

Abstract
The coronavirus disease 2019 (COVID-19) pandemic has caused over 600,000,000 infections globally thus far. Up to 30% of individuals with mild to severe disease develop long COVID, exhibiting diverse neurologic symptoms including dementias. However, there is a paucity of knowledge of molecular brain markers and whether these can precipitate the onset of Alzheimer’s disease (AD). Herein, we report the brain gene expression profiles of severe COVID-19 patients showing increased expression of innate immune response genes and genes implicated in AD pathogenesis. The use of a mouse-adapted strain of SARS-CoV-2 (MA10) in an aged mouse model shows evidence of viral neurotropism, prolonged viral infection, increased expression of tau aggregator FKBP51, interferon-inducible gene Ifi204, and complement genes C4 and C5AR1. Brain histopathology shows AD signatures including increased tau-phosphorylation, tau-oligomerization, and α-synuclein expression in aged MA10 infected mice. The results of gene expression profiling of SARS-CoV-2-infected and AD brains and studies in the MA10 aged mouse model taken together, for the first time provide evidence suggesting that SARS-CoV-2 infection alters expression of genes in the brain associated with the development of AD. Future studies of common molecular markers in SARS-CoV-2 infection and AD could be useful for developing novel therapies targeting AD.
Keywords: SARS CoV-2, COVID-19, Alzheimer’s disease, neuroinflammation
Graphical abstract
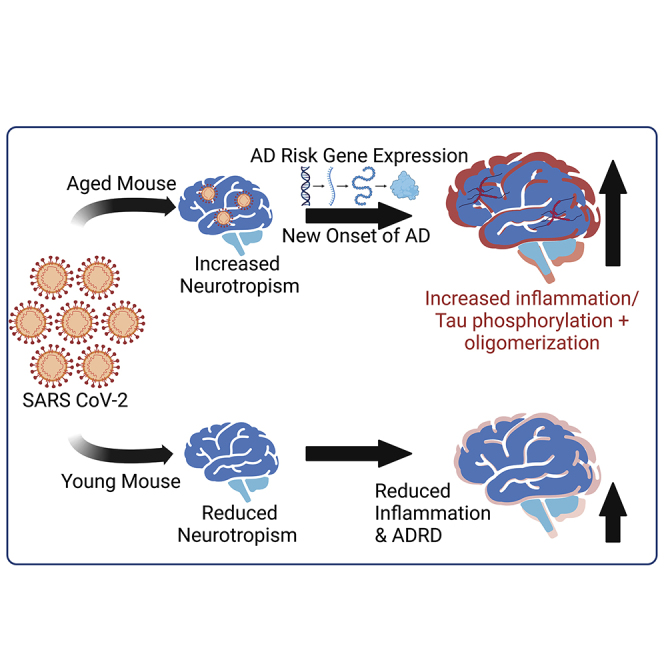
The brain gene expression profile of severe COVID-19 patients shows increased expression of several genes implicated in Alzheimer’s disease pathogenesis. The use of a mouse-adapted strain of SARS CoV-2 (MA10) in a novel aged mouse model reveals evidence of viral neurotropism and Alzheimer’s pathology including tau phosphorylation and oligomerization.
Introduction
Alzheimer’s disease and related dementias (ADRDs) are the most crippling cognitive threat to our aging population. By 2050, it is expected that the United States will spend $1.2 trillion to maintain the constantly deteriorating quality of life of 16 million Americans with AD,1 including 5.5 million Americans age 65 and older. There is no effective treatment or cure for ADRDs, and this is partly due to poor understanding of the underlying mechanisms and the diverse risk factors. Clinically, AD manifests as progressive cognitive decline and worsening memory deficits,2 which have been critically linked to the aberrant accumulation of tau protein in neurons.3, 4, 5
Inflammaging is another common hallmark of ADRD, which can be exacerbated by the state of chronic inflammation triggered by pathological microbes, including viruses and their toxic metabolites. Previously, the “infectious AD hypothesis” was proposed based on the reactivation of neurotropic viruses such as herpes simplex virus (HSV), cytomegalovirus (CMV), and human herpes virus (HHV)6, which caused the deposition of protein aggregates leading to cognition impairment.6 Also, the 1918 influenza outbreak reportedly resulted in a significant increase in Parkinson’s disease cases in years following.7 Epidemiological and laboratory rodent studies show the potential for respiratory viruses to have a neurological impact, sometimes in the absence of direct viral infection in the central nervous system (CNS).8 Although the precise mechanisms are poorly understood, it has been suggested that infections induce innate immune activation, causing neuroinflammation, which, in turn, promotes tau pathogenesis.9, 10, 11, 12 Microglia can become activated by tau oligomers, thus promoting a feedforward cycle of inflammation and neurotoxicity.13 Once a positive regulatory loop between tau production and inflammation is established, the progression to ADRDs is no longer dependent on the initial cause of inflammation.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused the coronavirus disease 19 (COVID-19) pandemic with ∼600 million confirmed infections thus far14 and with long-term sequelae (long COVID) presenting in a significant proportion of infected individuals.15 A subgroup of severe long-COVID cases show a major neurological component with evidence of neuroinvasion correlating with death.16 About 80% of CoV-2-infected patients show CNS manifestations including dizziness, headache, loss of smell/taste, impaired consciousness, ataxia, epilepsy, encephalitis, and acute cerebrovascular disease, which are 40% more common in severe COVID-19 cases than in mild disease.17, 18, 19, 20 COVID-19 is also associated with ischemic stroke,21,22 encephalopathy,19,23, syncope,24 fluid-attenuated inversion recovery (FLAIR) hyperintensities,25 and brain stem infections.26 A comparison of magnetic resonance images (MRIs) in 51- and 81-year-old subjects before and after CoV-2 infections revealed significantly decreased orbitofrontal cortex thickness.27 Moderate and severe COVID-19 have been associated with changes in brain structure and blood flow.27,28 Despite progress made, there is a paucity of knowledge regarding the cellular and molecular mechanisms by which the CoV-2 infection impacts the brain. There is an urgent unmet need to identify whether these changes lead to ADRD so that effective therapeutic interventions can be developed to ameliorate risk for ADRD following CoV-2 infection.
Previously, bioinformatic analyses of gene expression changes in COVID-19 patients identified significant immune dysregulation involving complement activation.29 Also, CoV-2 contains 16 non-structural proteins and 8 open reading frame accessory genes, of which NSP1, ORF3a/3b, ORF6, ORF7a/7b, ORF8, and ORF9b are known to dysregulate the anti-viral response, creating the immuno-pathology observed in COVID-19.30,31 We reasoned that these innate immune changes could play key roles in ADRD onset and/or progression. To investigate this idea, we analyzed gene expression in the brains of deceased CoV-2 patients and uninfected patients (NCBI Gene Expression Omnibus [GEO] database Series GEO: GSE188847) and compared these data with AD brains versus controls (GEO: GSE118553). These analyses identified several “hub” genes that are similarly regulated in both diseases and led us to hypothesize that CoV-2 can become neurotropic in aged brains causing innate immune activation, neuroinflammation, and neurodegeneration leading to tauopathy and the cognition impairment of AD. To test this, we established CoV-2 infection in a mouse model using a mouse-adapted strain of SARS-CoV-2 (MA10) and examined how susceptibility to this viral infection changes with age. We further examined the association of gradual aging with inflammation, neurodegeneration, and infection-driven expression of ADRD genes/proteins with potential to cause the onset and/or rapid progression of ADRD.
Results
Gene expression changes in brains of patients with COVID-19 and AD
We obtained whole-transcriptome expression data from COVID-19 versus control and AD versus control frontal cortex patient samples from the NCBI GEO database. Differential gene analysis generated 2,446 genes significantly up- or downregulated in the brains of COVID-19 patients and 856 genes significantly up- or downregulated in the brains of patients with AD (Figures 1A and 1B). Dimensionality reduction and clustering yielded only moderate separation between disease and control groups, highlighting the impact of patient-to-patient variation in gene expression (Figures S1A and S1B). Sorting the differentially expressed genes into GO pathways revealed several common pathways affected by both diseases, although the absolute gene counts were skewed by the difference in size of each dataset (Figure 1C).
Figure 1.

