

Abstract
Nucleus–mitochondria crosstalk is essential for cellular and organismal homeostasis. Although anterograde (nucleus-to-mitochondria) pathways have been well characterized, retrograde (mitochondria-to-nucleus) pathways remain to be clarified. Here, we found that mitochondrial dysfunction triggered a retrograde signaling via unique transcriptional and chromatin factors in hepatic cells. Our transcriptomic analysis revealed that the loss of mitochondrial transcription factor A led to mitochondrial dysfunction and dramatically induced expression of amphiregulin (AREG) and other secretory protein genes. AREG expression was also induced by various mitochondria stressors and was upregulated in murine liver injury models, suggesting that AREG expression is a hallmark of mitochondrial damage. Using epigenomic and informatic approaches, we identified that mitochondrial dysfunction-responsive enhancers of AREG gene were activated by c-JUN/YAP1/TEAD axis and were repressed by chromatin remodeler BRG1. Furthermore, while mitochondrial dysfunction-activated enhancers were enriched with JUN and TEAD binding motifs, the repressed enhancers possessed the binding motifs for hepatocyte nuclear factor 4α, suggesting that both stress responsible and cell type-specific enhancers were reprogrammed. Our study revealed that c-JUN and YAP1-mediated enhancer activation shapes the mitochondrial stress-responsive phenotype, which may shift from metabolism to stress adaptation including protein secretion under such stressed conditions.
Graphical Abstract
Graphical Abstract.
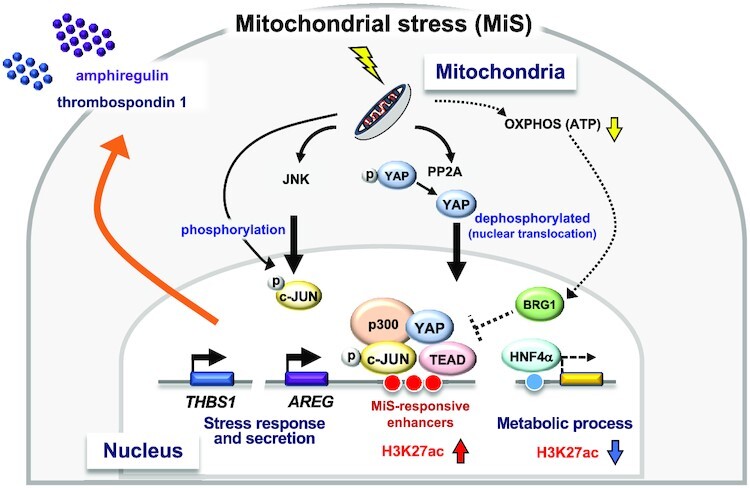
Proposed model for mitochondrial stress-induced epigenomic remodeling and AREG expression.
INTRODUCTION
Nucleus–mitochondria crosstalk is essential for cell maintenance and environmental responses (1). While mitochondria rely most of their function on nuclear genome-encoded proteins, mitochondria supplies the nucleus with metabolites such as acetyl-CoA and S-adenosylmethionine, which are essential for DNA and histone modifications (2,3). Thus, cells need to regulate both anterograde (nucleus-to-mitochondria) and retrograde (mitochondria-to-nucleus) pathways in a highly ordered manner to properly maintain cell function.
In the anterograde pathway, nuclear transcription factors such as peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and nuclear respiratory factor 1 (NRF1) promote the expression of mitochondria-related genes including mitochondrial transcription factor A (TFAM), which plays a pivotal role in regulating transcription, replication and organization of the mitochondrial genome (4–6). Therefore, TFAM is involved in metabolic activities in the mitochondria as an essential regulator between cellular organelles. In addition, chromatin regulators in the nucleus significantly contribute to mitochondrial functions. For example, lysine specific demethylase-1 (LSD1) inhibits oxidative phosphorylation (OXPHOS) and fatty acid oxidation by repressing the PGC-1α gene via flavin adenine dinucleotide (FAD)-dependent demethylation of histone H3 at lysine 4 (7,8). SETD8 (also known as PR-Set7) protects cellular senescence by limiting protein synthesis and energy metabolism through the mono-methylation of histone H4 at lysine 20 (9). These previous studies have established that mitochondrial function is controlled by the nuclear factors (10,11).
Mitochondria are important regulators of a variety of metabolites, including acetyl-CoA, S-adenosylmethionine, ATP, NAD+/NADH, FAD/FADH2 and second messengers such as free calcium and reactive oxygen species in mammalian cells (1,3). Because the production of these metabolites is influenced by mitochondria damage, they directly or indirectly constitute the retrograde pathway, also known as mitochondrial stress signaling. In addition, some transcription factors/cofactors such as SIRT3 contribute to retrograde signaling (12,13). Recently, G-protein pathway suppressor 2 (GPS2) was found to mediate the retrograde pathway via the mitochondria-to-nucleus translocation (14). However, not much is known about how damaged mitochondria transfer their signal to the nuclear machinery and change gene expression. In particular, mechanisms by which retrograde signal can induce specific epigenomic changes at specific loci remain unclear.
Human cases and mouse models of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) are associated with fat accumulation in the liver, mitochondrial dysfunction and cellular senescence in hepatocytes (15–18). These chronic fatty liver conditions widely affect the global adult population and are potentially severe because of the increased risk of metabolic syndrome and the development of hepatocellular carcinoma. Although mitochondrial stresses play crucial roles in disease development, it is unclear whether abnormal crosstalk between the nucleus and mitochondria coexists with NAFLD/NASH pathophysiology.
In this study, we identified a TFAM depletion-induced retrograde pathway that involves multiple signaling and transcription factors in hepatic cells. Our data reveal the molecular mechanism of mitochondrial stress-induced phenotype, which might explain how cells adapt and survive under stressful conditions.
MARERIALS AND METHODS
Cell culture
HepG2 cells were purchased from the Japanese Collection of Research Bioresources (JCRB) Cell Bank and cultured in Dulbecco’s modified Eagle’s medium (DMEM)-high glucose (Sigma) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) and penicillin/streptomycin (P/S). HeLa cells (JCRB) were cultured in DMEM/F-12 Ham medium (Sigma) supplemented with 10% FBS and P/S. IMR90 cells (ATCC) were cultured in Eagle’s minimum essential medium with Earle (Sigma) supplemented with 200 mM L-glutamine, 10% FBS, and P/S. To establish ρ0 cells, HepG2 cells were cultured in the above-mentioned DMEM-high glucose containing 1 μg/ml ethidium bromide, 100 μg/ml sodium pyruvate and 50 μg/ml uridine for 10 days (19). Mitochondrial DNA (mtDNA) was extracted using DNAzol Reagent (Thermo Fisher Scientific), and the tRNA-Leu and β2 microglobulin contents were quantified by quantitative real-time PCR. The amount of mtDNA was calculated using the tRNA-Leu/β2 microglobulin (20). For inhibition of oxidative phosphorylation, HepG2 cells were treated with 0.5 μg/ml of oligomycin (O4876, Sigma) for 24 h. For the knockdown experiments, siRNAs were transfected using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. For c-JUN knockdown, siGENOME Human JUN siRNA SMARTpool (Dharmacon) was used. Target sequences of siRNAs are listed in Supplementary Table S1. Unless otherwise stated, knockdown experiments were conducted after 72 h. An inhibitor of the c-JUN N-terminal kinase (JNK), SP600125 (420119, Sigma-Aldrich), was added to the medium at the indicated concentrations until 4–72 h after siRNA transfection.
