

Abstract
Background
Myeloid cells dominate metabolic disease-associated inflammation (metaflammation) in mouse obesity, but contributions of myeloid cells to the peripheral inflammation that fuels sequelae of human obesity are untested. We used unbiased approaches to rank contributions of myeloid and T cells to peripheral inflammation in people with obesity across the spectrum of metabolic health.
Methods
We stimulated PBMCs from people with obesity +/− prediabetes or type 2 diabetes with T cell targeting CD/CD28 or myeloid-targeting LPS for 20-72hrs to assess cytokine production using bio-plex. Bioinformatic modeling ranked cytokines with respect to their predictive power for metabolic health. We quantitated intracellular TNF-α as a classical indicator of metaflammation.
Results
Cytokines increased over 72hrs following T cell- but not myeloid-targeted stimulation to indicate acute myeloid inflammation may shift to T cell inflammation over time. T cells contributed more TNF-α to peripheral inflammation regardless of metabolic status. Bioinformatic combination of cytokines from all cohorts, stimuli, and time points, indicated T cell-targeted stimulation was most important for differentiating inflammation in diabetes, consistent with previous identification of a mixed Th1/Th17 cytokine profile in diabetes.
Conclusion
T cells dominate peripheral inflammation in obesity; thus, targeting T cells may be an effective approach for prevention/management of metaflammation.
1. Introduction
Inflammatory sequelae of obesity are induced by peripheral changes in immunomodulatory lipids, immune cell expansion in classical metabolic regulators like adipose tissue (AT), and pro-inflammatory hormones (1-4). Our current understanding of low-grade systemic inflammation, or metainflammation, in obesity and type 2 diabetes (herein “diabetes”) is largely based on studies using mouse models that highlight the importance of pro-inflammatory macrophages in AT and other metabolic tissues (5, 6). Although macrophages are irrefutably important to metabolic immunopathogenesis, the frequency of T cells in visceral AT is higher in non-obese primates/humans compared to mice (7) suggesting the hypothesis that the contribution of T cells to human metabolic disease is underappreciated.
Peripheral inflammation in human obesity and diabetes, which is likely influenced by recirculation of immune cells through the expanded AT (8), remains less understood than mouse AT inflammation (9). Both the serum and AT of lean people and mice have higher concentrations of cytokines characteristic of non-inflammatory immune cells (IL-10, IL-13, and IL-33) (10), which maintain metabolic and immunological homeostasis within insulin-sensitive tissues. Conversely, IL-1β, IL-6, and TNF-α are higher in people with obesity and clinically predict diabetes risk (11, 12). Additionally, PBMCs from people with obesity/diabetes have higher competency to produce pro-inflammatory cytokines generally attributed to CD4+ T cells (TNF-β, IL-17, and IL-21) that overlap with the T cell profile in human AT (13-15). Given that both CD4+ and CD8+ T cells are relatively abundant in human blood and increase IFN-γ production in response to obesity/diabetes (16, 17), these data suggest that an insufficient understanding of the contribution of T cells to peripheral inflammation in obesity may explain why clinical trials of immunomodulatory drugs for diabetes have generally not been successful and thus not broadly adopted (18).
Crosstalk amongst immune cells adds further complexity to the search for modifiable cell types that disproportionately contribute to diabetes-associated inflammation. Many investigations indicate that myeloid cells are the source of TNF-α in obesity and diabetes; our work showed that CD4+ and CD8+ T cell TNF-α is activated by IL-17F, a product of Th17s (14). Closer scrutiny of our own work and others’ demonstrated both a Th17 profile and an underlying Th1 profile (including TNF-α) characterizes diabetes-associated inflammation, in concordance with murine and human analyses (13, 14, 17, 19, 20). Methods used in these studies often did not allow attribution of cytokines to myeloid or T cells, and thus the relative contribution of these two major sources of peripheral metaflammation remains unclear.
