

Abstract
C-terminally modified peptides are important for the development and delivery of peptide-based pharmaceuticals because they impact peptide activity, stability, hydrophobicity, and membrane permeability. Additionally, the vulnerability of C-terminal esters to cleavage by endogenous esterases makes them excellent pro-drugs. Methods for post-SPPS C-terminal functionalization potentially enable access to libraries of modified peptides, facilitating tailoring of their solubility, potency, toxicity, and uptake pathway. Apparently minor structural changes can significantly impact the binding, folding, and pharmacokinetics of the peptide. This review summarizes developments in chemical methods for C-terminal modification of peptides published since the last review on this topic in 2003.
Graphical Abstract
A go-to compilation of recent strategies to access C-terminally modified peptides contextualized by a discussion of the major synthetic challenges that have historically hampered progress in this area.
1 |. INTRODUCTION
Peptides are becoming increasingly important pharmaceutical targets as classically “druggable” targets dwindle and methods for peptide synthesis, delivery, and penetration through the cell membrane continue to improve. Relatively minor structural changes can significantly impact the peptide’s folding, binding, and pharmacokinetics. For example, roughly half of biologically active peptides possess an α-amidated C-terminal moiety, and this post-translational modification (PTM) is often essential for recognition of the peptides by their receptors.1 Because of their importance, many strategies to access synthetic C-terminally modified peptides have been developed. In recent years, the peptide community has made significant strides in this area, largely due to the need to access bench stable thioesters and thioester surrogates for use in native chemical ligation (NCL) reactions, which are used primarily in academic laboratories for the assembly of complex peptide and protein targets.2 During Bocsolid-phase peptide synthesis (SPPS), the thioester is installed via incorporation of a mercaptopropionate linker before attachment of the C-terminal amino acid.3 Over recent decades, Fmoc-SPPS has increased in popularity because it uses the safer and operationally more simple trifluoroacetic acid (TFA) to cleave the peptide from resin instead of gaseous HF. Unfortunately, thioesters are not stable to nucleophiles like piperidine, which is used to remove Fmoc after each amino acid coupling.4 Furthermore, C-terminal thioesters are more prone to epimerization in presence of basic species than carboxylic acids and amides. Many strategies have been developed to access to C-terminal thioesters via Fmoc-SPPS. However, these methods have been reviewed extensively elsewhere and will not be discussed further here.5 This review will discuss synthetic approaches to access non-thioester C-terminal modifications of peptides published since 2003. The advantages and limitations of these strategies will be highlighted.
1.1 |. Structural Effects of C-Terminal Carboxamides
Modification of the C terminus from carboxylic acid to carboxamide directly impacts peptide conformation, which can influence biological activity. For example, α-conotoxin ImI (1) is a carboxamide-terminated, two-disulfide peptide composed of 12 amino acids that is isolated from the venom of conus snails (Figure 1A). This peptide and its analogs bind to Aplysia and Bulinus acetylcholine binding proteins (AChBP).6 The overall structure is primarily dictated by the two disulfides in the sequence. However, as illustrated in Figure 1B, the acid analog deviates noticeably from the native carboxamide secondary structure.7 Furthermore, the reduced linear form of native α-conotoxin ImI and the carboxylic acid analog produce different major disulfide isoforms upon oxidative folding (Figure 1C). The ImI carboxamide folds predominantly into the globular isoform (2, 54%), while the ImI carboxylic acid folds primarily into the ribbon structure (3, 67%).7,8
Figure 1.

A) Primary structure of native α-conotoxin ImI, B) backbone overlay of both amide and acid analogs of α-conotoxin ImI determined by Nuclear Magnetic Resonance (NMR), C) the three isoforms of α-conotoxin ImI.
Most C-terminal modifications result in neutralization of the negative charge that the carboxylic acid derivative displays at physiological pH. Without this charge, the peptide’s hydrophobicity increases, changing the receptor binding properties of the peptide.9,10 For example, replacing the carboxy terminus of human parathyroid hormone (hPTH) with the carboxamide increases its activity. Willick and co-workers hypothesized that the C-terminal amide of hPTH-NH2 participates in a hydrogen bond with the PTH-receptor and that this interaction mimics the peptide bond present in the natural hormone fragment.11 They postulate that PTH analog effectiveness may be the result of the conformation of the analog and analog-receptor complex and that the amide analog may be stable to some carboxypeptidases,12 resulting in a longer overall lifetime than the parent carboxylic acid.
Many inflammatory peptides bear C-terminal carboxamides. In addition to the carboxamide’s decreased polarity in relations to carboxylates, this modification changes intramolecular hydrogen bonding patterns, altering the structure of the α-helix. The installation of the C-terminal carboxamide increases its ability to disrupt the cell membrane.13 Amphiphilic α-helices are found near the C terminus of most hormones that react with type II G-protein-coupled receptors (GPCRs).13 GPCRs are common targets for broad-spectrum therapeutics due to their role in human physiological responses to stimulants like hormones and neurotransmitters.14 The ability of the C-terminal functionality to hydrogen bond with a suitable group, such as the peptide backbone or amino acid sidechain, is integral to the helical stability.11 The potency of proline-rich antimicrobial peptides towards Gram negative bacteria increases upon C-terminal modification, specifically, hydrazide, alcohol, and amide terminated peptides are more active than the carboxylate.15 Further enhancement of antibacterial activity was observed through multimerization of these C-terminally modified peptides.16 Mechanisms of action for these peptides include cell wall disruption,17 inhibition of the protein DnaK,18 and inhibition of bacterial ribosomal protein expression.19
1.2 |. Ester and Secondary Amide Modifications
Several other representative linear C-terminally modified peptides are illustrated in Figure 2. CΑ−074 methyl ester (5) is a cathepsin B inhibitor. Cathepsin B is a biomarker in several cancers and is associated with tumorigenesis.20 The carboxylic acid analog, CΑ−074, is also active, but the C-terminal acid substantially reduces the membrane permeability, making it an ineffective drug.21 α-Factor peptide (6), is a pheromone produced by the yeast Saccharomyces cerevisiae. The C-terminal cysteine thiol is farnesylated and bears a methyl ester moiety that is essential for its activity. Other alkyl ester analogs of α-factor peptide were also active, provided the alkyl group remained small enough to allowing the peptide C terminus to fit within the binding pocket.22 Amide-modified C-terminal peptide ABT-510 (7) mimics endogenous protein thrombospondin-1 (TSP-1) and inhibits pro-angiogenic growth factors associated with tumor neovascularization.23 ABT-510 was briefly in a phase II clinical trial treating patients with stage IV melanoma.24 Auristatin PYE (8) is a derivative of dolastatin 10 bearing a pyridine-tethered C-terminal amide.25 Auristatin PYE is a potent antimitotic agent, and the alkyl-linked pyridyl group at the C terminus increases the peptide’s solubility in water while still maintaining potency towards certain tumor lines.26
Figure 2.

Selected C-terminally modified peptides.
Another example of a C-terminal amide is the attachment of glycosylphosphatidylinositol (9) to the C terminus of a peptide, which is a post-translational modification that enables membrane anchoring of peptides and proteins in eukaryotic cells (Figure 3).9,27 A phosphoethanolamine moiety links the inositol core to the C terminus of the peptide. The phospholipid portion of GPI is responsible for membrane anchoring. This PTM has also been observed on glycoprotein surfaces. A deficiency in this PTM is commonly associated with a form of anemia called paroxysmal nocturnal hemoglobinuria (PNH).28
Figure 3.

Membrane anchoring of peptides via C-terminal glycosylphosphatidylinositol attachment.
1.3 |. β-Amino alcohols
Peptaibols are a class of bioactive peptides that have a characteristic C-terminal β-amino alcohol such as phenylalaninol (10), homoleucinol (11), or 2-[(2′-aminopropyl)methylamino]ethanol (12, Figure 4A). Peptaibols were first isolated from Trichoderma viride in 1967.29,30 Peptaibol peptides are known to permeate the cell membrane. These membrane-active peptides (MAP) are hypothesized to form pores in lipid bilayers and display a wide range of biological activities including antibacterial, antifungal, and other activities.31 One example is culicinin D (13, Figure 4B), a linear peptaibol isolated from Culicinomyces clavisporus, which exhibits anti-cancer activity, in addition to its analogs.32,33
Figure 4.

A) C-terminal β-amino alcohols commonly found in peptaibols, B) the linear peptaibol culicinin D.
1.4 |. Macrocyclic Peptides
Macrocyclic peptides involving linkage to the C terminus can be viewed as C-terminally modified peptides. Specifically, sidechain heteroatoms or the N-terminal amine can be used as nucleophiles to attack the activated carbonyl (14), which allows access to sidechain-to-tail (15) or head-to-tail (16) macrocycles, respectively (Scheme 1).34 These targets are plagued by a host of challenges unique to them, and they have recently been reviewed. Thus, we direct the reader to those references and acknowledge here that some of the strategies described in this review have been employed in macrocyclization chemistries as well.35
Scheme 1.

Access to macrocyclic peptides using activated carbonyl chemistry.
1.5 |. Self-Assembly Properties
Modification of the C-terminal moiety can enable modulation of peptide self-assembly. Indeed, this strategy that has been used to target and kill cancer cells.36 In 2016, Xu and co-workers investigated the impact of a tetrapeptide’s C-terminal moiety on its ability to self-assemble.37 All modified peptides displayed higher self-assembly tendencies versus the parent carboxylic acid substrate, a discovery that is consistent with the repulsive nature of negatively charged groups brought into close proximity. Following this simple modification, certain tripeptides had IC50 values comparable to that of the widely used chemotherapy drug cisplatin.38 With relatively small molecular weight increases, significant benefits to self-assembly behavior were observed. Taken together, these examples provide strong evidence for the importance of both naturally occurring and synthetic C-terminally modified peptides.
1.6 |. Biosynthetic Pathways for Naturally Occurring C-Terminally Modified Peptides
C-terminal amidation and esterification reactions that occur in nature are potentially inspirational for chemical synthesis efforts. Thus, we briefly outline here the biosynthetic pathways for C-terminal carboxamide and methyl ester peptides. A bifunctional dimeric enzyme called peptidylglycine α-amidating monooxygenase (PAM) is responsible for removal of the carbon-containing portion of a C-terminal glycine to unmask the C-terminal carboxamide of a contracted peptide sequence (Scheme 2). The two subunits of PAM are peptidylglycine α-hydroxylating monooxygenase (PHM) and peptidyl-α-hydroxyglycine-α-amidating lyase (PAL). Interaction of the precursor peptide (17) with PHM in combination with Cu(II), molecular oxygen, and ascorbate leads to α-radical formation on glycine (18). Further oxidation by PHM proceeds with facial selective atom transfer to afford hydroxyacetate 19.39 Subsequently, PAL in combination with Zn(II) induces C–N bond cleavage to afford 2-oxoacetate (21) and reveal the C-terminal carboxamide 20. Oxytocin and vasopressin are examples of peptides that undergo this oxidative cleavage to unmask the bioactive C-terminal carboxamide.40
Scheme 2.

Two-step biosynthetic amidation of peptidylglycine
The biosynthetic pathway for α-factor peptide, a C-terminal farnesylated cysteine methyl ester, is illustrated in Scheme 3. The initial biosynthetic intermediate for α-factor peptide (22) contains an N-terminal extension, the mature fragment, and a C-terminal CAAX domain.41 The farnesylation of cysteine is mediated by Ram1/Ram2, which is a heterodimeric farnesyltranferase, and converts peptide 22 to 23. The CAAX portion of the peptide is then cleaved from the mature fragment by either Ste24 or Rce1 to unmask the C terminus of the mature fragment (24). The C terminus is then esterified by Ste14 to produce methyl ester peptide 25. The N-terminal portion of the N-terminal extension is cleaved by Ste24 to generate penultimate intermediate 26. Cleavage of the remainder of the N-terminal extension fragment is mediated by Axl1, a putative zinc metalloprotease, yielding α-factor peptide 6. Peptide 6 is subsequently exported by an ATP binding cassette (ABC) protein transporter, Ste6, which allows for the peptide to interact with its receptor, Ste3.
Scheme 3.

