

Summary
Neoadjuvant immune checkpoint blockade has shown promising clinical activity. Here, we characterized early kinetics in tumor-infiltrating and circulating immune cells in oral cancer patients treated with neoadjuvant anti-PD-1 or anti-PD-1/CTLA-4 in a clinical trial (NCT02919683). Tumor-infiltrating CD8 T cells that clonally expanded during immunotherapy expressed elevated tissue-resident memory and cytotoxicity programs, which were already active prior to therapy, supporting the capacity for rapid response. Systematic target discovery revealed that treatment-expanded tumor T cell clones in responding patients recognized several self-antigens, including the cancer-specific antigen MAGEA1. Treatment also induced a systemic immune response characterized by expansion of activated T cells enriched for tumor-infiltrating T cell clonotypes, including both pre-existing and emergent clonotypes undetectable prior to therapy. The frequency of activated blood CD8 T cells, notably pretreatment PD-1-positive KLRG1-negative T cells, was strongly associated with intra-tumoral pathological response. These results demonstrate how neoadjuvant checkpoint blockade induces local and systemic tumor immunity.
In brief
Analysis of immune cell kinetics in oral cancer patients responding to neoadjuvant immune checkpoint blockade identifies diverse features in circulating and tumor-infiltrating T cell subpopulations that underlie rapid response to therapy and correlate with pathological outcome.
Introduction
Immune checkpoint blockade (ICB) therapy induces durable clinical responses in a diverse range of human cancers by enhancing T cell-mediated immunity (Baumeister et al., 2016; Sharma and Allison, 2015). Recent clinical trials have highlighted the potential of neoadjuvant ICB for resectable tumors (Topalian et al., 2020; Versluis et al., 2020), demonstrating several conceptual advantages. First, the integrity of lymphatics connecting the tumor to draining lymph nodes is maintained, enabling priming of new T cell responses as demonstrated in murine models (Fransen et al., 2018). Second, ICB-induced expansion of T cells within the tumor and its infiltrative margin may enhance local control. Third, immune function is not compromised by prior chemotherapy and radiation therapy (Friedman et al., 2020; Liu et al., 2016).
Early-stage neoadjuvant ICB trials in resectable melanoma, lung, colon, and head and neck cancer have shown significant clinical activity, although results need to be confirmed in phase 3 trials (Amaria et al., 2018; Blank et al., 2018; Chalabi et al., 2020; Forde et al., 2018; Huang et al., 2019; Lee and Brady, 2021; Menzies et al., 2021). A recent meta-analysis of four neoadjuvant melanoma trials demonstrated a two-year relapse-free survival of 100% and 94–96% for patients with complete or major/partial pathological responses, respectively (Menzies et al., 2021). In patients with resectable lung cancer treated with neoadjuvant PD-1 blockade, a major pathological response was observed in nine of 20 patients (Forde et al., 2018). A high response rate was also observed in mismatch repair-deficient colon cancer with neoadjuvant anti-PD-1 plus CTLA-4 therapy (Chalabi et al., 2020). A recent phase 3 clinical trial in patients with resectable lung cancer demonstrated that neoadjuvant PD-1 mAb plus chemotherapy resulted in longer event-free survival than chemotherapy and a higher frequency of complete pathological responses (24% for combination therapy, 2.2% for monotherapy) (Forde et al., 2022).
Recent single cell RNA-Seq (scRNA-seq) studies have revealed the diversity of T cell states within the TME, including a prominent population of CD8 T cells that express key markers of tissue-resident memory (Trm) T cells (van der Leun et al., 2020). Trm play an essential role in immunity to pathogens, persist long-term, and act as early responders to pathogen re-entry (Gueguen et al., 2021; Szabo et al., 2019b). Trm express key molecules that enforce a tissue-resident state, including integrin αEβ7 (αE chain also designated CD103), CD69 and HOBIT (encoded by ZNF683) (Kumar et al., 2017). Integrin αEβ7 binds to E-cadherin on epithelial cells, including epithelial cancer cells, and promotes the polarized release of T cell cytotoxic granules to target cells (Le Floc’h et al., 2007). CD69 and HOBIT promote tissue residency by inhibiting expression of molecules required for T cell egress. Trm are poised for rapid response but are highly regulated to prevent immunopathology. Intra-tumoral Trm express several inhibitory receptors, including PD-1 and CTLA-4, and may therefore be responsive to ICB therapy (Clarke et al., 2019; Park et al., 2019). Several clinical studies have correlated Trm infiltration with clinical outcomes (Banchereau et al., 2021; Duhen et al., 2018; Ganesan et al., 2017; Krishna et al., 2021). For example, a Trm gene expression signature correlated with longer survival in patients with early-stage triple-negative breast cancer (Savas et al., 2018). However, increased CD8 T cell infiltration is a strong positive prognostic indicator even in the absence of immunotherapy, and many associated CD8 T cell markers in pre-treatment biopsies correlate with longer survival following ICB (Galon et al., 2006; Tumeh et al., 2014), complicating the relationship of individual Trm genes to response correlates.
In squamous cell head and neck cancers (HNSCC), early studies of neoadjuvant ICB have demonstrated promising pathological response rates and the potential for significant tumor downstaging (Ferrarotto et al., 2020; Ferris et al., 2021; Uppaluri et al., 2020). Here, we leveraged the unique opportunity offered by this clinical trial design to investigate early therapy-induced changes in tumor-infiltrating and circulating immune cells. Comprehensive scRNA-seq and T cell receptor (TCR) repertoire analysis showed that CD8 T cells with a tissue residency program were early responders to ICB. Kinetic analysis of tumor-infiltrating and circulating immune cells revealed striking treatment-induced expansion of emergent T cell clones in tumors and the systemic circulation that were undetectable prior to therapy. Neoadjuvant ICB thus enhanced local and systemic tumor immunity in oral SCC.
Results
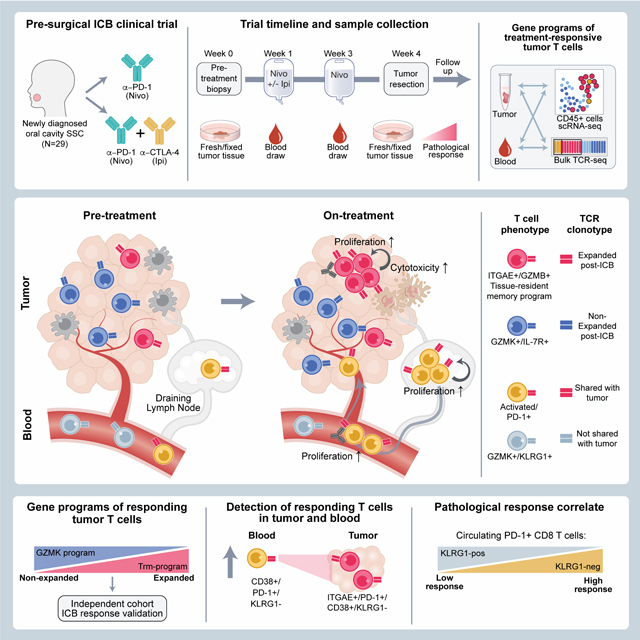
HNSCC neoadjuvant ICB clinical trial and study design
We recently reported the clinical findings from a phase 2 trial of neoadjuvant ICB in untreated patients with HNSCC of the oral cavity (Schoenfeld et al., 2020). Patients (mean [SD] age, 62 [12] years; 18 men [62%] and 11 women [38%]) were randomized to receive monotherapy with a PD-1 monoclonal antibody (mAb) (nivolumab, 3mg/kg) weeks 1 and 3 (n=14 patients) or combination therapy with two doses of PD-1 mAb and a single infusion of a CTLA-4 mAb (ipilimumab, 1mg/kg in week 1) (n=15 patients); surgery was performed 3–7 days following the second dose (Figure 1A). Several indicators provided evidence of response despite the short timeframe of treatment, with greater than 50% of patients across cohorts achieving a reduction in tumor volume prior to surgery (Figure 1B). Four patients had a major/complete pathological response of >90% based on histological analysis of surgical specimens (one monotherapy and three combination therapy patients) (Figure S1A). Updated analysis with three-year follow-up demonstrated overall survival in >80% of enrolled patients, despite detection of lymph node metastases in 58.6% of patients at initial presentation (Figure 1C). These survival data compare favorably to historical datasets, including a randomized trial of neoadjuvant chemotherapy versus no chemotherapy in resectable HNSCC with 3-year overall survival rates of 61.9% and 60.2% in the experimental and control arms, respectively (Licitra et al., 2003). Oral cavity tumors are not commonly associated with human papillomavirus (HPV); all six tested patients were found to be negative for p16/HPV. Pre-treatment tumor mutational burden was not correlated with response in the subset of analyzed patients (Figure S1B).
Figure 1: Study design and clinical response data.

A) Tumor and blood sample collection from HNSCC patients for single-cell RNA-seq and TCR repertoire analysis.
B) Clinical metadata and analyses performed for each patient, including treatment cohort and clinical response metrics (volumetric response based on bidirectional tumor measurements, clinical and pathological downstaging at time of surgery, pathological response based on histological assessment).
C) Overall and progression-free survival of patients for a median follow-up period of 36 months. See also Figure S1 and Table S1.
The neoadjuvant ICB trial provided an opportunity to investigate early immune responses during ICB without interference by therapies that compromise immune function (Figure 1A-B, Table S1). We performed sc(TCR+RNA)-seq of tumor-infiltrating CD45+ immune cells collected post-treatment during surgery (n=19 patients); six of these patients (three per treatment arm) also had a pre-treatment research biopsy (Figures 1B, S1C). The combined tumor scRNA-seq dataset contained 74,557 high-quality cells from well-defined immune lineages (Figure S1D-I). ScRNA-seq data were also generated from circulating immune cells at three timepoints (pre-treatment, two week on-treatment, 10 to 12-week follow-up). Additionally, we performed deep sequencing of the T cell receptor (TCR) repertoire (TCR β chain) from pre-and post-treatment tumor and blood specimens. These TCR sequences were used to annotate sc(TCR+RNA)-seq data for sensitive and quantitative detection of treatment-responsive T cell clonotypes (Figure 1A-B).
Identification of treatment-expanded CD8 and CD4 T cell populations
Tumor-infiltrating CD8 T cells (CD8 TILs) partitioned to seven sub-clusters (Figure 2A), with two major populations distinguished by expression of GZMK (clusters 1 & 2) versus GZMB (clusters 3 & 4) (Figure 2B), similar to scRNA-seq studies of other epithelial tumors (Hornburg et al., 2021). Cells in clusters 3 & 4 expressed Trm hallmark genes ITGAE (CD103) and ZNF683 (HOBIT) (Figure 2B, S2A, Table S2) implicated in protective tumor immunity (Park et al., 2019; Savas et al., 2018). ITGAE+ clusters 3 & 4 cells expressed similar cytotoxicity and inhibitory receptor gene programs, although cells in cluster 4 were distinguished by a cell cycle signature (Figure 2B, S2A). We validated the presence of CD8 Trm cells within tumors from a subset of patients by flow cytometry using the CD103 and CD69 surface markers. CD103+ CD69+ CD8 T cells expressed higher levels of granzyme B and PD-1 compared to CD103- cells (Figure 2C; S2B-C). Comparison of matched pre- and post-treatment specimens demonstrated a post-treatment increase in the percentage of cells in clusters 4 and 1. Cluster 1 was increased in both treatment cohorts, whereas a significant increase in cluster 4 (cycling cells) was observed only with combination therapy (Figure 2D).
Figure 2: Identification of treatment-responsive CD8 and CD4 T cell populations.

A) UMAP embedding of tumor CD8 T cells. Inset shows the normalized expression level of CD3D gene (n=19 post-Tx patients, n=6 pre-Tx patients).
B) Normalized expression of selected markers defining tumor CD8 T cell clusters.
C) Identification of CD103+C69+ Trm cells by flow cytometry, pre-gated on live/CD45+/CD3+/CD8+ T cells; quantification of PD-1 and granzyme B protein level for indicated populations (patient P32).
D) Tumor CD8 T cell cluster frequency comparing paired pre-Tx and post-Tx samples (n = 6 patients). Right, fraction of tumor CD8 T cells in clusters 1 and 4 for pairs of pre-Tx and post-Tx samples split by cohort (two-sided paired t-test, * p<0.05).
E) Single-cell TCR clonotype size projected onto a UMAP embedding for pre- and post-Tx samples. Bar plot displays the average clonotype size in each pre-Tx and post-Tx CD8 T cell cluster (C1&2 versus C3&4 paired two-sided Wilcoxon rank-sum test p=0.016).
F) Sharing of expanded TCR clonotypes across tumor CD8 T cell clusters in post-Tx tumors. Top, clonotypes from tumor CD8 clusters 1 & 2 or clusters 3 & 4 and sharing across clusters. Bottom, heatmap indicating the number of shared expanded TCR clonotypes for each cluster pair in aggregated patients. Bold boxes indicate statistically significant sharing of expanded clonotypes between clusters, accounting for cluster size (FDR < 1e-05, one-sided Fisher’s exact test followed by Benjamini-Hochberg correction).
G) Fraction of clonotypes from tumor CD8 T cell clusters 1–4 shared with blood bulk TCR repertoire; TCRs aggregated from individual patients (n=10, paired two-sided Wilcoxon rank-sum test, *p<0.05, **p<0.01).
H) UMAP embedding showing sub-clusters of total tumor CD4 T cells (n=19 post-Tx samples and n=6 paired pre-Tx samples).
I) Normalized expression of selected marker genes in tumor CD4+ T cells.
J) Left, average tumor CD4 T cell cluster frequency in paired pre-Tx and post-Tx samples. Right, tumor CD4 cluster 5 frequency for paired pre-Tx and post-Tx tumor samples, split by cohort (paired two-sided t-test, * p<0.05)
K) Comparison of tumor CD4 T cell cluster frequency between treatment cohorts in tumor post-Tx samples. Average and SEM are shown for each patient group (n=10 mono patients, n=9 combo patients, two-sided t-test, * p<0.05).
L) Single-cell TCR clonotype size projected onto tumor CD4 UMAP visualization. Bar plot displays the average clonotype size for each pre-Tx and post-Tx CD4 T cell cluster.
M) Sharing of expanded TCR clonotypes across tumor post-Tx CD4 T cells as in (F). Bold boxes indicate statistically significant clonotype sharing between clusters (FDR < 1e-05, one-sided Fisher’s exact test followed by Benjamini-Hochberg correction).
Pseudotime analysis identified two major trajectories, composed primarily of cells from GMZK+ clusters 1 & 2 (trajectory 1) and ITGAE+ clusters 3 & 4 (trajectory 2) (Figure S2D). Further analysis showed that cells from clusters 3 & 4 formed a single cluster following removal of cell cycle genes, further supporting the dynamic relationship of T cells in both clusters (Figure S2E). We then examined the frequency and cluster distribution of TCR clonotypes, defined as T cells with identical TCR sequences. Pre-treatment samples showed similar average clonotype sizes across clusters 1–4, whereas in post-treatment samples, ITGAE+ clusters 3–4 were enriched for larger clonotype sizes (Figure 2E). We observed significant clonotype sharing between cells in ITGAE+ clusters 3 & 4 and separately between GZMK+ clusters 1 & 2 (Figure 2F, S2F-G), consistent with the two major trajectories defined by pseudotime analysis (Figure S2D).
Among tumor-infiltrating CD4 T cells, the percentage of cycling CD4 T cells was increased following combination therapy (Figure 2H-K, S2H, Table S2). Substantial TCR clonotype sharing between effector and cycling CD4 T cell clusters (clusters 4 & 5, respectively) was observed in post-treatment samples (Figure 2L-M). Effector CD4 T cell cluster 4 showed higher expression of CXCL13, IFNG and inhibitory receptor genes as corresponding CD8 clusters (Figure 2I).
Treatment-induced clonal expansion of tissue-resident memory T cells
T cell activation following recognition of MHC-bound peptide antigens results in substantial proliferative expansion, and TCR-seq enables sensitive detection of clonal expansion based on the unique TCR gene rearrangements of each T cell. We identified TCR clonotypes that expanded in response to therapy (Tx-E TCRs) and investigated the corresponding gene expression programs (Figure 3A). The following considerations were important for our strategy. First, we integrated scRNA-seq data with bulk TCR β chain repertoire-seq for sensitive and quantitative detection of treatment-expanded (Tx-E) TCRs. From tumor tissue, we obtained an average of >19,000 and 31,000 productive bulk TCR β sequences in pre- and post-treatment samples, respectively (Figure S3A-B). Second, we excluded TCRs that were more abundant in blood to focus the analysis on tumor-associated TCRs (Figure S3C,D). Third, we identified TCRs enriched in tumors relative to blood that did not clonally expand during ICB (Tx-NE TCRs). TCR sequences detected only once and potentially marking naïve blood T cells were excluded. We compared the expression profiles of these Tx-E and treatment non-expanded T cells (Tx-NE) T cells (Figure 3A), thereby isolating gene programs specific to treatment-related expansion. To identify significantly expanded Tx-E clonotypes, we used a model accounting for differences in sampling depth and temporal variability (Figure S3E). Overall, Tx-E clonotypes comprised less than 1% of total clonotypes, explaining the absence of overall clonality changes at these early treatment timepoints (Figures S3F, G) (Amaria et al., 2018).
Figure 3: Treatment-induced clonal expansion of tissue-resident memory T cells.

A) Strategy for discovery of Tx-E and Tx-NE tumor TCRs.
B) Classification of Tx-E TCR origin. Fraction of each clonotype category is shown for individual patients (pre-existing vs emergent, paired two-sided t-test).
C) Projection of TCRs with significant Tx-E or Tx-NE onto UMAP embeddings of tumor CD8 TIL. Bar plots show fraction of Tx-E and Tx-NE cells in CD8 T cell clusters (paired two-sided t-test).
D) Projection of Tx-E TCRs onto post-treatment scRNA-seq data, grouped by pre-existing or emergent clonotypes.
E) Quantification of Tx-E cell fraction per cluster in pre-Tx versus post-Tx samples (average and SEM for each patient group, n=5 pre-Tx patients, n=13 post-Tx patients, two-sided t-test, *p < 0.05).
F) Quantification of Tx-E cell fraction per cluster in post-Tx samples comparing treatment cohorts (average and SEM for each patient group, n=7 mono patients, n=6 combo therapy patients, two-sided t-test, *p < 0.05).
G) UMAP embedding of post-Tx CD4 clusters highlighting tumor Tx-NE/Tx-E TCRs; bar plots show fraction of Tx-NE and Tx-E cells in tumor CD4 T cell clusters (paired two-sided t-test). See also Figure S3.
The majority of Tx-E TCRs (mean of 59.1%) were detected within tumors prior to therapy (‘pre-existing TCRs’), yet a substantial fraction of ‘emergent TCRs’ (mean of 40.9%) were identified only in post- but not pre-treatment tumors (Figure 3B). Classification of pre-existing versus emergent clonotype status was consistent even with substantial dataset down-sampling (Figure S3H). We mapped the bulk TCR β chain sequences to the scRNA-seq data from CD8 TILs. Cells mapped to Tx-E clonotypes were significantly enriched in CD8 T cell cluster 3 (ITGAE+) and cluster 4 (cycling) (Figures 3C-F, S3I), whereas Tx-NE cells were enriched in GZMK+ clusters 1 & 2. This differential distribution of Tx-E and Tx-NE CD8 T cells was particularly notable at the pre-treatment timepoint, suggesting that CD8 T cells in ITGAE+ clusters 3 & 4 were poised to respond to ICB therapy (Figure 3C). Both pre-existing and emergent TCRs were enriched in ITGAE+ clusters 3 & 4. (Figure 3D). Notably, the fraction of Tx-E cells in cluster 3 was significantly higher post-treatment in patients on combination therapy, likely explained by high expression of both PD-1 and CTLA-4 receptors among cells in this cluster (Figures 3E-F). Although treatment-related clonal expansion was less marked in CD4 T cells, CD4 Tx-E could also be readily identified among effector (CXCL13+) and cycling clusters (clusters 4 & 5) (Figures 3G, S3J). These data identified T cells with tissue-residency and effector programs as early responders to neoadjuvant ICB therapy.
Shared gene programs of treatment-expanded CD8 and CD4 T cells
Next, we identified differentially expressed (DE) genes between Tx-E and Tx-NE tumor-infiltrating T cells. Effector, tissue-residency, and inhibitory receptor genes were upregulated in Tx-E, while GZMK was the top gene upregulated in Tx-NE cells (Figure 4A, S4A, Table S3). Upregulated genes in Tx-E cells included genes previously identified in human Trm isolated from epithelia, including ITGAE, ZNF683, GPR25 and CXCR6 (Kumar et al., 2017; Szabo et al., 2019a) (Figure S4A). Tx-E cells had higher scores for tissue-resident memory, cytotoxicity, NK cell receptor, and inhibitory receptor gene signatures, consistent with their enrichment in ITGAE+ clusters 3 & 4 (Figure 4B,C, Figure S4B, Table S3, Table S4). Pre- and post-treatment T cells with Tx-E TCRs shared many DE genes, indicating that T cells with this gene expression program were poised to respond to ICB treatment (Figure S4C-D). A similar analysis of Tx-E versus Tx-NE genes for CD4 T cells revealed differential expression of genes encoding effector molecules and inhibitory receptors in Tx-E cells, while Treg signature genes (FOXP3 and IL2RA) were notably enriched in Tx-NE cells (Figure S4E-F, Table S3). Comparison of CD8 and CD4 T cells identified significant overlap in the set of top Tx-E genes, including CXCL13, IFNG, LAG3 and GZMA (Figure 4D).