Transcriptomic changes observed in the frontal cortex of severe COVID and AD brains
(A) Mean diff plot showing significantly upregulated (red) and downregulated (green) genes in AD brains versus control brains. (B) MA plot showing significantly upregulated (red) and downregulated (green) genes in COVID-19 versus control brains. (C) Differentially expressed genes in each dataset sorted into Gene Ontology (GO) pathways (Panther-db). Pathways containing at least 10 differentially expressed genes from each dataset are shown. Please see supplemental information for complete gene list.
COVID-19 brains exhibit an AD risk gene expression signature
In order to compile a set of AD risk genes, we searched relevant literature for studies describing the molecular etiology of AD and providing evidence for the specific function of individual genes/proteins in AD onset or progression. We have compiled our findings into five functional categories of inflammation, protein folding/trafficking, complement activation, calcium homeostasis, and amyloid/tau processing (Table 1). We imported this gene list into Ingenuity Pathway Analysis (IPA) and plotted experimentally observed interactions as a network. Fold change values (Figure S2) from the COVID-19 brain dataset were overlaid onto the network, revealing increased inflammatory cytokines IL-18, CXCL8, and IL-6 receptor along with the transcription factors STAT3 and KLF4 and the marker of neuroinflammation GFAP. Although specific cell types responsible for promoting inflammation in these patients is not known, we did observe an increase in gene signatures associated with monocytes, neutrophils, and activated dendritic cells in the COVID-19 brains; however, these changes were not statistically significant (Figure S3). Hub gene analysis using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database of protein interactions reinforced the evidence of neuroinflammation yielding canonical mediators and regulators of inflammation (CCL2, CXCL8, NF-κB, and STAT3) as some of the differentially expressed genes with the most interactors in the dataset (Figure S4). Complement activation was also evident from increased C4a and C5AR1. Decreased expression of HSP chaperones HSP90AB1, HSP90AA1, and HSPA8 was observed, while FKBP5 was increased. Calcium signaling components (CAMKK2 and calbindin) and the receptor for cholecystokinin, CCKBR, were all decreased in expression (Figure 2).
Table 1.
ADRD risk/pathology genes
| Gene | Expression in COVID-19 | Function | Reference |
|---|---|---|---|
| CCL20 | predicted activation | recruitment of leukocytes | Goldeck et al.32 |
| CTCFL | increased | controls gene expression in myeloid cells | Corces et al.,33 Novikova et al.34 |
| CXCL8 | increased | neuroinflammation | Zuena et al.35 |
| EGFR | increased | astrocyte activation, inflammation | Mansour et al.36 |
| GFAP | increased | astroglial activation | Cicognola et al.37 |
| IFI16 | increased | interferon-induced DNA sensor | Velez et al.38 |
| IL-17 | predicted activation | neuroinflammation | Brigas et al.39 |
| IL-18 | increased | neuroinflammation/amyloid processing | Ojala et al.40 |
| IL-6R | increased | neuroinflammation | Haddick et al.41 |
| KLF4 | increased | regulates neuronal apoptosis and axon regeneration | Cheng et al.42 |
| LGALS3 | increased | microglia-mediated inflammation, amyloid aggregation | Tan et al.,43 Tao et al.44 |
| TAC1 | decreased | vasodilator | Dharshini et al.45 |
| CAV1 | increased | endocytosis and protein trafficking | Gaudreault et al.46 |
| FKBP5 | increased | tau phosphorylation, tau oligomerization, glucose metabolism | Wang et al.,47 Fuji et al.,48 Blair et al.49 |
| HSP90 | decreased | protein folding | Blair et al.50 |
| HSPA8 | decreased | autophagy/tau regulation | Loeffler et al.51 |
| IFITM3 | increased | modulates γ-secretase | Hur et al.52 |
| C3/4 | increased | complement activation, association with plaques | Tenner et al.53 |
| C5AR1 | increased | C5a receptor on myeloid cells | Hernandez et al.54 |
| CR1 | predicted activation | phagocytosis of immune complexes, inflammation | Zhu et al.55 |
| CALB1 | decreased | calcium sequestration | Sanfillipo et al.56 |
| CAMKK2 | decreased | regulatory kinase Ca-dependent signaling | Sabbir et al.,57 Mairet-Coello et al.58 |
| BDNF | decreased | protects against tau-related neurodegeneration | Elliott et al.59 |
| CCK/BR | decreased/ predicted decrease | hormone, maintaining memory | Plagman et al.60 |
| PLAT | increased | tissue plasminogen activator, cleaves pro-BDNF, cleaves amyloid plaques | Shibata et al.61 |
Figure 2.

Severe COVID alters expression of key Alzheimer’s risk genes in the human brain
Network of AD risk genes depicting experimentally observed interactions (IPA database) and fold change of gene expression observed in COVID frontal cortex samples versus control, GEO: GSE188847 (green/red).
SARS-CoV-2 MA10 neurotropism is increased in aged mice
Since age is one of the crucial risk factors for the severity of infection, we investigated the neurological consequences associated with CoV-2 infection in both young and old mice at both acute (4 days post-infection [DPI]) and post-acute (18 DPI) time points. Thus, 3-, 6-, and 20-month-old C57BL/6 mice were infected with 1 × 105 of MA-10 intranasally. The infected mice were sacrificed 4 and 18 DPI, and viral titer and gene expression analyses were performed. A plaque assay was performed from the brain samples to quantitate the viral titer (Figures 3A and 3B). No plaques were detected in the infected young mice (3 months), but a significant number of plaques were detected in the infected adult (6 months) and in aged mice (20 months) groups. Also, the plaques were detected at only 18 DPI, and no plaques were detected in 4 DPI brain samples. qPCR analysis of RNA isolated from three brain regions (olfactory bulb, cortex, and hippocampus) confirmed the presence of CoV-2 N-protein transcript (Figure 3C). Samples from 3-month-old mice did not contain any transcript. Six-month-old mice contained transcript in the cortex at 4 DPI and the cortex plus olfactory bulb at 18 DPI. Only the 20-month-old mice contained CoV-2 transcript in all brain regions at 18 DPI, additionally testing positive in the olfactory bulb and hippocampus at 4 DPI.
Figure 3.

Infection with SARS-CoV-2 MA10 virus induces neuroinflammation in aged mice
(A) Representative images of plaque assays performed on homogenized brain tissue collected from MA10-infected 3-, 6-, and 20-month-old mice. (B) Histogram depicting quantification of viral plaque assay to determine the concentration of infectious particles present in brain tissue (PFU/mL). (C) Histogram depicting mRNA fold change of CoV-2 nucleoprotein (N) expression in brains collected from MA10-infected mice compared with mock. (D) Histograms depicting mRNA fold change of proinflammatory genes (Il6, Tnfα, and Ccl20), and inflammasome genes (Nlrp3, Ifi204, Ilβ), in infected brain. n = 3; data expressed as mean ± SEM; ∗ compared with respective mock; # 18 DPI compared with 4 DPI of the same brain region; ∗,#p < 0.05 by ANOVA with Holm-Sidak test.
Neuroinflammation is increased in old versus young MA10-infected mice
A qPCR analysis showed that the pro-inflammatory cytokines Il-6, Tnf-α, and Ccl20 were all generally increased in the 6- and 20-month-old infected mice, with the highest expression observed in the 20-month-old mice at 18 DPI (Figure 3D). Only Il-6 and Tnf-α were significantly increased in the 3-month-old mice, and this increase was only observed in the olfactory bulb. The inflammasome component encoding genes Nlrp3 and IFI-16 (Ifi204 mouse homolog) were also significantly upregulated in 6- and 20-month-old mice along with Il1β but were unchanged in 3-month-old mice. An increase in active caspase-1 (p20) was observed in all brain regions of the 20-month-old infected mice (Figure S5). Overall, these data indicate that the intensity and pervasiveness of inflammation caused by CoV-2 infection in the brain increased with age and that inflammation may persist long term for at least 18 days.