Antibodies
For immunofluorescence, western blot analysis or ChIP-qPCR, the following antibodies were used in these experiments: TFAM (ab89818 or ab176558, Abcam), amphiregulin (AF262, R&D Systems), beta tubulin (66240–1-Ig, Proteintech), c-JUN (H-79) (sc-1694, Santa Cruz Biotechnology) or c-JUN (G-4) (sc-74543), phospho-c-Jun (Ser73) (D47G9) XP rabbit mAb (#3270, Cell Signaling Technology), YAP/TAZ (D24E4) (#8418) or YAP (D8H1X) XP® rabbit mAb (#14074), phospho-YAP (Ser127) (D9W2I) rabbit mAb (#13008), purified mouse anti-TEF-1 (610923, BD Biosciences), TFIID (TBP) (58C9) (sc-421), BRG-1 (G-7) (sc-17796), anti-FLAG M2 monoclonal, affinity purified (F1804, Sigma-Aldrich), anti-histone H3 (mono methyl K4) - ChIP grade (ab8895) or anti-histone H3 (acetyl K27) - ChIP grade (ab4729).
Transcriptome analysis
Genome-wide expression analysis was conducted using the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array. mRNA from HepG2 and HeLa cells was extracted using the RNeasy Mini Kit (QIAGEN), and cDNA synthesis, cRNA amplification, hybridization and scanning were provided by Takara Bio Inc. The raw data were normalized with the MAS5 algorithm, and data annotation analysis was conducted using the GeneSpring GX software (Agilent). The GeneChip array data have been deposited in the GEO under ID codes GSE196511. In addition, HNF4α-regulated genes were analyzed using the expression array data from HNF4α-KD HepG2 cells (GSE29084) (21). The raw data were normalized using the RMA algorithm.
Quantitative RT-PCR
Total RNA was extracted from cells using TRIzol reagent (Thermo Fisher Scientific). Complementary DNA was synthesized using ReverTra Ace qPCR RT Master Mix or ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO). Relative gene expression was quantified using THUNDERBIRD SYBR qPCR Mix or THUNDERBIRD Next SYBR qPCR Mix (TOYOBO) reagent and StepOnePlus or QuantStudio 3 Real Time PCR System (Thermo Fisher Scientific). Fold changes were determined using the 2-ΔΔCt method, and were calculated as the difference between the experimental and the respective control groups. Primer sequences are listed in Supplementary Table S2.
Immunofluorescence
Immunofluorescence of the cell lines was conducted as previously described (22). Briefly, cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature, washed with PBS, permeabilized with 0.2% Triton X-100 and 0.5% BSA in PBS for 5 min on ice, blocked with 0.5% bovine serum albumin in PBS, incubated with specific primary antibodies for 1 h at room temperature or overnight at 4°C, and then incubated with appropriate fluorescent-labeled secondary antibodies for 1 h at room temperature. The cells were counterstained with DAPI (1 μg/ml) before mounting. Images were obtained with a microscope IX-71 (Olympus) and the image acquisition software Lumina Vision Version 2.4 (Mitani).
Western blot analysis
At 72 h after siRNA transfection, cells were washed in PBS and lysed with ice-cold RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% (v/v) Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate) supplemented with a protease inhibitor and phosphatase inhibitor cocktails (Nacalai Tesque). For AREG detection, cells were treated with 5 μg/ml brefeldin A (022–15991, FUJIFILM Wako Chemicals)-containing medium for 6 h prior to harvesting. Lysates were subjected to SDS–PAGE, followed by immunoblotting using the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked with 5% skim milk and 0.3% Tween20 in PBS for 30 min at room temperature, incubated with primary antibody overnight at 4°C, and incubated with horseradish peroxidase-labeled secondary antibody for 1 h at room temperature. ECL Prime western blotting Detection Reagent (GE Healthcare) or Western Lightning Plus-ECL (PerkinElmer Japan) were used as the substrate. Band detection and quantification were conducted using ImageQuant LAS 4000 and ImageQuant TL (GE Healthcare).
Luciferase vector construction
To construct the pGL4.15-RE-AREG promoter, the BamHI-SalI fragment containing the SV40 early enhancer/promoter was deleted from pGL4.15. Additionally, 2.3 kb of the AREG promoter, amplified by PCR from the HepG2 genome, was inserted into the HindIII site. RE1, RE2 or RE3 fragments were also amplified from the HepG2 genome and inserted into the NheI-BglII site. The primers, amplicon size and restriction enzyme sites at both ends were as follows:
RE 1; RE 1_EcoRV-F, RE 1_BglII-R, 1.3 kb, EcoRV and BglII
RE 2; RE 2_XhoI-F, RE 2_EcoRV-R, 0.5 kb, XhoI and EcoRV
RE 3; RE 3_NheI-F, RE 3_XhoI-R5, 0.5 kb, NheI and XhoI
Luciferase reporter assay
Luciferase reporter analyses were conducted using a dual-luciferase reporter assay system (Promega) following the manufacturer’s protocol. For AREG enhancer analysis, the pGL4.15-RE-AREG promoter reporter vector was co-transfected with the reference vector pGL4.74[hRluc/TK] into the HepG2 cells. After 48 h of culture, the cells were collected for luciferase measurements. Data are presented as mean ± SD. Tukey’s honestly significant difference test was conducted with a significance level of 0.05, using JMP 14 software (SAS).
Cytoplasmic and nuclear extraction
Trypsinized cells were resuspended in nuclear extraction buffer (5 mM PIPES, pH 8.0, 85 mM KCl, 0.5% NP-40) supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail (Nacalai Tesque) by pipetting. After incubation on ice for 10 min, the cells were centrifuged at 830 × g for 5 min, and the supernatant was collected as the cytoplasmic fraction. The pellet was washed with ice-cold wash buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA) containing protease or phosphatase inhibitor cocktail, then resuspended in 2× sample buffer, centrifuged at 18 000 × g for 10 min at 4°C, and the supernatant was collected as the nuclear fraction.
Lentiviral transduction
To generate the CSII-EF-3xFLAG-IRES2-Bsd mock vector, the IRES2-Bsd fragment from CSII-CMV-MCS-IRES2-Bsd (RDB04385, RIKEN BRC DNA BANK) and an oligo with XhoI and EcoRI sites, added to both ends of the 3xFLAG sequence, were inserted into the BamHI-XbaI sites of CSII-EF-MCS (RDB04378). For overexpression of c-JUN, CSII-EF-3xFLAG-c-JUN-IRES2-Bsd was generated by inserting a 1 kb PCR amplicon from HepG2 cDNA into the EcoRI-BamHI sites of CSII-EF-3xFLAG-IRES2-Bsd. The PCR primers used were c-JUN_EcoRI-F and c-JUN_BamHI-R. For overexpression of YAP1 or YAP1(S127A) mutant, 1.5 kb fragments were amplified by PCR from p2xFlag CMV2-YAP2 (#19045, Addgene) or p2xFlag CMV2-YAP2-S127A mutant (#19050, Addgene) using YAP1_EcoRI-F and YAP1_NotI-R. Amplicons (1.5 kb) were inserted into the EcoRI-NotI sites of CSII-EF-3xFLAG-IRES2-Bsd. Primer sequences are listed in Supplementary Table S2. Each plasmid was co-transfected with pCAG-HIVgp (RDB04394) and pCMV-VSV-G-RSV-Rev (RDB04393) into the Lenti-X 293T cells (Takara Bio). After 48 h, the supernatant containing the virus was collected, filtered, and infected with HepG2 cells in the presence of 8 μg/ml polybrene. Infected cells were selected with 3 μg/ml blasticidin S (029–18701, FUJIFILM Wako Chemicals). For co-expression of c-JUN and YAP1, HepG2 cell lines established by mock, YAP1, or YAP1(S127A) lentivirus infection were infected with the c-JUN virus for 48 h.
Chromatin immunoprecipitation (ChIP)-qPCR
ChIP experiments for detecting modified histones were conducted as previously described (8). Briefly, cells were crosslinked with 1% formaldehyde for 10 min at room temperature. Following cell lysis, cells were sonicated by Picoruptor (Diagenode) for 10 min or treated with 10 U of Micrococcal nuclease per 1 × 106 nuclei at 37°C for 20 min. Chromatin fragments were incubated at 4°C overnight with the appropriate antibodies, followed by a pull-down assay using protein A/G-conjugated beads. Purified DNA was subjected to quantitative PCR (qPCR) using the primer sets listed in Supplementary Table S2. For ChIPs targeting transcription factors, Dynabeads M-280 sheep anti-rabbit or anti-mouse IgG (Thermo Fisher Scientific) were used for pull-down.
