

SUMMARY
Chromatin is organized into multiscale three-dimensional structures, including chromosome territories, A/B compartments, topologically associating domains, and chromatin loops. This hierarchically organized genomic architecture regulates gene transcription, which in turn is essential for various biological processes during brain development and adult plasticity. Here we review different aspects of spatial genome organization and their functions in regulating gene expression in the nervous system as well as their dysregulation in brain disorders. We also highlight new technologies to probe and manipulate chromatin architecture and discuss how investigating spatial genome organization can lead to a better understanding of the nervous system and associated disorders.
Keywords: central nervous system, 3D genome, genomic architecture, chromatin, brain disorders, genetic risk factors, neuroepigenetics
Chromatin is organized into multiscale hierarchical three-dimensional structures. In this review, Fujita et al. discuss different aspects of spatial genome organization and their functions in regulating gene expression in the nervous system and their dysregulation in brain disorders.
INTRODUCTION
The completion of the human genome sequencing project in 2001 provided a blueprint of the genome at the linear sequence level (Lander et al., 2001; Venter et al., 2001). The human genome is encoded by approximately 3.2 billion nucleotides of DNA, and most human cells, which are diploid, contain about 6 billion base pairs, divided into 46 chromosomes. These 6 billion base pairs, equivalent to about 2 meters of linear DNA, are folded into three-dimensional (3D) structures and packaged into nuclei, which are about 5–10 μm in diameter. Histone proteins compact DNA to form nucleosomes, which allows packaging of the DNA into the microscopic nuclear space. Recent technical advances such as chromosome conformation capture (3C) analysis, which detects DNA-DNA interactions between close genomic loci within the 3D space of the nucleus, have led to new insights into the spatial organization of chromatin. Interphase chromosomes are spatially organized at hierarchical levels, from chromatin loops that allow associations between promoters and other regulatory elements, such as enhancers, over short- and long-range linear genomic distances, to chromatin domains, topologically associating domains (TADs), and A/B compartments; moreover, entire chromosomes themselves occupy defined regions in the nucleus, termed chromosome territories (Figure 1A). These hierarchical structures are essential for gene expression control and disturbances in these structures have been implicated in various human disorders (Yu and Ren, 2017). While traditionally investigated in cancer cell lines, recent studies have started to profile 3D genomes in the nervous system and address their functions and mechanisms of action. Here, we review recent studies on 3D genome organization and its dynamics underlying neural development and neuronal plasticity. We further highlight the disorganization of spatial chromosome architectures in brain disease states and discuss how 3D genome knowledge can advance our understanding of contributions of genome variants to different brain disorders. Finally, we discuss future perspectives and questions to be addressed.
Figure 1. Hierarchical chromatin structure.
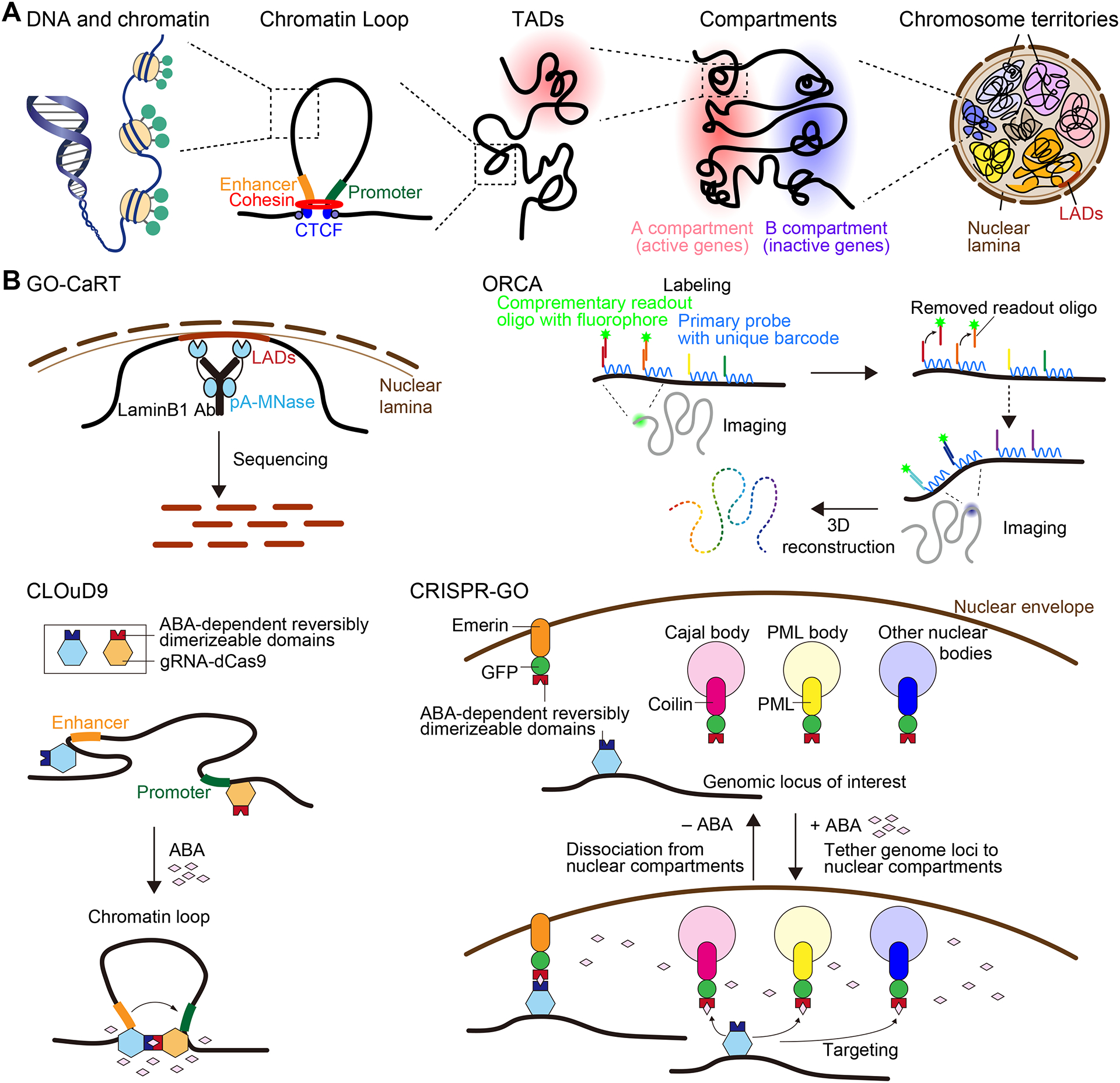
(A) Illustration of multiple layers of chromosome structural organization in the nucleus.
(B) Examples of technologies to profile different forms of chromosome structural organization and induce chromatin looping. Genome Organization using CUT&RUN Technology (GO-CaRT) uses LaminB1 tethered proteinA-MNase and enables the cleavage of nuclear lamina proximal DNA, followed by sequencing and LAD calling. Chromatin loop reorganization using CRISPR-dCas9 (CLOuD9) induces abscisic acid (ABA)-dependent reversible chromatin looping. Optical reconstruction of chromatin architecture (ORCA) enables the reconstruction of the trajectory of a genomic region of interest by tiling the region in short sections with an average resolution of 17 kb. CRISPR-genome organizer (CRISPR-GO) induces ligand-mediated dimerization of CRISPR-dCas9 and proteins specific for versatile nuclear compartments.
Hierarchical chromatin structure
Multiple layers of chromatin structural organization
There are multiple layers of chromosome hierarchical structure, including epigenetic modifications to the linear genome such as DNA methylation and histone modifications, as well as chromatin loops, TADs, A/B compartments, and 3D genomic locations in the nucleus (Figure 1A).
When cells are not in mitosis, chromosomes tend not to intermingle but instead occupy distinct chromosome territories (Cremer and Cremer, 2010). Hi-C, a 3C-derived technology that allows the complete detection of “all versus all” long-distance chromatin interactions across the entire genome via sequencing, confirmed the presence of chromosome territories and further revealed intra-chromosomal compartmentalization into regions of open and closed chromatin, termed “A” and “B” compartments, respectively (Lieberman-Aiden et al., 2009). “A” compartments include genomic loci that are generally gene rich, transcriptionally active, and DNase I hypersensitive; conversely, loci found in “B” compartments are relatively gene poor, transcriptionally silent, and harbor heterochromatic sequences. The spatial segregation of A/B compartments was confirmed by imaging-based methods (Su et al., 2020; Wang et al., 2016).