Expression of ADRD risk/pathology genes is increased in aged MA10-infected mice
We next examined the expression of five key AD risk genes identified by the network analysis in Figure 2 (FKBP5, IFITM3, IFI16, CR1, and C5AR1). FKBP5 is a polyfunctional chaperone protein known to be associated with cognitive abnormalities and dysregulation of calcium homeostasis in AD.62 IFITM3 is induced by the interferon-driven inflammatory response and is known to increase the activity of γ-secretase for Aβ production.63 Similarly, the interferon-stimulated gene IFI16 has been identified as a possible driver of neuroinflammation, and synaptic loss, in the AD brain.64 IFI16 is an inflammasome component that serves as a viral nucleic acid sensor and promotes production of interleukin-1β (IL-1β).65 CR1 is the receptor for complement factors C3b and C4b, which are known to be hyperactivated in COVID-19.55 It is expressed on antigen-presenting cells including microglia as well as on neurons. C5AR1 is a receptor for C5a expressed primarily on myeloid cells and has been used as a biomarker for AD.54 None of these genes were found to be upregulated in 3-month-old mice; however, all of them were significantly upregulated in 6- and 20-month-old mice, although Ifitm3 expression was lower in the hippocampus than in the olfactory bulb and cortex (Figure 4A). An increase in Fkbp5 protein expression was confirmed in all CoV-2-infected brain regions by immunohistochemistry (Figure 4B). A similar increase in expression of inflammatory and ADRD risk genes was also recapitulated in 17-month-old mice (data not shown).
Figure 4.

Infection with SARS-CoV-2 MA10 virus induces ADRD risk genes and complement activation in aged mice
(A) Histograms depicting mRNA fold change of ADRD risk genes (Fkbp5, Ifitm3), and complement activation genes (Cr1, C5ar1), in brains collected from MA10-infected mice. (B) Bright-field images and respective quantification showing FKBP5 expression in the olfactory bulb, cortex, and hippocampus of MA10-infected 20-month-old mice brains at 18 DPI. (C) Ingenuity Pathway Analysis (IPA) network showing genes that are differentially expressed in cortex of infected aged mice (20 months old) 18 DPI as assayed by nCounter neuroinflammatory and glial profiling panel. n = 3; data expressed as mean ± SEM; ∗ compared with respective mock; # compared with respective 4 DPI; ∗p < 0.05 by ANOVA with Holm-Sidak test.
Inflammation and ADRD gene signature are correlated with tau pathology, α-synuclein, and demyelination
We analyzed pooled CoV-2- or mock-infected mouse cortex samples for the expression of gene transcripts of additional AD risk genes by Nanostring nCounter Glial Profiling panel and Neuroinflammatory panel. These panels include key processes and pathways regulated under diseased conditions, such as cell stress and damage response (134 genes), glial regulation (180 genes), inflammation, and peripheral immune invasion (188 genes). The IPA analysis of Nanostring data (MA10-infected mouse brain samples) also showed a similar signature as that of the data collected from the GEO database (human brain tissue from CoV-2-infected patients). Upregulated genes include Lgals3, Egfr, C4a, Fkbp5, Gfap, Mapt, Ifitm3, Stat3, C5ar1, and Il6r. Downregulated genes include Hsp90ab1, Bdnf, and App (Figure 4C).
Our data and others have suggested that CoV-2 infection may promote aberrant tau accumulation.66 Hence, we examined the tau pathology in MA10-infected mice by immunohistochemical staining using pT231 tau, an AD-relevant tau phosphorylation.67 Brains from each group were sectioned and stained at 18 DPI. p-tau-positive cells were not present in the mock-infected mice or in the 3-month-old MA10-infected mice (Figure 5A). However, the number of p-tau-positive cells was significantly elevated in all examined brain regions of the 6- and 20-month-old mice, with significant increases in the olfactory bulb and cortex of the 20-month-old group compared with the 6-month-old group. We also examined the accumulation of tau oligomers, which data support as a toxic form of tau,68, 69, 70 using an oligomeric tau-specific antibody and again found a significant increase in the 6- and 20-month-old MA10-infected groups compared with mock as well as a significant increase in the 20-month-old MA10 group compared with the 6-month-old MA10 group in all brain regions (Figure 5B). Similar increases in oligo-tau and p-tau were also recapitulated in 17-month-old mice (data not shown). Furthermore, p-tau was observed specifically in brain regions adjacent to CD31+ endothelial cells (Figure 5C), which, in prior studies, was linked with blood-brain barrier dysfunction.71 Increased von Willebrand factor (vWF) staining in infected brains was observed as evidence of this blood-brain barrier (BBB) damage (Figure S6). In addition, immunohistochemical analysis of CoV-2-infected brain tissue shows elevated GFAP expression (astroglial activation) and IBA1 expression (microglial activation) in the olfactory bulb, cortex, and hippocampus (Figures S7 and S8). A significantly increased grade of demyelination (MBP staining) in the striatum of infected aged brain tissues was observed along with significantly increased α-synuclein staining in the 6- and 20-month-old infected mice (Figures S9 and S10).
Figure 5.

SARS-CoV-2 MA10 infection induces p-tau expression in olfactory bulb, cortex, and hippocampus of 6- and 20-month-old mice
(A) Bright-field images showing immunoperoxidase staining of p-tau (pT231) in olfactory bulb (top panel), cortex (middle panel), and hippocampus (bottom panel) of 3-, 6-, and 20-month-old mice at 18 DPI. Histogram showing ImgaeJ quantification of p-tau expressing cells. (B) Bright-field images showing immunoperoxidase staining of tau (T22), oligomeric expression in olfactory bulb (top panel), cortex (middle panel), and hippocampus (bottom panel) of 3-, 6- and 20-month old mice, at 18 DPI. Histogram showing ImgaeJ quantification of tau (T22)-expressing cells. (C) Co-localization of CD31-p-tau expression in olfactory bulb, cortex, and hippocampus. CD31: red, DAPI: blue, and p-tau: green. Arrows indicate areas of p-tau/CD31 co-localization. n = 3; data expressed as mean ± SEM; ∗ compared with respective mock; # compared between 6- and 20-month-old infected mice, ∗,#p < 0.05.
Discussion
This article provides evidence for two major findings. First, a comparison of gene expression changes in brains of human patients with AD or COVID-19 versus their respective controls along with brain pathological studies in the CoV-2 MA10 aged mouse model has led to identification of several common hub genes and pathways that are similar in pathogenesis of AD in mice and humans. To the best of our knowledge, our analysis is the first of its kind wherein transcript expression in the brain (cortex) has been compared between elderly patients with severe COVID-19 and patients with AD. Previously, a solely bioinformatic study compared the AD brain transcript profile (GEO: GSE147507; n = 97) with COVID lung gene expression (GEO: GSE132903; n = 2).72 Second, the mouse studies using CoV-2- MA10 virus revealed that CoV-2 neurotropism was dependent upon the age of the mice. Thus, replication of MA10 virus was evident in the brains of aged (6- and 20-month-old) mice but not in young 3-month-old mice. Notably, despite evidence of neurotropism for SARS-CoV and Middle East respiratory syndrome (MERS)-CoV and affirmative CoV-2 neurotropism studies in vitro in human cells,73 in vivo neurotropism of CoV-2 has remained unclear due to the lack of virus detection in many post-mortem patient brain and CNS samples. Our establishment of CoV-2-MA10 infection in an aged mouse model has provided the first in vivo evidence for CoV-2 neurotropism in wild-type mice. The MA10 aged mouse model has provided a unique opportunity to investigate the connection between viral infection and ADRDs including the mechanistic underpinnings of this relationship. With millions of patients infected by CoV-2 having mild, moderate, and severe infections and 15%–30% of those also suffering from long COVID, potential increased risk of ADRDs would have significant clinical impact.15
The major evidence for SARS-CoV-2 neurotropism (N-gene transcript expression and viral plaque assay) definitively demonstrates viral replication in the brain tissue, though the cellular localization of the virus within the mouse brain (endothelial, microglia, neuron, etc.) and the extent of viral replication within these cells remain unknown. Our findings are consistent with the previous epidemiological and laboratory reports of severe viral infections leading to a preponderance of CNS diseases including AD, Parkinson's disease, and encephalopathy.6, 7, 8 Notably, we observed increased vWF expression in aged, infected brains as evidence of vascular damage along with increased p-tau in regions adjacent to the endothelial cell marker CD31, suggesting there may be a vascular etiology to this pathology. This is consistent with primate data that suggested that CoV-2 replication mainly within vascular endothelial cells in the brain.74 Further, we found that changes in expression of genes in COVID-19 brains included the Wnt signaling pathway. This pathway is dysregulated in AD, with beta catenin (CTNNB1) aggregation linked to proteosomal dysfunction and tau phosphorylation by GSK3β,75 thus contributing to AD risk.
