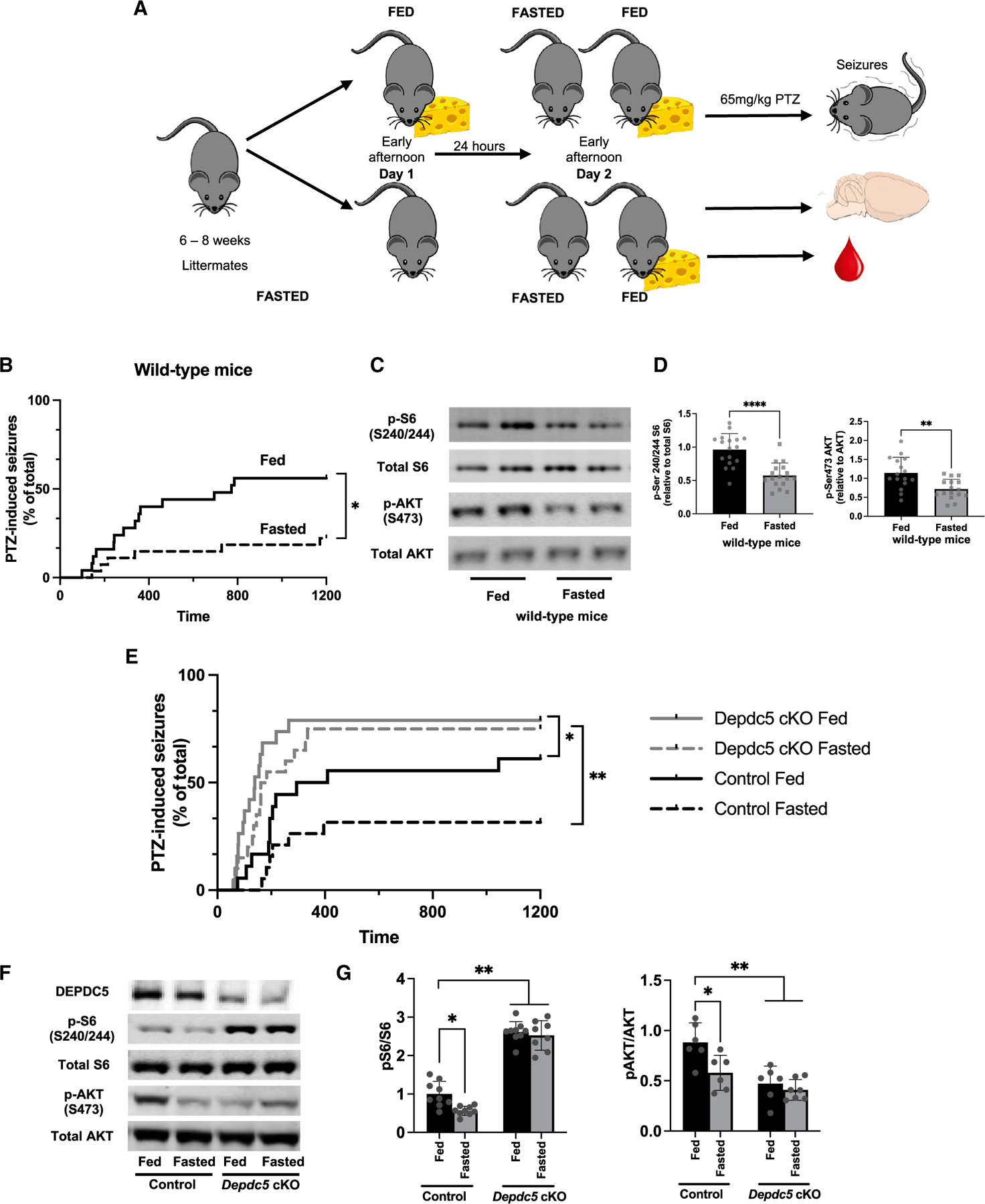
(A) Fasting protocol for 24 h of 6- to 8-week-old mice randomized to either fast or fed condition. Studies were performed at the same time of day. Separate cohorts were either euthanized for blood and brain collection or administered the chemoconvulsant pentylenetetrazol (PTZ) to induce seizures.
(B) Compared with fed mice, those fasted for 24 h had a reduction in seizures after PTZ challenge (65 mg/kg intraperitoneal injection) (n = 25 fed [14 M, 11 F]; 27 fasted mice [14 M, 13 F]; *p = 0.0126, log rank test).
(C) Immunoblots from cortical brain lysates and (D) relative quantifications demonstrate a reduction in pSer240/244 S6 and pSer473 AKT after 24-h fasting. Individual symbols represent each independent animal tested with mean ± SD. **p < 0.01, ****p < 0.0001, Student’s t test.
(E) After PTZ (65 mg/kg intraperitoneal injection), there was no difference between seizure susceptibility between fed or fasted Depdc5cc+ mice, which were significantly increased compared with littermate control mice (log rank test, *p < 0.05, **p < 0.01). Six- to 8-week-old mice randomized to either fast or fed condition; n = 20 control fed (8 M, 10 F), 19 control fasted (7 M, 12 F), 19 Depdc5cc+ fed (8 M, 10 F), and 120 Depdc5cc+ fasted (9 M, 11 F) littermate mice.
(F) Immunoblots and (G) relative quantifications from cortical brain lysates demonstrate a reduction in pSer240/244 S6 after 24-h fasting in only controls; hyperactive mTORC1 in Depdc5cc+ mouse brains was unchanged by fasting. Individual symbols indicate individual animals for the above measurements, mean ± SD with *p < 0.05, **p < 0.01, ****p < 0.0001. Two-way ANOVA with Tukey’s post hoc analysis.
DEPDC5 is required for protection from seizures by fasting in mice
Nutrient sensing through mTOR may be responsible for the protective effect of fasting on seizures. We test the impact of DEPDC5 loss on fasting and seizure susceptibility using a neuron-specific Depdc5 conditional knockout mouse model (Depdc5cc+) (Yuskaitis et al., 2018). Fasting for 24 h failed to protect Depdc5cc + mice from PTZ-induced seizures, whereas littermate controls continued to demonstrate a protective effect from fasting (Figure 1E). Seizure onset occurred earlier in Depdc5cc+ mice compared with controls but was unaffected by fasting (Figure S1E). Seizure duration was unaffected by genotype or diet (Figure S1F). Post-fasting weight loss and reduced blood glucose after fasting were similar between Depdc5cc+ and control mice (Figures S1G and S1H). Fasting induced ketosis as measured by elevated 3-hydroxybutanonic acid (e.g., beta-hydroxybutyrate) in both genotypes with higher levels in Depdc5cc+ mice compared with controls (Figure S1I).
Next, we measured mTOR-specific signaling response to fasting in the brains of mice with selective loss of DEPDC5 in neurons and littermate controls. In contrast to the fasting-induced reduction of mTORC1 in control brains, fasting for 24 h had no impact on mTORC1 activity in Depdc5cc+ brains (Figures 1F and 1G). Fasting reduced mTORC2 activity in control brains, but again fasting had no impact on mTORC2 activity in Depdc5cc+ brains (Figures 1F and 1G). Although Depdc5cc+ mice have an enlarged brain phenotype, which is reversed by chronic rapamycin treatment (Yuskaitis et al., 2019), acute fasting had no effect on brain weight in either genotype (Figure S1J). These results suggest that defects in sensing of amino acids upstream of DEPDC5 in Depdc5cc+ neurons might explain these findings.
mTORC1 response to amino acid and/or growth factor withdrawal in primary neurons
To assess mTOR signaling in neurons, we examined the response of the mTOR signaling pathway to amino acid and growth factor manipulation in primary rat cortical neurons and two non-neuronal immortalized cell lines, HEK293T and HeLa, commonly used to study mTORC1 signaling. The mTORC1 signaling in HEK293T cells was sensitive to amino acid withdrawal but insensitive to growth factor withdrawal, whereas primary cortical neurons were responsive to both growth factors and amino acid withdrawal (Figures 2A, S2A, S2B, and S2C). Insensitivity of HEK293T cells to growth factors is a result of a genetic manipulation that resulted in constitutive PI3K-AKT signaling (Fonseca et al., 2011; Yu et al., 2005). The HeLa cell line is known to be responsive to both upstream signals (Nicklin et al., 2009). A reduction in mTORC1 signaling was noted after growth factor and/or amino acid withdrawal in both HeLa and primary neurons (Figure 2B). Interestingly, the mTORC1 response to growth factors was more prominent in HeLa cells, whereas the neurons appeared to have equivalent mTORC1 response to reduction of growth factor or amino acids. We conducted amino acid withdrawal in the presence or absence of growth factors at multiple time points and determined that both 60 and 90 min of amino acid withdrawal are equivalent (Figures S2D, S2E, S2F, and S2G). We determined that adding back amino acids after 60-min withdrawal does not fully restore mTORC1 activity until 80 min (Figures S2D and S2G). Taken together, the mTORC1 response in HeLa cells and neurons was in line with the current model of signal integration by mTORC1 (Valvezan and Manning, 2019).
Figure 2. mTOR signaling in primary cortical neurons after amino acid and/or growth factor withdrawal.