Another major type of chromatin organization involves megabase-size folding entities termed TADs (Nora et al., 2012; Rao et al., 2014). TADs were characterized using Hi-C maps at 40-kb resolution and are markedly smaller than A/B compartments - the median sequence size is 800 kb for TADs and 3 Mb for compartments. It has been proposed that restricting interactions between genes and their regulatory sequences is one major function of TADs. In mammals, TAD boundaries are usually demarcated by zinc-finger binding protein CCCTC-binding factor (CTCF) and the cohesin complex (Rao et al., 2014). The cohesin complex forms a ring-like structure comprised of four core subunits: SMC1, SMC3, RAD21 and SA1/2 (Figures 1A and 2A). Cohesin cooperates with CTCF to form a chromatin loop and functions with the general transcriptional co-activator, the Mediator complex (Wendt et al., 2008). Some studies proposed that CTCF and cohesin promote “loop extrusion”, which contributes to TAD formation (Ganji et al., 2018; Sanborn et al., 2015). In this model, cohesin is loaded onto DNA by cohesin loaders, including nipped-B-like protein (NIPBL) and MAU2, then slides along the chromatin and extrudes it outwards until it reaches the chromatin boundaries that are often formed by CTCF. Real-time visualization has recently confirmed that cohesin and its loaders induce genomic interphase DNA into loops by extrusion (Bauer et al., 2021). Direct interaction of the N-terminal segment of CTCF with cohesin contributes to loop stabilizing activity. DNA is spontaneously translocated by cohesin’s hinge, and NIPBL separates from the hinge and clamps DNA onto the head of SMC3 in an ATP-dependent manner.
Figure 2. Molecular machineries regulating chromosome structural organization.

(A) A schematic diagram of the Cohesin complex.
(B) A model of SMC structure and the locations of SMC3 mutations found in human patients.
(C) Impact of mutations of cohesin complex components on the nervous system, including gene expression, dendritic growth, dendritic spine morphologies, and behavior in mouse models.
In most cell types, heterochromatin normally resides at the nuclear periphery, whereas euchromatin situates toward the nuclear interior. In one exception, this organization is inverted in rod photoreceptor neurons of nocturnal retinas, in which the dense heterochromatin localized in the nuclear center may serve as collecting lenses to enhance light transduction efficiency (Falk et al., 2019; Sofueva et al., 2013; Solovei et al., 2009). Lamina-associated domains (LADs), which are genomic regions that are in close contact with the nuclear lamina, are also thought to help organize chromosomes inside the nucleus and have been largely associated with gene repression (Guerreiro and Kind, 2019; van Steensel and Belmont, 2017).
Advanced technologies to probe 3D genome and investigate its function
Since the initial 3C technology, various methodologies have been developed to investigate chromatin architecture. First, there have been significant improvements in the mapping resolution and in the reduction of cell numbers required, even to the single-cell level, along with advances in assays for chromatin-protein interactions and general chromatin architecture, such as single-cell Hi-C, CUT&Tag, and single-cell ATAC-seq. Advances in multi-omics analyses are expected to provide a comprehensive understanding of the association between 3D genome organization and gene transcription. These sequencing-based approaches have also been adapted to new technologies to investigate the genomic distribution in the 3D space of the nucleus. For example, Genome Organization using CUT&RUN Technology (GO-CaRT) is an application of CUT&RUN for mapping genomic interactions within nuclear compartments and enables profiling of the LAD architecture (Figure 1B) (Ahanger et al., 2021).
Second, advances in labeling and imaging technologies have provided novel insights regarding chromatin structure in the nuclear space (Boettiger and Murphy, 2020; Chen et al., 2016; Girelli et al., 2020; Quinodoz et al., 2018; Ricci et al., 2017). New strategies for fluorescence in situ hybridization, such as Oligopaints technologies, signal amplification by exchange reaction (SABER) (Jungmann et al., 2014), and labeling using genome editing tools, such as zinc-finger nuclease, transcription activator-like effector nuclease, and the clustered regularly interspersed short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system (Hilton et al., 2015), have enabled innovative chromatin imaging. In particular, the Oligopaint optical reconstruction of chromatin architecture (ORCA) approach allows the visualization of nanoscale DNA paths in single-cells and measurement of kilobase-scale interactions among nearby cis-regulatory elements (Mateo et al., 2019) (Figure 1B). Further improvements in live cell imaging will enable the tracking of dynamic changes in spatial chromatin structures throughout development and in response to stimulation.
Third, various genome engineering technologies have been developed for dynamic manipulation of 3D chromatin structures. Tethering repressed genes to active cis-regulatory elements by using artificial zinc fingers substantially activates their expression in both human and mouse cells (Deng et al., 2012). CRISPR-dCas9 (CLOuD9), which uses dCas9 protein in combination with a reversible chemically-induced proximity system that utilizes the plant phytohormone S-(+)-abscisic acid (ABA) and modified components of the plant ABA signaling pathway, induces targeted and reversible chromatin looping upon ABA treatment (Hao et al., 2017; Morgan et al., 2017) (Figure 1B). CRISPR-genome organizer (CRISPR-GO) is a technology that expands applications of CRISPR into regulation of the spatial positioning of gene loci in the cell nucleus (Wang et al., 2018). CRISPR-GO induces ligand-mediated chemical dimerization of CRISPR-dCas9 and nuclear fraction-specific proteins to artificially rearrange the genome structure, which can be used to elucidate the causal relationship between the macroscale (~ μm) spatial structure and cellular functions (Figure 1B). In another method, light-activated dynamic looping enables the formation of long-range contacts between distal genomic loci in response to blue light over short time scales (Kim et al., 2019). Future application of these tools will allow assessment of the causal role of chromatin looping in controlling target gene expression under physiological conditions.
3D genome organization during nervous system development
The nervous system development involves neural patterning, generation of different cell types from neural progenitors, and maturation and integration of various neural cell types. Dynamic physiological changes in the chromatin structure through epigenetic mechanisms enable spatio-temporal regulation of gene expression during these complex processes (Yao et al., 2016; Yoon et al., 2018).
Chromocenter: compaction of the chromatin state towards neural differentiation
The global nuclear structure undergoes dynamic changes during sequential differentiation from embryonic stem cells (ESCs) to neural progenitor cells (NPCs), and then to terminally differentiated neurons. The 3D positioning of genes in the nucleus plays an important role during brain development (Ito and Takizawa, 2018; Takizawa and Meshorer, 2008). Evidence from several studies has indicated that the chromatin state is globally open in ESCs and becomes increasingly condensed during differentiation into NPCs and neurons (Kishi and Gotoh, 2018). Chromocenters, which are heterochromatin foci that can be stained strongly with DNA-interacting dyes, exhibit considerable diversity in their size, number, and distribution in various differentiation stages and cell types (Billia et al., 1992; Billia and de Boni, 1991; Manuelidis, 1984; Martou and De Boni, 2000; Solovei et al., 2004). Preformed chromocenters in ESCs disperse into smaller foci during differentiation into NPCs (Aoto et al., 2006; Meshorer et al., 2006; Williams et al., 2006). During NPC differentiation into post-mitotic neurons, the number of chromocenters decreases, and they converge into larger clusters located in the center of the nucleus. The findings that ESCs have more active chromatin marks and fewer heterochromatin domains suggest that these changes in chromocenters may be associated with the expansion and segregation of heterochromatin and the deposition of active histone marks during cellular differentiation (Hawkins et al., 2010; Kishi et al., 2012; Xie et al., 2013).