Figure 4: Gene programs of Tx-E T cells and relation to clinical response.

A) Top 20 differentially expressed (DE) genes between tumor CD8 Tx-NE and Tx-E cells in pre-Tx and post-Tx samples; averaged scaled gene expression of all cells in each group.
B-C) Comparison of averaged activity scores for indicated gene signatures between Tx-NE and Tx-E T cells in pre-Tx and post-Tx patient samples (n=4 pre-Tx patients, n= 13 post-Tx patients, two-sided t-test, *p<0.05, **p<0.01). For two pre-Tx patients we were unable to calculate scores due to missing bulk TCR timepoint/lack of pre-Tx cells.
D) Top Tx-E versus Tx-NE DE genes shared by tumor CD4 and CD8 T cells in post-Tx samples.
E-F) Signature gene sets from E used to analyze bulk RNA-seq data from an independent ICB-treated patient cohort (metastatic urothelial cancer patients treated with anti-PD-L1). F: Left, activity scores for patients with complete or partial response (CR/PR) versus stable disease or progressive disease (SD/PD), two-sided t-test. Right, Kaplan-Meier curves showing overall survival (likelihood ratio test).
We next asked whether gene signatures of Tx-E T cells were associated with outcomes in a large independent ICB treatment cohort, as the tumor scRNA-seq cohort size in the present trial was not powered for such an assessment. A large bulk RNA-seq dataset was not available from ICB-treated HNSCC patients. However, urothelial cancer has a mutational burden and ICB-response rate comparable to HNSCC (Miao et al., 2018) with an available RNA-seq dataset from a phase 3 urothelial cancer clinical trial evaluating a PD-L1 blocking mAb (Mariathasan et al., 2018). We analyzed specific gene signatures from both Tx-E and Tx-NE T cells, the latter serving as a control signature (Table S4). These signatures did not include general T cell lineage genes such as CD8A, CD8B, and CD3D, which considered alone were not predictive of response or survival in this cohort (Figure S4G). The Tx-E CD8 T cell gene signature from pre-treatment samples, correlated with response and overall survival, in contrast to the Tx-NE signature (Figures 4E-F). Tx-E signatures from post-treatment samples again identified a response correlation for Tx-E but not Tx-NE gene sets (Figure S4G). These findings demonstrate that CD8 T cells with gene programs of tissue residency, cytotoxicity and inhibitory receptors were early responders to neoadjuvant ICB.
Expanded T cell clones recognize diverse shared target antigens
Investigation of the antigen specificity of Tx-E T cells could uncover novel targets for adoptive T cell therapy. We used T-Scan, a comprehensive, genome-wide screening method (Kula et al., 2019) to investigate the specificity of 80 CD8 T cell-derived TCRs from five patients (P13, P19, P20, P21 and P32), four of whom had a substantial pathological response of >50%. Genes encoding each TCR were synthesized, and primary CD8 T cells expressing these exogenous TCRs were co-cultured with an epitope library in HEK293 target cells expressing the relevant MHC class I alleles (Table S5). The library included 559,359 protein fragments spanning all wildtype human proteins, common SNPs (>1% frequency), 2,903 cancer mutations, and 293 human endogenous retroviral sequences. Target cells also expressed a granzyme B-activated fluorescence reporter, enabling enrichment of library cells targeted by CD8 T cells (Figure 5A).
Figure 5. Identification of tumor-associated targets of expanded TCRs.

A) Representation of results from a genome-wide T-scan screen. Each circle represents a single 90-aa protein fragment; y-axis represents the fold-change of each peptide in relation to the unsorted input library (eight sorted replicates). Protein fragments with fold enrichment >2.5 that share overlapping peptide sequences from same target are highlighted.
B) HLA, TCR expansion and epitope information.
C) Treatment-related expansion of clonotypes with validated target antigens. Frequency calculated from bulk TCR β chain dataset.
D) Validation of the T-scan screen hits. Target cells (HEK293T) that expressed the relevant HLAs (indicated in B) were pulsed with the indicated peptides; non-pulsed cells were used as controls. IFNγ release by CD8+ T cells expressing the patient TCRs following incubation with target cells (average and SD of n = 3, ** P < 0.01; *** P < 0.001 Student’s t-test).
E) Projection of TCRs with validated target antigens onto tumor CD8 UMAP embedding. Three low-frequency TCRs were not identified in filtered CD8 T cell sub-clusters, although single-cell TCR pairs could be identified in un-filtered data.
F) Cancer cell reactivity of primary T cells expressing MAGEA1-specific TCRs. CD8+ T cells expressing P32-39 or P32-41 TCRs were co-cultured for 18h with a panel of C*07:02 positive cancer cell lines that expressed MAGEA1 at variable levels; HEK293T cells engineered to express C*07:02 ± MAGEA1 protein fragment were included as controls. IFNγ concentration in the supernatant was measured by ELISA assay. Each condition compared to C*07:02 HEK293 control (average and SD of n = 3, ****P < 0.0001 one-way ANOVA and Bartlett’s post-test).
G) Cytotoxic activity of T cells expressing MAGEA1-specific TCRs. A101D and A2058 tumor cells were co-cultured with P32-39 TCR, P32-41, or with non-transduced control donor T cells at indicated E:T ratios. Red fluorescence was measured over time and normalized to timepoint 0. Circles represent an average of n = 2.
This screen identified six target antigens for seven TCRs (Figure 5B) that represented pre-existing or emergent clonotypes that expanded substantially during treatment (Figures 5B-C). Patterns of antigen specificity included either broad expression in cancer and healthy tissues (DST, AHNAK2) or overexpression in a subset of cancers with limited expression in healthy tissues (endogenous retroviral antigen ERVMER34-1 and MAGEA1) (Figures 5B, S5A-B). HEK293 cell clones expressing single HLA alleles and 90-aa protein fragments were used to deconvolve TCRs, define HLA restriction, and map peptide epitopes (Figures 5D, S5C-D). TCRs with identified cancer-associated antigens nearly exclusively mapped to cells from clusters 3 & 4, despite selection of TCRs based on treatment-expansion rather than cluster assignment (Figures 5E, S5E).
Of particular interest was a previously unidentified C*07:02-restricted epitope of the cancer-testis antigen MAGEA1. The MAGEA1 gene was previously shown to be overexpressed in HNSCC but not in healthy adult tissues except testis (Figueiredo et al., 2006). Two distinct TCR clonotypes, P32-39 and P32-41, mapped to the same epitope, indicative of a polyclonal response to MAGEA1 (Figure S5D). Both TCRs recognized cancer cell lines with endogenous expression of both C*07:02 and MAGEA1, and the degree of T cell activation (IFNγ secretion) correlated with MAGEA1 expression levels (Figure 5F). T cells with these TCRs efficiently killed cancer cell lines with endogenous expression of HLA-C*07:02 and MAGEA1 (Figure 5G). These results indicated that novel targets for TCR-engineered T cell therapy could be identified by investigating T cell clonotypes that expanded early during ICB.
Neoadjuvant ICB modulates T cell – myeloid interactions
Diverse populations of myeloid cells shape T cell-mediated tumor immunity through expression of PD-1 and CTLA-4 ligands and additional cell surface and secreted molecules (Engblom et al., 2016). Investigation of tumor myeloid populations revealed 13,060 cells that partitioned into nine subclusters (Figure S5F-G Table S2). The CD14+ monocyte cluster significantly increased post-treatment (cluster 1), whereas dendritic cells decreased post-treatment (Figure S5H-I). We then examined putative interactions between myeloid cell populations and T cells, based on expression of cognate receptor-ligand pairs. The CD80 and CD86 ligands for the inhibitory CTLA-4 receptor were preferentially expressed by LAMP3+ DCs (cluster 8, mregDCs) (Maier et al., 2020), although CD86 exhibited broader expression than CD80. PDL1 (CD274) expression was also enriched among LAMP3+ DCs as well as CXCL8+ TAMs (Figure S5J). Systematic analysis of receptor-ligand interactions between CD8 T cell clusters 3 & 4 and myeloid clusters showed many post-treatment changes, including CXCL16 – CXCR6 and IFNG-inducible chemokines CXCL9 and CXCL10 (Figures S5K,L).
Neoadjuvant ICB enhances systemic tumor immunity
We investigated whether neoadjuvant ICB could also enhance systemic tumor immunity, which may be of particular importance for protecting against distant metastases (Fairfax et al., 2020; Kamphorst et al., 2017; Wu et al., 2020). We performed sc(TCR+RNA)-seq on peripheral blood mononuclear cells (PBMCs) collected pre- and on-treatment from 27 patients (Figure 1B; Figure S6A) and integrated these data with bulk TCR-seq for sensitive detection of treatment-induced clonal expansion (Figure 1B).
Among blood CD8 T cells, only cluster 6 changed in frequency over the course of treatment. These cells exhibited an activated, proliferative state based on HLA-DRA, CD38, and MKI67 expression (Figures 6A-C, S6B, Table S6). Two of nine CD4 T cell subclusters increased on-treatment, including effector and Treg populations (Figures S6C-E, Table S6). Multiple DC populations significantly decreased in frequency during therapy (Figures S6F-H, Table S6), potentially due to migration to tumor-draining lymph nodes.
Figure 6: Neoadjuvant ICB enhances systemic tumor immunity.

A) UMAP embedding showing sub-clusters of total blood CD8 T cells across three timepoints (B1 pre-treatment, B2 on-treatment, B3 post-treatment/post-surgery, n=27 B1, n=24 B2, n=18 B3).
B) Violin plots showing normalized scRNA-seq expression of selected genes for cluster 6 of blood CD8 T cells.
C) Longitudinal comparison of blood CD8 T cell cluster frequency (partially paired two-sided t-test,**p<0.01; average and SEM for each patient group).
D) Mapping of blood Tx-E TCRs (based on bulk TCR β repertoire sequencing) to blood CD8 T cell scRNA-seq data.
E) Mapping of total tumor TCR clonotypes (left) and tumor Tx-E clonotypes (right), identified by bulk TCR β repertoire sequencing, to blood on-treatment (B2) CD8 T cell scRNA-seq data. Bar plots show distribution of tumor Tx-NE and Tx-E TCRs among blood CD8 T cell clusters (color code for clusters as in A).
F) Strategy for sorting of Tx-E T cells from B1 and B2 blood samples based on surface markers inferred from scRNA-seq data.
G) Representative flow cytometry plots for sorting of activated blood CD8 T cells.
H) Comparison of sorted T cells as determined by flow cytometry (% live CD38+ HLA-DR+ cells) versus scRNA-seq (% CD8 cluster 6 T cells); fit with a linear regression model.
I) Projection of sorted blood CD8 T cells to reference total blood CD8 T cells; 63.5% of sorted T cells projected to CD8 C6 cluster. Contour lines reflect the density of projected cells on reference UMAP embedding.
J) Sub-clusters of sorted blood CD8 T cells (n=4 B1 and B2).
K) Top DE genes among subclusters of sorted blood CD8 T cells, ranked by averaged ln (fold change) compared to all other clusters and z-score normalized.
L) Fraction of total and sorted blood CD8 TCRs that could be mapped to respective tumor bulk TCR dataset.
M) Mapping of tumor Tx-E and Tx-NE TCRs to sorted blood CD8 T cells . Left, total sorted CD8 T cells; right, sorted CD8 T cells at B1 and B2 timepoints (n=2 patients, P13, P32 for whom we could calculate tumor Tx-E/NE TCRs; n=2 patients did not have pre/post treatment bulk TCR data). See also Figure S6 and Table S6.
We next analyzed dynamic changes in circulating TCR clonotypes and their relationship to intra-tumoral T cell populations. Blood TCRs that expanded during treatment based on bulk TCR-seq were enriched in CD38+ CD8 T cell cluster 6 (Figure 6D). TCRs that expanded in tumors during therapy (Tx-E) were also enriched in the CD38+ CD8 T cell cluster 6, whereas Tx-NE TCRs mapped to multiple clusters (Figure 6E). Identification of circulating T cells with tumor-associated and treatment-responsive TCRs suggested an important connection to intra-tumoral immune responses, which we further investigated by sorting of CD38+ and HLA-DR+ cells reflecting markers of PBMC CD8 T cell cluster 6 (Figure 6F). Sorted CD38+ HLA-DR+ T cells showed increased levels of activation markers CXCR3 and PD-1 compared to total CD8 T cells, similar to previous findings in lung cancer (Figure 6G-H) (Kamphorst et al., 2017). We then performed scRNA-seq of sorted blood CD38+ HLA-DR+ CD8 cells from pre- and on-treatment (B1 and B2) blood samples from four patients. The expression profiles of these sorted cells strongly overlapped with CD8 cluster 6, confirming successful enrichment of this population (Figures 6I-K, S7A, Table S6).
A majority of TCR sequences from sorted blood CD38+ HLA-DR+ CD8 T cells could be matched to tumor-associated TCRs, representing a significant enrichment compared to the total CD8 blood dataset (Figure 6L, Figure S7B). Strikingly, tumor Tx-E TCRs were almost exclusively present in subclusters 3 & 4 of sorted blood CD38+ HLA-DR+ CD8 T cells from on-treatment samples (Figure 6M) which overexpressed chemokine receptor (CXCR6), cytotoxicity (GNLY) and inhibitory receptor (LAG3, HAVCR2) genes (cluster 3) as well as cell cycle genes (cluster 4) (Figure 6K). These data showed that tumor-associated CD8 T cells could be identified in increased numbers at an early 2-week on-treatment timepoint in the blood based on key activation markers.
Novel TCRs emerge in tumor and blood during treatment
We further investigated the relationship between blood and tumor TCR clonotype expansion over the course of ICB-treatment using the bulk TCR-seq datasets. In addition to pre-existing clones (>1 sequence in pre-treatment samples) that expanded during treatment, we identified a substantial number of emergent clonotypes that were undetectable in pre-treatment tumors, both of which were also detected in blood samples (Figures 7A-B). For example, patient P32 showed a strong cellular response to combination therapy and high pathological response. A majority of tumor Tx-E clonotypes were also detected in on-treatment blood samples, including a large number of emergent clonotypes, despite the short 2-week timeframe of neoadjuvant ICB. Treatment-responsive clonotypes tended to peak in blood samples at the 2-week on-treatment timepoint (B2) but contracted to low/undetectable levels at the follow-up timepoint (B3, ~10–12 weeks) (Figure 7A-B), potentially due to homing to tissues where these T cells could provide protective immunity. Quantification of blood Tx-E clonotypes as percentage of total clonotypes showed a significant increase in patients on combination compared to monotherapy. This increase was only statistically significant for emergent, but not pre-existing, Tx-E TCRs (Figure 7C). Patients on combination therapy also had a higher percentage of blood-expanded clonotypes shared with the tumor (Figures 7D, S7C), consistent with the finding that CTLA-4 blockade boosts T cell priming (Kvistborg et al., 2014; Sharma and Allison, 2015). These data demonstrated that neoadjuvant ICB enhanced systemic tumor immunity.
Figure 7: Activation state of circulating T cells as biomarkers of tumor pathological response.

A) Frequencies of tumor Tx-E TCR clonotypes and their associated frequencies in the blood for a patient with an exceptional cellular response (P32).
B) Frequencies of tumor Tx-E TCR clonotypes and their associated frequencies in the blood. Data for four patients with notable Tx-E TCR abundance are shown. Each row represents one tumor Tx-E clonotype grouped as indicated; grey cell means TCR was not detected for given sample.
C) Frequency of blood Tx-E TCR clonotypes plotted as percentage of total blood clonotypes within treatment cohorts. Shown are all Tx-E TCRs (left) or split into emergent (middle) and pre-existing (right) TCRs. Each dot represents an individual patient (n=15 mono patients, n=12 combo patients; two-sided Wilcoxon rank-sum test).
D) Frequency of total or emergent blood Tx-E clonotypes shared with tumor, plotted as percentage of total blood clonotypes. Each point represents an individual patient for whom frequencies could be calculated (n= 12 mono, n=10 combo; two-sided Wilcox test).
E) Correlation of blood CD8 T cell cluster fraction with tumor pathological response at B1 . Difference between low and high pathological response groups was determined (n = 7 for high response, n = 9 for low response, two-sided Wilcoxon rank-sum test, *p<0.05, **p<0.01).
F) Kinetic changes in blood CD4 T cells. Left, frequency of CD4+ CD38+ HLA-DR+ cells, pre-gated on live/CD3+/CD4+ cells shown across timepoints (n=23 B1, n=24 B2, n=19 B3). Right, frequency of CD4+ CD38+ HLA-DR+ cells at B2 comparing cohorts (n= 14 B2 Mono, n = 10 B2 Combo; two-sided unpaired Wilcoxon rank-sum test).
G) Kinetic changes of blood CD8 T cells. Left, Frequency of CD8+ CD38+ HLA-DR+ cells, pre-gated on live/CD3+/CD8+ cells, shown for each patient across timepoints and colored by treatment (n=23 B1, n=24 B2, n=19 B3, two-sided Wilcoxon rank-sum test). Right, quantification of Ki-67+ frequency (%) among CD38+ HLA-DR+ and CD38- HLA-DR- cells for samples with ≥ 100 cells per population (n = 14 B1, n = 17 B2, paired two-sided Wilcoxon rank-sum test, ****p<0.0001).
H) Correlation of pathological response with frequency of CD8+ CD38+ HLA-DR+ T cells in pre-Tx and on-Tx blood samples (B1: n= 8 low, n = 9 medium, n= 5 high; B2: n= 8 low, n= 8 medium, n = 6 high; two-sided Wilcoxon rank-sum test).
I-J) Correlation of pathological response with KLRG1 status of PD-1+ CD8 T cells.
I: Representative flow cytometry plots identifying PD-1+ CD8 T cells (top) and KLRG1 expression by PD-1+ cells (bottom) from low and high pathological response patients at B1 timepoint.
J: Frequency of PD-1+ cells (left) and PD-1+ KLRG1- cells (right) at B1 and B2 timepoints; at B2 on-treatment timepoint, PD-1 was detected with anti-IgG4 (bound nivolumab) (B1: n= 8 low, n = 9 medium, n= 5 high; B2: n= 8 low, n= 8 medium, n = 6 high; two-sided Wilcoxon rank-sum test).
Biomarkers of intra-tumoral pathological response
The identification of TCR clonotypes that expanded in both blood and tumor during treatment suggested that blood T cell populations could serve as early biomarkers of response to ICB, supported by the correlation of blood CD8 cluster 6 frequency with pathological response (Figure 7E). We further investigated the relationship between blood T cell populations and clinical response by analyzing serial blood samples from 28 trial patients by flow cytometry (Figure 1B). Activated CD4 and CD8 T cell populations (CD38+ HLA-DR+) substantially expanded from pretreatment (B1) to the 2-week on-treatment timepoint (B2) (Figures 7F-G, S7D-E, Table S7). A large proportion of CD38+ HLA-DR+ cells were in a proliferative state based on Ki-67 expression, consistent with the scRNA-seq data (Figure 7G). Importantly, the frequency of activated CD8 T cells (CD38+ HLA-DR+) in pre-treatment (B1) blood samples was associated with the degree of pathological response in the tumor (Figure 7H, S7I), but was not associated with response at the on-treatment (B2) blood samples due to a broad increase of CD38+HLA-DR+ T cells across response groups (Figures 7H, S7F). Expression of CD38 and HLA-DR activation markers was strongly associated with expression of PD-1, which is also upregulated following T cell activation (Figure S7G). However, the frequency of PD-1+ T cells in pre- or on-treatment blood samples did not correlate with pathological response (Figures 7I-J). Further analysis showed that PD-1+ CD8 T cells comprised two populations defined by expression of KLRG1, a marker of terminal differentiation (Figure 7I, S7H). Importantly, in both pre- and on-treatment blood samples (B1 and B2) the percentage of PD-1+ CD8 T cells that was KLRG1 negative was strongly associated with pathological response, highlighting the importance of the differentiation state of circulating PD-1+ T cells in their response to ICB (Figures 7J, S7H-I, Table S7).
Finally, we examined expression of these markers in tumor CD8 T cell clusters 3 & 4 that responded to pre-surgical ICB by clonal expansion. These clusters shared high expression of CD38, HLA-DR, and PDCD1 (PD-1), while expressing minimal KLRG1. (Figure S7J). Therefore, blood Tx-E and response-correlated T cell populations shared phenotypic markers with clonally expanded T cells within the tumor. The frequency of activated blood CD4 T cells (CD38+ HLA-DR+) positively correlated with the percentage of expanded TCR clonotypes in tumors (Figure S7K). These data identified activated blood T cell populations as important biomarkers of pathological response in pre-treatment and 2-week on-treatment blood samples.
Discussion
Here we identified CD8 TILs with a tissue-residency program as early responders to neoadjuvant ICB. We found that Trm responsive to neoadjuvant ICB were characterized by gene programs related to activation, cytotoxicity, and effector function, as well as inhibitory receptors including PD-1. The Trm program may be important in neoadjuvant ICB by promoting adhesion of CD8 T cells to tumor cells (ITGAE), rapid T cell-mediated effector function (GZMB, PRF1, IFNG) and recruitment of additional immune cell populations (CXCL13). Our findings can be distinguished from previous studies that correlated Trm infiltration or gene sets in pre-treatment biopsies to cancer prognosis or subsequent ICB clinical response (Banchereau et al., 2021; Clarke et al., 2019; Krishna et al., 2021; Savas et al., 2018). While such correlations are informative, many CD8 T cell markers also correlate with longer survival, even in the absence of immunotherapy (Galon et al., 2006; Jiang et al., 2018; Tumeh et al., 2014). Through utilization of pre- and post-treatment datasets and integrated TCR-seq approaches, we explicitly identified Trm as the predominant T cell population responding to neoadjuvant ICB.
