

Abstract
BACKGROUND AND AIMS:
Evidence supports a carcinogenic role of Escherichia coli carrying the polyketide synthase (pks) island that encodes enzymes for colibactin biosynthesis. We hypothesized that the association of western-style diet (rich in red and processed meat) with colorectal cancer incidence might be stronger for tumors containing higher amounts of pks+ E. coli.
METHODS:
Western diet score was calculated using food frequency questionnaire data obtained every four years during follow-up of 134,775 participants in two U.S.-wide prospective cohort studies. Using quantitative polymerase chain reaction, we measured pks+ E. coli DNA in 1,175 tumors among 3,200 incident colorectal cancer cases that had occurred during the follow-up. We utilized the 3,200 cases and inverse probability weighting (to adjust for selection bias due to tissue availability), integrated in multivariable-adjusted duplication-method Cox proportional hazards regression analyses.
RESULTS:
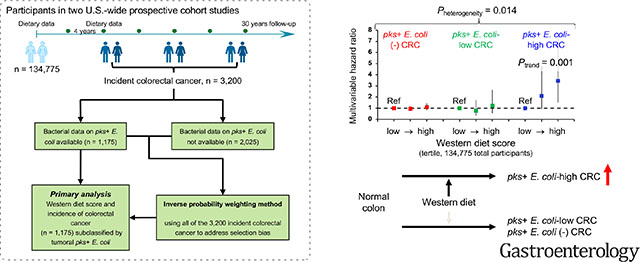
The association of the western diet score with colorectal cancer incidence was stronger for tumors containing higher levels of pks+ E. coli (Pheterogeneity = 0.014). Multivariable-adjusted hazard ratios (with 95% confidence interval) for the highest (vs. lowest) tertile of the western diet score were 3.45 (1.53–7.78) (Ptrend = 0.001) for pks+ E. coli-high tumors, 1.22 (0.57–2.63) for pks+ E. coli-low tumors, and 1.10 (0.85–1.42) for pks+ E. coli-negative tumors. The pks+ E. coli level was associated with lower disease stage but not with tumor location, microsatellite instability, or BRAF, KRAS, or PIK3CA mutations.
CONCLUSIONS:
Western-style diet is associated with higher incidence of colorectal cancer containing abundant pks+ E. coli, supporting a potential link between diet, the intestinal microbiota, and colorectal carcinogenesis.
Keywords: immunology, microbiome, molecular pathological epidemiology
Graphical Abstract
Lay Summary
We found that western-style diet (rich in red and processed meat and sugar) increased risk of colorectal cancer containing high amounts of specific toxinproducing E. coli bacterium.
INTRODUCTION
Accumulating evidence indicates that certain intestinal microorganisms influence colorectal tumor development through DNA damage, inflammation, and other mechanisms1–5. Among intestinal bacteria, Escherichia coli (E. coli) strains of the B2 phylotype commonly harbor the 54-kb polyketide synthase (pks) pathogenicity island that encodes enzymes for colibactin biosynthesis1,3,6. Experimental studies have shown that colibactin can alkylate DNA, induce DNA double-strand breaks, and cause a specific somatic mutational pattern in human cells7–9. However, human population studies are needed to better understand the role of pks island-carrying E. coli (hereafter referred to as pks+ E. coli) in colorectal cancer.
Diet and nutrition are considered crucial factors for colorectal cancer development. A meta-analysis has shown a weak-to-modest relationship between western dietary patterns and colorectal cancer risk10. An experimental study indicates that western-style diet (characterized by high intake of red and processed meat, sugar, and refined grains and low intake of in vegetables and legumes) can induce systemic and intestinal inflammation11. Considering the possible interplay between diet and pathogenic bacteria, it is of particular interest to study western-style diet in relation to pks+ E. coli within colorectal tumor tissue. Such analyses may contribute to the development of cancer prevention strategies targeting diet and microbiota.
In this study, we tested the hypothesis that the association of western-style diet with colorectal cancer incidence might be stronger for tumors containing higher amounts of pks+ E. coli. We utilized a molecular pathological epidemiology database of two U.S.-wide longitudinal prospective cohort studies with incident colorectal cancer cases. This comprehensive dataset offered a unique opportunity to examine long-term dietary patterns of individuals (who had not known whether they would develop cancers or not) in relation to colorectal cancer incidence subclassified by pks+ E. coli levels, while adjusting for potential confounders and selection bias due to tissue availability. In addition, we comprehensively assessed clinical, pathological, molecular, and prognostic features according to the amount of pks+ E. coli in colorectal carcinoma tissue.
MATERIALS AND METHODS
Study Population and Dietary Assessment
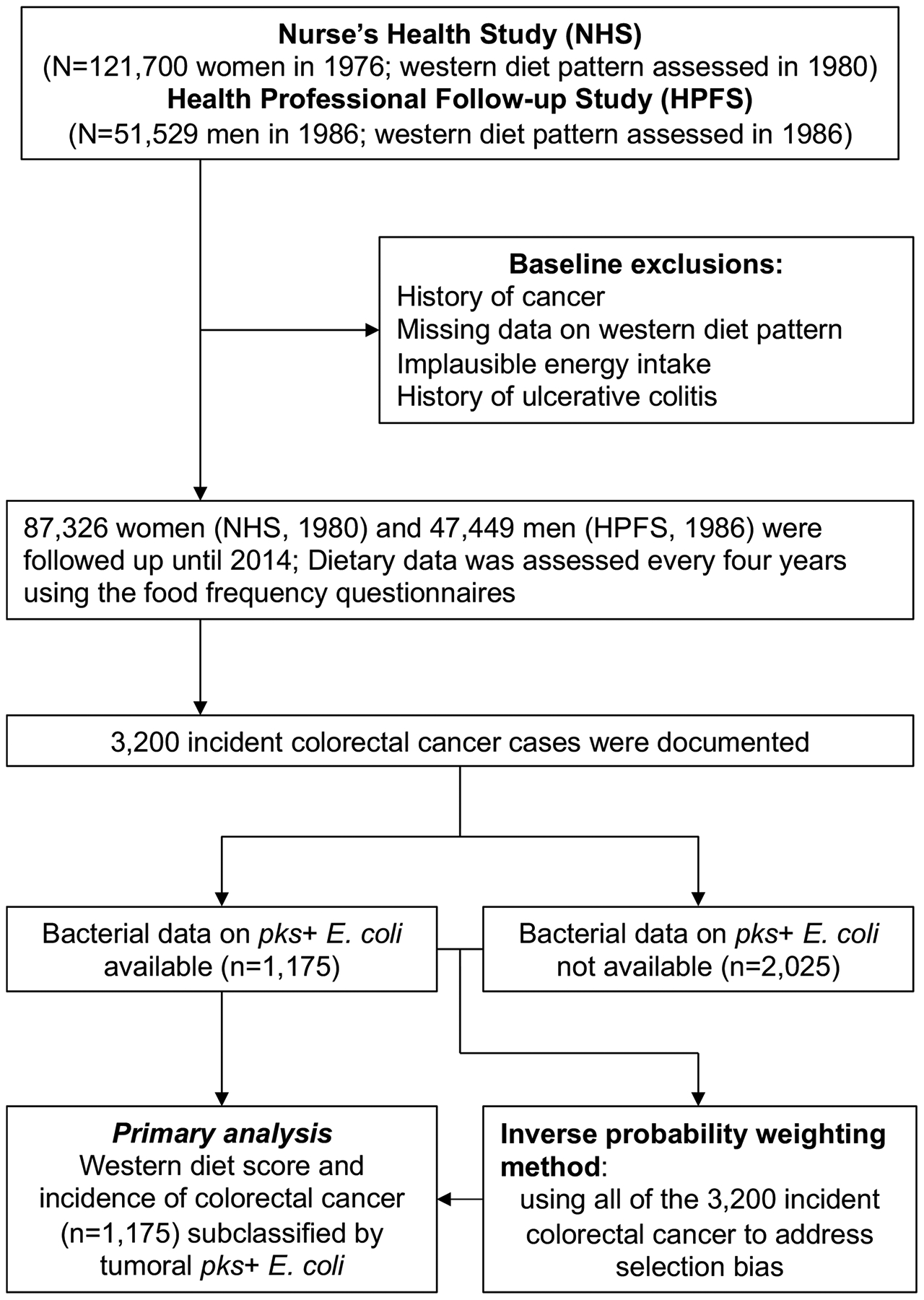
We used two prospective cohort studies in the U.S., namely the Nurses’ Health Study (NHS, 121,700 women aged 30–55 years at enrollment in 1976)12,13 and the Health Professionals Follow-up Study (HPFS, 51,529 men aged 40–75 years at enrollment in 1986)13,14 (Figure 1). Study participants had been followed by use of questionnaires every two years on lifestyle and diagnoses of major diseases. The response rate has exceeded 90% for each follow-up questionnaire cycle in both cohorts. Dietary data were collected using self-administrated semi-quantitative food frequency questionnaires in 1984, 1986, and every four years thereafter in the NHS, and every four years since 1986 in the HPFS. Validity of semiquantitative food frequency questionnaires in the assessment of dietary intake was extensively assessed and documented in studies using diet records and plasma nutrients15–17. Total nutrient intakes were calculated by summing intakes from all foods and adjusted for total energy intake by the residual method. In this study, we used data from 134,775 participants who provided sufficient longitudinal dietary information.