Genome-wide analyses of chromatin accessibility using techniques such as assay for transposase-accessible chromatin using sequencing (ATAC-seq), formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-seq), and DNAse I hypersensitive site sequencing (DNase-seq), have also revealed dynamic changes in chromatin openness during neural differentiation processes (de la Torre-Ubieta et al., 2018; Frank et al., 2015; Thakurela et al., 2015). Together, these studies provide evidence suggesting that an “open” chromatin structure is crucial for pluripotency during sequential differentiation from ESCs to differentiated neurons.
Compartments and TADs: Fewer chromatin interactions in active-domain and more chromatin interactions in inactive-domain during neural differentiation
A/B compartments undergo dynamic switching during transitions from ESCs to NPCs and then to neurons. High resolution Hi-C analyses have revealed the progressive changes in differentiation stage-specific chromatin architecture both in mouse and human cultured cells. Interactions between active TADs become weaker, whereas interactions in inactive TADs become stronger as mouse ESCs differentiate into NPCs and then to neurons (Bonev et al., 2017). Similarly, progressive changes in chromatin architecture have been observed during neuron and astrocyte differentiation from human induced pluripotent stem cells (iPSCs) (Rajarajan et al., 2018). About 36% of A and B compartments throughout the genome switch during ESC differentiation (Dixon et al., 2015). Intriguingly, compartment switching affects the expression of only a certain subset of genes, suggesting that compartmental status may influence the accessibility of genomic regions for only limited subsets of genes. Consistent with global compaction of the chromatin structure during ESC differentiation into NPCs, the total size of A compartments in differentiated cells, including NPCs, was reduced by 5% compared with that in ESCs. Collectively, the compaction of nuclear chromatin domains seems to be a general feature of neural differentiation and may contribute to the stable silencing of genes unnecessary for differentiated neurons.
Phase separation, which describes the concept of un-mixing of a liquid into two different liquid phases, has recently emerged as an important phenomenon in the formation of biomolecular condensates. Phase-separated multi-molecular assemblies have been suggested as a general regulatory mechanism to compartmentalize biochemical reactions within cells (Hnisz et al., 2017). Analysis of A/B compartments at a finer resolution of 500 bp demonstrated the likelihood of a much finer A/B compartment organization emerging with phase separation at the scale of small regulatory sequences, contrary to the conventional findings obtained as large (Mb-scale) blocks of sequence by coarse-resolution compartment profiles (Gu et al., 2021). This study also distinguished between loops that are formed by extrusion and those that are formed through a non-extrusion mechanism.
Compared with A/B compartments, TADs appear to be relatively more stable during neural differentiation. TAD boundaries are stable during cell divisions and conserved among various cell types or lineages (Dixon et al., 2012; Ho et al., 2014), although inter-TAD interactions and chromatin interactions within TADs can occur during cell differentiation (Dixon et al., 2016; Dixon et al., 2015; Fraser et al., 2015; Hansen et al., 2018; Nora et al., 2012; Rao et al., 2014; Schmitt et al., 2016). These findings suggest that chromatin is regionalized at somewhat stable boundaries, producing restricted regions in which chromatin interactions occur more often.
Loops and local interactions
Within TADs, chromatin structures seem to be more extensively reorganized locally thorough chromatin looping, including interactions between promoters and enhancers, which often occurs in a developmental stage- and cell type- specific manner.
Chromatin interaction analysis with paired-end tag sequencing has revealed local chromosomal structures linked to the control of cell identity in ESCs (Dowen et al., 2014). Super-enhancers, which are cell type-specific and play key roles in cell fate determination, are marked by high levels of H3K27ac (Hnisz et al., 2013; Parker et al., 2013). Super-enhancer-driven cell identity genes are generally localized within chromatin loops formed by CTCF and cohesin. These looped structures form insulated neighborhoods whose integrity is important for the proper expression of local genes. The phase separation model was recently used to explain the features of transcriptional control, including the formation of super-enhancers and heterochromatin domains (Hnisz et al., 2017).
During the course of neural differentiation, dynamic alterations of CTCF-mediated loops occur in both mouse and human developing brains (Bonev et al., 2017; de la Torre-Ubieta et al., 2018; Lu et al., 2020; Rajarajan et al., 2018). During the NPC to neuron transition, loops associated with genes related to cell proliferation, morphogenesis, and neurogenesis are lost, which is consistent with the commitment to a lineage change from the precursor stage towards a postmitotic neuronal identity (Bonev et al., 2017) (Figure 3A). Similarly, during the NPC to glia transition, loops associated with genes related to neuron-specific functions are lost, which is consistent with a non-neuronal lineage commitment (Rajarajan et al., 2018). In addition, the loss of many shorter-range contacts and loops during NPC differentiation into neurons is associated with concomitant increases in longer range (>100–200 kb) contacts in both humans and mice. Together, these results provide insights into the relationship between transcriptional control of cell identity and local chromosome structures mediated by chromatin looping.
Figure 3. Dynamic 3D genome re-organization during development and in response to stimulation in neurons.

(A) Features of 3D genome re-organization during embryonic stem cell – neural progenitor – neuron/glia differentiation.
(B) Dynamic chromatin looping in response to neuronal activation, a model of signal integration for immediate early gene (IEG) and secondary response genes (SRG).
(C) A potential model for multiple inputs/activity-inducible enhancers to regulate the same downstream gene. Invariant and activity-induced loops form the hub and enable multiple enhancers to contact and regulate the same downstream gene. This process may precede transcriptional activation.
(D) A model of epigenetic priming during sequential phases of learning and memory.
Proximity ligation-assisted chromatin immunoprecipitation with sequencing (PLAC-seq) and HiChIP are both powerful tools for studying long-range chromatin contacts anchored at genomic regions bound by specific proteins or histone modifications. Promoter-enriched trimethylation of lysine 4 on histone H3 (H3K4me3) mark-based PLAC–seq analysis of purified radial glia neural stem cells, intermediate progenitor cells, excitatory neurons, and interneurons from the developing human cortex has shown that a subset of promoters with increased levels of chromatin interactivity are enriched for cell type-specific genes (Song et al., 2020), suggesting that interactions at these loci may drive cell type-specific differences during human corticogenesis. Furthermore, during differentiation of human iPSCs into neurons, the number of enhancer and promoter interactions increases in NPCs and neurons compared to those in iPSCs. The genes involved in NPC and neuron chromatin loops are strongly associated with neuronal differentiation functions (Lu et al., 2020). Many neural enhancer-promoter aggregates are substantially strengthened in primary brain tissue (Lu et al., 2020). In addition, multi-omics analysis among human, macaque, and mouse uncovered a human-specific loop and showed that enhancer-promoter interactions and regulation of EPHA7 affect neuronal dendrite development and spine maturation (Luo et al., 2021). Single-cell analyses have showed that both the 3D genome structural types and transcriptome are fundamentally changed between postnatal (P) 7 and P28 in the mouse brain, and furthermore, adult neurons, but not other adult neural cell types such as astrocytes and oligodendrocytes, undergo large-scale gene positioning changes (Tan et al., 2021). These observations suggest that enhancers and promoters continue to aggregate and stabilize, even after differentiation into neurons, which can regulate neuronal maturation.
LADs
Gene expression is often associated with lamina-genome interactions. In general, genes that move away from the lamina are concomitantly activated, while genes that are located within LADs are transcriptionally inactive (van Steensel and Belmont, 2017). The 3D positioning of genes in the nucleus plays an important role in neuronal function during brain development (Ito and Takizawa, 2018; Takizawa and Meshorer, 2008). A study on DNA adenine methyltransferase identification (DamID) of lamin B1 to map LADs revealed that in at least four cell types, ESCs, NPCs, terminally differentiated astrocytes, and mouse 3T3 embryonic fibroblasts, LADs are mostly repressive chromatin features (Peric-Hupkes et al., 2010). At a finer level, the dynamic reshaping of nuclear lamina-chromatin interactions of hundreds of genes over ESC to NPC and NPC to astrocyte differentiation has been detected. Associations with nuclear lamina are tightly linked to gene repression, and reorganization of these interactions during differentiation involves many genes that are important for cellular identity. Furthermore, promoter transplantation experiments showed that promoters become active when they are systematically moved from their native LAD location to a more neutral chromatin environment, suggesting a causal relationship between local chromatin state and gene activity (Leemans et al., 2019).