It is important to consider whether these treatment-responsive populations exhibited programs of exhaustion. The paradigm of T cell exhaustion was first established in murine models and defined by sequential loss of proliferative capacity, cytokine production and cytotoxicity (Blank et al., 2019; Philip and Schietinger, 2021). Although exhaustion is defined by functional criteria, T cells are frequently considered to be exhausted based on expression of inhibitory receptors, including PD-1 and CTLA-4. However, inhibitory receptors are also upregulated during T cell activation and are constitutively expressed by tissue-resident T cells, including the treatment-responsive cells identified here (Mami-Chouaib et al., 2018; Singer et al., 2016). We hypothesize that CD8 T cells which clonally expanded in response to neoadjuvant ICB were not exhausted, for the following reasons: 1) Exhaustion was originally defined by an early loss of proliferative capacity; however, these cells underwent substantial clonal expansion within a short timeframe in response to neoadjuvant ICB, and one of the two clusters enriched for treatment-responsive cells represented actively cycling cells; 2) Exhaustion is also defined by reduced effector responses, yet treatment-responsive cells identified here were characterized by high expression of cytotoxicity, cytokine, and chemokine genes. Rather, we propose that the activity of such tumor-infiltrating Trm (and Trm populations more generally) is highly regulated by multiple inhibitory receptors. This conclusion is consistent with findings from multiple human cancer types, including breast, lung and ovarian cancers, where Trm are marked by proliferation signatures and clonal expansion, despite PD-1 expression (Bassez et al., 2021; Clarke et al., 2019; Gueguen et al., 2021; Hornburg et al., 2021). We do, however, also note that a substantial fraction of tumor-infiltrating T cells failed to respond to neoadjuvant ICB by clonal expansion, including a fraction of T cells in Trm clusters, and such T cells may indeed be in an exhausted state. The failure of a subset of Trm to respond to ICB may also be related to TCR specificity and/or their location within the tumor.
We identified two divergent CD8 T-cell trajectories in tumors based on TCR clonotype sharing and pseudotime modeling (trajectory 1: GZMK; trajectory 2: ITGAE, GZMB). These distinct trajectories may be related to patterns of T cell antigen specificity in human tumors. Analysis of tumor-infiltrating T cells in human lung cancer following neoadjuvant PD-1 blockade showed that transcriptional programs were related to patterns of antigen specificity: neoantigen-specific T cells expressed elevated Trm markers (ITGAE, HOBIT) and inhibitory receptors (PDCD1, CD39) while virus-specific cells differentially expressed GZMK and NKG7 (EBV-specific TCRs) or TCF7 and IL7R (influenza-specific TCRs) (Caushi et al., 2021). A recent study of melanoma-infiltrating CD8 T cells supports this conclusion (Oliveira et al., 2021). The transcriptional programs of neoantigen-specific CD8 T cells described in these two studies have similarities to the Trm program identified here (trajectory 2). Clonally expanded TCRs for which we identified tumor antigens using the T-Scan platform also mapped to clusters 3 & 4, consistent with this hypothesis.
We found that the majority of Tx-E clonotypes (range of 37.5% to 85.7% per patient) pre-existed in the tumor prior to treatment, consistent with a recent study of neoadjuvant PD-1 blockade for breast cancer (Bassez et al., 2021). Conversely, a substantial fraction of such clonotypes were only detectable in tumors following therapy (emergent clonotypes); emergent clones were also previously identified in basal carcinomas treated with a PD-1 mAb (Yost et al., 2019). Integration of tumor and blood TCR repertoire datasets revealed that a large fraction of such emergent clones was present in both on-treatment blood samples (B2 timepoint) and surgical tumor specimens. These data suggest that early intra-tumoral ICB responses are mediated primarily by pre-existing T cells with a Trm gene program; in parallel, new T cell populations may be primed in tumor-draining lymph nodes that then traffic through the blood into the tumor. Thus, neoadjuvant ICB enhances both local and systemic tumor immunity which could contribute to control of residual local and metastatic disease.
Systematic analysis of blood T cells populations by scRNA-seq demonstrated enrichment of activated CD8 T cells (cluster 6) at the 2-week on-treatment timepoint. Activated CD4 and CD8 T cells (CD38+ HLA-DR+) expanded significantly on-treatment, and a large fraction of these cells were in a proliferative state based on the Ki-67 marker (Kamphorst et al., 2017). PD-1 positive CD8 T cells were enriched in this activated population (CD38+ HLA-DR+). While PD-1+ CD8 T cell frequency did not correlate with response, the percentage of PD-1+ CD8 T cells that were KLRG1-negative was strongly associated with response in both pre-treatment and 2-week on-treatment blood samples. KLRG1 is a marker of terminal differentiation, highlighting the importance of the differentiation state of circulating PD-1+ CD8 T cells. We hypothesize that PD-1+ KLRG1-negative CD8 T cells are less terminally differentiated than KLRG1-positive cells and may therefore have greater proliferative capacity and potential to acquire protective cellular states (such as Trm) in the tumor. Indeed, in a murine model of viral infection, skin Trm cells exclusively originated from KLRG1-negative CD8 T cells (Mackay et al., 2013). Pre-treatment blood biomarkers could therefore be valuable to identify patients who may benefit from pre-surgical ICB.
Limitations of this study
A limitation of this study is that paired pre- and on-treatment tumor specimens were only available from six patients because pre-treatment research biopsies were voluntary per the clinical protocol. However, we were able to perform scRNA-seq and flow cytometry analyses on serial blood samples from most patients which provided important biomarkers of response that were consistent with major findings for tumor-infiltrating T cells. The favorable survival outcomes in both treatment cohorts also precluded assessment of clinical benefit of combination therapy versus monotherapy. Future trials will be required to determine whether this combination therapy improves progression-free and overall survival within the neoadjuvant setting and to further study the incidence of treatment-related adverse events with a low, single dose of anti-CTLA-4.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kai W. Wucherpfennig (Kai_Wucherpfennig@dfci.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Processed, de-identified scRNA-seq gene expression, TCR, and bulk TCR datasets generated during this study are publicly available at the Gene Expression Omnibus (GEO) as of the date of publication. The accession number for these datasets is listed in the key resources table. The raw scRNA-seq gene expression sequencing data cannot be deposited in a public repository because of restrictions in informed consent. Raw data may be requested from kai_wucherpfennig@dfci.harvard.edu with appropriate institutional approvals. This paper analyzes existing, publicly available data. The accession number for this dataset is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human TruStain FcX™ (Fc Receptor Blocking Solution) | Biolegend | Cat# 422302; RRID:AB_2818986 |
| Anti-human CD3 PE-Dazzle 594 | Biolegend | Cat#300450; RRID:AB_2563618 |
| Anti-human CD45 Pacific Blue | Biolegend | Cat#304022 RRID:AB_493655 |
| Anti-human CD66b PE-Cy7 | Biolegend | Cat#305116; RRID:AB_2566605 |
| Anti-human CD15 APC | Biolegend | Cat#301908; RRID:AB_314200 |
| TotalSeq™-C0251 anti-human Hashtag 1 Antibody | Biolegend | Cat# 394661; RRID:AB_2801031 |
| TotalSeq™-C0252 anti-human Hashtag 2 Antibody | Biolegend | Cat#394663; RRID:AB_2801032 |
| TotalSeq™-C0253 anti-human Hashtag 3 Antibody | Biolegend | Cat#394665; RRID:AB_2801033 |
| TotalSeq™-C0254 anti-human Hashtag 4 Antibody | Biolegend | Cat#394667; RRID:AB_2801034 |
| TotalSeq™-C0255 anti-human Hashtag 5 Antibody | Biolegend | Cat#394669; RRID:AB_2801035 |
| TotalSeq™-C0256 anti-human Hashtag 6 Antibody | Biolegend | Cat#394671; RRID:AB_2820042 |
| TotalSeq™-C0257 anti-human Hashtag 7 Antibody | Biolegend | Cat#394673; RRID:AB_2820043 |
| Anti-human CXCR3 BV421 | Biolegend | Cat#353716; RRID:AB_2561448 |
| Anti-human CD8a BV570 | Biolegend | Cat#301038; RRID:AB_2563213 |
| Anti-human CD4 BV650 | Biolegend | Cat#317436; RRID:AB_2563050 |
| Anti-human CD3e BV785 | Biolegend | Cat#300472; RRID:AB_2687178 |
| Anti-human CD38 Alexa Fluor 488 | Biolegend | Cat#356634; RRID:AB_2734381 |
| Anti-human CD27 PerCP-Cy5.5 | Biolegend | Cat#356407; RRID:AB_2561906 |
| Anti-human HLA-DR PE/Dazzle594 | Biolegend | Cat#307654; RRID:AB_2563646 |
| Anti-human KLRG1 PE/Cy7 | Invitrogen | Cat#25-9488-42; RRID:AB_2573546 |
| Anti-human PD-1 APC | Biolegend | Cat#329908; RRID:AB_940475 |
| Anti-human γδ TCR BV421 | Biolegend | Cat#331218; RRID:AB_2562317 |
| Anti-human CD59 V450 | BD | Cat#560740; RRID:AB_1727512 |
| Anti-human HLA-DR BV570 | Biolegend | Cat#307638; RRID:AB_2650882 |
| Anti-human CD25 BV605 | Biolegend | Cat#302632; RRID:AB_11218989 |
| Anti-human CD103 BV785 | Biolegend | Cat#350230; RRID:AB_2734364 |
| Anti-human CD8a Alexa Fluor 488 | Biolegend | Cat#300916; RRID:AB_756152 |
| Anti-human CD3 Alexa Fluor 532 | eBioscience | Cat#58-0038-42; RRID:AB_11218675 |
| Anti-human PD-1 PE | eBioscience | Cat#12-9969-42; RRID:AB_10736473 |
| Anti-human CD45 PE/Cy7 | Biolegend | Cat#304010; RRID:AB_314398 |
| Anti-human CD45 Per-CpVio700 | Miltenyi | Cat#130-097-527; RRID:AB_2660425 |
| Anti-human CTLA-4 PE/Cy7 | Biolegend | Cat#349914; RRID:AB_2563098 |
| Anti-human CD4 Alexa Fluor 700 | eBioscience | Cat#56-0049-42; RRID:AB_11219085 |
| Anti-human GranzymeB PE/CF594 | BD | Cat#562462; RRID:AB_2737618 |
| Anti-human FOXP3 APC | eBioscience | Cat#17-4777-42 |
| Anti-human IL-7R BV421 | Biolegend | Cat#351310; RRID:AB_10960140 |
| Anti-human CD45RO Pacific Blue | Biolegend | Cat#304216; RRID:AB_493659 |
| Anti-human HLA-DR BV510 | Biolegend | Cat#307646; RRID:AB_2561948 |
| Anti-human CXCR3 BV605 | Biolegend | Cat#353728; RRID:AB_2563157 |
| Anti-human CD4 BV711 | Biolegend | Cat#317440; RRID:AB_2562912 |
| Anti-human PD-1 PE | Biolegend | Cat#329906; RRID:AB_940483 |
| Anti-human IgG4 Fc PE | Southern Biotech | Cat#9200-09; RRID:AB_2796693 |
| Anti-human KLRG1 Alexa Fluor 647 | Biolegend | Cat#367704; RRID:AB_2565981 |
| Anti-human FOXP3 PE/CF594 | BD | Cat#563955; RRID:AB_2738507 |
| Anti-human Ki-67 PerCp/Cy5.5 | Biolegend | Cat#350520; RRID:AB_2562295 |
| Anti-human/mouse GranzymeB PE/Cy7 | Biolegend | Cat#396410; RRID:AB_2801079 |
| Anti-mouse TCRβ biotin | Biolegend | Cat#109204; RRID:AB_313427 |
| Anti-human CD3e | eBioscience | Cat# 16-0037-85; RRID:AB_468855 |
| Biological samples | ||
| Fresh tumor tissue biopsies or surgical resections | Dana-Farber Cancer Institute/Brigham and Women’s Hospital | IRB# DC/HCC 16-284 |
| Paraffin-embedded formalin fixed tumor slides, prepared from tumor biopsies or tumor resections | Dana-Farber Cancer Institute/Brigham and Women’s Hospital | IRB# DC/HCC 16-284 |
| Peripheral blood mononuclear cells, prepared from whole blood draws | Dana-Farber Cancer Institute |
IRB# DC/HCC 16-284 |
| Chemicals, peptides, and recombinant proteins | ||
| ACK red blood cell lysis buffer | Thermo Fisher Scientific |
Cat#A1049201 |
| Ultrapure Bovine Serum Albumin, RNase-free | Thermo Fisher Scientific |
Cat#AM2616 |
| Recombinant Human IL-2 | Peprotech | Cat#200-02 |
| Recombinant Human IL-7 | R&D Systems | Cat#207-IL-025 |
| Recombinant Human IL-15 | R&D Systems | Cat#247-ILB-025 |
| DMEM | ThermoFisher Scientific |
Cat#11995073 |
| RPMI 1640 | ThermoFisher Scientific |
Cat#21875034 |
| ACK Lysing Buffer | ThermoFisher Scientific |
Cat#A1049201 |
| Critical commercial assays | ||
| 10X Chromium Single Cell 5′ Library & Gel Bead Kit | 10X Genomics | Cat#1000006 |
| 10X Chromium Single Cell A Chip Kit | 10X Genomics | Cat#1000152 |
| 10X Chromium Single Cell V(D)J Enrichment Kit, Human T Cell |
10X Genomics | Cat#1000005 |
| 10X Chromium Single Cell 5′ Library Construction Kit | 10X Genomics | Cat#1000020 |
| 10X Chromium Single Cell 5’ Feature Barcode Library Kit |
10X Genomics | Cat#1000080 |
| 10X Chromium i7 Multiplex Kit | 10X Genomics | Cat#120262 |
| Chromium i7 Multiplex Kit N, Set A | 10X Genomics | Cat#1000084 |
| CD45 MicroBeads, human | Miltenyi Biotec | Cat#130-045-801 |
| Zombie NIR Fixable Viability Kit | Biolegend | Cat# 423106 |
| Zombie Aqua™ Fixable Viability Kit | Biolegend | Cat#423102 |
| StraightFrom® Leukopak® CD8 Microbead Kit | Miltenyi Biotec | Cat#130-117-019 |
| Incucyte® Nuclight Red Lentivirus | Satorius | Cat#4476 |
| CryoStor® CS10 | Stem Cell Technologies |
Cat#07959 |
| Anti-Biotin MicroBeads | Miltenyi Biotec | Cat#130-090-485 |
| Annexin V MicroBead Kit | Miltenyi Biotec | Cat#130-090-201 |
| Human IFN-gamma (3rd Gen) | Protein Simple | Cat# SPCKA-PS002574 |
| Deposited data | ||
| Single cell RNA-seq data and bulk TCR sequencing data | This paper | GSE200996 |
| Bulk TCR-sequencing data from urothelial cancer dataset | (Mariathasan et al., 2018) |
http://research-pub.gene.com/IMvigor210CoreBiologies/
; EGAS00001002556 |
| Experimental models: Cell lines | ||
| MHC-null HEK293T engineered to express a fluorescent reporter activated by T cell killing | (Ferretti et al., 2020; Kula et al., 2019) |
N/A |
| U266B1 Plasma cell myeloma | ATCC | Cat#TIB-196 |
| A101D Melanoma | ATCC | Cat#CRL-7898 |
| A2058 Melanoma | ATCC | Cat#CRL-11147 |
| SKMEL5 Melanoma | ATCC | Cat#HTB-70 |
| 786-O Renal cell carcinoma | ATCC | Cat#CRL-1932 |
| Lenti-X™ 293T Cell Line | Takara | Cat#632180 |
| Software and algorithms | ||
| FlowJo V10 | TreeStar | RRID:SCR_008520 |
| Prism V9 | Graph Pad | RRID:SCR_002798 |
| Cellranger mkfastq v3.0.0 | 10X Genomics | http://software.10xgenomics.com |
| Cellranger v4.0.0 | 10X Genomics | http://software.10xgenomics.com |
| Cumulus | (Li et al., 2020) | https://cumulus.readthedocs.io/en/latest/i ndex.html |
| R (v4.0.0 and v4.1.2) | The R Foundation | https://www.rproject.org |
| DropletUtils (v1.8.0, R package) | (Lun et al., 2019) | Bioconductor |
| DoubletFinder (v2.0.2) | (McGinnis et al., 2019) | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Seurat (v3.2.0, R package) | (Hao et al., 2021) | https://satijalab.org/seurat/index.html |
| SingleR (v1.2.4, R package) | (Aran et al., 2019) | Bioconductor |
| Slingshot (v1.6.1, R package) | (Street et al., 2018) | Bioconductor |
| AUCell (v1.13.1, R package) | (Aibar et al., 2017) | Bioconductor |
| fgsea (v1.14.0, R package) | (Korotkevich et al., 2021) | Bioconductor |
| Molecular Signatures Database | (Liberzon et al., 2015) | http://software.broadinstitute.org/gsea/msigdb/index.jsp |
| CellChat (v1.1.0, R package) | (Jin et al., 2021) | https://github.com/sqjin/CellChat |
| LymphoSeq (v1.16.0, R package) | (Coffey, 2021) | Bioconductor |
| Immunarch (v0.6.5, R package) | (Nazarov, 2020) | https://immunarch.com/ |
| Genome Analysis Toolkit | Broad Institute | https://gatk.broadinstitute.org/hc/en-us |
| FACETS | (Shen and Seshan, 2016) | https://github.com/vanallenlab/facets |
| tradeSeq (v1.2.01, R package) | (Van den Berge et al., 2020) | Bioconductor |
| beta-binomial model for bulk TCR expansion analysis | (Rytlewski et al., 2019) | https://github.com/jrytlewski/beta_binomial_paper |
| IMvigor210CoreBiologies (v1.0.0, R package) | (Mariathasan et al., 2018) | http://researchpub.gene.com/IMvigor210CoreBiologies/ |
| ProjecTILs (v1.0.0, R package) | (Andreatta et al., 2021) | https://github.com/carmonalab/ProjecTILs |
| GMM-demux | (Xin et al., 2020) | https://github.com/CHPGenetics/GMM-Demux |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients and sample collection
This study investigated specimens from patients with oral cavity squamous cell carcinoma enrolled in a phase 2 clinical trial that investigated neoadjuvant immune checkpoint blockade, as previously described in detail (Schoenfeld et al., 2020). Briefly, newly diagnosed, previously untreated patients with oral cavity squamous cell carcinoma were eligible for enrollment if they presented with locally advanced disease (T2b4) or had local lymph node metastases, without evidence of distant metastases. This protocol (16–284) was approved by the Dana-Farber Harvard Cancer Center (DF- HCC) institutional review board and written informed consent was obtained per institutional guidelines. Patients were an average age of 62 years (+/−12 years, SD) and comprised 62% men (n=18) and 38% women (n=11) (Schoenfeld et al., 2020). We analyzed specimens from all patients who were initially treated in this trial (n=15 mono, n=15 combo), although one patient (P02, monotherapy group) was later removed from the formal clinical trial and excluded from response analyses due to subsequent determination of pre-treatment metastatic disease.
Tumor tissue from pre-treatment biopsies or post-treatment surgical resections was collected in DMEM media (ThermoFisher Scientific # 11995073) for rapid analysis of live cells by scRNA-seq, while another part of the tissue was fixed in formalin. For patients with a complete or near-complete response to treatment, surgical specimens from the necrotic tumor bed were collected for immune cell profiling. FFPE-embedded diagnostic pre-treatment biopsies were also investigated.
Blood was collected in BD Vacutainer® CPT™ tubes (Becton Dickinson) with sodium heparin and mixed to prevent clotting. Tubes were centrifuged at 1500 g for 15 minutes, the PBMC layer was collected and washed with PBS. PBMCs were frozen in a solution of 80% RPMI/10% FBS/10% DMSO (1–15×106 cells/mL) and placed in a CoolCell™ (Corning) cell-freezing container at −80° C for up to one week, before transfer to liquid nitrogen for long-term storage.
METHOD DETAILS
Tumor cell suspension preparation
Fresh tumor tissue was transferred to a 6 cm tissue culture dish in a small volume of DMEM media and cut into <2 mm pieces with a razor blade. Tissue pieces were centrifuged (400 g for 5 minutes at 4°C for all centrifuge steps) and resuspended in 2–5 mL of a collagenase-containing enzymatic digestion solution (Miltenyi Human Tumor Dissociation Kit #30-095-929), based on the weight of collected tissue, according to manufacturer’s instructions. Biopsies were incubated at 37°C for 20–30 minutes with mixing using a 5–10 mL serological pipet and intermittent gentle vortexing at 2–5-minute intervals. The extent of tumor tissue dissociation was variable and likely dependent on extracellular matrix abundance. The resulting cell suspension was passed through a 70 μm nylon mesh filter and washed with an additional 5–10 mL of RPMI media.