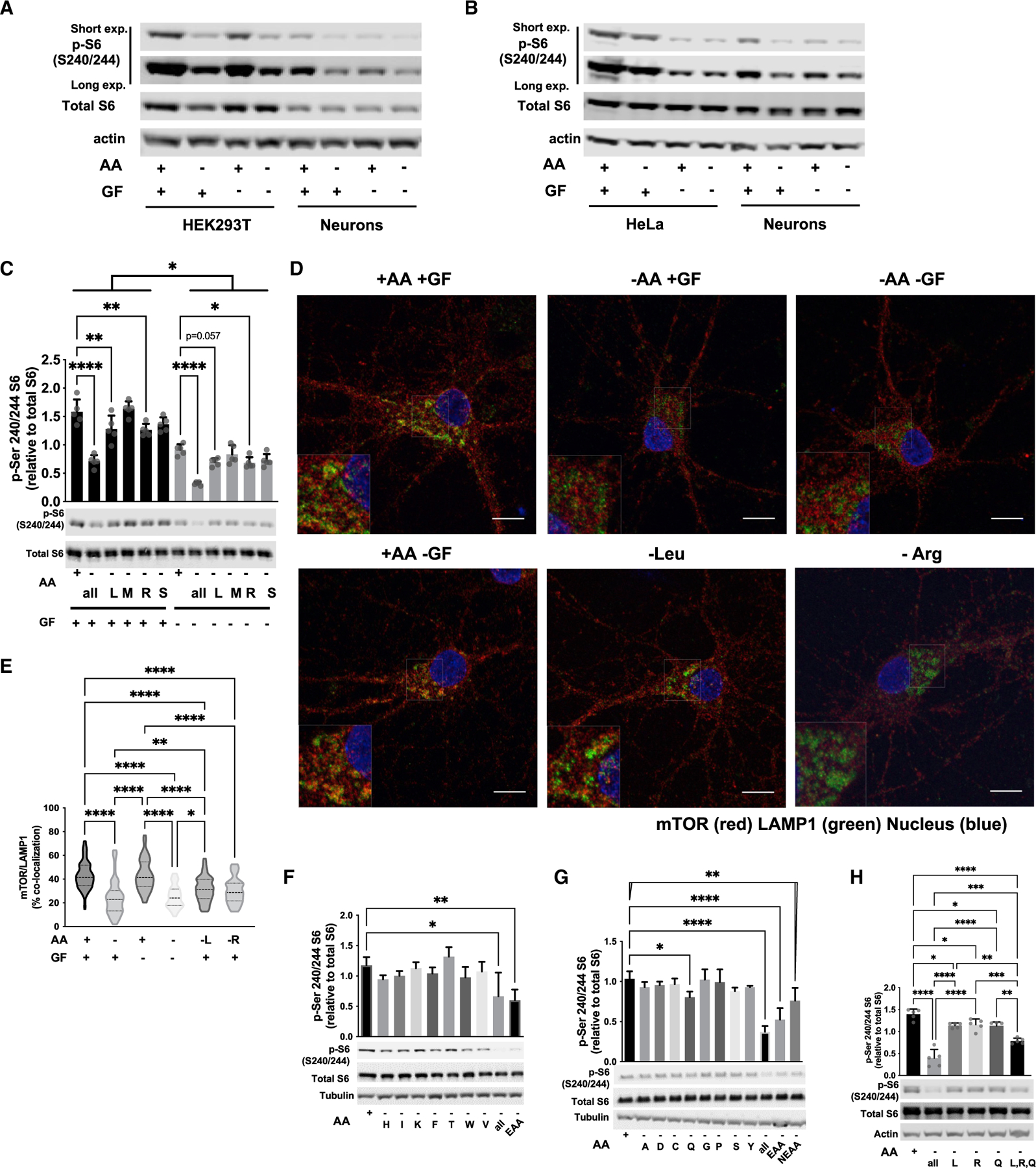
(A) Representative immunoblots of mTORC1 activity by pS6 phosphorylation from 20 μg protein from HEK293T or DIV12 primary cortical neurons treated for 60 min after withdrawal of amino acids (AAs) and/or growth factors (GF), n = 4.
(B) Representative immunoblots of mTORC1 activity by pS6 phosphorylation from 20 μg protein from HeLa or DIV12 primary cortical neurons treated for 60 min in the indicated conditions, n = 2.
(C) Leucine (L) and arginine (R) withdrawal for 90 min is able to reduce mTORC1 activity in primary cortical neurons in the presence or absence of GF, while methionine (M) and serine (S) are not.
(D) Representative images of mTOR lysosomal localization in primary neurons stained with mTOR (red) and the lysosomal marker, LAMP1 (green) after incubation with or without AA and/or GF for 90 min. Scale bars = 10 μm, 2× inset.
(E) Mander’s colocalization of mTOR and LAMP1 indicate a decrease in mTOR lysosomal localization by L, R, or all AA withdrawal, but not GF alone. At least 50 cells quantified across three independent experiments.
(F) The combination of all essential amino acids (EAAs), but not individual EAAs, reduces mTORC1 activity after 90-min treatment in primary neuronal cultures.
(G) Withdrawal of individual or combined non-essential amino acids (NEAA) for 90 min, glutamine (Q) alone, all EAAs, all NEAAs, or all AAs reduces mTORC1 in primary neuronal cultures.
(H) Immunoblots and quantification of mTORC1 activity in primary neuronal cultures demonstrating that the combination of leucine (L), arginine (R), glutamine
(Q) withdrawal leads to an additive reduction in mTORC1 activity. Graph of mean ± SD with symbols representing individual replicates from at least three independent experiments, one-way ANOVA with Tukey’s post hoc analysis. *p < 0.05, **p < 0.01, ****p < 0.001, ****p < 0.0001.
We tested if mTORC1 was sensitive to the withdrawal of individual amino acids. Leucine, arginine, and methionine have known sensors upstream of mTORC1 identified in HEK293T cells, many of which rely on the GATOR1 complex (Chantranupong et al., 2016; Gu et al., 2017; Wolfson et al., 2016, 2017). In neurons, 90-min withdrawal of leucine or arginine resulted in a reduction in mTORC1 activity (Figure 2C). Methionine or serine withdrawal did not have a significant impact on mTORC1 activity in neurons. Across all conditions, the sensitivity to individual amino acid withdrawal was blunted in the absence of growth factors.
We evaluated lysosomal localization of mTOR in response to amino acid withdrawal in primary neurons similar to studies in other cell types (Bar-Peled et al., 2013; Iffland et al., 2018, 2020; Sancak et al., 2010). In the absence of amino acids, lysosomal localization of mTOR is reduced in neurons and independent of growth factors (Figures 2D and 2E). Withdrawal of either leucine or arginine alone also reduced lysosomal localization of mTOR.
We then evaluated the remainder of essential (H, I, K, F, T, W, and V) and non-essential amino acids (A, D, C, Q, G, P, S, and Y) for their effects on mTORC1 signaling in the presence of growth factors. Individual essential amino acid withdrawal had no effect on mTORC1 activity, but withdrawal of all essential amino acids led to a significant reduction in mTORC1 activity (Figure 2F). Of the non-essential amino acids, only glutamine withdrawal led to a significant reduction in mTORC1 activity (Figure 2G). Glutamine sensing by mTORC1 has been reported in other cell types (Jewell et al., 2015; Meng et al., 2020). An additive effect of all non-essential amino acids reducing mTORC1 activity was noted, but not as robust as the effect of essential amino acids. We then tested select combinations of amino acids. Withdrawal of branch-chain amino acids (L, I, V) and amino acids with known mTORC1 sensors (L, M, R) did not lead to further decrease in mTORC1 activity beyond withdrawal of leucine alone (Figure S2H). The three amino acids we identified that selectively regulated mTORC1 activity (L, R, Q), demonstrated an additive effect on mTORC1 activity (Figure 2H). The additive response was not equivalent to withdrawal of all amino acids, suggesting that additional amino acids likely contribute to mTORC1 signaling; however, leucine, arginine, and glutamine are the predominant amino acids that regulate mTORC1 activity in neurons.
DEPDC5 and TSC2 modulate the mTORC1 response to nutrients in primary neurons
We addressed whether amino acid or growth factor signaling through mTOR was GATOR1 dependent. In complete media conditions, DEPDC5 knockdown resulted in a significant increase in mTORC1 activity (Figures 3A and S3A), but no change in mTORC2 activity as measured by pAKT (Figures 3A and S3B). In response to either amino acid or growth factor withdrawal, mTORC1 activity was decreased in DEPDC5 knockdown neurons but remained elevated relative to the scrambled controls. Measuring the ratio of pS6 levels in the amino acid-deficient compared with replete conditions, mTORC1 signaling in DEPDC5-deficient neurons was more resistant to amino acid withdrawal than controls (Figure 3B). Direct inhibition of mTORC1 by rapamycin abolished the hyperactive mTORC1 signaling in DEPDC5 knockdown at baseline and amino acid-deficient conditions (Figure S3C).
Figure 3. In neurons, DEPDC5 is essential for leucine, arginine, and glutamine signaling through mTORC1, whereas TSC2 is essential for growth factor signaling through mTORC1.
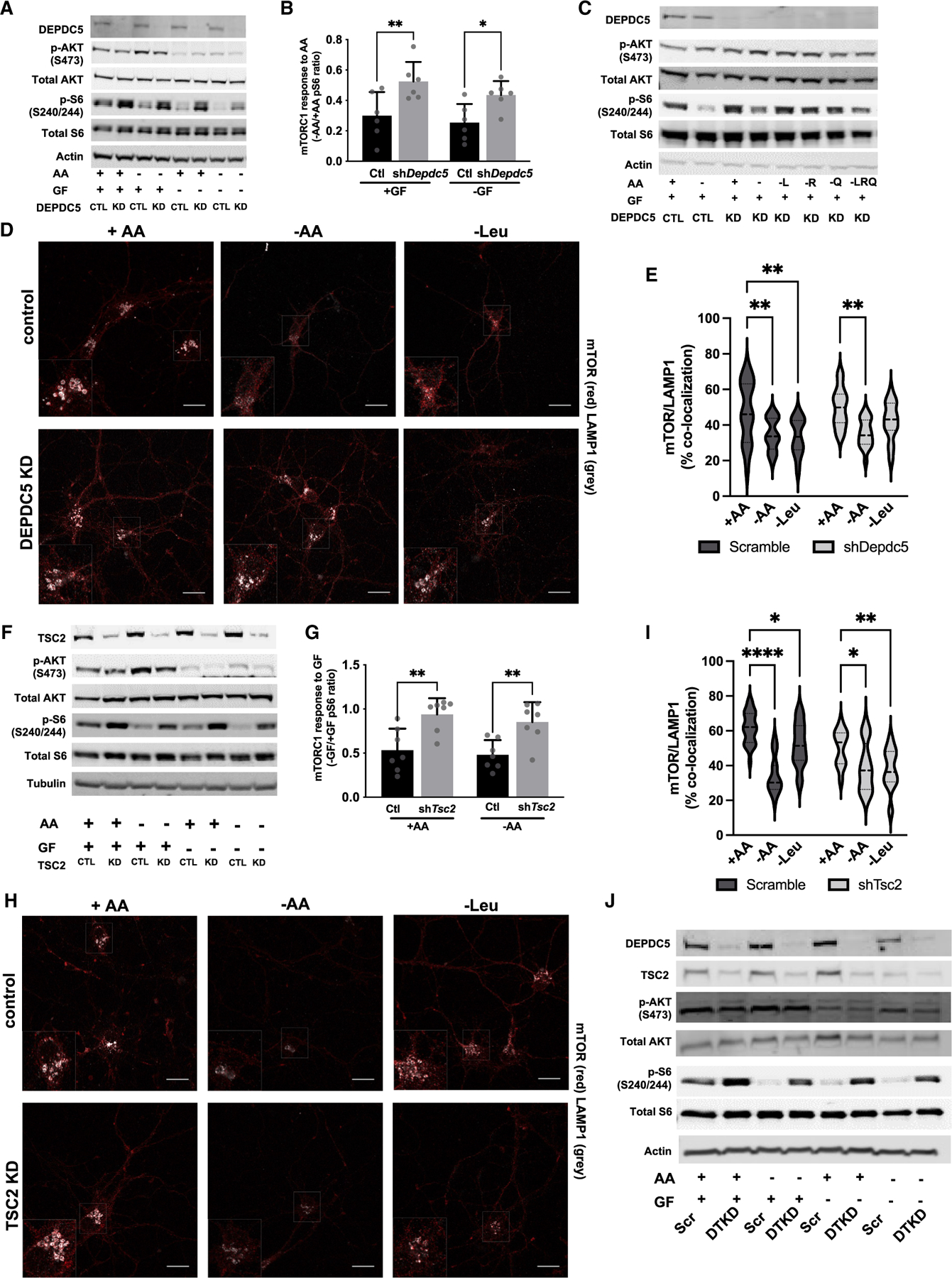
(A) Immunoblots from scramble control (CTL) and DEPDC5 knockdown (KD) primary cortical neurons after 90-min amino acid (AA) and/or growth factor (GF) withdrawal.