However, other studies over the last decade have demonstrated that some chromatins in proximity to the lamina, especially those portions in proximity to nuclear pores, are euchromatic with some highly expressed genes (Fiserova et al., 2017; Krull et al., 2010). Moreover, a recent study with GO-CaRT has found some highly transcribed genes in LADs characterized by H3K9me2 depletion that are localized close to the repressive nuclear lamina, suggesting that even in the repressive context of genome-lamina interactions, depletion of H3K9me2 may promote transcriptional activation (Ahanger et al., 2021). Through comparison among the genomic interactions with nuclear lamina in the developing mouse, macaque, and human brain, it was found that evolutionarily conserved LADs are enriched for transcriptionally active neuronal genes associated with synapse function. Interestingly, LAD architecture observed in NPCs varies in vitro and in the brain in vivo. Moreover, region-specific differences of LAD architecture correspond to region-specific gene expression in both the developing human and mouse brain, suggesting that the variety of LAD architecture can control the regional identity of brain cells.
While it has become clear that dynamic changes in chromosome architecture at various scales play critical roles in brain development, many questions remain to be answered. For example, how is chromosome architecture regulated in space and time throughout the transition from proliferation to differentiation, and in postmitotic cells? What exactly are the causal and non-causal roles of 3D genome alterations in neural development? What are the mechanisms underlying transcriptional regulation by chromatin architecture? For instance, several studies reported that transcription can occur without obvious TADs and CTCF-mediated loops at particular stages of early embryogenesis (Du et al., 2017; Ke et al., 2017). Elucidating the detailed mechanisms involved would be a milestone in addressing fundamental questions in neural development.
Dynamic changes in 3D genome organization upon stimulation
Neuronal activation and resulting synaptic activities drive transcriptional programs involving genes critical for proper neuronal circuit formation, maturation, and plasticity. Many immediate early genes (IEGs) are upregulated by neuronal activity and are defined as genes that are rapidly and transiently induced (within 5 to 10 minutes of stimulation) by extracellular stimuli without the requirement for de novo protein synthesis (Yap and Greenberg, 2018). Since IEGs often encode transcription factors, they can regulate a set of secondary response genes (SRGs), which are expressed in the order of hours in response to signaling and require new protein synthesis. While there are probably several hundred IEGs, SRGs are far more numerous and are involved in various and cell-type specific functions in neurons. c-Fos, an neuronal activity-induced IEG, can serve as a pioneering factor in opening chromatin accessibility across the genome at c-Fos-binding sites to regulate SRG expression in the adult mouse hippocampus in vivo (Su et al., 2017). In addition, recent studies used high-resolution Chromosome Conformation Capture Carbon Copy (5C) and Hi-C methods have revealed how activity-dependent enhancers are temporally connected via long-range chromatin loops to regulate gene expression during a wide range of neuronal activity paradigms (Beagan et al., 2020; Fernandez-Albert et al., 2019). IEGs, including Fos and Arc, connect to activity-dependent enhancers via singular short-range loops that form within 20 minutes of neuronal stimulation, whereas the SRG Bdnf engages with both pre-existing and activity-induced long-range loops that form within 1–6 hours (Beagan et al., 2020) (Figure 3B). Moreover, activity-dependent loops form prior to the peak of mRNA levels of IEGs, suggesting a mechanism to parse multiple inputs for gene expression regulation (Joo et al., 2016). However, the precise mechanism of this delayed peak of transcription after activity-induced loop formation remains unclear. One potential mechanism is the recruitment of a transcriptional activator after epigenetic priming as proposed in the study discussed below (Figure 3C) (Marco et al., 2020). The dynamics of the 3D genome structure might allow the sequential control of neuronal activity-induced gene expression, induction of IEGs following by SRGs, resulting in changes in cellular function.
A recent genetic study has shown that chromatin loop formation can be causally linked to activity-dependent gene expression and such regulation can impact brain function. PLAC-seq identified long-distance interactions between activity-dependent gene promoters and enhancers upon neuronal stimulation (Yamada et al., 2019). The core cohesin subunit Rad21 is required for activity-dependent transcription, and the occupancy of Rad21 at enhancers and promoters is correlated with changes in H3K27ac upon neural stimulation. Furthermore, conditional knockout of Rad21 in adult mouse cerebellar granule neurons significantly reduces enhancer–promoter interactions and delays motor learning in a behavioral test.
Memory formation and recall involve coordinated neuronal activity and the role of 3D genome organization has been examined in the context of engram ensembles. A multi-omics study, including RNA-seq, ATAC-seq, and Hi-C, in neurons that were activated during memory encoding, consolidation, or recall in the adult mouse brain has shown sequential changes of 3D genome architecture during different phases of learning and memory (Figure 3D) (Marco et al., 2020). First, memory encoding via neuronal activation increases chromatin accessibility, predominantly on enhancers. At this phase, these primed loci lack the interaction with promoters and corresponding transcriptional changes, but they engage in de novo functional enhancer-promoter interactions during later phases of memory formation, suggesting epigenetic priming. After memory encoding, memory consolidation corresponds with the reorganization of large chromatin segments and increased enhancer-promoter interactions, which enables increased gene expression that presumably allows stabilization of the memory. Furthermore, in the context of memory recall, reactivated engram neurons use a subset of de novo long-range enhancer-promoter interactions where primed enhancers are linked to their respective promoters to upregulate genes involved in mRNA transport and local protein translation in synaptic compartments. This study illustrates how the dynamics of 3D genome architecture can support engram formation.
Taken together, the alteration of spatial chromosome structure can be an epigenetic priming event in response to external stimulation, and these changes in chromatin structure potentially control downstream functions, such as gene expression changes, the maintenance of neuronal activity, and plasticity. This epigenetic priming can contribute to the link between environmental changes and brain functions. Since chromatin architecture is cell-type specific and context-dependent, it will be interesting in the future to investigate how stimulation-induced changes in the 3D genome may regulate gene expression in other neural cell types, such as glia and microglia, and during other biological processes, such as injury and aging (Fujita and Yamashita, 2020).
Dysregulation of spatial chromatin architecture in brain disorders
Dysregulation of chromatin architecture has been frequently linked to diseases. For example, deleterious mutations in the genes encoding chromatin architectural proteins, such as CTCF and cohesion, cause various developmental abnormalities. Deleterious mutations in cohesin core subunit- or cohesin-related genes cause the multisystem developmental disorder Cornelia de Lange syndrome (CdLS). Mutations in the gene for cohesin loader, NIPBL, were first identified in CdLS, followed by mutations in the genes for the cohesin subunits SMC1A, SMC3, and RAD21. Mutations in the regulatory factors BRD4, ANKRD11, and HDAC8, which regulate SMC3 acetylation and cohesion cycling (Deardorff et al., 2012), were also detected for CdLS (Kline et al., 2018). Individuals with CdLS show diverse symptoms, including intellectual disabilities, anxiety, attention deficit hyperactivity disorder, and autism-like characteristics. In animal models, Smc3-deficient heterozygous mice show increased dendritic complexity and decreased spine density in cortical neurons (Fujita et al., 2017). Neuron-specific Smc3 deletion mice exhibit the same phenotype as the global heterozygous knockout mice, indicating that cohesin function in postmitotic neurons is required for proper neuronal functions (Figure 2B). These mice also demonstrate increased anxiety-related behaviors that are consistent with the symptoms of CdLS. Since Smc3 is an important subunit of cohesin for its recycling and function, these observations suggest that 3D genome organization through cohesin is required to establish neural circuits and expression of specific brain functions.
Causal mutations for some brain diseases have also been shown to impact the spatial genome architecture, such as in Fragile X syndrome (Sun et al., 2018). 5C in B-lymphocytes and brain tissue derived from individuals with Fragile X syndrome showed severe disruption of TAD boundaries and loss of CTCF occupancy at the specific site adjacent to FMR1 disease-associated short tandem repeats. Future studies will address whether other disease-related large copy number variants (CNVs) and specific genomic variants located in critical chromatin architecture regulatory regions, such as CTCF binding sites and TAD boundaries, may impact chromatin structure, leading to dysregulation of gene expression.