Isolation of tumor-infiltrating CD45+ cells for scRNA-seq
For patients P14-P32, tumor CD45+ cells were isolated by fluorescence-activated cell sorting (FACS). Filtered tumor cell suspensions were centrifuged and resuspended in 3 mL ACK red blood cell lysis buffer (Gibco #A1049201) for 1 minute at RT, then immediately diluted with 10 mL of PBS (Life Technologies #10437028) to halt osmotic stress. Cells were pelleted and resuspended in Zombie NIR (Biolegend) viability dye solution (1:500 in PBS), followed by a 15 minute incubation at RT in the dark. Cells were counted with a hemocytometer, yielding on average 1.3×104 live cells/mg of tumor tissue. Up to 500,000 live cells were then resuspended in 100 μL of FACS wash buffer (FWB; PBS, 2% FBS, 1 mM EDTA) with the addition of the following antibodies: TruStain FcX (Biolegend 422302), CD3-PE/Dazzle (Biolegend 300450), CD45-Pacific Blue (Biolegend 304022), CD66b-PE/Cy7 (Biolegend 305116) and CD15-APC (Biolegend 301908). Cells were incubated on ice for 20 minutes, washed with 1 mL FWB, filtered through 40 μm flow-cap strainers, and then resuspended in 500 μL FWB. Cells were sorted using a FACS Aria II or III instrument (BD Biosciences). For most samples, 12–15 × 103 live CD45+CD66b-mononuclear cells (MNC) cells were sorted into T cell media (RPMI + 10% FBS) and immediately transferred to ice. CD66b+ granulocytes were excluded because these cells tend to yield low-quality scRNA-seq data using the 10x Genomics protocol.
For patient P13, tumor-infiltrating immune cells were isolated with anti-CD45 magnetic beads. Following filtering of dissociated tumor suspension as above, cells were centrifuged and then resuspended in 80 μL of MACS buffer (0.5% FBS, 2 mM EDTA in PBS). Human CD45 MicroBeads (20 μL, Miltenyi) were mixed with cells, and then incubated at 4°C for 15 minutes per manufacturer’s protocol. After washing with MACS buffer, beads were applied to a MS magnetic column (Miltenyi) placed in a magnetic MACS separator and washed with 3×500 μL of MACS buffer. To elute cells, the column was removed from the magnet and flushed with 1 mL of MACS buffer.
All scRNA-seq was performed using the 10x Genomics 5’ V1 assay and protocols (10x Genomics). CD45+ cells isolated as above were centrifuged and washed with PBS containing 0.05% RNase-free BSA (ThermoFisher Scientific #AM2616), leaving a volume of <32 μL after the final wash. Up to 15,000 cells based on cell sorter count were loaded into a Single Cell Chip A channel, along with reverse transcriptase reagent mixture and 5’ gel beads according to the manufacturer’s protocol. Chips were loaded into the 10x Genomics Chromium Controller for single-cell partitioning. Emulsions were immediately recovered from the chip and incubated in a deep-well block Thermocycler (Bio-Rad Laboratories) for the reverse transcription and template-switch reaction. Emulsions containing synthesized cDNAs were stored at 4°C for a maximum of 48 hours.
Preparation of total PBMC for 5’ barcoded scRNA-seq
PBMCs isolated as described above were thawed in groups in 5–6 vials at a time, with each vial representing an individual blood draw. Thawed PBMCS were immediately diluted into 10 mL PBS, centrifuged (400 g for 5 minutes at 4°C for all centrifugation steps) and resuspended in PBS. Cells were counted with a hemacytometer; viability of thawed cells averaged 55%. Live cells were sorted to ensure the quality of scRNA-seq libraries. Between 0.5–1×106 cells were stained with Zombie NIR (Biolegend) diluted 1:500 in PBS for 15 minutes at RT. Cells were centrifuged and then resuspended in 90 μL of FWB, to which the following were added: 5 μL of TruStain Fx FcBlock (Biolegend), 2 μL of αCD45-Pacific Blue (Biolegend), 2 μL of αCD3-PE/Dazzle594 (Biolegend) and 2 μL (1 μg) of an individual TotalSeqC hashing antibody (Hashtags 1–7, Biolegend). For each group of up to six PBMC vials, unique hashtags were used for subsequent identification of cells from individual samples. Cells were incubated for 20 minutes at 4°C, washed twice with 500 μL FWB, and filtered through 40 μM flow-cap strainers. Live CD45+ cells (50,000 to 100,000 cells) were then separately sorted using a FACS Aria II instrument for each hashtag-labeled sample. Cells were sorted into 500 μL 0.1% BSA in 1.5 mL DNA LoBind microcentrifuge tubes (Eppendorf). Following sorting, cells were centrifuged, washed with 0.25% BSA, and resuspended in a final volume of 30–50 μL 0.25% BSA. Live cells were counted with a hemacytometer.
For generation of single-cell emulsions, 6000 live cells from each sample (up to six samples) were pooled and loaded into a single Chip A channel, along with the reverse transcriptase reagent mixture and 5’ gel beads, according to the 10x Genomics 5’ V1 manufacturer’s protocol. Chips were loaded into the 10x Genomics Chromium Controller for single-cell partitioning. Emulsions were immediately recovered from the chip and incubated in a deep-well block Thermocycler (Bio-Rad Laboratories) for the reverse transcription and template-switch reaction. Emulsions containing synthesized cDNAs were stored at 4°C for a maximum of 48 hours.
Sorting of activated blood T cells for scRNA-seq
Frozen PBMC samples from four patients (P13, P19, P26, and P32) at two timepoints (B1 and B2) were sorted using T cell activation markers identified in scRNA-seq data. Cells were thawed in two groups (B1 or B2), diluted in FWB, and counted with a hemacytometer. Up to 5×106 cells were then centrifuged and resuspended in 1:500 Zombie Aqua (Biolegend) in PBS and incubated at RT for 15 minutes. Cells were resuspended in TruStainFx FcBlock (1:20 dilution) in FWB, incubated at 4°C for 15 minutes, after which additional antibodies for sorting and analysis were added as indicated in a total volume of 500 μL FWB:
| Reagent | Dilution | Supplier | Catalog Number |
|---|---|---|---|
| BV421 anti-CXCR3 | 1:20 | Biolegend | 353716 |
| BV570 anti-CD8a | 1:50 | Biolegend | 301038 |
| BV650 anti-CD4 | 1:40 | Biolegend | 317436 |
| BV785 anti-CD3e | 1:50 | Biolegend | 300472 |
| Alexa Fluor 488 anti-CD38 | 1:40 | Biolegend | 356634 |
| PerCP-Cy5.5 anti-CD27 | 1:40 | Biolegend | 356407 |
| PE/Dazzle594 anti-HLA-DR | 1:20 | Biolegend | 307654 |
| PE/Cy7 anti-KLRG1 | 1:20 | Invitrogen | 25-9488-42 |
| APC anti-PD-1 | 1:20 | Biolegend | 329908 |
Unique sample-specific hashtag TotalSeqC antibodies (Biolegend, #1–4) were added to each sample (10 μL per sample) for identification of cells from individual specimens. Cells were stained for 20 minutes at 4°C, washed twice with FWB and filtered through a 40 μm flow-cap strainer. Using a BD FACS Aria II instrument (BD Biosciences), live CD3+CD8+ cells were sorted based on CD38+HLA-DR+ expression. Approximately 5,000 to 125,000 cells could be sorted for different samples and cell populations. Sorted cells were washed with 0.25% BSA and counted if numbers permitted (>30,000 cells).
Before loading into 10x Genomics Single Cell chips, sorted cells from each patient were separately pooled, using all sorted cells or up to 10,000 cells. Each pool of cells was loaded into single Chip A channel, along with the reverse transcriptase reagent mixture and 5’ gel beads according to the 10x Genomics 5’ V1 assay 5’ V1 protocol. Emulsions were immediately recovered from the chip and incubated in a deep-well block Thermocycler (Bio-Rad Laboratories) for the reverse transcription and template-switch reactions.
Library preparation and sequencing for 5’ barcoded scRNA-seq
All steps from cDNA isolation to library preparation were completed as per manufacturer’s protocol for the 10x Genomics 5’ V1 assay (10x Genomics). For libraries of tumor-infiltrating CD45+ cells, isolated cDNA was amplified (14 cycles) using standard cDNA Primer Mix, after which cDNA was divided for TCR enrichment/library preparation with the Chromium Single Cell V(D)J TCR kit, or for preparation of a gene expression library.
For blood (PBMC) libraries, cDNA was amplified (12 cycles) using SC5’ Feature cDNA Primers from the Chromium Single Cell 5’ Feature Barcode Library Kit (10x Genomics). Gene expression, TCR, and hashtag libraries were prepared from cDNA according to the manufacturer’s protocols.
Quality of cDNAs and resulting libraries was evaluated using the D5000 and D1000 high-sensitivity kits on a 2200 TapeStation system (Agilent).
Tumor-infiltrating CD45+ scRNA-seq gene expression libraries were sequenced on the Illumina HiSeq X platform with a Read2 only (98 bp) configuration. Tumor TCR libraries were sequenced on either the Illumina Miseq (V2 flowcell), Hiseq 2500, or HiSeq 4000 platform, all utilizing a 150 bp paired-end read configuration. All PBMC libraries (including gene expression, TCR, and hashtag barcodes) were sequenced on the Illumina HiSeq 4000 platform using a 150 bp paired-end read configuration. Data were demultiplexed by the appropriate i7 SI-GA sample index (Cellranger mkfastq, 10x Genomics) and fastq files were transferred for downstream processing.
Bulk-TCR β chain repertoire sequencing
All bulk TCR sequencing was performed using the Adaptive Biotechnologies ImmunoSeq deep TCR β assay. Formalin-fixed paraffin embedded (FFPE) sections were used for tumor TCR sequencing. For PBMC TCR sequencing, between 0.5–2.0 ×106 PBMCs were pelleted and snap-frozen. DNA extraction, library preparation, and sequencing was performed by Adaptive Biotechnologies according to standard protocols. TCR V-Jβ segment annotation, CDR3β loop sequences, and counts were delivered and analyzed as described below.
Flow cytometry analysis of tumor-infiltrating T cell populations
Up to 1×106 dissociated cells from tumors were resuspended in Zombie NIR (Biolegend) viability dye solution (1:500 in PBS), followed by 15 minutes incubation at RT in the dark. Cells were then centrifuged and stained in FWB (50–100 μL total volume) for 30 minutes at 4° C in the dark using antibodies to the following cell surface markers:
| Reagent | Dilution | Supplier | Catalog Number |
|---|---|---|---|
| TruStain Fx FcBlock | 1:20 | Biolegend | 422302 |
| BV421 anti-γδ TCR | 1:20 | Biolegend | 331218 |
| V450 anti-CD69 | 1:40 | BD | 560740 |
| BV510 anti-CD14, CD15, CD64, CD163 (Dump mixture) | 1:80 each | Biolegend, BD |
301842, 323028, 563459, 333628 |
| BV570 anti-HLA-DR | 1:20 | Biolegend | 307638 |
| BV605 anti-CD25 | 1:20 | Biolegend | 302632 |
| BV785 anti-CD1Q3 | 1:40 | Biolegend | 350230 |
| Alexa Fluor 4BB anti-CDBa | 1:40 | Biolegend | 300916 |
| Alexa Fluor 532 anti-CD3 | 1:40 | eBioscience | 58-0038-42 |
| PE anti-PD-1 | 1:40 | eBioscience | 12-9969-42 |
| PE-Cy5 anti-CD45* | 1:40 | Biolegend | 304010 |
| Per-CpVio700 anti-CD45* | 1:20 | Miltenyi | 130-097-527 |
| PE/Cy-7 anti-CTLA-4 | 1:20 | Biolegend | 349914 |
| Alexa Fluor 700 anti-CD4 | 1:20 | eBioscience | 56-0049-42 |
only one of these was used for a given sample
Following incubation, cells were washed twice with 150–200 μL FWB before proceeding to intracellular staining. Cells were then fixed and permeabilized with the FoxP3/transcription factor buffer staining set (eBioscience) for 20 min at RT. Cells were washed with 1x permeabilization buffer before proceeding to intracellular staining with the following antibodies diluted in 1X permeabilization buffer:
| Reagent | Dilution | Supplier | Catalog Number |
|---|---|---|---|
| PE/CF594 anti-Granzyme B | 1:50 | BD | 562462 |
| APC anti-FoxP3 | 1:20 | eBioscience | 17-4777-42 |
Cells were stained for 20 minutes at RT in the dark, and then washed twice with 1X permeabilization buffer. Stained cells were resuspended in 300 μL FWB and stored at 4°C protected from light until analysis within 48 hours.
Flow cytometry analysis of blood T cell populations
Up to 2×106 dissociated PBMCs were resuspended in Zombie NIR (Biolegend) viability dye solution (1:500 in PBS), followed by 15 minutes incubation at RT in the dark. Cells were then centrifuged, resuspended in FWB and stained at 0.5–1×106 cells/mL concentration with the following antibodies
| Reagent | Dilution | Supplier | Catalog Number |
|---|---|---|---|
| TruStain Fx FcBlock | 1:20 | Biolegend | 422302 |
| BV421 anti-IL-7R | 1:25 | Biolegend | 351310 |
| Pacific Blue anti-CD45RO | 1:80 | Biolegend | 304216 |
| BV510 anti-HLA-DR | 1:35 | Biolegend | 307646 |
| BV570 anti-CDBα | 1:50 | Biolegend | 301038 |
| BV605 anti-CXCR3 | 1:25 | Biolegend | 353728 |
| BV711 anti-CD4 | 1:25 | Biolegend | 317440 |
| BV7B5 anti-CD103 | 1:50 | Biolegend | 350229 |
| Alexa Fluor 488 anti-CD38 | 1:25 | Biolegend | 356634 |
| Alexa Fluor 532 anti-CD3 | 1:50 | Invitrogen | 58-0038-42 |
| PE anti-PD-1* | 1:40 | Biolegend | 329906 |
| PE anti-hIgG4* | 1:20 | Southern Biotech | 9200-09 |
| Alexa Fluor 647 anti-KLRG1 | 1:20 | Biolegend | 367704 |
anti-PD-1 was used for pre-treatment B1 samples; anti-IgG4 was used for on- and post-treatment B2 and B3 samples.
Following incubation, cells were washed twice with 150–200 μL FWB before proceeding to intracellular staining. Cells were then fixed and permeabilized with the FoxP3/transcription factor buffer staining set (eBioscience) for 20 min at RT. Cells were washed with 1x permeabilization buffer before proceeding to intracellular staining with the following antibodies diluted in 1X permeabilization buffer:
| Reagent | Dilution | Supplier | Catalog Number |
|---|---|---|---|
| Alexa Fluor 532 anti-CD3 | 1:50 | Invitrogen | 58-0038-42 |
| PE-CF594 anti-FoxP3 | 1:25 | BD | 563955 |
| PerCP-Cy5.5 anti-Ki-67 | 1:25 | Biolegend | 350520 |
| PE-Cy7 anti-Granzyme B | 1:50 | Biolegend | 396410 |
Cells were stained for 20 minutes at RT in the dark, and then washed twice with 1X permeabilization buffer. Stained cells were resuspended in 300 μL FWB and analyzed as below.
Spectral flow cytometry analysis of tumor and blood T cells
Stained cells were passed through a 40 μm filter and analyzed using a Sony SP6800 Spectral Analyzer. Single-color controls for spectral unmixing, using the identical antibodies as for analysis, were acquired using OneComp eBeads (ThermoFisher 01-1111-41) and fixed when applicable to match experimental conditions. The single-color control for Zombie NIR viability dye was acquired with PBMCs. Data were analyzed using FlowJo (Tree Star) version 10.4. Dead cells and aggregates were excluded from analyses. Gating was confirmed with fluorescence-minus-one (FMO) controls.
T cell engineering for T-scan antigen discovery screen
Primary CD8+ T cells from healthy donors were isolated using the StraightFrom® Leukopak® CD8 Microbead Kit (Miltenyi Biotec) according to manufacturer’s protocol. Isolated cells were frozen in CryoStor® CS10 (Stem Cell Technologies) and stored in liquid nitrogen until use. Individual paired TCR alpha and TCR beta sequences were cloned into a single construct expressing mouse TRBC and TRAC regions to create human-mouse TCRs separated by a P2A skip sequence. The TCR constructs were packaged with Lenti-X™ cells (Takara Bio USA, Mountain View, CA).
CD8+ T cells were thawed, activated, plated in 24-well plates and transduced with lentiviral particles to express a unique orphan HNSCC TCR (per well) or a control TCR with known target recognition. Four days after transduction, cells were harvested and either pooled prior to isolation (for T-Scan screens) or isolated individually (for validation and functional assays). Cells were isolated using anti-mTCR biotin antibody (BioLegend), anti-biotin microbeads (Miltenyi) in MACS running buffer for manual magnetic separation using a QuadroMACS Separator and LS columns (Miltenyi). Isolated cells were washed and resuspended in fresh complete T cell medium and expanded in G-Rex®10 flasks (Wilson Wolf). Cells were then frozen in CryoStor® CS10 and stored in liquid nitrogen until used for experiments.
Reporter cell line engineering for T-Scan antigen discovery screen
MHC-null HEK293T cells were engineered to express a fluorescent reporter activated by T cell killing as described (Ferretti et al., 2020; Kula et al., 2019). HLA alleles relevant to each patient were introduced into reporter cells (Table S5) which were then transduced with a genome-wide library of 90-aa protein fragments, as described previously (Kula et al., 2019).
T-Scan screen
To stimulate T cells for antigen screens, CD8+ TCR-T cell pools were thawed, and re-stimulated by co-culturing with irradiated allogenic PBMCs in the presence of 0.1 μg/mL anti-CD3 (OKT3, eBioscience) and 50 U/mL recombinant IL-2 (Peprotech). After expansion for 7 days, the T cells were added to library-transduced reporter cells and incubated at 37°C for 4h. After incubation, cells were harvested by trypsinization and labeled with Annexin V magnetic microbeads (Miltenyi) according to the manufacturer’s instructions. Annexin-labeled cells were isolated using an AutoMACS Pro (Miltenyi), and reporter-positive cells were sorted using a MoFlo Astrios EQ cell sorter (Beckman Coulter).
Genomic DNA (gDNA) was extracted from sorted cells and processed as described previously (Ferretti et al., 2020). Nucleotide sequences were mapped to the reference nucleotide library. The proportion of read counts for each tile was calculated for each screen replicate (n = 8) and for the input for each pool of transduced reporter cells. Enrichment of each tile was calculated by dividing the proportion of the tile in the screen replicate by the proportion of the tile in the input library. A modified geometric mean of the enrichment of an identical tile across the 8 screen replicates (calculated by adding 0.1 to all enrichment values and taking the geometric mean) was used to identify reproducible screen hits. Specific MHC-binding epitopes for each tile above the threshold of 2-fold enrichment were predicted using NetMHC4.0. Candidate epitopes for each tile were selected by identifying predicted strong binding epitopes shared across overlapping adjacent and redundant tiles that were enriched in the screen.
Peptide validation assay
HEK293T cells expressing the target-relevant HLA alleles were seeded in a 96 well plate. The HEK293T cells were pulsed with the target peptide (Genscript) at 1ug/mL for 1 hour. HEK293T cells for all conditions were then co-cultured with the TCR-transduced T cells at an E:T ratio of 1:1 for 18 hours. Cells were then harvested by pipetting, transferred to V-bottom 96-well plates, and centrifuged at 2,000 rpm for 2 minutes. The supernatant was collected for IFNγ measurement using an ELISA-based human IFNγ 3rd generation single-plex assay (Protein Simple) according to manufacturer instructions.
Cancer Cell Line Reactivity
Public databases were evaluated to identify cancer cell lines that expressed C*07:02 (TCLP- Tron Cell Line Portal; (Scholtalbers et al., 2015)) and variable levels of MAGEA1 transcript (CCLE-Cancer Cell Line Encyclopedia, Broad Institute). The selected cell lines, their MAGEA1 expression levels in transcripts per million (TPM), and cell culture conditions are listed:
| Cell line | Origin | MAGEA1 TPM | Media |
|---|---|---|---|
| U266B1 | Plasma cell myeloma | 657 | RPMI 15% FBS |
| A101D | Melanoma | 359 | DMEM 10% FBS |
| A2058 | Melanoma | 128 | DMEM 10% FBS |
| SKMEL5 | Melanoma | 8 | EMEM 10% FBS |
| 786-O | Renal cell carcinoma | 0.4 | RPMI 10% FBS |
Cancer cell lines were purchased from ATCC (Manassas, VA) and HEK293T cells were purchased from Takara Bio (Shiga, Japan). All media were supplemented with 1% antibiotics and incubated at 37°C in 5% CO2.
Cells were seeded in a 96-well plate overnight in appropriate media. On the following day, CD8+ T cells were thawed, washed in complete RPMI-1640 medium, and added to the wells for an effector to target (E:T) ratio of 1:1 for IFNγ measurement and incubated overnight. Cells were then harvested by pipetting, transferred to V-bottom 96-well plates, and centrifuged at 2,000 rpm for 2 minutes. The supernatant was collected for IFNγ measurement using a human IFNγ 3rd generation single-plex assay (Protein Simple) according to the manufacturer’s instruction.