In a broad sense, the CoV-2 neuropathology contains many elements overlapping with AD. Neuroinflammation is a major driver of both diseases; however, additional pleiotropic signaling pathways are also implicated, including EGFR, Wnt, and immune cell activation. Indeed, we have observed gene expression changes within these pathways in both diseases, albeit with only moderate overlap between individual genes. Increased oxidative stress and dysregulated ribosomal function are known features of both diseases as well. Through pathway analysis of known AD-gene relationships in the IPA and STRING databases and literature review, we have grouped ADRD risk genes into 5 categories: inflammation, protein folding/trafficking, complement activation, calcium homeostasis, and amyloid/tau processing (Table 1). Many changes in expression of these risk genes and specific gene mutations have been associated with late-stage AD through sequencing of brain tissue from post-mortem patients. However, due to the complexity of the disease and the inability to collect longitudinal samples, direct causes of AD remain poorly defined. Recently, the once-controversial “infectious hypothesis” of AD has been gaining more attention.76,77 Although it seems unlikely that an infectious agent such as a prion or virus will emerge as the direct cause of AD, it is possible that the molecular signaling changes brought on by inflammation due to aging and followed by infection (“double-hit model”) could serve as a trigger for disease progression in individuals who are already genetically predisposed. The data presented herein support this hypothesis.
Consistent with our previous report,29 the results of our gene expression profiles in COV-2-infected and AD brains showed similarly increased activation of complement/coagulation signaling and the inflammasome, which are known hallmarks of moderate and severe COVID-19. We have presented evidence of this same activation in both human and mouse brains (IL-18, CCL20, NLRP3, C4a, and C5AR1) (Figures 2A and 4A–4C). Furthermore, we have shown how increased expression of these genes may activate downstream AD risk genes (IFI16, IFITM3, FKBP5, GFAP) ultimately leading to tau and α-synuclein pathology as observed in the mouse model (Figures 5 and S9). Indeed, some of these genes play dual roles in innate immunity and AD. IFITM3 is an interferon-stimulated gene that prevents viral entry into cells by disrupting cholesterol synthesis and shuttling viral particles to lysosomes. However, it is also known to have secondary activities including activation of PI3K signaling and activation of γ-secretase to increase Aβ production. α synuclein-GSK3β activity could exacerbate this AD pathology by promoting a feedback loop of tau phosphorylation, and aggregation of both tau and Aβ, with a simultaneous increase in Aβ production.78 Presenilin-1 (PSEN1) plays a role in proteolytic cleavage of APP to Aβ, which interacts with α-synuclein, forming complexes as witnessed in patients with AD with PSEN1 mutations. Altogether, there are a number of in vitro and in vivo studies supporting the leading role that α-synuclein plays in several mechanisms and processes linked to AD.79, 80, 81
Perhaps the most significant connection between CoV-2 infection and AD observed in both human tissue and the mouse model is the potential for CoV-2 infection to promote tau phosphorylation and oligomerization. This phenomenon has been observed at the protein level in the brain tissue of both elderly demetia patients and patients who have died from COVID-19.66 In this case, a leaky calcium channel driven by virus-induced inflammasome activation and oxidative stress is hypothesized to promote tau pathology through a reduction in calbindin. We observed a similar decrease in calbindin expression in COVID-19 brains along with a decrease in CAMKK2 and evidence of inflammasome activation (increased IL-18, IFI16, and STAT3). In the mouse model, we observed increased Nlrp3, Tnfα, and active cleaved caspase-1 (p20), indicating inflammasome activation, but here we were also able to observe the tau pathology directly, including significant increases in tau phosphorylation and oligomerization. Studies are ongoing to determine the timeline of inflammasome induction in the CoV-2-infected brain and whether this induction is dependent on recognition of viral RNA or host DNA damage.
One limitation of our study is the inability to reliably discern between the causes and effects of AD since patient tissues are collected post-mortem at late-stage AD. These patients typically exhibit an upregulation of protein synthesis in response to the high degree of protein misfolding and aggregation characteristic of AD. Therefore, the upregulation of protein synthesis in viral infection to mediate production of viral particles may be a coincidence rather than a promotor of AD development. Another limitation of our study is that despite the similarities we observed at the pathway level in brains of patients with COVID-19 versus AD, as expected there were dissimilarities at the level of individual gene expression. The latter was expected primarily due to the two following reasons. First, the COVID-19 group had a severe active viral infection at the time of sample collection, the AD group did not. Second, the AD brains were collected from patients with late-stage AD pathology, while the COVID-19 patients were beginning to express molecular markers of AD but were not diagnosed with AD. Lastly, our studies do not address whether gene expression changes in SARS-CoV-2-infected brains can cause AD-related changes in cognition; this is due to the fact that our study focused on acute SARS-CoV-2 infection following a single inoculation event and was not followed up beyond 18 DPI. This is beyond the scope of our present study, and it remains to be elucidated in future studies. Furthermore, respiratory viruses other than SARS-CoV-2 such as influenza and respiratory syncytial virus have also been known to present with CNS complications. Future studies will be needed to compare their infections in the aged mouse model to determine whether the ADRD pathology we observed is specific to SARS-CoV-2, although some changes to the model may be required to accommodate the differing mechanisms/pathologies of infection by these viruses in mice.
Overall, we have shown that CoV-2 infection induces gene expression changes in multiple pathways linked to AD susceptibility including inflammation, protein chaperones, amyloid processing, ubiquitin mediated protein degradation, FKBPs in tau processing, endoplasmic reticulum (ER) protein transport, and calcium homeostasis. While the complexity of AD pathology does not lend itself to discovery of a single definitive link suggesting CoV-2 as a causative agent of AD, the significant overlap in the gene expression profiles of the two diseases across multiple functional pathways in combination with the ADRD pathology observed in the brains of aged, infected mice warrants further mechanistic studies.
Materials and methods
Differential gene expression analysis
Gene expression data from severe COVID-19 (n = 12) or COVID-19-unaffected (n = 12) individuals were obtained from the NCBI GEO database (GEO: GSE188847). In the associated study, frontal cortex tissues were collected within a post-mortem interval of less than 48-h. Unaffected patients were selected to be age and sex matched with COVID-19 patients. Salmon transcript quantification files for each patient were retrieved from GEO. A transcript to gene map file was exported from Ensembel Bio-Mart using GRCh38.p.13. Pooled differential gene analysis (12 COVID-19 versus 12 control samples) was performed using DESeq 2. The fold change output for each gene was filtered to only include significant changes (p ≤ 0.05). Gene expression data from frontal cortex samples from patients with AD (n = 40) and normal controls (n = 22) were obtained from the GEO database (GEO: GSE118553). The GEO2R web tool (NCBI) was used to compare gene expression between AD and control groups, generating a list of differentially expressed genes with fold change and adjusted82 p value.
Protein-protein network and hub gene analysis
The lists of significantly differentially expressed genes in AD and COVID-19 were uploaded into the STRING database, and networks of gene interactions were obtained. Local network clusters matching genes upregulated in COVID-19 were ranked for their fit to the STRING database according to enrichment score. This entire network was imported into Cytoscape for hub gene analysis using the Cytohubba tool. The top 20 genes in 4 ranked algorithms; maximal clique centrality (MCC), density of maximum neighborhood component (DMNC), maximum neighborhood component (MNC), and Degree were identified, and the intersections between the four algorithms where two or more algorithms agreed were considered as the hub genes. This analysis was repeated for the AD versus control dataset. AD-related gene clusters were determined by taking the list of genes from the AD “disease” category in the IPA database (1,016 genes) and importing them into STRING as a network. Markov cluster algorithm (MCL) clustering was performed on the network with an inflation parameter of 2, and the top 8 clusters were selected for analysis.
IPA
Alzheimer’s genes were selected from literature (see Table 1). IPA (Qiagen) was used to plot known connections between the genes creating a molecular network. RNA sequencing (RNA-seq) profiling of human frontal cortex in severe COVID-19 (n = 12) or unaffected (n = 12) patients was obtained from the NCBI GEO database (GEO: GSE188847). Pooled differential gene analysis of COVID-19 versus unaffected patients was performed using DESeq2. Significantly differentially expressed genes (p < 0.05) were uploaded into IPA, and their fold change information was overlaid onto the Alzheimer’s gene network.