Figure 1.
Flow diagram of the study population in the Nurses’ Health Study and the Health Professionals Follow-up Study.
Abbreviation: HPFS, Health Professionals Follow-up Study; NHS, Nurses’ Health Study.
The participants had been followed since the baseline questionnaire return until colorectal cancer diagnosis, loss to follow-up, end of follow-up (June 1, 2014 for the NHS, January 1, 2014 for the HPFS), or death, whichever came first. Participants who had major illnesses including colorectal cancer reported those through questionnaires. Unreported lethal colorectal cancer cases were ascertained through use of the National Death Index. Clinical information such as tumor location and disease stage based on the American Joint Committee on Cancer (AJCC) classification was extracted from medical record by a study physician18. We included both colon and rectal carcinomas based on the colorectal continuum model19. We gathered formalin-fixed paraffin-embedded (FFPE) tissue blocks from pathology files of hospitals throughout the U.S. where the patients’ tumors were resected. Histopathological features including tumor differentiation, extracellular mucin, and signet ring cells were evaluated by the study pathologist (S.O.)20. In this study, the inverse probability weighting (IPW) method using both cases with available tissue bacterial data (n = 1,175) and those without tissue bacterial data (n = 2,025) was integrated into duplication-method Cox proportional hazards regression analysis to adjust for selection bias due to tissue bacterial data availability (Figure 1). Characteristics of the cases with tissue bacterial data were similar to those without tissue bacterial data (Table S1). In addition, during an assay validation step, we utilized tissues from 21 anonymized colorectal cancer patients who had surgical resections performed at the Brigham and Women’s Hospital or Kumamoto University.
Informed consent was obtained from all participants at enrollment and consent for tissue specimen use was additionally obtained before tissue collection. This study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital (Boston, MA) and Kumamoto University (Kumamoto, Japan), and those of participating registries as required.
Tumor Tissue Analyses
Genomic DNA was extracted from archival FFPE tissue sections of colorectal carcinoma using the QIAamp DNA FFPE Tissue Kit and GeneRead DNA FFPE Kit (Qiagen, Hilden, Germany). We used custom TaqMan primer-probe sets (Applied Biosystems, Foster City, CA) for the clbB gene DNA sequence of pks+ E. coli4 and for the reference human gene SLCO2A1 that has been used in other bacterial assays on FFPE tissue-derived DNA21 (the names used follow the recommendations for standardized nomenclature of genes and their products by an expert panel22). Genomic DNA concentration derived from samples was measured by Qubit Fluorometer (Thermo Fisher Scientific, Waltham, MA). Each reaction contained 20 ng of genomic DNA and was assayed in 20 μl reactions containing 1x final concentration TaqMan Environmental Master Mix 2.0 (Applied Biosystems), in a 96-well optical polymerase chain reaction (PCR) plate. Amplification and detection of DNA were performed with a QuantStudio 3 Real-time PCR System (Thermo Fisher Scientific) using the following reaction conditions; 10 minutes at 95°C and 45 cycles of 15 seconds at 95°C, 30 seconds at 57°C, and 30 seconds at 72°C. The primer and probe sequences for each TaqMan Gene Expression Assay were as follows: pks+ E. coli forward primer, 5’-GCAACATACTCGCCCAGACT-3’; pks+ E. coli reverse primer, 5’-TCTCAAGGCGTTGTTGTTTG-3’; pks+ E. coli FAM probe, 5’-CAAGGTGCGCGCTAGGCTGT-3’; SLCO2A1 forward primer, 5’-ATCCCCAAAGCACCTGGTTT-3’; SLCO2A1 reverse primer, 5’-AGAGGCCAAGATAGTCCTGGTAA-3’; SLCO2A1 VIC probe, 5’-CCATCCATGTCCTCATCTC-3’. To validate our PCR assay, Sanger dideoxy sequencing was performed on the PCR product from three anonymized colorectal carcinoma patients in which the PCR assay detected pks+ E. coli DNA. The PCR product (165 bp) using the forward and reverse primer sets was isolated by agarose gel electrophoresis. The isolated PCR product was amplified by subcloning and sequenced by Sanger dideoxy sequencing using BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). We used Competent Quick DH5a (Toyobo, Japan) and QIAprep Spin Miniprep Kit (Qiagen) in the transformation and extraction. We confirmed that the PCR product had sequence of the clbB gene of pks+ E. coli in all of the three cases. In two colorectal carcinoma cases with detectable pks+ E. coli, the cycle threshold (Ct) values for pks+ E. coli and SLCO2A1 decreased linearly with the amount of input DNA (in a log scale) from the same specimens (r2 > 0.95) (Figure 2A). We also confirmed that the Ct values (for pks+ E. coli) decreased linearly with the amount of input DNA (in a log scale) from pks+ E. coli DNA (American Type Culture Collection, Manassas, VA) (r2 > 0.99) (Figure 2B) and that there was no amplification of DNA from E. coli without the pks island (DH10B) (Thermo Fisher Scientific) as a negative control. These positive and negative controls were used in each PCR run on the HPFS and the NHS specimens. Furthermore, in six colorectal carcinoma cases (three positive and three negative cases for pks+ E. coli DNA), the interassay coefficient of variation of Ct values from each specimen was <1% for both targets in repeated assays of five different batches (Table S2). In the cohort cases, each specimen was analyzed in duplicate for each target in a single batch, and we used the mean of the two Ct values for each target. The amount of pks+ E. coli was calculated as a relative unitless value normalized with SLCO2A1 using the 2−ΔCt method (where ΔCt = the average Ct value of pks+ E. coli – the average Ct value of SLCO2A1) as previously described23. Cases with detectable pks+ E. coli were dichotomized into high-level vs. low-level based on the median cut-off point. Microsatellite instability (MSI) status was determined based on PCR of 10 microsatellite markers (D2S123, D5S346, D17S250, BAT25, BAT26, BAT40, D18S55, D18S56, D18S67, and D18S487) as previously described18. CpG island methylator phenotype (CIMP) was determined using MethyLight assays24 of the 8 promoter CpG islands (CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3, and SOCS1)25. Methylation level of long-interspersed nucleotide element-1 (LINE-1) was measured using bisulfite PCR and Pyrosequencing as previously described26. PCR and pyrosequencing were performed for KRAS (codons 12, 13, 61, and 146)27,28, BRAF (codon 600), and PIK3CA (exons 9 and 20), as previously described29.
Figure 2.
