

Abstract
Telomerase reverse transcriptase (TERT) (catalytic subunit of telomerase) is linked to the development of coronary artery disease (CAD); however, whether the role of nuclear vs. mitchondrial actions of TERT is involved is not determined. Dominant-negative TERT splice variants contribute to decreased mitochondrial integrity and promote elevated reactive oxygen species production. We hypothesize that a decrease in mitochondrial TERT would increase mtDNA damage, promoting a pro-oxidative redox environment. The goal of this study is to define whether mitochondrial TERT is sufficient to maintain nitric oxide as the underlying mechanism of flow-mediated dilation by preserving mtDNA integrity.Immunoblots and quantitative polymerase chain reaction were used to show elevated levels of splice variants α- and β-deletion TERT tissue from subjects with and without CAD. Genetic, pharmacological, and molecular tools were used to manipulate TERT localization. Isolated vessel preparations and fluorescence-based quantification of mtH2O2 and NO showed that reduction of TERT in the nucleus increased flow induced NO and decreased mtH2O2 levels, while prevention of mitochondrial import of TERT augmented pathological effects. Further elevated mtDNA damage was observed in tissue from subjects with CAD and initiation of mtDNA repair mechanisms was sufficient to restore NO-mediated dilation in vessels from patients with CAD. The work presented is the first evidence that catalytically active mitochondrial TERT, independent of its nuclear functions, plays a critical physiological role in preserving NO-mediated vasodilation and the balance of mitochondrial to nuclear TERT is fundamentally altered in states of human disease that are driven by increased expression of dominant negative splice variants.
Keywords: coronary artery disease, mitochondria, telomerase, microcirculation, endothelial function, mitochondrial DNA damage
ABBREVIATIONS
- ACh
acetylcholine
- AGS 499
small molecule TERT activator
- ANG II
angiotensin II
- BIBR 1532
small molecule inhibitor of TERT activity
- CAD
coronary artery disease
- c-PTIO
carboxy-PTIO potassium salt
- ENDO III
recombinant endonuclease III
- FL
full length
- FMD
flow mediated dilation
- GFP
green fluorescent protein
- H2O2
hydrogen peroxide
- L-NAME
Nω-nitro-L-arginine methyl ester
- LV
left ventricular
- mENDO III
mutant ENDO III
- MitoPY1
mitochondrial specific H2O2 probe
- mtDNA
mitochondrial DNA
- mtH2O2
mitochondrial derived hydrogen peroxide
- mtROS
mitochondrial reactive oxygen species
- MTS
mitochondrial target sequence
- NLS
nuclear target sequence
- NO
nitric oxide
- non-CAD
noncoronary artery disease (not clinical diagnosis and ≤1 co-morbidities/risk factors)
- Pap
papaverine
- RMRP
mitochondrial RNA processing endoribonuclease
- TERT
telomerase reverse transcriptase
- α-del
α-deletion
- β-del
β-deletion
Introduction
Cardiovascular disease remains the single greatest cause of mortality in the United States and represents a growing global public health threat. The microcirculation is increasingly recognized as a critical organ in the development of a broad range of cardiovascular diseases.3–8 Endothelial dysfunction, which manifests as reduced nitric oxide (NO) bioavailability, is considered an early marker for many cardiovascular diseases, including coronary artery disease (CAD). Indeed, many studies have demonstrated endothelial dysfunction prior to onset of CAD using acetylcholine or flow-mediated dilation (FMD) stimulation.10 In contrast to conduit arteries, the human microcirculation of subjects with CAD maintains FMD, despite the loss of endothelial NO release.8,11 In these subjects, the mechanism of dilation shifts to mitochondria-derived hydrogen peroxide (H2O2).5,12 This switch in the mediator of dilation preserves FMD but creates a potentially toxic environment in which vascular and parenchymal tissue are exposed to proinflammatory H2O213 instead of anti-inflammatory and antioxidative NO.14 Understanding the pathways and mechanisms responsible for this switch in the mediator of FMD may be the key to minimizing tissue stress or injury from vascular paracrine oxidative toxicity.
We have previously shown that telomerase reverse transcriptase (TERT), the catalytic subunit of telomerase,15 plays a noncanonical role in preventing the increase of mitochondrial reactive oxygen species (mtROS) in coronary arterioles from subjects with CAD. Specifically, activation of TERT can reverse the mechanism of flow-induced, endothelium-dependent dilation from H2O2 to NO, restoring the phenotype observed in subjects without CAD.16 Furthermore, our previous work showed that TERT-deficient mice displayed a H2O2-mediated FMD, which mimics the CAD phenotype.17 Transgenic overexpression of TERT protects against Angiotensin II (ANG II)-induced endothelial dysfunction18,19 in a mechanism that is partly mediated by suppression of mtROS levels. Taken together, these findings suggest that TERT protects endothelial integrity and maintains NO-dependent vasodilation, while the loss of TERT results in an H2O2-dependent, CAD-like vasodilator phenotype. However, the mechanisms by which TERT confers endothelial protection remain under examination.
mtROS is associated with the development of arteriolosclerosis and endothelial dysfunction, which predisposes individuals to vascular complications.20 It remains unclear if elevated levels of mtROS represent a causative event or is simply a result of decreased mitchondrial integrity, including elevated mitochondrial DNA (mtDNA) damage. As a number of cardiovascular pathologies have been associated with changes in mitochondrial structure and function,21,22 yet a large body of evidence of failed antioxidant trials23 implies a more complex relationship between mitchondrial defects and disease progression/development. In rodent models of heart failure, targeted mtDNA repair is sufficient to improve microvascular endothelial function in the coronary and systemic microcirculation and improves cardiac function.24,25 This evidence suggests a strong correlation between mtDNA damage and endothelial dysfunction that may represent a valid target for new therapytics. To date, targeting mtDNA damage as means to restore physiological function has not been tested in the human microcircuilation.
The pleiotropic functions of TERT in a range of cellular processes have been linked to TERT's subcellular localization (eg, in the nucleus, mitochondria, or cytosol). In fact, prior evidence suggests a close connection of mitochondrial TERT and development of mtDNA damage.26,27 TERT roles in transcriptional regulation and telomere extension have been extensively studied,28–30 the regulation of TERT through mRNA splicing events and the functions of the different splice variants are less well understood. TERT pre-mRNA is alternatively spliced in many species and, to date, more than 20 human TERT transcript variants have been detected.31–34 The 2 most-studied TERT alternative splice variants are spliced at the α and β sites. Skipping 36 nucleotides in exon 6, deletes the α site, while alternate splicing at the β site in exon 7 and 8 results in a 183-nucleotide deletion and generates a transcript containing a premature termination codon32 and leads to a loss of enzymatic activity. Interestingly, studies have demonstrated that when overexpressed, the β-deletion (β-del) TERT isoform competes with the full length (FL) TERT isoform for binding to the telomerase RNA component, thereby inhibiting endogenous telomerase activity.35 Furthermore, β-del TERT is also localized to the mitochondria where it prevents cells from undergoing stress-induced apoptosis.35 Despite these pleiotropic functions of TERT, associated with TERT's subcellular localization (eg, in the nucleus, mitochondria, or cytosol) and a wide range of cellular processes, mechanisms and disease-causing events remain ill defined. In other words, however, whether localization of TERT to the mitochondria plays a role in maintaining NO-dependent vasodilation is unclear.
In the present study, we aimed to investigate whether nuclear or mitochondrial TERT is necessary to promote the vascular phenotype observed in CAD patients and to define the contribution of mtDNA damage to the pathological changes observed in the microcirculation of patients with CAD. Furthermore, we aimed to define whether the dominant negative splice variant, β-del TERT, is critical in the phenotypic change in dilator mechanism observed in the microvasculature of CAD patients. Our central hypothesis is that an increase in β-del TERT leads to a competes with full-length TERT, increases mtROS, and consequently increases mtDNA damage.
Methods
Tissue Acquisition and General Protocol
All protocols were approved by the Institutional Review Board of the Medical College of Wisconsin and Froedtert Hospital. Deidentified surgical discard sections of human atrial and adipose (visceral and subcutaneous) tissue were obtained and placed in cold 4°C HEPES (NaCl 275 mmol/L, KCL 7.99 mmol/L, MgSO4 4.9 mmol/L, CaCl2·2H2O 3.2 mmol/L, KH2PO4 2.35 mmol/L, EDTA 0.07 mmol/L, glucose 12 mmol/L, HEPES acid 20 mmol/L, and adipose) or cardioplegic (atrial) buffer solution. Tissue was obtained from subjects with either (1) one or fewer cardiovascular risk factors (“non-CAD”), or (2) a clinical diagnosis of CAD for all functional studdies. For evaluation of gene expression, non-CAD was defined as 3 or less trattional risk factors due to limited avadibility of atrial vessels with only 1 risk factor. Table 1 summarizes patient characteristics from whom tissue was obtained.
Table 1.