Biosynthetic pathway for α-factor peptide.
2 |. KEY SYNTHETIC CHALLENGES
2.1.1 |. Epimerization of the α-carbon – oxazalone formation
Modification of the C terminus can be performed on resin during SPPS when the synthesis is performed in the N-terminal to C-terminal direction (N to C), also known as inverse solid phase peptide synthesis (ISPPS).42 However, the N to C approach remains limited by epimerization via oxazolone formation, as illustrated in Scheme 4.42 During a coupling step, the C-terminal amino acid of the linear peptide (27) is activated toward nucleophilic attack (28). However, intramolecular attack of the adjacent backbone amide carbonyl can lead to oxazolone formation (29). Once the oxazolone is formed, deprotonation at the α-position is driven by aromatization to form 5-hydroxyoxazole 30, which planarizes the α-position. Non-selective protonation of the α-position gives a mixture of diastereomers (31), resulting in epimerized peptide. The nucleophile then opens the oxazalone to give the target peptide (32) with loss of stereointegrity at the C-terminal residue. This is the major limitation of N to C peptide synthesis, and it also plagues many C-terminal modification/diversification approaches.
Scheme 4.

Mechanism of C-terminal amino acid epimerization initiated by oxazolone formation.
2.1.2 |. Epimerization-Prone Residues – Histidine and Cysteine
A long-standing challenge in C-terminal modification is the synthesis and modification of C-terminal cysteine and histidine peptides.43,44 Cysteine in particular constitutes a major chemical problem because unlike other amino acids, Cys is prone to both oxazalone formation and direct deprotonation at the α-position. Thus, it is susceptible to epimerization during multiple steps in the synthetic process.45 Specialized conditions are needed to avoid epimerization of cysteine during its attachment to the linker.22,45,46 Repeated exposure to piperidine during elongation of the peptide via Fmoc SPPS can induce epimerization,45,47 and extensive epimerization can occur during activation of the C terminus of peptides.48
2.2 |. Instability of Activated C-Terminal Groups
The most straightforward approach, from a synthetic perspective, to access C-terminally modified peptides would be simple activation of the carboxylic acid via one of the many known strategies,49 followed by displacement with an appropriate nucleophile. In addition to their tendency to undergo epimerization, activated C-terminal carboxylic acids are vulnerable to hydrolysis, which can interfere with desired nucleophilic displacement reactions, lowering yields and hampering purification efforts. Additionally, some nominally stable C-terminal modifications are prone to hydrolysis. Thioesters (33) suffer degradation upon storage and must be handled under acidic conditions to avoid hydrolysis. C-Terminal thioesters are particularly important to academic peptide chemists because of their utility in the de novo synthesis of large peptides and proteins via native chemical ligation (Scheme 5).2,5 In an NCL reaction, thioester 33 can undergo transthioesterification reactions with thioester additives (e.g., MPAA) and, eventually, a peptide fragment bearing an N-terminal cysteine (34). Cysteine thioester 35 then undergoes a rapid S to N acyl transfer to form the more thermodynamically stable amide bond (36). Thioesters such as 33 can be obtained using several strategies. These C-terminal thioester syntheses have been reviewed extensively elsewhere and will not be further discussed here.5,50
Scheme 5.

Native chemical ligation.
3 |. CHEMICAL STRATEGIES TO ACCESS A SINGLE NON-THIOESTER C-TERMINAL DERIVATIVE
3.1 |. Strategic Selection of Linker Prior to Solid-Phase Peptide Synthesis
A variety of linkers have been developed by the peptide community for the direct synthesis of particular C-terminal functionalities via C to N SPPS. As previous reviews have summarized, an appropriate linker can be selected prior to SPPS to afford the desired C-terminal moiety upon cleavage.50,51 For example, Wang (37) and Rink Amide (39) resins are used to access C-terminal acids (38) or carboxamides (40), respectively (Scheme 6). Analogously, solid-supports are available for the synthesis of N-alkyl or N-aryl amides, aldehydes, alcohols, and other functionalities.50,52 These solutions are typically limited to situations where a single C-terminal modification is desired rather than when access to several different functionalities is required.
Scheme 6.

Generation of C-terminal acids and amides from Wang and Rink amide linkers.
In some cases, linkers have been repurposed to access alternative functionalities. For example, Dawson and co-workers demonstrated use of the hydrazide-2-chlorotrityl linker to access thioesters via conversion of the peptide C-terminal hydrazide to a pyrazole leaving group that could be displaced in-situ by aromatic thiols.53 Amblard and co-workers cleverly appropriated the 2-chlorotrityl linker, which typically affords sidechain-protected Cterminal carboxylic acids, to access C-terminal β-amino alcohols by masking the C-terminal residue as an N-linked O-acyl isopeptide (Scheme 7).54 The desired C-terminal β-amino alcohol was attached to 2-chlorotrityl resin (41) via the amine, and then the alcohol was coupled to the next desired amino acid to produce the ester linkage. After Fmoc-SPPS (42), the peptide was cleaved from resin by protonation of the amine (43). Upon neutralization, the resulting peptide (44) undergoes O to N acyl transfer via the tetrahedral intermediate 45 to produce the C-terminal β-amino alcohol peptide (46). This strategy avoids the need to treat the peptide with a reducing agent, but the formation of the O-acyl linkage did induce epimerization of the amino acid coupled to alcohol 41. When Cys(Trt) (47) was the incoming amino acid in the synthesis of octreotide, an 8-mer somatostatin analog,55 30% epimerization of cysteine was observed. This result is not surprising because cysteine is prone to epimerization. This outcome could be avoided by performing a Mitsunobu reaction using the incoming amino acid as a nucleophile and avoiding activation of the cysteine carboxylic acid. After this modified strategy was employed, octreotide was isolated in 80% yield after HPLC purification. This approach is conceptually analogous to Melnyk’s bis(2-sulfanylethyl)amino (SEA) linker method56 and Liu’s thiol-appended O-methylhydroxylamine rearrangement57 for accessing C-terminal thioesters.
Scheme 7.

Ester linked peptides undergo O to N acyl shifts to access C-terminal alcohols.
3.2 |. Sidechain Anchoring of a Pre-Functionalized C-Terminal Residue
Attachment of the peptide to the solid support through the sidechains of select amino acids can allow functionalization of the C-terminal carboxylic acid either before or after immobilization. For modification after SPPS, the protected C-terminal acid is deprotected and the desired functional group coupled using traditional activation strategies. However, oxazolone formation during activation is a significant risk, and epimerization must be closely monitored.58 Alternatively, a pre-functionalized amino acid is directly attached to the resin via the sidechain (Scheme 8). This approach is particularly important for C-terminal cysteines, which are prone to epimerization during activation (vide supra). Alternatively, the cysteine can be pre-functionalized with different alcohols or amines to give the corresponding esters and amides (48).22 The thiol sidechain is subsequently attached to 2-chlorotrityl chloride (CTC) resin via an SN1 reaction (forming 49). After completion of the SPPS (50), the peptide can be cleaved from the resin using a mild cleavage cocktail to access protected peptide 51.59
Scheme 8.

Utilizing pre-functionalized cysteine to access C-terminal cysteine peptide derivatives.
The primary advantage to this strategy is that performing the functionalization prior to attachment and elongation avoids oxazolone formation, a major contributor to epimerization in peptide synthesis.60 However, repeated or prolonged exposure of 2CT-linked cysteine to piperidine causes significant epimerization via direct deprotonation because of the increased acidity of the α-hydrogen (Table 1).
Table 1.
Epimerization of 2-chlorotrityl-bound Cys upon exposure to piperidine.
| entry | 49 or peptide 50 treated with 20% plperldlne/DMF | R | final peptide | time (h) | epimerization (% D-Cys) |
|---|---|---|---|---|---|
| 1 | C (49) | Me | GFC | 0.17 | 1.4 ± 0.2 |
| 2 | C (49) | Me | GFC | 2 | 3.3 ± 0.6 |
| 3 | C (49) | Me | GFC | 4 | 5.0 ± 0.4 |
| 4 | C (49) | Me | GFC | 16 | 4.6 ± 0.2 |
| 5 | GFC (50) | Me | GFC | 0.17 | 0.5 ± 0.1 |
| 6 | GFC (50) | Me | GFC | 2 | 2.8 ± 0.3 |
| 7 | GFC(50) | Me | GFC | 4 | 5.5 ± 0.1 |
| 8 | GFC(50) | Me | GFC | 16 | 17.8 ± 0.3 |
Significant epimerization is observed during traditional Fmoc-SPPS of C-terminal cysteine peptides linked via 2-chlorotrityl group. Fang and co-workers recently reported a multi-step process for the conversion of a C-terminal cysteine hydrazide to the corresponding C-terminal cysteine-containing peptide acid with minimal epimerization of cysteine (Scheme 9).61 The target peptide C-AhPDF 1.1b (protonated 55) was synthesized on an acid-bound 2-chlorotrityl linker leading to unacceptable levels of epimerization (30%, entry 1, Table 2). Elongation of the peptide on a hydrazide-bound 2-chlorotrityl linker significantly lowered the epimerization during SPPS 3% (55, entry 2). Thus, the approach in Scheme 9 was devised: after resin cleavage to access the protected C-terminal hydrazide (52), treatment with NaNO2 at pH 3 afforded the corresponding peptide azide (53). A water-soluble thiol was added in pH 8 buffer to afford thioester 54, which could then be hydrolyzed to carboxylate 55. Acidic workup would afford the targeted peptide acid.
Scheme 9.

General strategy to avoid C-terminal Cys(Trt) epimerization typically observed during SPPS.
Table 2.
Epimerization observed during various strategies.
| entry | strategy (when epimerization observed) | C-AhPDF 1.1b C terminus (Sequence: ACRNQCIRLEK-ARHGSCNYVFPAHKCICYFC) | epimerization (% D-Cys) |
|---|---|---|---|
| 1 | Fmoc-SPPS with 2-CT (during elongation) | OH | 30% |
| 2 | Fmoc-SPPS with hydrazine 2-CT (during elongation) | NHNH 2 | <3% |
| 3 | β-mercaptoethanol, pH 8 (during acyl transfer) | OH | 5% |
| 4 | mercaptophenyl acetic acid, pH 8 (during acyl transfer) | OH | <1% |
Use of 200 equiv sodium 2-mercaptoethanesulfonate (MesNa) led to poor conversion in the hydrolysis of 54 to 55, while use of 200 equiv MPAA resulted in formation of guanydyl amide by-products (from the guanidine in the buffer) and poor yields. In the presence of 200 equiv β-mercaptoethanol (BME), transthioesterification to thioester 56 was observed, and subsequent S to O acyl transfer led to ester 57 (Scheme 10). The strong nucleophilicity of alkyl thiols combined with the elongation of C–S bonds relative to C–O bonds enabled episulfide formation with release of the C-terminal carboxylate 55, which was protonated to generate C-AhPDF 1.1b with 5% epimerization of the C-terminal residue (entry 3, Table 2).
Scheme 10.

Acyl transfer strategy for formal hydrolysis of thioester.
Despite a reaction time of 1 h, Fang and co-workers hypothesized that the slow conversion in this reaction increased the opportunity for epimerization. In a final experiment, excess MPAA (20 equiv) was added to carboxylic azide 53 while still at pH 3. After 5 min, BME (200 equiv) was added and the pH adjusted to 8. Presumably, intermediate thioester 54 is formed followed by transthioesterification to 56. Fragmentation of 57 ultimately affords the carboxylic acid (55) with 98% conversion and no observed epimerization (entry 4, Table 2).
Fang and co-workers mention the increased reactivity of aryl thiols vs. alkyl thiols under weakly acidic conditions. This statement is potentially misleading. The aryl thiol is certainly less nucleophilic than an alkyl thiol. However, increased reactivity will be observed due to the increased acidity of the aryl thiol, which will be partially deprotonated at pH ~5.2 and up. The first portion of this formal hydrolysis reaction is at pH 3 (strongly acidic), and the second reaction is conducted at pH 8 (weakly basic). Given that the thiol pKa of MPAA is 6.6,62 the Henderson-Hasselbach equation quickly reveals that the ratio of MPAA thiolate to thiol is ~25:1. The pKa of BME is 9.6,63 so the ratio of thiolate to thiol is ~1:40. It follows that the rapid displacement of the azide relative to BME is a result of the increased nucleophilicity of a thiolate relative to a thiol.
Inclusion of 200 equiv BME avoids formation of the side products associated with the MPAA only reaction. Presumably, this is simply a kinetic trick analogous to the role of MPAA in preventing hydrolysis during NCL reactions. Additionally, it is conceivable that the MPAA ester, which possesses a pendant carboxylate, serves to facilitate delivery of β-mercaptoethanol via an interaction such as adduct 58, depicted in Scheme 11. Nucleophilic attack and deprotonation would afford tetrahedral intermediate 59, which would collapse to generate the β-mercaptoethanol thioester. Regardless, the combination of MPAA and BME allowed for epimerization-free conversion of 52 to 55. This multi-step process was effective in providing C-terminal cysteine acids with epimerization limited to that observed during the SPPS itself.
Scheme 11.