(B) Reduced mTORC1 sensitivity of DEPDC5 KD neurons to AA withdrawal as measured by the ratio of quantified pS6 from AA withdrawal versus AA replete conditions from (A).
(C) Immunoblots demonstrate that mTORC1 (pS240/244-S6), but not mTORC2 (pS473-AKT), is insensitive to withdrawal of leucine (L), arginine (R), and/or glutamine (Q) in DEPDC5 KD primary cortical neurons.
(D) Representative images of mTOR lysosomal localization in primary neurons transduced with scramble or shDepdc5 lentivirus stained with mTOR (red) and the lysosomal marker LAMP1 (gray) after incubation with or without AA and/or GF for 90 min. Scale bars = 20 μm, 2× inset. (E) mTOR colocalization with LAMP1 after 90-min AA or leucine withdrawal is reduced by all AA withdrawal but insensitive to leucine alone withdrawal in DEPDC5 KD neurons by Mander’s colocalization index from at least 50 cells quantified across three independent experiments.
(F) Immunoblots from scramble control (CTL) and TSC2 knockdown (KD) primary cortical neurons after 90-min amino acid (AA) and/or growth factor (GF) withdrawal.
(G) Ratio of quantified pS6 from GF withdrawal versus GF replete conditions in (F) demonstrates that TSC2 KD neurons are insensitive to GF withdrawal independent of AAs.
(H) Representative images of mTOR lysosomal localization in primary neurons transduced with scramble or TSC2 knockdown lentivirus stained with mTOR (red) and the lysosomal marker LAMP1 (gray) after incubation with or without AA and/or GF for 90 min. Scale bars, 20 μm, 2× inset.
(I) mTOR colocalization with LAMP1 is reduced by 90-min AA or leucine withdrawal in both TSC2 KD and control neurons by Mander’s colocalization index from at least 50 cells quantified across three independent experiments.
(J) Immunoblots from scramble control (CTL) and double-knockdown of DEPDC5 and TSC2 (DTKD) primary cortical neurons after 90-min AA and/or GF withdrawal. For all experiments, graphs of mean ± SD, with symbols representing individual replicates from at least three independent experiments, two-way ANOVA with Tukey’s post hoc analysis. *p < 0.05, **p < 0.01, ****p < 0.001, ****p < 0.0001.
We tested if DEPDC5-deficient neurons are responsive to the amino acids with the greatest impact on mTORC1 function we previously identified (i.e., leucine, arginine, and glutamine). mTORC1 signaling in DEPDC5-deficient neurons is insensitive to single amino acid withdrawal or the combined withdrawal of all three amino acids (Figures 3C, S3D, and S3E). After amino acid withdrawal, DEPDC5 neurons responded with increased mTORC1 signaling by addition of all amino acids but were resistant to stimulation with the combination of leucine, arginine, and glutamine (Figures S3F and S3G). Next, we measured colocalization of mTOR and the lysosomal marker LAMP1, by immunofluorescence staining in DEPDC5-deficient and scramble control cortical neurons. Control neurons display a significant reduction in mTOR/LAMP1 colocalization in response to withdrawal of all amino acids or leucine alone (Figures 3D, 3E, and S3H). DEPDC5-deficient neurons were not responsive to leucine withdrawal but retained partial response to complete amino acid withdrawal. Collectively, these data demonstrate DEPDC5-dependent neuronal sensing of leucine, arginine, and glutamine via mTORC1 and suggest potential DEPDC5-independent amino acid sensing mechanisms also functioning in neurons.
We asked if TSC may play a role in nutrient sensing in neurons by assessing the mTOR response in TSC2-deficient neurons to nutrient and growth factor withdrawal. In TSC2-deficient neurons, mTORC1 response was sensitive to amino acid withdrawal but completely insensitive to growth factor withdrawal (Figures 3F and S4A). Measuring the ratio of pS6 levels in the growth factor-deficient compared with replete conditions, mTORC1 signaling in TSC2-deficient neurons was completely resistant to growth factor withdrawal unlike controls (Figure 3G). mTORC2 response was reduced in both TSC2-deficient and control neurons after growth factor but not amino acid withdrawal (Figures 3F and S4B). In response to withdrawal of either all amino acid or leucine alone, lysosomal localization of mTOR was reduced similarly in both TSC2-deficient and control neurons (Figures 3H, 3I, and S4E). These data provide evidence that TSC2 is not involved in the amino acid-regulated localization of mTORC1 to lysosomes in neurons and primarily inhibits mTORC1 in response to growth factor withdrawal, as reported in non-neuronal cells (Menon et al., 2014).
mTORC1 signaling retained partial sensitivity to amino acids in either DEPDC5- or TSC2-deficient neurons. Therefore, we evaluated the response of mTOR to nutrient withdrawal in DEPDC5 and TSC2 double-knockdown neurons (DTKD). In cortical neurons serially transduced with shDepdc5 on DIV1 followed by shTsc2 on DIV5, an almost complete absence of both DEPDC5 and TSC2 proteins were noted by DIV12 (Figure 3J). mTORC1 activity was partially reduced in DTKD neurons after amino acid withdrawal, indicating DTKD neurons retain some sensitivity to amino acids (Figures 3J, S4C, and S4D). In DTKD neurons, mTORC1 activity was insensitive to growth factor withdrawal and mTORC2 was decreased at baseline but retained response to growth factor withdrawal (Figures 3J, S4C, and S4D). Collectively, we identified the main amino acids regulating mTORC1 in neurons (i.e., leucine, arginine, and glutamine), which are GATOR1-dependent, but that GATOR1-independent signaling mechanisms also exist.
Metabolomics in DEPDC5-deficient mouse brain and implications for fasting-induced protection from seizures and seizure-induced death
We next assessed whether specific metabolic processes might play a role in the DEPDC5-dependent seizure protection by fasting. We performed whole tissue metabolomic analysis on cortical brain samples from Depdc5cc+ and control mice in both the fed and 24-h fasted states (Table S1). The top 20 metabolites differentially altered by diet and genotype are presented (Figure 4A). Elevation of 3-hydroxybutanonic acid (e.g., beta-hydroxybutyrate) confirmed a state of ketosis after fasting in both genotypes (Figure 4B). Among the top 20 identified metabolites, several amino acids were identified as being reduced by fasting including asparagine, histidine, glutamine, tyrosine, and methionine. Not only was glutamine reduced in the fasted state, but glutamine was further reduced in the Depdc5cc+ brains compared with controls regardless of the fed or fasted state (Figure 4C). We also examined arginine levels and identified a reduction in levels in the fasting state, which was further exacerbated in Depdc5cc+ brains (Figure 4D). Leucine was increased in the fasted mice but less so in Depdc5cc+ brains compared with controls (Figure 4E). Despite metabolic alterations from fasting, our results indicate loss of DEPDC5 sensing mechanisms render neurons defective in amino acid sensing upstream of mTORC1 regulation and insensitive to the protective effects of fasting on seizures.
Figure 4. Brain metabolomic alterations after fasting does not alter seizure-induced death of Depdc5 neuronal knockout mice.
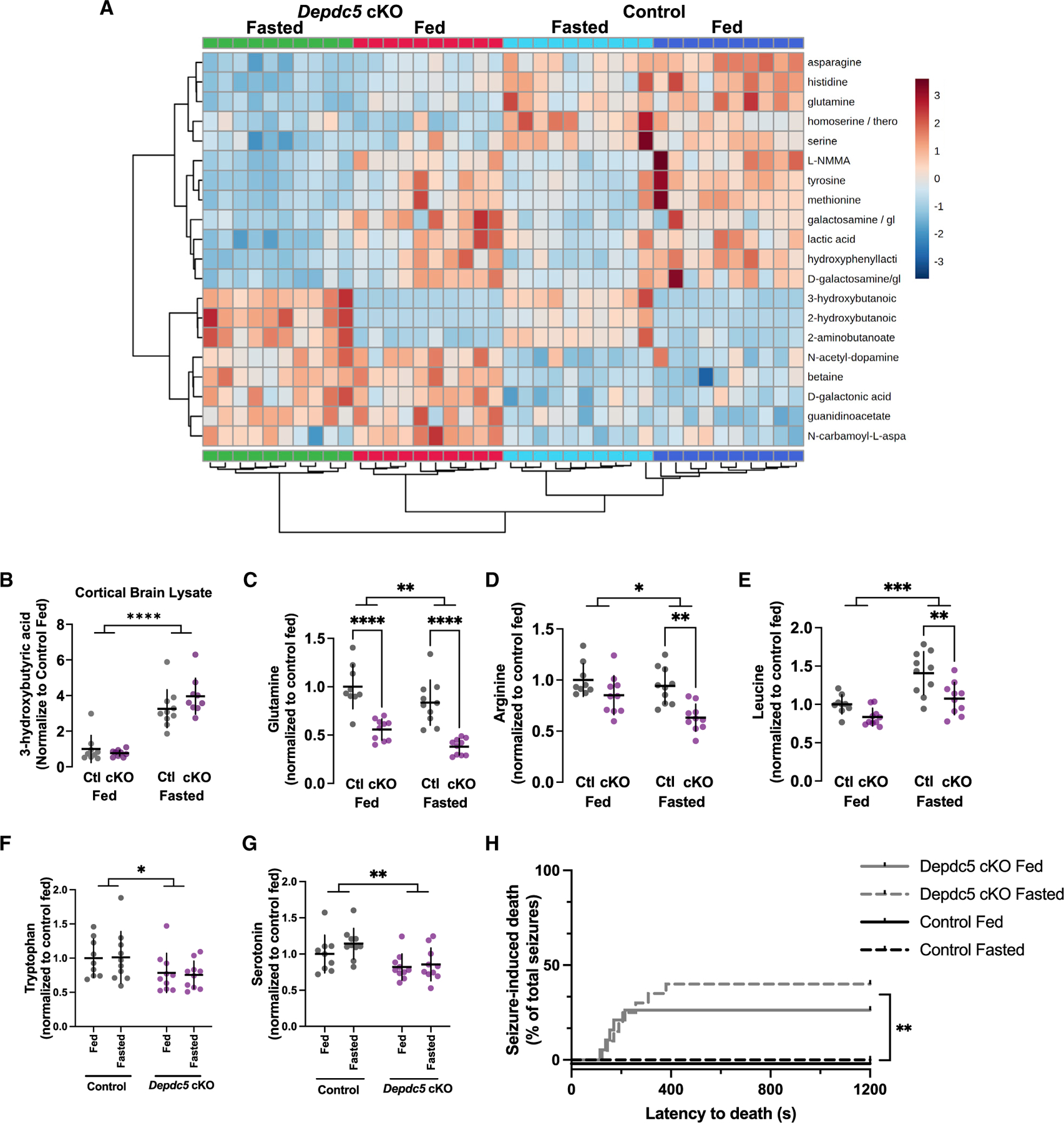
(A) Top 20 metabolites from metabolomic analysis of cortical lysates from Depdc5cc+ and control mice either fed or fasted.