Patient Characteristics
| Non-CAD | CAD | |
|---|---|---|
| Total Samples | 101 | 72 |
| Gender | ||
| F (% of total) | 70 (69.3) | 55 (76.4) |
| M (% of total) | 30 (29.7) | 17 (23.6) |
| Unknown (% of total) | 1 (0.99) | |
| Age | ||
| Years | 48.9 ± 14.6 | 65.6 ± 10.0* |
| Race/Ethnicity | ||
| AA | 12 | 6 |
| Cauc | 83 | 59 |
| Hisp | 4 | 2 |
| Asian | 1 | 1 |
| Unknow | 1 | 4 |
| Underlying Diseases/Risk Factors | ||
| Coronary artery disease | 0 | 72 |
| Tobacco | 9 | 19 |
| Hypertension | 19 | 55 |
| Hypercholesterolemia | 14 | 49 |
| Congestive heart failure | 5 | 8 |
| Myocardial Infarction | 3 | 13 |
| Sample Usage Breakdown | ||
| Isolated vessel peparations# | 79 | 43 |
| Molecular work& | 33 | 33 |
Mean ± SD. * P < 0.05 vs. non-CAD.
CAD, coronary artery disease; HTN, hypertension; AA, African-American; Cauc, Caucasian; Hisp, Hispanic.
# Total number of vessels used in each category and not the total number of tissues (some tissues were used for multiple experiments). All IHC staining was performed in a subset of same tissues as functional studdies.
& Total number of tissues used in each category and is a sum of LV tissue and isolated microvessels.
Some samples were used for molecular and isolated vessel preparations in parallel.
Materials
All chemicals were purchased at pharmaceutical grade. Endothelin-1 (ET-1, CAS No.: 117399–94–7 Sigma) was prepared in 1% bovine serum albumin. Lentiviral constructs were produced by the Blood Center of Wisconsin Hybridoma Core Laboratory and dissolved in distilled water. Mitochondria Peroxy Yellow 1 (MitoPY1, Tocris Cat. No. 4428), telomerase inhibitor BIBR 1532 (Table 2), and activator AGS 499 (Table 2) were prepared in DMSO. All other chemicals, Nω-nitro-L-arginine methyl ester (L-NAME, Cayman Chemical #80 210) and polyethylene glycol-catalase (PEG-catalase; Sigma #C4963 or Quanta Biodesign #22 501), carboxy-PTIO potassium salt (c-PTIO: Cayman Chemical #81 540), acetylcholine chloride (ACh, Sigma, CAS No.: 60–31–1), papaverine hydrochloride (Pap, Sigma, CAS Number: 61–25–6), NO Detection Kit (Enzo Life Sciences Cat. No 51013–200), and ENDO III (gift from Dr. Mark N. Gillespie—University of South Alabama) were prepared in distilled water. Decoy peptides were produced by Genemed Synthesis, Inc. and dissolved in PBS.
Table 2.
Telomerase Modulators
| Name | Order Infomation | Effect |
|---|---|---|
| AGS 499 | Gift from Dr. Ester Priel, B.G. NEGEV TECHNOLOGIES | Telomerase transcriptional activator1 |
| Control siRNA | Ambion, Silencer Negative Control siRNA #1, Cat. No. AM4611 | Control siRNA without specific target in human genome |
| siRNA TERT | Ambion Cat. No. 4427037-s372 | Genetic knock down of telomerase reverse transcriptase FL protein |
| siRNA β-del TERT | Ambion Custom siRNA Sense—UCAAGAGCCACGUCCUACGttAntisense—CGUAGGACGUGGCUCUUGAag | Genetic knock down of telomerase reverse transcriptase β-del TERT splice variant specific |
| BIBR 1532 | Tocris Bioscience Cat. No. 2981 | Selective, small molecule telomerase inhibitor |
| Lenti β-del TERT | Generated by MCW/Versity Hybridoma Core | Dominant negative splice variant of TERT lacking exon 7–8 and containing premature stop codon. |
| Lenti MTS mutant TERT-GFP | Mutation of mitochondrial target sequence R3E/R6E resulting in loss of TERT mitochondrial localization2 | |
| Lenti NLS mutant TERT-GFP | Deletion of nuclear localization signal (mutant 7A) resulting in loss of TERT nuclear localization9 | |
| NLS mimic | RRRGGSASRSLPLPKRPRR | Mimics nuclear localization signal around amino acid S227 |
| NLS mimic—A | RRRGGAASRSLPLPKRPRR | Negative control for NLS mimic; mutated equivalent of S227 to A (not phosphorlatable) |
| MTS mimic | MPRAPRCRAVRSLLRSHYRE | Mimics mitochondrial localization signal amino acid R3 and R6 |
Cannulated Artery Preparation
Coronary and adipose arterioles (approximately 50–200 μm inner diameter) were cleaned of adipose and connective tissue and prepared for pressure myography experiments. Briefly, both ends of the vessel were cannulated with glass micropipettes in an organ chamber filled with physiological saline solution. The vessel was then pressurized (60 mm Hg), and changes in vessel diameter in response to mechanical (ie, flow) and pharmacological stimuli were assessed via videomicroscopy, as we have described previously.36
Vascular Response to Flow and Pharmacological Interventions
Microvascular function was assessed as described previously37–39 using pipettes with matched impedance. Data are reported as % max diameter at a given pressure gradient. Pressure gradients of 5–100 cm H2O were generated, assessing steady-state diameter and flow after each change, which represents estimated shear rates of 5–25 dynes/cm2. Two flow-response curves were generated for each vessel comparing no treatment (vehicle) to effects of pharmacological inhibitors (L-NAME; 100 μmol/L and PEG-catalase; 500 U/mL). Vessels were incubated for 15–20 h in endothelial cell growth medium containing 5% serum (EBM-2 medium CC-3156 with the EGM-2 Microvascular SingleQuot Kit Supplement CC-4147, Lonza) with and without genetic or pharmacological treatments. All agents were added to culture media or organ bath at <1% of total volume. Concentrations are stated as final concentrations. Vessels were constricted with ET-1 (0.1–1 nmol/L) to achieve a 20–50% stable reduction in passive diameter. Dose response curves to the endothelial-dependent vasodilators flow (FMD, 5–100 cm H2O) or acetylcholine (ACh; 1–100 µmol/L) were performed to evaluate specific endothelial-dependent dilation. At the end of each experiment, papaverine (100 µmol/L) was used to determine the maximal (passive) diameter at 60 mm Hg. Vessels that failed to constrict to ET-1 for a minimum of 20% and/or failed to dilate to minimum of 75% to papaverine were excluded from analysis.
Telomerase Modulation and mtDNA Repair
To ascertain the importance of subcellular TERT localization and the importance of β-del TERT splice variants in microvascular pathology, vessels were treated using pharmacological (AGS 499, 25 n m; BIBR 1532, 10 µm) or genetic tools (Table 2) to modulate telomerase activity and localization. Lentiviral constructs were used at a concentration of ∼108 TU/mL for 15–20 h.2,40–42 After 24 h (siRNA and pharmacological modulators) or 48 h (viral overexpression), vessels were used for microvascular studies or cells for molecular experiments.2,41 TERT expression was verified via IHC for GFP or TERT. Supplementary Figure S9 shows the efficacy of Lentiviral TERT overexpression. Life imaging for GFP expression in isolated vessels using different viral constructs employed specificity of the observed phenotype due to TERT modulation was determined by co-treatment with BIBR 1532.
A recombinant endonuclease III (ENDO III, 25 µg/mL) or a mutant inactive version of the protein (mENDO III) was added to culture medium for 15–20 h43,24 to initiate mtDNA damage repair. Endo III has a HIV-tat tag to enhance cell permeability and a mitochondrial targeting sequence to specifically target and repair mtDNA.
Quantitative Real-time Polymerase Chain Reaction
Total mRNA was harvested from human CAD and non-CAD tissue lysates using an Ambion PureLink RNA Kit (left ventricular (LV) tissue) or a NEB Monarch RNA kit (microvessel). Approximately 1500 ng (left ventricule) or 50 ng (microvessel) of RNA was used to synthesize cDNA using the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit. Gene expression was quantified by quantitative real-time polymerase chain reaction using primers (FL-, α-deletion (α-del), β-del TERT) and SYBR green from Qiagen in a BioRad CFX96 Touch Real-time PCR Detection System. Expression levels were normalized to 18S rRNA [Cat# QT00199367 Qiagen (QuantiTech)]. Splice variant specific primers for TERT were purchased from IDT as previously published44: Exo6 (F): TTG TCA AGG TGG ATG TGA CG, α-del TERT (F): CTT TGT CAA GGA CAG GCT CA, Exo7 (R): ATG TAC GGC TGG AGG TCT GT, β-del -TERT (R): GGA CGT AGG ACG TGG CTC T, FL TERT (F): GCG TTT GGT GGA TGA TTT CT, FL TERT (R): CAG GGC CTC GTC TTC TAC AG. Primers for PGC1α, catalase, SOD2, PARP1, OGG1, and SIRT3 were ordered (Qiagen validated Quantitech Primer Assays). Complete list of primers with catalogue numbers are shown in Supplementary Table S1.