Hydrogen bonding model may explain improved reactivity and reduced epimerization during BME displacement of MPAA ester.
Aspartic64 and glutamic acid65 sidechains can be attached to Wang or Rink amide-linked resins (Scheme 12).66 In the latter case, treatment with TFA cocktails would afford an asparagine or glutamine residue. After attachment of the aspartic and glutamic acid sidechains to the solid support followed by subsequent SPPS, the C-terminal allyl protected peptide 60 was treated with de-allylation conditions to afford the C-terminal carboxylic acid 61. The acid was coupled to amino acids pre-functionalized with a C-terminal thioester moiety. This coupling resulted in resin-bound C-terminal elongated thioesters. The fully deprotected C-terminally elongated thioesters (62) were obtained after trifluoroacetic acid cleavage cocktail treatments. Additionally, lysine,67 serine,68 threonine,68b arginine,69 and tyrosine68b,70 have been linked via their sidechain moieties.71 This strategy has proven useful for on-resin macrocyclization reactions.72
Scheme 12.

Carboxylic acid sidechain anchoring to access C-terminal elongated thioesters.
3.3 |. Removable Solubilizing Tag Strategy for Solution-Phase Synthesis
Chiba and co-workers reported a solution-phase strategy to access secondary amides using a solubilizing C-terminal auxiliary.73 Previously, this moiety was used for in-solution peptide synthesis and the tag was saponified to unmask the carboxylic acid, followed by amidation; however, this approach led to significant epimerization.74 In the new approach, the solubilizing tag (aldehyde 63) undergoes reductive amination with an amine to access secondary amine 64 (Scheme 13). The tag is then used to enable solution-phase peptide synthesis to give the C-terminally tagged peptide 65. The auxiliary is removed under acidic conditions, giving the C-terminal ethyl amide peptide 66. No epimerization was observed for Arg, the second amino acid coupled (67). However, the epimerization of the C-terminal amino acid, Pro, was not investigated.
Scheme 13.

Pre-functionalization of a hydrophobic tag for solution phase peptide synthesis.
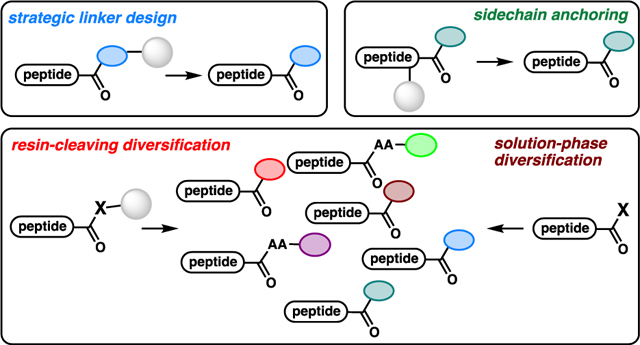
4 |. STRATEGIES FOR C-TERMINAL DIVERSIFICATION OF A SINGLE PEPTIDE PRECURSOR
Recent work in the community has largely focused on methods to access multiple C-terminal moieties from a single peptide precursor, a process that we describe as C-terminal diversification. The ability to access a diverse library of C-terminally modified peptides from a single peptide substrate is potentially powerful. Once the Fmoc solid-phase peptide synthesis (Fmoc-SPPS) is performed on unfunctionalized resin, the resin-bound peptide can be divided into different reaction vessels (Scheme 14). These individual vials can be treated with different conditions to access a library of C-terminally modified peptides. This can expedite the synthesis of these different modifications by only requiring one on-resin synthesis, minimizing solvent waste and synthesis time.
Scheme 14.

C-Terminal diversification of resin-bound peptides.
A similar concept can be envisioned for post-cleavage diversification in solution. Selectivity is a major concern during C-terminal modification of unprotected peptides. Selective activation and functionalization of the C-terminal carboxylic acid in the presence of other reactive groups is challenging. To avoid undesired reactions, enzymatic approaches are usually used, or the modification is performed on protected peptides. We will discuss methods for diversification of resin-bound peptides and then of peptides in solution.
4.1 |. Nucleophilic Cleavage of Linkers – Displacement of Esters and Aryl Diazenes
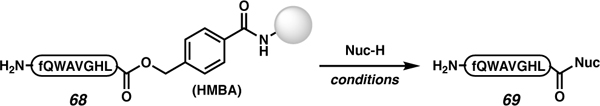
In 2007, Lever and co-workers demonstrated that the 4-hydroxymethylbenzoic acid (HMBA) linker could be used towards the synthesis of C-terminally modified peptides (Table 3).75 The HMBA linker was initially developed to allow on-resin sidechain deprotection with TFA.76 The ester-linked HMBA is acid stable, but it is susceptible to nucleophilic attack. Specifically, an 8-mer peptide on HMBA resin (68) was treated with amines, alcohols, and hydrazine to produce an amide, ester, and hydrazide terminated peptide (69), respectively.
Table 3.
Lever and co-worker’s utilization of ester-linked peptides to access C-terminally modified peptides via nucleophilic displacement. All amino acids have L-stereochemistry, with the exception of the lowercase letter f, which indicates D-Phe.
| |||
|---|---|---|---|
|
| |||
| entry | Nuc-H | Conditions | % isolated yield (purity) |
| 1 | EtNH2 | NMP, EtNH2(g) added at −78 °C then 18 h at 23 °C | 16 (93) |
| 2 | BuNH2 | TEA/BuNH2/THF (1:5:5), 18 h, 50 °C | 13 (91.5) |
| 3 | MeOH | NMP, Hünig’s base/MeOH/DMF (1:5:5), 50 °C | 32 (94) |
| 4 | PrOH | TEA/PrOH/THF (1:5:5), KCN, 18 h, 50 °C | 25 (97) |
| 5 | H2NNH2 | 5% v/v H2NNH2/DMF, N2, 18 h | 23 (91) |
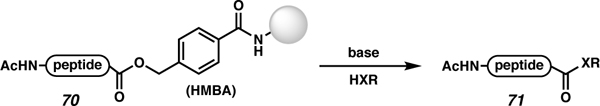
Further expansion on the utility of the HMBA linker towards C-terminal modifications was performed by Meldal and co-workers in 2016.77 In this work, transesterification, hydrolysis, and thiolysis reactions were performed to yield C-terminal esters, acids, and thioesters, respectively (Table 4). The HMBA linker (70) undergoes transesterification with potassium alkoxides in alcoholic solutions to access C-terminal esters (71). Neat amines react to form C-terminal amides, and thiolates (generated from a mixture of thiol with KOtBu) react to form thioesters. The observed reaction conversions vary depending on the steric bulk of both the C-terminal amino acid and the nucleophile. For example, C-terminal isoleucine behaved poorly with bulky nucleophiles like iBuNH2, while glycine fully converted to the desired product with all nucleophiles, regardless of their bulk. The epimerization of the C-terminal residue was investigated for the thiolysis reaction using propanethiol and KOtBu. As previously mentioned, the KOtBu is used to generate the reactive thiolate. However, KOtBu has a pKa of ~16, which could readily deprotonate both propanethiol (pKa ~10)78 and the α-position (pKa 9). Thus, it is not surprising that the stereointegrity at that C-terminal amino acid is completely lost, giving a 1:1 ratio of both diastereomers. The advantage of this approach is that the resin is readily available; however, this approach generates high amounts of epimerization due to the use of KOtBu, leading Meldal and co-workers to state that, in practice, this chemistry is limited to C-terminal glycine.
Table 4.
Accessing C-terminally modified peptides via a nucleophilic displacement of ester linkages.
| ||||
|---|---|---|---|---|
|
| ||||
| entry | peptide | Nuc-H | conversion (%) | epimerization (% D-AA)a |
| 1 | WI | PrNH2 | 5 | n.i. |
| 2 | WI | HO-(CH2)2-NH2 | 50 | n.i. |
| 3 | WG | PrNH2 | 100 | n.i. |
| 4 | WG | IBuNH2 | 100 | n.i. |
| 5 | WA | PrNH2 | 99 | n.i. |
| 6b | WA | PrSH | 70 | >99 |
N.i. = not investigated
KOtBu was used to form the thiolate.
The HMBA linker has also been used to access macrocyclic peptides. 79 Although C-terminally modified peptides were not the ultimate goal, this strategy produced activated C-terminal amides, which then underwent head-to-tail cyclization to produce macrocyclic peptides in a single reaction vessel.
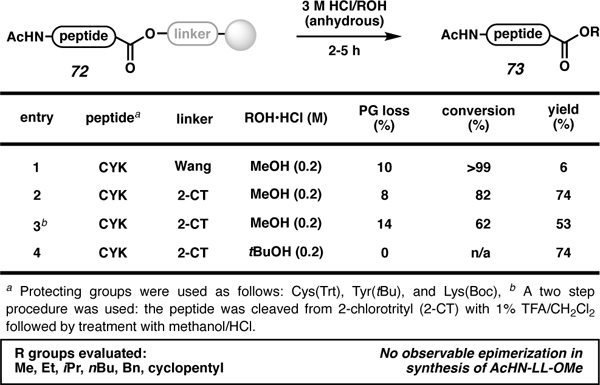
In 2010, Lokey and co-workers converted resin-bound peptides to C-terminal esters (Table 5).80 They used peptides attached to 2-chlorotrityl and Wang resins (72) in the presence of anhydrous HCl in various alcohols to achieve this transformation. Due to the acidic nature of the reaction, loss of up to 35% of protecting groups was observed. Overall, higher yields were obtained with peptides (73) attached to 2-chlorotrityl resin (Table 5, entries 2–4) compared with Wang resin (entry 1).
Table 5.
Acid-catalyzed C-terminal esterification using the HMBA linker.
|
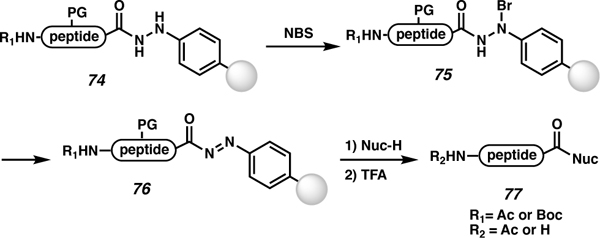
In 2004, Camarero and co-workers developed an aryl hydrazine linker for C-terminal modifications (Table 6).81,82,83 The aryl hydrazine is stable to Fmoc-SPPS, and after peptide synthesis, the hydrazine linker (74) can be oxidized with N-bromosuccinimide (NBS) to afford N-bromo-N-phenylacetohydrazide linker 75, which forms diazenyl bound peptide 76 after loss of HBr. Peptide 76 can be readily displaced with different nucleophiles to access the C-terminally modified peptides (77). This strategy has been utilized to access C-terminal thioesters without epimerization, which were ultimately used for native chemical ligation. However, NBS can be problematic for oxidation-prone peptides. Residues such as tryptophan, tyrosine, methionine, and cysteine were investigated. When peptides containing protected tryptophan and tyrosine were subjected to NBS, there was no observable oxidation (Table 6, entries 2–5). However, when the unprotected forms were exposed to NBS there was a complicated mixture of oxidized residues. Methionine and protected cysteine were completely oxidized in the presence of NBS, but they can be reduced in the subsequent TFA deprotection step if EtSH is added (entries 2,4,5). In the effort to avoid cysteine oxidation, Cys(Npys) was incorporated and no oxidation was observed (entry 5). The limitation with this protecting group is that it can only be coupled on the N-terminal residue, since Npys is labile to Fmoc-deprotection conditions. Camarero and co-workers recommend Cys(StBu) for any internal cysteine residues. Other laboratories have displaced the C-terminal diazene to access individual C-terminal modifications using various nucleophiles, as reviewed in 2007.83 In 2010, this linker strategy was applied to C-terminal cysteine targets that could not be accessed by use of a Wang linker due to extensive piperidinylalanine formation.84 While this route afforded the target peptides according to low-resolution ESI-MS data, no evidence was provided to establish purity or extent of epimerization, if any.
Table 6.
Aryl hydrazine oxidation to access C-terminal thioesters and amides.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | peptide | X(PG) | Nuc-H | yield (%) | epimerization (% D-AA)a |
| 1 | FA | – | H2N-C6H4-pNO2 | 46 | <1 |
| 2 | LMYKA | – | H-Gly-SEt | 85 | n.i. |
| 3 | LWA | Boc | H-Gly-SEt | 80 | n.i. |
| 4 | LCYKA | Trt | H-Ala-SEt | 70 | n.i. |
| 5 | CYAVTGKDSPAA | Npys | H-Gly-SEt | 75 | n.i. |
n.i. = not investigated.
4.2 |. Nucleophilic Cleavage of Linkers – Displacement of N-Acyl Carbamates, Ureas, and Triazoles
An elegant strategy to activate the C terminus of the peptide using a serine cyclic urethane, was developed by Raj and co-workers in 2016 (Scheme 15).85 The approach uses serine as the activatable linker (78). After deprotection of trityl (Trt) from the hydroxyl group of serine (79) using TFA, the cyclic urethane (81) is generated using DSC and DMAP in DMF. The activated DMAP species readily reacts with the free hydroxyl group to first generate the succinimide carbonate intermediate (80), which undergoes cyclization with the neighboring amide (80 to 81). Once activated, the nucleophile displaces the carbamate to access the C-terminally modified peptide 82. This approach was epimerization-free when tested on C-terminal alanine peptides with both water and a thiol as nucleophiles. Histidine/cysteine epimerization was not reported. Additional nucleophiles that were tested with this strategy was an alcohol, benzyl amine, and a hydride source (NaBH4) to produce a C-terminal ester, amide, and β-amino alcohol, respectively. The key advantage of using serine as the activatable linker is that Fmoc-Ser(Trt)-OH is commercially available and its implementation is straightforward.86 A practical drawback to this strategy is that the carbonate (80) is slow to cyclize to the carbamate (81), requiring overnight agitation.
Scheme 15.