To quantify the relative contributions of circulating T and mononuclear myeloid (“myeloid” herein) cells to peripheral inflammation in obesity and metabolic decline, we measured cytokine production by stimulated PBMCs to assess cytokine competency, the ability of cells to contribute to overall inflammation. In diseases likes obesity, which lack a definitive single PBMC stimulant despite convincing evidence of immune cell activation, pan-stimulation of the large number of circulating immune cells available from blood, which recirculate through the expanded AT (8), has been critical for our understanding of mechanisms driving metaflammation and comparison amongst studies. New work establishes that stimulation duration impacts PBMC responses to T cell (CD3/CD28) but not myeloid (LPS) targeted stimulation, regardless of the donor’s metabolic health. Thus, the acute response of myeloid cells is diluted by the sustained contribution of T cells to overall “inflammation”. CD4+ T cells also have the competency to contribute more TNFα to peripheral metaflammation than myeloid cells, regardless of time point. Finally, models that combine cytokine production from both cell types showed that cytokines generated by T cell stimulation rank higher for defining peripheral metaflammation. These results indicate a major role of T cells in peripheral inflammation in human obesity and diabetes to significantly extend our understanding of immune cells as dominant sources of metaflammation.
2. Research Design and Methods
2.1. Patients and Study Approval
The University of Kentucky Institutional Review Board approved this cross-sectional study in accordance with the Declaration of Helsinki. Donors were recruited from the community by the University of Kentucky Center for Clinical and Translational Sciences from fall 2018-winter 2022. Donors were divided into an overweight/obese (herein designated “obese” for simplicity) non-diabetes (ND), prediabetes, or diabetes cohort by %HbA1c, fasting blood glucose, and/or 2hr oral glucose tolerance as categorized by the American Diabetes Association (Table S1). Exclusions included inflammatory or auto-immune diseases (IBD, type 1 diabetes, etc.), NSAIDs <72hrs before blood draw, heart failure, CKD (eGFR<45), liver disease, and cancer <5yrs past. Some people with diabetes used insulin, but none used >100 U/day. People with stable coronary disease taking statins, 81mg aspirin/day, ACE inhibitors and/or diabetes control medications (for diabetes subjects only) were included (medications listed in Table S2). Study size was appropriate to generate models with precision, recall, area under the curve (AUC) and F1 >0.7.
2.2. Cell Manipulation and Cytokine Measurements
PBMCs were archived as published (21), then thawed at 37°C before dilution in RPMI1640 with 2mM L-glutamine, 25mM HEPES, 1% 100X Penicillin/Streptomycin (Gibco), 10% FBS (all from Gibco), and 1mM pyruvate (Corning). Cells were resuspended in RPMI-1640 at 106 cells/mL, seeded into 24-well plates (CellStar) at 500,000 cells/well, then stimulated with E. Coli O111:B4 LPS (25 ng/mL, Millipore Sigma) to primarily target myeloid cells or human αCD3/αCD28 Dynabeads (1 bead/cell, Gibco) to primarily target T cells, for 20-72 hours. Aliquots of supernatants were stored at −80°C for analysis after ≤2 freeze/thaw cycles. Cytokines were quantified using a 25-plex Th17 magnetic bead kit (Millipore Sigma) and a Bio-Rad FLEXMAP 3D with Luminex xPONENT 4.2 and Bio-plex Manager (Bio-Rad) softwares with 1:10 dilutions as appropriate. Values below the level of detection were replaced with a value that was 1/10 of the minimum standard curve value (specific to each cytokine) for analysis.
2.3. Intracellular Staining
LPS- or CD3/CD28-stimulated PBMCs were treated with Brefeldin A (Invitrogen eBioscience) during the last 5h of stimulation. LPS treated cells were incubated on ice with 2.5mM EDTA (Gibco) for 30min. Cells remaining adherent were removed with a rubber scraper (CytOne). Alternatively, CD3/CD28-stimulated cells were pelleted, and beads were removed with a magnet. All cells were resuspended in Zombie NIR Viability stain (Biolegend) at 1:250 in 1X DPBS at 4°C for 30min. Cells were washed, pelleted, and resuspended with anti-CD4 (FITC; Biolegend), CD8 (BV785; Biolegend), and CD14 (PECy7; Biolegend; diluted 1:200); CD19 (BV510; BD Biosciences) and CD161 (BB700; BD Biosciences; diluted 1:100); and CD11b (BV605; BD Biosciences; diluted 1:50), then incubated (4°C, 30min). Cells were then treated with fixation buffer (Invitrogen eBioscience) at RT for 30mins, washed in 1X permeabilization buffer (Invitrogen eBioscience), and pelleted. Cells were resuspended in anti-TNFα (PE; BD Pharmingen; 1:50 dilution in 1X permeabilization buffer) and incubated (4°C, 30min) before pelleting and resuspension in 200μL 1% paraformaldehyde (ChemCruz) in 1X DPBS and analysis on a Beckman Coulter CytoFLEX. UltraComp eBeads (Invitrogen) allowed compensation. Fluorescence minus one (FMO) controls used LPS- or CD3/CD28-stimulated PBMCs (20 or 40hrs, respectively) to validate gating.