ChIP-seq analysis
For ChIP-seq analyses, 5 × 106 HepG2 cells per immunoprecipitation (IP) were collected 72 h after siRNA transfection. Crosslinking and fragmentation were conducted as described for the ChIP-qPCR. Chromatin fragments were incubated at 4°C overnight with 2 μg of appropriate antibodies and immunoprecipitated with Dynabeads protein A/G (Life Technologies). After decrosslinking, the DNA was purified using a QIAquick PCR Purification Kit (Qiagen). The ChIP-seq library was prepared using a NEBNext Ultra DNA Library Prep Kit and sequenced using the Illumina NextSeq 500/550 system (Illumina), according to the manufacturer’s protocol. The sequenced reads were mapped to the human reference genome (hg19) using the Burrows-Wheeler alignment algorithm with default parameters (23). Peak detection was performed using the MACS2 algorithm with an FDR cutoff value of 0.05, a composite broad region option (24), and selected peaks of enrichment greater than five times the control KD. The ChIP-seq data have been deposited in the GEO under ID codes GSE197242.
Transcription factors co-localized with the modified active enhancer mark regions were predicted using ChIP-Atlas (25) and the ChIP-seq data for c-JUN (GSM1700784) (26), YAP1 (GSM1614029) (27) and HNF4α (GSM469863) (28) were used for subsequent analyses. Peak annotations and motifs were analyzed using Homer (29), and the co-localization of ChIP-seq data was visualized using the compute matrix of deep tools (30). Functional enrichment analysis was conducted using clusterProfiler (31,32) with an FDR cutoff of 0.05. The results were visualized via dot plot or gene concept-network (cnet) plot.
For analyzing the active enhancer mark and transcription factor enrichment on enhancer regions of secretory protein genes induced by mitochondrial stress, enhancers of the genes were predicted using GeneHancer (33). ChIP-seq data of TFAM-depleted HepG2 cells, c-JUN (GSM1700784), YAP1 (GSM1614029) and TEAD1 (GSM1667161) were integrated using the Integrative Genomics Viewer (34).
Data analysis of murine NAFLD/NASH model
To analyze the in vivo mitochondrial stress in mice, RT-qPCR was conducted using liver samples. For the NAFLD model, 7-week-old male C57BL/6J mice were fed with normal-diet (ND), high-fat diet (HFD), or methionine and choline-deficient (MCD) diet for 4 weeks (35). For the NASH model, we reanalyzed the transcriptome dataset GSE137449 (36). Briefly, row count data obtained from RNA sequences in the liver of wild-type (WT) mice treated with ND or a choline-deficient L-amino acid-defined HFD (CDAHFD) were normalized using Deseq2 (37). Equality of variance was examined using with a significance level of 0.05.
Real-time measurement of OXPHOS activity
Cellular OXPHOS activity was monitored in real time using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience), according to the manufacturer’s instructions. HepG2 or HeLa cells were transfected with siRNAs 3 days prior to the assay, and 5 × 104 or 4 × 104 cells per well of HepG2 or HeLa cells were seeded on the Seahorse assay plate a day before the assay. The maximum OXPHOS capacity was determined as described previously (8).
Determination of intracellular ATP
To determine intracellular ATP concentrations, HepG2 cells were collected 48 h after siRNA transfection. ATP concentrations were determined using an ATP Bioluminescence Assay Kit CLS II (Roche), according to the manufacturer's instructions. ATP concentrations were normalized to the number of cells in the same samples.
Formaldehyde-assisted isolation of regulatory elements (FAIRE)-qPCR
FAIRE experiments to detect open chromatin sites were conducted according to a previously published protocol (38). Briefly, 9 × 105 HepG2 cells were seeded the day before siRNA transfection, and the cells were harvested 24 h after transfection. Cells were crosslinked with 1% formaldehyde for 10 min at room temperature, and after cell lysis, the DNA was sheared using a Bioruptor (Diagenode) for 10 min. The samples were de-crosslinked by overnight incubation at 65°C and purified by ethanol precipitation. The samples were subsequently treated with RNase A, purified using a QIAquick PCR purification kit (Qiagen), and used for qPCR analysis. The primer sets are listed in Supplementary Table S2.
Identification of super-enhancers
Super-enhancers were identified by Homer’s findPeaks tool based on the algorithm of Whyte et al. (39) using acetylated H3K27 peak data from control- and TFAM-knockdown HepG2 cells.
Analysis of genome-wide long-range interactions
To analyze the interaction of RE sites and distal genomic regions, we used ChromContact, a web tool to utilize the information obtained by Hi-C (40). Regions that interact with RE1, RE2 or RE3 were examined in AREG-expressing (NHEK, HMEC) and non-AREG-expressing (IMR90, HUVEC) cells at a resolution of 5 kb.
RNA sequencing data analysis of human NAFLD patients
Transcriptome data (GSE160016) from the livers of donors who underwent organ donation after cardiac death (6 non-NAFLD donors and 5 NAFLD donors) (41) were reanalyzed as human controls and mitochondrial stress conditions, respectively.
Motif Analysis of c-JUN and TEAD1
Potential binding sequences of AP-1 and TEAD (TEA domain transcription factor) near the RE1, RE2, and RE3 sites were predicted with MatInspector (http://www.genomatix.de) (42).
Definition of secretory protein
The 2641 genes predicted by The Human Protein Atlas (HPA) (www.proteinatlas.org) (43) according to the definition of ‘all ensemble genes with at least one predicted secretory transcript’ were used as secretory proteins.
RESULTS
Elevated expression of AREG is a hallmark of mitochondrial dysfunction
TFAM is a nuclear-encoded protein and indispensable for the function of mitochondrial genome. To understand the nuclear response against mitochondrial dysfunction (Figure 1A), we conducted transcriptome analyses in cultured human cells (HepG2 and HeLa) under TFAM knockdown (KD), using two siRNAs against the coding and 3′-untranslated sequences of TFAM mRNA (Supplementary Figure S1A). Under TFAM-KD for 72 h, both oxidative phosphorylation (OXPHOS) and glycolytic activities decreased, together with a reduction in intracellular ATP levels (Supplementary Figure S1B,C), indicating that metabolic dysfunction occurred in mitochondria. We found that 52 nuclear genes were commonly upregulated by ≥1.5-fold by TFAM-KD, while 23 genes were downregulated (Figure 1B and Supplementary Table S3). Among the genes upregulated by TFAM-KD, Amphiregulin (AREG) showed the highest induction ratio (Figure 1C), in addition to an enrichment of secretory protein genes such as thrombospondin-1 (THBS1) and CD44. AREG upregulation was verified by quantitative RT-PCR analyses (Figure 1D,E). We also found a dramatic increase of AREG expression in diploid fibroblast IMR90 under TFAM-KD (Figure 1E). By immunostaining analysis, we confirmed that the AREG protein was abundantly expressed in TFAM-KD cells (Figure 1F). Intracellular AREG in TFAM-KD cells was increased by the treatment with brefeldin A (BFA), an ER-Golgi trafficking inhibitor (Figure 1G), indicating that AREG was actively secreted. Notably, AREG upregulation was also found under two different mitochondrial stresses in HepG2 cells, OXPHOS inhibition by oligomycin treatment and mtDNA depletion (rho0 or ρ0 state) by long-term ethidium bromide treatment (Figure 1H,I). These data indicated that the induced expression of AREG is a hallmark of mitochondrial dysfunction, and thus a key target of the retrograde signaling.
Figure 1.