Site-selective cleavage and functionalization of peptides using serine activation.
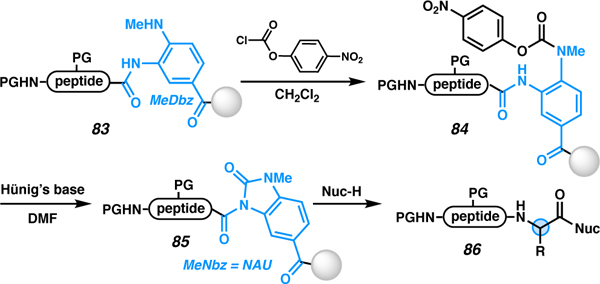
In 2017, our laboratory reported the direct displacement of the activated MeDbz linker (83, Table 7) by a variety of nucleophiles.87 The first-generation diaminobenzoyl (Dbz) linker was developed by Dawson and co-workers in 2008 to enable access to a C-terminal N-acyl urea as a thioester surrogate for NCL.88 However, some users have observed undesired side reactions during either the SPPS89,90 or the acid-mediated cleavage/deprotection step.91 Thus, a second-generation linker (MeDbz) was developed that avoids branched products during amino acid coupling through a steric effect providing a more user-friendly approach.92 These linkers are displaced by thiolates in situ during NCL and with hydrazine to form C-terminal hydrazides. Both thiols and hydrazine are exceptional nucleophiles, and so the question of whether other nucleophiles might prove capable of displacing the Nbz or MeNbz groups, particularly while avoiding epimerization, was left unanswered. Thus, our laboratory established the scope of nucleophiles and C-terminal residues compatible with direct conversion of an N-acyl urea (NAU) to a range of C-terminal modifications.
Table 7.
Nucleophilic cleavage of N-acyl ureas (activated N-methyldiaminobenzoyl linker)
| ||||
|---|---|---|---|---|
|
| ||||
| entry | Peptidea | Nuc-H | % conversion (% isolated yield) | epimerization (% D-AA)b |
| 1 | AWG | NH3 | >99 (41) | n.a. |
| 2 | AWG |

|
>99 (30) | n.a |
| 3 | AWG | MeOH | >99 (41) | n.a |
| 4 | AWI | NH3 | >99 (68) | n.i. |
| 5 | AWA | BuNH2 | >99 | <1 |
| 6c | AWA | H2O | 70 | <1 |
| 7 | AWH(Trt) | NH3 | – | <1 |
| 8 | AWC(Trt) | NH3 | >99 | <1 |
| 9 | AWC(Trt) | BuNH2 | >99 | <1 |
| 10 | AWC(Trt) | PhCH2NH2 | >99 | <1 |
| 11d | AWC(Trt) | MeOH/Na2H PO4 | >99 | <1 |
| 12c | AWC(Trt) | H2O | 56 | <1 |
Trp (W) was Boc protected
n.d. = not determined, n.i. = not investigated
Hünig’s base was used to form the hydroxide species
an aqueous buffer of NaH2PO4/Na2HPO4 at pH 8 was used as co-solvent.
To access the activated C termini, the diamine linker is activated with 4-nitrophenyl chloroformate to generate an NAU (Table 7). The secondary amine (83) reacts with the p-nitrophenyl chloroformate to form 84 which undergoes a base-mediated cyclization to form the NAU (MeNbz)-appended peptide (85). Once activated, the peptide can be displaced with a variety of nucleophiles (86). Initial work was developed on C-terminal Gly to establish the scope of eligible nucleophiles without complications related to epimerization (Table 7, entries 1–3). Subsequent work investigated the ability of nucleophiles to displace a bulky C-terminal amino acid, Ile, which resulted in excellent conversion and a moderate yield (entry 4). Additionally, C-terminal Ala epimerization was investigated by displacing with an amine (entry 5) and water (entry 6). Importantly, when Hünig’s base was used, no epimerization was observed via this approach, even for epimerization-prone amino acids such as histidine and cysteine (entries 7–12).93 This strategy was the first to demonstrate the synthesis of C-terminal Cys peptides and their subsequent modification without any detectable epimerization, even when protected with the problematic trityl group. Other highlights of this approach include the mild activating conditions employed, the wide diversity of nucleophiles investigated, the ability to utilize both protected peptides on resin and unprotected peptides in solution (vide infra), and the utilization of a commercially available resin. Sidechain macrocyclization products are also accessible via this approach in the absence of an exogenous nucleophile. In the presence of an N-terminal Cys or Gly, macrocyclization readily occurs in a self-cleaving manner to afford head-to-tail macrocycles.94
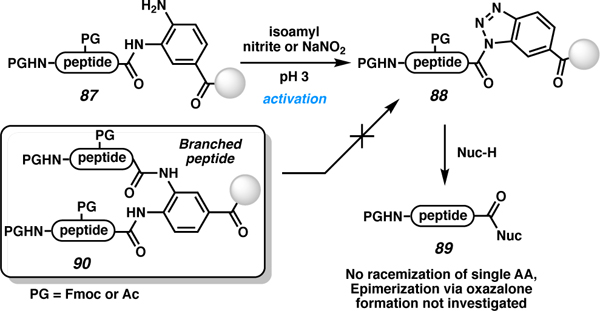
A related approach published by Kao and co-workers in 2018 employs a triazole activation method for the diaminobenzoyl (Dbz) linker (Table 8).88,95 Here, the aniline nitrogen (87) reacts with nitric oxide to form a nitrosyl derivative, and then intramolecular attack by the amide N lone pair leads to loss of water and forms the reactive benzotriazole leaving group (88). The activated peptide can be displaced by various nucleophiles (Table 8, entries1–6), including dendrimers (entry 7) and peptides (entry 8), to access C-terminally modified and elongated peptides (89). An exciting application in this work was use of linear pentapeptides as nucleophiles. A limitation of this method is that the Dbz linker, when not handled correctly, has several known pitfalls. When Dbz is incorporated, the aniline amine is free during the entire synthesis. The amine is susceptible to acylation resulting in branched peptides (90) and unintended capping products. Kao and co-workers assert that this branching is tolerable because these undesired peptides will not be cleaved from the resin with a nucleophile, since the linker is not activated. However, this leads to significantly reduced peptide recovery. While less reactive than 87, even the more user-friendly MeDbz (83) linker92 is susceptible to acylation with more reactive electrophiles like acetic anhydride.90 In related work, Dbz has also been coupled to the side chain of Asn/Gln. The linker was used to attach a trityl-solubilizing tag to aid in NCL. Once the peptide ligation was performed, the Dbz tag was subsequently removed by activation to Nbz and treatment with a H2O or NH3 source.96
Table 8.
Nucleophilic cleavage of N-acyl triazoles (activated diaminobenzoyl linker).
| |||
|---|---|---|---|
|
| |||
| entry | peptide | Nuc-H | yield (%) |
| 1 | H | EtOH | 82 |
| 2 | C | EtOH | 84 |
| 3 | HSSKLQL | H2o | 82 |
| 4 | HSSKLQL | propargylamine | 78 |
| 5 | LMYKA | aniline | 66 |
| 6 | AYRGA | mercaptopropionic acid | 80 |
| 7a | HSSKLQL | dendrimer | 70 |
| 8 | VPGVG | VPGVG | 92 |
The nucleophilic dendrimer has a structure of (G:5)-dendri-PAMAM-(NH2)128, which resulted in the product (G:5)-dendri-PAMAM-(LQLKSSH)32.
4.3 |. Solution-Phase Diversification of Peptide C-Termini
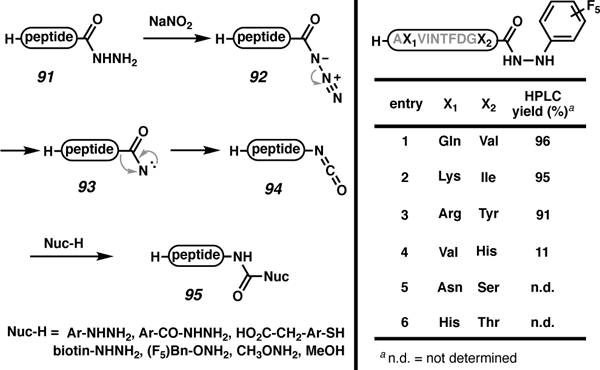
Solution-phase diversification can be necessary when issues of peptide solubility or resin incompatibility with activation conditions are encountered. Pentelute and co-workers recently developed a stereoretentive Curtius rearrangement strategy, which allows diversification in solution without activation of the C-terminal carbonyl (Table 9).97 This functional group interconversion method uses a C-terminal peptide hydrazide (91). Activation with NaNO2 forms the azide (92), which then undergoes the Curtius rearrangement. Initial formation of nitrene 93 followed by rearrangement leads to isocyanate 94, which can be directly functionalized by a nucleophile to give non-amide C-terminally modified peptides (95). The resulting modifications, including ureas and carbamates, are excellent handles for peptidomimetics.98 This strategy is compatible with unprotected peptides, which allows for easier manipulations in terms of solubility. Although traditional Curtius rearrangements retain stereointegrity,99 the enantiopurity of the C-terminal amino acid was still investigated. As expected, the amino acid retained its stereochemistry. This transformation showed excellent reactivity with C-terminal amino acids, valine, isoleucine, and tyrosine (Table 9, entries 1–3); however, this reaction was challenging for C-terminal amino acids like serine, threonine, and histidine (0–11% conversion, entries 4–6). Aspartic acid derived (aspartic acid, asparagine, and glutamine) C-terminal hydrazides were not tested.
Table 9.
Isocyanate functionalization.
|
In addition to the previously discussed displacement of N-acyl ureas on solid support (Table 7), the Stockdill laboratory developed a solution-phase approach to functionalizing N-acyl urea-appended peptides (96) to access diverse unprotected peptides (Table 10). Various amines and alcohols were used as nucleophiles to diversify the C terminus (97).87 It is important to note that when substrates contain appropriately positioned nucleophilic sides chains (e.g. Lys), sidechain macrocyclization (98) and/or hydrolysis (99) were observed (entries 3–5).
Table 10.
Solution-phase, nucleophilic displacement of NAU-appended peptides to access unprotected C-terminally modified peptides.
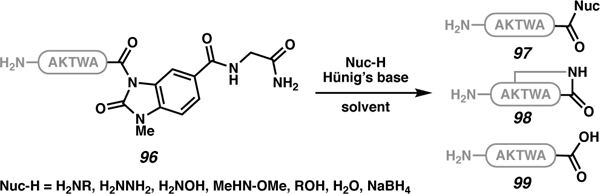
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | Nuc-H | base (equiv) | solvent | ratio of | ||
| 97 | 98 | 99 | ||||
| 1 | BuNH2 | - | MeCN/H20 | 93 | - | 7 |
| 2 | BuNH2 | - | MeCN | >99 | - | - |
| 3 | aniline | Hünig’s base (5) | MeCN | 8 | 92 | - |
| 4 | MeOH | Hünig’s base (5) | MeCN | 42 | 58 | - |
| 5 | NaBH4 | - | THF/MeCN | 77 | - | 23 |
Recently, Geyer and co-workers expanded on the auxiliary used for C(sp3)-H arylation towards the synthesis of C-terminally modified peptides (Scheme 16).100 After the arylation with the tethered quinoline auxiliary (100), reduction of the pyridine ring to tetrahydroquinoline (101) allows for the C terminus to be activated. The activation is achieved using triphosgene in the presence of Hünig’s base to produce the C-terminal NAU (102). Similar to NAU work previously described, this functionality is readily displaced with a variety of nucleophiles to produce the C-terminal peptide 103. In this work, H2O, MeOH, and a hydride source (NaBH4) were used as nucleophiles. Additionally, single amino acids (H-Gly-OMe) and the N-terminal amine of peptides on resin were well tolerated.
Scheme 16.