(B–F) Brain 3-hydroxybutyrate values from metabolomic analysis were significantly increased in fasted mice compared with fed mice. Metabolomic analysis, normalized peak intensity values were baseline corrected to the average of the control fed mice. Metabolomic analysis of individual amino acid levels in the brain demonstrated the following: (C) glutamine decreased by fasting in both genotypes but consistently lower in Depdc5cc+ brains compared with controls,
(D) arginine decreased in fasting state in both genotypes and only significantly lower in Depdc5cc+ brains compared with controls, (E) leucine increased after fasting but less in Depdc5cc+ brains compared with controls, and (F) tryptophan was unchanged by fasting but decreased in Depdc5cc+ brains compared with controls.
(G) Serotonin levels correlated with tryptophan levels as they were also decreased in Depdc5cc+ brains compared with controls. Individual symbols indicate individual animals for the above measurements with *p < 0.05, **p < 0.01, ****p < 0.0001, two-way ANOVA with Tukey’s post hoc analysis.
(H) PTZ-seizure-induced death was increased in Depdc5cc+ mice and not changed by fasting, n > 16 mice per group (log rank test, **p < 0.01 in both fed and fasted Depdc5cc+ mice groups compared individually with fed and fasted littermate controls).
DEPDC5 is a risk gene for sudden unexplained death in epilepsy (SUDEP) and Depdc5 mouse models exhibit seizure-induced death (Klofas et al., 2020b; Yuskaitis et al., 2018). Tryptophan is the sole precursor of serotonin, and lower brain levels of serotonin are associated with seizures and seizure-induced death (Petrucci et al., 2020). We asked if tryptophan or serotonin levels were altered by fasting or are reduced in Depdc5cc+ brains. No change was noted in tryptophan with fasting; however, there was a significant reduction in brain tryptophan levels in Depdc5cc+ brains compared with controls (Figure 4F). Cortical serotonin levels were significantly reduced in Depdc5cc+ brains compared with controls and fasting had no effect (Figure 4G). Although fasting did not protect from seizures in Depdc5cc+ mice, we asked whether seizure-induced death might have been prevented with fasting. Only Depdc5cc+ mice died from PTZ-induced seizures and fasting had no effect on seizure-induced death (Figure 4H). Fasting does not alter the reduced brain serotonin and tryptophan levels and predisposition to seizure-induced death of Depdc5cc+ mice.
DISCUSSION
Caloric restriction protects from seizures in rodent models of epilepsy; however, the mechanisms have remained unclear (DeVivo et al., 1975; Greene et al., 2001; Viggiano et al., 2016). Our data reveal a connection between fasting effects on mTOR and seizure propensity. We demonstrate that loss of the upstream regulators of mTOR (i.e., DEPDC5 and TSC2) leads to hyperactive mTORC1 in neurons, and a decrease in sensitivity to detect nutrient or growth factor withdrawal. Our in vitro data provide important insights into differences between neuronal and non-neuronal mTORC1 signaling. We established that mice were unable to respond to fasting and reduce mTORC1 without intact DEPDC5 signaling. Thus, DEPDC5 loss renders mice insensitive to the seizure-protective effects of fasting. Collectively, we provide evidence that nutrient-sensing mechanisms upstream of mTOR are a necessary component of fasting-mediated reduction of seizure susceptibility.
The upstream signals and underlying mechanism have remained unclear about the association between seizure susceptibility and mTOR. We and others have demonstrated a critical role for hyperactive mTORC1 signaling in epilepsy. Neuronal loss of upstream regulators of mTORC1 (e.g., DEPDC5, TSC2, or TSC1) leads to seizures in mice (Meikle et al., 2007; Yuan et al., 2012; Yuskaitis et al., 2018). The level of mTOR hyperactivity has been shown to correlate with seizure severity (Nguyen et al., 2019), whereas inhibition of mTORC1 reduces seizures in hyperactive mTORC1 models (Switon et al., 2017; Yuskaitis et al., 2019).
In addition to hyperactive mTOR increasing seizure susceptibility, seizure induction alone is sufficient to increase mTOR activity (Buckmaster et al., 2009; Zeng et al., 2009). Caloric restriction and the ketogenic diet reduce seizures and reduce mTORC1 activity in animal models (McDaniel et al., 2011; Phillips-Farfan et al., 2015). However, direct mTOR inhibition by rapamycin is insufficient to protect from seizures when mTOR is not hyperactive (Hartman et al., 2012). These data suggest that the upstream signals and regulators of mTOR activity may play a larger role in neuronal function and seizure susceptibility than previously recognized. Our results provide critical insights into the mechanisms of nutrient signaling in neurons and evidence that DEPDC5 is a critical mediator of the protective effects of fasting on seizure susceptibility. However, the interplay between hyperactive mTOR prior to seizure onset and the increase in mTOR activity after seizures is still not fully understood.
To better understand the mechanisms of fasting, we directly compared the mTOR signaling in response to amino acid and growth factor withdrawal in neurons. Most studies defining mTOR signaling were performed in immortalized and proliferating cell lines (Liu and Sabatini, 2020). The well-defined canonical mTOR signaling integrates amino acid and growth factor signaling (Liu and Sabatini, 2020; Valvezan and Manning, 2019). However, cell-type differences in the mTORC1 response to serum starvation (Pirkmajer and Chibalin, 2011) or amino acids (Meng et al., 2020) have been described. We determined that mTORC1 activity was equally sensitive to amino acid and growth factor withdrawal in neurons. The difference in growth factor to amino acid-mediated mTOR signaling response in HeLa compared with neurons may be due to a variety of factors. mTOR is essential for regulating cell cycle and proliferation, which are necessary for immortalized cell lines (Fingar et al., 2004), whereas rat cortical neurons are post-mitotic in standard culture conditions (Brewer, 1999). Autophagy response due to TSC loss differs between neurons and mouse embryonic fibroblasts (Di Nardo et al., 2014). Our results suggest that the balance of upstream mTOR regulation through growth factors and amino acids may explain these differences in downstream mTOR function.
In neurons, we found that leucine, arginine, and glutamine modulate the mTORC1 response in a DEPDC5-dependent manner. Sensors for leucine, arginine, and S-adenosyl methionine (SAM) have been identified predominantly in HEK293T cells (Gu et al., 2017; Wolfson et al., 2016, 2017). However, mTORC1 responds to manipulation of other amino acids depending on the cell type (Carroll et al., 2016; Meng et al., 2020). From our study, methionine does not appear to be a major driver of neuronal mTORC1 signaling. In contrast, reductions in leucine and arginine are readily sensed by neurons in a DEPDC5-dependent manner. We also identified glutamine as a driver of mTORC1 signaling in a DEPDC5-dependent manner. The actual sensor for glutamine upstream of mTOR is not known, but in other cell types it is sensed in a Rag-independent manner (Jewell et al., 2015; Meng et al., 2020). Further study is necessary to fully elucidate the mechanisms of glutamine signaling upstream of mTORC1 in neurons. Moreover, we demonstrated that DEPDC5-deficient neurons partially retained the ability to sense complete amino acid withdrawal. These results suggest that there may be some amino acids that act on mTOR in a DEPDC5-independent manner.
In addition to identifying specific amino acids regulated through DEPDC5 in neurons, we also demonstrated that neuronal-specific DEPDC5-deficient mice were insensitive to the protective effects of fasting on seizures. We sought to identify additional metabolic pathways altered in the setting of fasting and mediated by DEPDC5. Metabolomic analysis identified amino acids among the top metabolic changes between fed and fasted states. Glutamine was decreased in the fasted state with a larger decrease in DEPDC5-deficient brains. Brain glutamate is primarily generated from glutamine through the glutamine-glutamate cycle rather than transported from the blood (Yudkoff et al., 2006). Elevated glutamine is associated with persistent epileptiform activity in brain slices (Tani et al., 2010). DEPDC5-deficient mice are not protected from seizures despite lower glutamine levels. The lower amino acid levels in DEPDC5 brains likely reflects increased translation due to hyperactive mTOR (Kelly et al., 2018). Therefore, simply lowering glutamine levels is unlikely to be a viable therapeutic strategy to treat seizures.
In addition to glutamine, we found that DEPDC5-deficient neurons are insensitive to leucine withdrawal, suggesting that they lack sensing fluctuation in leucine content. In mice, we identified brain leucine was increased after fasting, although it has been demonstrated that in the fasted state the relative amount of leucine in the brain is reduced compared with blood by 35% (Yudkoff et al., 2006). The role of leucine and epilepsy is conflicting. Branch-chain amino acids, including leucine, have been shown to protect against seizures in mouse models (Dallerac et al., 2017; Hartman et al., 2015). In contrast, a leucine-enriched diet had no effect on a rat model of seizures but did enhanced the anti-seizure effects of the ketogenic diet (Takeuchi et al., 2021). While the mechanisms of how amino acid levels may alter seizure susceptibility remain unclear, loss of DEPDC5-mediated sensing of amino acids abolishes any protective effects provided by fasting. Other suggested mechanisms for seizure protection by fasting include directly lowering glucose or increasing ketone bodies (Greene et al., 2001; Kim et al., 2015). Depdc5 neuronal-specific knockout mice had reductions in blood glucose, several amino acids, and increases in ketones upon fasting, yet the mice were not protected from seizures. Therefore, changes in glucose, ketones, or amino acid levels were insufficient to overcome the loss of DEPDC5 and mTOR dysfunction.
DEPDC5 has been identified as a risk gene for SUDEP (Nascimento et al., 2015). Depdc5 knockout animal models are associated with seizure-induced death (Klofas et al., 2020b; Yuskaitis et al., 2019). Cardiac dysfunction is unlikely to be the underlying cause for SUDEP in DEPDC5-related epilepsy (Bacq et al., 2022). Serotonergic dysfunction is implicated in the pathogenesis of SUDEP (Devinsky et al., 2016; Petrucci et al., 2020). We report a reduction in serotonin and the precursor amino acid tryptophan in Depdc5-neuronal-specific knockout mice. Low serotonin brain levels are associated with seizures and seizure-induced death in rodent models (Dailey et al., 1992). Post-seizure reductions in serotonin levels are associated with post-seizure apnea in patients (Murugesan et al., 2019). Fasting did not alter the levels of serotonin or tryptophan, nor did it protect from seizure-induced death in Depdc5 neuronal-specific knockout mice. These results suggest that increasing serotonin levels might prevent seizure-induced death in Depdc5 neuronal-specific knockout mice, as has been reported in other models (Tupal and Faingold, 2019; Zeng et al., 2015). Future directions include understanding the mechanisms for how neuronal DEPDC5 loss leads to a reduction in brain serotonin levels.