Incucyte® NucLight™ Red transduction and cytotoxicity assay
Cancer cell lines were transduced with Incucyte® NucLight™ Red lentivirus reagent (Sartorius, Göttingen, Germany). Labeled cells were selected using 1.5 μg/mL of puromycin over 72 hours. Cancer cells expressing Incucyte® NucLight™ were seeded into 96-well plates (Corning, Flintshire, UK). On the same day, T cells were thawed, washed in complete RPMI-1640, and allowed to rest overnight in medium with 50 U/mL IL-2 (PeproTech), 5 ng/mL IL-7, and 5 ng/mL IL-15 (R&D Systems). All cells were rested overnight at 37°C in 5% CO2. Serial dilutions of the T cells were prepared for differing effector to target ratios and co-cultured with the plated cancer cells. One image per well from two technical replicates was taken every 2 hours over a 72 hour time course. Imaging used a 4x objective lens and data were analyzed using the Incucyte® Basic Software. Red channel acquisition time was 400 ms, and its background was subtracted by enabling Top-Hat segmentation. Edge split was applied so that the software could recognize closely spaced cells as multiple objects, rather than one object.
Exome sequencing for determination of tumor mutational burden
DNA was extracted from FFPE tumor sections and from corresponding PBMC frozen pellets (0.5×106 cells) by the Broad Institute Genomics platform. Tumor and germline DNA were sequenced using Illumina NovaSeq 6000 and Twist bait and target capture sets. Paired-end reads were aligned to hg19 human reference genome.
QUANTIFICATION AND STATISTICAL ANALYSIS
Flow cytometry data analysis
Population frequencies exported from FlowJo were analyzed for statistical significance using Prism 8.4.1 (GraphPad) or R (v4.1.2). Summary plots were prepared using Prism 8.4.1 (GraphPad) or the ggplot2 R package (v3.3.5). The statistical method, statistics, and sample numbers for each analysis are shown in the figure legends.
Processing of blood single cell RNA-seq data
Raw scRNA-sequencing reads were demultiplexed and aligned to the human reference genome (GRCh38-2020-A from 10x Genomics) for each pooled library using Cell Ranger (v4.0.0). Each pooled library contained multiple patient samples and timepoints; cells from each sample were labelled with distinct Hashtag oligonucleotides (HTO, Cell Hashing). Sequencing files for Gene Expression (measure mRNA level of cells) and Antibody Capture (measure HTO levels of cells) were fed to the “cellranger count” function with the –libraries flag, and the sequences of all HTO used in the library were fed to this function with –feature-ref flag. This generated a digital gene expression matrix, which measured the number of unique molecular identifier (UMI) for each gene and each cell barcode. The raw gene-by-barcode count matrix was then processed to filter potential empty drops using emptyDrops function from DropletUtils (v1.8.0, R package) (Lun et al., 2019) with the parameters of “lower = 500, ignore = 5”. We initially selected droplets as containing a cell if total UMI counts of this droplet exceed the 99th percentile of the top 10,000 barcodes divided by 10. Finally, only cells with FDR≤0.01 were retained as real cells for the following analysis. The raw HTO-by-barcode matrix was subsequently filtered based on identified real cells.
Demultiplexing samples in each blood library based on hashtags
The filtered HTO-by-barcode matrix for each pooled library was processed using GMM-demux, which is a Gaussian mixture model-based multiplet identification method with high performance, distinguishing singlets from doublets or multiplets based on detection of HTOs with default parameters (Xin et al., 2020). Predicted singlets with a confidence score ≥0.8 were defined as true singleT cells. Cell barcodes were then classified and allocated to respective patient samples based on hashtag identities. After processing, the gene-by-barcode count matrix was refined based on these identified singlets and used for the following analyses. On average, 83.46% of cells from filtered matrix were predicted as singlets, 15.53% were predicted as doublets or multiplets, and 0.28% were negative.
Quality control of filtered blood scRNA-seq data
We applied the following steps to filter out low-quality cells: (1) built a median-centered median absolute deviation (MAD)-variance normal distribution based on the fraction of mitochondrial genes of all single cells in each dataset and removed the cells with a mitochondrial fraction larger than the upper end of this distribution (determined by Benjamini-Hochberg corrected P < 0.01, for blood dataset: 10.03%) (Pijuan-Sala et al., 2019); (2) removed cells with less than 300 identified genes.
The cell hashing method could efficiently remove sample to sample doublets, although potential doublets within each patient sample sharing the same hashtag could have remained. Utilizing TCR sequences as barcodes of individual T cells, we sequentially applied the following steps to exclude potential doublets: (1) clustering analysis was performed based on all CD45+ cells to identify the major immune cell populations, such as T cells, B cells and myeloid cells; (2) among T cells, cells containing two TCR β chain sequences were removed (unless detected multiple times as a clonally expanded population); (3) DoubletFinder (v2.0.2) was applied to calculate the doublet scores for cells within each patient sample (McGinnis et al., 2019). We compared doublet scores of all cells and confirmed that TCR-identified doublets showed significantly higher doublets scores compared to the average of remaining cells. Doublet score of 80% quantile of all TCRderived doublets was then set as cutoff value to further filter out any remaining doublets. After these processing steps, we obtained 145,584 single cells for the blood dataset.
Processing of tumor single cell RNA-seq data
Raw scRNA-sequencing reads were demultiplexed to FASTQ files using Cell Ranger mkfastq (v3.0.0, 10x Genomics), and the Cumulus cellranger-workflow implementation of Cell Ranger count (v4.0.0) on Terra (https://terra.bio/) was used to align reads to the human reference genome (GRCh38-2020-A) and generate a raw counts matrix (Li et al., 2020). Cumulus was used to filter cells from the Cell Ranger raw counts matrix with fewer than 400 UMIs, 200 genes, or greater than 20% of UMIs mapped to mitochondrial genes. Count matrices from each sample were pooled and initial clusters were identified based on the Seurat clustering analysis pipeline (described below). Two clusters corresponding to endothelial and stromal cells were excluded. Then, the potential doublets included in tumor were also filtered out by using the same TCR-related strategies as used in the blood data. Finally, the remaining 74,557 tumor-infiltrating immune cells were used for the following analysis.
Clustering analysis of blood and tumor-infiltrating immune cells
The obtained UMI count matrix from the above process was fed into Seurat (v3.2.0, R package) for further processing (Hao et al., 2021). ‘NormalizeData’ function with default parameters was applied to normalize the expression level of genes in each single cell for both blood and tumor datasets. Then, 2,000 highly variable genes were identified using the ‘FindVariableFeatures’ function with ‘vst’ method. To avoid the dominant effect of T cell receptor variable genes in T cell clustering analysis, all TRAV, TRBV, TRDV and TRGV genes were removed from the high variable gene list. Immunoglobulin variable (IG[X]V genes) genes were also removed from the highly variable gene list for clustering of the whole CD45+ dataset. The ‘ScaleData’ function was used to scale and center gene expression matrices after regressing out heterogeneity associated with mitochondrial contamination. To perform clustering, the first 20 principal components were selected to construct the shared nearest neighbor (SNN) graph with ‘FindNeighbors’ function, and then clusters were determined using the Louvain algorithm. Cluster resolution was based on the principle that each cell cluster should express a unique group of genes.
To evaluate whether the transcriptome profile of cells from cluster 3 (ITGAE+ CD8) are associated with cells from cluster 4 (Cycling CD8) in tumor CD8 T cells after mitigating the cycling effect, we subsetted aggregated cells from these two clusters and then re-performed clustering analysis based on the above procedure, with or without regressing out the effects of cell cycle heterogeneity in ‘ScaleData’ function. If the cell cycle effect was not mitigated, cells from these two clusters were again clearly separated even with newly calculated highly variable genes, but mitigating the cell cycle effect through removal of cycle-associated variable genes showed cell mixing. Therefore, cells from cluster 4 are the cycling counterpart of cluster 3, and these two clusters show a very similar transcription profile.
Annotation of cell clusters, data visualization and differential expression analysis
The following two complementary approaches were used to annotate single cell clusters and sub-clusters: (1) we applied SingleR (v1.2.4, R package), which leverages reference transcriptomic datasets of known cell types to predict the identities of cell clusters in an unbiased way (Aran et al., 2019); (2) we checked whether well-studied marker genes of immune cell types were ranked among the top differentially expressed genes of query clusters, and then assigned the most likely identity for each cell cluster (Luoma et al., 2020). We first applied SingleR to determine if the predicted annotations for a query cluster based on different reference datasets were consistent (Hao et al., 2021), and then assigned the predicted cell type annotation to this cluster. If the prediction was not reliable (primarily for sub-cluster annotation), we checked top-ranked differentially expressed genes of this cluster. The ‘FindAllMarkers’ Seurat function with default parameters but with set min.pct= 0.3 was applied to identify differentially expressed genes of different clusters and sub-clusters. The uniform manifold approximation and projection (UMAP) was finally applied based on the above described SNN graph to visualize the single cell transcriptional profile in 2D space (Becht et al., 2019).
Statistical tests for partially paired samples from pre-Tx and post-Tx group was performed by optimal pooled t-test (Guo and Yuan, 2017). Other bar plots, box plots, violin plots and heatmaps were generated using customized R code through ggplot2 (v3.3.2, R package).
Prediction of CD4 and CD8 T cells based on a published reference dataset
As some T cell and NK cell populations can share similar transcriptomic profiles, it is difficult to confidently distinguish T cells from NK cells solely based on the clustering analysis described above. To improve T cell prediction, we identified potential T cells based on the expression of T cell marker genes (CD3D, CD3E and CD3G) and expression of rearranged αβ TCR genes. We then performed unsupervised analysis by mapping these identified T cells to previously annotated CITE-seq reference datasets in Seurat, which unambiguously identified CD4 and CD8 T cell subpopulations (Hao et al., 2021). This annotated reference dataset contained 162,000 PBMC labeled with 228 CITE-seq antibodies and provided different layers of cell annotation. We first normalized the expression matrix of our T cells with ‘SCTransform’ function to match the format of the downloaded reference dataset. Only ‘CD4 T’, ‘CD8 T’ and Other T’ cell populations in the reference dataset were selected for the prediction. Then, ‘FindTransferAnchors’ function was applied to identify anchors between reference and query datasets; the precomputed supervised PCA (spca) transformation and other default parameters were used for the processing. Finally, the ‘MapQuery’ function was used to transfer different T cell identities and protein data from reference to the query T cells. CD4 T and CD8 T cells were selected based on a prediction score >0.6. A small number of CD8A and CD8B positive cells in the CD4 T cell group and CD4 positive cells in CD8 T cells were also removed. For the blood dataset, 33,446 CD4 T cells and 14,207 CD8 T cells were annotated; for the tumor dataset, 24,032 CD4 T cells and 18,126 CD8 T cells were identified. This clustering and annotation analysis was subsequently applied to CD4 and CD8 T cell subsets.
Single cell pseudotime analysis
To define imputed pseudotime trajectories in tumor pre-Tx CD8 T cells, we applied the slingshot (v1.6.1, R package) (Street et al., 2018). The UMAP matrix obtained from above clustering analysis was fed into ‘slingshot’ funtion, and the IL7R+ CD8 T cell cluster (cluster 5) was set as the root state for evaluating the cell trajectories. Then, tradeSeq (v1.2.01, R package) was used to analyze the genes along the trajectories (Van den Berge et al., 2020). The negative binomial generalized additive model (NB-GAM) was fitted by ‘fitGAM’ function to smooth each gene’s expression; nknots parameter set as 6 was evaluated by ‘evaluateK’ function. Then, ‘predictCells’ function was used to extract fitted expression values for each cell. Finally, the expression profile of interested gene set was visualized by a heatmap, in which cells from each individual trajotory were ordered based on predicted pseudotime.
Calculation of gene set activity score
We applied AUCell (v1.13.1, R package), incorporated in SCENIC pipeline, to evaluate the activity score of a gene set in each single cell based on transcriptome profiles (Aibar et al., 2017). For query gene set, AUCell first builds the area under the recovery curve based on the ranking of top 5% of all genes in each single cell; the x axis represents ranking of these highly expressed genes based on their expression values, and y axis represents the number of genes recovered from the query gene set. The gene set activity score is measured as the proportion of expressed genes in the query gene set and their relative expression levels compared to the other genes in each cell. The tissue-resident memory T (Trm) cell signature genes were derived from a previous publication (Kumar et al., 2017). The cytotoxicity signature contained PRF1, GZMB, GZMA, GZMH, NKG7, and GNLY as derived from a previous publication (Mathewson et al., 2021). The inhibitory receptor signature contained: PDCD1, CTLA4, HAVCR2, LAG3, TIGIT. The NK cell receptor signature contained: KLRD1, FGFBP2, FCGR3A, S1PR5, KLRC1, KLRC3, KLRB1, KLRC2.
Functional enrichment analysis
The fgsea (v1.14.0, R package) was used to perform fast gene set enrichment analysis on clonal expansion associated genes (Tx-E vs Tx-NE) (Korotkevich et al., 2021). Pathway gene sets were downloaded from hallmark gene sets from the Molecular Signatures Database (MsigDB, http://software.broadinstitute.org/gsea/msigdb/index.jsp) (Liberzon et al., 2015). These hallmark pathways, as well as genes with averaged log-transformed fold-change (Tx-E vs. Tx-NE cells), were then fed into the ‘fgsea’ function (minSize=2, maxSize=1000, eps=0). Finally, only hallmark gene sets with an adjusted P value <0.05 were selected as the functionally enriched biological states or processes.
T cell – myeloid cell interaction analysis
To evaluate cell – cell interactions between T cell and myeloid cell clusters in tumors, we applied CellChat (v1.1.0, R package) to infer and visualize intercellular communication based on the expression of known ligand-receptor pairs (Jin et al., 2021). Here we mainly focused on interactions between tumor CD8 T cells of cluster 3 & 4 (C3/C4), given the TCR overlap and lineage relationship for these two groups, as well as all myeloid clusters (M1-M9), with a focus on changes in predicted cell-cell communication between pre-Tx and post-Tx samples. We first inferred T cell - myeloid cell interactions in pre-Tx and post-Tx groups by following CellChat’s recommended analysis of individual datasets with default parameters (except that thresh.p = 0.05 in ‘identifyOverExpressedGenes’ function, raw.use = TRUE and population.size = TRUE in ‘computeCommunProb’ function, min.cells = 10 in ‘filterCommunication’ function). All categories of ligand-receptor interactions in the database were used in the analysis. Subsequently, pre-Tx and post-Tx objects were merged by ‘mergeCellChat’ function for comparison. Finally, the ‘netVisual_bubble’ function was applied to visualize differences of communication probabilities by ligand-receptor pairs in both directions, T cells to myeloid cells as well as myeloid cells to T cells.
Processing of T cell receptor (TCR) scRNA-seq data from blood and tumor specimens
To generate the single-cell V(D)J sequences and annotate each library, we aligned TCR FASTQ reads to human GRCh38 V(D)J reference (v4.0.0 from 10X Genomics, modified to remove TRDD segments) using “cellranger vdj” function from Cell Ranger (v4.0.0). The filtered contig annotations, which contained high-level annotations of each high-confidenT cellular contig, were further filtered by removing records with ‘raw_consensus_id’ as ‘none’. For blood single-cell V(D)J analysis, the identities of different patient samples in each pooled library were labelled based on demultiplexing of the gene expression matrix. For all clonotypes detected in patient samples, there were 79.37% (blood) and 75.73% (tumor) of clonotypes containing one TRA and one TRB sequences as well as 8.20% (blood) and 8.18% (tumor) of clonotypes containing two TRA and one TRB sequences. Only 1.44% (blood) and 2.49% (tumor) of clonotypes contained one TRA and two TRB sequences, and only 0.53% (blood) and 2.34% (tumor) of clonotypes contained two TRA and two TRB sequences. In total, the TCR sequences for 98.20% of CD4+ T cells and 93.38% of CD8+ T cells could be detected in the blood dataset as well as for 87.05 % of CD4+ T cells and 86.84 % of CD8+ T cells in the tumor dataset.
Processing of bulk TCR repertoire sequencing data from tumor and blood samples
Raw TCR sequences and counts were pre-processed with ‘productiveSeq’ function (set aggregate as “aminoAcid”) from LymphoSeq (v1.16.0, R package) to filter out unproductive TRB rearrangements (Coffey, 2021). Matrices of blood and tumor clonotype frequencies were then generated for each patient.
To account for the possibility of blood contamination of tumor samples, we first identified potential blood clonotypes from tumor data based on the frequency (𝑓) of each individual clonotype in both blood and tumor, with the assumption that highly expanded clonotypes in blood would show a higher probability of being detected in tumor. For a patients with pre-Tx and post-Tx bulk TCR datasets in tumor and blood, a clonotype satisfying the criteria and was considered a blood-contaminated clonotype in tumor. For patients with pre-Tx and post-Tx bulk TCRs data in tumor but only a post-Tx TCR dataset in blood, we considered clonotypes with as potential blood-contaminated clonotypes. On average, approximately 1.87% of TCRs were identified as blood contaminated clonotypes in tumor bulk TCR data and removed for downstream analyses.
The clonality metric of the TCR repertoire was calculated based on the frequencies of all productive sequences as follows (Khunger et al., 2019):
where pi is the frequency of clone i from a patient sample with n unique clones. This metric is normalized to the number of unique clones and ranges from 0 to 1. A higher clonality score indicates a more uneven repertoire structure characterized by more highly expanded clones.
The Morisita index (MI), measuring the repertoire similarity between pre-Tx and post-Tx samples in each patient, was defined by the following equation (Khunger et al., 2019):
where ai and bi are the counts of clone i in pre-Tx sample A and post-Tx sample B from the same patient, An and Bn are the total number of clones in samples A and B, and n is the total number of unique clones in the union of A and B samples. The Morisita Index ranges from 0 to 1, with values closer to 1 indicating higher similarity and less repertoire turnover.
Differences in clonality and Morisita indices between different stages and cohorts were evaluated by two-sided t-test. The Immunarch (v0.6.5, R package) was used to calculate the size distribution of tumor bulk TCR clonotypes by using ‘repClonality’ function (Nazarov, 2020).
Identification of differentially expanded clonotypes in blood and tumor samples
To identify significantly expanded clonotypes between pre-Tx and post-Tx samples for a given patient, we applied a beta-binomial model, accounting for variance due to random sampling and incorporating time-dependent variance from a highly diverse repertoire (Rytlewski et al., 2019). Differentially expanded clonotypes after treatment (Tx-E) were further divided into emergent and pre-existing clones based on their fractions in pre-Tx and post-Tx samples. If a Tx-E clonotype could only be detected in the post-Tx sample it was defined as emergent clonotype, but if it was found in both pre- and post-Tx samples it was defined as a pre-existing clonotype. The percentage of Tx-E among total clonotypes between patients from treatment cohorts was compared to evaluate the association between treatment type and clonal expansion.
Tx-E and Tx-NE clonotypes identified from bulk TCR data were mapped to T cells in scRNA-seq datasets based on sharing of identical TRB sequences. Only clonotype with >1 sequence within a tumor were included in the Tx-NE set for mapping to avoid inclusion of naïve T cells. The fraction of mapped Tx-E cells and non-expanded (Tx-NE) within different CD4 or CD8 T cell sub-clusters was compared to identify which T cell clusters were associated with T cell expansion following ICB. Then, we used ‘FindAllMarkers’ function and Wilcoxon Rank Sum test from Seurat to identify differentially expressed genes between Tx-E and Tx-NE cells in tumor-infiltrating CD4+ and CD8+ T cells (Hao et al., 2021). To cross-validate individual genes differentially expressed between Tx-E and Tx-NE cells, we applied a pseudobulk strategy aggregating gene expression among single cells in each patient, and then used a two-sided Wilcoxon rank-sum test to evaluate statistical significance.
Evaluation of bulk TCR repertoire saturation for identification of emergent clonotypes
To evaluate whether the sequencing of TCR repertoire was sufficient to reliably distinguish emergent versus pre-existing expanded clonotypes, we performed down-sampling analysis of the tumor bulk TCR repertoire, under the assumption that some pre-existing expanded clonotypes identified from the whole TCR dataset would switch to emergent clonotypes in down-sampled datasets if the sequencing of bulk TCR dataset is not saturated. Specifically, we subsampled the 10% to 100% (with interval equal to 10%) of TCRs from the whole tumor TCR dataset for each patient and re-identified expanded pre-existing and emergent clonotypes from this down-sampled TCR dataset (Coffey, 2021). Each down-sampling step was repeated for 10 times to evaluate the variation. Then, we calculated the fraction of pre-existing clonotypes from the original pre-existing expanded clonotypes that switched to emergent clonotypes upon down-sampling. Finally, we generated a saturation curve for each patient, with the x-axis as the down sampled datasets from 10% to 100% of the whole TCR dataset, and y-axis as the fraction of remaining pre-existing clonotypes. On average, only ~4% of pre-existing expanded clonotypes switched to the emergent state in the 0.80 subsampled dataset.
Validation of Tx-E gene signatures in a published dataset
Because of the limited number of patients enrolled in this clinical trial, assessing potential statistical correlations between Tx-E associated genes and response metrics was not feasible. Furthermore, it is best practice to probe an independent validation dataset with the discovery gene signature. We utilized a published bulk RNA-seq dataset of pre-treatment biopsies from 298 metastatic urothelial cancers (mUC) patients treated with a PD-L1 mAb (Mariathasan et al., 2018), and obtained the public data from IMvigor210CoreBiologies (v1.0.0, R package).