2.4. Statistical Analysis
We used GraphPad Prism for a mixed-effects analysis (restricted maximum likelihood) with Tukey’s multiple comparisons to assess temporal differences in cytokines. One-way ANOVAs with Tukey’s multiple comparisons (Gaussian data) or a Kruskal-Wallis test with Dunn’s multiple comparisons (non-Gaussian data), both with a 95% confidence interval, determined cohort-specific differences in stimuli responses. P <0.05 indicated differences.
2.5. Bioinformatics
Maximum Relevance and Minimum Redundancy (mRMR) (22) and extreme gradient bosting (XGBoost) (23) were used to perform feature engineering and classification tasks, respectively. mRMR ranks features based on their relevance to outcome variable and feature redundancy. Top-5 features selected by mRMR trained XGBoost models. The XGBoost algorithm uses an additive manner to optimize the objective function in Equation (1)
| (1) |
Where is the prediction, xi is the input, K is the number of additive functions and is the regression tree space. Scikit-learn (24) and XGBoost (23) libraries were used to implement predictive models. Leave one out cross validation trained/tested the models; averaged values across all samples is reported. Precision, recall, F1-score and Area Under the Curve (AUC) assessed model performances as follows.
| (2) |
| (3) |
| (4) |
For the partial least square discriminant analysis (PLSDA) (25), cytokine data were log-transformed to mitigate skewing. Cytokines with a small range of values had the option to be as/more important than cytokines with a large range of values. We thus scaled each cytokine to zero mean and unit variance before applying PLSDA. After all data preprocessing (based on exploratory analysis), PLSDA was applied using the R function splsda() in the “mixOmics” package (26). Data was analyzed using each of two options: 1. two components each with all 25 cytokines or 2. using the R function tune.splsda() to automatically find the optimal number of cytokines that separated groups. The top ranked cytokines are shown. Analysts were blinded to subject group identity.
3. Results
3.1. PBMCs increase cytokine production with time following T cell but not myeloid stimulation
To determine the relative contributions of myeloid and T cells to obesity-associated inflammation in the periphery, we first established the importance of stimulation time for cytokine production. We used CD3/CD28 Dynabeads to primarily stimulate T cells or E. coli LPS to primarily stimulate myeloid cells in PBMCs from donors with obesity and classified as ND, prediabetes, or diabetes for 20-72 hours, then used PLSDA constrained by donor type to ask how time impacts cytokine profiles in PBMCs responding to T cell- or monocyte-targeted stimulation. The shift in inflammation defined by a mathematical combination of all cytokines projected in 2 dimensions showed that CD3/CD28-activated inflammation shifted over time (Figs. 1A-C, top panels). A focus on the 5 cytokines that were most important for distinguishing profile shifts indicated cohort-dependent differences in cytokines that distinguished the CD3/CD28 response (bracket on bar graphs, Figs. 1 A-C). A mixed Th17/Th1 profile (IL-17F/A/IFN-γ/TNF-β) generated at 72 hrs dominated “inflammation” in PBMCs from donors with diabetes (Fig. 1A, lower panel, red bars; note lack of importance of cytokines produced at 20- and 40-hrs, grey and black bars). A similar 72 hr Th17/Th1 profile (TNF-β, IL-17F/A/GMCSF) dominated inflammation in prediabetes (Fig. 1B, lower panel), although 72 hr. IFN-γ was somewhat less important for defining inflammation in prediabetes compared to diabetes cultures (compare Figs. 1A&B where IFN-γ is ranked 2nd or 8th most important for diabetes or prediabetes, respectively, *). Cytokines related to type 2 immune responses (IL-9,-13,-21) were more important in CD3/CD28-elicited inflammation in cultures from donors with obesity and normal glucose tolerance (ND) (Fig. 1C), suggesting that loss of a type 2 profile associates with metabolic decline in obesity. Cytokine production at 40 hrs post-CD3/CD28 was over-represented as a secondary component for defining inflammation for all subjects’ cells (Fig. S1A). Mixed-effects analysis of individual cytokines over time for each cohort supported the PLSDA demonstration that cytokines produced by PBMCs stimulated with CD3/CD28 (but neither LPS nor lack of stimulating ligands) increases over time (Fig S2).