The IPA (Qiagen) database was queried for all genes known to contribute to or be altered in AD. The sub-set of these genes differentially expressed in COVID-19 were selected. Significantly differentially expressed genes (p < 0.05) were uploaded into STRING, and the STRING database of protein-protein interactions was used to generate an interaction network. This network was imported into Cytoscape, and hub genes were identified within the network using Cytohubba. The IPA database was queried for genes known to increase susceptibility for Alzheimer’s and to play a role in the early development of Alzheimer’s. These Alzheimer’s genes were connected to the hub genes using known relationships in the IPA database.
SARS-CoV-2 MA10 virus stock preparation and titration with plaque-based assays
All experiments utilizing replication-competent SARS-CoV-2 were performed in a biosafety level 3 (BSL-3) laboratory at the University of South Florida. All viral stocks were produced and isolated from supernatants of Vero-E6 cells expressing human ACE2 and TMPRSS2 (BEI Resources NR-54970), cultured in T175 flasks to a confluency of 80%–90%, and infected with an original passage 2 (P2) MA10, at an MOI of 0.05 for 4 h, in 8 mL serum-free OptiMEM. Media was then replaced with 20 mL DMEM media supplemented with 5% FBS and 1% penicillin-streptomycin antibiotic and incubated for 72 h until clear cytopathic effect (CPE) was present. MA10 was obtained from BEI Resources (BEI Resources NR-55329). Supernatants were harvested, cleared of cell debris by centrifugation (500 × g, 10 min) and filtration (0.45 μm), mixed with 10% SPG buffer (ATCC #MD9692), aliquoted, and stored at −80°C. Viral titers were quantified by determining the number of individual plaque-forming units (PFUs) after 72 h of infection on confluent Vero-E6 ACE2 TMPRSS2 cells. In brief, 25 μL viral stock was added to 225 μL OptiMEM (a10−1 dilution) and was subsequently serially diluted (10-fold) in serum-free medium and inoculated on 7.5 × 105 Vero E6 ACE2 TMPRSS2 cells in triplicates in a 48-well plate for 3 h. The inoculum was then removed and replaced with 0.8% carboxymethylcellulose in OptiMEM +2%FBS+1% penicillin-streptomycin antibiotic as overlay media for 72 h. The plates were then fixed with 80% methanol in water overnight, rinsed with 1× PBS, and stained with 0.1% crystal violet solution in ethanol. Plaques were then counted and calculated as PFU/mL.
Animal experiments
All animal procedures were conducted in accordance with the NIH guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the University of South Florida. 3-, 6- and, 20-month-old male C57BL/6J mice were housed in a BSL-3 animal facility on a 12 h light-12 h dark cycle with food and water available ad libitum. Mice of each age group were infected with 100,000 PFUs of MA10 or mock ultraviolet light-inactivated MA10 that was generated and titrated using the previously described method. Mice under deep isoflurane anesthesia were intranasally inoculated with 50 μL (25 μL per nostril) of virus diluted in OptiMEM media. Mice were observed after inoculation for regain of consciousness and lucidity. Each subsequent day proceeding viral inoculation, the mice were individually examined for signs of infection including body weight change and alterations in body temperature, respiration, and lethargy. Mice were sacrificed 4 and 18 DPI. The brains and lungs were harvested and processed for plaque assay, RNA isolation, and immunostaining. Brain and lung tissue from mice were longitudinally sectioned in half, with half of one lung or brain sample going for RNA processing and the other half for plaque assay, while the second, intact lung was fixed with formalin for sectioning.
Plaque assay from organ samples
Half of a longitudinally section organ (∼214 mg brain tissue or ∼50 mg lung) was homogenized in 200 μL OptiMEM media containing 10% penicillin-streptomycin-amphotericin antibiotic. The homogenate was then cleared of cell debris by centrifugation (500 × g, 10 min). Viral titers from infected organs were quantified by determining the number of individual PFUs after 72 h of infection on confluent Vero-E6 ACE2 TMPRSS2 cells. 25 μL supernatant from the organ homogenate was added to 225 μL OptiMEM (10−1 dilution) and then serially diluted (10-fold) in serum-free medium and inoculated on 7.5 × 105 Vero E6 ACE2 TMPRSS2 cells in triplicates in a 48-well plate for 3 h. The inoculum was then removed and replaced with 0.8% carboxymethylcellulose in OptiMEM +2%FBS+1% penicillin-streptomycin antibiotic as overlay media for 72 h. The plates were then fixed with 80% methanol overnight, rinsed with 1× PBS, and stained with 0.1% crystal violet solution in ethanol. Plaques were then counted and calculated as PFU/mL of homogenate.
RNA isolation and PCR
Total RNA was isolated using Trizol (Life Technologies). The extracted RNA was subjected to nCounter gene expression analysis (NanoString Technologies) according to the manufacturer’s instructions. Also, RNA was treated with DNAse I (Invitrogen, cat. no. 18068) to remove the residual genomic DNA. 1 μg RNA was used to prepare cDNA using Maxima Enzyme 5× reaction mix (Thermo Fisher Scientific). Quantitative real-time PCR reaction was performed using the cDNA in CFX384 TouchTM Real-Time PCR detection system (Bio-Rad). The reaction mixture was set up to 5 μL containing 1 μL 5× qPCR master mix, 0.5 μL forward primer and reverse primer (see Table S11), 1 μL water, and 1 μL cDNA. The reaction was performed using the following program: 95°C or 3 min, followed by 45 cycles of 95°C for 10 s, 60°C for 1 min, and 72°C for 15 s. All experiments were run in triplicate for three individual experiments. Gene expression from each age group was normalized to its respective mock control.
Immunofluorescence staining
Slide-mounted 30 μm brain sections were heated with antigen retrieval solution (1:100; Vector Laboratories, Burlingame, CA, USA) for 45 min at 90°C, cooled, and washed with PBS. The sections were permeabilized, and non-specific antigens were blocked for 1 h with serum-blocking solution (10% host serum, 0.2%Triton in PBS). Next, sections were incubated with primary antibody solution (5% host serum, 0.1% Triton X-100 in PBS; see Table S12) overnight at 4°C. After washing with PBS, sections were incubated with fluorescent tagged secondary antibody, washed again, dried, and mounted with DAPI containing anti-fade mounting medium.
Immunoperoxidase staining
For immunoperoxidase staining, after heat antigen retrieval, sections were treated with 3% hydrogen peroxide in water for 20 min, incubated with serum-blocking solution, and incubated overnight at 4°C with primary antibody (see Table S12). Following washing, sections were then sequentially incubated with biotinylated secondary antibody for 2 h at room temperature, avidin-biotin peroxidase (ABC, 1:100 Vector Laboratories) for 1 h at room temperature, and 3,3′-Diaminobenzidine (DAB) substrate solution (Vector Laboratories) for 5 min. Sections were washed, dried, and cover slipped with DPX mounting medium. Bright-field or fluorescence images were taken with an Olympus X71 microscope using appropriate filters.
Image analysis and quantitation
All quantitation was performed using NIH ImageJ Software. For immunohistochemical analysis, images were acquired using an Olympus IX71 microscope controlled by DP70 manager software (Olympus America, Melville, NY, USA). The images were taken at the same exposure and digital gain settings for a given magnification to minimize the differential background. Three sagittal brain sections were stained with each antibody and 3 images (20×) per region per section were captured, and analysis was performed. The images were converted to grayscale before quantification. The grayscale images were adjusted to exclude noise pixels. The same settings were used for quantifying all images. Positive cells were counted for p-tau, T22 oligomeric tau, IBA1, and α-synuclein staining using ImageJ software. Similarly, integrated density/unit area (immunoreactivity) was measured for FKBP5, MBP, and GFAP staining using ImageJ software. The results were expressed as mean number of positive cells or mean area of immunoreactivity ± standard error of mean (SEM).
Statistical analysis
All data are presented as mean ± SEM. Statistical significance was evaluated by ANOVA with Holm-Sidak test for multiple comparisons if not mentioned otherwise. A p value of less than 0.05 was considered statistically significant for all comparisons.