In conclusion, we demonstrate that DEPDC5 is required to mediate the protective mTOR-related effects of fasting on seizure susceptibility. We demonstrate the critical role for DEPDC5 in mTORC1 sensing the withdrawal of leucine, glutamine, and arginine in neurons. Neuronal sensing of these amino acids may be critical for the protective effects of fasting on seizures. Fasting was insufficient to protect Depdc5 neuronal-specific knockout mice from seizures or seizure-induced death. While DEPDC5-related epilepsy may be insensitive to nutrient manipulation, the nutrient-sensing pathways through mTOR may be a potential therapeutic strategy for treating seizures of other causes.
Limitations of the study
While our in vitro data are compelling, there are still large gaps between primary neuronal cultures and the mature brain. The functional role of single amino acids on mTORC1 activity in vivo were not examined in this study. In addition, our biochemical readout for mTORC1 activity was pS6. A recent study evaluated the amino acid response in a neuroblastoma cell line using the fluorescence resonance energy transfer (FRET)-based TORCAR based on 4EBP1 activity (Iffland et al., 2020). That study concluded that DEPDC5-deficient neuron-like cell lines were completely insensitive to amino acid withdrawal. We found the expression and sensitivity of 4EBP1 to be low. It is also suggested that 4EBP2, not 4EBP1, controls mTORC1 signaling, at least in parvalbumin-positive neurons (Sharma et al., 2021). Although our results are largely consistent with prior studies, the differences may be due to methodological or cell-type differences. An additional limitation of our study is the use of short hairpin RNA (shRNA) knockdown of DEPDC5. Although our approach has been used to extensively to characterize the role of TSC in neurons (Di Nardo et al., 2014, 2020a; Nie et al., 2015), a small amount of DEPDC5 protein may remain and may be sufficient to retain minimal function. It remains to be seen if the mTORC1 response in neuronal cultures compared with a neuronal-like cell line and how these results relate to the intact brain.
We did not investigate the role of mTOR after seizure onset, rather we studied the mTOR activity and the metabolomic state prior to seizure onset to determine changes associated with seizure susceptibility only prior to seizures. Studies demonstrate that mTOR activity also changes after seizures, highlighting the complexity of the temporal dynamics of mTOR activity before and after seizures (Zhang and Wong, 2012). Finally, a limitation of our metabolomic data is that we only analyzed brain samples. Future studies analyzing brain and serum metabolomics in the context of known behavioral differences in Depdc5cc+ mice (e.g., hyperactivity) will provide insight into whether the metabolomic changes are intrinsic to the neurons or indirect from either behavioral or systemic changes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mustafa Sahin (Mustafa.Sahin@childrens.harvard.edu).
Materials availability
Unique materials generated in this study are available from the lead contact without restriction.
Data and code availability
The metabolomics data obtained in this study have been deposited at the NIH Common Fund’s NMDR (supported by NIH grant, U01-DK097430) website, the Metabolomics Workbench, https://www.metabolomicsworkbench.org. Accession number is listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| phospho-S6 (Ser240/244) | Cell Signaling Technology | Cat# 5364; RRID:AB_10694233 |
| S6 | Santa Cruz Biotechnology | Cat# sc-74459; RRID:AB_1129205 |
| phospho-AKT (Ser473) | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| AKT | Cell Signaling Technology | Cat# 4691; RRID:AB_915783 |
| NPRL2 | Cell Signaling Technology | Cat# 37344; RRID:AB_2799113 |
| mTOR | Cell Signaling Technology | Cat# 2983; RRID:AB_2105622 |
| phospho-mTOR (Ser 2448) | Cell Signaling Technology | Cat# 5536; RRID:AB_10691552 |
| beta-actin | Cell Signaling Technology | Cat# 3700; RRID:AB_2242334 |
| phospho-p70 S6 Kinase (Thr389) | Cell Signaling Technology | Cat# 9234; RRID:AB_2269803 |
| DEPDC5 | Sigma-Aldrich | Cat# HPA055619; RRID:AB_2682864 |
| TSC2 | Santa Cruz Biotechnology | Cat# sc-271314; RRID:AB_10611435 |
| p70 S6 Kinase | Santa Cruz Biotechnology | Cat# sc-8418; RRID:AB_628094 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9644; RRID:AB_2097841 |
| 4E-BP2 | Cell Signaling Technology | Cat# 2845; RRID:AB_10699019 |
| alpha-tubulin | Cell Signaling Technology | Cat# 3873; RRID:AB_1904178 |
| LAMP1 | Enzo Life Sciences | Cat# ADI-VAM-EN001; RRID:AB_10630197 |
| Goat anti-mouse IRDye 680RD | LI-COR Biosciences | Cat# 925–68070; RRID:AB_2651128 |
| Donkey anti-rabbit IRDye 800CW | LI-COR Biosciences | Cat# 926–32213; RRID:AB_621848 |
| Goat anti-mouse Alexa Fluor 488 | Invitrogen | Cat# A32723; RRID:AB_2633275 |
| Donkey anti-mouse Cy5 | Jackson ImmunoResearch | Cat# 715–175-150; RRID:AB_2340819 |
| Donkey anti-rabbit Cy3 | Jackson ImmunoResearch | Cat# 711–165-152; RRID:AB_2307443 |
|
Bacterial and virus strains | ||
| Tsc2-sh-GFP | Di Nardo et al., 2009 | N/A |
| Control sh-GFP for Tsc2 | Flavell et al., 2006 | N/A |
| Depdc5-sh-GFP | Origene | TL508165 |
| Control-sh-GFP | Origene | TR30021 |
|
Chemicals, peptides, and recombinant proteins | ||
| Pentylenetetrazol | Sigma-Aldrich | P6500–25G |
| Poly-D-Lysine Hydrobromide | MP Biomedical | Cat# 102694 |
| Laminin | ThermoFisherScientific | 23017–015 |
| Rapamycin | EMD Millipore | Cat# 53123–88-9 |
| DAPI | ThermoFisherScientific | Cat# 28908 |
| Glycine | Sigma | G8790–100G |
| L-Alanine | Sigma | A7469–25G |
| L-Arginine | Sigma | A8094–25G |
| L-Asparagine monohydrate | Sigma | A7094–25G |
| L-Cysteine | Sigma | C7352–25G |
| L-Glutamine | Sigma | G3126–100G |
| L-Histidine | Sigma | H6034–25G |
| L-Isoleucine | Sigma | I7403–25G |
| L-Leucine | Sigma | L8912–25G |
| L-Lysine Monohydrochloride | Sigma | L8662–25G |
| L-Methionine | Sigma | M5308–25G |
| L-Phenylalanine | Sigma | P5482–25G |
| L-Proline | Sigma | P5607–25G |
| L-Serine | Sigma | S4311–25G |
| L-Threonine | Sigma | T8441–25G |
| L-Tryptophan | Sigma | T8941–25G |
| L-Tyrosine | Sigma | T8566–25G |
| L-Valine | Sigma | VO513–25G |
|
Critical commercial assays | ||
| Beta hydroxybutyrate assay kit | Abcam | ab83390 |
| One touch ultra 2 glucometer | One touch | 024046 |
|
Deposited data | ||
| Metabolomic analysis | This paper; Metabolomics Workbench |
Table S1, Metabolomics Workbench: https://doi.org/10.21228/M88D9X |
|
Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat# CRL-3216, RRID: CVCL_0063 |
| HeLa | Generated in BD Manning lab | Huang et al., 2008 |
|
Experimental models: Organisms/strains | ||
| Sprague-Dawley Rat | Charles River Laboratories | N/A |
| Mouse: Depdc5f/f-SynCre+ | Generated in M Sahin lab | Yuskaitis et al., 2018 |
|
Software and algorithms | ||
| Fiji Software v2.0.0 | Open Source | http://fiji.sc/;RRID:SCR_002285 |
| Image Studio Lite analysis software v5.2.5 | LI-COR | https://www.licor.com/ |
| GraphPad PRISM v9.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| MetaboAnalyst v5.0. | MetaboAnalyst Software | https://www.metaboanalyst.ca/ |
|
Recombinant DNA | ||
| psPAX2 | Addgene | RRID:Addgene_12260 |
| pMD2.G | Addgene | RRID:Addgene_12259 |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mouse procedures were performed in accordance with the Guide for the Humane Use and Care of Laboratory Animals, and the study was approved by the Animal Care and Use Committee of Boston Children’s Hospital. All mice were housed in a 12-h light-dark cycle, climate-controlled room, with access to food and water ad lib. All surgeries were performed under isoflurane anesthesia, and all efforts were made to minimize pain. Neuron-specific Depdc5 conditional knockout mice (Depdc5cc+) and littermate control mice (Depdc5cw- and Depdc5cc-) were generated as previously described (Yuskaitis et al., 2018). Experiments were performed between 6 and 9 weeks on male and female littermate mice as indicated in the figure legends.
Cultured primary neurons
Cultures were prepared as we previously described (Di Nardo et al., 2020b). Briefly, hippocampi and cortices from 18-day-old rat embryos (Charles River CD1) were isolated under the microscope and collected in Hank’s Balanced Salt Solution containing 10mM MgCl2, 1mM kynurenic acid, 10mM HEPES and penicillin/streptomycin. After 5min dissociation at 37°C in 30U/mL of papain, neurons were mechanically triturated and plated in Neurobasal (NB) medium containing B27 supplement (Thermofisher), 2mM L-glutamine, penicillin/streptomycin and primocin (NB/B27). Biochemical analysis was performed on cortical cultures plated at 750,000 cells/well onto six-well plates coated with 20 mg/mL poly-D-lysine (PDL) and laminin. Immunofluorescent analysis was performed on hippocampal cultures plated at 50,000 cells/well onto coverslips in 24-well plates coated with 20 mg/mL poly-D-lysine (PDL).
Cell lines
HEK293T cells (ATCC) or HeLa control cells (Huang et al., 2008) were cultured in Dulbecco’s MEM (DMEM; Gibco) supplemented with 10% fetal calf serum (Gibco), 1% L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco) and maintained at 37°C in a 5% CO2 humidified atmosphere.
METHOD DETAILS
Acute fasting paradigm
Group housed mice, 6–9 weeks old from Depdc5cc + mice or littermate controls were randomized to fed or fasted conditions. Mice were weighed followed by food removal at 1pm for 24hr with ad lib water access. Twenty-four hours post-fasting, mice were weighed followed by experimentation for biochemistry or seizure susceptibility. For biochemistry, mice were sacrificed by guillotine with blood and brain rapidly collected. Mouse cerebral cortex was rapidly dissected and flash frozen.
Fasting blood glucose and beta-hydroxybutyrate measurement
Mice were fasted for 24 h during the day and blood collected at the time of decapitation. Blood glucose was measured immediately by Onetouch Ultra2 glucometer (LifeScan). Serum was collected by centrifugation at 1,000 x g for 10-min at 4°C and stored at −80°C. Serum beta-hydroxybutyrate measurement was performed following the manufactures protocol using beta Hydroxybutyrate (beta HB) Assay Kit (Abcam).
Acute PTZ seizure paradigm
PTZ (Sigma-Aldrich, #P6500) was dissolved fresh before each experiment in 0.9% saline at 10 mg/mL. Group housed mice, 6–9 weeks old from a stable mixed background line, were intraperitoneally (i.p.) injected with 65 mg/kg PTZ during the light phase of the light-dark cycle. Mice were monitored by an investigator blinded to genotype and treatment for generalized tonic-clonic seizures over a 20-min period, as previously described (Yuskaitis et al., 2021). After the 20-min observation period, mice were sacrificed.