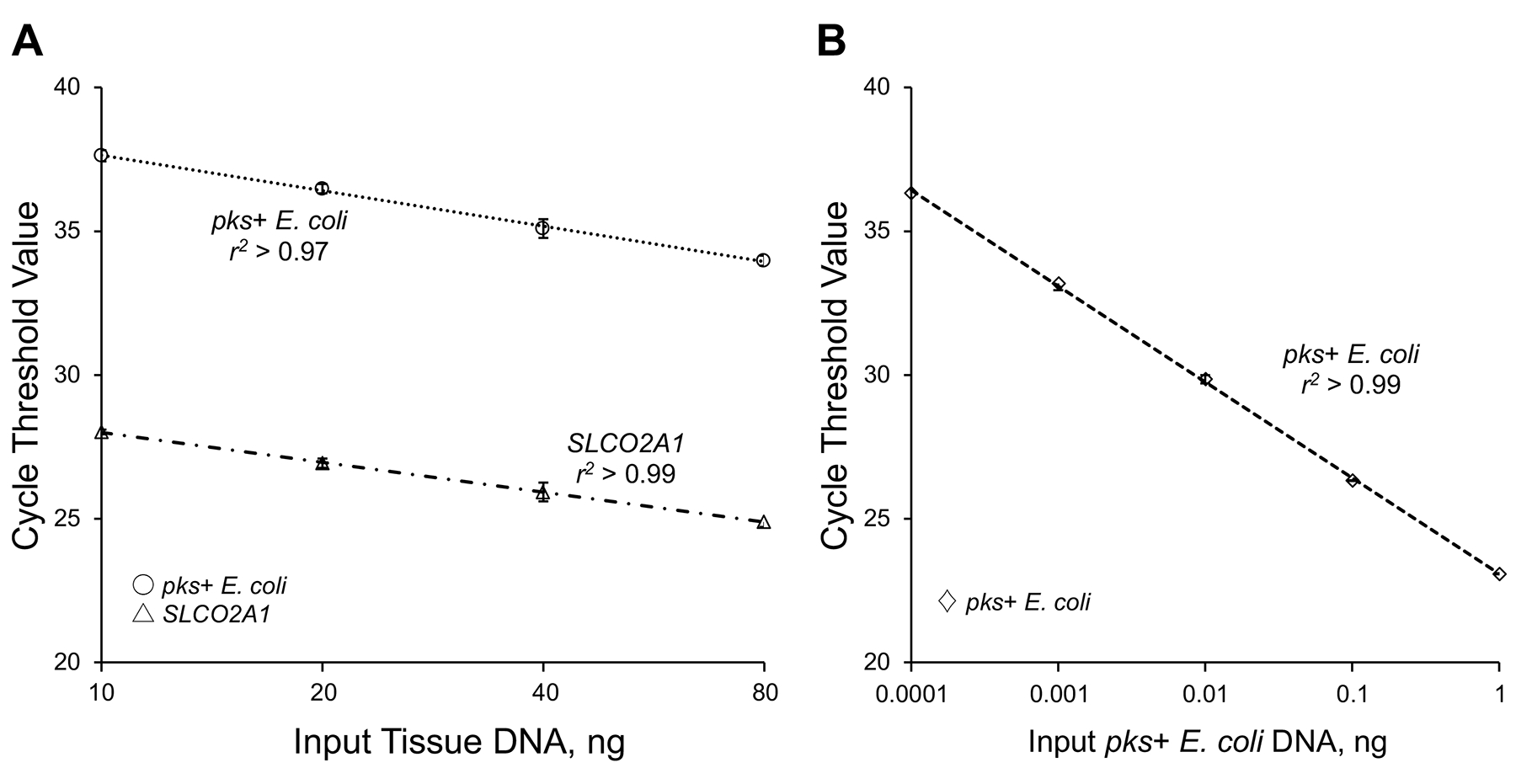
Assessment of linearity in quantitative real-time PCR assay. (A): Quantitative real-time PCR assay for pks+ E. coli DNA and the human reference gene SLCO2A1 using 2-fold dilution series (10, 20, 40, and 80 ng) from the same DNA specimen from formalin-fixed paraffin-embedded tissue. (B): Quantitative real-time PCR assay for pks+ E. coli DNA using 10-fold dilution series (0.0001, 0.001, 0.01, 0.1, and 1 ng) from DNA from cultured pks+ E. coli. Symbols indicate mean, error bars, standard deviation of cycle threshold values of quadruplicate runs. The coefficient of determination (r2) in the assays for pks+ E. coli DNA and SLCO2A1 is shown.
Abbreviation: PCR, polymerase chain reaction.
Statistical Analysis
Detailed statistical analysis methods are described in Supplementary Methods. All statistical analyses were performed using SAS software (version 9.4, SAS Institute, Cary, NC), and all P values were two-sided. We adjusted the two-sided α level to 0.012 (approximately 0.05/4) for multiple hypothesis testing by Bonferroni correction, considering our use of one heterogeneity trend test (for levels of pks+ E. coli) and three stratum-specific (high, low, and negative pks+ E. coli) statistical trend tests.
The western-style diet was derived by principal component analyses of the extensive diet data, as previously described and validated12,14. Each participant was assigned a factor score, determined by adding the reported frequencies of food item intakes weighted by the factor loadings (Table 1). To capture long-term habitual consumption, we calculated the cumulative mean of the western diet scores from all data-available preceding food frequency questionnaires up to each questionnaire cycle. Table 1 shows the distribution of western diet score in each cohort.
Table 1.
Distribution of Western Diet Scores and Factor Loading Matrix for Western-style Diet in Each Cohort
| Health Professionals Follow-up Study | Nurses’ Health Study | |
|---|---|---|
| Distribution (percentile) | ||
| Minimum | −3.58 | −3.98 |
| 1% | −1.51 | −1.45 |
| 5% | −1.17 | −1.06 |
| 10% | −0.97 | −0.85 |
| 25% | −0.60 | −0.47 |
| 50% | −0.10 | 0.015 |
| 75% | 0.49 | 0.56 |
| 90% | 1.14 | 1.12 |
| 95% | 1.59 | 1.50 |
| 99% | 2.52 | 2.31 |
| Maximum | 10.0 | 9.66 |
| Food item* | ||
| Unprocessed red meat | 0.66 | 0.61 |
| Processed meat | 0.61 | 0.58 |
| High fat dairy food | 0.51 | 0.50 |
| French fries | 0.49 | 0.46 |
| Eggs | 0.47 | 0.41 |
| Desserts§ | 0.43 | 0.45 |
| Condiments‖ | 0.39 | 0.36 |
| Refined grains | 0.38 | 0.38 |
| Butter | 0.38 | 0.50 |
| Mayonnaise | 0.36 | 0.34 |
| Margarine | 0.34 | 0.32 |
| Snacks** | 0.34 | |
| Pizza | 0.33 | 0.36 |
| Creamy soups | 0.31 | 0.32 |
| Sugar-sweetened beverages | 0.31 | 0.33 |
| Potatoes | 0.34 |
Only items with correlation coefficients >0.30 are presented. With the orthogonal rotation used, correlations are identical to factor loading matrix.
Desserts include chocolate, candy bars, cookies, brownies, cake, pie, and pastries.
Conditions include soy sauce, non-dairy creamer, Worcestershire sauce, red chili sauce, and pepper.
Snacks include chips, popcorn, and crackers.
To limit the number of primary hypotheses, our primary hypothesis testing was the assessment of heterogeneity of the association of the western diet score with the incidence of colorectal cancer subclassified by tissue bacterial amount. We examined heterogeneity across the ordinal tumor subtypes (by the one degree-of-freedom statistical trend test for negative vs. low vs. high) in the multivariable-adjusted duplication-method Cox proportional hazards model using the meta-regression method with a subtype-specific random effect term30. For statistical trend tests, the diet score was used as a continuous variable with cohort-specific ceilings at the 10th and 90th percentiles to eliminate outlier effects. We also examined hazard ratios for each cancer subgroup by comparing dietary score tertiles as secondary analyses. To control for selection bias due to tissue bacterial data availability in the 1,175 cases, we used the 3,200 incident colorectal cancer cases and inverse probability weighting (IPW) method31 combined with Cox proportional hazards regression models. Multivariable Cox regression models were stratified by age, sex (cohort), and questionnaire year and additionally adjusted for body mass index (continuous with 35 kg/m2 ceiling), pack-years smoked (continuous with 50 pack-years ceiling), first-degree relative family history of colorectal cancer (yes vs. no), previous colonoscopy/sigmoidoscopy (yes vs. no), physical activity (continuous with 50 metabolic-equivalent-task-score hours/week ceiling), aspirin or nonsteroidal anti-inflammatory drug use (≥2 tablets/week: yes vs. no), multivitamin use (yes vs. no), and alcohol consumption (continuous with 30 g/day ceiling). In the NHS (female)-only analyses, we additionally adjusted for postmenopausal hormone use (yes vs. no). For individuals with missing data in one questionnaire, data from preceding questionnaires were used.