Transcriptome analyses identify AREG as a mitochondrial stress-induced secretory factor. (A) Study of mitochondrial stress-induced cellular response. Mitochondrial stresses stimulate signaling, transcriptional and epigenomic changes for nuclear gene control. (B) Transcriptome analyses of TFAM knockdown (KD) cells. The Venn diagrams show the number of genes whose expression levels changed by ≥1.5-fold after the distinct TFAM-KD experiments (siTFAM #1 or #2) in HepG2 and HeLa cells. (C) The top-ranked genes commonly upregulated by TFAM-KD include AREG and THBS1 (Supplementary Table S3). Among them, secretory protein genes are shown in the heatmap. (D) Inverse correlation of TFAM and AREG mRNAs after TFAM-KD. RT-qPCR analysis was conducted in the time course. siTFAM #1 was indicated as ‘siTFAM’ in most experiments throughout the study. (E) Induction of AREG mRNA by TFAM-KD. TFAM-KD for 72 h in HepG2, HeLa cells and IMR90 fibroblasts. mRNA levels were normalized by the RPLP0 (ribosomal protein lateral stalk subunit P0) gene and were indicated by mean ± SD (n = 3). **P < 0.01, *P < 0.05. (F) Immunofluorescence and western blot analyses of AREG protein in HepG2 cells. TFAM-KD was carried out for 72 h (left); scale bar: 10 μm. The cells were then treated with 5 μg/ml of brefeldin A (BFA) for 6 h to store intracellular AREG without being secreted (right). (G) Accumulation of AREG in IMR90 cells by TFAM-KD and BFA treatment. (H,I) Induction of AREG mRNA by various mitochondrial stresses. HepG2 treated with 0.5 μg/ml oligomycin for 24 h (H), HepG2-derived ρ0 cells established by ethidium bromide treatment for 10 days (I). mRNA levels were analyzed as described in panel (E).
Tfam downregulation and Areg upregulation in mouse liver tissues under mitochondrial stress
We then investigated whether the expression of TFAM and AREG are altered under mitochondrial stress conditions in vivo. When fed a high-fat diet (HFD) combined with methionine and choline deficiency (HFD/MCD), mice developed liver injury due to mitochondrial damage (44). By comparing the transcriptome profile of HFD/MCD-fed mouse liver with that of normal diet (ND)-fed mice (35), we found a clear downregulation of Tfam expression in the HFD/MCD mice, together with a significant inverse correlation with that of Areg (P < 0.01) (Figure 2A). Next, we analyzed the transcriptome dataset from another liver injury model, in which mice were fed either ND or L-amino acid-defined/choline-deficient HFD (CD) (36). In comparison with the ND-fed mice, Areg and other TFAM-KD induced genes (Figure 1C), including Thbs1 and Cd44, were significantly increased in the CD mice, in the presence of a decrease in Tfam expression (Figure 2B and Supplementary Figure S2A). In addition to mouse liver injury model, we evaluated human transcriptome datasets of liver specimens from non-alcoholic fatty liver disease (NAFLD) and non-NAFLD groups (41). AREG and TFAM transcripts tended to increase and decrease in the NAFLD group, respectively, in comparison with the non-NAFLD group (Supplementary Figure S2B). Collectively, the data suggest that the induction of Areg is a hallmark of liver injury, in which TFAM is inactivated.
Figure 2.

Tfam downregulation and Areg upregulation in mouse liver tissues under mitochondrial stresses in vivo. (A) Diet-induced murine NASH model corresponding to mitochondrial stresses. Seven-week-old male C57BL/6J mice were fed with normal diet (ND), high-fat diet (HFD) and HFD in combination with methionine and choline deficiency (MCD) for 4 weeks. RT-qPCR analysis of Tfam and Areg mRNAs from liver sections (n = 6). Box plot data are shown by the median and minimum/maximum whiskers. Inverse correlation between Tfam and Areg expression in the MCD group (right). Pearson product-moment correlation coefficient and P values are indicated. (B) Expression of mitochondrial stress-related genes such as Tfam, Areg, Thbs1 and Cd44 in L-amino acid-defined HFD/choline deficiency (CD)-fed mice that had moderate liver injury (n = 8). Data are from GSE137449; *P < 0.05, **P < 0.01.
Unique transcriptional regulators act on AREG gene enhancers under mitochondrial stress
Two tandem AREG genes, separated by a 170 kb-genomic sequence, are located within the EGF family gene cluster at human chromosome 4q13.3 (45) (Figure 3A). To evaluate the impact of mitochondrial dysfunction on the epigenome in HepG2 cells, we performed chromatin immunoprecipitation-sequencing (ChIP-seq) of mono-methylated H3K4 (H3K4me1) and acetylated H3K27 (H3K27ac), which are histone marks characteristic of enhancer activity (46,47). Based on the enrichment of these active enhancer marks by TFAM-KD, we identified three putative regulatory elements (REs) between AREG and betacellulin (BTC) genes, named RE1, RE2 and RE3, in this region (Figure 3A). Consistent with the upregulation of the AREG gene, TFAM-KD elevated the levels of both H3K4me1 and H3K27ac at these sites (Figure 3B). Further, these RE1-3 sites were selectively detected as a super-enhancer that showed the accumulation of H3K4me1 and H3K27ac in TFAM-KD cells (Supplementary Figure S3A). Because enhancer activation is often accompanied by chromatin opening, we performed a FAIRE-quantitative PCR to analyze the chromatin structure around the RE sites. We found constitutively accessible chromatin structure at RE1, RE2 and RE3 in both the control and TFAM-KD cells (Supplementary Figure S3B). Using the chromatin interaction database (ChromoContact) (40), we found that RE3 interacted with the promoter region of the 5′-located AREG gene, which formed an approximately 285 kb chromatin loop harboring two AREG genes in AREG-positive cells but not in AREG-negative cells (Supplementary Figure S3C). To directly assess the enhancer activity, functional reporter assays were conducted in which the human AREG promoter was fused to the luciferase reporter gene with various combinations of the RE sequences (Figure 3C). As expected, AREG/luciferase activities were induced by each RE and were synergistically augmented by combinations of RE under the TFAM-KD condition, suggesting that RE enhancers cooperatively regulate AREG induction.
Figure 3.

Epigenomic analyses reveal multiple transcriptional regulators acting on AREG gene enhancers under mitochondrial stress. (A) ChIP-seq analyses of active enhancer marks (H3K4me1 and H3K27ac) in HepG2 cells. After TFAM-KD for 72 h, enhancer marks-enriched sites were found between AREG and betacellulin (BTC) genes, named as RE1, RE2 and RE3. (B) Verification of H3K4me1 and H3K27ac enrichments at RE sites. Three independent ChIP-qPCR analyses were conducted under conditions similar to those in (A). The bars indicate the relative changes of modified histones (TFAM-KD versus control-KD) on a log2 scale. (C) Enhancer activity assay of single or multiple combinations of RE fused to AREG promoter-driven luciferase gene. Control and TFAM-KD HepG2 cells were analyzed 48 h after reporter plasmid transfection (n = 3). Differences in letters above the bars (a-h) indicate significant differences (Tukey’s HSD test, P < 0.05). (D) ChIP-Atlas analysis of transcriptional regulators at mitochondrial stress-responsive RE enhancers. The datasets were extracted from the top-ranked fold enrichment (Supplementary Table S4). The ChIP-seq data are from GSM1835989 for BRG1, GSM1240110 for p300, GSM1240109 for MLL4, GSM1700784 for c-JUN, GSM777644 for c-FOS, GSM1010772 for TEAD4, GSM1614029 for YAP1 and GSM1667161 for TEAD1. (E, F) Knockdown effects of RE-associated transcriptional regulators on AREG induction by TFAM depletion. RT-qPCR and immunofluorescence analyses were performed using indicated combinations of siRNAs against TFAM and each factor for 72 h in HepG2 cells; scale bar: 100 μm; **P < 0.01.
To identify transcriptional regulators that directly target these AREG enhancers, we compared our ChIP-seq data with published datasets using ChIP-Atlas (http://chip-atlas.org/) (25). The RE sites showed high enrichment of c-JUN, c-FOS, YAP1 and TEAD proteins in various AREG-expressing cells (Figure 3D and Supplementary Table S4). In addition, BRG1/SMARCA4, p300 and MLL4/KMT2D were significantly co-localized in the RE enhancers.