Western Blot Analysis
Frozen human hearts or isolated vessels (3–5 microvessels/experiment) were homogenized in an ice-cold lysis buffer (4% SDS, 0.1.2 m Tris, 20% glycerol, 0.4 m m DTT, 2 m m EDTA), which contained protease and phosphatase inhibitors (5872S; cell signaling), centrifuged, and the supernatant was extracted. Total protein concentration was determined using a BCA protein assay kit according to the manufacturer's instructions (23 225; Thermo Scientific). Lysates were loaded and separated using SDS-PAGE and transferred onto a PVDF membrane. The following antibodies were used: A custom β-del TERT antibody (1:500; Open Biosystems) generated from rabbit against a unique human β-TERT antigen sequence-RPVPGDPAGLHPLHAALQPVLRR and anti-GAPDH (1:1000; ab8245; Abcam). β-TERT antibody validation is shown in Supplementary Figure S10.
mtDNA Damage Analysis
Quantitative PCR (qPCR) was used to assay mtDNA damage as previously described.45 Total mtDNA was extracted using QIAGEN Genomic Tip and Genomic DNA Buffer Set Kit (QIAGEN, Valencia, CA, USA). Purified genomic DNA was quantified fluorometrically using Pico Green dsDNA reagent (Molecular Probes, Life Technologies, USA). Lambda (λ)/Hind-III DNA (Gibco Invitrogen, Paisley, UK) was used to generate a standard curve and adjust the final DNA concentration to 3 ng/μL. The “hot start” PCR used the Gene Amp XL PCR Kit (Applied Biosystems, Foster City, CA, USA) with 15 ng DNA, 1X buffer, 100 ng/μL BSA, 200 μm dNTPs, 20 pmol of each primer (Small fragment: Sense 5′-CCC CAC AAA CCC CAT TAC TAA ACC CA-3′, Antisense: 5′-TTT CAT CAT GCG GAG ATG TTG GAT GG-3′; Large fragment: Sense: 5′-TCT AAG CCT CCT TAT TCG AGC CGA-3′, Antisense: 5′-TTT CAT CAT GCG GAG ATG TTG GAT GG-3′), 1.3 m m Mg++ and water. The reaction was brought to 75°C then 1 U/reaction enzyme was added. Specific primers were used to amplify a large fragment of mtDNA (8.9 kb) to determine mtDNA integrity. A small fragment (221 bp) of the mitochondrial genome was used to monitor changes in mtDNA copy number and to normalize the amplified data. Relative amplifications were calculated to compare CAD arterioles to non-CAD. These values were then used to estimate, assuming a Poisson distribution, the number of lesions present in mtDNA as previously described.45
Measurement of mtROS and NO Production in Isolated Vessels
MitoPY146 was used to evaluate microvessel generation of mtH2O2. As previously described,2,42 after cannulation in a warmed chamber (37°C) containing HEPES buffer at pH 7.4, arterioles were perfused intraluminally with MitoPY1 (5 μmol/L, 1 h) using flow levels below the threshold of causing dilation until the luminal surface was bathed in MitoPY1 containing buffer. Next, the trans-vascular pressure gradient was changed from 0 to 100 cm H2O and fluorescence was measured after 2 min. Fluorescence was evaluated at an excitation wavelength of 488 nm and emission was measured between 530 and 590 nm (GFP filter). NO production in response to flow was measured using an NO-specific probe47 under similar conditions as MitoPYI. Fluorescence readings were performed at excitation wavelength of 561–594 nm and emission measured at 615 nm. All data are normalized to pre flow values and expressed as fold increase at 100 cm H2O (5 min) pressure gradient.
All comparisons were made to no flow conditions and expressed as fold change. The response to the NOS inhibitor L-NAME (prevented increase in NO levels) or Peg-Cat (scavenges H2O2)2,42,48 was measured. Then the specificity of the probe was established using the NO scavenger c-PTIO (1 μm).
Immunohistochemistry (IHC)
Isolated vessels from surgically discarded tissue were fixed in 10% zinc formalin, embedded in paraffin wax, cut into 4 µm sections, and rehydrated. A DAKO autostainer was then used for antigen retrieval and antibody staining. Sections were incubated with secondary antibody alone or normal serum IgG as negative controls. The MCW/CHW Pathology Core facility was used for all IHC studies. Antibodies to both TERT (Bioss, bs-1411R), β-del TERT (custom antibody Open Biosystems), and GFP (Abcam # ab290) were used.
Statistical Methods
Data presented as mean ± SD. For all flow response curves, differences between groups at each concentration were determined using a 2-way repeated-measures analysis of variance (ANOVA). A post hoc Tukey's test was used for individual comparisons. A probability value of P < 0.05 was statistically significant. Fluorescence studies, western blots, or RNA expression analysis used either a paired t-test or 1-way ANOVA with post hoc Tukey's test to identify individual differences within groups. Statistical outliers were identified via Thompson-tau method and excluded from analysis.
Results
Increased Levels of Dominant Negative TERT Splice Variants Are Observed in Patients With CAD
FL-, α-del, and β-del are all naturally occurring splice variants of TERT. Expression of FL-, α-del, and β-del TERT mRNA was measured in LV tissue from CAD and non-CAD subjects by quantitative real-time PCR. Compared to non-CAD, tissue from CAD subjects showed decreased FL-TERT mRNA expression and increased α-del and β-del TERT expression (Figure 1A–C). IHC and immunoblots for β-del TERT showed elevated protein levels in microvessels and LV in CAD subjects compared to non-CAD (Figure 1D–F). Supplementary Figure S1 shows representative images of western blots and IHC supporting these findings.
Figure 1.

Increased levels of β-del TERT expression in CAD subjects. Relative levels of FL (A), α-del (B), and β-del (C) TERT mRNA expression were measured in LV tissue from non-CAD vs. CAD subjects (N = 6–9). (D) β-del TERT expression was measured by IHC in coronary arteries isolated from non-CAD vs. CAD subjects (representative of 3 replicates). (E + F) Western blot analyses of β-del TERT levels in LV tissue of patients with and without CAD. *P < 0.05 vs. non-CAD t-test. Values are means ± SEM, N = 5–8.
Increased β-del TERT Promotes Pathological while Decreased β-del TERT Promotes Physiological Mechanism of FMD
β-del TERT has been shown to behave in a dominant negative fashion to FL-TERT due to the deletion in the reverse transcriptase domain.35 To investigate the effect of the β-del TERT isoform on microvascular function, microvessels extracted from adipose tissue of non-CAD subjects were treated with lentivirus expressing β-del TERT-GFP fusion protein. Expression of β-del TERT-GFP was confirmed via IHC (Figure 2A–D). FMD in microvessels from non-CAD adipose is normally mediated by endothelial production of NO.49 β-del TERT overexpression switched the mediator of FMD from NO to H2O2 (Figure 2E), which is similar to what is observed in microvessels from CAD patients. Supplementary Figure S3 shows expression and subcellular localization of FL and β-del TERT. Smooth muscle vasodilator function, assessed as the response to papaverine, was unaltered (Figure 2F). Based on our prior work, empty lentiviral expression alone has no impact on dilator capacity or underlying mechanisms.41
Figure 2.

Overexpression of β-del TERT in non-CAD adipose microvessels confers a CAD phenotype. Lentiviral transfection of β-del TERT-GFP (107 TU/mL intraluminal) increased expression of β-del TERT and GFP compared to negative controls. Representative image of IHC β-del TERT (A + B) or GFP (C + D). Lenti-GFP has no impact on dilator capacity or mechanism (ie, remains NO-mediated) (E), whereas β-del TERT is sufficient to induce loss of NO-mediated dilation to flow (ie, no longer inhibited by L-NAME) while triggering a compensatory increase in flow-induced H2O2 (inhibited by PEG-catalase) dilation (F). (G) Smooth muscle-dependent dilation to papaverine was not impaired. * P < 0.05 2-way ANOVA RM, N = 7–8. Lenti-GFP, N = 4 historic data from Kadlec et al. (2017).41
Knockdown of β-del TERT and total-TERT (all splice variants) levels in vessels transfected with the siRNA against β-del TERT compared to the negative control siRNA was verified by IHC (Figure 3A and B). Silencing β-del TERT in CAD vessels partially restored the mediator of FMD from H2O2 to NO-mediated vasodilation (Figure 3C). Smooth muscle vasodilator function assessed as the response to papaverine was unaltered (Figure 3D). Evaluation of flow-induced NO and mtH2O2 production in vessels isolated from subjects with CAD confirmed the functional findings. Following transfection with β-del TERT siRNA, flow-induced NO levels increased, which was accompanied by decreased mtH2O2 production (Figure 3F).
Figure 3.