Lentivirus production and transduction
Viral stocks for lentiviral infection were prepared by co-transfection of the two packaging plasmids psPAX2 and pMD2.G into HEK293T cells with the plasmid to be co-expressed using PEI. Viral particles were collected 48hrs and 72hrs after transfection and filtered through a 0.45 mm membrane. Rat neurons were infected at 1 day in vitro (DIV1) for 6 h then replaced by fresh NB/B27 medium. Depdc5 knockdown was achieved by (sh) RNAs (Origene, TL705799) and a non-effective 29-mer scrambled shRNA (Scr) construct (Origene, TR30021) in a pGFP-C-Lenti vector. Sequences used were shDepdc5: TGTCCGA CCTGGAGGATACACGCCTCAGA and shScramble: GCACTACCAGAGCTAACTCAGATAGTACT. The lentiviral Tsc2 shRNA construct was previously described (Di Nardo et al., 2009). The target sequence for the Tsc2 gene was the following: GGTGAAGAGAGCCGTATCACA. For Tsc2 knockdown, GFP-tagged control shRNA construct (here referred as ctrl-sh) was against the luciferase gene was previously described (Flavell et al., 2006). For double-knockdown of Depdc5 and Tsc2 (DTKD), lentivirus with shDepdc5 was introduced on DIV1 followed by lentivirus with shTsc2 on DIV5. All experiments were performed between DIV11–12. Only confluent neuronal cultures with at least 80% transduction, as detected by GFP + fluorescence, were used for subsequent experiments.
Neuronal culturing and nutrient manipulation
All quantifications were performed in a blinded fashion. Rat cortical neurons were plated at a density of 750,000 cells/well onto six-well plates and 1,000,000 cells/well coated with 20 mg/mL poly-D-lysine (PDL). Transduction of the neurons was performed with lentivirus as previously described (Di Nardo et al., 2020b). For immunoblotting, neurons were washed with cold PBS on ice and lysates collected in RIPA buffer with protease and phosphatase inhibitors.
Nutrient manipulation
Neurons were starved by washing them twice with amino acid free neurobasal (US Scientific) media without B27 supplement followed by treating with different amino acid solutions at physiological concentration (1X) prepared from 100x stocks of alanine (A), cysteine (C), phenylalanine (F), glycine (G), histidine (H), isoleucine (I), lysine (K), leucine (L), methionine (M), asparagine (N), proline (P), glutamine (Q), arginine (R), serine (S), threonine (T), valine (V) tryptophan (W), and tyrosine (Y) from 100X stock incubated at the indicated times. HEK293T cells were starved by washing them twice with DMEM media deprived of fetal bovine serum (FBS) and amino acids (AA) (Invitrogen) followed by treatment with +/−AA or +/−FBS. Growth factor withdrawal was performed by removing growth supplementation from the media.
Immunoblotting
Protein lysates were collected on ice in RIPA Buffer (ThermoFisher Scientific) with protease and phosphatase inhibitors (Roche Diagnostics) followed by BCA protein assay (ThermoFisher Scientific). Western blotting was performed using equal amounts of protein extract per lane (20–30 μg of protein) under reducing conditions in Bolt Bis-Tris 4–12% SDS polyacrylamide precast gels (ThermoFisher) and 4–20% Criterion™ TGX™ Precast Midi Protein Gels (Biorad). Gels were transferred onto Immobilon-FL PVDF membranes (Millipore), blocked with Odyssey TBS blocking buffer (LI-COR) for 1 h at room temperature, followed by primary antibody in Odyssey blocking buffer +0.2% Tween 20 (Sigma) overnight at 4°C. Antibodies used for western blotting were as follows: NPRL2 (Cell Signaling, cat.no. 37344, 1:500), Total mTOR (Cell Signaling, cat.no. 2983, 1:1000), Phospho-mTOR Ser 2448 (Cell Signaling, cat.no. 5536, 1:1000), Beta Actin (Cell Signaling, cat.no.3700, 1:5000), Phospho-S6 Ribosomal Protein (Ser 240/244) (Cell Signaling, cat.no. 5364, 1:1000), Anti-Ribosomal Protein S6 (Santa Cruz Biotechnology, cat.no. sc-74459, 1:1000), Phospho-p70 S6 Kinase Thr389 (Cell Signaling cat.no.9234, 1:500), Total Akt (Cell Signaling, cat.no.4691, 1:1000), Phospho-Akt Ser473 (Cell Signaling, cat.no. 4060, 1:1000), Anti-DEPDC5(Sigma-Aldrich, cat.no. HPA055619, 1:1000), Anti-Tuberin(Santa Cruz Biotechnology, cat.no. sc-271314, 1:1000), Anti-p70 S6 Kinase (Santa Cruz Biotechnology, cat.no. sc-8418,1:1000), 4E-BP1 (Cell Signaling, cat.no. 9644, 1:1000), Phospho-4E-BP1(Thr37/46) (Cell Signaling, cat.no. 2855, 1:1000), and 4E-BP2 (Cell Signaling, cat.no. 2845, 1:1000), alpha-tubulin (Abcam, cat.no. 15246, 1:10,000). After washing blots three times for 5 min each in Tris-Buffered Saline-Tween 20 (TBST), IRDye 800CW- or 680RD-conjugated secondary antibodies (1:10,000; LI-COR) in Odyssey blocking buffer, 0.2% Tween 20, and 0.01% SDS was added for 1 h at room temperature. To visualize bands, Odyssey Fc Imager was used, and densitometry analysis was performed using Image Studio Software (LI-COR). Quantifications were performed by protein normalization using actin or tubulin as loading control. Level of phosphorylated proteins was expressed as the ratio of phosphorylated/total level.
Lysosomal localization and immunofluorescence
Lysosomal colocalization to mTOR was performed by immunofluorescence on rat hippocampal neurons plated in 24-well plates at a density of 50,000/well onto 20 mg/mL Poly-D-Lysine (PDL) coated coverslips. DIV12 neurons were fixed in 4% PFA for 15 min at room temperature. Fixed primary hippocampal neurons were permeabilized and blocked in Intercept PBS blocking buffer (Licor #927–70001) with 0.1% Triton for 1hr at room temperature. Coverslips were incubated in blocking buffer containing primary antibodies 1hr at room temperature. Following three 5-min washes with PBS, coverslips were incubated in blocking buffer with secondary antibodies for 30-min at room temperature. Following three 10-min washes with PBS, with DAPI (Thermo #62248, 1:10.000) added to the last wash, coverslips were mounted to slides using Fluoromount-G (Southern Biotech #0100–01). Primary antibodies used: GFP (Invitrogen #A10262, 1:250), mTOR (CST #2983, 1:500), LAMP1 (Enzo #ADI-VAM-EN001, 1:250); secondary antibodies used: Chicken-Alexa Fluor488 (Abcam # ab150169, 1:1000), mouse-Alexa Fluor488 (Invitrogen #A32723, 1:1000), rabbit-Cy3 (Jackson ImmunoResearch #711–165-152, 1:500) and mouse-Cy5 (Jackson ImmunoResearch #715–175-150, 1:500). The slides were imaged with a Nikon Ti-E inverted microscope (Nikon Instruments, Melville, NY), using a 60× objective lens with a Zyla 4.2 cMOS camera and NIS elements software for acquisition parameters, shutters, filter positions and focus control. mTOR and LAMP1 images were imaged as z-stacks, summarized to maximum projection images and background-subtracted using the rolling ball method in Fiji with a diameter of 50 pixels. The soma area was determined by manually selecting the region of interest in the mTOR images. Quantitative co-localization analyses was generated by using the Coloc2 plug-in in Fiji. Representative cells are shown in all figures at the same exposure and magnification. All quantifications were repeated from 3 independent biological experiments with 2 technical replicates each time, taking 20 images of each condition and measured in a blinded fashion.
Metabolomic analysis
Metabolomic analysis was performed as previously described (McElroy et al., 2020). Briefly, mouse cortical lysate samples were rapidly isolated, and flash frozen in liquid nitrogen. Soluble metabolites were extracted directly from tissue using cold methanol/water (80/20, v/v) at approximately 1μL per 50μg of tissue followed by ultrasonication (Branson Sonifier 250) for 15 s. Homogenized samples were incubated at −80°C to precipitate proteins and subsequently centrifuged at 18,000xg for 20 min at 4°C to pellet the debris. The supernatants containing soluble metabolites were collected in new tubes and evaporated to dryness using a SpeedVac concentrator (Thermo Savant). Next, the metabolites were reconstituted in acetonitrile/water (60/40, v/v), vortex-mixed, and centrifuged at 18,000xg for 30 min at 4°C to remove debris. Samples were analyzed by High-Performance Liquid Chromatography and High-Resolution Mass Spectrometry and Tandem Mass Spectrometry (HPLC-MS/MS). Specifically, system consisted of a Thermo Q-Exactive in line with an electrospray source and an Ultimate3000 (Thermo) series HPLC consisting of a binary pump, degasser, and auto-sampler outfitted with a Xbridge Amide column (Waters; dimensions of 3.0 mm × 100 mm and a 3.5 μm particle size). The mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 10 mM ammonium hydroxide, 10 mM ammonium acetate, pH = 9.0; B was 100% Acetonitrile. The gradient was as following: 0 min, 15% A; 2.5 min, 30% A; 7 min, 43% A; 16 min, 62% A; 16.1–18 min, 75% A; 18–25 min, 15% A with a flow rate of 150 μL/min. The capillary of the ESI source was set to 275°C, with sheath gas at 35 arbitrary units, auxiliary gas at 5 arbitrary units and the spray voltage at 4.0 kV. In positive/negative polarity switching mode, an m/z scan range from 60 to 900 was chosen and MS1 data was collected at a resolution of 70,000. The automatic gain control (AGC) target was set at 1 × 106 and the maximum injection time was 200 ms. The top 5 precursor ions were subsequently fragmented, in a data-dependent manner, using the higher energy collisional dissociation (HCD) cell set to 30% normalized collision energy in MS2 at a resolution power of 17,500. Besides matching m/z, metabolites are identified by matching either retention time with analytical standards and/or MS2 fragmentation pattern. Data acquisition and analysis were carried out by Xcalibur 4.1 software and Tracefinder 4.1 software, respectively (both from Thermo Fisher Scientific). The peak area for each detected metabolite was normalized by the total ion current which was determined by integration of all of the recorded peaks within the acquisition window. Downstream analysis was carried out using Microsoft Excel and R. The list of the top 20 candidate metabolites for differential abundance was created by sorting metabolites by p value of a t test comparing 10 mice (5 males and 5 females between 7 and 10 weeks of age) from Depdc5cc+ and Tsc2cc + mice with respective littermate controls subjected to either 24hr fasting or fed states. Pathway analysis of KEGG compound IDs, principal components analysis score plots, and multiple comparisons adjusted T-tests were performed using MetaboAnalyst 5.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 9. Data are presented as mean ± standard deviation (SD), as indicated. All data was collected in blinded manner without knowledge of groups. Unblinding occurred at the time of analysis, but statistical tests were determined a priori to avoid bias. Chi-square test was performed for categorical variables. Continuous variables were analyzed using two-tailed student’s t-test for two groups. One-way or two-way analysis of variance (ANOVA) tests with post-hoc Tukey’s multiple-comparison tests and post hoc analysis were performed. Alpha was set at 0.05 for significance and ranges for p-values are reported for significant results.