Fig. 1: Cytokine responses to T cell-targeting, but not myeloid-targeting stimuli, change over time and are impacted by diabetes.

(A-C) PLSDA of inflammatory profiles generated by combining all cytokines measured after PBMC stimulation with CD3/CD28 for 20 (gray), 40 (black), or 72 (red) hrs as indicated. PBMCs were from subjects with BMI in the high overweight/obese BMI range (“obesity” herein to simplify) and (A) diabetes, (B) prediabetes, or (C) normoglycemia (ND). Top panels show 2-dimensional projections of the cytokine profiles in the first (most distinguishing) component, colored and grouped by time point; bottom panels rank cytokines produced at all times from most (at bottom) to least (at top) important for defining the responses that are most highly characteristic, as a group, of cells from the subject cohort indicated. (D) The R program automatically identified the optimal number of cytokine values/time points that distinguished CD3/CD28 elicited inflammation for cells from subject cohorts as indicated to focus on distinguishing cytokines while eliminating background “noise”. (E) Cytokines that, as a group, are the second most distinguishing components of inflammation for cells from subject cohorts as indicated. (F) PLSDA of inflammatory profiles generated by combining all cytokines measured after PBMC stimulation with LPS for 20 (green), 40 (blue), or 72 (purple) hrs as indicated. PBMCs were from subjects with obesity as indicated. (G) The R program automatically identified the optimal number of cytokine values/time points that distinguished LPS- elicited inflammation for cells from subjects with obesity and prediabetes (blue) or diabetes (red). Cytokines are ranked in the first, second and third components as most (bottom) to least (top) important for differentiating responses from prediabetes and diabetes cells. Ns are per Table S1.
Follow-up analysis used R to automatically find the optimal number of cytokine values/time points that distinguished inflammation for cells from each of the three subject cohorts to focus on important distinguishing cytokines while eliminating background “noise”. Automated cytokine selection showed that 72 hr production of IL-17F and IFN-γ dominated the first component of CD3/CD28-elicited cytokines in diabetes, consistent with our previous identification of a mixed Th17/Th1 profile in diabetes (14, 19), while TNF-β (lymphotoxin A; a Th1 cytokine (29)) at 72 hrs was most distinguishing for prediabetes. The auto-selected first component for CD3/CD28-stimulated ND cells included cytokines from all major CD4+ T effector subsets (Th1,2,9,17). (Fig. 1D). The second component profiles for CD3/CD28-stimulated PBMCs included cytokine production at 40 hrs in both diabetes and ND donors (Fig. 1E, left; black/gray bars and data not shown), but remained dominated by 72-hr cytokine amounts in prediabetes (Fig. 1E, right; red bars). Taken together these data indicate shifts in CD3/CD28-elicited responses over time are impacted by metabolic status of the cell donor and/or diabetes drugs. In sharp contrast, treatment time had little effect on cytokine production by both LPS-stimulated and unstimulated PBMCs (Fig. 1F,S1B, and S2A&C). Although LPS responses insignificantly changed over time, automated selection of cytokines elicited from PBMCs by LPS showed significant separation of profiles between donors with diabetes and prediabetes (but not ND), and highlighted 3 components that differentially supported diabetes. CCL20 (with IL-22 dominating prediabetes responses in the first component) was most prominent, with lesser contributions to diabetes-specific inflammation by IL-23 and IL-1β (components 2 and 3 respectively; Fig. 1G). We conclude from both CD3/CD28 and LPS-elicited profiles that metabolic status impacts cytokine competency in cells from donors with obesity, consistent with our work on purified CD4+ T cells (30).