siRNA mediated knockdown of β-del TERT partially restores NO bioavailability in CAD. Representative images showing siRNA targeted β-del TERT (A) or total TERT (recognized all isoforms) from (B) knockdown and control siRNA treated vessels. β-del TERT in part restores NO-mediated dilation to flow (L-NAME inhibitable) in vessels from subjects with CAD am H2O2 (Peg-catalase inhibitable) dilation was reduced (C). Endothelium-independent dilation to papaverine was not impaired (E). siRNA to β-del TERT increased flow-induced NO production while reducing H2O2 (D). CTRL siRNA had no effect on dilator function or NO/H2O2 production (not shown). * P < 0.05 2-way ANOVA RM, N = 7–8 for C.* P < 0.05 unpaired t-test, N = 5–8 for D and E.
Silencing total TERT in non-CAD vessels switched the mediator of FMD from NO to H2O2 and decreased vasodilation to ACh, which is similar to what we previously reported with the pharmacological TERT inhibitor BIBR 1532 (Supplementary Figure S2).2 Because H2O2 is already the predominant mechanism of dilation in CAD vessels, the effect of total-TERT knockdown was not tested in CAD vessels. Control siRNA, as previously published,50 had no effect of dilator function or its underlying mechanism (Supplementary Figure S2).
Mitochondrial, but not Nuclear, TERT Regulates the Mediator of FMD in Health and Disease
To differentiate the effects of mitochondrial vs. nuclear TERT, we utilized lentiviral vectors expressing WT- or mutant versions of TERT. WT-TERT-GFP and MTS-mutant TERT-GFP (mutation of the MTS)2 localize primarily to the nucleus with a small amount of diffuse cytoplasmic localization, whereas NLS-mutant TERT (deletion of the NLS)9 primarily localizes to the cytoplasm and mitochondria (Supplementary Figure S4).
In vessels from non-CAD patients, overexpression of WT- or NLS-mutant TERT did not alter dilator function. In contrast, overexpression of MTS-mutant TERT resulted in a CAD-like phenotype by switching the mediator of dilation from NO to H2O2 (Figure 4). This corresponds with findings in microvessels from CAD patients, where MTS-mutant TERT overexpression did not alter dilation, while overexpression of NLS-mutant TERT restored NO-mediated dilation (Figure 5). Thus, NLS-mutant TERT overexpression in CAD vessels mimics targeted β-del TERT knockdown (Figure 5A) and promotes a dilator phenotype like non-CAD vessels. Collectively, these observations suggest that extranuclear TERT (eg, TERT localized to mitochondria) promotes NO-mediated FMD, whereas TERT localized to the nucleus does not impact FMD. Moreover, TERT overexpression rescues NO-mediated FMD in CAD vessels, which is independent of nuclear localization, while preventing mitochondrial localization of TERT induces a CAD-like dilator phenotype in non-CAD vessels.
Figure 4.

Increased nuclear but not mitochondrial TERT causes pathological switch form NO to H2O2 mediated dilation. (A) Representative images showing lentiviral overexpression of WT, MTS, or NLS mutant TERT compared to negative controls. WT-TERT had no effect on the underlying mechanism of FMD (B), while overexpression of NLS mutant TERT shifted FMD from NO (L-NAME inhibitable) to H2O2 (Peg-catalase inhibitable) (C). Similar to WT-TERT overexpression, MTS mutant TERT had no effect on the mechanism of dilation (D). Smooth muscle dependent dilation to papaverine was not impaired (E). * P < 0.05 2-way ANOVA RM, N = 6–8.
Figure 5.

Increased mitochondrial but not nuclear TERT restores physiological NO in vessels from patients with CAD. (A) In vessels from patients with CAD, overexpression of the MTS mutant TERT has no affect on the mechanism of vasodilation. (B) Overexpression of the NLS mutant TERT changed the mechanism of FMD from H2O2 (Peg-catalase inhibitable) to NO (L-NAME inhibitable). (C) Smooth muscle dependent dilation to papaverine was not impaired. * P < 0.05 2-way ANOVA RM, N = 6–8.
mtDNA Integrity is Critical to Preserve NO-mediated Dilation
Prior evidence from our lab2,19,42 and that of other groups26,27,51,52 suggest a critical role of telomerase activity in preventing mtDNA damage. To our knowledge, increase in mtDNA damage or its functional relevance has not been directly investigated in the human microcirculation. We observed increased mitochondrial lesion frequency (reduced amplification) in vessels from subjects with CAD compared to non-CAD vessels (Figure 6A). To confirm this observation, vessels from subjects with and without CAD were stained with an antibody specific for 8-oxo-dG aggregates as a marker of DNA damage. Although this is not a quantifiable measure and is not specific to mtDNA (nuclear DNA and RNA also detected), a qualitative increase in staining intensity was observed in vessels from subjects with CAD (Supplementary Figure S5). To establish the functional relevance of mtDNA damage, vessels from atrial or adipose tissue from subjects with CAD were treated with mitochondria-tagged endonuclease III (Endo III) to initiate mtDNA repair machinery, which restored NO as the mediator of FMD (Figure 6B). In a control experiment, treating CAD vessels with the catalytically inactive mEndo III did not restore NO-driven FMD (Figure 6E). Thus, mtDNA lesions and DNA damage are elevated in CAD tissue and initiating mtDNA repair with Endo III rescues NO-mediated FMD.
Figure 6.

mtDNA damage repair in CAD microvessels induces NO-mediated FMD. (A) Isolated microvessels from showed higher levels of mtDNA damage in patients with CAD than non-CAD. Quantitative end-point PCR was used to measure the relative amplification of an 8.8 kbp segment and a 221 bp segment of the mitochondrial genome and calculate the relative DNA lesion frequency (N = 10–12). (B) NO-driven FMD was restored in microvessels from subjects with CAD after treatment with Endo III (N = 6–8) but not after (C) treatment with inactive mEndo III (N = 3–5). (D) In vessels from subjects without CAD, Endo III prevented the BIBR 1532-induced switch from NO to pathological H2O2 (N = 3–4). (E) Smooth muscle response to papaverine was not altered (N = 4–8). * P < 0.05 A unpaired t-test; B–D 2-way ANOVA RM.
Supplementary Figure S2 and our previously published work2 demonstrate that the decreased TERT is sufficient to trigger the switch in the mediator of FMD from NO to H2O2. To confirm the relationship between mtDNA damage and TERT-associated changes in dilator mechanisms, vessels from subjects without CAD were co-incubated with the specific telomerase inhibitor BIBR 1532 and Endo III. Although BIBR alone switches the mediator of FMD from NO to H2O2,2 co-incubation with Endo III prevented the pathological dilator switch (Figure 6D) and induced decrease in ACh mediated dilation (Supplementary Figure S6).
Mimeitc Peptides as Means to Manipulate Mitochondrial and Nuclear TERT-related Vascular Function
Both the NLS and MTS of TERT have been defined previously.9,27,53 We used these amino acid sequences to design peptides that will serve as decoys of the actual enzyme to inhibit or decrease the post-transcriptional modifications to TERT that are necessary for nuclear import (phosphorylation of S227) or mitochondrial import (nature of post-translational modifications to date not defined). Peptides are displayed in Table 2.
To test if these novel decoy peptides have any effect on vascular function, we first confirmed cell permeability and uptake by overnight treatment with 5-FAM labeled NLS mimetic peptide. Both isolated microvessels and cultured cells showed unassisted uptake of the peptide as demonstrated by an increase in 5-FAM fluorescence (Supplementary Figure S7).
Next, vessels from subjects with and without CAD were incubated with decoy peptides designed to prevent nuclear import (NLS mimetic) or mitochondrial translocation (MTS mimetic). The MTS mimetic resulted in a switch of vasodilator mechanism from NO to H2O2, mimicking the phenotype of subjects with CAD (Figure 7A). The NLS mimetic did not alter FMD in vessels from patients without CAD; however, it was sufficient to restore NO-mediated FMD in vessels from patients with CAD (Figure 7B and C). Neither of the decoy peptides had any effect on papaverine-induced dilation (Figure 7D).
Figure 7.

MTS mimetic induces pathological H2O2 mediated dilation, while NLS mimetic restores physiological NO-mediated dilation. (A) Treatment of vessels from non-CAD subjects with the TERT MTS mimetic peptide induces a CAD like phenotype for FMD (Peg-catalase inhibitable), (B) while treatment with TERT NLS mimetic has no effect. (C) Treatment of vessels from subjects with CAD with TERT NLS mimetic is sufficient to restore FMD to NO (L-NAME mediated). (D) Endothlial indepedent dilation to papaverine was not alterd in any groups. * P < 0.05 2-way ANOVA RM, N = 6–11.