Data availability
All whole-transcriptome gene expression datasets analyzed during the current study are available in the NCBI Gene Expression Omnibus repository (https://www.ncbi.nlm.nih.gov/geo/) under the accession numbers GEO: GSE188847 and GSE118553. All other data generated or analyzed during this study are included in this published article (and its supplemental information).
Acknowledgments
This work is supported by the Research Career Scientist (RCS) awards IK6BX004214 to P.C.B., IK6BX004212 to S.M., and to S.S.M.; Veterans Affairs Merit Review grants BX004626 to L.J.B. and BX005490A and BX005757 to S.M. and S.S.M.; Public Service grants R01 CA180758 to B.C. and NS073899 to L.J.B,; FDOH- James & Esther King Biomedical Research Program grant 22K09 to S.M.; and USF-PRRN funding to S.M. and S.S.M. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs, the United States government, or the National Institutes of Health. All animal studies were conducted at the University of South Florida, and all study protocols were reviewed and approved by the USF Institutional Animal Care and Use Committee (IACUC). All experiments were performed in accordance with IACUC-approved guidelines and regulations. All experiments using replication competent SARS-CoV-2 were performed in the USF BSL-3/ABSL-3 (GHIDR) facility.
Author contributions
R.G. processed and analyzed transcriptomic data, contributed to analysis of data, prepared figures, and was a major contributor in writing the manuscript. K.M. performed mouse studies, prepared figures, and contributed to writing of the manuscript. A.R.M. contributed to virus production and purification and to writing and editing the manuscript. T.M. contributed to analysis and interpretation of data and editing the manuscript. B.C. contributed to analysis and interpretation of results and editing the manuscript. L.J.B. contributed to analysis and interpretation of results and editing the manuscript. P.C.B. contributed to analysis and interpretation of results and editing the manuscript. S.S.M. contributed to experiment design, analysis and interpretation of data, and writing the manuscript. S.M. contributed to experiment design, analysis and interpretation of data, and writing the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors have no competing interests to declare.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.09.007.
Supplemental information
References
- 1.Thies W., Bleiler L., Alzheimer's Association 2013 Alzheimer's disease facts and figures. Alzheimers Dement. 2013;9:208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Knopman D.S., Amieva H., Petersen R.C., Chételat G., Holtzman D.M., Hyman B.T., Nixon R.A., Jones D.T. Alzheimer disease. Nat. Rev. Dis. Primers. 2021;7:33. doi: 10.1038/s41572-021-00269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson P.T., Alafuzoff I., Bigio E.H., Bouras C., Braak H., Cairns N.J., Castellani R.J., Crain B.J., Davies P., Del Tredici K., et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bejanin A., Schonhaut D.R., La Joie R., Kramer J.H., Baker S.L., Sosa N., Ayakta N., Cantwell A., Janabi M., Lauriola M., et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain. 2017;140:3286–3300. doi: 10.1093/brain/awx243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia C., Makaretz S.J., Caso C., McGinnis S., Gomperts S.N., Sepulcre J., Gomez-Isla T., Hyman B.T., Schultz A., Vasdev N., et al. Association of in vivo [18F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol. 2017;74:427–436. doi: 10.1001/jamaneurol.2016.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wainberg M., Luquez T., Koelle D.M., Readhead B., Johnston C., Darvas M., Funk C.C. The viral hypothesis: how herpesviruses may contribute to Alzheimer's disease. Mol. Psychiatr. 2021;26:5476–5480. doi: 10.1038/s41380-021-01138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sulzer D., Antonini A., Leta V., Nordvig A., Smeyne R.J., Goldman J.E., Al-Dalahmah O., Zecca L., Sette A., Bubacco L., et al. COVID-19 and possible links with Parkinson's disease and parkinsonism: from bench to bedside. NPJ Parkinsons Dis. 2020;6:18. doi: 10.1038/s41531-020-00123-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai J.P., Baker A.J. Influenza-associated neurological complications. Neurocrit. Care. 2013;18:118–130. doi: 10.1007/s12028-012-9796-8. [DOI] [PubMed] [Google Scholar]
- 9.Li H., Liu C.C., Zheng H., Huang T.Y. Amyloid, tau, pathogen infection and antimicrobial protection in Alzheimer's disease -conformist, nonconformist, and realistic prospects for AD pathogenesis. Transl. Neurodegener. 2018;7:34. doi: 10.1186/s40035-018-0139-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sy M., Kitazawa M., Medeiros R., Whitman L., Cheng D., Lane T.E., Laferla F.M. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am. J. Pathol. 2011;178:2811–2822. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee D.C., Rizer J., Selenica M.L.B., Reid P., Kraft C., Johnson A., Blair L., Gordon M.N., Dickey C.A., Morgan D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ising C., Venegas C., Zhang S., Scheiblich H., Schmidt S.V., Vieira-Saecker A., Schwartz S., Albasset S., McManus R.M., Tejera D., et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669–673. doi: 10.1038/s41586-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morales I., Jiménez J.M., Mancilla M., Maccioni R.B. Tau oligomers and fibrils induce activation of microglial cells. J. Alzheimers Dis. 2013;37:849–856. doi: 10.3233/JAD-131843. [DOI] [PubMed] [Google Scholar]
- 14.World Health Organization WHO COVID-19 dashboard. 2020. https://covid19.who.int/
- 15.Malkova A., Kudryavtsev I., Starshinova A., Kudlay D., Zinchenko Y., Glushkova A., Yablonskiy P., Shoenfeld Y. Post COVID-19 syndrome in patients with asymptomatic/mild form. Pathogens. 2021;10:1408. doi: 10.3390/pathogens10111408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang R.D., Liu M.Q., Chen Y., Shan C., Zhou Y.W., Shen X.R., Li Q., Zhang L., Zhu Y., Si H.R., et al. Pathogenesis of SARS-CoV-2 in transgenic mice expressing human Angiotensin-converting Enzyme 2. Cell. 2020;182:50–58.e8. doi: 10.1016/j.cell.2020.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao L., Jin H., Wang M., Hu Y., Chen S., He Q., Chang J., Hong C., Zhou Y., Wang D., et al. Neurologic manifestations of hospitalized patients with Coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77:683–690. doi: 10.1001/jamaneurol.2020.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao L., Wang M., Chen S., He Q., Chang J., Hong C., Zhou Y., Wang D., Li Y., Jin H., Hu B. Neurological manifestations of hospitalized patients with COVID-19 in Wuhan, China: a retrospective case series study. medRxiv. 2020 doi: 10.1101/2020.02.22.20026500. Preprint at. [DOI] [Google Scholar]
- 19.Ye M., Ren Y., Lv T. Encephalitis as a clinical manifestation of COVID-19. Brain Behav. Immun. 2020;88:945–946. doi: 10.1016/j.bbi.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim G.U., Kim M.J., Ra S.H., Lee J., Bae S., Jung J., Kim S.H. Clinical characteristics of asymptomatic and symptomatic patients with mild COVID-19. Clin. Microbiol. Infect. 2020;26:948.e1–948.e3. doi: 10.1016/j.cmi.2020.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beyrouti R., Adams M.E., Benjamin L., Cohen H., Farmer S.F., Goh Y.Y., Humphries F., Jäger H.R., Losseff N.A., Perry R.J., et al. Characteristics of ischaemic stroke associated with COVID-19. J. Neurol. Neurosurg. Psychiatry. 2020;91:889–891. doi: 10.1136/jnnp-2020-323586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benussi A., Pilotto A., Premi E., Libri I., Giunta M., Agosti C., Alberici A., Baldelli E., Benini M., Bonacina S., et al. Clinical characteristics and outcomes of inpatients with neurologic disease and COVID-19 in Brescia, Lombardy, Italy. Neurology. 2020;95:e910–e920. doi: 10.1212/WNL.0000000000009848. [DOI] [PubMed] [Google Scholar]