We then examined the contribution of these RE-associated factors to AREG induction by TFAM depletion. Under TFAM-KD, the inhibition of c-JUN, YAP1 or TEAD1 resulted in the reduction of AREG at both the RNA and protein levels (Figure 3E and Supplementary Figure S3D), indicating that these transcription factors promoted AREG induction. In contrast, the loss of BRG1 upregulated AREG expression (Figure 3F and Supplementary Figure S3D,E), indicating that this ATP-dependent chromatin remodeler suppressed AREG expression. Since BRG1 was decreased under TFAM-KD (Supplementary Figure S3F), in parallel with a decline in intracellular ATP (Supplementary Figure S1C), BRG1 activity might have been suppressed by mitochondrial dysfunction. Collectively, these results indicate that AREG expression is induced by dynamic activities of positive and negative regulators under mitochondrial stress.
c-JUN is upregulated by mitochondrial stress and directly promotes AREG expression
We next sought to gain mechanistic insights into how mitochondrial stress-responsive AREG enhancers are regulated. Among the factors involved in the upregulation of AREG genes, we first focused on c-JUN, since c-JUN expression was dramatically induced under TFAM-KD (Figure 1C), and all three RE enhancers contains the consensus motif for AP1 complex (c-FOS/c-JUN) (Supplementary Table S5). We found that phosphorylated c-JUN (p-c-JUN) at Ser73, a functionally active form of this protein (35,48–50), was increased in the nuclear fraction of the TFAM-KD cells (Figure 4A). Phosphorylated c-JUN was also increased in TFAM-depleted HeLa or IMR90 cells and in oligomycin-treated HepG2 cells (Supplementary Figure S4A). Consistently, p-c-JUN abundantly bound to the RE enhancers under TFAM inhibition, indicating its dynamic participation in the mitochondrial stress response (Figure 4B). To further test the involvement of c-JUN in AREG expression, we introduced a lentiviral vector to force c-JUN expression (Figure 4C). Interestingly, c-JUN overexpression strongly induced AREG expression (Figure 4D). Under these conditions, we observed no evident cell damage (Supplementary Figure S4B), or no changes in the expression of TFAM and mtDNA-coded genes such as cytochrome c oxidase subunit 1 (CO1), NADH dehydrogenase 1 (ND1) and NADH dehydrogenase 6 (ND6), or in the relative copy numbers of mtDNAs (Supplementary Figure S4C). Interestingly, the use of the JNK inhibitor, SP600125, did not significantly inhibit c-JUN phosphorylation and AREG induction under the TFAM-KD (Supplementary Figure S4D) (see the Discussion). These results suggest that mitochondrial dysfunction upregulates c-JUN, which in turn strongly enhance AREG expression.
Figure 4.

c-JUN and activated YAP1/TEAD1 induce AREG expression under mitochondrial stress. (A) Western blot analysis of c-JUN and p-c-JUN (phosphorylated c-JUN at Ser73) in cytoplasmic and nuclear fractions from HepG2 cells (control and TFAM-KD for 72 h). (B) ChIP-qPCR analysis of p-c-JUN at RE sites in the AREG gene region. Control and TFAM-KD cells for 72 h were tested (n = 3). Transcription start sites (TSSs) of AREG and JUN genes were used as negative and positive controls, respectively (left). Data are represented as mean ± SD. ** P < 0.01. Based on Supplementary Table S5, fold difference bars indicate the relative enrichments of p-c-JUN for the three REs (TFAM-KD vs. control-KD) on a log2 scale (right). (C) Overexpression of c-JUN using lentivirus gene transfer and blasticidin S selection in HepG2 cells. Representative images of mock and c-JUN-expressing virus infected cells (day 7) (Supplementary Figure S4B). (D) Expression of c-JUN and AREG in c-JUN overexpressing cells (n = 3); ** P < 0.01, *P< 0.05. (E) Western blot analysis of YAP1, TAZ, and TEAD1 in cytoplasmic and nuclear fractions from HepG2 cells (control and TFAM-KD for 72 h). (F, G) ChIP-qPCR analysis of YAP1 (F) and TEAD1 (G) at RE sites in the AREG gene region. Control and TFAM-KD cells for 72 h were tested (n = 3). TSS of AREG gene and other sites were used as negative controls (left). Data are represented as mean ± SD. **P < 0.01. Based on Supplementary Table S5, fold difference bars indicate the relative enrichments of YAP1 and TEAD1 at RE2/3 enhancers (TFAM-KD versus control-KD) on a log2 scale (right). (H) RT-qPCR analysis of AReG mRNA; n = 3, **P < 0.01, *P < 0.05.
Activated YAP1 targets AREG gene enhancers
We then examined the involvement of YAP1 and TEAD1 in the mitochondrial stress response, since YAP1 expression was upregulated in parallel with AREG and c-JUN in TFAM-KD HepG2 cells (Supplementary Figure S4E). YAP1 is phosphorylated and stabilized by the LAT kinases, whereas dephosphorylated YAP1 translocates to the nucleus, regulates gene expression together with TEAD1, and is then degraded by the proteasome pathway (51). TFAM depletion evidently increased cytoplasmic YAP1 in HepG2 cells (Figure 4E), but nuclear YAP1 was undetectable probably due to its degradation. The amount of TEAD1 in the nuclei was not affected by TFAM-KD. Similarly, TFAM depletion increased the total YAP1 as well as phosphorylated c-JUN in HeLa and IMR90 cells (Supplementary Figure S4A). The RE2/3 enhancers of the AREG genes, which contains the TEAD-binding motifs (Supplementary Table S5), were targeted by YAP1 and TEAD1 under TFAM-KD, indicating that YAP1 was actively involved in AREG induction (Figure 4F,G). To validate this event, we used lentivirus gene transfer to examine the overexpressing effect of wild-type YAP1 and constitutively active YAP1 (S127A; phosphorylation-defective) on AREG expression (Supplementary Figure S5A). Overexpression of YAP1 caused no evident cell damage. FLAG-tagged YAP1 (S127A) was short-lived at the protein level and was predominantly localized to the nucleus (Supplementary Figure S5B). Interestingly, YAP1 (S127A) overexpression moderately increased AREG expression (Figure 4H and Supplementary Figure S5B), possibly via the enrichment of YAP1 (S127A) and TEAD1 at RE2 (Supplementary Figure S5C), suggesting that YAP1 functions for AREG induction.
c-JUN and YAP1 cooperatively upregulate AREG and THBS1 genes
To investigate the cooperative role of c-JUN and YAP1, we examined the effect of co-expression of c-JUN with either YAP1 or YAP1 (S127A) in HepG2 cells (Figure 5A). We did not observe any mutual influence on the expression levels. Co-expression of c-JUN and YAP1 (S127A) upregulated the expression of AREG (Figure 5B), with an amplitude similar to TFAM-KD (Figure 1E). Notably, the combination of c-JUN and YAP1 did not affect the expression of TFAM, and mtDNA-coded CO1, ND1 and ND6 genes, suggesting that c-JUN and YAP1 enhanced the expression of AREG without inducing mitochondrial dysfunction.
Figure 5.

(A) Preparation of HepG2 cells that co-overexpressed c-JUN and YAP1 by lentivirus gene transfer. Expression levels of c-JUN, YAP1 and YAP1(S127A) mRNAs. (B) RT-qPCR analysis of AREG and THBS1, and mitochondria-related genes. n = 3, data are represented as mean ± SD; **P < 0.01, *P< 0.05. (C) Coexistence of active marks (H3K4me1 and H3K27ac), c-JUN and YAP1/TEAD1 in the proximal and distal enhancers of THBS1 gene. ChIP-seq data are from GSM1700784 for c-JUN, GSM1614029 for YAP1 and GSM1667161 for TEAD1.