To select genes from our CD8+Tx-E/NE gene sets to examine in the external dataset, we first filtered Tx-NE and Tx-E lists genes present in CD8 clusters 1–2 and 3, respectively, to ensure genes are well-represented in the main clusters associated with Tx-NE/Tx-E states, and not in cluster 4, which is dominated by a cell cycle signature. We then removed housekeeping genes, including ribosomal genes, mitochondrial genes, and EEF family translation machinery, to enrich for genes specific to T cells. We then performed additional filtering and removal of cell cycle genes as obtained from published signatures (Tirosh et al., 2016); this was important as the bulk RNAseq dataset contains cycling tumor cells and associated cell cycle genes that could be anti-correlated with T cell infiltration and proliferation. Finally, we determined whether the resulting gene lists were specific to T and NK cells, rather than represented in other immune lineages or in tumor itself. This step ensured that both Tx-NE and Tx-E lists equally represented T cell-specific genes, correcting for general T cell infiltrate that is known to correlate with response and outcome in bulk RNA-seq datasets. To evaluate T/NK specificity, we utilized lung SSC scRNA-seq datasets containing immune, tumor, and stromal populations (10X Genomics). We evaluated the average expression of Tx-NE/Tx-E genes among different lineages, and selected genes specifically expressed in T/NK cells. The final gene lists are as follows:
| Signature | Genes |
|---|---|
| Tx-E, pre-Tx | KLRD1, CXCL13, GNLY, KLRC2, CD7, ITGAE, GZMB, CTLA4, ZNF683, TNFRSF18, CD226, SIRPG, KLRB1, PRF1, TIGIT, LAG3 |
| Tx-NE, pre-Tx |
GZMK, CST7, EOMES, GZMM, SAMD3, LIME1, CXCR3, SH2D1A,
TRAT1, CD5 |
| TX-E, post-Tx |
GZMB, CTLA4, CXCL13, CD7, ITGAE, GZMA, GNLY, SIRPG, KLRD1,
IFNG, ZNF683 |
| Tx-NE, post-Tx | GZMK, IL7R, CST7, LEPROTL1, SAMD3, PDCD4 |
We then calculated the activity score (as described above) based on the expression profiles of these genes in this mUC dataset. We then applied a two-sided t-test to compare the statistical difference between response and non-response groups based on the activity score of these gene signatures. Finally, we divided patients into two groups based on the activity score of each gene signature (top and bottom 50%) and used the likelihood ratio test to statistically compare survival probabilities.
Sharing of TCR sequences across T cell clusters
We applied a similar strategy as previously developed (Luoma et al., 2020) to statistically evaluate the relationship between tumor T cell clusters based on sharing of TCR clonotypes across clusters. Among CD4+ cells, we identified 145 and 442 shared clonotypes (detected in at least one cell in two or more different clusters) in pre-Tx and post-Tx patient groups, respectively. In CD8+ T cells, 363 and 985 shared clonotypes were detected in pre-Tx and post-Tx patient groups, respectively. We then counted the number of shared clonotypes among different clusters and applied Fisher’s exact test on 2 × 2 cluster-by-cluster contingency tables to evaluate the significance of TCR clonotype sharing. Clonotypes shared at a frequency higher than occurring in a randomized matrix were identified as statistically significant, based on a one-sided Fisher’s exact test followed by Benjamini-Hochberg correction.
To statistically evaluate the sharing of TCR clonotypes across different clusters between pre-Tx and post-Tx datasets, we also built a 2×2 contingency table for each cluster pair across time in pre- and post-Tx samples. Briefly, we counted the number of clonotypes from each pre-Tx cluster that are shared with cells from the same or other post-Tx clusters, and then applied one-sided Fisher’s exact test followed by Benjamini-Hochberg correction to evaluate significance. Only cluster pairs with FDR<0.05 were highlighted.
Projection of sorted, activated blood T cells to total blood T cell scRNA-seq data
scRNA-seq data from sorted activated blood T cells underwent pre-processing, demultiplexing, quality control, and clustering as performed based on the above-described steps. We subsequently used similar strategies as in ProjecTILs (Andreatta et al., 2021) to project sorted, activated blood CD4 and CD8 T cells to total blood CD4 and CD8 T cells, respectively. In brief, we first recomputed PCA and UMAP embeddings by using ‘prcomp’ function from basic R package of stats and ‘umap’ function from umap (v0.2.7.0, R package), respectively. Then, pre-calculated PCA coefficients from the reference blood dataset were applied to the normalized sorted blood scRNA-seq data. Finally, the ‘predict’ function from the umap package was used to transform PCA embeddings into UMAP coordinates. After processing, the sorted blood data were embedded into the original UMAP space of the reference blood dataset, enabling joint visualization as well as classification of the sorted cells into reference cells. To confidentially calculate the percentage of projected T cells in each reference cluster, we first applied the ‘cellstate.predict’ function in ProjecTILs (v1.0.0, R package) to assign a confidence score for each cell using only selected cells with score>0.8 for evaluation. Lineage connections between sorted T cells, reference blood T cells, and bulk TCRs were established based on identical TRB sequences for different datasets.
Calculation of tumor mutation burden
The quality of the sequencing data was further assessed using several methods included in the Getz Lab CGA WES Characterization Pipeline (https://github.com/broadinstitute/CGA_Production_Analysis_Pipeline) developed at the Broad Institute. Samples were determined to be poor quality if the mean target coverage was < 25X (tumor) or < 15X (matched normal) determined by PicardMultipleMetrics (https://software.broadinstitute.org/gatk/documentation/tooldocs/4.0.0.0/picard_analysis_Collect MultipleMetrics.php), the cross-sample contamination of samples estimation ≥ 5% determined by ContEst (Cibulskis e , tumor purity ≥ 20% determined by FACETS (Shen and Seshan, 2016) and ABSOLUTE (Carter et al., 2012), and tumor-in-normal contamination ≥ 20% determined by deTiN (Taylor-Weiner et al., 2018). All pre-treatment tumor and normal samples were found to have sufficient quality.
Somatic variants were called, filtered, and annotated using a modified version of the Getz Lab CGA WES Characterization Pipeline. Somatic single-nucleotide variants were identified by MuTect1 (Cibulskis et al., 2011) and insertions and deletions (indels) were identified by both Strelka (Saunders et al., 2012) and MuTecT2 (McKenna et al., 2010). To filter false positive germline and common artifacts from somatic variant calls, variants with less than 2% allele frequency were excluded and a Panel of Normal (PoN) filter was employed (Lawrence et al., 2014). Filters were applied to exclude artifactual variants arising from oxidation of guanine to 8-Oxoguanine during sequencing, formaldehyde deamination of cytosines during DNA fixation, and DNA sequence misalignment to the reference genome (Costello et al., 2013). FFPE artifacts were filtered with an Artifact-Q cut-off value of 35 and poorly mapped reads were filtered by BLAST-like Alignment Tool (BLAT).
Tumor mutational burden was calculated as previously described (Miao et al., 2018). Briefly, the total number of mutations was divided by the number of megabases covered at a sufficient depth of coverage to detect variants with 80% power given an estimated tumor purity. Tumor purity estimates and cancer cell fractions (CCFs) were inferred from FACETS (Shen and Seshan, 2016). Clonal mutations were defined as mutations with a CCF equal to or greater than 0.8. Comparisons between clinical outcomes and mutational burden were performed using the Mann-Whitney test in R version 3.6.1.
Gene Expression Analysis of T-scan targets
Data from TCGA and Gtex were downloaded from the UCSC Xena browser (Goldman et al., 2020). To represent the expression profile of all the genes of interest in one plot, the TPM value for each gene was normalized to the maximum and minimum for each dataset where 1 represented the maximum expression for each gene dataset and 0 represented the minimum for each gene dataset. The mean of the top quartile for each tissue type was calculated to represent the highest expressing samples as the most therapeutically relevant for tumor tissue, or the tissue that pose the highest off tumor risk for normal tissues.
Supplementary Material
A: Pathological response for each patient shown in descending order and colored by response bins and cohort.
B: Tumor mutational burden (TMB) of pre-treatment samples stratified by volumetric response (n = 10, p = n.s., two-sided Mann-Whitney U test).
C: Gating strategy used for sorting of CD45+ mononuclear cells (MNC) from HNSCC tumor cell suspensions for scRNA-seq. CD66b+ neutrophils were excluded from the analysis due to poor compatibility with the 10X Genomics platform.
D: Quality metrics for CD45+ MNC dataset from tumor (left) and blood (right) specimens. Average for patients in each group with standard error of the mean are shown.
E-F: UMAP embedding of major immune cell lineages (E) and patient ID (F) (n=19 post-Tx samples and six matched pre-Tx samples).
G: UMAP embedding showing normalized expression of immune lineage marker genes.
H: Representation of major immune cell populations in scRNA-seq data from pre-tx (n=6) and post-tx (n=19) tumor samples.
I: Cell cluster frequency shown as a fraction of total cells for each patient grouped by treatment stage (six paired pre-Tx and post-Tx patients, partially paired two-sided t-test, *p<0.05).
A: Heatmap showing differentially expressed genes across tumor CD8 T cell sub-clusters. The top 10 cluster-associated genes, among highly variable genes, are shown and ranked by averaged ln (fold change) compared to all other clusters. Each row was z-score normalized.
B: Gating strategy for flow cytometric analysis of tumor CD8+ T cells.
C: Granzyme B+ CD8 T cells shown as a percentage of indicated populations determined by flow cytometry. Shown are seven patients for whom this analysis was performed (paired t-test).
D: Pseudotime trajectories of pre-treatment tumor CD8 T cells showing two potential differentiation trajectories (left); cells are colored based on tumor CD8+ T cell clusters as in Figure 2A. Heatmaps display gene expression dynamics of key marker genes (top) and inhibitor receptor genes (bottom) for trajectories 1 and 2. The colored bar plot on the top of each heatmap indicates cluster identities along the trajectory.
E: Relationship between CD8 clusters 3 and 4. Left, clusters 3 & 4 from main tumor CD8 T cell UMAP embedding were selected. Right, re-clustering and UMAP embedding of selected cells based on highly variable genes. Exclusion of cell cycle genes reveals mixing of cells originating from clusters 3 &4, indicative of shared gene programs.
F: Fraction of cells among CD8 T cell clusters that share a TCR with indicated clusters C2 (left) and C3 (right) at the post-Tx timepoint shown as a fraction of total cells in each cluster. Statistical comparisons relevant to C1-C4 TCR sharing are shown (two-sided t-test, *p < 0.05, *p < 0.0001).
G: Sharing of expanded TCR clonotypes across tumor pre- and post-treatment CD8 T cell clusters from aggregated patients (n=5 with pre/post pairs). Values indicate the -log10(FDR) of shared TCR clonotypes for each cluster pair across time, values with FDR<0.05 are labelled as 0.
H. Violin plots showing the normalized top two differentially expressed genes for tumor CD4 T cell clusters. The y-axis of violin plots represents the normalized gene expression value.
A: Number of productive bulk TCR β chain sequences for blood and tumor samples from each patient.
B: Distribution of tumor TCR clonotype size determined by bulk TCR β chain sequencing of pre-Tx and post-Tx samples. Values on the top of bar plot represent p values between pre-Tx and post-Tx samples in each category.
C: Cartoon showing different categories of tumor TCR clonotypes and examples for each of these categories with TCR frequencies (%) in tumor and blood.
D: Fraction of tumor TCR clonotypes classified as ‘blood-contaminated TCRs’ (% of TCRs higher in blood than tumor sequences) and removed from subsequent tumor Tx-E/Tx-NE analysis.
E: Scatter plots of TCR clonotype frequencies for selected patients comparing pre-Tx and post-Tx samples. TCR clonotypes with count >5 (red dotted line) were tested for differential abundance using a beta-binomial model. Orange clonotypes increased post-Tx; cyan clonotypes decreased post-Tx; changes in dark grey clones were not statistically significant.
F: Frequency of total tumor Tx-E (left) and emergent (right) TCR clonotypes shown as percentage of total clonotypes, compared between treatment cohorts (n=7 mono patients, n=8 combo patients, p values are calculated by two-sided t-test).
G: Overall assessment of clonality using tumor bulk TCR β repertoire sequencing comparing pre-Tx and post-Tx samples (left, two-sided t-test) or treatment cohorts (middle, post-Tx patient samples, two-sided t-test). Morisita similarity between pre- and post-treatment samples shown for treatment cohorts (right, two-sided t-test).
H. Downsampling of bulk TCR β datasets for indicated patients and analysis of pre-existing versus emergent TCR clonotype status. For each subsampling interval, the fraction of pre-existing clonotypes that retain their status (not re-classified as emergent due to loss of sequencing depth in pre-Tx dataset) is shown (average and SEM are shown for each subsampling interval).
I: Integration of Tx-E TCRs identified by deep TCR repertoire sequencing with scRNA-seq data. Heatmap displaying the percentage of Tx-E TCRs among tumor CD8+ T cell clusters (Figure 2A) from pre-Tx and post-Tx tumor samples for each patient. Grey color indicates absence of Tx-E TCRs in each cluster; white box indicates that no sample was available.
J: Heatmap displaying the percentage of Tx-E TCRs in pre-Tx and post-Tx samples among CD4+ T cell clusters (Figure 2H) for the indicated patients. Grey color indicates absence of Tx-E TCRs in each cluster; white boxes indicate no sample collected.
A: Ranking of differentially expressed genes between tumor CD8 Tx-E and Tx-NE cells in post-Tx samples, colored by p-value (Wilcoxon rank-sum test). Selected genes are highlighted by indicated categories.
B: Normalized expression (log-transformed) of key genes comparing tumor CD8 Tx-NE and Tx-E CD8 T cells aggregated by patient in a pseudo-bulk approach (n=13 patients, p values calculated by two-sided Wilcoxon rank-sum test).
C: Differentially expressed genes between Tx-NE or Tx-E cells shared by CD8 T cells in pre-Tx and post-Tx samples. Color bar representing averaged scaled gene expression level is shown for each gene across all cells in each group.
D: Functional enrichment analysis of hallmark gene sets comparing Tx-E cells to Tx-NE tumor CD8 T cells. NES, normalized enrichment score.
E: Heatmap showing top 20 differentially expressed genes between tumor Tx-NE and Tx-E CD4 T cells in post-Tx samples, shown as averaged scaled gene expression of all cells in each group. Marker genes belonging to indicated gene programs are colored as in S4A.
F: Normalized expression (log-transformed) of key genes comparing tumor CD4 Tx-NE and Tx-E CD8 T cells aggregated by patient in a pseudo-bulk approach (n=13 patients, p values calculated by two-sided Wilcoxon rank-sum test).
G: Association of CD8 T cell gene signatures with clinical response in an independent ICB-treated patient cohort (metastatic urothelial cancer patients treated with anti-PD-L1). General T cell, Tx-E (post-Tx), and Tx-NE (post-tx) CD8 T cell gene signatures are shown. Left: signature scores for patients with complete or partial response (CR/PR) versus stable disease or progressive disease (SD/PD), two-sided t-test. Right: overall survival (likelihood ratio test).
A. Heat map representing the mRNA expression profile for targets recognized by Tx-E TCRs. Values depicts the mean of the top quartile of normalized TPM for each indicated tissue type from TCGA (left, indicated cancer types) or GTEx (right, normal tissues).
B. Expression profile of MAGEA1. Expression of MAGEA1 is plotted for HNSCC (TCGA) or normal tissues (GTEx).
C. Heat maps illustrating TCR and HLA deconvolution. For TCR deconvolution (left), each row represents a single TCR co-cultured with non-transduced reporter cells or reporter cells transduced with MAGEA1 protein fragment; reporter activity (% IFP) was measured in triplicates (columns). For HLA deconvolution (right), each row represents a reporter cell line engineered to express a single HLA, as indicated, ± MAGEA1 protein fragment.
D. Dose-response of P32-39 and P32-41 TCRs against minimal MAGEA1 peptide epitope (VRFFFPSL) using IFNγ secretion as a readout.
E. Distribution of TCRs associated with validated targets in 5B among tumor CD8 T cell clusters.
F: UMAP embedding showing sub-clustering and annotation of tumor myeloid cells (13,060 cells, n=19 post-Tx samples, including 6 paired pre-Tx and post-Tx samples).
G: Normalized expression profiles of selected key marker genes for different myeloid clusters.
H: Comparison of the averaged cell frequency of each myeloid cell cluster between paired pre-Tx and post-Tx samples. Left, clusters labelled with red (blue) asterisk indicates significant increase (decrease) in post-Tx samples (n= 6, paired two-sided t-test, *p<0.05, **p<0.01). Right, line plots show paired pre-Tx and post-Tx samples from individual patients (n=6).
I: Comparison of tumor myeloid cell cluster frequency between treatment cohorts in tumor post-Tx samples. Average and SEM are shown for each patient group (n=10 mono patients, n=9 combo patients, two-sided t-test, * p<0.05).
J: UMAP projection of normalized expression profiles of indicated genes.
K: Expression of receptor-ligand pairs that can mediate interaction between tumor CD8 T cell clusters C3/C4 and myeloid cell clusters (top: ligand from T cells and receptor on myeloid cells; bottom: ligand from myeloid cells and receptor on T cells). Dot color reflects communication probabilities and dot size represents computed p-values (one-sided permutation test) for interactions that increased in post-treatment samples; empty space indicates no increase posttreatment.
L: Violin plots showing the normalized scRNA-seq expression distribution of selected ligand-receptor pairs for pre-Tx and post-Tx samples, compared across tumor CD8 T cell and myeloid cell clusters.
ScRNA-seq was performed for blood CD45+ immune cells at three timepoints, pretreatment (B1, n=27 samples), two-week on-treatment (B2, n=24 samples) and post-treatment (B3, n=18 samples).
A: UMAP embedding showing major CD45+ immune cell populations (left) and distribution of cells from individual patients (right) (145,584 cells, 27 patients).
B: UMAP embedding colored by patient showing the distribution of blood CD8+ T cells across clusters (left). Normalized top two differentially expressed genes in blood CD8+ T cell clusters (right). The y-axis of violin plots represents the normalized gene expression value.
C: UMAP embedding showing sub-clusters and annotations of blood CD4 T cells from all samples (33,446 cells, 27 patients).
D: Violin plots showing the normalized top two differentially expressed genes and other key marker genes for blood CD4 T cell clusters. The y-axis of violin plots represents the normalized gene expression value.
E: Comparison of CD4+ blood T cell cluster frequency across time (top) and between treatment cohorts (bottom). Average and SEM are shown for treatment timepoints (top, partially paired two-sided t-test, *p<0.05, **p<0.01, ***p<0.001) and treatment cohorts (bottom, n=14 mono patients, n=10 combo patients, two-sided t-test, *p<0.05, **p<0.01).
F: UMAP embedding showing sub-clusters and annotations of blood myeloid cells (top) and distribution of cells from each individual patient (bottom).
G: UMAP embedding colored by normalized expression profiles of selected marker genes in blood myeloid cells.
H: Comparison of myeloid blood cell cluster frequency across time (left) and between treatment cohorts (right). Average and SEM are shown for all treatment timepoints (partially paired two-sided t-test, *p<0.05, **p<0.01, ***p<0.001) and treatment cohorts (right, n=14 mono patients, n=10 combo patients, two-sided t-test, N.S p>0.05).
A: Validation of strategy for sorting blood CD8 cells reflective of cluster 6. Comparison of normalized average expression for selected markers between blood reference CD8 T cell cluster 6 (CD8-C6) and sorted blood CD8 T cells.
B: Mapping of sorted blood single-cell TCRs to tumor CD8 single-cell data (n=4 patients). Bar plots showing the fraction of cells mapped to each tumor CD8 T cell cluster.
C: Frequency of total or emergent tumor Tx-E clonotypes that could also be found in the blood, plotted as a percentage of total tumor clonotypes for both monotherapy and combination therapy cohorts. Each point represents an individual patient for whom these frequencies could be calculated (n= 7 mono, n=4 combo). Not significant by two-sided t-test (p values shown).
D: Left, representative flow cytometry plots of indicated blood CD4+ T cell populations, pre-gated on live CD4+ T cells. Right, frequency (%) of CD4+ Treg cells across timepoints. Statistical comparison between B1 and B2 is shown (n=23 B1, n=24 B2, n=19 B3, unpaired two-sided Wilcoxon rank-sum test).
E: Summary of blood T cell populations that statistically increased on-treatment (B1 versus B2, Wilcoxon rank-sum test).
F: Comparison of indicated CD8+ T cell population frequencies (%) across time and within pathological response groups. Paired samples are shown where available.
G: Flow cytometry analysis comparing markers of activated versus non-activated blood CD8+ T cells. Left, Representative flow cytometry plots at B1 timepoint showing PD-1 and KLRG1 expression gated on CD38- HLA-DR- and CD38+ HLA-DR+ populations. Right, summary of frequencies for each patient and statistical comparison for samples with ≥ 100 cells in each population (n = 14 B1, n = 17 B2, paired Wilcoxon rank-sum test, ****p<0.0001).
H: Flow cytometry analysis comparing markers of KLRG1- versus KLRG1+ cells pre-gated on PD-1+ CD8+ T cells. Top, representative flow cytometry plots at B1 timepoint showing CD38 and Ki-67 expression gated as shown. Bottom, summary of frequencies for each patient and statistical comparison for samples with ≥ 100 cells of each population (n = 11 B1, n = 15 B2, paired Wilcoxon rank-sum test, P values indicated).