To further establish the effects of NLS mimetic and MTS mimetic on endothelial function, vessels were treated with vehicle or ANG II (10–8 m) to mimic physiologically relevant stress. ANG II caused a significant decrease in endothelium-dependent dilation. The global telomerase inhibitor BIBR 1532 was used to establish the specificity of the decoy peptides to telomerase activity. The NLS mimetic was sufficient to preserve ACh- and flow-induced endothelium-dependent dilation after an acute ANG II challenge (Supplementary Figure S8A and C). However, the protective effect was abolished by either co-treatment with BIBR 1532 or when using a peptide with a mutation in the AA equivalent to S227 of full length TERT (NLS mimetic-A) (Supplementary Figure S7B and C). The MTS mimetic did not have any effect on baseline dilation but augmented ANG II-induced endothelial dysfunction with FMD and ACh, alike (data not shown). Smooth muscle function in response to papaverine was not impaired with either of the treatments (Supplementary Figure S7D).
Decoy Peptides as Means to Manipulate Mitochondrial and Nuclear TERT-related Vascular Function
To elucidate molecular changes in pathways involved in redox regulation, mtDNA integrity, and general mitochondrial function, we used qPCR to investigate the expression of genes from isolated vessels from non-CAD and CAD subjects from both atrial and adipose tissues. Table 3 shows the summary of the results. While some trends were observed, to our surprise, few changed where significant when comparing all samples. However, when separating the data by vascular bed and sex, additional significant changes were uncovered. Changes in SOD2 levels are in line with previous resports as a downstream target of TERT.54 In relation to our past observations, these findings are somewhat surprising due to the fact that to date we have not observed any significant differences comparing isolated vessel function or molecular changes from male vs. female or between adipose peripheral vs. atrial microvessels. Future investigations are necessary to define the physiological relevance of these changes.
Table 3.
Changes in Gene Expression in Isolated Microvesses From Subjects With CAD vs. Non-CAD Diagniosis
| Atrial | Adipose | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Both Genders | Male | Female | Both Genders | Male | Female | Both Genders | Male | Female | ||
| PGC1α | Non-CAD | 1.00 ± 0.79 | 1.00 ± 0.71 | 1.00 ± 0.94 | 1.00 ± 0.38 | 1.00 ± 0.37 | 1.00 ± 0.44 | 1.00 ± 0.70 | 1.00 ± 0.65 | 1.00 ± 0.81 |
| CAD | 0.66 ± 0.62 | 1.52 ± 1.03 | 0.26 ± 0.25$ | 0.89 ± 0.36 | 0.62 ± 0.28 | 0.98& | 0.73 ± 0.57 | 1.05 ± 0.58 | 0.35 ± 0.33 | |
| CAT | Non-CAD | 1.00 ± 0.36 | 1.00 ± 0.24 | 1.00 ± 0.54 | 1.00 ± 0.43 | 1.00 ± 0.31 | 1.00 ± 0.52 | 1.00 ± 0.39 | 1.00 ± 0.26 | 1.00 ± 0.53 |
| CAD | 0.97 ± 0.42 | 1.03 ± 0.35 | 0.80 ± 0.35 | 0.97 ± 0.38 | 1.28 ± 0.46 | 0.70& | 0.97 ± 0.41 | 1.07 ± 0.40 | 0.78 ± 0.32 | |
| SOD2 | Non-CAD | 1.00 ± 0.53 | 1.00 ± 0.59 | 1.00 ± 0.64 | 1.00 ± 0.48 | 1.00 ± 0.06 | 1.00 ± 0.50 | 1.00 ± 0.51 | 1.00 ± 0.52 | 1.00 ± 0.59 |
| CAD | 0.61 ± 0.55* | 1.49 ± 1.75 | 0.43 ± 0.27* | 0.77 ± 0.38* | 0.44 ± 0.29* | 0.94& | 0.66 ± 0.51* | 0.75 ± 0.59 | 0.50 ± 0.31* | |
| PARP1 | Non-CAD | 1.00 ± 0.37 | 1.00 ± 0.16 | 1.00 ± 0.56 | 1.00 ± 0.57 | 1.00 ± 0.38 | 1.00 ± 0.74 | 1.00 ± 0.45 | 1.00 ± 0.24 | 1.00 ± 0.64 |
| CAD | 0.85 ± 0.31 | 0.97 ± 0.33 | 0.73 ± 0.38 | 0.90 ± 0.29 | 0.99 ± 0.35 | 0.75& | 0.86 ± 0.30 | 0.98 ± 0.34 | 0.73 ± 0.36 | |
| OGG1 | Non-CAD | 1.00 ± 0.61 | 1.00 ± 0.59 | 1.00 ± 0.67 | 1.00 ± 0.37 | 1.00 ± 0.22 | 1.00 ± 0.14 | 1.00 ± 0.54 | 1.00 ± 0.52 | 1.00 ± 0.55 |
| CAD | 0.79 ± 0.33 | 1.23 ± 1.07 | 0.87 ± 0.45 | 0.87 ± 0.47 | 0.62 ± 0.43 | 0.90& | 0.78 ± 0.41 | 1.02 ± 0.95 | 0.87 ± 0.42 | |
| SIRT3 | Non-CAD | 1.00 ± 0.51 | 1.00 ± 0.32 | 1.00 ± 0.46 | 1.00 ± 0.54 | 1.00 ± 0.24 | 1.00 ± 0.70 | 1.00 ± 0.52 | 1.00 ± 0.31 | 1.00 ± 0.57 |
| CAD | 0.87 ± 0.46 | 1.00 ± 0.45 | 0.73 ± 0.29 | 1.11 ± 0.62 | 1.17 ± 0.64 | 0.87& | 0.95 ± 0.53 | 1.04 ± 0.54 | 0.75 ± 0.27 | |
Data presented as fold change to non-CAD mean ± SD; N = 4–17.
& N = 1 (not statistical evaluation possible) * P < 0.05 unpaired t-test; $P < 0.1 unpaired t-test.
Discussion
Novel data presented here support the following significant findings: (1) Patients with CAD have altered TERT splice variant distribution in cardiac tissue and microvessels compared to subjects without CAD. Specifically, both α- and β-del TERT levels are elevated, while FL-TERT mRNA levels are reduced in CAD patients. (2) Overexpression of β-del TERT in non-CAD vessels confers a CAD phenotype, while silencing of β-del TERT in vessels from CAD subjects partially restores NO-mediated FMD, increases flow-induced NO production, and suppresses H2O2 levels. (3) Increasing mitochondrial TERT but not nuclear TERT restores physiological mechanisms of FMD, while nuclear TERT promotes pathological H2O2-mediated dilation. (4) Elevated mtDNA damage is observed in CAD tissues. mtDNA repair initiated by administration of Endo III restores NO-mediated dilation and overcomes the effect of global pharmacological TERT inhibition. (5) Lastly, we introduce novel decoy peptides with the potential to manipulate subcellular TERT localization that show potential to restore physiological vasodilation and protect against ANG II-induced endothelial dysfunction. Figure 8 illustrates proposed mechaims supported by the new findings of this manuscript.
Figure 8.
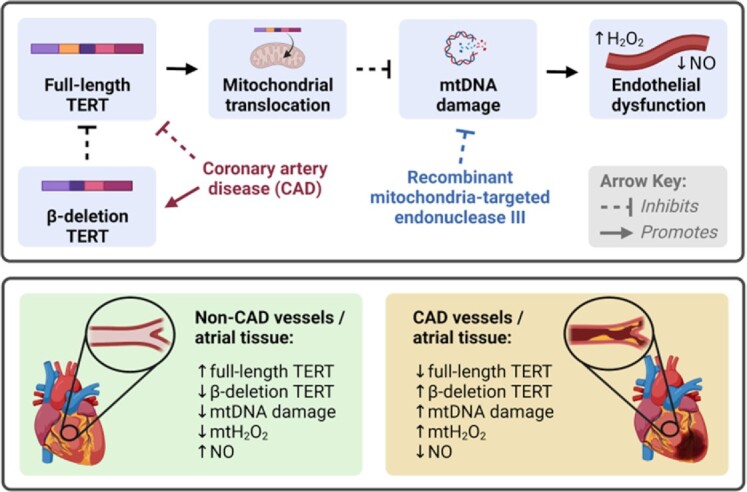
Proposed mechanism of TERT/β-Del TERT in regulating mechanism of flow-mediated dilation in the human microcirculation. CAD, coronary artery disease; H2O2, hydrogen peroxide; mt, mitochondrial; NO, nitric oxide; and TERT, catalytic subunit of human telomerase complex.
More than 20 alternate splice variants of TERT have been previously described.31–34 Of these, the most highly expressed splice variants, aside from full-length TERT, are those with deletions in the catalytic domain, namely α-del and β-del TERT.55 Expression patterns of these splice variants have been widely described during embryogenesis56,57 and for various malignancies.58–61 However, to our knowledge, the role of alternate splicing of TERT in cardiovascular disease has not been investigated. We have previously shown that TERT is necessary for normal endothelial function in the microvasculature. In the present study, we expanded on these findings. Understanding how alternate splicing of TERT is regulated may provide new therapeutic targets for the treatment of cardiovascular disease.