In secondary analyses to assess clinical, pathological, and molecular features according to pks+ E. coli status (negative, low, and high), we used the chi-square test (for categorical variables), an analysis of variance (for continuous variables), or Spearman correlation analysis (for ordinal variables). In secondary analyses to assess patient survival, we used IPW-adjusted Kaplan-Meier analysis and multivariable Cox proportional hazards regression models (see details in Supplementary Methods). In secondary analyses of the association of red meat variables (total, unprocessed, and processed red meat intake) with the incidence of colorectal cancer subclassified by pks+ E. coli status, we examined heterogeneity across the ordinal tumor subtypes (by the one degree-of-freedom statistical trend test for pks+ E. coli negative vs. low vs. high) in the multivariable-adjusted duplication-method Cox proportional hazards model using the meta-regression method with a subtype-specific random effect term.
RESULTS
We utilized data from 134,775 participants of the Health Professionals Follow-up Study (HPFS) and the Nurses’ Health Study (NHS) (Table 2 and Figure 1). During 3,766,179 person-years of follow-up, we documented 3,200 incident colorectal cancer cases. In multivariable analyses using each cohort, the western diet score was weakly associated with colorectal cancer incidence (Table S3). Because the results were similar in the two cohorts (P for heterogeneity > 0.6), we combined the two cohorts for further analyses to maximize statistical power while adjusting for cohort (i.e., sex).
Table 2.
Age-standardized Characteristics According to Western Diet Score Tertiles in the Health Professionals Follow-up Study (Men, 1986–2014) and the Nurses’ Health Study (Women, 1980–2014)
| Characteristic* | Health Professionals Follow-up Study | Nurses’ Health Study | ||||
|---|---|---|---|---|---|---|
| Western diet score | Western diet score | |||||
| Tertile 1 | Tertile 2 | Tertile 3 | Tertile 1 | Tertile 2 | Tertile 3 | |
| Participants, No. | 17,429 | 14,217 | 15,803 | 27,645 | 27,702 | 31,979 |
| Mean age, years | 65.1 | 64.7 | 63.3 | 62.8 | 61.3 | 60.0 |
| Mean body mass index, kg/m2 | 25.3 | 25.9 | 26.3 | 24.9 | 25.3 | 25.9 |
| Mean physical activity, METS-hours/week† | 29.6 | 27.2 | 26.2 | 12.4 | 10.5 | 9.3 |
| Mean pack-years smoked | 9.0 | 11.3 | 15.2 | 11.6 | 12.5 | 13.8 |
| Family history of colorectal cancer, % | 15.1 | 14.7 | 14.5 | 19.1 | 18.8 | 18.9 |
| History of previous endoscopy, % | 25.1 | 24.9 | 22.6 | 28.6 | 27.6 | 26.3 |
| Current multivitamin use, % | 48.5 | 47.8 | 44.3 | 55.1 | 52.3 | 48.2 |
| Regular aspirin or NSAID use, %‡ | 43.7 | 48.4 | 49.4 | 57.7 | 60.6 | 60.8 |
| Postmenopausal, % | 74.8 | 74.0 | 72.7 | |||
| Current hormone use, %§ | 47.1 | 44.8 | 42.4 | |||
| Dietary intake (means) | ||||||
| Total calorie intake, kcal/day | 1,610 | 1,900 | 2,420 | 1,430 | 1,620 | 1,990 |
| Unprocessed red meat, servings/day | 0.30 | 0.56 | 0.88 | 0.45 | 0.63 | 0.82 |
| Processed red meat, servings/day | 0.12 | 0.28 | 0.56 | 0.15 | 0.28 | 0.47 |
| Poultry, servings/day | 0.40 | 0.39 | 0.39 | 0.34 | 0.31 | 0.31 |
| Fruit, servings/day | 2.82 | 2.39 | 2.25 | 2.49 | 2.18 | 2.07 |
| Vegetable, servings/day | 3.31 | 3.15 | 3.26 | 2.88 | 2.62 | 2.63 |
| Alcohol, g/day | 8.0 | 11.2 | 13.9 | 6.5 | 6.1 | 5.5 |
| Folate, μg/day | 619 | 545 | 481 | 494 | 424 | 371 |
| Calcium, mg/day | 1,030 | 928 | 860 | 1,070 | 927 | 819 |
| Vitamin D, IU/day | 515 | 428 | 368 | 429 | 355 | 299 |
| Dietary fiber, g/day | 25.7 | 21.5 | 19.1 | 18.9 | 16.1 | 14.3 |
Updated information throughout follow-up was used to calculate the mean for continuous variables and percentage for categorical variables. All variables are age-standardized except age.
Physical activity is represented by the product sum of the metabolic equivalent task score (METS) of each specific recreational activity and hours spent on that activity per week.
Regular users are defined as ≥2 standard (325-mg) tablets of aspirin or ≥ 2 tablets of NSAIDs per week.
Proportion of current menopausal hormone use is calculated among postmenopausal women only.
Abbreviations: METS, metabolic equivalent task score; NSAIDs, non-steroidal anti-inflammatory drugs.
We developed and validated the assay to quantify pks+ E. coli DNA in tumor tissue. The assay, which was successfully conducted in duplicate in 1,175 cases among the 3,200 colorectal cancer patients, detected pks+ E. coli in 111 patients while 1,064 patients were negative for this bacterium. Clinical, pathological, and molecular features according to the amount of pks+ E. coli in colorectal carcinoma tissue are shown in Table 3. The amount of pks+ E. coli DNA was inversely associated with AJCC stage (P = 0.008) but not with the other features examined.
Table 3.
Clinical, Pathological, and Molecular Characteristics of Colorectal Cancer Cases According to the Amount of pks+ E. coli DNA in Colorectal Cancer Tissue
| Characteristic* | All cases (N = 1175) | Amount of pks+ E. coli DNA in colorectal cancer tissue | P value† | ||
|---|---|---|---|---|---|
| Negative (N = 1064) | Low (N = 55) | High (N = 56) | |||
| Sex | 0.28 | ||||
| Female (NHS) | 656 (56%) | 588 (55%) | 31 (56%) | 37 (66%) | |
| Male (HPFS) | 519 (44%) | 476 (45%) | 24 (44%) | 19 (34%) | |
| Mean age ± SD (years) | 69.0 ± 8.8 | 68.9 ± 8.8 | 69.6 ± 10.0 | 69.9 ± 8.1 | 0.61 |
| Year of diagnosis | 0.09 | ||||
| 1995 or before | 399 (34%) | 371 (35%) | 12 (22%) | 16 (29%) | |
| 1996–2000 | 375 (32%) | 333 (31%) | 18 (33%) | 24 (43%) | |
| 2001–2008 | 401 (34%) | 360 (34%) | 25 (45%) | 16 (29%) | |
| Family history of colorectal cancer in first-degree relative(s) | 0.17 | ||||
| Absent | 935 (80%) | 854 (81%) | 42 (76%) | 39 (71%) | |
| Present | 235 (20%) | 206 (19%) | 13 (24%) | 16 (29%) | |
| Tumor location | 0.70 | ||||
| Cecum | 202 (17%) | 182 (17%) | 11 (20%) | 9 (16%) | |
| Ascending to transverse | 364 (31%) | 334 (32%) | 16 (29%) | 14 (25%) | |
| Descending to sigmoid | 355 (30%) | 314 (30%) | 19 (35%) | 22 (39%) | |
| Rectum | 249 (21%) | 229 (22%) | 9 (16%) | 11 (20%) | |
| AJCC disease stage | 0.008 | ||||
| I | 262 (24%) | 230 (23%) | 12 (24%) | 20 (42%) | |
| II | 354 (33%) | 319 (32%) | 19 (37%) | 16 (33%) | |
| III | 311 (29%) | 288 (29%) | 14 (27%) | 9 (19%) | |
| IV | 157 (14%) | 148 (15%) | 6 (12%) | 3 (6.3%) | |
| Tumor size ± SD (cm) | 4.4 ± 2.0 | 4.4 ± 2.0 | 4.7 ± 2.0 | 4.5 ± 2.1 | 0.39 |
| Tumor differentiation | 0.42 | ||||
| Well to moderate | 1053 (90%) | 954 (90%) | 47 (85%) | 52 (93%) | |
| Poor | 118 (10%) | 106 (10%) | 8 (15%) | 4 (7.1%) | |
| MSI status | 0.49 | ||||
| Non-MSI-high | 947 (83%) | 860 (83%) | 40 (78%) | 47 (87%) | |
| MSI-high | 188 (17%) | 170 (17%) | 11 (22%) | 7 (13%) | |
| CIMP status | 0.63 | ||||
| Low/negative | 885 (82%) | 803 (82%) | 36 (82%) | 46 (87%) | |
| High | 197 (18%) | 182 (18%) | 8 (18%) | 7 (13%) | |
| Mean LINE-1 methylation level ± SD | 63.0 ± 9.8 | 63.0 ± 9.8 | 63.4 ± 11.5 | 63.5 ± 7.2 | 0.88 |
| KRAS mutation | 0.50 | ||||
| Wild-type | 645 (59%) | 586 (59%) | 30 (64%) | 29 (53%) | |
| Mutant | 443 (41%) | 400 (41%) | 17 (36%) | 26 (47%) | |
| BRAF mutation | 0.55 | ||||
| Wild-type | 942 (84%) | 852 (84%) | 41 (82%) | 49 (89%) | |
| Mutant | 177 (16%) | 162 (16%) | 9 (18%) | 6 (11%) | |
| PIK3CA mutation | 0.51 | ||||
| Wild-type | 878 (84%) | 795 (84%) | 39 (85%) | 44 (90%) | |
| Mutant | 168 (16%) | 156 (16%) | 7 (15%) | 5 (10%) | |
Percentage indicates the proportion of patients with a specific clinical, pathological, or molecular characteristic among all patients or in strata of the amount of pks+ E. coli DNA in colorectal cancer tissue.