Recent studies have also revealed spatial chromatin alterations in neurological diseases including schizophrenia (SCZ), bipolar disorder (BD), and Alzheimer’s disease (AD) (Bendl et al., 2021; Girdhar et al., 2022; Nott et al., 2019). SCZ and BD risk loci have been reported to be enriched with active neuronal promoters and enhancers in the adult human frontal lobe (Fullard et al., 2018; Girdhar et al., 2018; Hauberg et al., 2020; Roussos et al., 2014). Promoters and enhancers are enriched with trimethyl-H3K4 (H3K4me3) and acetyl-H3K27 (H3K27ac), and these histone modifications are tightly associated with higher-order chromatin structures, including megabase-scale TADs and A/B compartments. Analysis of histone modification landscapes from SCZ, BD, and control brains revealed that widespread disease-associated alterations affect the neuronal H3K27ac acetylome, but not the H3K4me3 methylome (Girdhar et al., 2022). In addition, the authors examined chromosomal domains (known as mini-TADs) as cis-regulatory domains (CRDs), which span the spatial clustering of chromatin peaks that extend across 104–106-bp chromosomal organization, and showed that CRDs are tightly linked to the structures of self-interacting domains. Furthermore, dysregulated acetylation of mini TAD-level chromosomal domains in the prefrontal cortex of SCZ and BD subjects shows cell type specificity, with domain hyperacetylation consistently linked to regulatory sequences that are specifically important for glutamatergic neurons, and hypoacetylation to GABAergic inhibitory interneurons and oligodendrocytes and myelination. Similar approaches have been successfully applied to examine open chromatin regions in the postmortem brains of AD patients (Bendl et al., 2021). AD-associated changes in chromatin accessibility varied considerably according to the cell type and brain region. Interactome maps from PLAC-seq identified several parameters, such as AD-risk variants that were linked to more distal active promoters rather than the closest promoter, enhancers harboring AD-risk variants that were PLAC-linked to active promoters of both GWAS-assigned genes and an extended subset of genes not assigned to GWAS loci, and microglia-specific enhancers harboring AD risk variants linked to genes expressed in multiple cell types, suggesting cell type-specific disease susceptibility (Gosselin et al., 2017; Nott et al., 2019). Together, these studies suggest that histone modifications and spatial chromatin organization in neurological diseases track the underlying genetic risk architecture and the possible cell types in which they function.
Linking disease-associated genetic variants to target gene regulation via the 3D genome
Over the past decade, a large number of genome-wide association studies (GWAS) have identified many genetic variants associated with different brain disorders. In comparison to causal genetic variants for Mendelian disorders that lie mostly within protein-coding regions of the genome, human neurological and neuropsychiatric disease-associated genetic variants are largely found within non-coding regions of the genome which could serve as regulatory elements (Lu et al., 2020; Song et al., 2020; Song et al., 2019). A challenge for the field is to interpret the functional consequence of these disease-associated genetic variants on disease risk. These non-coding variants are hypothesized to regulate the level of gene expression in the developing and adult human brain in cell type-specific epigenomic contexts given the unique composition of DNA-binding machinery present within spatiotemporally-defined and heterogeneous cell states, such as basal and activity-dependent states in neurons, but the direct target genes and the impact on their expression are not always clear.
Expression quantitative trait locus (eQTL) mapping has provided a powerful cis-regulatory logic to link disease-associated genetic variants to target genes in cell type-specific contexts (Consortium, 2020). However, a reliance on large human population sample sizes to identify statistically significant variants or cis-eQTLs associated with target gene expression differences is a primary limitation of such eQTL studies. Cell type-specific 3D genome maps provide an alternative framework to link disease-associated genetic variants residing within candidate regulatory elements to target genes (Figure 4A). Mapping diseaseassociated genetic variants to cell type-specific 3D genomes obtained from healthy control samples, such as primary human brain tissue or human iPSC-derived neural cell types, allows for the creation of a predictive framework to link disease-associated genetic variants to candidate target genes via enhancer-promoter chromatin contacts in the absence of acquiring primary tissue from patients with neurological or neuropsychiatric disease or creating human disease-specific patient iPSC lines (Rajarajan et al., 2019; Won et al., 2016). Development of computational tools such as H-MAGMA to predict the target genes of disease-associated genetic variants have leveraged Hi-C datasets from primary human developing cortex and adult dorsolateral prefrontal cortex to show that non-coding variants often interact with distal genes, and assigning variants simply to the nearest gene may miss important neurobiological insights into the genetic architecture of neurological and neuropsychiatric diseases (Sey et al., 2020). Simultaneously mapping variants in linkage disequilibrium with GWAS associations to cell type-specific 3D genomes allows for the creation of a more comprehensive predictive framework to identify putative genetic variation conferring disease risk. Incorporation of larger cohorts of human population genetic variation (Genomes Project et al., 2015), including single nucleotide variants (SNVs) from the recent Genome Aggregation Database (gnomAD) consortium (Karczewski et al., 2020), expands the scope of such a 3D genome predictive framework. Indeed, similar to analyses conducted using the gnomAD datasets (Karczewski et al., 2020) suggesting protein-coding genes intolerant to inactivation, large-scale cohorts of whole genomes could be explored to identify critical non-coding regions of the human genome intolerant to inactivation, which may be important for human neurobiological phenotypes.
Figure 4. Linking brain disorder-associated noncoding genomic variants to target genes via chromatin looping and testing their functional impacts.
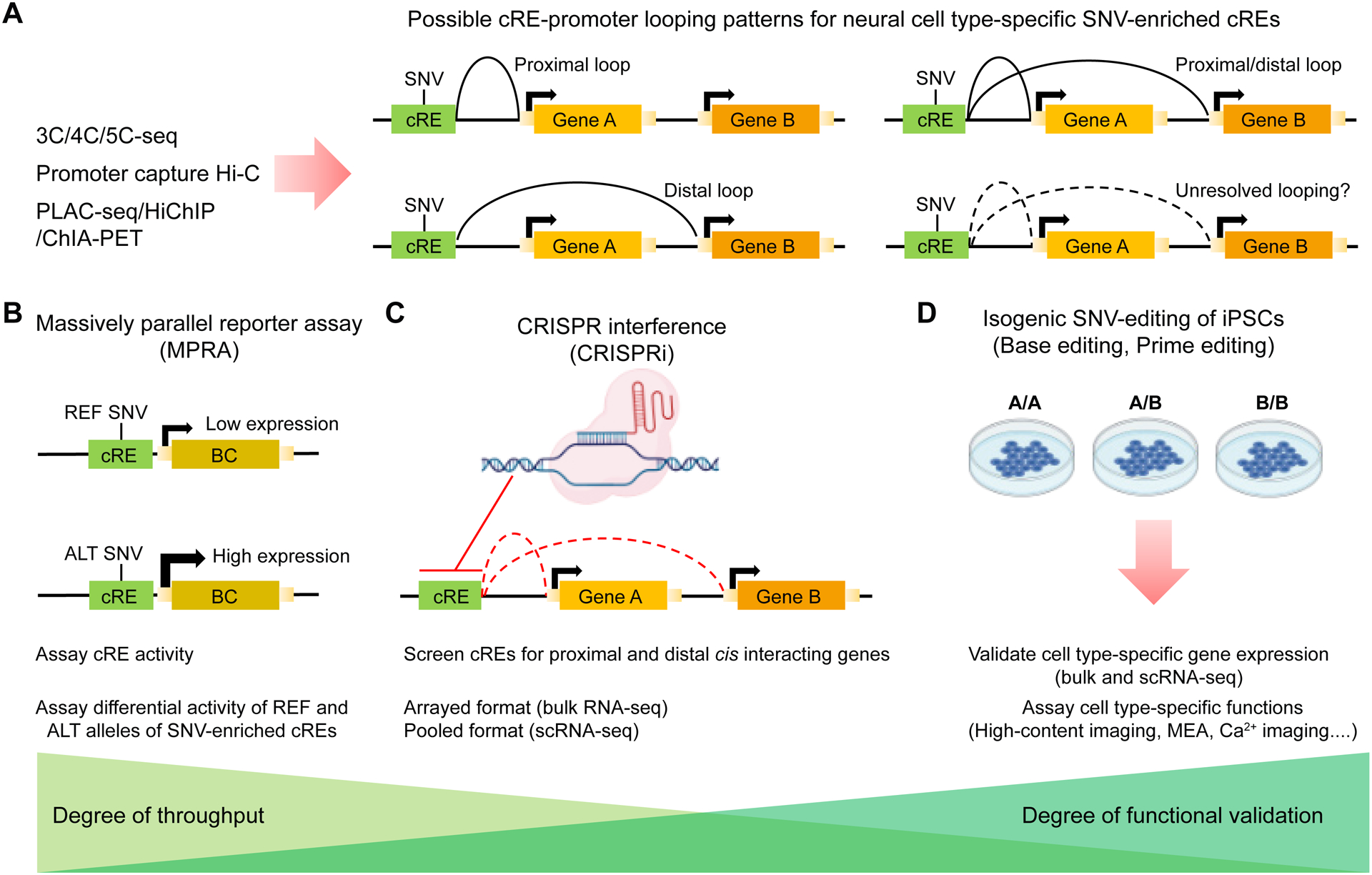
(A) Various methods to profile the 3D genome allow for the high-throughput prioritization of GWAS disease-associated single nucleotide variants (SNVs) in neural cell type-specific contexts. SNV-enriched candidate regulatory elements (cREs) defined with biochemical epigenetic marks and accessible chromatin integrated with cRE-promoter looping patterns identity putative SNV-target gene pairs dysregulated in brain disorders. Proximal, distal, proximal/distal, and unresolved cRE-promoter looping patterns are four possible mechanisms of SNV-mediated dysregulation of gene expression via the 3D genome.