3.2. Changes in T cell and myeloid cell cytokines accompany metabolic decline
Our work indicated that combinatorial cytokine profiles differentiated ND from diabetes inflammation (14, 19). To better understand the unique cytokine competency of PBMCs from obese prediabetes subjects and put our combinatorial findings (Fig. 1) into the more traditional context of the literature, we compared amounts of individual cytokines produced by stimulated PBMCs amongst all subject groups. Consistent with previous findings (14, 15), CD3/CD28 stimulation for 40-72 hr elicited more IL-17A, IFN-γ and/or IL-17F, though not additional cytokines, by cells from donors with prediabetes or diabetes compared to donors with normoglycemia (Fig. 2A&B and data not shown). Although a direct comparison of cytokine production by CD3/CD28-stimulated PBMCs from our past data to outcomes herein are problematic due to equipment changes, sample storage time and other technical differences, comparisons between outcomes from newly recruited diabetes subjects from our historical and present sites (Boston-metro and Kentucky, respectively) showed trends toward less production of Th17 signature cytokines by cultures from clinically and demographically similar Kentucky subjects (Fig. 2C). Cultures from Kentuckians with T2D had higher amounts of the Th1 cytokine IFN-γ and trends of others like IL-6 and IL-12p70, similar to our findings in the Boston cohort (Fig. 2A&B; (14, 19). The dominance of IFN-γ is unlikely caused by the high prevalence of smokers in Kentucky, which supports Th1 polarization (31), as we recruited zero versus one active smoker in the KY or Boston-metro diabetes cohort, respectively. Regardless of location-associated difference, these analyses confirm the conclusion that time-dependent changes in cytokine competency are shaped by metabolic status in CD3/CD28-stimulated PBMCs.
Fig. 2: Loss of glycemic control associates with changes in T cell cytokine production.
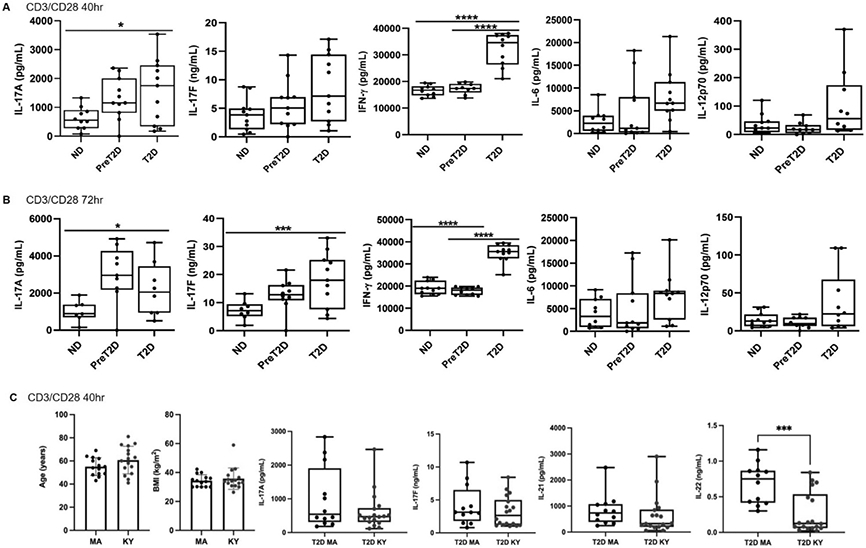
PBMCs from ND (left), prediabetes (middle) and diabetes (right; as indicated on X axis) subjects were activated with T cell-targeted stimuli and cytokine production was quantified after (A) 40 or (B) 72 hrs. Differences in cytokine production based on glycemic control of the PBMC donor were identified by 2-Way ANOVA. P values <0.05 are indicated. Ns are listed in Table S1. (C) Cytokine production by PBMCs from diabetes subjects recruited from the Boston MA metro area (left, N=17; 12 males) or at the Univ. of Kentucky (right, N=12; 10 males). Panels at left show subjects were matched for BMI and age. Trends towards differential production of multiple Th17-signature cytokines are shown, with IL-22 reaching statistical difference as tested by unpaired two tailed t tests. Boxes indicate average (line) and bottom/top quartiles, and whiskers indicate minimum to maximum.