To assess associations between the categories (negative, low, and high) of pks+ E. coli DNA in colorectal cancer tissue and categorical data, the chi-square test was performed. To compare age, and LINE-1 methylation level, an analysis of variance was performed. To compare AJCC disease stage, Spearman analysis was performed.
Abbreviations: AJCC, American Joint Committee on Cancer; CIMP, CpG island methylator phenotype; HPFS, Health Professionals Follow-up Study; LINE-1, long-interspersed nucleotide element-1; MSI, microsatellite instability; NHS, Nurses’ Health Study; SD, standard deviation.
We examined the association of the western diet score with colorectal cancer incidence, using all 3,200 incident cases, the 1,175 cases with bacterial data, and the remaining 2,025 cases without bacterial data (Table S4). There was no substantial difference in the results from these three analyses. To adjust for selection bias due to bacterial data availability, we used the inverse probability weighting (IPW) method31 on the 3,200 cases for further analyses.
Our analysis showed that the association of western diet scores with colorectal cancer incidence differed by tissue pks+ E. coli levels (P for heterogeneity = 0.014; Table 4), and was stronger for tumors containing higher-level pks+ E. coli. Multivariable hazard ratios in individuals with scores in the highest (vs. the lowest) tertile of western diet scores were 3.45 [95% confidence interval (CI), 1.53–7.78; P for trend = 0.001 across the tertiles] for colorectal cancer with high-level pks+ E. coli, 1.22 (95% CI, 0.57–2.63) for cancer with low-level pks+ E. coli, and 1.10 (95% CI, 0.85–1.42) for cancer without detectable pks+ E. coli. In a sensitivity analysis, we confirmed that the analysis without IPW yielded results (Table S5) similar to the IPW-adjusted analysis.
Table 4.
Incidence of Colorectal Cancer by pks+ E. coli Status in Relation to Cumulative Average Western Diet Score in the Combined Cohorts of the Health Professionals Follow-up Study (1986–2014) and the Nurses’ Health Study (1980–2014)
| Western diet score | P for trend* | P for heterogeneity† | |||
|---|---|---|---|---|---|
| Tertile 1 | Tertile 2 | Tertile 3 | |||
| Person-years | 1,255,030 | 1,254,558 | 1,256,591 | ||
| Overall colorectal cancer | |||||
| Cases, No. (total n=1,175) | 392 | 391 | 392 | ||
| Age-adjusted HR (95% CI)§ | 1 (referent) | 1.04 (0.93–1.15) | 1.28 (1.14–1.44) | <0.001 | |
| Multivariable HR (95% CI)‖ | 1 (referent) | 0.98 (0.88–1.09) | 1.14 (1.01–1.29) | 0.010 | |
| pks+ E. coli status | 0.014 | ||||
| pks+ E coli negative | |||||
| Cases, No. (total n=1,064) | 364 | 354 | 346 | ||
| Age-adjusted HR (95% CI)§ | 1 (referent) | 1.00 (0.82–1.21) | 1.23 (0.96–1.58) | 0.068 | |
| Multivariable HR (95% CI)‖ | 1 (referent) | 0.95 (0.78–1.15) | 1.10 (0.85–1.42) | 0.40 | |
| pks+ E. coli low | |||||
| Cases, No. (total n=55) | 18 | 15 | 22 | ||
| Age-adjusted HR (95% CI)§ | 1 (referent) | 0.82 (0.36–1.83) | 1.37 (0.64–2.97) | 0.51 | |
| Multivariable HR (95% CI)‖ | 1 (referent) | 0.77 (0.35–1.73) | 1.22 (0.57–2.63) | 0.76 | |
| pks+ E. coli high | |||||
| Cases, No. (total n=56) | 10 | 22 | 24 | ||
| Age-adjusted HR (95% CI)§ | 1 (referent) | 2.24 (1.00–5.04) | 3.83 (1.69–8.66) | <0.001 | |
| Multivariable HR (95% CI)‖ | 1 (referent) | 2.11 (0.94–4.73) | 3.45 (1.53–7.78) | 0.001 | |
The trend test was performed using the western diet score as a continuous variable with cohort-specific ceilings at 10th and 90th percentiles in the regression model. The 90th and 10th percentile values were used for scores above 90th percentile and those below the 10th percentile, respectively, to eliminate outlier effects.
The meta-regression method with a subtype-specific random effect term was used to test whether the association has a trend across the ordinal subtypes in the multivariable-adjusted model, where the western diet score was used as a continuous variable with cohort-specific ceilings at 10th and 90th percentiles.
Duplication-method Cox proportional hazards model weighted by inverse probabilities based on tissue bacterial data availability for competing risks data was used with total caloric intake adjusted and stratification by age (in months), sex (i.e., cohort) and year of questionnaire return.
Additionally adjusted for body mass index (continuous with 35 kg/m2 ceiling), cumulative pack-years smoked (continuous with 50 pack-years ceiling), family history of colorectal cancer in any first-degree relative (yes vs. no), previous lower gastrointestinal endoscopy (yes vs. no), physical activity (continuous with a ceiling at 50 metabolic equivalent task score-hours per week), regular use of aspirin or nonsteroidal anti-inflammatory drugs (>2 tablets/week: yes vs. no), multivitamin use (yes vs. no), and alcohol consumption (continuous with 30 g/day ceiling).
Abbreviations: CI, confidence interval; HR, hazard ratio.
In secondary subgroup analyses, we found similar differential associations by pks+ E. coli status in men and women (Table 5). In analyses using patients stratified by tumor microsatellite instability (MSI) status (Table S6), the differential association by pks+ E. coli status was apparent for the non-MSI-high subtype while statistical power was limited for the MSI-high subtype.
Table 5.