- 23.Poyiadji N., Shahin G., Noujaim D., Stone M., Patel S., Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: CT and MRI features. Radiology. 2020;296:201187. doi: 10.1148/radiol.2020201187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tape C., Byrd K.M., Aung S., Lonks J.R., Flanigan T.P., Rybak N.R. COVID-19 in a patient presenting with syncope and a normal chest X-ray. R. I. Med. J. 2020;103:50–51. [PMC free article] [PubMed] [Google Scholar]
- 25.Poyiadji N., Shahin G., Noujaim D., Stone M., Patel S., Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology. 2020;296:E119–E120. doi: 10.1148/radiol.2020201187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Weyhern C.H., Kaufmann I., Neff F., Kremer M. Early evidence of pronounced brain involvement in fatal COVID-19 outcomes. Lancet. 2020;395:e109. doi: 10.1016/S0140-6736(20)31282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douaud G., Lee S., Alfaro-Almagro F., Arthofer C., Wang C., McCarthy P., Lange F., Andersson J.L.R., Griffanti L., Duff E., et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature. 2022;604:697–707. doi: 10.1038/s41586-022-04569-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin Y., Wu J., Chen T., Li J., Zhang G., Wu D., Zhou Y., Zheng N., Cai A., Ning Q., et al. Long-term microstructure and cerebral blood flow changes in patients recovered from COVID-19 without neurological manifestations. J. Clin. Invest. 2021;131:147329. doi: 10.1172/JCI147329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howell M.C., Green R., McGill A.R., Kahlil R.M., Dutta R., Mohapatra S.S., Mohapatra S. Activation of intracellular complement in lungs of patients with severe COVID-19 disease decreases T-cell activity in the lungs. Front. Immunol. 2021;12:700705. doi: 10.3389/fimmu.2021.700705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGill A.R., Kahlil R., Dutta R., Green R., Howell M., Mohapatra S., Mohapatra S.S. SARS-CoV-2 immuno-pathogenesis and potential for diverse vaccines and therapies: opportunities and challenges. Infect. Dis. Rep. 2021;13:102–125. doi: 10.3390/idr13010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu D., Biswal M., Neal A., Hai R. Review Devil's tools: SARS-CoV-2 antagonists against innate immunity. Curr. Res. Virol. Sci. 2021;2:100013. doi: 10.1016/j.crviro.2021.100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldeck D., Larbi A., Pellicanó M., Alam I., Zerr I., Schmidt C., Fulop T., Pawelec G. Enhanced chemokine receptor expression on leukocytes of patients with Alzheimer's disease. PLoS One. 2013;8:e66664. doi: 10.1371/journal.pone.0066664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corces M.R., Shcherbina A., Kundu S., Gloudemans M.J., Frésard L., Granja J.M., Louie B.H., Eulalio T., Shams S., Bagdatli S.T., et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer's and Parkinson's diseases. Nat. Genet. 2020;52:1158–1168. doi: 10.1038/s41588-020-00721-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novikova G., Kapoor M., Tcw J., Abud E.M., Efthymiou A.G., Chen S.X., Cheng H., Fullard J.F., Bendl J., Liu Y., et al. Integration of Alzheimer's disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 2021;12:1610. doi: 10.1038/s41467-021-21823-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuena A.R., Casolini P., Lattanzi R., Maftei D. Chemokines in Alzheimer's disease: new insights into prokineticins, chemokine-like proteins. Front. Pharmacol. 2019;10:622. doi: 10.3389/fphar.2019.00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mansour H.M., Fawzy H.M., El-Khatib A.S., Khattab M.M. Repurposed anti-cancer epidermal growth factor receptor inhibitors: mechanisms of neuroprotective effects in Alzheimer's disease. Neural Regen. Res. 2022;17:1913–1918. doi: 10.4103/1673-5374.332132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cicognola C., Janelidze S., Hertze J., Zetterberg H., Blennow K., Mattsson-Carlgren N., Hansson O. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer's Res. Ther. 2021;13:68. doi: 10.1186/s13195-021-00804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vélez J.I., Lopera F., Sepulveda-Falla D., Patel H.R., Johar A.S., Chuah A., Tobón C., Rivera D., Villegas A., Cai Y., et al. APOE∗E2 allele delays age of onset in PSEN1 E280A Alzheimer's disease. Mol. Psychiatr. 2016;21:916–924. doi: 10.1038/mp.2015.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brigas H.C., Ribeiro M., Coelho J.E., Gomes R., Gomez-Murcia V., Carvalho K., Faivre E., Costa-Pereira S., Darrigues J., de Almeida A.A., et al. IL-17 triggers the onset of cognitive and synaptic deficits in early stages of Alzheimer's disease. Cell Rep. 2021;36:109574. doi: 10.1016/j.celrep.2021.109574. [DOI] [PubMed] [Google Scholar]
- 40.Ojala J.O., Sutinen E.M. The role of interleukin-18, oxidative stress and metabolic syndrome in Alzheimer's disease. J. Clin. Med. 2017;6:E55. doi: 10.3390/jcm6050055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haddick P.C.G., Larson J.L., Rathore N., Bhangale T.R., Phung Q.T., Srinivasan K., Hansen D.V., Lill J.R., Alzheimer’s Disease Genetic Consortium ADGC Alzheimer’s Disease Neuroimaging Initiative ADNI. Pericak-Vance M.A., et al. A common variant of IL-6R is associated with elevated IL-6 pathway activity in Alzheimer's disease brains. J. Alzheimers Dis. 2017;56:1037–1054. doi: 10.3233/JAD-160524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Z., Zou X., Jin Y., Gao S., Lv J., Li B., Cui R. The role of KLF4 in Alzheimer's disease. Front. Cell. Neurosci. 2018;12:325. doi: 10.3389/fncel.2018.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan Y., Zheng Y., Xu D., Sun Z., Yang H., Yin Q. Galectin-3: a key player in microglia-mediated neuroinflammation and Alzheimer's disease. Cell Biosci. 2021;11:78. doi: 10.1186/s13578-021-00592-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao C.C., Cheng K.M., Ma Y.L., Hsu W.L., Chen Y.C., Fuh J.L., Lee W.J., Chao C.C., Lee E.H.Y. Galectin-3 promotes Abeta oligomerization and Abeta toxicity in a mouse model of Alzheimer's disease. Cell Death Differ. 2020;27:192–209. doi: 10.1038/s41418-019-0348-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dharshini S.A.P., Taguchi Y.H., Gromiha M.M. Investigating the energy crisis in Alzheimer disease using transcriptome study. Sci. Rep. 2019;9:18509. doi: 10.1038/s41598-019-54782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaudreault S.B., Dea D., Poirier J. Increased caveolin-1 expression in Alzheimer's disease brain. Neurobiol. Aging. 2004;25:753–759. doi: 10.1016/j.neurobiolaging.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 47.Wang L., Pavlov P.F., Kumar R., Winblad B. FKBP51-Hsp90 complex as a novel therapeutic target for Alzheimer’s disease. Alzheimers Dement. 2020;16 [Google Scholar]
- 48.Fujii T., Ota M., Hori H., Hattori K., Teraishi T., Matsuo J., Kinoshita Y., Ishida I., Nagashima A., Kunugi H. The common functional FKBP5 variant rs1360780 is associated with altered cognitive function in aged individuals. Sci. Rep. 2014;4:6696. doi: 10.1038/srep06696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blair L.J., Nordhues B.A., Hill S.E., Scaglione K.M., O'Leary J.C., 3rd, Fontaine S.N., Breydo L., Zhang B., Li P., Wang L., et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Invest. 2013;123:4158–4169. doi: 10.1172/JCI69003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blair L.J., Sabbagh J.J., Dickey C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer's disease. Expert Opin. Ther. Targets. 2014;18:1219–1232. doi: 10.1517/14728222.2014.943185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loeffler D.A., Klaver A.C., Coffey M.P., Aasly J.O., LeWitt P.A. Age-related decrease in heat shock 70-kDa protein 8 in cerebrospinal fluid is associated with increased oxidative stress. Front. Aging Neurosci. 2016;8:178. doi: 10.3389/fnagi.2016.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hur J.Y. Innate immunity protein IFITM3 in Alzheimer's disease. DNA Cell Biol. 2021;40:1351–1355. doi: 10.1089/dna.2021.0585. [DOI] [PubMed] [Google Scholar]