In addition to AREG, we found a number of secretory protein-encoding genes, whose expression was significantly upregulated by TFAM-KD (Figure 1C). Among them, THBS1 was also upregulated by the co-expression c-JUN and YAP1 (Figure 5B). Our ChIP-seq analyses revealed that TFAM-KD increased the levels of both H3K4me1 and H3K27ac at the proximal and upstream distal enhancers of THBS1, which were occupied by c-JUN and YAP1/TEAD1 (Figure 5C). Similar data were obtained in genes encoding other secretory proteins, including CD44, C1orf54 (hypothetical) and ERLIN2 (Supplementary Figure S6), indicating that c-JUN and YAP1 play central roles in shaping mitochondrial stress-associated transcriptome. Further, this finding was emphasized by our data that AREG, THBS1, p-c-JUN and YAP1 were upregulated in ρ0 cells, while TFAM clearly disappeared (Supplementary Figure S7).
The loss of TFAM triggers epigenetic remodeling at enhancers associated with stress response and metabolism pathways
To explore whether c-JUN/YAP1/TEAD1 axis is prevalent in the chromatin regulation under mitochondrial stress, we analyzed the enrichment of transcription factor-binding motifs in regions with altered enhancer marks (H3K4me1 and H3K27ac) by TFAM-KD. JUN/AP-1 and TEAD consensus sequences were listed at the top-ranked motifs in regions with increased active enhancer marks (Figure 6A). Consistently, under TFAM-KD, enhancer marks were accumulated near the c-JUN- or YAP1-bound sites which were defined using publicly available ChIP-seq data (Figure 6B). By a functional enrichment analysis using clusterProfiler, we found that genes associated with various stress response pathways were enriched in 309 genes which were bound by c-JUN and upregulated by TFAM-KD with enriched enhancer marks (Figure 6C,D and Supplementary Table S6). In addition, both similar and unique pathways were enriched in 195 genes which were bound by YAP1 and upregulated by TFAM-KD with enriched enhancer marks (Figure 6D,E and Supplementary Table S6). Actually, 175 of the 195 YAP1-targeted genes were merged with the c-JUN-targeted genes (Supplementary Figure S8A). These results indicate that TFAM depletion triggers a c-JUN- and YAP-mediated epigenetic remodeling leading to stress responses. Notably, gene concept-network (cnet) plot analyses showed that these c-JUN- and YAP1-targeted genes encoded many secretory proteins such as AREG and THBS1 (Supplementary Figure S8B,C).
Figure 6.

Genome-wide gene regulation mediated by c-JUN, YAP1 and HNF4α under mitochondrial stress. (A) Enriched motifs in the regions with increased or decreased enhancer marks (H3K4me1 and H3K27ac) in TFAM depleted-HepG2 cells. Motifs for transcription factor binding were identified using the HOMER algorithm. (B) Enhancer marks near c-JUN, YAP1 or HNF4α binding sites (–3 to + 3 kb) in control and TFAM-KD cells. Normalized enrichment values are from GSM1700784 for c-JUN, GSM1614029 for YAP1 and GSM469863 for HNF4α. (C,E) Venn diagram of 309 genes merged with transcriptional upregulation (≥1.5-fold in TFAM-KD), increased enhancer marks by TFAM-KD and c-JUN binding (C), and 195 genes merged with transcriptional upregulation (≥1.5-fold in TFAM-KD), increased enhancer marks by TFAM-KD and YAP1 binding (E) (listed in Supplementary Table S6). (D) Top five Molecular Signatures Database (MSigDB) Hallmark pathways enriched in 309 c-JUN target genes (C) and 195 YAP1 target genes (E). The gene ratios of Peroxisome and TGF-β signaling in c-JUN targets and TGF-β signaling in YAP1 targets were 0.08, 0.05 and 0.06, respectively. FDR < 0.05. (F) Venn diagram of 325 genes merged with transcriptional downregulation (≥1.5-fold in TFAM-KD), decreased enhancer marks by TFAM-KD, and HNF4α binding (listed in Supplementary Table S6). (G) The MSigDB Hallmark pathways enriched in 325 overlapped genes (F). The gene ratios for Xenobiotic metabolism and Bile acid metabolism were 0.16 and 0.10, respectively; FDR < 0.05.
On the other hand, we found enriched binding motifs for cell type-specific transcription factors such as hepatocyte nuclear factor-4α (HNF4α) in regions with reduced enhancer marks by TFAM-KD (Figure 6A). Consistently, HNF4α-bound enhancers showed decreased H3K27ac levels in the TFAM-KD HepG2 cells (Figure 6B). 325 HNF4α-bound genes were overlapped with transcriptional downregulation and decreased enhancer marks under TFAM-KD (Figure 6F and Supplementary Table S6). Functional enrichment analysis of 325 overlapping genes revealed the pathways involved in liver-specific metabolic processes (Figure 6G), suggesting that multiple metabolic genes were influenced by HNF4α under mitochondrial stress. A large portion of such genes were positively regulated by HNF4α under normal condition (Supplementary Figure S8D), suggesting that HNF4α function may be compromised under mitochondrial dysfunction. Taken together, our study highlighted that TFAM depletion triggers a unique retrograde signaling to transcriptional and epigenetic reprogramming, possibly resulting in a shift from metabolism to stress responses including protein secretion.
DISCUSSION
Mitochondria-to-nucleus signaling profoundly contributes to cellular functions and homeostasis. Although key players for such retrograde signaling including metabolites, kinases and transcriptional regulators have been identified, not much is known about how these players dynamically communicate to generate specific gene expression patterns. Here, we found that the loss of TFAM caused mitochondrial dysfunction, stimulated cooperation of c-JUN and YAP1, and induced expression of the AREG gene with transcriptionally active marks at newly identified RE enhancers. Further, investigations of murine NAFLD/NASH models showed that Areg gene was upregulated, while the Tfam gene was downregulated in the liver tissues, suggesting that AREG expression is a hallmark of mitochondrial dysfunction in the liver. Thus, multiple signaling pathways cooperate to specifically induce transcriptional and epigenetic alterations when mitochondria are damaged.
Communication between the nucleus and mitochondria consists of anterograde and retrograde pathways (1). TFAM is a well-known transcription factor acting on mtDNA in the anterograde pathway (3,6,52). We found that TFAM depletion directly initiated mitochondrial stress or dysfunction, followed by a subsequent retrograde pathway. Thus, these bidirectional pathways are continuously linked to each other as a biological cycle. We also found that mitochondrial stress is transmitted to the nucleus via activation of c-JUN and YAP-mediated signaling, and that the resulting cells reprogram gene expression, allowing unique protein production and secretion. Although c-JUN is known to be functionally activated via its phosphorylation by the JNK (35,48–50), we observed that the JNK inhibitor, SP600125, did not diminish the increase in phosphorylated c-JUN under the TFAM-KD (Supplementary Figure S4D), indicating that other kinases may operate for this mitochondrial stress response. In addition, YAP proteins translocate to the nucleus through dephosphorylation mediated by protein phosphatase 2A (PP2A) (51,53). The use of the PP2A inhibitor, LB-100, increased the phosphorylated form of YAP1 under TFAM-KD (data not shown), suggesting that PP2A is involved in the mitochondrial stress response. However, our experimental condition using TFAM-KD may represent the early stage of mitochondrial dysfunction, as gene sets involved in the mitochondrial unfolded protein response (UPRmt) (54,55) did not vary in the cells tested (data not shown). If c-JUN and YAP1 are more essential for acute response to mitochondrial stress, it may be reasonable that we observed no evident upregulation of c-Jun and Yap1 mRNAs in murine NAFLD/NASH models (data not shown). In addition, the loss of Tfam might have distinct effects on gene control including Areg expression, depending on cell types and experimental conditions (56).