Incidence of Colorectal Cancer by pks+ E. coli Status in Relation to Cumulative Average Western Diet Score in the Health Professionals Follow-up Study (1986–2014) and the Nurses’ Health Study (1980–2014)
| Western diet score | P for trend* | P for heterogeneity† | |||
|---|---|---|---|---|---|
| Tertile 1 | Tertile 2 | Tertile 3 | |||
| Health Professionals Follow-up Study (men) | |||||
| Person-Years | 365,506 | 365,602 | 365,680 | ||
| pks+ E. coli status | 0.71 | ||||
| pks+ E. coli negative | |||||
| Cases, No. (total n=476) | 156 | 158 | 162 | ||
| Age-adjusted HR (95% CI)§ | 1 (referent) | 1.19 (0.95–1.50) | 1.41 (1.09–1.82) | <0.001 | |
| Multivariable HR (95% CI)‖ | 1 (referent) | 1.12 (0.89–1.42) | 1.26 (0.96–1.65) | 0.015 | |
| pks+ E. coli positive | |||||
| Cases, No. (total n=43) | 10 | 15 | 18 | ||
| Age-adjusted HR (95% CI)§ | 1 (referent) | 1.77 (0.80–3.95) | 2.33 (1.05–5.14) | 0.042 | |
| Multivariable HR (95% CI)‖ | 1 (referent) | 1.62 (0.72–3.61) | 2.05 (0.93–4.50) | 0.10 | |
| Nurses’ Health Study (women) | |||||
| Person-years | 889,524 | 888,957 | 890,911 | ||
| pks+ E. coli status | 0.018 | ||||
| pks+ E. coli negative | |||||
| Cases, No. (total n=588) | 208 | 196 | 184 | ||
| Age-adjusted HR (95% CI)§ | 1 (referent) | 0.95 (0.75–1.20) | 1.18 (0.87–1.61) | 0.28 | |
| Multivariable HR (95% CI)‖ | 1 (referent) | 0.89 (0.70–1.13) | 1.03 (0.75–1.41) | 0.90 | |
| pks+ E. coli positive | |||||
| Cases, No. (total n=68) | 18 | 22 | 28 | ||
| Age-adjusted HR (95% CI)§ | 1 (referent) | 1.17 (0.59–2.31) | 2.09 (1.07–4.09) | 0.019 | |
| Multivariable HR (95% CI)‖ | 1 (referent) | 1.10 (0.55–2.18) | 1.81 (0.92–3.54) | 0.058 | |
The trend test was performed using the western diet score as a continuous variable with cohort-specific ceilings at 10th and 90th percentiles in the regression model. The 90th and 10th percentile values were used for scores above 90th percentile and those below the 10th percentile, respectively, to eliminate outlier effects.
The meta-regression method with a subtype-specific random effect term was used to test whether the association has a trend across the ordinal subtypes (negative vs. low vs. high) in the multivariable-adjusted model, where the western diet score was used as a continuous variable with cohort-specific ceilings at 10th and 90th percentiles.
Duplication-method Cox proportional hazards model weighted by inverse probabilities based on tissue bacterial data availability for competing risks data was used with total caloric intake adjusted and stratification by age (in months) and year of questionnaire return.
Additionally adjusted for body mass index (continuous with 35 kg/m2 ceiling), cumulative pack-years smoked (continuous with 50 pack-years ceiling), family history of colorectal cancer in any first-degree relative (yes vs. no), previous lower gastrointestinal endoscopy (yes vs. no), physical activity (continuous with a ceiling at 50 metabolic equivalent task score-hours per week), regular use of aspirin or nonsteroidal anti-inflammatory drugs (>2 tablets/week: yes vs. no), multivitamin use (yes vs. no), and alcohol consumption (continuous with 30 g/day ceiling). We additionally adjusted for postmenopausal hormone use (yes vs. no) for the Nurses’ Health Study analysis.
Abbreviations: CI, confidence interval; HR, hazard ratio.
In secondary analyses to assess the prognostic association of the amount of pks+ E. coli, we conducted survival analysis using Kaplan-Meier method and Cox proportional hazard regression (Figure S1 and Table S7). In univariable analyses of colorectal cancer-specific survival, there was a statistically-insignificant favorable prognostic association of the amount of pks+ E. coli (Ptrend = 0.028, with the α level of 0.012), which did not persist in multivariable analyses (Ptrend > 0.16).
We further examined whether red meat variables (total, unprocessed, and processed red meat intake amounts), which was the largest component of the western diet score, might be differentially associated with colorectal cancer by pks+ E. coli status (Table S8). We found that the differential association by the amount of pks+ E. coli was not statistically significant for any of these red meat variables (Pheterogeneity > 0.05).
DISCUSSION
Colorectal cancer is a heterogeneous group of neoplastic diseases influenced by many factors including diet, lifestyle, and intestinal microbiota32–37. Using two prospective cohort studies in the U.S. with three decades of follow-up, we discovered a stronger association of western-style diet with the incidence of colorectal carcinoma containing higher amounts of pks+ E. coli. Our findings provide evidence for western-style diet characterized by high intake of red and processed meat, sugar, and refined grains as a risk factor for colorectal cancer, especially its subtype containing a high amount of pks+ E. coli. Our novel data can inform research efforts devoted to developing cancer prevention strategies that modify diet and the intestinal microbiome.
Previous metagenomic studies have cast light on the role of the intestinal microbiome in colorectal carcinogenesis38–40. Molecular pathological analyses of colorectal cancer have also supported the role of specific intestinal microbes such as colibactin-producing pks+ E. coli in tumor development41,42. A recent study has elucidated the structure of colibactin and enabled the synthesis of colibactin6. Experimental studies indicate that the genotoxic colibactin can alkylate DNA on adenine residues7 and induce double-strand breaks, leading to a specific mutational signature8,9. Another study has shown that organoids that recovered from short-term infection with pks+ E. coli reveal characteristics of colorectal carcinoma cells, such as enhanced proliferation, WNT-independence, and impaired differentiation, at least in part through alterations in TP53-signaling43. In addition, evidence suggests that pks+ E. coli suppresses the host immune response in the tumor microenvironment44. Taken together, although pks+ E. coli likely plays a role in colorectal carcinogenesis, it currently remains uncertain when and how pks+ E. coli exerts an effect on tumor development. Investigating the detailed mechanism and the associations of this bacterium with lifestyle and dietary risk factors is of particular interest.
Dietary influences on the microbiome in stool and colonic tissue have been investigated. Experimental studies have shown that daily microbiome variation is related to food group choices45, and that a high-fat diet can alter intestinal bacterial composition11 and lead to the development of systemic inflammation46,47. Observational studies have found relationships between low quality diet and inflammatory diet with intestinal dysbiosis48,49 as well as between western-style diet and high plasma soluble CD14 level, a biomarker of mucosal barrier dysfunction50. These lines of evidence suggest that dietary factors can influence intestinal microbial composition and inflammatory status. The prior principal component analysis on diet data in the population revealed two dominant dietary patterns, namely the western-style pattern and prudent dietary pattern51. A meta-analysis indicates a weak-to-moderate association between western-style diet and colorectal cancer risk10. In contrast, the prudent dietary pattern characterized by high intake of fruits, vegetables, fish, poultry, and whole-grains have been inversely associated with colorectal cancer risk10. Nonetheless, the strength of the association remains uncertain due to residual or unmeasured confounding by other healthy or unhealthy behaviors associated with the dietary patterns.
Utilizing the molecular pathological epidemiology approach33,52–54, we found a strong association between western diet and the colorectal cancer subgroup containing high levels of pks+ E. coli. This specific link between western diet and pks+ E. coli suggests potentially interactive carcinogenic effects. In further analysis of red meat variable, we did not observe a statistically significant association of any red meat variable with the incidence of colorectal cancer by pks+ E. coli status. Our data suggest that red meat intake by itself is unlikely the sole factor that contributed to the differential association of the western diet with colorectal cancer by pks+ E. coli status. One possibility is that western diet may promote the proliferation and activity of pks+ E. coli and/or strengthen the carcinogenic effects of pks+ E. coli through alteration of the local tissue microenvironment. It is evident that the molecular pathological epidemiology approach allows for the generation of intriguing hypotheses based on human population data. Although our analyses showed the correlation between western diet and the incidence of colorectal cancer containing high abundance of pks+ E. coli, a replication using additional independent cohorts and experimental research is necessary.
In addition, we found that the association of western diet with colorectal cancer incidence according to pks+ E. coli might be different by sex (Table 5). Although intriguing, those results were obtained by our secondary subgroup analyses, and as such, generalizability needs to be tested in independent datasets. If replicated, our findings may inform differential interactive influences of western diet and pks+ E. coli in male vs. female. While the mechanisms underlying these sex-specific effects remain to be elucidated, differences in biological features of colorectal cancer between men and women have been demonstrated55–57. Additional studies are warranted to investigate how western diet and pks+ E. coli may exert interactive carcinogenic effects, and which specific food items might contribute to the observed differential associations between western diet and colorectal cancer incidence according to pks+ E. coli status.