Nuclear TERT vs. a Noncanonical Role of Mitochondrial TERT
The first role of TERT discovered was telomere elongation, the absence of which results in gradual telomere shortening that has been implicated in aging and cancer-related pathologies. While a significant body of evidence suggests that telomere shortening contributes to development of cardiovascular disease, the underlying mechanisms are complex. Different tissue- and cell-specific roles of TERT have been described that make interpretation of existing data challenging [reviewed by Hoffmann et al. (2021)51]. In addition with our current knowledge, pinpointing a causative event in humans (reduced telomerase activity should preceed telomere shortening) is difficult. Evidence from rodent models suggests that gradual telomere shortening over 3 to 4 generations is necessary to provoke cellular stress responses to critically short telomeres.62,63 Our previous work suggests that the onset of CAD results in changes of microvascular physiology/pathology, which can be overcome acutely by a transcriptional activator of TERT (AGS-499).2,42 Similarly, acute telomerase inhibition provokes a CAD-like phenotype in isolated vessels from patients without cardiovascular disease.64,65 In addition, genetic deletion of TERT, the protein subunit of telomerase, but not TERC, the RNA subunit of telomerase, causes a telomere-independent microvascular phenotype in mice.19 This evidence suggests that extranuclear TERT serves as a response to cellular stress and is critical in maintaining normal physiological function. Recent work by Chatterjee et al. (2021)66 demonstrates that overexpression of TERT in a mitochondrial-dependent manner is sufficient to overcome doxorubicin-induced heart failure in mice. This is relevant because, in nongenetically modified models, the onset of phenotypes such as chemotherapy-induced heart failure or ANG II-induced hypertension is too short to be fully explained by telomere shortening. In the present study, we show for the first time that mitochondrial/extranuclear, but not nuclear, translocation of TERT is able to restore physiological responses. In particular, we demonstrate that viral overexpression of novel decoy peptides restores NO-mediated dilation in vessels from subjects with CAD, where FMD is otherwise mediated by H2O2. Previous work by Radam et al. (2014)67 has described that different levels of extracellular O2 are able to alter splice variant distribution of TERT, which is in line with our findings of elevated β-del TERT in microvessels and cardiac tissue of subjects with CAD, as CAD is a form of ischemic heart disease.
A number of studies have demonstrated a direct link between low levels of TERT and increased mtDNA damage in response to various stressors.26,27,53,68 The underlying mechanisms by which TERT protects mitochondrial integrity is not fully understood and are likely multifactorial. As TERT is an RNA-dependent DNA polymerase, binding of TERT to mtDNA is a logical assumption. However, given that the mitochondrial genome is circular and, hence, does not contain telomeres, telomere elongation does not explain the protective effects. Several studies have demonstrated that, in the nucleus, TERT regulates gene expression of key proteins involved in maintaining mitochondrial health (eg, MnSOD or catalase).69–71 Other possible mechanisms are supported by the literature, including regulation of mitochondrial encoded RNAs, a critical RNA component of mitochondrial RNA processing endoribonuclease (RMRP), which is required for cellular development and/or mtDNA replication.72
The rapid timeline of experiments employing TERT modulation (<24 h for pharmacological manipulation and ∼48 h for viral overexpression), combined with our prior work showing that changes in dilator phenotype are independent of transcription and translation,2 further underlines a noncanonical role of TERT. Recently introduced overexpression models that target FL-TERT specifically to the mitochondria or nucleus show a protective effect of mitochondrial TERT, while nuclear TERT protects from myocardial ischemia/reperfusion injury by improving complex I composition and function.73 These findings are in line with the phenotype observed in our own rat model of global loss of TERT function that have increased susceptibility to ischemia/reperfusion injury.18 In summary, data presented in this manuscript underline the possible efficacy of increased mitochondrial TERT as a new target for cardiovascular disease, including CAD. Current means to increase TERT in patients rely on lifestyle interventions or dietary supplements (eg, TA-65) that increase total TERT, which includes nuclear TERT and may therefore potentially contribute to pathological cell proliferation. To our knowledge, the decoy peptides presented in the current study are the first pharmacological tools that allow separation of mitochondrial vs. nuclear actions of TERT.
Limitations of Current Study
The present study has a number of limitations that are difficult to address in human studies or with use of human tissue. Due to the relatively short window of opportunity to study functional changes in isolated human vessels (∼24–36 h for atrial derived vessels and 48–72 h for adipose-derived vessels), long-term manipulations, and functional evaluations are not possible. Future mechanistic studies in preclinical models are necessary to establish the long-term physiological relevance of subcellular manipulation of TERT.
The current data and past publications by us and others clearly indicate a role of TERT in regulation of cellular redox environment. Mitochondrial TERT suppresses ROS production, while the lack of mitochondrial TERT is associated with elevated free radicals. Unfortunately, in large part due to small sample size of human microvessels, we are not able to differentiate the underlying mechanism and define if ROS, mtDNA damage, or lack of ATP is causative for functional phenotypes. Further, in our studies, acute inhibition of TERT does not cause an increase in mtDNA damage (data not shown). While vessels from patients with CAD have been exposed to many stressors in vivo (eg, increases in ROS, elevated glucose, or reduced O2) that promotes mtDNA damage, ex vivo exposure of vessels to the TERT inhibitor BIBR 1532 only directly impact of TERT without further stressors that may cause mtDNA damage in the absence of TERT activity. TERT activity is hypothesized to protect against mtDNA damage, but the loss of TERT activity will not necessarily cause mtDNA damage without further stressors. These findings would suggest that TERT is sufficient to protect from mtDNA damage in vivo but not causative for mtDNA damage in ex vivo experiments.
While the current study directly links the cellular localization of TERT with mtDNA damage and elevated levels of ROS, proximal and distal signaling components remain unexplored. Previous work by our lab and others indicates key pathways (eg, autophagy,50 Sirtuin signaling,74 PGC1α,41 ceramide,75 Lysophosphatidic acid,76 acute exposure to elevated intraluminal pressure,48,77 and ANG 1–742) are involved in protecting mitochondrial integrity and will require additional studies in animal models. Similar changes in gene expression presented in Table 3 show differnees between CAD and non-CAD but do not necessarily correlate or are a result of changes in TERT splice variant distribution.
For the first time, we introduce a novel tool to manipulate subcellular TERT levels via decoy peptides that mimic NLS or MTS sequences. To our knowledge, these are the first pharmacological tools that allow separation of mitochondrial vs. nuclear actions of TERT, and our current data show expected functional changes as evidenced by similar effects as targeted overexpression of TERT mutants that lack MTS or NLS. However, we are unable to confirm the mechanism of action of these peptides. We propose that NLS and MTS mimetics prevent post-transcriptional modifications of TERT necessary for nuclear or mitochondrial import; we are not able to recapitulate this idea in cultured endothelial cells and do not have sufficient material in isolated vessels to perform cell franctionaltion experiments. We have tested multiple commercial TERT antibodies in cultured endothelial cells and failed to obtain images with sufficient background to noise ratio that allows us to definitively conclude that NLS mimetic in fact prevents nuclear import, while MTS mimetic prevents mitochondrial import after exposure to H2O2. Similarly, we do not see changes in p-TERT S227 levels after treatment with NLS mimetic. The exact mechanism of post-translational modification at the MTS sequence is, to date, not defined, and hence could not be tested in immunoblots. Presented data with viral mediated overexpression construct back up the general idea of a critical role of mitchondrial but not nuclear TERT; however, we cannot determine if the balance between nuclear vs. mitcondrial TERT is driving the phenotype or if the overexpression constructs silence any naturally occurring TERT effects. Future studies need to identify how these decoy peptides affect TERT localization.
Conclusions and Future Outlook
The subcellular distribution of TERT and its splice variants modulate microvascular endothelial function and determine whether FMD is mediated by NO or H2O2, which has important implications for CAD. Cumulatively, our data suggest that mitochondrial localization/activity of TERT is a key factor in the development of this phenotype. The ability to manipulate splice variants and the intracellular distribution of TERT presents intriguing possibilities for both cardiovascular protection and anticancer therapy via a noncanonical role of TERT without promoting proliferative capacity (eg, that observed in cancer cells and atherosclerosis). Indeed, ongoing efforts utilizing splice-blocking oligonucleotides for TERT or other molecular means show promising results in cell culture models or embryonic development.72 The introduction of cell-permeable peptides that can alter cellular localization of TERT represents another promising direction to target TERT as a treatment for chronic disorders ranging from cardiovascular disease to cancer and neurodegenerative disease where TERT has been described as a key component of disease pathology.
Supplementary Material
ACKNOWLEDGEMENTS
We like to thank Drs Mark N. Gillespie and Geln Wilson, both from the University of South Alabama, Department of Pharmacology, for providing us with recombinant ENDO III protein for our studdies.
We like to thank Dr Johnathan D. Ebben University of Wisconsin, Madison, WI for his contibution to development and desing of TERT decoy peptides.
Contributor Information
K Ait-Aissa, Department of Internal Medicine, University of Iowa, Iowa City, IA 52242, USA.
L E Norwood-Toro, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
J Terwoord, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
M Young, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
L A Paniagua, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Innovation Institute, University of Louisville, Louisville, KY 40292, USA.