- 53.Tenner A.J. Complement-mediated events in Alzheimer's disease: mechanisms and potential therapeutic targets. J. Immunol. 2020;204:306–315. doi: 10.4049/jimmunol.1901068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hernandez M.X., Jiang S., Cole T.A., Chu S.H., Fonseca M.I., Fang M.J., Hohsfield L.A., Torres M.D., Green K.N., Wetsel R.A., et al. Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol. Neurodegener. 2017;12:66. doi: 10.1186/s13024-017-0210-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu X.C., Yu J.T., Jiang T., Wang P., Cao L., Tan L. CR1 in Alzheimer's disease. Mol. Neurobiol. 2015;51:753–765. doi: 10.1007/s12035-014-8723-8. [DOI] [PubMed] [Google Scholar]
- 56.Sanfilippo C., Castrogiovanni P., Imbesi R., Di Rosa M. CHI3L2 expression levels are correlated with AIF1, PECAM1, and CALB1 in the brains of Alzheimer's disease patients. J. Mol. Neurosci. 2020;70:1598–1610. doi: 10.1007/s12031-020-01667-9. [DOI] [PubMed] [Google Scholar]
- 57.Sabbir M.G. Loss of Ca(2+)/calmodulin dependent protein kinase kinase 2 leads to aberrant transferrin phosphorylation and trafficking: a potential biomarker for Alzheimer's disease. Front. Mol. Biosci. 2018;5:99. doi: 10.3389/fmolb.2018.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mairet-Coello G., Courchet J., Pieraut S., Courchet V., Maximov A., Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron. 2013;78:94–108. doi: 10.1016/j.neuron.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elliott E., Atlas R., Lange A., Ginzburg I. Brain-derived neurotrophic factor induces a rapid dephosphorylation of tau protein through a PI-3 Kinase signalling mechanism. Eur. J. Neurosci. 2005;22:1081–1089. doi: 10.1111/j.1460-9568.2005.04290.x. [DOI] [PubMed] [Google Scholar]
- 60.Plagman A., Hoscheidt S., McLimans K.E., Klinedinst B., Pappas C., Anantharam V., Kanthasamy A., Willette A.A., Alzheimer's Disease Neuroimaging Initiative Cholecystokinin and Alzheimer's disease neuroimaging. Neurobiol. Aging. 2019;76:201–207. doi: 10.1016/j.neurobiolaging.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shibata N., Kawarai T., Meng Y., Lee J.H., Lee H.S., Wakutani Y., Shibata E., Pathan N., Bi A., Sato C., et al. Association studies between the plasmin genes and late-onset Alzheimer's disease. Neurobiol. Aging. 2007;28:1041–1043. doi: 10.1016/j.neurobiolaging.2006.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao W., Konsolaki M. FKBP immunophilins and Alzheimer's disease: a chaperoned affair. J. Biosci. 2011;36:493–498. doi: 10.1007/s12038-011-9080-7. [DOI] [PubMed] [Google Scholar]
- 63.Hur J.Y., Frost G.R., Wu X., Crump C., Pan S.J., Wong E., Barros M., Li T., Nie P., Zhai Y., et al. The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer's disease. Nature. 2020;586:735–740. doi: 10.1038/s41586-020-2681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roy E.R., Wang B., Wan Y.W., Chiu G., Cole A., Yin Z., Propson N.E., Xu Y., Jankowsky J.L., Liu Z., et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 2020;130:1912–1930. doi: 10.1172/JCI133737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coulon P.G., Dhanushkodi N., Prakash S., Srivastava R., Roy S., Alomari N.I., Nguyen A.M., Warsi W.R., Ye C., Carlos-Cruz E.A., et al. NLRP3, NLRP12, and IFI16 inflammasomes induction and caspase-1 activation triggered by virulent HSV-1 strains are associated with severe corneal inflammatory herpetic disease. Front. Immunol. 2019;10:1631. doi: 10.3389/fimmu.2019.01631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reiken S., Sittenfeld L., Dridi H., Liu Y., Liu X., Marks A.R. Alzheimer's-like signaling in brains of COVID-19 patients. Alzheimers Dement. 2022;18:955–965. doi: 10.1002/alz.12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luna-Viramontes N.I., Campa-Córdoba B.B., Ontiveros-Torres M.Á., Harrington C.R., Villanueva-Fierro I., Guadarrama-Ortíz P., Garcés-Ramírez L., de la Cruz F., Hernandes-Alejandro M., Martínez-Robles S., et al. PHF-core tau as the potential initiating event for tau pathology in Alzheimer's disease. Front. Cell. Neurosci. 2020;14:247. doi: 10.3389/fncel.2020.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ward S.M., Himmelstein D.S., Lancia J.K., Binder L.I. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 2012;40:667–671. doi: 10.1042/BST20120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinisi A., Flach M., Sprenger F., Frank S., Tolnay M., Winkler D.T. Severe oligomeric tau toxicity can be reversed without long-term sequelae. Brain. 2021;144:963–974. doi: 10.1093/brain/awaa445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shafiei S.S., Guerrero-Muñoz M.J., Castillo-Carranza D.L. Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017;9:83. doi: 10.3389/fnagi.2017.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blair L.J., Frauen H.D., Zhang B., Nordhues B.A., Bijan S., Lin Y.C., Zamudio F., Hernandez L.D., Sabbagh J.J., Selenica M.L.B., Dickey C.A. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 2015;3:8. doi: 10.1186/s40478-015-0186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang F., Xu J., Xu S.J., Guo J.J., Wang F., Wang Q.W. Analysis and identification genetic effect of SARS-CoV-2 infections to Alzheimer's disease patients by integrated bioinformatics. J. Alzheimers Dis. 2022;85:729–744. doi: 10.3233/JAD-215086. [DOI] [PubMed] [Google Scholar]
- 73.Das M., Penn C., Martinez T., Mayilsamy K., McGill A., Wiling A., Mohapatra S.S., Mohapatra S. COVID-19 neurotropism and implications for therapy. Neuroimmunol. Neuroinflamm. 2020;7:141–149. [Google Scholar]
- 74.Rutkai I., Mayer M.G., Hellmers L.M., Ning B., Huang Z., Monjure C.J., Coyne C., Silvestri R., Golden N., Hensley K., et al. Neuropathology and virus in brain of SARS-CoV-2 infected non-human primates. Nat. Commun. 2022;13:1745. doi: 10.1038/s41467-022-29440-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Palomer E., Buechler J., Salinas P.C. Wnt signaling deregulation in the aging and Alzheimer's brain. Front. Cell. Neurosci. 2019;13:227. doi: 10.3389/fncel.2019.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seaks C.E., Wilcock D.M. Infectious hypothesis of Alzheimer disease. PLoS Pathog. 2020;16:e1008596. doi: 10.1371/journal.ppat.1008596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abbott A. Are infections seeding some cases of Alzheimer's disease? Nature. 2020;587:22–25. doi: 10.1038/d41586-020-03084-9. [DOI] [PubMed] [Google Scholar]
- 78.Twohig D., Nielsen H.M. alpha-synuclein in the pathophysiology of Alzheimer's disease. Mol. Neurodegener. 2019;14:23. doi: 10.1186/s13024-019-0320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haass C., Kaether C., Thinakaran G., Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Winslow A.R., Moussaud S., Zhu L., Post K.L., Post K.L., Dickson D.W., Berezovska O., McLean P.J. Convergence of pathology in dementia with Lewy bodies and Alzheimer's disease: a role for the novel interaction of alpha-synuclein and presenilin 1 in disease. Brain. 2014;137:1958–1970. doi: 10.1093/brain/awu119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaneko H., Kakita A., Kasuga K., Nozaki H., Ishikawa A., Miyashita A., Kuwano R., Ito G., Iwatsubo T., Takahashi H., et al. Enhanced accumulation of phosphorylated alpha-synuclein and elevated beta-amyloid 42/40 ratio caused by expression of the presenilin-1 deltaT440 mutant associated with familial Lewy body disease and variant Alzheimer's disease. J. Neurosci. 2007;27:13092–13097. doi: 10.1523/JNEUROSCI.4244-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All whole-transcriptome gene expression datasets analyzed during the current study are available in the NCBI Gene Expression Omnibus repository (https://www.ncbi.nlm.nih.gov/geo/) under the accession numbers GEO: GSE188847 and GSE118553. All other data generated or analyzed during this study are included in this published article (and its supplemental information).