I: Summary of blood T cell populations that were statistically related to degree of pathological response (low versus high response, Wilcoxon rank-sum test p<0.05).
J: Violin plots of tumor CD8+ T cell clusters, showing markers discriminating blood T cell populations associated with pathological response. The y-axis of violin plots represents the normalized gene expression value.
K: Correlation between blood T cell populations and TCR expansion in tumors. Frequency (%)of tumor Tx-E clonotypes among total tumor TCR clonotypes is plotted versus the indicated blood T cell populations measured by flow cytometry. Statistical association determined by Pearson correlation (n = 11).
Three major clinical readouts were used: clinical to pathological downstaging at time of surgery (compared to pretreatment evaluation), volumetric response based on tumor measurements along two axes as well as pathological response evaluated by histological analysis (indicated as % viable tumor post-treatment, also summarized as a tumor regression score and near complete/complete response). Radiologic assessment was attempted (RECIST response) but imaging artifacts (bone, teeth) precluded assessment in the majority of patients.
Differentially expressed genes are shown for tumor-infiltrating immune cell populations (combined samples) for the indicated datasets: CD8+ T cell clusters, CD4+ T cell clusters and myeloid cell clusters.
Genes differentially expressed between tumor Tx-E and Tx-NE T cells are shown for the indicated datasets: CD8+ T cell subset (Figure 4A, pre-Tx and post-Tx), , and CD4+ T cell subset (Figure 4E). The top 20 DE genes, and additional key genes, identified by single-cell methods were additionally validated by pseudo-bulk method as indicated.
Gene signatures used to calculate activity scores in Tx-E/NE T cells (Figure 4C-D) and for assessing response and survival in an independent cohort of patients with urothelial cancer treated with a PD-L1 mAb (Figure 4F, S4F).
Total numbers of TCRs screened and HLA alleles screened for each patient.
Differentially expressed genes are shown for blood cell immune cell clusters (combined samples) for the indicated datasets: CD8+ T cell clusters, CD4+ T cell clusters, myeloid cell clusters, and sorted CD38+HLA-DR+ blood CD8+ T cells.
Median frequencies and P values of blood T cell populations that were significantly different between timepoints or response groups.
Highlights.
Neoadjuvant ICB induces local and systemic T cell responses in oral cancer patients
Treatment-responsive T cell clones recognize self-antigens including tumor antigens
Responding T cells in tumors display a tissue-resident memory program
Circulating PD-1+KLRG1-CD8 T cell frequency correlates with pathological response
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, Cormier JN, Lewis C, Hwu WJ, Hanna E, et al. (2018). Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med 24, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M, Corria-Osorio J, Muller S, Cubas R, Coukos G, and Carmona SJ (2021). Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat Commun 12, 2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D., Looney AP., Liu L., Wu E., Fong V., Hs A., Chak S., Naikawadi RP., Wolters PJ., Abate AR., et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau R, Chitre AS, Scherl A, Wu TD, Patil NS, de Almeida P, Kadel Iii EE, Madireddi S, Au-Yeung A, Takahashi C, et al. (2021). Intratumoral CD103+ CD8+ T cells predict response to PD-L1 blockade. J Immunother Cancer 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassez A, Vos H, Van Dyck L, Floris G, Arijs I, Desmedt C, Boeckx B, Vanden Bempt M, Nevelsteen I, Lambein K, et al. (2021). A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat Med 27, 820–832. [DOI] [PubMed] [Google Scholar]
- Baumeister SH, Freeman GJ, Dranoff G, and Sharpe AH (2016). Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol 34, 539–573. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. (2019). Defining ‘T cell exhaustion’. Nat Rev Immunol 19, 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank CU, Rozeman EA, Fanchi LF, Sikorska K, van de Wiel B, Kvistborg P, Krijgsman O, van den Braber M, Philips D, Broeks A, et al. (2018). Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med 24, 1655–1661. [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EH, Mog BJ, Hou W, Justesen S, Blosser R, et al. (2021). Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 596, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, Lopez-Yurda M, Grootscholten C, Beets GL, Snaebjornsson P, et al. (2020). Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 26, 566–576. [DOI] [PubMed] [Google Scholar]
- Cibulskis K, McKenna A, Fennell T, Banks E, DePristo M, and Getz G (2011). ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 27, 2601–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, Chee SJ, Eschweiler S, King EV, Awad AS, et al. (2019). Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med 216, 2128–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey D (2021). LymphoSeq: Analyze high-throughput sequencing of T and B cell receptors. R package version 3.13. [Google Scholar]
- Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, et al. (2013). Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res 41, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, et al. (2018). Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 9, 2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom C, Pfirschke C, and Pittet MJ (2016). The role of myeloid cells in cancer therapies. Nat Rev Cancer 16, 447–462. [DOI] [PubMed] [Google Scholar]
- Fairfax BP., Taylor CA., Watson RA., Nassiri I., Danielli S., Fang H., Mahe EA., Cooper R., Woodcock V., Traill Z., et al. (2020). Peripheral CD8(+) T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat Med 26, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarotto R, Bell D, Rubin ML, Hutcheson KA, Johnson JM, Goepfert RP, Phan J, Elamin YY, Torman DK, Warneke CL, et al. (2020). Impact of Neoadjuvant Durvalumab with or without Tremelimumab on CD8(+) Tumor Lymphocyte Density, Safety, and Efficacy in Patients with Oropharynx Cancer: CIAO Trial Results. Clin Cancer Res 26, 3211–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti AP, Kula T, Wang Y, Nguyen DMV, Weinheimer A, Dunlap GS, Xu Q, Nabilsi N, Perullo CR, Cristofaro AW, et al. (2020). Unbiased Screens Show CD8(+) T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity 53, 1095–1107 e1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris RL, Spanos WC, Leidner R, Goncalves A, Martens UM, Kyi C, Sharfman W, Chung CH, Devriese LA, Gauthier H, et al. (2021). Neoadjuvant nivolumab for patients with resectable HPV-positive and HPV-negative squamous cell carcinomas of the head and neck in the CheckMate 358 trial. J Immunother Cancer 9, e002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo DL, Mamede RC, Proto-Siqueira R, Neder L, Silva WA Jr., and Zago MA (2006). Expression of cancer testis antigens in head and neck squamous cell carcinomas. Head Neck 28, 614–619. [DOI] [PubMed] [Google Scholar]
- Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Zahurak M, Yang SC, Jones DR, Broderick S, et al. (2018). Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med 378, 1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, Awad MM, Felip E, Broderick SR, Brahmer JR, Swanson SJ, et al. (2022). Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N Engl J Med 386, 1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen MF, Schoonderwoerd M, Knopf P, Camps MG, Hawinkels LJ, Kneilling M, van Hall T, and Ossendorp F (2018). Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 3, e124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Moore EC, Zolkind P, Robbins Y, Clavijo PE, Sun L, Greene S, Morisada MV, Mydlarz WK, Schmitt N, et al. (2020). Neoadjuvant PD-1 Immune Checkpoint Blockade Reverses Functional Immunodominance among Tumor Antigen-Specific T Cells. Clin Cancer Res 26, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, et al. (2017). Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol 18, 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. (2020). Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38, 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguen P., Metoikidou C., Dupic T., Lawand M., Goudot C., Baulande S., Lameiras S., Lantz O., Girard N., Seguin-Givelet A., et al. (2021). Contribution of resident and circulating precursors to tumor-infiltrating CD8(+) T cell populations in lung cancer. Sci Immunol 6, eabd5778. [DOI] [PubMed] [Google Scholar]
- Guo B, and Yuan Y (2017). A comparative review of methods for comparing means using partially paired data. Stat Methods Med Res 26, 1323–1340. [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornburg M, Desbois M, Lu S, Guan Y, Lo AA, Kaufman S, Elrod A, Lotstein A, DesRochers TM, Munoz-Rodriguez JL, et al. (2021). Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 39, 928–944 e926. [DOI] [PubMed] [Google Scholar]
- Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, Manne S, Kraya AA, Wubbenhorst B, Dorfman L, et al. (2019). A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 25, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat Commun 12, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. (2017). Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A 114, 4993–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khunger A, Rytlewski JA, Fields P, Yusko EC, and Tarhini AA (2019). The impact of CTLA-4 blockade and interferon-alpha on clonality of T-cell repertoire in the tumor microenvironment and peripheral blood of metastatic melanoma patients. Oncoimmunology 8, e1652538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. bioRxiv, 060012. [Google Scholar]
- Krishna C., DiNatale RG., Kuo F., Srivastava RM., Vuong L., Chowell D., Gupta S., Vanderbilt C., Purohit TA., Liu M., et al. (2021). Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 39, 662–677 e666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kula T, Dezfulian MH, Wang CI, Abdelfattah NS, Hartman ZC, Wucherpfennig KW, Lyerly HK, and Elledge SJ (2019). T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 178, 1016–1028 e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, et al. (2017). Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep 20, 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, Welters MJ, van der Burg S, Kapiteijn E, Michielin O, et al. (2014). Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Science translational medicine 6, 254ra128. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Floc’h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, Chouaib S, and Mami-Chouaib F (2007). Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med 204, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, and Brady MS (2021). Neoadjuvant immunotherapy for melanoma. J Surg Oncol 123, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Gould J, Yang Y, Sarkizova S, Tabaka M, Ashenberg O, Rosen Y, Slyper M, Kowalczyk MS, Villani AC, et al. (2020). Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq. Nat Methods 17, 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licitra L, Grandi C, Guzzo M, Mariani L, Lo Vullo S, Valvo F, Quattrone P, Valagussa P, Bonadonna G, Molinari R, et al. (2003). Primary chemotherapy in resectable oral cavity squamous cell cancer: a randomized controlled trial. J Clin Oncol 21, 327–333. [DOI] [PubMed] [Google Scholar]
- Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K, Young A, O’Donnell JS, Allen S, Smyth MJ, et al. (2016). Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov 6, 1382–1399. [DOI] [PubMed] [Google Scholar]
- Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, participants in the 1st Human Cell Atlas, J., and Marioni JC (2019). EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol 20, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoma AM., Suo S., Williams HL., Sharova T., Sullivan K., Manos M., Bowling P., Hodi FS., Rahma O., Sullivan RJ., et al. (2020). Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 182, 655–671 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. (2013). The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14, 1294–1301. [DOI] [PubMed] [Google Scholar]
- Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, et al. (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mami-Chouaib F, Blanc C, Corgnac S, Hans S, Malenica I, Granier C, Tihy I, and Tartour E (2018). Resident memory T cells, critical components in tumor immunology. J Immunother Cancer 6, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathewson ND, Ashenberg O, Tirosh I, Gritsch S, Perez EM, Marx S, Jerby-Arnon L, Chanoch-Myers R, Hara T, Richman AR, et al. (2021). Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184, 1281–1298 e1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies AM, Amaria RN, Rozeman EA, Huang AC, Tetzlaff MT, van de Wiel BA, Lo S, Tarhini AA, Burton EM, Pennington TE, et al. (2021). Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat Med 27, 301–309. [DOI] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Vokes NI, Liu D, Taylor-Weiner A, Wankowicz SM, Adeegbe D, Keliher D, Schilling B, Tracy A, et al. (2018). Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet 50, 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarov V.i.b.R., E. (2020). immunarch: An R Package for Painless Bioinformatics Analysis of T-Cell and B-Cell Immune Repertoires. Zenodo, 10.5281/zenodo.3367200. [DOI] [Google Scholar]
- Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, Forman J, Huang T, Li S, Zhang W, et al. (2021). Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 596, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SL, Gebhardt T, and Mackay LK (2019). Tissue-Resident Memory T Cells in Cancer Immunosurveillance. Trends in immunology 40, 735–747. [DOI] [PubMed] [Google Scholar]
- Philip M, and Schietinger A (2021). CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijuan-Sala B, Griffiths JA, Guibentif C, Hiscock TW, Jawaid W, Calero-Nieto FJ, Mulas C, Ibarra-Soria X, Tyser RCV, Ho DLL, et al. (2019). A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytlewski J., Deng S., Xie T., Davis C., Robins H., Yusko E., and Bienkowska J. (2019). Model to improve specificity for identification of clinically-relevant expanded T cells in peripheral blood. PLoS One 14, e0213684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, et al. (2018). Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 24, 986–993. [DOI] [PubMed] [Google Scholar]
- Schoenfeld JD, Hanna GJ, Jo VY, Rawal B, Chen YH, Catalano PS, Lako A, Ciantra Z, Weirather JL, Criscitiello S, et al. (2020). Neoadjuvant Nivolumab or Nivolumab Plus Ipilimumab in Untreated Oral Cavity Squamous Cell Carcinoma: A Phase 2 Open-Label Randomized Clinical Trial. JAMA Oncol 6, 1563–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtalbers J, Boegel S, Bukur T, Byl M, Goerges S, Sorn P, Loewer M, Sahin U, and Castle JC (2015). TCLP: an online cancer cell line catalogue integrating HLA type, predicted neo-epitopes, virus and gene expression. Genome Med 7, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Shen R, and Seshan VE (2016). FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 44, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2016). A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 166, 1500–1511 e1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, Cheng YL, Bush EC, Dogra P, Thapa P, et al. (2019a). Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nature communications 10, 4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo PA, Miron M, and Farber DL (2019b). Location, location, location: Tissue resident memory T cells in mice and humans. Sci Immunol 4, eaas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Weiner A., Stewart C., Giordano T., Miller M., Rosenberg M., Macbeth A., Lennon N., Rheinbay E., Landau DA., Wu CJ., et al. (2018). DeTiN: overcoming tumor-in-normal contamination. Nat Methods 15, 531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Taube JM, and Pardoll DM (2020). Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 367, eaax0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppaluri R, Campbell KM, Egloff AM, Zolkind P, Skidmore ZL, Nussenbaum B, Paniello RC, Rich JT, Jackson R, Pipkorn P, et al. (2020). Neoadjuvant and Adjuvant Pembrolizumab in Resectable Locally Advanced, Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicenter, Phase II Trial. Clin Cancer Res 26, 5140–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berge K, Roux de Bezieux H, Street K, Saelens W, Cannoodt R, Saeys Y, Dudoit S, and Clement L (2020). Trajectory-based differential expression analysis for single-cell sequencing data. Nat Commun 11, 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Leun AM, Thommen DS, and Schumacher TN (2020). CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer 20, 218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versluis JM, Long GV, and Blank CU (2020). Learning from clinical trials of neoadjuvant checkpoint blockade. Nat Med 26, 475–484. [DOI] [PubMed] [Google Scholar]
- Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, Chiang EY, Iftikhar H, O’Gorman WE, Au-Yeung A, et al. (2020). Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278. [DOI] [PubMed] [Google Scholar]
- Xin H, Lian Q, Jiang Y, Luo J, Wang X, Erb C, Xu Z, Zhang X, Heidrich-O’Hare E, Yan Q, et al. (2020). GMM-Demux: sample demultiplexing, multiplet detection, experiment planning, and novel cell-type verification in single cell sequencing. Genome Biol 21, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Pathological response for each patient shown in descending order and colored by response bins and cohort.
B: Tumor mutational burden (TMB) of pre-treatment samples stratified by volumetric response (n = 10, p = n.s., two-sided Mann-Whitney U test).
C: Gating strategy used for sorting of CD45+ mononuclear cells (MNC) from HNSCC tumor cell suspensions for scRNA-seq. CD66b+ neutrophils were excluded from the analysis due to poor compatibility with the 10X Genomics platform.
D: Quality metrics for CD45+ MNC dataset from tumor (left) and blood (right) specimens. Average for patients in each group with standard error of the mean are shown.
E-F: UMAP embedding of major immune cell lineages (E) and patient ID (F) (n=19 post-Tx samples and six matched pre-Tx samples).
G: UMAP embedding showing normalized expression of immune lineage marker genes.
H: Representation of major immune cell populations in scRNA-seq data from pre-tx (n=6) and post-tx (n=19) tumor samples.
I: Cell cluster frequency shown as a fraction of total cells for each patient grouped by treatment stage (six paired pre-Tx and post-Tx patients, partially paired two-sided t-test, *p<0.05).
A: Heatmap showing differentially expressed genes across tumor CD8 T cell sub-clusters. The top 10 cluster-associated genes, among highly variable genes, are shown and ranked by averaged ln (fold change) compared to all other clusters. Each row was z-score normalized.
B: Gating strategy for flow cytometric analysis of tumor CD8+ T cells.
C: Granzyme B+ CD8 T cells shown as a percentage of indicated populations determined by flow cytometry. Shown are seven patients for whom this analysis was performed (paired t-test).
D: Pseudotime trajectories of pre-treatment tumor CD8 T cells showing two potential differentiation trajectories (left); cells are colored based on tumor CD8+ T cell clusters as in Figure 2A. Heatmaps display gene expression dynamics of key marker genes (top) and inhibitor receptor genes (bottom) for trajectories 1 and 2. The colored bar plot on the top of each heatmap indicates cluster identities along the trajectory.
E: Relationship between CD8 clusters 3 and 4. Left, clusters 3 & 4 from main tumor CD8 T cell UMAP embedding were selected. Right, re-clustering and UMAP embedding of selected cells based on highly variable genes. Exclusion of cell cycle genes reveals mixing of cells originating from clusters 3 &4, indicative of shared gene programs.
F: Fraction of cells among CD8 T cell clusters that share a TCR with indicated clusters C2 (left) and C3 (right) at the post-Tx timepoint shown as a fraction of total cells in each cluster. Statistical comparisons relevant to C1-C4 TCR sharing are shown (two-sided t-test, *p < 0.05, *p < 0.0001).
G: Sharing of expanded TCR clonotypes across tumor pre- and post-treatment CD8 T cell clusters from aggregated patients (n=5 with pre/post pairs). Values indicate the -log10(FDR) of shared TCR clonotypes for each cluster pair across time, values with FDR<0.05 are labelled as 0.
H. Violin plots showing the normalized top two differentially expressed genes for tumor CD4 T cell clusters. The y-axis of violin plots represents the normalized gene expression value.
A: Number of productive bulk TCR β chain sequences for blood and tumor samples from each patient.
B: Distribution of tumor TCR clonotype size determined by bulk TCR β chain sequencing of pre-Tx and post-Tx samples. Values on the top of bar plot represent p values between pre-Tx and post-Tx samples in each category.
C: Cartoon showing different categories of tumor TCR clonotypes and examples for each of these categories with TCR frequencies (%) in tumor and blood.
D: Fraction of tumor TCR clonotypes classified as ‘blood-contaminated TCRs’ (% of TCRs higher in blood than tumor sequences) and removed from subsequent tumor Tx-E/Tx-NE analysis.
E: Scatter plots of TCR clonotype frequencies for selected patients comparing pre-Tx and post-Tx samples. TCR clonotypes with count >5 (red dotted line) were tested for differential abundance using a beta-binomial model. Orange clonotypes increased post-Tx; cyan clonotypes decreased post-Tx; changes in dark grey clones were not statistically significant.
F: Frequency of total tumor Tx-E (left) and emergent (right) TCR clonotypes shown as percentage of total clonotypes, compared between treatment cohorts (n=7 mono patients, n=8 combo patients, p values are calculated by two-sided t-test).
G: Overall assessment of clonality using tumor bulk TCR β repertoire sequencing comparing pre-Tx and post-Tx samples (left, two-sided t-test) or treatment cohorts (middle, post-Tx patient samples, two-sided t-test). Morisita similarity between pre- and post-treatment samples shown for treatment cohorts (right, two-sided t-test).
H. Downsampling of bulk TCR β datasets for indicated patients and analysis of pre-existing versus emergent TCR clonotype status. For each subsampling interval, the fraction of pre-existing clonotypes that retain their status (not re-classified as emergent due to loss of sequencing depth in pre-Tx dataset) is shown (average and SEM are shown for each subsampling interval).
I: Integration of Tx-E TCRs identified by deep TCR repertoire sequencing with scRNA-seq data. Heatmap displaying the percentage of Tx-E TCRs among tumor CD8+ T cell clusters (Figure 2A) from pre-Tx and post-Tx tumor samples for each patient. Grey color indicates absence of Tx-E TCRs in each cluster; white box indicates that no sample was available.
J: Heatmap displaying the percentage of Tx-E TCRs in pre-Tx and post-Tx samples among CD4+ T cell clusters (Figure 2H) for the indicated patients. Grey color indicates absence of Tx-E TCRs in each cluster; white boxes indicate no sample collected.
A: Ranking of differentially expressed genes between tumor CD8 Tx-E and Tx-NE cells in post-Tx samples, colored by p-value (Wilcoxon rank-sum test). Selected genes are highlighted by indicated categories.
B: Normalized expression (log-transformed) of key genes comparing tumor CD8 Tx-NE and Tx-E CD8 T cells aggregated by patient in a pseudo-bulk approach (n=13 patients, p values calculated by two-sided Wilcoxon rank-sum test).
C: Differentially expressed genes between Tx-NE or Tx-E cells shared by CD8 T cells in pre-Tx and post-Tx samples. Color bar representing averaged scaled gene expression level is shown for each gene across all cells in each group.
D: Functional enrichment analysis of hallmark gene sets comparing Tx-E cells to Tx-NE tumor CD8 T cells. NES, normalized enrichment score.
E: Heatmap showing top 20 differentially expressed genes between tumor Tx-NE and Tx-E CD4 T cells in post-Tx samples, shown as averaged scaled gene expression of all cells in each group. Marker genes belonging to indicated gene programs are colored as in S4A.