Nucleophilic cleavage of an N-acyl urea via quinoline auxiliary reduction followed by activation with triphosgene.
4.4 |. Single Amino Acid C-Terminal Elongation
Inspired by native chemical ligation, Aucagne and co-workers developed a single amino acid elongation strategy to access C-terminal cysteine peptides (Scheme 17).101 The work was developed to circumvent C-terminal cysteine epimerization, which was a major issue in the synthesis of their desired target, the reduced form of mini-protein AhPDF1.1b. Using classical Fmoc-SPPS with cysteine on the C terminus, 30% epimerization was observed, resulting in significant loss of yield. To circumvent this epimerization, the peptide was synthesized on a pre-functionalized resin with a bound N-alkylated cysteine auxiliary (104, Scheme 17A). The disulfide-protected cysteine auxiliary is stable to both Fmoc-SPPS and peptide cleavage with TFA. Once in solution, the disulfide is reduced to the free-thiol (105), and then an in-situ N to S acyl transfer affords C-terminal alkyl thioester 106. This intermediate then undergoes NCL to give the elongated peptide 107. This approach requires heating to 50 °C at pH 5.8 and 48 h of reaction time. Aucagne and co-workers attribute this sluggish reactivity to C-terminal proline, which is known to be a poor NCL participant.102 A drawback to these conditions is that the long reaction time and higher temperatures cause both hydrolysis (108) and aspartimide (109) by-products (Scheme 17B).
Scheme 17.

A) In solution, single cysteine elongation of mini-protein AhPDF1.1.b and AhPDF1.5 (PDB file of AhPDF1: 2M8B). B) Undesired by-products observed with this method.
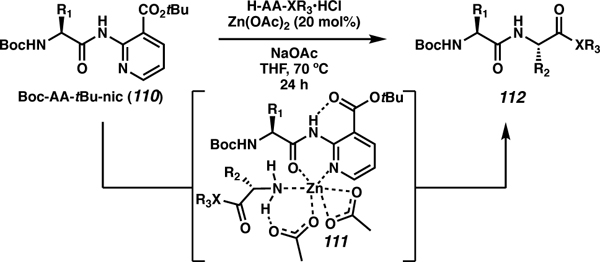
C-terminal elongation of pre-functionalized amino acids was recently achieved by Ballet and co-workers by using a tert-butyl nicotinate (tBu-nic) directing group on the C terminus of amino acids and peptides (Table 11).103 This transamidation was catalyzed using Zn, which chelates to the tBu-nic of the starting amino acid (110) and the amine of the incoming functionalized amino acid (111). This chemistry was initially performed with single amino acids to form dipeptides (112). Then side-chain elongation of a tetra- and pentapeptide was explored allowing for coupling to the side chain of carboxylic acid amino acids. Further utility of this work was shown when the N terminus of a heptapeptide was deprotected, macrocyclization was achieved to access a head-to-tail macrocycle.
Table 11.
Zn-mediated transamidation of C-terminal peptides.
| |||
|---|---|---|---|
|
| |||
| entry | Boc-AΑ-tBu-nic (110) | H-AΑ-XR3•HCl | isolated yield (%) |
| 1 | Boc-Phe-NH-tBu-nic | H-Phe-OMe•HCl | 95 |
| 2 | Boc-Alα-NH-tBu-nic | H-Leu-OAll•HCl | 99 |
| 3 | Boc-Tyr(tBu)-NH-tBu-nic | H-Val-OtBu•HCl | 85 |
| 4 | Boc-Pro-NH-tBu-nic | H-Leu-OMe•HCl | 95 |
| 5 | Boc-Met-NH-tBu-nic | H-Trp-OBn•HCl | 80 |
| 6 | Boc-Cys(tBu)-NH-tBu-nic | H-Lys(Cbz)-OMe•HCl | 57 |
| 7 | Boc-Pro-Leu-Gly-NH-tBu-nic | H-Phe-OMe•HCl | 88 |
Stockdill and co-workers used free and sidechain protected amino acids to elongate the C terminus of N-acyl urea-activated peptides (113, Scheme 18).93,104 In analogy to the Aucagne approach above, if C-terminal cysteine epimerization, dehydroalanine formation, or piperidinylalanine formation are issues, this approach can be used to access post-SPPS C-terminally elongated cysteine peptides (114) without by-product formation. This elongation is achieved by harnessing the thiol of an exogenous unprotected cysteine to cleave peptides at the C terminus from an NAU linker followed by an S to N acyl transfer to generate the peptide backbone amide (114b). The chemistry can be performed in solution or on resin to access diverse short or lipophilic peptides. Modifications pre-installed at the C terminus of the incoming cysteine can generate the native carboxamide or ester functionality upon displacement of the peptide. This approach allows direct functionalization without the need to utilize a poor or hindered nucleophile for C-terminal modifications and utilizes milder conditions and shorter reaction times to generate the native peptide. In a subsequent manuscript, Stockdill and co-workers utilized non-cysteine amino acids (114a) for direct displacement of peptides from N-acyl urea linkages using unprotected amino acids with non-nucleophilic or protected sidechain groups.104
Scheme 18.