Supplementary Material
Highlights.
mTORC1 integrates amino acid and growth factor signaling in neurons
Knockdown of DEPDC5 and/or TSC2 leads to sustained mTORC1 signaling with fasting
Neuronal DEPDC5 loss reduces serotonin levels and provokes seizure-induced death
Acute fasting protects from seizures by DEPDC5-mediated amino acid sensing mechanisms
ACKNOWLEDGMENTS
C.J.Y. receives grant support from NIH 1K08NS107637, the Hearst Foundation, the Boston Children’s Hospital Translational Research Program, and Boston Children’s Office of Faculty Development Career Development Fellowship. This study was supported by the BCH/Harvard Medical School Intellectual and Developmental Disabilities Research Center (NIH P50HD105351), and the BCH Rosamund Stone Zander Translational Neuroscience Center. R.P.C. was supported by a Northwestern University Pulmonary and Critical Care Department Cugell predoctoral fellowship. N.S.C. receives grant support from NIH1R35CA197532 and NIH1PO1AG049665 with metabolomics services provided by the Metabolomics Core Facility at Robert H. Lurie Comprehensive Cancer Center of Northwestern University.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111278.
DECLARATION OF INTERESTS
M.S. reports grant support from Novartis, Biogen, Astellas, Aeovian, Bridgebio, and Aucta. He has served on scientific advisory boards for Novartis, Roche, Regenxbio, SpringWorks Therapeutics, Jaguar Therapeutics, and Alkermes.
REFERENCES
- Ahmed MM, Carrel AJ, Cruz Del Angel Y, Carlsen J, Thomas AX, González MI, Gardiner KJ, and Brooks-Kayal A (2021). Altered protein profiles during epileptogenesis in the pilocarpine mouse model of temporal lobe epilepsy. Front. Neurol 12, 654606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacq A, Roussel D, Bonduelle T, Zagaglia S, Maletic M, Ribierre T, Adle-Biassette H, Marchal C, Jennesson M, An I, et al. (2022). Cardiac investigations in sudden unexpected death in DEPDC5-related epilepsy. Ann. Neurol 91, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, and Sabatini DM (2013). A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bough KJ, and Eagles DA (1999). A ketogenic diet increases the resistance to pentylenetetrazole-induced seizures in the rat. Epilepsia 40, 138–143. [DOI] [PubMed] [Google Scholar]
- Brewer GJ (1999). Regeneration and proliferation of embryonic and adult rat hippocampal neurons in culture. Exp. Neurol 159, 237–247. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Ingram EA, and Wen X (2009). Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J. Neurosci 29, 8259–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll B, Maetzel D, Maddocks OD, Otten G, Ratcliff M, Smith GR, Dunlop EA, Passos JF, Davies OR, Jaenisch R, et al. (2016). Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. Elife 5, e11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervenka M, Pascual JM, Rho JM, Thiele E, Yellen G, Whittemore V, and Hartman AL (2021). Metabolism-based therapies for epilepsy: new directions for future cures. Ann. Clin. Transl. Neurol 8, 1730–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, and Sabatini DM (2016). The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Sgritta M, Mays J, Zhou H, Lucero R, Park J, Wang IC, Park JH, Kaipparettu BA, Stoica L, et al. (2019). Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat. Med 25, 1684–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings NE, and Lamming DW (2017). Regulation of metabolic health and aging by nutrient-sensitive signaling pathways. Mol. Cell. Endocrinol 455, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey JW, Mishra PK, Ko KH, Penny JE, and Jobe PC (1992). Serotonergic abnormalities in the central nervous system of seizure-naive genetically epilepsy-prone rats. Life Sci 50, 319–326. [DOI] [PubMed] [Google Scholar]
- Dallérac G, Moulard J, Benoist JF, Rouach S, Auvin S, Guilbot A, Lenoir L, and Rouach N (2017). Non-ketogenic combination of nutritional strategies provides robust protection against seizures. Sci. Rep 7, 5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deCampo DM, and Kossoff EH (2019). Ketogenic dietary therapies for epilepsy and beyond. Curr. Opin. Clin. Nutr. Metab. Care 22, 264–268. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, and Richerson G (2016). Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 15, 1075–1088. [DOI] [PubMed] [Google Scholar]
- DeVivo DC, Malas KL, and Leckie MP (1975). Starvation and seizures. Observation on the electroconvulsive threshold and cerebral metabolism of the starved adult rat. Arch. Neurol 32, 755–760. [DOI] [PubMed] [Google Scholar]
- Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, and Sahin M (2009). Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J. Neurosci 29, 5926–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo A, Lenoël I, Winden KD, Rühmkorf A, Modi ME, Barrett L, Ercan-Herbst E, Venugopal P, Behne R, Lopes CAM, et al. (2020a). Phenotypic screen with TSC-deficient neurons reveals heat-shock machinery as a druggable pathway for mTORC1 and reduced cilia. Cell Rep 31, 107780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo A, Vasquez S, and Sahin M (2020b). A cell-based assay optimized for high-content cilia imaging with primary rat hippocampal neurons. STAR Protoc 1, 100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo A, Wertz MH, Kwiatkowski E, Tsai PT, Leech JD, Greene-Colozzi E, Goto J, Dilsiz P, Talos DM, Clish CB, et al. (2014). Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum. Mol. Genet 23, 3865–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, Finan PM, Kwiatkowski DJ, Murphy LO, and Manning BD (2012). TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 47, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagles DA, Boyd SJ, Kotak A, and Allan F (2003). Calorie restriction of a high-carbohydrate diet elevates the threshold of PTZ-induced seizures to values equal to those seen with a ketogenic diet. Epilepsy Res 54, 41–52. [DOI] [PubMed] [Google Scholar]
- Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, and Blenis J (2004). mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell Biol 24, 200–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, and Greenberg ME (2006). Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Fonseca BD, Alain T, Finestone LK, Huang BPH, Rolfe M, Jiang T, Yao Z, Hernandez G, Bennett CF, and Proud CG (2011). Pharmacological and genetic evaluation of proposed roles of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK), extracellular signal-regulated kinase (ERK), and p90(RSK) in the control of mTORC1 protein signaling by phorbol esters. J. Biol. Chem 286, 27111–27122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, Curatolo P, de Vries PJ, Dlugos DJ, Berkowitz N, et al. (2016). Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 388, 2153–2163. [DOI] [PubMed] [Google Scholar]
- Greene AE, Todorova MT, McGowan R, and Seyfried TN (2001). Caloric restriction inhibits seizure susceptibility in epileptic EL mice by reducing blood glucose. Epilepsia 42, 1371–1378. [DOI] [PubMed] [Google Scholar]
- Griffith JL, and Wong M (2018). The mTOR pathway in treatment of epilepsy: a clinical update. Future Neurol 13, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, Scaria SM, Harper JW, Gygi SP, and Sabatini DM (2017). SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358, 813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, and Avruch J (1998). Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem 273, 14484–14494. [DOI] [PubMed] [Google Scholar]
- Hartman AL, Santos P, Dolce A, and Hardwick JM (2012). The mTOR inhibitor rapamycin has limited acute anticonvulsant effects in mice. PLoS One 7, e45156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman AL, Santos P, O’Riordan KJ, Stafstrom CE, and Marie Hardwick J (2015). Potent anti-seizure effects of D-leucine. Neurobiol. Dis 82, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Dibble CC, Matsuzaki M, and Manning BD (2008). The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell Biol 28, 4104–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iffland PH, 2nd, Barnes AE, Baybis M, and Crino PB (2020). Dynamic analysis of 4E-BP1 phosphorylation in neurons with Tsc2 or Depdc5 knockout. Exp. Neurol 334, 113432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iffland PH, 2nd, Baybis M, Barnes AE, Leventer RJ, Lockhart PJ, and Crino PB (2018). DEPDC5 and NPRL3 modulate cell size, filopodial outgrowth, and localization of mTOR in neural progenitor cells and neurons. Neurobiol. Dis 114, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, and Guan KL (2015). Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E, Schaeffer SM, Dhamne SC, Lipton JO, Lindemann L, Honer M, Jaeschke G, Super CE, Lammers SH, Modi ME, et al. (2018). mGluR5 modulation of behavioral and epileptic phenotypes in a mouse model of tuberous sclerosis complex. Neuropsychopharmacology 43, 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Simeone KA, Simeone TA, Pandya JD, Wilke JC, Ahn Y, Geddes JW, Sullivan PG, and Rho JM (2015). Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol 78, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klofas LK, Short BP, Snow JP, Sinnaeve J, Rushing GV, Westlake G, Weinstein W, Ihrie RA, Ess KC, and Carson RP (2020a). DEPDC5 haploinsufficiency drives increased mTORC1 signaling and abnormal morphology in human iPSC-derived cortical neurons. Neurobiol. Dis 143, 104975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klofas LK, Short BP, Zhou C, and Carson RP (2020b). Prevention of premature death and seizures in a Depdc5 mouse epilepsy model through inhibition of mTORC1. Hum. Mol. Genet 29, 1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH (2004). More fat and fewer seizures: dietary therapies for epilepsy. Lancet Neurol 3, 415–420. [DOI] [PubMed] [Google Scholar]