(B) Massively parallel reporter assays (MPRAs) are high-throughput screening techniques to assess the differential activity of SNV-enriched cREs between reference (REF) and alternate (ALT) disease-associated alleles in cell type-specific contexts. Pooled MPRA libraries are assembled with SNV-enriched cREs cloned in front of an expressed barcode (BC) for high-throughput analysis.
(C) CRISPR interference (CRISPRi) targeted to noncoding cREs allow for medium-throughput screening of cRE-target gene pairs. The assay will screen cREs for proximal and distal cis-interacting genes and can include SNV-enriched cREs identified in (A). The assay can be performed in an arrayed format with bulk RNA-seq readout or a pooled format with single-cell RNA-seq (scRNA-seq) readout.
(D) Isogenic SNV-editing of iPSCs via CRISPR base editing or prime editing coupled with functional assays are low-throughput methods to validate the function of disease-associated SNVs in cell type-specific contexts. Isogenic SNV-edited iPSCs with three possible genotypes (A/A, A/B, and B/B) can be differentiated to desired cell types to assay transcriptomic consequences of SNVs via bulk RNA-seq and scRNA-seq and functional consequences of SNVs such as neural morphology via high-content imaging, electrophysiology via multi-electrode arrays (MEAs), or neuronal signaling via calcium imaging.
While the 3D genome can be used to link disease-associated variants to candidate target genes, these approaches are largely descriptive and correlative and require further validation via in situ and synthetic perturbational approaches to provide deeper neurobiological mechanistic insights into how disease-associated variants drive disease phenotypes. Enhancer-promoter proximity is hypothesized to regulate gene expression, but proximity alone may not explain all cases of gene expression regulation. Proximity may be maintained even for inactive genes or enhancers (Ghavi-Helm et al., 2014), and it is also possible that enhancer transcription in neurons may allow enhancer sequences to exert functional consequences in the absence of direct enhancer-promoter proximity (Kim et al., 2010). Therefore, a variety of synthetic approaches differing in their degree of throughput and functional validation are required to validate how SNVs may exert their impact on disease-relevant phenotypes. As a high-throughput approach with a low degree of functional validation, massively parallel reporter assays (MPRAs) can assess the differential activity of SNV-enriched candidate regulatory elements (cREs) between reference (REF) and alternate (ALT) disease-associated alleles on activating gene expression in cell type-specific contexts (Figure 4B). A medium-throughput in situ perturbational approach with a medium degree of functional validation employs CRISPR interference (CRISPRi) to epigenetically silence cREs to measure the downstream consequences on gene expression in the local genomic area (proximal and distal cis-interacting genes) or genome-wide (Figure 4C). A low-throughput approach with the highest degree of functional validation involves directly installing SNVs into the genome with techniques, such as CRISPR base editing or prime editing, to directly assess the functional consequence of disease-associated genetic variants on cell type-specific target gene expression and cell type-specific neurobiological functions through comparisons between isogenic conditions (Figure 4D). The advances of human iPSC-derived 2D neural cultures and 3D brain organoids (Qian et al., 2019) provide platforms for these analyses and furthermore for downstream functional analyses of cellular functions (Figure 4D). For further review of these approaches, readers are directed to an elegant recent review (Townsley et al., 2020).
Future Perspectives
Our understanding of the role and regulatory mechanisms of spatial chromatin structure and its effects on gene expression and biological functions has progressed significantly. However, as new findings emerge, they raise further questions regarding the mechanism and function of the 3D genome organization in gene expression and physiological functions.
First, we need technological advances for more detailed profiling of 3D genome organization in different cell types and especially from different in vivo conditions, such as different developmental stages and basal/stimulated states. Single-cell analysis has led to deeper and more robust classification of brain cell types and advances in technical approaches to understand 3D genome organization at the single-cell level, such as single-cell Hi-C, CUT & Tag, single-cell ATAC-seq, oligopaint imaging approaches, will help to specifically address how 3D genome organization affects neural circuit formation and disease pathogenesis in distinct cell types. Future studies to allow for increased resolution, even at the single-cell level, in in vivo conditions will help to decipher how chromatin alterations in different contexts link to gene expression changes and functional outcomes.
Second, discerning cause and consequence of the spatial alteration of chromatin architecture is of particular importance. Several studies have shown that the ablation of CTCF or cohesin has limited effects on gene expression and histone modifications even though it causes TAD loss, suggesting that transcriptional regulation might be preserved even if TADs are almost completely disrupted (Nora et al., 2017; Schwarzer et al., 2017). However, deletion of the genes encoding for CTCF or cohesin complex components causes severe deficits in neural circuit formation and animal behavior. The reasons why behavioral changes occur with minimal differences in gene expression remain unclear. It is possible that disorganization of chromatin domain interactions may not affect the basal level of gene expression, but impact stimulation-induced gene expression. It is also possible that defects in gene expression is cell type specific. Indeed, in a single-cell ATAC-seq study of the developing human forebrain, cell type-specific changes in chromatin accessibility during corticogenesis were detected, and cell type distinctions beyond transcriptional definitions could be resolved (Ziffra et al., 2020). The recent technical advance of Simultaneous High-throughput ATAC and RNA Expression with sequencing (SHARE-seq) enabled combinatorial measurements of chromatin accessibility and gene expression within the same individual cell, which showed that chromatin accessibility at domains of regulatory chromatin precedes gene expression during lineage commitment (Ma et al., 2020). These studies highlight the importance of investigating the temporal order and casual relationships among spatial chromatin organization, gene expression and functional outcomes. Moreover, functional, and casual relationship analyses may reveal the fundamental logic underlying the regulation of gene expression by spatial genome organization. Several studies have investigated the alteration of spatial chromosome architecture in different contexts (Marco et al., 2020; Yamada et al., 2019) and demonstrated that spatial chromatin changes serve as a platform for parsing multiple inputs for gene expression regulation and epigenetic priming during learning and memory. Future studies will reveal more basic principles.