Our ChIP-seq investigations revealed that mitochondrial stress frequently activated distal enhancers of target genes, which were enriched with c-JUN and YAP1/TEAD1 binding (Figures 3D, 5C and Supplementary Figure S6). Additionally, such distal enhancers were accompanied by the accumulation of chromatin remodeling factors such as p300, MLL4, and BRG1 in the AREG gene (Figure 3D and data not shown in other genes). It has been previously reported that AP-1 (c-FOS/c-JUN) and YAP/TAZ/TEAD are colocalized at distal enhancers of proliferative genes and promote growth via chromatin loop formation between enhancers and promoters in breast cancer cells (27). In fibroblasts, the AP-1 complex, together with TEAD and lineage-specific transcription factors, binds to nucleosome-occluded enhancers and recruits the BAF chromatin remodeling complex to establish an accessible chromatin state (57). These results suggest that c-JUN and YAP/TEAD collaborate with the chromatin remodeling complex at distal enhancers to induce selective expression of target genes. In other words, combinations of c-JUN and YAP/TEAD may be an enhancer repertoire strategy.
For the YAP-driven cell growth and tumorigenesis, robust YAP binding was restricted to a relatively small number of hyperactive distal enhancers. YAP interacted with the transcriptional mediator complexes at the enhancers, which allowed the recruitment of cyclin-dependent kinase 9, which regulated the promoter-proximal polymerase II (Pol II) pause release (58). This enabled rapid and regulatory gene induction, similar to the immediate early genes (59,60). Consistently, we found that the mediator complex subunit 1 (MED1) and cyclin-dependent kinase 8 accumulated at the YAP1-enriched RE enhancers of the AREG gene (data not shown). Thus, acute mitochondrial stress responses may utilize the Pol II pause release to support rapid gene control.
Interestingly, mitochondrial stress induced production or secretion of newly synthesized proteins such as AREG and THBS1. AREG is an autocrine or paracrine growth modulatory factor while THBS1 plays a role in extracellular matrix and cell-to-cell interactions. Both proteins have been demonstrated to control the proliferation of various cells under developmental, inflammatory, senescent and cancerous conditions (61–63). There is the possibility that this secretion is a compensatory growth-promoting alert from mitochondrial dysfunction cells to acceptor cells in the tissue.
Cellular senescence is induced by various stresses, such as oncogene induction, telomere attrition, DNA damage, and deregulation of chromatin. Senescent cells have been found to cause mitochondrial dysfunction, either activation or decline, and a proinflammatory program referred to as the senescence-associated secretory phenotype (SASP) (64,65). Wiley et al. have reported that knockdown of sirtuin 3 (SIRT3), a mitochondrial sirtuin, led to senescence in human fibroblasts which was not accompanied by the expression of proinflammatory cytokines (66). Interestingly, SIRT3 depletion-induced senescent cells upregulated the AREG and IL-10 genes. This may be consistent with our results that cells undergoing mitochondrial dysfunction acquired the unique production of AREG and THBS1 in hepatic cells and liver tissues.
Chronic liver diseases such as NAFLD/NASH represent hepatic triglyceride accumulation, mitochondrial dysfunction, and cellular senescence in the liver (15–18). Mitochondrial stress is deeply implicated in the development of these diseases, as the activities of OXPHOS and ATP synthesis decrease in liver tissues from these patients. Importantly, hepatic lipid overload is known to induce a c-JUN-mediated stress response, which is often associated with NAFLD pathogenesis (35). In the present study, murine NAFLD/NASH models showed upregulation of Areg and downregulation of Tfam gene in liver tissues (Figure 2). The human liver transcriptome of NAFLD patients exhibited a similar tendency of AREG and TFAM expression (Supplementary Figure S2). Our data suggest that alteration of anterograde and/or retrograde pathways contributes to NAFLD/NASH pathophysiology, which may be emerging diagnostic/therapeutic targets.
Taken together, our study highlights the molecular basis of mitochondria-to-nucleus signaling and the mitochondrial stress-induced transcriptional and epigenomic remodeling, and promotes our understanding of the mitochondrial dysfunction-driven cellular responses.
DATA AVAILABILITY
The sequencing data generated for this study are available in the Gene Expression Omnibus (GEO) database (GSE196511 and GSE197242).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members in our laboratory for helpful discussions, especially Dr. Tatsuro Yamamoto for assisting data analyses.
Contributor Information
Yuko Hino, Department of Medical Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University, Kumamoto 860-0811, Japan.
Katsuya Nagaoka, Department of Medical Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University, Kumamoto 860-0811, Japan.
Shinya Oki, Department of Drug Discovery Medicine, Kyoto University Graduate School of Medicine, Kyoto 606-8507, Japan.
Kan Etoh, Department of Medical Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University, Kumamoto 860-0811, Japan.
Shinjiro Hino, Department of Medical Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University, Kumamoto 860-0811, Japan.
Mitsuyoshi Nakao, Department of Medical Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University, Kumamoto 860-0811, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
JSPS KAKENHI [21H02686, 20KK0185 to M.N., 20H04108, 21K19513 to S.H.]; Takeda Science Foundation (to M.N., S.H.); Naito Foundation (to M.N.); Japan Agency for Medical Research and Development [21gk0210029h0101 to S.H.]; the program of the Inter-University Research Network for High Depth Omics, IMEG, Kumamoto University. Funding for open access charge: JSPS KAKENHI.
Conflict of interest statement. None declared.
REFERENCES
- 1. Quirós P.M., Mottis A., Auwerx J.. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016; 17:213–226. [DOI] [PubMed] [Google Scholar]
- 2. Mottis A., Herzig S., Auwerx J.. Mitocellular communication: shaping health and disease. Science. 2019; 366:827–832. [DOI] [PubMed] [Google Scholar]
- 3. Weinhouse C. Mitochondrial-epigenetic crosstalk in environmental toxicology. Toxicol. 2017; 391:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larsson N.-G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G.S., Clayton D.A.. Mitochondrial transcription factor a is necessary for mtDNA maintance and embryogenesis in mice. Nat. Genet. 1998; 18:231–236. [DOI] [PubMed] [Google Scholar]
- 5. Parisi M.A., Clayton D.A.. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. 1991; 252:965–969. [DOI] [PubMed] [Google Scholar]
- 6. Scarpulla R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008; 88:611–638. [DOI] [PubMed] [Google Scholar]
- 7. Anan K., Hino S., Shimizu N., Sakamoto A., Nagaoka K., Takase R., Kohrogi K., Araki H., Hino Y., Usuki S.et al.. LSD1 mediates metabolic reprogramming by glucocorticoids during myogenic differentiation. Nucleic Acids Res. 2018; 46:5441–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hino S., Sakamoto A., Nagaoka K., Anan K., Wang Y., Mimasu S., Umehara T., Yokoyama S., Kosai K.I., Nakao M.. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 2012; 3:758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanaka H., Takebayashi S., Sakamoto A., Igata T., Nakatsu Y., Saitoh N., Hino S., Nakao M.. The SETD8/PR-Set7 methyltransferase functions as a barrier to prevent senescence-associated metabolic remodeling. Cell Rep. 2017; 18:2148–2161. [DOI] [PubMed] [Google Scholar]
- 10. Nakao M., Anan K., Araki H., Hino S.. Distinct roles of the NAD+-Sirt1 and FAD-LSD1 pathways in metabolic response and tissue development. Trends Endocrinol. Metab. 2019; 30:409–412. [DOI] [PubMed] [Google Scholar]
- 11. Nakao M., Tanaka H., Koga T.. Cellular senescence variation by metabolic and epigenomic remodeling. Trends Cell Biol. 2020; 30:919–922. [DOI] [PubMed] [Google Scholar]
- 12. Scher M.B., Vaquero A., Reinberg D.. SirT3 is a nuclear NAD + -dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007; 21:920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwer B., North B.J., Frye R.A., Ott M., Verdin E.. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide–dependent deacetylase. J. Cell Biol. 2002; 158:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cardamone M.D., Tanasa B., Cederquist C.T., Huang J., Mahdaviani K., Li W., Rosenfeld M.G., Liesa M., Perissi V.. Mitochondrial retrograde signaling in mammals is mediated by the transcriptional cofactor GPS2 via direct mitochondria-to-nucleus translocation. Mol. Cell. 2018; 69:757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loomba R., Friedman S.L., Shulman G.I.. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021; 184:2537–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mansouri A., Gattolliat C.H., Asselah T.. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterol. 2018; 155:629–647. [DOI] [PubMed] [Google Scholar]