To similarly challenge the bioinformatic indication that metabolic status impacts an early peak of LPS-activated cytokines (Fig. 1G), we compared production of single cytokines following 20 hr LPS stimulation in PBMCs from all subject cohorts. About half the cytokines were produced in higher amounts as disease progressed, although some, like IL-1β and TNF-α, failed to meet statistical criteria for increases (Fig. 3 and data not shown). These data independently indicate the potential contribution of myeloid inflammation increases with obesity-associated metabolic decline independent of culture time.
Fig. 3: Loss of glycemic control associates with changes in LPS-stimulated cytokine production.
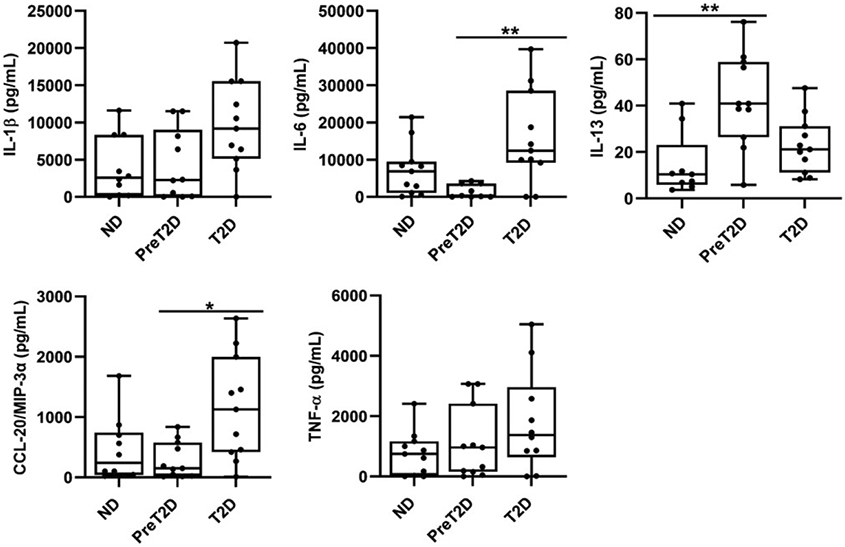
PBMCs from ND (left), prediabetes (middle) and diabetes (right; as indicated) subjects were activated with myeloid-targeted stimuli (LPS) and cytokine production was quantified after 20hrs. Cytokines that were detectable in a majority of the samples are shown. Differences in cytokine production based on glycemic control of the PBMC donor were identified by 2-Way ANOVA with trends for IL-1β and TNF-α failing to reach statistical significance. Box and whisker plots are as described in Fig. 2 with Ns in Table S1.
3.3. T cell- compared to myeloid-stimulated PBMCs produce more TNF-α
Given the unexpected evidence that T cells may contribute more to metaflammation in the long run (Fig. 1), we focused our analysis on production of TNF-α, the first cytokine linked to obesity-associated metabolic disease (32, 33). Both T cells and monocytes from donors with obesity make TNF-α (14), but myeloid cells are often the sole focus of TNF-α production in obesity as first recognized in mouse AT (34). We compared production of TNF-α by 20 hr LPS-stimulated and 20, 40 or 72 hr-CD3/CD28-stimulated PBMCs amongst cohorts. Irrespective of time point or metabolic status, CD3/28 stimulation yielded more TNF-α than LPS, although IL-6, another diabetes-associated cytokine, was produced at higher concentrations by myeloid stimulation of diabetes cells, regardless of the comparator T cell time point (Fig. 4A,B and S3). Cytokines made predominantly by myeloid cells (IL-1β; Fig. 4C) or by T cells (IFN-γ and IL-12p70; Fig. 4D,E) were elicited by the expected stimulus, while some cytokines were similarly produced (CCL-20; Fig. 4F and S3).
Fig. 4: T cell stimulation elicits more TNF-α than myeloid stimulation of PBMCs from all obese subjects.