Finally, aberrant 3D genome organization has been detected in neurological and psychiatric disorders such as SCZ, BP, and autistic spectrum disorders (Girdhar et al., 2022; Rajarajan et al., 2018). In addition, mutations in the chromosomal architectural proteins, such as CTCF and cohesion, impair neural development and brain functions. The majority of disease-associated SNVs (~93%) are localized within non-coding sequences, suggesting the importance of regulatory elements such as promoters and enhancers in normal development and risk for diseases (Maurano et al., 2012). Addressing the functional link between GWAS variants concerning regulatory DNA variation in common human disease to target genes will contribute to a better understanding of the etiology and pathogenesis of neurological and psychiatric disorders. Furthermore, techniques for the manipulation of 3D genome organization, such as forced chromatin looping, can help to unveil the principles of 3D genome organization and its functions, and assist in the development of novel approaches to repair 3D genome disorganization in disease states.
Acknowledgements
We thank members of Song and Ming laboratories for comments and suggestions. The research in the authors’ laboratories were supported by grants from the National Institutes of Health (R35NS116843 and R01AG057497 to H.S, and R35NS097370 and R01MH125528 to G-l.M.) and Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to G-l.M.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
H.S. is on the advisory board of Neuron. The author declares no other conflicts of interest.
References
- Ahanger SH, Delgado RN, Gil E, Cole MA, Zhao J, Hong SJ, Kriegstein AR, Nowakowski TJ, Pollen AA, and Lim DA (2021). Distinct nuclear compartment-associated genome architecture in the developing mammalian brain. Nat Neurosci 24, 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto T, Saitoh N, Ichimura T, Niwa H, and Nakao M (2006). Nuclear and chromatin reorganization in the MHC-Oct3/4 locus at developmental phases of embryonic stem cell differentiation. Dev Biol 298, 354–367. [DOI] [PubMed] [Google Scholar]
- Bauer BW, Davidson IF, Canena D, Wutz G, Tang W, Litos G, Horn S, Hinterdorfer P, and Peters JM (2021). Cohesin mediates DNA loop extrusion by a “swing and clamp” mechanism. Cell 184, 5448–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagan JA, Pastuzyn ED, Fernandez LR, Guo MH, Feng K, Titus KR, Chandrashekar H, Shepherd JD, and Phillips-Cremins JE (2020). Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat Neurosci 23, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl J, Hauberg ME, Girdhar K, Im E, Vicari JM, Rahman S, Dong P, Misir R, Kleopoulos SP, Reach SM, et al. (2021). The three-dimensional landscape of chromatin accessibility in Alzheimer’s disease. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billia F, Baskys A, Carlen PL, and De Boni U (1992). Rearrangement of centromeric satellite DNA in hippocampal neurons exhibiting long-term potentiation. Brain Res Mol Brain Res 14, 101–108. [DOI] [PubMed] [Google Scholar]
- Billia F, and de Boni U (1991). Localization of centromeric satellite and telomeric DNA sequences in dorsal root ganglion neurons, in vitro. J Cell Sci 100 (Pt 1), 219–226. [DOI] [PubMed] [Google Scholar]
- Boettiger A, and Murphy S (2020). Advances in Chromatin Imaging at Kilobase-Scale Resolution. Trends Genet 36, 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572 e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Guan J, and Huang B (2016). Imaging Specific Genomic DNA in Living Cells. Annu Rev Biophys 45, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, and Cremer M (2010). Chromosome territories. Cold Spring Harb Perspect Biol 2, a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, and Geschwind DH (2018). The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, et al. (2012). HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, and Blobel GA (2012). Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149, 1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Gorkin DU, and Ren B (2016). Chromatin Domains: The Unit of Chromosome Organization. Mol Cell 62, 668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schujiers J, Lee TI, Zhao K, et al. (2014). Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Zheng H, Huang B, Ma R, Wu J, Zhang X, He J, Xiang Y, Wang Q, Li Y, et al. (2017). Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547, 232–235. [DOI] [PubMed] [Google Scholar]
- Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, et al. (2019). Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Albert J, Lipinski M, Lopez-Cascales MT, Rowley MJ, Martin-Gonzalez AM, Del Blanco B, Corces VG, and Barco A (2019). Immediate and deferred epigenomic signatures of in vivo neuronal activation in mouse hippocampus. Nat Neurosci 22, 1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiserova J, Efenberkova M, Sieger T, Maninova M, Uhlirova J, and Hozak P (2017). Chromatin organization at the nuclear periphery as revealed by image analysis of structured illumination microscopy data. J Cell Sci 130, 2066–2077. [DOI] [PubMed] [Google Scholar]
- Frank CL, Liu F, Wijayatunge R, Song L, Biegler MT, Yang MG, Vockley CM, Safi A, Gersbach CA, Crawford GE, et al. (2015). Regulation of chromatin accessibility and Zic binding at enhancers in the developing cerebellum. Nat Neurosci 18, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J, Ferrai C, Chiariello AM, Schueler M, Rito T, Laudanno G, Barbieri M, Moore BL, Kraemer DC, Aitken S, et al. (2015). Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol Syst Biol 11, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Masuda K, Bando M, Nakato R, Katou Y, Tanaka T, Nakayama M, Takao K, Miyakawa T, Tanaka T, et al. (2017). Decreased cohesin in the brain leads to defective synapse development and anxiety-related behavior. J Exp Med 214, 1431–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, and Yamashita T (2020). Alterations in Chromatin Structure and Function in the Microglia. Front Cell Dev Biol 8, 626541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullard JF, Hauberg ME, Bendl J, Egervari G, Cirnaru MD, Reach SM, Motl J, Ehrlich ME, Hurd YL, and Roussos P (2018). An atlas of chromatin accessibility in the adult human brain. Genome Res 28, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, and Dekker C (2018). Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, and Furlong EE (2014). Enhancer loops appear stable during development and are associated with paused polymerase. Nature 512, 96–100. [DOI] [PubMed] [Google Scholar]
- Girdhar K, Hoffman GE, Bendl J, Rahman S, Dong P, Liao W, Hauberg ME, Sloofman L, Brown L, Devillers O, et al. (2022). Chromatin domain alterations linked to 3D genome organization in a large cohort of schizophrenia and bipolar disorder brains. Nat Neurosci 25, 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdhar K, Hoffman GE, Jiang Y, Brown L, Kundakovic M, Hauberg ME, Francoeur NJ, Wang YC, Shah H, Kavanagh DH, et al. (2018). Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat Neurosci 21, 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girelli G, Custodio J, Kallas T, Agostini F, Wernersson E, Spanjaard B, Mota A, Kolbeinsdottir S, Gelali E, Crosetto N, et al. (2020). GPSeq reveals the radial organization of chromatin in the cell nucleus. Nat Biotechnol. 38, 1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Harris H, Olshansky M, Eliaz Y, Krishna A, Kalluchi A, Jacobs M, Cauer G, Pham M, Rao SSP, et al. (2021). Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. [Google Scholar]
- Guerreiro I, and Kind J (2019). Spatial chromatin organization and gene regulation at the nuclear lamina. Curr Opin Genet Dev 55, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Cattoglio C, Darzacq X, and Tjian R (2018). Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 9, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N, Shearwin KE, and Dodd IB (2017). Programmable DNA looping using engineered bivalent dCas9 complexes. Nat Commun 8, 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauberg ME, Creus-Muncunill J, Bendl J, Kozlenkov A, Zeng B, Corwin C, Chowdhury S, Kranz H, Hurd YL, Wegner M, et al. (2020). Common schizophrenia risk variants are enriched in open chromatin regions of human glutamatergic neurons. Nat Commun 11, 5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. (2010). Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, and Gersbach CA (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, et al. (2014). Comparative analysis of metazoan chromatin organization. Nature 512, 449–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, and Takizawa T (2018). Nuclear Architecture in the Nervous System: Development, Function, and Neurodevelopmental Diseases. Front Genet 9, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JY, Schaukowitch K, Farbiak L, Kilaru G, and Kim TK (2016). Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat Neurosci 19, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann R, Avendano MS, Woehrstein JB, Dai M, Shih WM, and Yin P (2014). Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat Methods 11, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, Yao X, Li F, Zhu W, Gao L, et al. (2017). 3D Chromatin Structures of Mature Gametes and Structural Reprogramming during Mammalian Embryogenesis. Cell 170, 367–381 e320. [DOI] [PubMed] [Google Scholar]