S N Hader, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
W E Hughes, Department of Internal Medicine, University of Iowa, Iowa City, IA 52242, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
J C Hockenberry, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
J E Beare, Cardiovascular Innovation Institute, University of Louisville, Louisville, KY 40292, USA; Kentucky Spinal Cord Injury Research Center, University of Louisville, Louisville, KY 40292, USA.
J Linn, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
T Kohmoto, Department of Surgery, Division of Cardiothoracic Surgery, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
J Kim, Department of Physiology and Pharmacology, Schulich School of Medicine & Dentistry, The University of Western Ontario, London, ON N6A 5C1, Canada.
D H Betts, Department of Physiology and Pharmacology, Schulich School of Medicine & Dentistry, The University of Western Ontario, London, ON N6A 5C1, Canada.
A J LeBlanc, Cardiovascular Innovation Institute, University of Louisville, Louisville, KY 40292, USA; Department of Cardiovascular and Thoracic Surgery, School of Medicine, University of Louisville, Louisville, KY 40292, USA.
D D Gutterman, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
A M Beyer, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Department of Physiology, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
Funding
This work was supported by the National Institutes of Health and the American Heart Association under grant numbers 5T32HL134643 to (J.T.), HL133029 to (A.M.B.), HL157025 to (A.M.B.), OD018306 to (A.M.B.), HL135901 to (D.D.G.), T32GM089586 to (W.E.H. and A.H.A.), 20POST35050017 to (W.E.H.), and AG053585 to (A.J.L.) and the Canadian Institutes of Health Research (CIHR) to (B.D.H.).
Conflict of Interest
AMB holds U.S. Patent No. 10,858,397 "Peptide inhibitors of telomerase translocation and therapeutic uses thereof" The authors have no conflicts to disclose.
Data Availability
The data underlying this article are available in the article and in its online supplementary material. Patient information outside of the summary details provided cannot be shared for the privacy of individuals that participated in the study. Individual de-identified clinical information will be shared on reasonable request to the corresponding author. All data generated or analysed during this study are included in this published article (and its supplementary information files). Raw date for the current study are available from the corresponding author on reasonable request.
References
- 1. Eitan E, Tichon A, Gazit A, Gitler D, Slavin S, Priel E. Novel telomerase-increasing compound in mouse brain delays the onset of amyotrophic lateral sclerosis. EMBO Mol Med. 2012;4(4):313–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beyer AM, Freed JK, Durand MJet al. Critical role for telomerase in the mechanism of flow-mediated dilation in the human microcirculation. Circ Res. 2016;118(5):856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuo L, Davis MJ, Cannon MS, Chilian WM. Pathophysiological consequences of atherosclerosis extend into the coronary microcirculation. Restoration of endothelium-dependent responses by L-arginine. Circ Res. 1992;70(3):465–476. [DOI] [PubMed] [Google Scholar]
- 4. Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003;349(11):1027–1035. [DOI] [PubMed] [Google Scholar]
- 5. Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93(6):573–580. [DOI] [PubMed] [Google Scholar]
- 6. Trzeciak S, Dellinger RP, Parrillo JEMicrocirculatory Alterations in Resuscitation and Shock Investigators et al. Microcirculatory Alterations in Resuscitation and Shock Investigators . Early microcirculatory perfusion derangements in patients with severe sepsis and septic shock: relationship to hemodynamics, oxygen transport, and survival. Ann Emerg Med. 2007;49(1):88–98. [DOI] [PubMed] [Google Scholar]
- 7. Kuo L, Hein TW. Vasomotor regulation of coronary microcirculation by oxidative stress: role of arginase. Front Immunol. 2013;4:237. https://www.frontiersin.org/articles/10.3389/fimmu.2013.00237/full, (last accessed date on 19 August 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van de Hoef TP, van Lavieren MA, Damman Pet al. Physiological basis and long-term clinical outcome of discordance between fractional flow reserve and coronary flow velocity reserve in coronary stenoses of intermediate severity. Circ Cardiovasc Interv. 2014;7(3):301–311. [DOI] [PubMed] [Google Scholar]
- 9. Jeong SA, Kim K, Lee JHet al. Akt-mediated phosphorylation increases the binding affinity of hTERT for importin α to promote nuclear translocation. J Cell Sci. 2015;128(12):166132. [DOI] [PubMed] [Google Scholar]
- 10. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–844. [DOI] [PubMed] [Google Scholar]
- 11. Pepine CJ, Anderson RD, Sharaf BLet al. Coronary microvascular reactivity to adenosine predicts adverse outcome in women evaluated for suspected ischemia results from the National Heart, Lung and Blood Institute WISE (Women's Ischemia Syndrome Evaluation) study. J Am Coll Cardiol. 2010;55(25):2825–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beyer AM, Zinkevich N, Miller Bet al. Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease. Basic Res Cardiol. 2017;112(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110(8):1109–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim F, Pham M, Maloney Eet al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28(11):1982–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharma NK, Reyes A, Green Pet al. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2012;40(2):712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beyer AM, Freed JK, Durand MJet al. Critical role for telomerase in the mechanism of flow-mediated dilation in the human microcirculation. Circ Res. 2016;118(5):856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ait-Aissa K, Kadlec AO, Hockenberry J, Gutterman DD, Beyer AM. Telomerase reverse transcriptase protects against angiotensin II-induced microvascular endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2018;314(5):H1053–H1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ait-Aissa K, Heisner JS, Norwood Toro Let al. Telomerase deficiency predisposes to heart failure and ischemia-reperfusion injury. Front Cardiovasc Med. 2019;6:31. https://www.frontiersin.org/articles/10.3389/fcvm.2019.00031/full, (last accessed date on 02 April 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ait-Aissa K, Kadlec AO, Hockenberry J, Gutterman DD, Beyer AM. Telomerase reverse transcriptase protects against Angiotensin II induced microvascular endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2017;314(5):H1053–H1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng W, Cai G, Xia Yet al. Mitochondrial dysfunction in atherosclerosis. DNA Cell Biol. 2019;38(7):597–606. [DOI] [PubMed] [Google Scholar]
- 21. Opherk D, Zebe H, Weihe Eet al. Reduced coronary dilatory capacity and ultrastructural changes of the myocardium in patients with angina pectoris but normal coronary arteriograms. Circulation. 1981;63(4):817–825. [DOI] [PubMed] [Google Scholar]
- 22. Manolis AS, Manolis AA, Manolis TAet al. Mitochondrial dysfunction in cardiovascular disease: current status of translational research/clinical and therapeutic implications. Med Res Rev. 2021;41(1):275–313. [DOI] [PubMed] [Google Scholar]
- 23. Kiyuna LA, e Albuquerque RP, Chen C-H, Mochly-Rosen D, Ferreira JCB. Targeting mitochondrial dysfunction and oxidative stress in heart failure: challenges and opportunities. Free Radical Biol Med. 2018;129:155–168.. https://www.sciencedirect.com/science/article/pii/S0891584918311304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guarini G, Kiyooka T, Ohanyan Vet al. Impaired coronary metabolic dilation in the metabolic syndrome is linked to mitochondrial dysfunction and mitochondrial DNA damage. Basic Res Cardiol. 2016;111(3):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bradley JM, Li Z, Organ CL, Polhemus DJet al. A novel mtDNA repair fusion protein attenuates maladaptive remodeling and preserves cardiac function in heart failure. Am J Physiol Heart Circ Physiol. 2017;314(2):H311–H321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haendeler J, Drose S, Buchner Net al. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler Thromb Vasc Biol. 2009;29(6):929–935. [DOI] [PubMed] [Google Scholar]
- 27. Santos JH, Meyer JN, Van Houten B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum Mol Genet. 2006;15(11):1757–1768. [DOI] [PubMed] [Google Scholar]
- 28. Ramlee MK, Wang J, Toh WX, Li S. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes (Basel). 2016;7(8):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daniel M, Peek GW, Tollefsbol TO. Regulation of the human catalytic subunit of telomerase (hTERT). Gene. 2012;498(2):135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gladych M, Wojtyla A, Rubis B. Human telomerase expression regulation. Biochem Cell Biol. 2011;89(4):359–376. [DOI] [PubMed] [Google Scholar]