We acknowledge limitations in the current study. First, unmeasured and/or residual confounding might have substantially influenced our findings. We included most established risk factors in our analysis models with little evidence for substantial confounding by the included variables. Second, tissue bacterial data was unavailable for some incident cancer cases within the cohorts, which might have caused selection bias. However, by using all 3,200 incident colorectal cancers and the inverse probability weighting (IPW) method31, we were able to adjust for selection bias with the available covariates. Analyses with and without the IPW adjustment yielded similar results. Third, measurement errors were inherently present in the assessments of diet and tissue bacterial amounts, particularly with the use of FFPE tissue specimens. We utilized repeated assessments of diet every four years, which allowed us to estimate the effects of long-term dietary patterns. For bacterial analyses, we carefully optimized and validated our quantitative PCR assay for FFPE tissue specimens, to ensure high analytical sensitivity and specificity. Our validation study also demonstrated a high linearity (r2 > 0.95) and high precision (with <1% interassay coefficient of variation) of the assay. Fourth, our cohort populations mainly consisted of non-Hispanic Whites, and thus our findings need to be replicated in independent populations. Fifth, we utilized information on microbial contents in tumor tissue, which was not prospectively collected unlike dietary data. Therefore, establishing a cause-and-effect relationship between the microbial species and colorectal cancer requires additional studies. Finally, our findings were based on the observational cohort studies which had certain inherent limitations in data collections. Hence, additional epidemiological studies and experimental confirmation are ultimately needed.
There exist notable strengths in the current study. First, our dietary data had been prospectively and repeatedly collected for more than 30 years through validated food frequency questionnaires58. Second, our prospective cohort design enabled the collection of diet and other lifestyle data without knowing who would develop colorectal cancer later, thereby eliminating differential recall bias between cancer patients and cancer-free individuals. Third, the prospective study design also enabled us to leverage all 3,200 incident colorectal cancer cases with the IPW method to adjust for selection bias caused by tissue bacterial data availability. Fourth, we utilized molecular pathological epidemiology methods which can provide novel etiological insights into diet and bacterial species, thereby augmenting causal inference. Fifth, the cancer patient group was assembled from hundreds of hospitals located throughout the U.S., which increases the generalizability of our findings in contrast to studies based on only one or a few hospitals. Nonetheless, our findings should be replicated in independent populations.
In conclusion, we have found that the association of western diet with colorectal cancer incidence is stronger for tumors containing higher amounts of pks+ E. coli. Our findings provide evidence supporting the role of the gut microbiota in mediating the pathogenic link between diet and colorectal cancer. This study also underscores the importance of diet as a modifiable factor that may contribute to cancer prevention.
Supplementary Material
What You Need to Know
BACKGROUND AND CONTEXT
Western-style diet has been weakly associated with colorectal cancer risk; however, it remains unclear whether the association of western-style diet with colorectal cancer incidence varies by gut microbe.
NEW FINDINGS
Utilizing two U.S. longitudinal prospective cohort studies, we found that the association of western-style diet with colorectal cancer incidence was stronger for tumors containing higher amounts of pks+ E. coli.
LIMITATIONS
Our cohorts consisted predominantly of non-Hispanic Whites. Therefore, further studies using other populations are needed, as well as experimental confirmation to investigate the mechanisms.
IMPACT
Our findings provide evidence supporting the role of the specific bacterium in mediating a pathogenic link between diet and colorectal cancer and the importance of diet for cancer prevention.
Acknowledgments
We would like to thank the participants and staff of the Health Professionals Follow-up Study and the Nurses’ Health Study for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
Funding:
This work was supported by U.S. National Institutes of Health (NIH) grants (P01 CA87969 to M.J. Stampfer; UM1 CA186107 to M.J. Stampfer; P01 CA55075 to W.C. Willett; UM1 CA167552 to W.C. Willett; U01 CA167552 to W.C. Willett and L.A. Mucci; P50 CA127003 to C.S.F.; R01 CA118553 to C.S.F.; R01 CA169141 to C.S.F.; R01 CA137178 to A.T.C.; K24 DK098311 to A.T.C.; R35 CA197735 to S.O.; R01 CA151993 to S.O.; K07 CA190673 to R.N.; and K07 CA188126 to X.Z.); by Cancer Research UK Grand Challenge Award (UK C10674/A27140 to C.L.S., W.S.G., C.H., M.G., and S.O.); by the Stand Up to Cancer Colorectal Cancer Dream Team Translational Research Grant (SU2C-AACR-DT22-17 to C.S.F. and M.G.), administered by the American Association for Cancer Research, a scientific partner of SU2C; and by grants from the Project P Fund, The Friends of the Dana-Farber Cancer Institute, Bennett Family Fund, and the Entertainment Industry Foundation through National Colorectal Cancer Research Alliance. K.A. and T.U. were supported by a grant from Overseas Research Fellowship (201860083 to K.A., 201960541 to T.U.) from Japan Society for the Promotion of Science. R.Z. was supported by a fellowship grant from Huazhong University of Science and Technology. T.U., K.H., and K.F. were supported by fellowship grants from the Uehara Memorial Foundation. T.U. was supported by a grant from Yasuda Medical Foundation. K.H. was supported by the Mitsukoshi Health and Welfare Foundation. A.T.C. is a Stuart and Suzanne Steele MGH Research Scholar. J.A.M. research is supported by the Douglas Gray Woodruff Chair fund, the Guo Shu Shi Fund, Anonymous Family Fund for Innovations in Colorectal Cancer, Project P fund, and the George Stone Family Foundation. M.G. was supported by a Conquer Cancer Foundation of ASCO Career Development Award.
The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of Potential Conflicts of Interest:
A.T.C. previously served as a consultant for Bayer Healthcare and Pfizer Inc. J.A.M. has also served as an advisor/consultant to Ignyta, Array Pharmaceutical, and Cota Healthcare. C.S.F. is currently employed by Genentech, a subsidiary of Roche, and previously served as a consultant for Agios, Bain Capital, Bayer, Celgene, Dicerna, Five Prime Therapeutics, Gilead Sciences, Eli Lilly, Entrinsic Health, Genentech, KEW, Merck, Merrimack Pharmaceuticals, Pfizer Inc, Sanofi, Taiho, and Unum Therapeutics; C.S.F. also serves as a Director for CytomX Therapeutics and owns unexercised stock options for CytomX and Entrinsic Health. M.G. receives research funding from Bristol-Myers Squibb, Merck, Servier, and Janssen. This study was not funded by any of these commercial entities. No other conflicts of interest exist. The other authors declare that they have no conflicts of interest.
Abbreviations:
- AJCC
American Joint Committee on Cancer
- CI
confidence interval
- CIMP
CpG island methylator phenotype
- Ct
cycle threshold
- FFPE
formalin-fixed paraffin-embedded
- HPFS
Health Professionals Follow-up Study
- HR
hazard ratio
- IPW
inverse probability weighting
- LINE-1
long-interspersed nucleotide element-1
- MSI
microsatellite instability
- NHS
Nurses’ Health Study
- PCR
polymerase chain reaction
- pks
polyketide synthase
- SD
standard deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Use of Standardized Official Symbols: We use HUGO (Human Genome Organisation) Gene Nomenclature Committee-approved official symbols (or root symbols) for genes and gene products, including BRAF, CACNA1G, CD14, CDKN2A, CRABP1, IGF2, KRAS, MLH1, NEUROG1, PIK3CA, RUNX3, SLCO2A1, SOCS1, TP53, and WNT; all of which are described at www.genenames.org. Gene symbols are italicized whereas symbols for gene products are not italicized.