- Kim JH, Rege M, Valeri J, Dunagin MC, Metzger A, Titus KR, Gilgenast TG, Gong W, Beagan JA, Raj A, et al. (2019). LADL: light-activated dynamic looping for endogenous gene expression control. Nat Methods 16, 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi Y, Fujii Y, Hirabayashi Y, and Gotoh Y (2012). HMGA regulates the global chromatin state and neurogenic potential in neocortical precursor cells. Nat Neurosci 15, 1127–1133. [DOI] [PubMed] [Google Scholar]
- Kishi Y, and Gotoh Y (2018). Regulation of Chromatin Structure During Neural Development. Front Neurosci 12, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, Ishman SL, Kerr LM, Levin AV, Mulder PA, et al. (2018). Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet 19, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krull S, Dorries J, Boysen B, Reidenbach S, Magnius L, Norder H, Thyberg J, and Cordes VC (2010). Protein Tpr is required for establishing nuclear pore-associated zones of heterochromatin exclusion. EMBO J 29, 1659–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Leemans C, van der Zwalm MCH, Brueckner L, Comoglio F, van Schaik T, Pagie L, van Arensbergen J, and van Steensel B (2019). Promoter-Intrinsic and Local Chromatin Features Determine Gene Repression in LADs. Cell 177, 852–864 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Liu X, Huang WK, Giusti-Rodriguez P, Cui J, Zhang S, Xu W, Wen Z, Ma S, Rosen JD, et al. (2020). Robust Hi-C Maps of Enhancer-Promoter Interactions Reveal the Function of Non-coding Genome in Neural Development and Diseases. Mol Cell. 79, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Liu Y, Dang D, Hu T, Hou Y, Meng X, Zhang F, Li T, Wang C, Li M, et al. (2021). 3D Genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell 184, 723–740 e721. [DOI] [PubMed] [Google Scholar]
- Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Tay T, et al. (2020). Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183, 1103–1116 e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L (1984). Different central nervous system cell types display distinct and nonrandom arrangements of satellite DNA sequences. Proc Natl Acad Sci U S A 81, 3123–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Meharena HS, Dileep V, Raju RM, Davila-Velderrain J, Zhang AL, Adaikkan C, Young JZ, Gao F, Kellis M, et al. (2020). Mapping the epigenomic and transcriptomic interplay during memory formation and recall in the hippocampal engram ensemble. Nat Neurosci 23, 1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martou G, and De Boni U (2000). Nuclear topology of murine, cerebellar Purkinje neurons: changes as a function of development. Exp Cell Res 256, 131–139. [DOI] [PubMed] [Google Scholar]
- Mateo LJ, Murphy SE, Hafner A, Cinquini IS, Walker CA, and Boettiger AN (2019). Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. (2012). Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, and Misteli T (2006). Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell 10, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SL, Mariano NC, Bermudez A, Arruda NL, Wu F, Luo Y, Shankar G, Jia L, Chen H, Hu JF, et al. (2017). Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat Commun 8, 15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944 e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. (2019). Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Program NCS, et al. (2013). Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A 110, 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. (2010). Molecular maps of the reorganization of genomenuclear lamina interactions during differentiation. Mol Cell 38, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Song H, and Ming GL (2019). Brain organoids: advances, applications and challenges. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. (2018). Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757 e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajarajan P, Borrman T, Liao W, Espeso-Gil S, Chandrasekaran S, Jiang Y, Weng Z, Brennand KJ, and Akbarian S (2019). Spatial genome exploration in the context of cognitive and neurological disease. Curr Opin Neurobiol 59, 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajarajan P, Borrman T, Liao W, Schrode N, Flaherty E, Casino C, Powell S, Yashaswini C, LaMarca EA, Kassim B, et al. (2018). Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci MA, Cosma MP, and Lakadamyali M (2017). Super resolution imaging of chromatin in pluripotency, differentiation, and reprogramming. Curr Opin Genet Dev 46, 186–193. [DOI] [PubMed] [Google Scholar]
- Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, Stahl EA, Georgakopoulos A, Ruderfer DM, Charney A, et al. (2014). A role for noncoding variation in schizophrenia. Cell Rep 9, 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112, E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, and Ren B (2016). Genome-wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol 17, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L, et al. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sey NYA, Hu B, Mah W, Fauni H, McAfee JC, Rajarajan P, Brennand KJ, Akbarian S, and Won H (2020). A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat Neurosci 23, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofueva S, Yaffe E, Chan WC, Georgopoulou D, Vietri Rudan M, Mira-Bontenbal H, Pollard SM, Schroth GP, Tanay A, and Hadjur S (2013). Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J 32, 3119–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T, Guck J, and Joffe B (2009). Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137, 356–368. [DOI] [PubMed] [Google Scholar]
- Solovei I, Schermelleh L, During K, Engelhardt A, Stein S, Cremer C, and Cremer T (2004). Differences in centromere positioning of cycling and postmitotic human cell types. Chromosoma 112, 410–423. [DOI] [PubMed] [Google Scholar]
- Song M, Pebworth MP, Yang X, Abnousi A, Fan C, Wen J, Rosen JD, Choudhary MNK, Cui X, Jones IR, et al. (2020). Cell-type-specific 3D epigenomes in the developing human cortex. Nature 587, 644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Yang X, Ren X, Maliskova L, Li B, Jones IR, Wang C, Jacob F, Wu K, Traglia M, et al. (2019). Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat Genet 51, 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JH, Zheng P, Kinrot SS, Bintu B, and Zhuang X (2020). Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 182, 1641–1659 e1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat Neurosci 20, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JH, Zhou L, Emerson DJ, Phyo SA, Titus KR, Gong W, Gilgenast TG, Beagan JA, Davidson BL, Tassone F, et al. (2018). Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 175, 224–238 e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa T, and Meshorer E (2008). Chromatin and nuclear architecture in the nervous system. Trends Neurosci 31, 343–352. [DOI] [PubMed] [Google Scholar]
- Tan L, Ma W, Wu H, Zheng Y, Xing D, Chen R, Li X, Daley N, Deisseroth K, and Xie XS (2021). Changes in genome architecture and transcriptional dynamics progress independently of sensory experience during post-natal brain development. Cell 184, 741–758 e717. [DOI] [PubMed] [Google Scholar]
- Thakurela S, Sahu SK, Garding A, and Tiwari VK (2015). Dynamics and function of distal regulatory elements during neurogenesis and neuroplasticity. Genome Res 25, 1309–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley KG, Brennand KJ, and Huckins LM (2020). Massively parallel techniques for cataloguing the regulome of the human brain. Nat Neurosci 23, 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. (2001). The sequence of the human genome. Science 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- Wang H, Xu X, Nguyen CM, Liu Y, Gao Y, Lin X, Daley T, Kipniss NH, La Russa M, and Qi LS (2018). CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 175, 1405–1417 e1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, and Zhuang X (2016). Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. (2008). Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451, 796–801. [DOI] [PubMed] [Google Scholar]
- Williams RR, Azuara V, Perry P, Sauer S, Dvorkina M, Jorgensen H, Roix J, McQueen P, Misteli T, Merkenschlager M, et al. (2006). Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J Cell Sci 119, 132–140. [DOI] [PubMed] [Google Scholar]
- Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. (2013). Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Valnegri P, Juric I, Abnousi A, Markwalter KH, Guthrie AN, Godec A, Oldenborg A, Hu M, et al. (2019). Sensory experience remodels genome architecture in neural circuit to drive motor learning. Nature 569, 708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Christian KM, He C, Jin P, Ming GL, and Song H (2016). Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci 17, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap EL, and Greenberg ME (2018). Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, Vissers C, Ming GL, and Song H (2018). Epigenetics and epitranscriptomics in temporal patterning of cortical neural progenitor competence. J Cell Biol 217, 1901–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, and Ren B (2017). The Three-Dimensional Organization of Mammalian Genomes. Annu Rev Cell Dev Biol 33, 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziffra RS, Kim CN, Wilfert A, Turner TN, Haeussler M, Casella AM, Przytycki PF, Kreimer A, Pollard KS, Ament SA, et al. (2020). Single cell epigenomic atlas of the developing human brain and organoids. bioRxiv. [Google Scholar]