- 31. Avin BA, Umbricht CB, Zeiger MA. Human telomerase reverse transcriptase regulation by DNA methylation, transcription factor binding and alternative splicing (Review). Int J Oncol. 2016;49(6):2199–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu X, Wang Y, Chang G, Wang F, Wang F, Geng X. Alternative splicing of hTERT pre-mRNA: a potential strategy for the regulation of telomerase activity. Int J Mol Sci. 2017;18(3):567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yi X, Shay JW, Wright WE. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res. 2001;29(23):4818–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong MS, Wright WE, Shay JW. Alternative splicing regulation of telomerase: a new paradigm?. Trends Genet. 2014;30(10):430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Listerman I, Sun J, Gazzaniga FS, Lukas JL, Blackburn EH. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013;73(9):2817–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chabowski DS, Kadlec AO, Ait-Aissa Ket al. Lysophosphatidic acid acts on LPA1 receptor to increase H2O2 during flow-induced dilation in human adipose arterioles. Br J Pharmacol. 2018;175(22):4266–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuo L, Davis MJ, Chilian WM. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am J Physiol. 1990;259(4):H1063–1070. [DOI] [PubMed] [Google Scholar]
- 38. Kuo L, Chilian WM, Davis MJ. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol. 1991;261(6):H1706–1715. [DOI] [PubMed] [Google Scholar]
- 39. Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92(2):e31–40. [DOI] [PubMed] [Google Scholar]
- 40. Brand L, Hader SN, Sorci-Thomas M, Beyer AM. Adipose specific loss of PCPE2 promotes microvascular endothelial dysfunction and decreased angiogenic potential. FASEB J. 2019;33(S1):522–528. [Google Scholar]
- 41. Kadlec AO, Chabowski DS, Ait-Aissa Ket al. PGC-1α (peroxisome proliferator–activated receptor γ coactivator 1-α) overexpression in coronary artery disease recruits NO and hydrogen peroxide during flow-mediated dilation and protects against increased intraluminal pressure. Hypertension. 2017;70(1):166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Durand MJ, Zinkevich NS, Riedel Met al. Vascular actions of angiotensin 1-7 in the human microcirculation: novel role for telomerase. Arterioscler Thromb Vasc Biol. 2016;36(6):1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma C, Beyer AM, Durand Met al. Hyperoxia causes mitochondrial fragmentation in pulmonary endothelial cells by increasing expression of pro-fission proteins. Arterioscler Thromb Vasc Biol. 2018;38(3):622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khosravi-Maharlooei M, Jaberipour M, Tashnizi AH, Attar A, Amirmoezi F, Habibagahi M. Expression pattern of alternative splicing variants of human telomerase reverse transcriptase (hTERT) in cancer cell lines was not associated with the origin of the cells. Int J Mol Cell Med. 2015;4(2):109. [PMC free article] [PubMed] [Google Scholar]
- 45. Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. In: Henderson DS, ed. DNA Repair Protocols. Vol 314. Totowa, NJ: Springer, 2006:183–199.. https://link.springer.com/protocol/10.1007/978-1-62703-739-6_31. [DOI] [PubMed] [Google Scholar]
- 46. Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc. 2008;130(30):9638–9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahmoud AM, Hwang C-L, Szczurek MRet al. Low-fat diet designed for weight loss but not weight maintenance improves nitric oxide-dependent arteriolar vasodilation in obese adults. Nutrients. 2019;11(6):1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD. An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol Heart Circ Physiol. 2014;307(11):H1587–H1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beyer AM, Zinkevich N, Miller Bet al. Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease. Basic Res Cardiol. 2017;112(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hughes WE, Chabowski DS, Ait-Aissa Ket al. Critical interaction between telomerase and autophagy in mediating flow-induced human arteriolar vasodilation. Arterioscler Thromb Vasc Biol. 2021;41(1):446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hoffmann J, Richardson G, Haendeler J, Altschmied J, Andrés V, Spyridopoulos I. Telomerase as a therapeutic target in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2021;41(3):1047–1061. [DOI] [PubMed] [Google Scholar]
- 52. Ale-Agha N, Jakobs P, Goy Cet al. Mitochondrial telomerase reverse transcriptase protects from myocardial ischemia/reperfusion injury by improving complex I composition and function. Circulation. 2021;144(23):1876–1890. [DOI] [PubMed] [Google Scholar]
- 53. Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol. 2003;23(13):4598–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dikalov SI, Kirilyuk IA, Voinov M, Grigor'ev IA. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radical Res. 2011;45(4):417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kilian A, Bowtell DD, Abud HEet al. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet. 1997;6(12):2011–2019. [DOI] [PubMed] [Google Scholar]
- 56. Brenner CA, Wolny YM, Adler RR, Cohen J. Alternative splicing of the telomerase catalytic subunit in human oocytes and embryos. Mol Hum Reprod. 1999;5(9):845–850. [DOI] [PubMed] [Google Scholar]
- 57. Ulaner GA, Hu JF, Vu TH, Giudice LC, Hoffman AR. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998;58(18):4168–4172. [PubMed] [Google Scholar]
- 58. Li G, Shen J, Cao Jet al. Alternative splicing of human telomerase reverse transcriptase in gliomas and its modulation mediated by CX-5461. J Exp Clin Cancer Res. 2018;37(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alsiary R, Brownhill SC, Bruning-Richardson Aet al. Expression analysis of the MCPH1/BRIT1 and BRCA1 tumor suppressor genes and telomerase splice variants in epithelial ovarian cancer. Gene. 2018;672: 34–44.. https://www.sciencedirect.com/science/article/pii/S0378111918306243. [DOI] [PubMed] [Google Scholar]
- 60. Bojesen SE, Pooley KA, Johnatty SEet al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45(4):371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mavrogiannou E, Strati A, Stathopoulou A, Tsaroucha EG, Kaklamanis L, Lianidou ES. Real-time RT-PCR quantification of human telomerase reverse transcriptase splice variants in tumor cell lines and non-small cell lung cancer. Clin Chem. 2007;53(1):53–61. [DOI] [PubMed] [Google Scholar]
- 62. Perez-Rivero G, Ruiz-Torres MP, Rivas-Elena JVet al. Mice deficient in telomerase activity develop hypertension because of an excess of endothelin production. Circulation. 2006;114(4):309–317. [DOI] [PubMed] [Google Scholar]
- 63. Bar C, Bernardes de Jesus B, Serrano Ret al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat Commun. 2014;5(1):5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ait-Aissa K, Hockenberry J, Gutterman DD, Geurts A, Beyer AM. Critical role of telomerase in regulating cerebral vascular function and redox environment. FASEB J. 2016;30:953–955.. https://faseb.onlinelibrary.wiley.com/doi/abs/10.1096/fasebj.30.1_supplement.953.5. [Google Scholar]
- 65. Durand M, Gutterman D, Beyer A. Vasodilator and vasoprotective actions of angiotensin 1-7 in the human microcirculation—role of telomerase. FASEB J. 2015;29(S1):789–783. [Google Scholar]
- 66. Chatterjee S, Hofer T, Costa Aet al. Telomerase therapy attenuates cardiotoxic effects of doxorubicin. Mol Ther. 2021;29(4):1395–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Radan L, Hughes CS, Teichroeb JHet al. Microenvironmental regulation of telomerase isoforms in human embryonic stem cells. Stem Cells Dev. 2014;23(17):2046–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gordon DM, Santos JH. The emerging role of telomerase reverse transcriptase in mitochondrial DNA metabolism. J Nucleic Acids. 2010. https://www.hindawi.com/journals/jna/2010/390791/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Martens A, Schmid B, Akintola O, Saretzki G. Telomerase does not improve DNA repair in mitochondria upon stress but increases MnSOD protein under serum-free conditions. Int J Mol Sci. 2020;21(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Perez-Rivero G, Ruiz-Torres MP, Diez-Marques MLet al. Telomerase deficiency promotes oxidative stress by reducing catalase activity. Free Radical Biol Med. 2008;45(9):1243–1251. [DOI] [PubMed] [Google Scholar]
- 71. Muzykantov VR. Targeting of superoxide dismutase and catalase to vascular endothelium. J Controlled Release. 2001;71(1):1–21. [DOI] [PubMed] [Google Scholar]
- 72. Teichroeb JH, Kim J, Betts DH. The role of telomeres and telomerase reverse transcriptase isoforms in pluripotency induction and maintenance. RNA Biol. 2016;13(8):707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ale-Agha N, Jakobs P, Goy Cet al. Mitochondrial telomerase reverse transcriptase protects from myocardial ischemia/reperfusion injury by improving complex I composition and function. Circulation. 2021;144(23):1876–1890. [DOI] [PubMed] [Google Scholar]
- 74. Dikalova AE, Pandey A, Xiao Let al. Mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ Res. 2020;126(4):439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Freed JK, Beyer AM, LoGiudice JA, Hockenberry JC, Gutterman DD. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ Res. 2014;115(5):525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chabowski DS, Kadlec AO, Ait-Aissa Ket al. Lysophosphatidic acid acts on LPA1 receptor to increase H2O2 during flow-induced dilation in human adipose arterioles. Br J Pharmacol. 2018;175(22):4266–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Durand MJ, Dharmashankar K, Bian J-Tet al. Acute exertion elicits a H2O2-dependent vasodilator mechanism in the microvasculature of exercise-trained but not sedentary adults. Hypertension. 2015;65(1):140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material. Patient information outside of the summary details provided cannot be shared for the privacy of individuals that participated in the study. Individual de-identified clinical information will be shared on reasonable request to the corresponding author. All data generated or analysed during this study are included in this published article (and its supplementary information files). Raw date for the current study are available from the corresponding author on reasonable request.