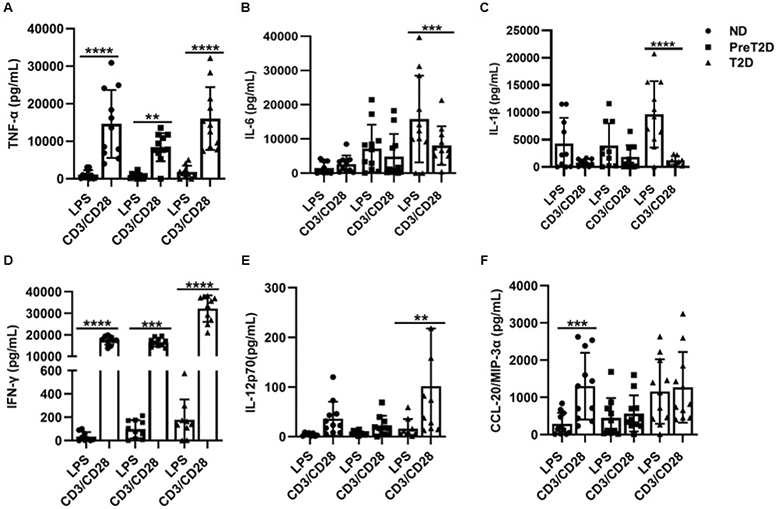
Direct comparison of cytokines elicited by 20 hr. stimulation of PBMCs from ND (circles), prediabetes (squares) or diabetes (triangles) subjects as indicated. Differences identified by a mixed effects analysis are indicated. Bars show average and SD with Ns shown in Table S1.
We used intracellular staining (Fig. S4A,B) to challenge the possibility that circulating CD4+ T cells produce more TNF-α than myeloid cells in response to canonical stimuli, and whether metabolic status or immune cell subset frequency affects this response. Control experiments showed that frequencies of CD11b+ myeloid cells, CD14+ myelo-monocytes, and CD4+ T cells, and frequencies of TNF-α+ cells within each subset were similar amongst the 3 cohorts and within expected frequencies for human PBMCs (Fig. 5A and S5A&B), consistent with previous analyses (19, 35). However, the frequency of TNFα-producing CD4+ cells (avg. ~30%) was higher than either TNFα-producing CD11b+ or CD14+ cells (avg. <10%; Fig. 5A). Follow-up work compared TNF-α production by 20hr LPS- and 40hr CD3/CD28-stimulated PBMCs to capture the earliest times of maximal TNF-α elicitation for each stimulus, and compared TNF-α production based on MFI, a measure of average cytokine production/cell. The average CD11b+ cell produced <10% of the TNF-α produced by CD14+ cells in LPS-responding PBMCs (Fig. 5B, top panel). Each CD4+ T cell from CD3/CD28-stimulated PBMCs produced ~12X or 1.3X more TNF-α on average than CD11b+ or CD14+ cells, respectively, in LPS-stimulated PBMCs and only a small fraction of CD11b+ and even fewer CD14+ cells produced a small amount of TNF-α+ (i.e., MFI) following CD3/CD28 stimulation, adding little to the overall TNF-α production in culture supernatants (Fig. 5B&C). CD4+s also trended to produce more TNF-α on average than each CD14+ cell in ND and diabetes samples, but this difference failed to reach statistical significance (Fig. 5D). Given the much higher frequency of TNF-α producing CD4+ cells and higher (or for CD14+, similar) TNF-α production/cell (i.e., MFI), intracellular staining supports the bioplex conclusion that activated peripheral blood CD4+ T lymphocytes produce more TNF-α than myeloid cells in obesity and obesity-associated diabetes, and thus T cells may contribute more to peripheral metaflammation than myeloid cells.
Fig. 5: Intracellular staining confirms the CD4+ T cells population produces more TNF-α than myeloid cells.