Amino acid displacement of N-acyl urea peptides to access elongated C-terminal peptides.
4.5 |. Recent Related Macrolactamization Methods
In near concurrent publications, the Albericio,34c–d Olsen,34b and Stockdill94 groups reported strategies for on-resin macrocyclization of C-terminal MeNbz peptides. Albericio and co-workers demonstrated the ability of amino acid side chains to displace MeNbz on resin, consistent with our in-solution Lys displacement work to access sidechain-to-tail macrocycles (Table 10). Specifically, they used the side chain amine of an unnatural amino acid, Dap, to displace MeDbz.34c Additionally, their laboratory was able to access Tyr-cyclodepsi- and cyclothiodepsipeptides via the displacement of MeNbz with the phenol of Tyr34d and the thiol of an internal Cys,105 respectively. Similar work was published by Olsen in 2017 and 2018, demonstrating the displacement of MeNbz with either an internal or N-terminal Cys to access cyclothiodepsipeptides106 or head-to-tail macrocycles,34b respectively. Of particular interest was their application of the macrocyclization followed by Cys desulfurization in the same reaction vessel to give clean natural product macrocycles in good isolated yields. Finally, in collaboration with members of the American Chemical Society Green Chemistry Institute’s Pharmaceutical Roundtable, we reported a self-cleaving macrocyclization using N-acyl ureas derived from Dawson’s MeDbz linker.94 This work was performed in green solvents and established the scope of C-terminal amino acids viable in this activation strategy. In a pentapeptide substrate, no epimerization could be observed, as confirmed by HPLC co-injection with an authentic sample. Additionally, this approach allowed access to cyclic tetrapeptides, unlike the Olsen conditions, which we hypothesize is related to reduced thiol nucleophilicity in the presence of residual trifluoroacetic acid.
A similar N-alkylated strategy to Aucagne’s work (Scheme 17) was recently employed using ethyl-L-cysteine to access macrocyclic peptides.107 In this work, the only nucleophile investigated was the thiol of the N-terminal cysteine, which resulted in the cyclized peptide. This transformation was achieved through an on-resin N to S acyl transfer followed by self-cleaving cyclization.
5 |. SUMMARY AND OUTLOOK
Recent years have seen a resurgence of interest in methods to access challenging C-terminally modified peptide substrates. These efforts have been hallmarked by several general approaches including access to single derivatives via linker-specific modifications, sidechain anchoring, and removable functionalizable tags or multiple derivatives from one SPPS effort via activatable or functionalizable linker strategies followed by reaction on solid support or in solution. Each of these approaches is advantageous depending on the context and the tendency for the target peptide to undergo epimerization. Using linker design to enable access to a specific moiety is the go-to method when a single C-terminal functionality is needed and when cleavage of the available linkers does not result in problematic by-products or peptide loss. For access to multiple C-terminal modifications, sidechain attachment through amino acids like cysteine and aspartic acid can be valuable, both for C-terminal modifications and on-resin macrocyclization. Incorporation of an activatable linker between the C-terminal amino acid and the solid-support to facilitate on-resin nucleophilic displacement is powerful because the functionalization step occurs concurrently with the cleavage from resin. For some applications, solution-phase modifications are advantageous because a fully deprotected, purified peptide can be employed. Simple distribution of a stock solution of the peptide to multiple reaction vessels then allows rapid access to a diverse library of peptide targets.
In summary, the methods discussed herein provide, in aggregate, outstanding options to the lay and expert peptide chemist alike with advantages and disadvantages depending on the target peptide and application goals. The most important considerations in our view are the stereointegrity of the method for the peptide of interest, the ease of application of the method given the resources available to the chemist in combination with their goals, and the stability of the residues within the peptide sequence to the target method. We have endeavored herein to provide a useful guide to the suite of tools available for C-terminal modification of peptides and anticipate that it will prove beneficial to the peptide community.
ACKNOWLEDGMENTS
The authors thank the National Institutes of Health (R01-GM129475 to JLS, IMSD Fellowship to LGM R25-GM058905), Wayne State University (Rumble-Schaap Fellowship to CAA), and Prof. Emeritus Paul Schaap (A. Paul and Carol C. Schaap Distinguished Graduate Fellowship to CAA) for generous financial support. The American Chemical Society Green Chemistry Institute (ACS GCI) is a not-for-profit organization whose mission is to catalyze and enable the implementation of green and sustainable chemistry and engineering throughout the global chemistry enterprise and the Society. This manuscript was developed with the support of the ACS GCI Pharmaceutical Roundtable (ACS GCI PR, http://www.acs.org/gcipharmaroundtable). The ACS GCI PR is composed of pharmaceutical, biopharmaceutical, contract manufacturing and research organizations, and supplier companies. This industrial roundtable was established to encourage innovation while catalyzing the integration of green chemistry and green engineering in the pharmaceutical industry. The activities of the Roundtable reflect its members’ shared belief that the pursuit of sustainable and green chemistry and engineering practices are imperative for business and environmental sustainability.
Funding information
National Institutes of Health, Grant/Award Numbers: R01-GM129475, R25-GM058905; Wayne State University; ACS GCI PR Grant
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Kim K-H; Seong BL Peptide Amidation: Production of Peptide Hormones in vivo and in vitro. Biotechnol. Bioprocess Eng. 2001, 6, 244–251. [Google Scholar]
- 2.a) Dawson PE; Muir TW; Clark-Lewis I; Kent SB Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779; [DOI] [PubMed] [Google Scholar]; b) Johnson ECB; Kent SBH Insights into the Mechanism and Catalysis of the Native Chemical Ligation Reaction. J. Am. Chem. Soc. 2006, 128, 6640–6646. [DOI] [PubMed] [Google Scholar]
- 3.a) Hackeng TM; Griffin JH; Dawson PE Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. USA 1999, 96, 10068–10073; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Camarereo J; Adeva A; Muir T. Chemical ligation of unprotected peptides directly from a solid support. Lett. Pept. Sci. 2000, 7, 17–21. [DOI] [PubMed] [Google Scholar]
- 4.Harris PWR; Brimble MA A comparison of Boc and Fmoc SPPS strategies for the preparation of C-terminal peptide α-thiolesters: NY-ESO-1 39Cys-68Ala-COSR. Pept. Sci. 2013, 100, 356–365. [DOI] [PubMed] [Google Scholar]
- 5.a) Zheng J-S; Tang S; Huang Y-C; Liu L. Development of New Thioester Equivalents for Protein Chemical Synthesis. Acc. Chem. Res. 2013, 46, 2475–2484; [DOI] [PubMed] [Google Scholar]; b) Li H; Dong S. Recent Advances in the Preparation of Fmoc- SPPS-Based Peptide Thioester and Its Surrogates for NCL-Type Reactions. Sci. China: Chem. 2017, 60, 201–213; [Google Scholar]; c) Yan B; Shi W; Ye L; Liu L. Acyl Donors for Native Chemical Ligation. Curr. Opin. Chem. Biol. 2018, 46, 33–40; [DOI] [PubMed] [Google Scholar]; d) Aqouridas V; Mahdi OE; Diemer V; Cargoët M; Monbaliu J-CM; Melnyk O. Native chemical ligation and extended methods: mechanisms, catalysis, scope, and limitations. Chem. Rev. 2019, 119, 7328–7443. [DOI] [PubMed] [Google Scholar]
- 6.a) Hansen SB; Sulzenbacher G; Huxford T; Marchot P; Taylor P; Bourne Y. Structures of Aplysia AChBP Complexes with Nicotinic Agonists and Antagonists Reveal Distinctive Binding Interfaces and Conformations. EMBO J. 2005, 24, 3635–3646; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Celie PH; Kasheverov IE; Mordvintsev DY; Hogg RC; van Nierop P; van Elk R; Rossum-Fikkert SE; Zhmak MN; Bertrand D; Tsetlin V; Sixma TK; Smit AB Crystal Structure of Nicotinic Acetylcholine Receptor Homolog AChBP in Complex with an α-Conotoxin PnIA Variant. Nat. Struct. Mol. Biol. 2005, 12, 582–588. [DOI] [PubMed] [Google Scholar]
- 7.Kang TS; Vivekanandan S; Jois SDS; Kini RM Effect of C-Terminal Amidation on Folding and Disulfide-Pairing of α-Conotoxin ImI. Angew. Chem. Int. Ed. 2005, 44, 6333–6337. [DOI] [PubMed] [Google Scholar]
- 8.Kini and co-workers note that when the folding is performed in the presence of a denaturant, the formation of the minor bead form (4) increases (from 3 to 11%), implying that the sidechains of the peptide also influence the folding tendencies.
- 9.Marino G; Eckhard U; Overall CM Protein Termini and Their Modifications Revealed by Positional Proteomics. ACS Chem. Biol. 2015, 10, 1754–1764. [DOI] [PubMed] [Google Scholar]
- 10.Prigge ST; Mains RE; Eipper BA; Amzel LM New Insights into Copper Monooxygenases and Peptide Amidation: Structure, Mechanism and Function. Cell. Mol. Life Sci. 2000, 57, 1236–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potetinova Z; Barbier J-R; Suen T; Dean T; Gardella TJ; Willick GE C-Terminal Analogues of Parathyroid Hormone: Effect of C terminus Function on Helical Structure, Stability, and Bioactivity. Biochemistry 2006, 45, 11113–11121. [DOI] [PubMed] [Google Scholar]
- 12.Mortensen UH, Raaschou-Nielsen M, and Breddam K. Recogni-tion of C-terminal amide groups by (serine) carboxypeptidase Y investigated by site-directed mutagenesis. J. Biol. Chem. 1994, 269, 15528–15532. [PubMed] [Google Scholar]
- 13.Sforça ML.; Oyama S.; Canduri F.; Lorenzi CCB.; Pertinhez TA.; Konno K.; Souza BM.; Palma MS.; Neto JR.; Azevedo WF.; Spisni A. How C-Terminal Carboxyamidation Alters the Biological Activity of Peptides from the Venom of the Eumenine Solitary Wasp. Biochemistry 2004, 43, 5608–5617. [DOI] [PubMed] [Google Scholar]
- 14.Rosenbaum DM; Rasmussen SGF; Kobilka BK The Structure and Function of G-Protein-Coupled Receptors. Nature 2009, 459, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W; Tailhades J; Hossain MA; O-Brien-Simpson NM; Reynolds EC; Otvos L Jr; Separavoic F; Wade JD C-Terminal modifications broaden activity of the Proline-rich antibicrobial peptide, Chex1-Arg20. Aust. J. Chem. 2015, 68, 1373–1378. [Google Scholar]
- 16.Li W; O’Brien-Simpson NM; Yao S; Tailhades J; Reynolds EC; Dawson RM; Otvos L Jr.; Hossain MA; Separovic F; Wade JD C-Terminal Modification and Multimerization Increase the Efficacy of a Proline-Rich Antimicrobial Peptide. Chem. Eur. J. 2017, 23, 390–396. [DOI] [PubMed] [Google Scholar]
- 17.Li W; Tailhades J; O’Brien-Simpson N; Separovic F; Otvos L; Hossain MA; Wade J. Proline-Rich Antimicrobial Peptides: Potential Therapeutics Against Antibiotic-Resistant Bacteria. Amino Acids 2014, 46, 2287–2294. [DOI] [PubMed] [Google Scholar]
- 18.a) DnaK is a chaperone protein with a heat shock response Kragol G; Lovas S; Varadi G; Condie BA; Hoffmann R; Otvos L. The Antibacterial Peptide Pyrrhocoricin Inhibits the ATPase Actions of DnaK and Prevents Chaperone-Assisted Protein Folding. Biochemistry 2001, 40, 3016–3026; [DOI] [PubMed] [Google Scholar]; b) Scocchi M; Lethy C; Decarli P; Mignogna G; Christen P; Gennaro R. The Proline-Rich Antibacterial Peptide Bac7 Binds to and Inhibits in vitro the Molecular Chaperone DnaK. Int. J. Pept. Res. Ther. 2009, 15, 147–155. [Google Scholar]
- 19.a) Krizsan A; Volke D; Weinert S; Strater N; Knappe D; Hoffmann R. Insect-Derived Proline-Rich Antimicrobial Peptides Kill Bacteria by Inhibiting Bacterial Protein Translation at the 70S Ribosome. Angew. Chem. Int. Ed. 2014, 53, 12236–12239; [DOI] [PubMed] [Google Scholar]; b) Seesfeldt AC; Nguyen F; Antunes S; Perebaskine N; Graf M; Arenz S; Inampudi KK; Douat C; Guichard G; Wilson DN; Innis CA The ProlineRich Antimicrobial Peptide Onc112 Inhibits Translation by Blocking and Destabilizing the Initiation Complex. Nat. Struct. Mol. Biol. 2015, 22, 470–475; [DOI] [PubMed] [Google Scholar]; c) Roy RN; Lomakin IB; Gagnon MG; Steitz TA The Mechanism of Inhibition of Protein Synthesis by the Proline-Rich Peptide Oncocin. Nat. Struct. Mol. Biol. 2015, 22, 466–469; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Seefeldt AC; Graf M; Pérébaskine N; Nguyen F; Arenz S; Mardirossian M; Scocchi M; Wilson DN; Innis CA Structure of the Mammalian Antimicrobial Peptide Bac7(1–16) Bound Within the Exit Tunnel of a Bacterial Ribosome. Nucleic Acids Res. 2016, 44, 2429–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Withana NP; Blum G; Sameni M; Slaney C; Anbalagan A; Olive MB; Bidwell BN; Edgington L; Wang L; Moin K; Sloane BF; Anderson RL; Bogyo MS; Parker BS Cathepsin B Inhibition Limits Bone Metastasis in Breast Cancer. Cancer Res. 2012, 72, 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steverding D. The Cathepsin B-Selective Inhibitors CΑ−074 and CΑ−074Me Inactivate Cathepsin L Under Reducing Conditions. Open Enzym. Inhib. J. 2011, 4, 11–16. [Google Scholar]