- 17. Samuel V.T., Shulman G.I.. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018; 27:22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sunny N.E., Bril F., Cusi K.. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 2017; 28:250–260. [DOI] [PubMed] [Google Scholar]
- 19. Nass M.M. Abnormal DNA patterns in animal mitochondria: ethidium bromide-induced breakdown of closed circular DNA and conditions leading to oligomer accumulation. Proc. Natl. Acad. Sci. USA. 1970; 67:1926–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rooney J., Ryde I., Sanders L., Howlett E., Colton M., Germ K., Mayer G., Greenamyre J., Meyer J.. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol. 2015; 1241:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolotin E., Liao H., Ta T.C., Yang C., Hwang-Verslues W., Evans J.R., Jiang T., Sladek F.M.. Integrated approach for the identification of human hepatocyte nuclear factor 4α target genes using protein binding microarrays. Hepatol. 2010; 51:642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saitoh N., Sakamoto C., Hagiwara M., Agredano-Moreno L.T., Jimeńez-Garciá L.F., Nakao M.. The distribution of phosphorylated SR proteins and alternative splicing are regulated by RANBP2. Mol. Biol. Cell. 2012; 23:1115–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H., Durbin R.. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nussbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of chip-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O.. ChIP-Atlas: a data-mining suite powered by full integration of public chip-seq data. EMBO Rep. 2018; 19:e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Z., Lan X., Wu D., Sunkel B., Ye Z., Huang J., Liu Z., Clinton S.K., Jin V.X., Wang Q.. Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat. Commun. 2015; 6:8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zanconato F., Forcato M., Battilana G., Azzolin L., Quaranta E., Bodega B., Rosato A., Bicciato S., Cordenonsi M., Piccolo S.. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015; 17:1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Daigo K., Kawamura T., Ohta Y., Ohashi R., Katayose S., Tanaka T., Aburatani H., Naito M., Kodama T., Ihara S.et al.. Proteomic analysis of native hepatocyte nuclear factor-4α (HNF4α) isoforms, phosphorylation status, and interactive cofactors. J. Biol. Chem. 2011; 286:674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu G., Wang L.G., Han Y., He Q.Y.. ClusterProfiler: an r package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L.et al.. 2021) clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov. 2:100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Stein T.I., Rosen N., Kohn A., Twik M., Safran M.et al.. GeneHancer: Genome-wide integration of enhancers and target genes in genecards. Database. 2017; 2017:bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nagaoka K., Hino S., Sakamoto A., Anan K., Takase R., Umehara T., Yokoyama S., Sasaki Y., Nakao M.. Lysine-specific demethylase 2 suppresses lipid influx and metabolism in hepatic cells. Mol. Cell. Biol. 2015; 35:1068–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heintz M.M., McRee R., Kumar R., Baldwin W.S.. Gender differences in diet-induced steatotic disease in Cyp2b-null mice. PLoS One. 2020; 15:e0229896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waki H., Nakamura M., Yamauchi T., Wakabayashi K.ichi, Yu J., Hirose-Yotsuya L., Take K., Sun W., Iwabu M., Okada-Iwabu M.et al.. Global mapping of cell type-specific open chromatin by FAIRE-seq reveals the regulatory role of the NFI family in adipocyte differentiation. PLoS Genet. 2011; 7:e1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sato T., Suyama M.. ChromContact: a web tool for analyzing spatial contact of chromosomes from Hi-C data. BMC Genomics. 2015; 16:1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hou J., Zhang J., Cui P., Zhou Y., Liu C., Wu X., Ji Y., Wang S., Cheng B., Ye H.et al.. TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J. Clin. Invest. 2021; 131:e135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Quandt K., Frech K., Karas H., Wingender E., Werner T.. Matlnd and matlnspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995; 23:4878–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A.et al.. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 44. Hebbard L., George J.. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2011; 8:35–44. [DOI] [PubMed] [Google Scholar]
- 45. Berasain C., Avila M.A.. Amphiregulin. Semin. Cell Dev. Biol. 2014; 28:31–41. [DOI] [PubMed] [Google Scholar]
- 46. Zhou V.W., Goren A., Bernstein B.E.. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011; 12:7–18. [DOI] [PubMed] [Google Scholar]
- 47. Calo E., Wysocka J.. Modification of enhancer chromatin: what, how, and why. Mol. Cell. 2013; 49:825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Czaja M.J. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol. Metab. 2010; 21:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seki E., Brenner D.A., Karin M.. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterol. 2012; 143:307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zeke A., Misheva M., Reményi A., Bogoyevitch M.A.. JNK signaling: regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev. 2016; 80:793–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meng Z., Moroishi T., Guan K.L.. Mechanisms of hippo pathway regulation. Genes Dev. 2016; 30:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Campbell C.T., Kolesar J.E., Kaufman B.A.. Mitochondrial transcription factor a regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta - Gene Regul. Mech. 2012; 1819:921–929. [DOI] [PubMed] [Google Scholar]
- 53. Schlegelmilch K., Mohseni M., Kirak O., Pruszak J., Rodriguez J.R., Zhou D., Kreger B.T., Vasioukhin V., Avruch J., Brummelkamp T.R.et al.. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell. 2011; 144:782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Melber A., Haynes C.M.. UPR mt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018; 28:281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Münch C., Harper J.W.. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature. 2016; 534:710–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fu Z., Ye J., Dean J.W., Bostick J.W., Weinberg S.E., Xiong L., Oliff K.N., Chen Z.E., Avram D., Chandel N.S.et al.. Requirement of mitochondrial transcription factor a in tissue-resident regulatory t cell maintenance and function. Cell Rep. 2019; 28:159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vierbuchen T., Ling E., Cowley C.J., Couch C.H., Wang X., Harmin D.A., Roberts C.W.M., Greenberg M.E.. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell. 2017; 68:1067–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Galli G.G., Carrara M., Yuan W.C., Valdes-Quezada C., Gurung B., Pepe-Mooney B., Zhang T., Geeven G., Gray N.S., de Laat W.et al.. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol. Cell. 2015; 60:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Adelman K., Kennedy M.A., Nechaev S., Gilchrist D.A., Muse G.W., Chinenov Y., Rogatsky I.. Immediate mediators of the inflammatory response are poised for gene activation through RNA polymerase II stalling. Proc. Natl. Acad. Sci. USA. 2009; 106:18207–18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Donner A.J., Ebmeier C.C., Taatjes D.J., Espinosa J.M.. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010; 17:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gutierrez L.S., Gutierrez J.. Thrombospondin 1 in metabolic diseases. Front. Endocrinol. (Lausanne). 2021; 12:638536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zaiss D.M.W., Gause W.C., Osborne L.C., Artis D.. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. 2015; 42:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Busser B., Sancey L., Brambilla E., Coll J.L., Hurbin A.. The multiple roles of amphiregulin in human cancer. Biochim. Biophys. Acta - Rev. Cancer. 2011; 1816:119–131. [DOI] [PubMed] [Google Scholar]
- 64. Coppé J.P., Patil C.K., Rodier F., Sun Y., Muñoz D.P., Goldstein J., Nelson P.S., Desprez P.Y., Campisi J.. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008; 6:2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kuilman T., Peeper D.S.. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer. 2009; 9:81–94. [DOI] [PubMed] [Google Scholar]
- 66. Wiley C.D., Velarde M.C., Lecot P., Liu S., Sarnoski E.A., Freund A., Shirakawa K., Lim H.W., Davis S.S., Ramanathan A.et al.. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016; 23:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated for this study are available in the Gene Expression Omnibus (GEO) database (GSE196511 and GSE197242).