- Krueger DA, Wilfong AA, Mays M, Talley CM, Agricola K, Tudor C, Capal J, Holland-Bouley K, and Franz DN (2016). Long-term treatment of epilepsy with everolimus in tuberous sclerosis. Neurology 87, 2408–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, and Sabatini DM (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol 21, 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Moehl K, Ghena N, Schmaedick M, and Cheng A (2018). Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci 19, 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MP, Cullen ER, Barrows CM, Shore AN, Tooke KI, Laprade KA, Stafford JM, and Weston MC (2020). Genetic inactivation of mTORC1 or mTORC2 in neurons reveals distinct functions in glutamatergic synaptic transmission. Elife 9, e51440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SS, Rensing NR, Thio LL, Yamada KA, and Wong M (2011). The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 52, e7–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy GS, Reczek CR, Reyfman PA, Mithal DS, Horbinski CM, and Chandel NS (2020). NAD+ regeneration rescues lifespan, but not ataxia, in a mouse model of brain mitochondrial complex I dysfunction. Cell Metab 32, 301–308.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, and Kwiatkowski DJ (2007). A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci 27, 5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng D, Yang Q, Wang H, Melick CH, Navlani R, Frank AR, and Jewell JL (2020). Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J. Biol. Chem 295, 2890–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, and Manning BD (2014). Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugesan A, Rani MRS, Vilella L, Lacuey N, Hampson JP, Faingold CL, Friedman D, Devinsky O, Sainju RK, Schuele S, et al. (2019). Postictal serotonin levels are associated with peri-ictal apnea. Neurology 93, e1485–e1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento FA, Borlot F, Cossette P, Minassian BA, and Andrade DM (2015). Two definite cases of sudden unexpected death in epilepsy in a family with a DEPDC5 mutation. Neurol. Genet 1, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LH, Mahadeo T, and Bordey A (2019). mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J. Neurosci 39, 2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie D, Chen Z, Ebrahimi-Fakhari D, Di Nardo A, Julich K, Robson VK, Cheng YC, Woolf CJ, Heiman M, and Sahin M (2015). The stress-induced atf3-gelsolin cascade underlies dendritic spine deficits in neuronal models of tuberous sclerosis complex. J. Neurosci 35, 10762–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padamsey Z, Katsanevaki D, Dupuy N, and Rochefort NL (2022). Neocortex saves energy by reducing coding precision during food scarcity. Neuron 110, 280–296.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucci AN, Joyal KG, Purnell BS, and Buchanan GF (2020). Serotonin and sudden unexpected death in epilepsy. Exp. Neurol 325, 113145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Farfán BV, Rubio Osornio MDC, Custodio Ramírez V, Paz Tres C, and Carvajal Aguilera KG (2015). Caloric restriction protects against electrical kindling of the amygdala by inhibiting the mTOR signaling pathway. Front. Cell. Neurosci 9, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirkmajer S, and Chibalin AV (2011). Serum starvation: caveat emptor. Am. J. Physiol. Cell Physiol 301, C272–C279. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, and Sabatini DM (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Sood R, Lou D, Hung TY, Lévesque M, Han Y, Levett JY, Wang P, Murthy S, Tansley S, et al. (2021). 4E-BP2-dependent translation in parvalbumin neurons controls epileptic seizure threshold. Proc. Natl. Acad. Sci. USA 118.e2025522118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, and Jaworski J (2017). Molecular neurobiology of mTOR. Neuroscience 341, 112–153. [DOI] [PubMed] [Google Scholar]
- Takeuchi F, Nishikata N, Nishimura M, Nagao K, and Kawamura M Jr. (2021). Leucine-enriched essential amino acids enhance the antiseizure effects of the ketogenic diet in rats. Front. Neurosci 15, 637288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Dulla CG, Huguenard JR, and Reimer RJ (2010). Glutamine is required for persistent epileptiform activity in the disinhibited neocortical brain slice. J. Neurosci 30, 1288–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupal S, and Faingold CL (2019). Fenfluramine, a serotonin-releasing drug, prevents seizure-induced respiratory arrest and is anticonvulsant in the DBA/1 mouse model of SUDEP. Epilepsia 60, 485–494. [DOI] [PubMed] [Google Scholar]
- Valvezan AJ, and Manning BD (2019). Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat. Metab 1, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viggiano A, Pilla R, Arnold P, Monda M, D’Agostino D, Zeppa P, and Coppola G (2016). Different calorie restriction treatments have similar antiseizure efficacy. Seizure 35, 45–49. [DOI] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, and Sabatini DM (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, et al. (2017). KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Kudchodkar SB, and Alwine JC (2005). Effects of simian virus 40 large and small tumor antigens on mammalian target of rapamycin signaling: small tumor antigen mediates hypophosphorylation of eIF4E-binding protein 1 late in infection. J. Virol 79, 6882–6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan E, Tsai PT, Greene-Colozzi E, Sahin M, Kwiatkowski DJ, and Malinowska IA (2012). Graded loss of tuberin in an allelic series of brain models of TSC correlates with survival, and biochemical, histological and behavioral features. Hum. Mol. Genet 21, 4286–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Nissim I, Horyn O, Lazarow A, Luhovyy B, Wehrli S, and Nissim I (2005). Response of brain amino acid metabolism to ketosis. Neurochem. Int 47, 119–128. [DOI] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Nissim I, Horyn O, Luhovyy B, Lazarow A, and Nissim I (2006). Short-term fasting, seizure control and brain amino acid metabolism. Neurochem. Int 48, 650–656. [DOI] [PubMed] [Google Scholar]
- Yuen AWC, and Sander JW (2014). Rationale for using intermittent calorie restriction as a dietary treatment for drug resistant epilepsy. Epilepsy Behav 33, 110–114. [DOI] [PubMed] [Google Scholar]
- Yuskaitis CJ, Jones BM, Wolfson RL, Super CE, Dhamne SC, Rotenberg A, Sabatini DM, Sahin M, and Poduri A (2018). A mouse model of DEPDC5-related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis 111, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuskaitis CJ, Rossitto LA, Groff KJ, Dhamne SC, Zhang B, Lalani LK, Singh AK, Rotenberg A, and Sahin M (2021). Factors influencing the acute pentylenetetrazole-induced seizure paradigm and a literature review. Ann. Clin. Transl. Neurol 8, 1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuskaitis CJ, Rossitto LA, Gurnani S, Bainbridge E, Poduri A, and Sahin M (2019). Chronic mTORC1 inhibition rescues behavioral and biochemical deficits resulting from neuronal Depdc5 loss in mice. Hum. Mol. Genet 28, 2952–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Long X, Cotten JF, Forman SA, Solt K, Faingold CL, and Feng HJ (2015). Fluoxetine prevents respiratory arrest without enhancing ventilation in DBA/1 mice. Epilepsy Behav 45, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, and Wong M (2009). The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J. Neurosci 29, 6964–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, and Wong M (2012). Pentylenetetrazole-induced seizures cause acute, but not chronic, mTOR pathway activation in rat. Epilepsia 53, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The metabolomics data obtained in this study have been deposited at the NIH Common Fund’s NMDR (supported by NIH grant, U01-DK097430) website, the Metabolomics Workbench, https://www.metabolomicsworkbench.org. Accession number is listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| phospho-S6 (Ser240/244) | Cell Signaling Technology | Cat# 5364; RRID:AB_10694233 |
| S6 | Santa Cruz Biotechnology | Cat# sc-74459; RRID:AB_1129205 |
| phospho-AKT (Ser473) | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| AKT | Cell Signaling Technology | Cat# 4691; RRID:AB_915783 |
| NPRL2 | Cell Signaling Technology | Cat# 37344; RRID:AB_2799113 |
| mTOR | Cell Signaling Technology | Cat# 2983; RRID:AB_2105622 |
| phospho-mTOR (Ser 2448) | Cell Signaling Technology | Cat# 5536; RRID:AB_10691552 |
| beta-actin | Cell Signaling Technology | Cat# 3700; RRID:AB_2242334 |
| phospho-p70 S6 Kinase (Thr389) | Cell Signaling Technology | Cat# 9234; RRID:AB_2269803 |
| DEPDC5 | Sigma-Aldrich | Cat# HPA055619; RRID:AB_2682864 |
| TSC2 | Santa Cruz Biotechnology | Cat# sc-271314; RRID:AB_10611435 |
| p70 S6 Kinase | Santa Cruz Biotechnology | Cat# sc-8418; RRID:AB_628094 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9644; RRID:AB_2097841 |
| 4E-BP2 | Cell Signaling Technology | Cat# 2845; RRID:AB_10699019 |
| alpha-tubulin | Cell Signaling Technology | Cat# 3873; RRID:AB_1904178 |
| LAMP1 | Enzo Life Sciences | Cat# ADI-VAM-EN001; RRID:AB_10630197 |
| Goat anti-mouse IRDye 680RD | LI-COR Biosciences | Cat# 925–68070; RRID:AB_2651128 |
| Donkey anti-rabbit IRDye 800CW | LI-COR Biosciences | Cat# 926–32213; RRID:AB_621848 |
| Goat anti-mouse Alexa Fluor 488 | Invitrogen | Cat# A32723; RRID:AB_2633275 |
| Donkey anti-mouse Cy5 | Jackson ImmunoResearch | Cat# 715–175-150; RRID:AB_2340819 |
| Donkey anti-rabbit Cy3 | Jackson ImmunoResearch | Cat# 711–165-152; RRID:AB_2307443 |
|
Bacterial and virus strains | ||
| Tsc2-sh-GFP | Di Nardo et al., 2009 | N/A |
| Control sh-GFP for Tsc2 | Flavell et al., 2006 | N/A |
| Depdc5-sh-GFP | Origene | TL508165 |
| Control-sh-GFP | Origene | TR30021 |
|
Chemicals, peptides, and recombinant proteins | ||
| Pentylenetetrazol | Sigma-Aldrich | P6500–25G |
| Poly-D-Lysine Hydrobromide | MP Biomedical | Cat# 102694 |
| Laminin | ThermoFisherScientific | 23017–015 |
| Rapamycin | EMD Millipore | Cat# 53123–88-9 |
| DAPI | ThermoFisherScientific | Cat# 28908 |
| Glycine | Sigma | G8790–100G |
| L-Alanine | Sigma | A7469–25G |
| L-Arginine | Sigma | A8094–25G |
| L-Asparagine monohydrate | Sigma | A7094–25G |
| L-Cysteine | Sigma | C7352–25G |
| L-Glutamine | Sigma | G3126–100G |
| L-Histidine | Sigma | H6034–25G |
| L-Isoleucine | Sigma | I7403–25G |
| L-Leucine | Sigma | L8912–25G |
| L-Lysine Monohydrochloride | Sigma | L8662–25G |
| L-Methionine | Sigma | M5308–25G |
| L-Phenylalanine | Sigma | P5482–25G |
| L-Proline | Sigma | P5607–25G |
| L-Serine | Sigma | S4311–25G |
| L-Threonine | Sigma | T8441–25G |
| L-Tryptophan | Sigma | T8941–25G |
| L-Tyrosine | Sigma | T8566–25G |
| L-Valine | Sigma | VO513–25G |
|
Critical commercial assays | ||
| Beta hydroxybutyrate assay kit | Abcam | ab83390 |
| One touch ultra 2 glucometer | One touch | 024046 |
|
Deposited data | ||
| Metabolomic analysis | This paper; Metabolomics Workbench |
Table S1, Metabolomics Workbench: https://doi.org/10.21228/M88D9X |
|
Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat# CRL-3216, RRID: CVCL_0063 |
| HeLa | Generated in BD Manning lab | Huang et al., 2008 |
|
Experimental models: Organisms/strains | ||
| Sprague-Dawley Rat | Charles River Laboratories | N/A |
| Mouse: Depdc5f/f-SynCre+ | Generated in M Sahin lab | Yuskaitis et al., 2018 |
|
Software and algorithms | ||
| Fiji Software v2.0.0 | Open Source | http://fiji.sc/;RRID:SCR_002285 |
| Image Studio Lite analysis software v5.2.5 | LI-COR | https://www.licor.com/ |
| GraphPad PRISM v9.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| MetaboAnalyst v5.0. | MetaboAnalyst Software | https://www.metaboanalyst.ca/ |
|
Recombinant DNA | ||
| psPAX2 | Addgene | RRID:Addgene_12260 |
| pMD2.G | Addgene | RRID:Addgene_12259 |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.