- 22.Diaz-Rodriguez V; Ganusova E; Rappe TM; Becker JM; Distefano MD Synthesis of Peptides Containing C-Terminal Esters Using Trityl Sidechain Anchoring: Applications to the Synthesis of C-Terminal Ester Analogs of the Saccharomyces cerevisiae Mating Pheromone α-Factor. J. Org. Chem. 2015, 80, 11266–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.National Cancer Institute. NCI Drug Dictionary. https://www.cancer.gov/publications/dictionaries/cancer-drug/def/abt-510 (accessed Nov 19, 2018).
- 24.Markovic SN; Suman VJ; Rao RA; Ingle JN; Kaur JS; Erickson LA; Pitot HC; Groghan GA; McWilliams RR; Merchan J; Kottschade LA; Nevala WK; Uhl CB; Allred J; Creagan ET A Phase II Study of ABT-510 (Thrombospondin-1 Analog) for the Treatment of Metastatic Melanoma. Am. J. Clin. Oncol. 2007, 30, 303–309. [DOI] [PubMed] [Google Scholar]
- 25.Doronina SO; Bovee TD; Meyer DW; Miyamoto JB; Anderson ME; Morris-Tilden CA; Senter PD Novel Peptide Linkers for Highly Potent Antibody-Auristatin Conjugate. Bioconjugate Chem. 2008, 19, 1960–1963. [DOI] [PubMed] [Google Scholar]
- 26.Newman DJ; Cragg GM Current Status of Marine-Derived Com-pounds as Warheads in Anti-Tumor Drug Candidates. Mar. Drugs 2017, 15, 98–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi T; Nishizaki R; Ikezawa H. The Presence of GPI-Linked Protein(s) in an Archaeobacterium, Sulfolobus acidocaldarius, Closely Related to Eukaryotes. Biochim. Biophys. Acta. 1997, 1334, 1–4. [DOI] [PubMed] [Google Scholar]
- 28.Krawitz PM; Höchsmann B; Murakami Y; Teubner B; Krüger U; Klopocki E; Neitzel H; Hoellein A; Schneider C; Parkhomchuk D; Hecht J; Robinson PN; Mundlos S; Kinoshita T; Schrezenmeier H. A Case of Paroxysmal Nocturnal Hemoglobinuria Caused by a Germline Mutation and a Somatic Mutation in PIGT. Blood 2013, 122, 1312–1315. [DOI] [PubMed] [Google Scholar]
- 29.Reusser F. Biosynthesis of antibiotic U-22,324, a cyclic polypeptide. J. Biol. Chem. 1967, 242, 243–247. [PubMed] [Google Scholar]
- 30.Daniel JFS; Filho ER Peptaibols of Trichoderma. Nat. Prod. Rep. 2007, 24, 1128–1141. [DOI] [PubMed] [Google Scholar]
- 31.a) Szekeres A; Leitgeb B; Kredics L; Antal Z; Hatvani L; Manczinger L; Vágvcölgyi C. Peptaibols and related peptaibiotics of Trichoderma. A review. Acta Microbiol. Immunol. Hung. 2005, 52, 137–168; [DOI] [PubMed] [Google Scholar]; b) Leitgeb B; Szekeres A; Manczinger L; Vágvölgyi C; Kredics L. The History of Alamethicin: A Review of the Most extensively studied peptaibol. Chem. Biodiversity 2007, 4, 1027–1051; [DOI] [PubMed] [Google Scholar]; c) Eid M; Rippa S; Castano S; Desbat B; Chpineau J; Rossi C; Béven L. Exploring the membrane mechanism of the bioactive peptaibol ampullosporin a using lipid monolayers and supported biomimetic membranes. J. Biophys. 2010. DOI: 10.1155/2010/179641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He H; Janso JE; Yang HY; Bernan VS; Lin SL; Yu K. Culicinin D, an antitumor peptaibol produced by the fungus Culicinomuces clavisporus, strain LL-12I252. J. Nat. Prod. 2006, 69, 736–741. [DOI] [PubMed] [Google Scholar]
- 33.Hung K.-y.; Harris PWR; Brimble MA Synthesis of the peptaibol framework of the anticancer agent culicinin D: stereochemical assignment of the AHMOD moiety. Org. Lett. 2012, 14, 5784–5787; b [DOI] [PubMed] [Google Scholar]
- 34.a) Frost JR; Wu Z; Lam YC; Owens AE; Fasan R. Side-chain-to-tail cyclization of ribosomally derived peptides promoted by aryl and alkyl amino-functionalized unnatural amino acids. Org. Biomol. Chem. 2016, 14, 5803–5812; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gless BH; Olsen CA Direct peptide cyclization and one-pot modification using the MeDbz linker. J. Org. Chem. 2018, 83, 10525–10534; [DOI] [PubMed] [Google Scholar]; c) Monaim SAH; Acosta GA; Royo M; El-Faham A; Torre BG; Albericio F. Solid-phase synthesis of homodetic cyclic peptides from Fmoc-MeDbz-resin. Tetrahedron Lett. 2018, 59, 1779–1782l [Google Scholar]; d) Acosta GA; Murray L; Royo M; de la Torre BG; Albericio F. Solid-phase synthesis of head to side-chain Tyr-cyclopepsipeptides through a cyclative cleavage from Fmoc-MeDbz/MeNbz-resins. Front. Chem. 2020, 8, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) White CJ; Yudin AK Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524; [DOI] [PubMed] [Google Scholar]; b) Yudin AK Macrocycles: lessons from the distant past, recent developments, and future directions. Chem. Sci. 2015, 6, 30–49; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chow HY; Zhang Y; Matheson E; Li X. Ligation technologies for the synthesis of cyclic peptides. Chem. Rev. 2019, 119, 9971–10001; [DOI] [PubMed] [Google Scholar]; d) Shinbara K; Liu W; van Neer RHP; Katoh T; Suga H. Methodologies for Backbone Macrocyclic Peptide Synthesis Compatible with Screening Technologies. Front. Chem. 2020, DOI: 10.3389/fchem.2020.00447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuang Y; Xu B. Nanofibers of Small Hydrophobic Molecules Dis-rupt Dynamics of Microtubules and Selectively Inhibit Glioblastoma Cell. Angew. Chem. Int. Ed. 2013, 52, 6944–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng Z; Wang H; Du X; Shi J; Li J; Xu B. Minimal C-Terminal Modification Boosts Peptide Self-Assembling Ability for Necroptosis of Cancer Cells. Chem. Commun. 2016, 52, 6332–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dasari S; Tschounwou PB Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 0, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheignon C.; Collin F.; Faller P.; Hureau C. Is Ascorbate Dr Jekyll or Mr Hyde in the Cu(Aβ) Mediated Oxidative Stress Linked to Alzheimer’s Disease? Dalton Trans. 2016, 45, 12627–12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eipper BA; Milgram SL; Husten EJ; Yun HY; Mains RE Peptidylglycine α-Amidating Monooxygenase: A Multifunctional Protein with Catalytic, Processing, and Routing Domains. Protein Sci. 1993, 2, 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michaelis S; Barrowman J. Biogenesis of the Saccharomyces cerevisiae Pheromone α-Factor, from Yeast Mating to Human Disease. Microbiol. Mol. Biol. Rev. 2012, 76, 626–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.a) Henkel B; Zhang L; Bayer E. Investigations on Solid-Phase Peptide Synthesis in N-to-C Direction (Inverse Synthesis). Liebigs Ann./Recl. 1997, 10, 2161–2186; [Google Scholar]; b) Thieriet N; Guibé F; Albericio F. Solid-phase peptide synthesis in the reverse (N to C) Direction. Org. Lett. 2000, 2, 1815–1817; [DOI] [PubMed] [Google Scholar]; c) Lipshutz BH; Shin Y-J A new silyl linker for reverse-direction solid-phase peptide synthesis. Tetrahedron Lett. 2001, 42, 5629–5633. [Google Scholar]; d) Sasubilli R; Gutheil WG General inverse solid-phase synthesis method for C-terminally modified peptide mimetics. J. Comb. Chem. 2004, 6, 911–915; [DOI] [PubMed] [Google Scholar]; e) Rai A; Gutheil WG A Dde resin based strategy for inverse solid-phase synthesis of amino terminated peptides, peptide mimetics and protected peptide intermediates. J. Pept. Sci. 2005, 11, 69–73; [DOI] [PubMed] [Google Scholar]; f) Harwood LM; Mountford SJ; Yan R. Morpholinone mediated oxazolone-free C-terminus amide coupling permitting a convergent strategy for peptide synthesis. J. Pept. Sci. 2009, 15, 1–4. [DOI] [PubMed] [Google Scholar]
- 43.Han Y; Albericio F; Barany G. Occurrence and Minimization of Cysteine Racemization during Stepwise Solid-Phase Peptide Synthesis. J. Org. Chem. 1997, 62, 4307–4312. [DOI] [PubMed] [Google Scholar]
- 44.Windridge G; Jorgensen EC 1-Hydroxybenzotriazole as a racemization-suppressing reagent for the incorporation of im-benzyl-L-histidine into peptide. J. Am. Chem. Soc. 1971, 93, 6318–6319. [DOI] [PubMed] [Google Scholar]
- 45.Ramos-Tomillero I; Rodríguez H; Albericio F. Tetrahydropyranyl, a nonaromatic acid-labile cys protecting group for Fmoc peptide chemistry. Org. Lett. 2015, 17, 1680–1683. [DOI] [PubMed] [Google Scholar]
- 46.Tsuda S; Masuda S; Yoshiya T. Epimerization-free preparation of c-terminal cys peptide acid by Fmoc SPPS using pseudoproline-type protecting group. J. Org. Chem. 2020, 85, 1674–1679. [DOI] [PubMed] [Google Scholar]
- 47.Nishiuchi Y; Hibino H. 4-Methoxybenzyloxymethyl group, a racemization-resistant protecting group for cysteine in Fmoc solid phase peptide synthesis. Org. Lett. 2012, 14, 1926–1929. [DOI] [PubMed] [Google Scholar]
- 48.Angell YM; Alsina J; Albericio F; Barany G. Practical protocols for stepwise solid-phase synthesis of cysteine-containing peptides. J. Peptide Res. 2002, 60, 292–299. [DOI] [PubMed] [Google Scholar]
- 49.a) El-Faham A; Albericio F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602; [DOI] [PubMed] [Google Scholar]; b) Albericio F; El-Faham A. Choosing the right coupling reagent for peptides: a twenty-five-year journey. Org. Process. Res. Dev. 2018, 22, 760–772. [Google Scholar]
- 50.Alsina J; Albericio F. Solid-Phase Synthesis of C-Terminal Modified Peptides. Pept. Sci. 2003, 71, 454–477. [DOI] [PubMed] [Google Scholar]
- 51.Moss JA Guide for resin and linker selection in solid-phase peptide synthesis. Curr. Protoc. Protein Sci. 2005, 18.7.1–18.7.19. [DOI] [PubMed] [Google Scholar]
- 52.a) Ferrer-Gago FJ; Koh LQ Methods and approaches for the solid-phase synthesis of peptide alcohols. ChemPlusChem 2020, 85, 641–652; [DOI] [PubMed] [Google Scholar]; b) Ferrer-Gago FJ; Koh LQ; Lane DP Functionalized resins for the synthesis of peptide alcohols. Chem. Eur. J. 2020, 26, 370–383. [DOI] [PubMed]
- 53.Flood DT; Hintzen JCJ; Bird MJ; Cistrone PA; Chen JS;l Dawson PE Leveraging the Knorr Pyrazole synthesis for the facile generation of thioester surrogates for use in native chemical ligation. Angew. Chem. Int. Ed. 2018, 57, 11634–11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tailhades J; Gidel M-A; Grossi B; Lécaillon J; Brunel L; Subra G; Martinez J; Amblard M. Synthesis of Peptide Alcohols on the Basis of an O-N Acyl transfer reaction. Angew. Chem. Int. Ed. 2010, 49, 117–120. [DOI] [PubMed] [Google Scholar]
- 55.Bauer W; Briner U; Doepfner W; Haller R; Huguenin R; Marbach P; Petcher TJ; Pless J. SDZ CO 611: A highly potent glycated analog of somatostatin with improved oral activity. Life Sci. 1982, 31, 1133. [DOI] [PubMed] [Google Scholar]
- 56.a) Ollivier N; Dheur J; Mhidia R; Blanpain A; Melnyk O. Bis(2-sulfanylethyl)amino native peptide ligation. Org. Lett. 2010, 12, 5238–5241; [DOI] [PubMed] [Google Scholar]; b) Hou W; Zhang X; Li F; Liu C-F Peptidyl N,N-Bis(2-mercaptoethyl)-amides as thioester precursors for native chemical ligation. Org. Lett. 2011, 13, 386–389; [DOI] [PubMed] [Google Scholar]; c) Dheur J; Ollivier N; Vallin A; Melnyk O. Synthesis of Peptide Alkylthioesters using the intramolecular N,S-acyl shift properties of Bis(2-sulfanylethyl)amido peptides. J. Org. Chem. 2011, 76, 3194–3202; [DOI] [PubMed] [Google Scholar]; d) Raibaut L; Seeberger P; Melnyk O. Bis(2-sulfanylethyl)amido peptides enable native chemical ligation at proline and minimize deletion side-product formation. Org. Lett. 2013, 15, 5516–5519; [DOI] [PubMed] [Google Scholar]; e) Desmet R; Pauzuolis M; Boll E; Drobecq H; Raibaut L; Melnyk O. Synthesis of unprotected linear or cyclic O-acyl isopeptides in water using bis(2-sulfanylethyl)amido peptide ligation. Org. Lett. 2015, 17, 3354–3357. [DOI] [PubMed] [Google Scholar]
- 57.Rao C; Liu C-F Peptide Weinreb amide derivatives as thioester precursors for native chemical ligation. Org. Biomol. Chem. 2017, 15, 2491–2496. [DOI] [PubMed] [Google Scholar]
- 58.For epimerization-free, in-solution activation followed by functionalization, see Maarseveen and co-worker’s (Ar-BOR)3, Cu(II) strategy: Popovic S; Bieräugel H; Detz RJ; Kluwer AM; Koole JAA; Streefkerk DE; Hiemstra H; Maarseveen JH. Epimerization-Free C-Terminal Peptide Activation. Chem. Eur. J. 2013, 19, 16934–16937. [DOI] [PubMed] [Google Scholar]
- 59.Barlos K; Gatos D; Kallitsis J; Papaphotiu G; Sotiriu P; Wenqing Y; Schäfer W. Representation of Protected Peptide Fragments using Substituted Triphenylmethyl Resins. Tetrahedron Lett. 1989, 30, 3943–3946. [Google Scholar]
- 60.Goodman M; Glaser CB Formation and Reactions of Amino Acids and Peptide Oxazolones. Tetrahedron Lett. 1969, 40, 3473–3475. [Google Scholar]