F: Normalized expression (log-transformed) of key genes comparing tumor CD4 Tx-NE and Tx-E CD8 T cells aggregated by patient in a pseudo-bulk approach (n=13 patients, p values calculated by two-sided Wilcoxon rank-sum test).
G: Association of CD8 T cell gene signatures with clinical response in an independent ICB-treated patient cohort (metastatic urothelial cancer patients treated with anti-PD-L1). General T cell, Tx-E (post-Tx), and Tx-NE (post-tx) CD8 T cell gene signatures are shown. Left: signature scores for patients with complete or partial response (CR/PR) versus stable disease or progressive disease (SD/PD), two-sided t-test. Right: overall survival (likelihood ratio test).
A. Heat map representing the mRNA expression profile for targets recognized by Tx-E TCRs. Values depicts the mean of the top quartile of normalized TPM for each indicated tissue type from TCGA (left, indicated cancer types) or GTEx (right, normal tissues).
B. Expression profile of MAGEA1. Expression of MAGEA1 is plotted for HNSCC (TCGA) or normal tissues (GTEx).
C. Heat maps illustrating TCR and HLA deconvolution. For TCR deconvolution (left), each row represents a single TCR co-cultured with non-transduced reporter cells or reporter cells transduced with MAGEA1 protein fragment; reporter activity (% IFP) was measured in triplicates (columns). For HLA deconvolution (right), each row represents a reporter cell line engineered to express a single HLA, as indicated, ± MAGEA1 protein fragment.
D. Dose-response of P32-39 and P32-41 TCRs against minimal MAGEA1 peptide epitope (VRFFFPSL) using IFNγ secretion as a readout.
E. Distribution of TCRs associated with validated targets in 5B among tumor CD8 T cell clusters.
F: UMAP embedding showing sub-clustering and annotation of tumor myeloid cells (13,060 cells, n=19 post-Tx samples, including 6 paired pre-Tx and post-Tx samples).
G: Normalized expression profiles of selected key marker genes for different myeloid clusters.
H: Comparison of the averaged cell frequency of each myeloid cell cluster between paired pre-Tx and post-Tx samples. Left, clusters labelled with red (blue) asterisk indicates significant increase (decrease) in post-Tx samples (n= 6, paired two-sided t-test, *p<0.05, **p<0.01). Right, line plots show paired pre-Tx and post-Tx samples from individual patients (n=6).
I: Comparison of tumor myeloid cell cluster frequency between treatment cohorts in tumor post-Tx samples. Average and SEM are shown for each patient group (n=10 mono patients, n=9 combo patients, two-sided t-test, * p<0.05).
J: UMAP projection of normalized expression profiles of indicated genes.
K: Expression of receptor-ligand pairs that can mediate interaction between tumor CD8 T cell clusters C3/C4 and myeloid cell clusters (top: ligand from T cells and receptor on myeloid cells; bottom: ligand from myeloid cells and receptor on T cells). Dot color reflects communication probabilities and dot size represents computed p-values (one-sided permutation test) for interactions that increased in post-treatment samples; empty space indicates no increase posttreatment.
L: Violin plots showing the normalized scRNA-seq expression distribution of selected ligand-receptor pairs for pre-Tx and post-Tx samples, compared across tumor CD8 T cell and myeloid cell clusters.
ScRNA-seq was performed for blood CD45+ immune cells at three timepoints, pretreatment (B1, n=27 samples), two-week on-treatment (B2, n=24 samples) and post-treatment (B3, n=18 samples).
A: UMAP embedding showing major CD45+ immune cell populations (left) and distribution of cells from individual patients (right) (145,584 cells, 27 patients).
B: UMAP embedding colored by patient showing the distribution of blood CD8+ T cells across clusters (left). Normalized top two differentially expressed genes in blood CD8+ T cell clusters (right). The y-axis of violin plots represents the normalized gene expression value.
C: UMAP embedding showing sub-clusters and annotations of blood CD4 T cells from all samples (33,446 cells, 27 patients).
D: Violin plots showing the normalized top two differentially expressed genes and other key marker genes for blood CD4 T cell clusters. The y-axis of violin plots represents the normalized gene expression value.
E: Comparison of CD4+ blood T cell cluster frequency across time (top) and between treatment cohorts (bottom). Average and SEM are shown for treatment timepoints (top, partially paired two-sided t-test, *p<0.05, **p<0.01, ***p<0.001) and treatment cohorts (bottom, n=14 mono patients, n=10 combo patients, two-sided t-test, *p<0.05, **p<0.01).
F: UMAP embedding showing sub-clusters and annotations of blood myeloid cells (top) and distribution of cells from each individual patient (bottom).
G: UMAP embedding colored by normalized expression profiles of selected marker genes in blood myeloid cells.
H: Comparison of myeloid blood cell cluster frequency across time (left) and between treatment cohorts (right). Average and SEM are shown for all treatment timepoints (partially paired two-sided t-test, *p<0.05, **p<0.01, ***p<0.001) and treatment cohorts (right, n=14 mono patients, n=10 combo patients, two-sided t-test, N.S p>0.05).
A: Validation of strategy for sorting blood CD8 cells reflective of cluster 6. Comparison of normalized average expression for selected markers between blood reference CD8 T cell cluster 6 (CD8-C6) and sorted blood CD8 T cells.
B: Mapping of sorted blood single-cell TCRs to tumor CD8 single-cell data (n=4 patients). Bar plots showing the fraction of cells mapped to each tumor CD8 T cell cluster.
C: Frequency of total or emergent tumor Tx-E clonotypes that could also be found in the blood, plotted as a percentage of total tumor clonotypes for both monotherapy and combination therapy cohorts. Each point represents an individual patient for whom these frequencies could be calculated (n= 7 mono, n=4 combo). Not significant by two-sided t-test (p values shown).
D: Left, representative flow cytometry plots of indicated blood CD4+ T cell populations, pre-gated on live CD4+ T cells. Right, frequency (%) of CD4+ Treg cells across timepoints. Statistical comparison between B1 and B2 is shown (n=23 B1, n=24 B2, n=19 B3, unpaired two-sided Wilcoxon rank-sum test).
E: Summary of blood T cell populations that statistically increased on-treatment (B1 versus B2, Wilcoxon rank-sum test).
F: Comparison of indicated CD8+ T cell population frequencies (%) across time and within pathological response groups. Paired samples are shown where available.
G: Flow cytometry analysis comparing markers of activated versus non-activated blood CD8+ T cells. Left, Representative flow cytometry plots at B1 timepoint showing PD-1 and KLRG1 expression gated on CD38- HLA-DR- and CD38+ HLA-DR+ populations. Right, summary of frequencies for each patient and statistical comparison for samples with ≥ 100 cells in each population (n = 14 B1, n = 17 B2, paired Wilcoxon rank-sum test, ****p<0.0001).
H: Flow cytometry analysis comparing markers of KLRG1- versus KLRG1+ cells pre-gated on PD-1+ CD8+ T cells. Top, representative flow cytometry plots at B1 timepoint showing CD38 and Ki-67 expression gated as shown. Bottom, summary of frequencies for each patient and statistical comparison for samples with ≥ 100 cells of each population (n = 11 B1, n = 15 B2, paired Wilcoxon rank-sum test, P values indicated).
I: Summary of blood T cell populations that were statistically related to degree of pathological response (low versus high response, Wilcoxon rank-sum test p<0.05).
J: Violin plots of tumor CD8+ T cell clusters, showing markers discriminating blood T cell populations associated with pathological response. The y-axis of violin plots represents the normalized gene expression value.
K: Correlation between blood T cell populations and TCR expansion in tumors. Frequency (%)of tumor Tx-E clonotypes among total tumor TCR clonotypes is plotted versus the indicated blood T cell populations measured by flow cytometry. Statistical association determined by Pearson correlation (n = 11).
Three major clinical readouts were used: clinical to pathological downstaging at time of surgery (compared to pretreatment evaluation), volumetric response based on tumor measurements along two axes as well as pathological response evaluated by histological analysis (indicated as % viable tumor post-treatment, also summarized as a tumor regression score and near complete/complete response). Radiologic assessment was attempted (RECIST response) but imaging artifacts (bone, teeth) precluded assessment in the majority of patients.
Differentially expressed genes are shown for tumor-infiltrating immune cell populations (combined samples) for the indicated datasets: CD8+ T cell clusters, CD4+ T cell clusters and myeloid cell clusters.
Genes differentially expressed between tumor Tx-E and Tx-NE T cells are shown for the indicated datasets: CD8+ T cell subset (Figure 4A, pre-Tx and post-Tx), , and CD4+ T cell subset (Figure 4E). The top 20 DE genes, and additional key genes, identified by single-cell methods were additionally validated by pseudo-bulk method as indicated.
Gene signatures used to calculate activity scores in Tx-E/NE T cells (Figure 4C-D) and for assessing response and survival in an independent cohort of patients with urothelial cancer treated with a PD-L1 mAb (Figure 4F, S4F).
Total numbers of TCRs screened and HLA alleles screened for each patient.
Differentially expressed genes are shown for blood cell immune cell clusters (combined samples) for the indicated datasets: CD8+ T cell clusters, CD4+ T cell clusters, myeloid cell clusters, and sorted CD38+HLA-DR+ blood CD8+ T cells.
Median frequencies and P values of blood T cell populations that were significantly different between timepoints or response groups.
Data Availability Statement
Processed, de-identified scRNA-seq gene expression, TCR, and bulk TCR datasets generated during this study are publicly available at the Gene Expression Omnibus (GEO) as of the date of publication. The accession number for these datasets is listed in the key resources table. The raw scRNA-seq gene expression sequencing data cannot be deposited in a public repository because of restrictions in informed consent. Raw data may be requested from kai_wucherpfennig@dfci.harvard.edu with appropriate institutional approvals. This paper analyzes existing, publicly available data. The accession number for this dataset is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human TruStain FcX™ (Fc Receptor Blocking Solution) | Biolegend | Cat# 422302; RRID:AB_2818986 |
| Anti-human CD3 PE-Dazzle 594 | Biolegend | Cat#300450; RRID:AB_2563618 |
| Anti-human CD45 Pacific Blue | Biolegend | Cat#304022 RRID:AB_493655 |
| Anti-human CD66b PE-Cy7 | Biolegend | Cat#305116; RRID:AB_2566605 |
| Anti-human CD15 APC | Biolegend | Cat#301908; RRID:AB_314200 |
| TotalSeq™-C0251 anti-human Hashtag 1 Antibody | Biolegend | Cat# 394661; RRID:AB_2801031 |
| TotalSeq™-C0252 anti-human Hashtag 2 Antibody | Biolegend | Cat#394663; RRID:AB_2801032 |
| TotalSeq™-C0253 anti-human Hashtag 3 Antibody | Biolegend | Cat#394665; RRID:AB_2801033 |
| TotalSeq™-C0254 anti-human Hashtag 4 Antibody | Biolegend | Cat#394667; RRID:AB_2801034 |
| TotalSeq™-C0255 anti-human Hashtag 5 Antibody | Biolegend | Cat#394669; RRID:AB_2801035 |
| TotalSeq™-C0256 anti-human Hashtag 6 Antibody | Biolegend | Cat#394671; RRID:AB_2820042 |
| TotalSeq™-C0257 anti-human Hashtag 7 Antibody | Biolegend | Cat#394673; RRID:AB_2820043 |
| Anti-human CXCR3 BV421 | Biolegend | Cat#353716; RRID:AB_2561448 |
| Anti-human CD8a BV570 | Biolegend | Cat#301038; RRID:AB_2563213 |
| Anti-human CD4 BV650 | Biolegend | Cat#317436; RRID:AB_2563050 |
| Anti-human CD3e BV785 | Biolegend | Cat#300472; RRID:AB_2687178 |
| Anti-human CD38 Alexa Fluor 488 | Biolegend | Cat#356634; RRID:AB_2734381 |
| Anti-human CD27 PerCP-Cy5.5 | Biolegend | Cat#356407; RRID:AB_2561906 |
| Anti-human HLA-DR PE/Dazzle594 | Biolegend | Cat#307654; RRID:AB_2563646 |
| Anti-human KLRG1 PE/Cy7 | Invitrogen | Cat#25-9488-42; RRID:AB_2573546 |
| Anti-human PD-1 APC | Biolegend | Cat#329908; RRID:AB_940475 |
| Anti-human γδ TCR BV421 | Biolegend | Cat#331218; RRID:AB_2562317 |
| Anti-human CD59 V450 | BD | Cat#560740; RRID:AB_1727512 |
| Anti-human HLA-DR BV570 | Biolegend | Cat#307638; RRID:AB_2650882 |
| Anti-human CD25 BV605 | Biolegend | Cat#302632; RRID:AB_11218989 |
| Anti-human CD103 BV785 | Biolegend | Cat#350230; RRID:AB_2734364 |
| Anti-human CD8a Alexa Fluor 488 | Biolegend | Cat#300916; RRID:AB_756152 |
| Anti-human CD3 Alexa Fluor 532 | eBioscience | Cat#58-0038-42; RRID:AB_11218675 |
| Anti-human PD-1 PE | eBioscience | Cat#12-9969-42; RRID:AB_10736473 |
| Anti-human CD45 PE/Cy7 | Biolegend | Cat#304010; RRID:AB_314398 |
| Anti-human CD45 Per-CpVio700 | Miltenyi | Cat#130-097-527; RRID:AB_2660425 |
| Anti-human CTLA-4 PE/Cy7 | Biolegend | Cat#349914; RRID:AB_2563098 |
| Anti-human CD4 Alexa Fluor 700 | eBioscience | Cat#56-0049-42; RRID:AB_11219085 |
| Anti-human GranzymeB PE/CF594 | BD | Cat#562462; RRID:AB_2737618 |
| Anti-human FOXP3 APC | eBioscience | Cat#17-4777-42 |
| Anti-human IL-7R BV421 | Biolegend | Cat#351310; RRID:AB_10960140 |
| Anti-human CD45RO Pacific Blue | Biolegend | Cat#304216; RRID:AB_493659 |
| Anti-human HLA-DR BV510 | Biolegend | Cat#307646; RRID:AB_2561948 |
| Anti-human CXCR3 BV605 | Biolegend | Cat#353728; RRID:AB_2563157 |
| Anti-human CD4 BV711 | Biolegend | Cat#317440; RRID:AB_2562912 |
| Anti-human PD-1 PE | Biolegend | Cat#329906; RRID:AB_940483 |
| Anti-human IgG4 Fc PE | Southern Biotech | Cat#9200-09; RRID:AB_2796693 |
| Anti-human KLRG1 Alexa Fluor 647 | Biolegend | Cat#367704; RRID:AB_2565981 |
| Anti-human FOXP3 PE/CF594 | BD | Cat#563955; RRID:AB_2738507 |
| Anti-human Ki-67 PerCp/Cy5.5 | Biolegend | Cat#350520; RRID:AB_2562295 |
| Anti-human/mouse GranzymeB PE/Cy7 | Biolegend | Cat#396410; RRID:AB_2801079 |
| Anti-mouse TCRβ biotin | Biolegend | Cat#109204; RRID:AB_313427 |
| Anti-human CD3e | eBioscience | Cat# 16-0037-85; RRID:AB_468855 |
| Biological samples | ||
| Fresh tumor tissue biopsies or surgical resections | Dana-Farber Cancer Institute/Brigham and Women’s Hospital | IRB# DC/HCC 16-284 |
| Paraffin-embedded formalin fixed tumor slides, prepared from tumor biopsies or tumor resections | Dana-Farber Cancer Institute/Brigham and Women’s Hospital | IRB# DC/HCC 16-284 |
| Peripheral blood mononuclear cells, prepared from whole blood draws | Dana-Farber Cancer Institute |
IRB# DC/HCC 16-284 |
| Chemicals, peptides, and recombinant proteins | ||
| ACK red blood cell lysis buffer | Thermo Fisher Scientific |
Cat#A1049201 |
| Ultrapure Bovine Serum Albumin, RNase-free | Thermo Fisher Scientific |
Cat#AM2616 |
| Recombinant Human IL-2 | Peprotech | Cat#200-02 |
| Recombinant Human IL-7 | R&D Systems | Cat#207-IL-025 |
| Recombinant Human IL-15 | R&D Systems | Cat#247-ILB-025 |
| DMEM | ThermoFisher Scientific |
Cat#11995073 |
| RPMI 1640 | ThermoFisher Scientific |
Cat#21875034 |
| ACK Lysing Buffer | ThermoFisher Scientific |
Cat#A1049201 |
| Critical commercial assays | ||
| 10X Chromium Single Cell 5′ Library & Gel Bead Kit | 10X Genomics | Cat#1000006 |
| 10X Chromium Single Cell A Chip Kit | 10X Genomics | Cat#1000152 |
| 10X Chromium Single Cell V(D)J Enrichment Kit, Human T Cell |
10X Genomics | Cat#1000005 |
| 10X Chromium Single Cell 5′ Library Construction Kit | 10X Genomics | Cat#1000020 |
| 10X Chromium Single Cell 5’ Feature Barcode Library Kit |
10X Genomics | Cat#1000080 |
| 10X Chromium i7 Multiplex Kit | 10X Genomics | Cat#120262 |
| Chromium i7 Multiplex Kit N, Set A | 10X Genomics | Cat#1000084 |
| CD45 MicroBeads, human | Miltenyi Biotec | Cat#130-045-801 |
| Zombie NIR Fixable Viability Kit | Biolegend | Cat# 423106 |
| Zombie Aqua™ Fixable Viability Kit | Biolegend | Cat#423102 |
| StraightFrom® Leukopak® CD8 Microbead Kit | Miltenyi Biotec | Cat#130-117-019 |
| Incucyte® Nuclight Red Lentivirus | Satorius | Cat#4476 |
| CryoStor® CS10 | Stem Cell Technologies |
Cat#07959 |
| Anti-Biotin MicroBeads | Miltenyi Biotec | Cat#130-090-485 |
| Annexin V MicroBead Kit | Miltenyi Biotec | Cat#130-090-201 |
| Human IFN-gamma (3rd Gen) | Protein Simple | Cat# SPCKA-PS002574 |
| Deposited data | ||
| Single cell RNA-seq data and bulk TCR sequencing data | This paper | GSE200996 |
| Bulk TCR-sequencing data from urothelial cancer dataset | (Mariathasan et al., 2018) |
http://research-pub.gene.com/IMvigor210CoreBiologies/
; EGAS00001002556 |
| Experimental models: Cell lines | ||
| MHC-null HEK293T engineered to express a fluorescent reporter activated by T cell killing | (Ferretti et al., 2020; Kula et al., 2019) |
N/A |
| U266B1 Plasma cell myeloma | ATCC | Cat#TIB-196 |
| A101D Melanoma | ATCC | Cat#CRL-7898 |
| A2058 Melanoma | ATCC | Cat#CRL-11147 |
| SKMEL5 Melanoma | ATCC | Cat#HTB-70 |
| 786-O Renal cell carcinoma | ATCC | Cat#CRL-1932 |
| Lenti-X™ 293T Cell Line | Takara | Cat#632180 |
| Software and algorithms | ||
| FlowJo V10 | TreeStar | RRID:SCR_008520 |
| Prism V9 | Graph Pad | RRID:SCR_002798 |
| Cellranger mkfastq v3.0.0 | 10X Genomics | http://software.10xgenomics.com |
| Cellranger v4.0.0 | 10X Genomics | http://software.10xgenomics.com |
| Cumulus | (Li et al., 2020) | https://cumulus.readthedocs.io/en/latest/i ndex.html |
| R (v4.0.0 and v4.1.2) | The R Foundation | https://www.rproject.org |
| DropletUtils (v1.8.0, R package) | (Lun et al., 2019) | Bioconductor |
| DoubletFinder (v2.0.2) | (McGinnis et al., 2019) | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Seurat (v3.2.0, R package) | (Hao et al., 2021) | https://satijalab.org/seurat/index.html |
| SingleR (v1.2.4, R package) | (Aran et al., 2019) | Bioconductor |
| Slingshot (v1.6.1, R package) | (Street et al., 2018) | Bioconductor |
| AUCell (v1.13.1, R package) | (Aibar et al., 2017) | Bioconductor |
| fgsea (v1.14.0, R package) | (Korotkevich et al., 2021) | Bioconductor |
| Molecular Signatures Database | (Liberzon et al., 2015) | http://software.broadinstitute.org/gsea/msigdb/index.jsp |
| CellChat (v1.1.0, R package) | (Jin et al., 2021) | https://github.com/sqjin/CellChat |
| LymphoSeq (v1.16.0, R package) | (Coffey, 2021) | Bioconductor |
| Immunarch (v0.6.5, R package) | (Nazarov, 2020) | https://immunarch.com/ |
| Genome Analysis Toolkit | Broad Institute | https://gatk.broadinstitute.org/hc/en-us |
| FACETS | (Shen and Seshan, 2016) | https://github.com/vanallenlab/facets |
| tradeSeq (v1.2.01, R package) | (Van den Berge et al., 2020) | Bioconductor |
| beta-binomial model for bulk TCR expansion analysis | (Rytlewski et al., 2019) | https://github.com/jrytlewski/beta_binomial_paper |
| IMvigor210CoreBiologies (v1.0.0, R package) | (Mariathasan et al., 2018) | http://researchpub.gene.com/IMvigor210CoreBiologies/ |
| ProjecTILs (v1.0.0, R package) | (Andreatta et al., 2021) | https://github.com/carmonalab/ProjecTILs |
| GMM-demux | (Xin et al., 2020) | https://github.com/CHPGenetics/GMM-Demux |