References
- 1.Nougayrede JP, Homburg S, Taieb F, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006;313:848–51. [DOI] [PubMed] [Google Scholar]
- 2.Wu S, Rhee KJ, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009;15:1016–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arthur JC, Perez-Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012;338:120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dejea CM, Fathi P, Craig JM, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018;359:592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett M, Hand CK, Shanahan F, et al. Mutagenesis by Microbe: the Role of the Microbiota in Shaping the Cancer Genome. Trends Cancer 2020;6:277–287. [DOI] [PubMed] [Google Scholar]
- 6.Xue M, Kim CS, Healy AR, et al. Structure elucidation of colibactin and its DNA cross-links. Science 2019;365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson MR, Jiang Y, Villalta PW, et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019;363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 2020;580:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dziubanska-Kusibab PJ, Berger H, Battistini F, et al. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat Med 2020;26:1063–1069. [DOI] [PubMed] [Google Scholar]
- 10.Magalhaes B, Peleteiro B, Lunet N. Dietary patterns and colorectal cancer: systematic review and meta-analysis. Eur J Cancer Prev 2012;21:15–23. [DOI] [PubMed] [Google Scholar]
- 11.O’Keefe SJ, Li JV, Lahti L, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun 2015;6:6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fung T, Hu FB, Fuchs C, et al. Major dietary patterns and the risk of colorectal cancer in women. Arch Intern Med 2003;163:309–14. [DOI] [PubMed] [Google Scholar]
- 13.Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med 2013;369:1095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu FB, Rimm EB, Stampfer MJ, et al. Prospective study of major dietary patterns and risk of coronary heart disease in men. Am J Clin Nutr 2000;72:912–21. [DOI] [PubMed] [Google Scholar]
- 15.Feskanich D, Rimm EB, Giovannucci EL, et al. Reproducibility and validity of food intake measurements from a semiquantitative food frequency questionnaire. J Am Diet Assoc 1993;93:790–6. [DOI] [PubMed] [Google Scholar]
- 16.Yuan C, Spiegelman D, Rimm EB, et al. Relative Validity of Nutrient Intakes Assessed by Questionnaire, 24-Hour Recalls, and Diet Records as Compared With Urinary Recovery and Plasma Concentration Biomarkers: Findings for Women. Am J Epidemiol 2018;187:1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Shaar L, Yuan C, Rosner B, et al. Reproducibility and Validity of a Semiquantitative Food Frequency Questionnaire in Men Assessed by Multiple Methods. Am J Epidemiol 2021;190:1122–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamauchi M, Morikawa T, Kuchiba A, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012;61:847–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamauchi M, Lochhead P, Morikawa T, et al. Colorectal cancer: a tale of two sides or a continuum? Gut 2012;61:794–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inamura K, Yamauchi M, Nishihara R, et al. Prognostic significance and molecular features of signet-ring cell and mucinous components in colorectal carcinoma. Ann Surg Oncol 2015;22:1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mima K, Sukawa Y, Nishihara R, et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol 2015;1:653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujiyoshi K, Bruford EA, Mroz P, et al. Opinion: Standardizing gene product nomenclature-a call to action. Proc Natl Acad Sci U S A 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008;3:1101–8. [DOI] [PubMed] [Google Scholar]
- 24.Ogino S, Kawasaki T, Brahmandam M, et al. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn 2006;8:209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nosho K, Irahara N, Shima K, et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One 2008;3:e3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irahara N, Nosho K, Baba Y, et al. Precision of pyrosequencing assay to measure LINE-1 methylation in colon cancer, normal colonic mucosa, and peripheral blood cells. J Mol Diagn 2010;12:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogino S, Kawasaki T, Brahmandam M, et al. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn 2005;7:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imamura Y, Lochhead P, Yamauchi M, et al. Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: cohort study and literature review. Mol Cancer 2014;13:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao X, Lochhead P, Nishihara R, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 2012;367:1596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang M, Spiegelman D, Kuchiba A, et al. Statistical methods for studying disease subtype heterogeneity. Stat Med 2016;35:782–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L, Nevo D, Nishihara R, et al. Utility of inverse probability weighting in molecular pathological epidemiology. Eur J Epidemiol 2018;33:381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamada T, Nowak JA, Milner DA Jr., et al. Integration of microbiology, molecular pathology, and epidemiology: a new paradigm to explore the pathogenesis of microbiome-driven neoplasms. J Pathol 2019;247:615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akimoto N, Ugai T, Zhong R, et al. Rising incidence of early-onset colorectal cancer - a call to action. Nat Rev Clin Oncol 2021;18:230–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gunter MJ, Alhomoud S, Arnold M, et al. Meeting report from the joint IARC-NCI international cancer seminar series: a focus on colorectal cancer. Ann Oncol 2019;30:510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tilg H, Adolph TE, Gerner RR, et al. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018;33:954–964. [DOI] [PubMed] [Google Scholar]
- 36.Rajpoot M, Sharma AK, Sharma A, et al. Understanding the microbiome: Emerging biomarkers for exploiting the microbiota for personalized medicine against cancer. Semin Cancer Biol 2018;52:1–8. [DOI] [PubMed] [Google Scholar]
- 37.Li J, Zhang AH, Wu FF, et al. Alterations in the Gut Microbiota and Their Metabolites in Colorectal Cancer: Recent Progress and Future Prospects. Front Oncol 2022;12:841552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yachida S, Mizutani S, Shiroma H, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med 2019;25:968–976. [DOI] [PubMed] [Google Scholar]
- 39.Thomas AM, Manghi P, Asnicar F, et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med 2019;25:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ternes D, Karta J, Tsenkova M, et al. Microbiome in Colorectal Cancer: How to Get from Meta-omics to Mechanism? Trends Microbiol 2020;28:401–423. [DOI] [PubMed] [Google Scholar]
- 41.Thakur BK, Malaise Y, Martin A. Unveiling the Mutational Mechanism of the Bacterial Genotoxin Colibactin in Colorectal Cancer. Mol Cell 2019;74:227–229. [DOI] [PubMed] [Google Scholar]
- 42.Arthur JC. Microbiota and colorectal cancer: colibactin makes its mark. Nat Rev Gastroenterol Hepatol 2020;17:317–318. [DOI] [PubMed] [Google Scholar]
- 43.Iftekhar A, Berger H, Bouznad N, et al. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat Commun 2021;12:1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopes A, Billard E, Casse AH, et al. Colibactin-positive Escherichia coli induce a procarcinogenic immune environment leading to immunotherapy resistance in colorectal cancer. Int J Cancer 2020;146:3147–3159. [DOI] [PubMed] [Google Scholar]
- 45.Johnson AJ, Vangay P, Al-Ghalith GA, et al. Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans. Cell Host Microbe 2019;25:789–802 e5. [DOI] [PubMed] [Google Scholar]
- 46.Wan Y, Wang F, Yuan J, et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: a 6-month randomised controlled-feeding trial. Gut 2019;68:1417–1429. [DOI] [PubMed] [Google Scholar]
- 47.Groschel C, Prinz-Wohlgenannt M, Mesteri I, et al. Switching to a Healthy Diet Prevents the Detrimental Effects of Western Diet in a Colitis-Associated Colorectal Cancer Model. Nutrients 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Ajami NJ, El-Serag HB, et al. Dietary quality and the colonic mucosa-associated gut microbiome in humans. Am J Clin Nutr 2019;110:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng J, Hoffman KL, Chen JS, et al. Dietary inflammatory potential in relation to the gut microbiome: results from a cross-sectional study. Br J Nutr 2020;124:931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabung FK, Birmann BM, Epstein MM, et al. Influence of Dietary Patterns on Plasma Soluble CD14, a Surrogate Marker of Gut Barrier Dysfunction. Curr Dev Nutr 2017;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mehta RS, Nishihara R, Cao Y, et al. Association of Dietary Patterns With Risk of Colorectal Cancer Subtypes Classified by Fusobacterium nucleatum in Tumor Tissue. JAMA Oncol 2017;3:921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogino S, Chan AT, Fuchs CS, et al. Molecular pathological epidemiology of colorectal neoplasia: an emerging transdisciplinary and interdisciplinary field. Gut 2011;60:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogino S, Nowak JA, Hamada T, et al. Insights into Pathogenic Interactions Among Environment, Host, and Tumor at the Crossroads of Molecular Pathology and Epidemiology. Annu Rev Pathol 2019;14:83–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mima K, Kosumi K, Baba Y, et al. The microbiome, genetics, and gastrointestinal neoplasms: the evolving field of molecular pathological epidemiology to analyze the tumor-immune-microbiome interaction. Hum Genet 2021;140:725–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lopes-Ramos CM, Kuijjer ML, Ogino S, et al. Gene Regulatory Network Analysis Identifies Sex-Linked Differences in Colon Cancer Drug Metabolism. Cancer Res 2018;78:5538–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berntsson J, Eberhard J, Nodin B, et al. Pre-diagnostic anthropometry, sex, and risk of colorectal cancer according to tumor immune cell composition. Oncoimmunology 2019;8:e1664275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abancens M, Bustos V, Harvey H, et al. Sexual Dimorphism in Colon Cancer. Front Oncol 2020;10:607909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rimm EB, Giovannucci EL, Stampfer MJ, et al. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am J Epidemiol 1992;135:1114–26; discussion 1127-36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.