- 61.Zuo C; Yan B-J; Zhu H-Y; Shi W-W; Xi T-K; Shi J; Fang G-M Robust synthesis of C-terminal cysteine-containing peptide acids through a peptide hydrazide-based strategy. Org. Biomol. Chem. 2019, 17, 5698–5702. [DOI] [PubMed] [Google Scholar]
- 62.Johnson ECB; Kent SBH Insights into the mechanism and catalysis of the native chemical ligation reaction. J. Am. Chem. Soc. 2006, 128, 6640–6646. [DOI] [PubMed] [Google Scholar]
- 63.Silberberg M. Chemistry: The Molecular Nature of Matter and Change (5th ed.). New York: McGraw-Hill Higher Education, 2008. ISBN 978–0-07–721650-4. [Google Scholar]
- 64.a) Rovero P; Quartara L; Fabbri G. Synthesis of cyclic peptides on solid support. Tetrahedron Lett. 1991, 32, 2639–2642; [Google Scholar]; b) Valero M-L; Giralt E; Andreu D. Solid phase-mediated cyclization of head-to-tail peptides: problems associated with side chain anchoring. Tetrahedron Lett. 1996, 37, 4229–4232; [Google Scholar]; c) Garcíα-Ramos Y; Giraud M; Tullα-Puche J; Albericio F. Optimized Fmoc solid-phase synthesis of thymosin α−1 by side-chain anchoring onto a PEG resin. Pept. Sci. 2009, 92, 565–572; [DOI] [PubMed] [Google Scholar]; d) Pelay-Gimeno M; García-Ramos Y; Martin MJ; Spengler J; Molina-Guijarro JM; Munt S; Francesch AM; Cuevas C; Tulla-Puche J; Albericio F. The first total synthesis of the cyclodepsipeptide pipecolidepsin A. Nat. Commun. 2013, 4:2352. [DOI] [PubMed] [Google Scholar]
- 65.Derbal S; Hensler M; Fang W; Nizet V; Ghedira K; Nefzi A. on resin amino acid side chain attachment strategy for the head to tail synthesis of new glutamine containing gramicidin-S analogs and their antimicrobial activity. Bioorg. Med. Chem. Lett. 2010, 20, 5701–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ficht S; Payne RJ; Guy RT; Wong C-H Solid-phase synthesis of peptide and glycopeptide thioesters through side-chain-anchoring strategies. Chem. Eur. J. 2008, 14, 3620–3629. [DOI] [PubMed] [Google Scholar]
- 67.a) Alsina J; Rabanal F; Giralt E; Albericio F. Solid-phase synthesis of “head-to-tail” cyclic peptides via lysine side-chain anchoring. Tetrahedron Lett. 1994, 35, 9633–9636; [Google Scholar]; b) Kappel JC; Barany G. Methionine anchoring applied to the solid-phase synthesis of lysine-containing ‘head-to-tail’ cyclic peptides. Lett. Pept. Sci. 2003, 10, 119–125. [Google Scholar]
- 68.a) Alsina J; Chiva C; Ortiz M; Rabanal F; Giralt E; Albericio F. Active carbonate resins for solid-phase synthesis through the anchoring of a hydroxyl function. Synthesis of cyclic and alcohol peptides. Tetrahedron Lett. 1997, 38, 883–886; [Google Scholar]; b) Yan LZ; Edwards P; Flora D; Mayer JP synthesis of cyclic peptides through hydroxyl side-chain anchoring. Tetrahedron Lett. 2004, 45, 923–925. [Google Scholar]
- 69.a) García O; Nicolás E; Albericio F. Solid-phase synthesis: a linker for side-chain anchoring of arginine. Tetrahedron Lett. 2003, 44, 5319–5321; [Google Scholar]; b) Beythien J; Barthélémy S; Schneeberger P; White PD A novel solid-phase linker strategy for the side-chain anchoring of arginine: an expeditious route to arginine 7-amido-4-methylcoumarins. Tetrahedron Lett. 2006, 47, 3009–3012. [Google Scholar]
- 70.a) Cabrele C; Langer M; Beck-Sickinger AG Amino acid side chain attachment approach and its application to the synthesis of tyrosine-containing cyclic peptides. J. Org. Chem. 1999, 64, 4353–4361; [Google Scholar]; b) Olsen CA; Jørgensen MR; Hansen SH; Witt M; Jaroszewski JW; Franzyk H. Side-chain-anchored N-Fmoc-Tyr-OPfp for bidirectional solid-phase synthesis. Org. Lett. 2005, 7, 1703–1706. [DOI] [PubMed] [Google Scholar]
- 71.Hanna CC; Kulkarni SS; Watson EE; Premdjee B; Payne RJ Solid-phase synthesis of peptide selenoesters via a side-chain anchoring strategy. Chem. Commun. 2017, 53, 5424–5427. [DOI] [PubMed] [Google Scholar]
- 72.De Vleeschouwer M; Sinnaeve D; Begin J. V. d.; Coenye T; Martins JC; Madder A. Rapid total synthesis of cyclic lipodepsipeptides as a premise to investigate their self-assembly and biological activity. Chem. Eur. J. 2014, 20, 7766–7775. [DOI] [PubMed] [Google Scholar]
- 73.Matsumoto E; Fujita Y; Okada Y; Kauppinen EI; Kamiya H; Chiba K. Hydrophobic Benzyl Amines as Supports for Liquid-Phase C-Terminal Amidated Peptide Synthesis: Application to the Preparation of ABT-510. Pept. Sci. 2015, 21, 691–695. [DOI] [PubMed] [Google Scholar]
- 74.Okada Y; Suzuki H; Nakae T; Fujita S; Abe H; Nagano K; Yamada T; Ebata N; Kim S; Chiba K. Tag-assisted liquid-phase peptide synthesis using hydrophobic benzyl alcohols as supports. J. Org. Chem. 2013, 78, 320–327. [DOI] [PubMed] [Google Scholar]
- 75.Abd-Elgaliel WR; Gallazzi F; Lever SZ Total solid-phase synthesis of bombesin analogs with different functional groups at the C-terminus. Pept. Sci. 2007, 13, 487–492. [DOI] [PubMed] [Google Scholar]
- 76.Atherton E; Logan CJ; Sheppard RC Peptide Synthesis. Part 2. Procedures for Solid-Phase Synthesis using N-Fluorenylmethoxycarbonylamino-acids on Polyamide Supports. Synthesis of Substance P and of Acyl Carrier Protein 65–74 Decapeptide. J. Chem. Soc., Perkin Trans. 1 1981, 0, 538–546. [Google Scholar]
- 77.Hansen J; Diness F; Meldal M. C-Terminally Modified Peptides via Cleavage of the HMBA Linker by O-, N- or S-nucleophiles. Org. Biomol. Chem. 2016, 14, 3238–3245. [DOI] [PubMed] [Google Scholar]
- 78.Thapa B; Schlegel HB Density Functional Theory Calculation of pKa’s of Thiols in Aqueous Solution Using Explicit Water Molecules and the Polarizable Continuum Model. J. Phys. Chem. A 2016, 120, 5726–5735. [DOI] [PubMed] [Google Scholar]
- 79.Hickey JL; Lin S. One-pot peptide cleavage and macrocyclization through direct amidation using triazabicyclodecene. Pept. Sci. 2020, DOI: 10.1002/pep2.24161 [DOI] [Google Scholar]
- 80.Turner RA; Weber RJ; Lokey RS Direct Conversion of Resin-Bound Peptides to C-Terminal Esters. Org. Lett. 2010, 12, 1852–1855. [DOI] [PubMed] [Google Scholar]
- 81.Kwon Y; Welsh K; Mitchell AR; Camarero JA Preparation of Peptide p-Nitroanilides Using an Aryl Hydrazine Resin. Org. Lett. 2004, 6, 3801–3804. [DOI] [PubMed] [Google Scholar]
- 82.Camarero JA; Hackel BJ; Yoreo JJ; Mitchell AR Fmoc-based synthesis of peptide α-thioesters using an aryl hydrazine support. J. Org. Chem. 2004, 69, 4145–4151. [DOI] [PubMed] [Google Scholar]
- 83.Woo Y-H; Mitchell AR; Camarero JA The use of aryl hydrazide linkers for the solid phase synthesis of chemically modified peptides. Int. J. Pept. Res. Ther. 2007, 13, 181–190. [Google Scholar]
- 84.Ni S; Zhang H; Huang W; Zhou J; Qian H; Chen W. The appli-cation of an aryl hydrazine linker prevents β-elimination side products in the SPPS of C-terminal cysteine peptides. J. Pept. Sci. 2010, 16, 309–313. [DOI] [PubMed] [Google Scholar]
- 85.a) Elashal HE; Sim YE; Raj M. Serine Promoted Synthesis of Peptide Thioester-Precursor on Solid Support for Native Chemical Ligation. Chem. Sci. 2017, 8, 117–123; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Elashal HE; Cohen RD; Raj M. Fmoc Solid-Phase Synthesis of C-Terminal Modified Peptides by Formation of a Backbone Cyclic Urethane Moiety. Chem. Commun. 2016, 52, 9699–9702. [DOI] [PubMed] [Google Scholar]
- 86.a) The price of 250 g of Fmoc-Ser(tBu)-OH is $210 and $1080 for Fmoc-Ser(Trt)-OH: Chem-Impex International, Inc. Fmoc-O-tert-butyl-L-serine. http://www.chemimpex.com/fmoc-i-o-i-i-tert-i-butyll-serine (accessed Aug 31, 2020);; b) Chem-Impex International, Inc. Fmoc-O-Trityl-L-serine. http://www.chemimpex.com/fmoc-i-o-itrityl-l-serine (accessed Aug 31, 2020).
- 87.Arbour CA; Saraha HY; McMillan TF; Stockdill JL Exploiting the MeDbz Linker to Generate Protected or Unprotected C-Terminally Modified Peptides. Chem. Eur. J. 2017, 23, 12484–12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.A) Blanco-Canosa JB; Dawson PE An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew. Chemie. Int. Ed. 2008, 47, 6851–6855; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weidmann J; Dimitrijević E; Hoheisel JD Dawson PE Boc-SPPS: Compatible Linker for the Synthesis of Peptide o-aminoanilides. Org. Lett. 2016, 18, 164–167. [DOI] [PubMed] [Google Scholar]
- 89.Mahto SK; Howard CJ; Shimko JC; Ottesen JJ A reversible protection strategy to improve Fmoc-SPPS of peptide thioesters by the N-acylurea approach. ChemBioChem 2011, 12, 2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arbour CA; Stockdill JL A Mild Capping Method for SPPS on the N-Methyl Diaminobenzoyl Linker: Synthesis of an N-Acyl Urea Appended C. elegans Neuropeptide. Tetrahedron Lett. 2018, 59, 3903–3906. [Google Scholar]
- 91.Bird MJ; Silvestri AP; Dawson PE Expedient on-resin synthesis of peptidic benzimidazoles. Bioorg. Med. Chem. Lett. 2018, 28, 2679–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Blanco-Canosa JB; Nardone B; Albericio F; Dawson PE Chemical Protein Synthesis Using a Second-Generation N-acylurea linker for the preparation of peptide-thioester precursors. J. Am. Chem. Soc. 2015, 137, 7197–7209. [DOI] [PubMed] [Google Scholar]
- 93.Arbour CA; Kondasinghe TD; Saraha HY; Vorlicek TL; Stockdill JL Epimerization-free access to C-terminal cysteine peptide acids, carboxamides, secondary amides, and esters via complimentary strategies. Chem. Sci. 2018, 9, 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Arbour CA; Belavek KJ; Tariq R; Mukherjee S; Tom JK; Isidro-Llobet A; Kopach ME; Stockdill JL Bringing Macrolactamization Full Circle: Self-Cleaving Head-to-Tail Macrocyclization of Unprotected Peptides via Mild N-Acyl Urea Activation. J. Org. Chem. 2019, 84, 1035–1041. [DOI] [PubMed] [Google Scholar]
- 95.Selvaraj A; Chen H-T; Huang AY-T; Kao C-L Expedient On-Resin Modification of a Peptide C terminus Through a Benzotriazole Linker. Chem. Sci. 2018, 9, 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsuda S; Masuda S; Yoshiya T. Solubilizing trityl-type tag to synthesize Asx/Glx-containing peptides. ChemBioChem 2019, 20, 2063–2069. [DOI] [PubMed] [Google Scholar]
- 97.Vinogradov AA; Simon MD; Pentelute BL C-Terminal Modification of Fully Unprotected Peptide Hydrazides via in Situ Generation of Isocyanates. Org. Lett. 2016, 18, 1222–1225. [DOI] [PubMed] [Google Scholar]
- 98.a) Burgess K; Ibarzo J; Linthicum DS; Russell DH; Shin H; Shitangkoon A; Totani R; Zhang A. J.Solid Phase Syntheses of Oligoureas. J. Am. Chem. Soc 1997, 119, 1556–1564; [Google Scholar]; b) Boeijen A; Liskamp RM J. Solid-Phase Synthesis of Oligourea Peptidomimetics. Eur. J. Org. Chem. 1999, 9, 2127–2135; [DOI] [PubMed] [Google Scholar]; c) Myers AC; Kowalski JA; Lipton MA Facile Incorporation of Urea Pseudopeptides into Protease Substrate Analogue Inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 5219–5222. [DOI] [PubMed] [Google Scholar]
- 99.Ghosh AK; Brindisi M; Sarkar A. The Curtius Rearrangement: Applications in Modern Drug Discovery and Medicinal Chemistry. Chem. Med. Chem. 2018, 13, 2351–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nicke L; Horx P; Harms K; Geyer A. Directed C(sp3)-H arylation of tryptophan: transformation of the directing group into an activated amide. Chem. Sci. 2019, 10, 8634–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lelièvre D; Terrier CP; Delmas AF; Aucagne V. Native chemical ligation strategy to overcome side reactions during Fmoc-Based Synthesis of C-terminal Cysteine-containing peptides. Org. Lett. 2016, 18, 920–923. [DOI] [PubMed] [Google Scholar]
- 102.Pollock SB; Kent SBH An investigation into the origin of the dramatically reduced reactivity of peptide-prolyl-thioesters in native chemical ligation. Chem. Commun. 2011, 47, 2342–2344. [DOI] [PubMed] [Google Scholar]
- 103.Hollanders K; Renders E; Gadais C; Masullo D; Raemdonck LV; Wybon CCD; Martin C; Herrebout WA; Maes BUW; Ballet S. Zn-catalyzed nicotinate-directed transamidations in peptide synthesis. ACS Catal. 2020, 10, 4280–4289. [Google Scholar]
- 104.Arbour CA; Stamatin RE; Stockdill JL Sequence Diversification by divergent C-Terminal Elongation of Peptides. J. Org. Chem. 2018, 83, 1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Acosta GA; Royo M; Torre BG; Albericio F. Facile solid-phase synthesis of head-side chain cyclothiodepsipeptides through a cyclative cleavage from MeDbz-resin. Tetrahedron Lett. 2017, 58, 2788–2791. [Google Scholar]
- 106.Gless BH; Peng P; Pedersen KD; Gotfredsen CH; Ingmer H; Olsen CA Structure-activity relationship study based on autoinducing peptide (AIP) from Dog Pathogen S. schleiferi. Org. Lett. 2017, 19, 5276–5279. [DOI] [PubMed] [Google Scholar]
- 107.Serra G; Posada L; Hojo H. On-resin synthesis of cyclic peptides via tandem N-to-S acyl migration and intramolecular thiol additive-free native chemical ligation. Chem. Commun. 2020, 56, 956–959. [DOI] [PubMed] [Google Scholar]