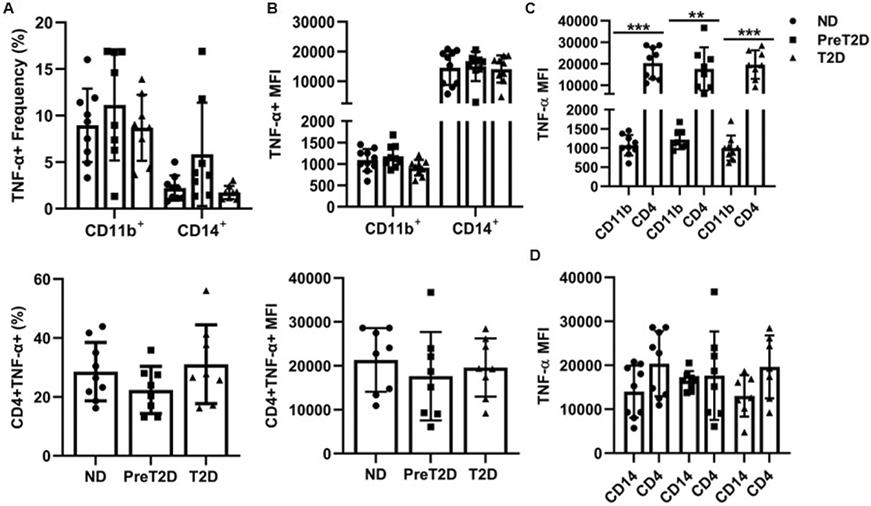
(A) Frequency of (top) CD11b+ myeloid cells and CD14+ monocytes or (bottom) CD4+ T cells from ND (circles), prediabetes (squares) or diabetes (triangles) PBMCs that stained positive for TNF-α. Frequency was relative to total PBMCs. (B) Mean fluorescence intensity (MFI) indicates the average number of TNF-α molecules produced on a per cell basis in the cell types indicated. A direct comparison of intracellular TNF-α as measured by MFI in CD4+ T cells versus (C) CD11b+ myeloid cells or (D) CD14+ monocytes. Frequencies in panel A x MFI in panel B approximate the contribution of each cell type to total TNF-α production. Differences were identified by one-way ANOVA or multiple-effects analysis with Tukey’s multiple comparisons. N=11/group.
2.4. T cell cytokine competency disproportionately contributes to diabetes-associated inflammation
Despite the demonstrated role of TNF-α in metaflammation (36), TNF-α is not a top cytokine for differentiating CD3/CD28 from LPS responses in obesity alone or in combination with metabolic decline (Fig. 1). We combined cytokine measures from all stimulation conditions (CD3/CD28, LPS and unstimulated for 20, 40 and 72 hrs) in PLSDA as an unbiased approach to address the relative importance of CD3/CD28- versus LPS-generated cytokines in metaflammation. CD3/CD28 cytokines elicited at 72 hrs (Fig. 6A, left; red bars) were most important for diabetes-associated inflammation, with dominance of LPS (20 hr)-elicited IL-1β, and to a lesser extent IL-23 and IL-6, as an underlying (second) component of diabetes inflammation (Fig. 6A, right; green bars). To independently challenge this indication that CD3/28 responses dominate peripheral inflammation in diabetes, we re-analyzed cytokine data by mRMR, a principal components method that combined cytokines produced by all subjects’ samples following each of the two stimuli for all time points. Separation of cytokine data from all time points and stimuli by subject types (ND, prediabetes, diabetes) produced a model with poor performance for differentiating cytokine values from ND compared to prediabetes subjects (0.500 precision) although accuracy of prediction of T2D from cytokines was 0.909 (Table S3). We therefore combined cytokine data from both types of non-T2D subjects (ND + prediabetes, designated ND+) to ask which cytokines elicited by which stimulus, and at which time post-stimulation, best differentiated diabetes from ND+ samples. Three of the top 5 cytokine measures that differentiated diabetes were IFN-γ produced by CD3/CD28 stimulation of PBMC for 72, 40 or 20 hrs (Fig. 6B). LPS elicited two of the top ranked cytokines (IL-33 at 40h and CCL20 at 20 hrs). Principal component analysis of these top 5 cytokines shows clear visual distinction of diabetes compared to ND+ inflammation, with confidence in the model boosted by very high precision (Fig. 6C-D). Based on both PLSDA and mRMR, we conclude that T cell cytokines, perhaps fueled in part by the Th1 cytokine IFN-γ and a lesser contribution by TNF-α, ranked highly as an indicator of diabetes in several analyses (Fig. 1A, 1D, 4, 5, 6) to contribute a greater amount of disease-defining peripheral inflammation to human diabetes than do myeloid cells.
Fig. 6: Combinatorial models indicate T cells cytokine are more important than myeloid cytokines for diabetes-associated inflammation.

(A) PLSDA model that combined cytokines produced by PBMCs stimulated as indicated in the Outcome key, then ranked cytokines produced by each condition/sample from most (bottom) to least (top) important for defining inflammation in diabetes. Left panel shows cytokines in the first component (dimension) that distinguishes diabetes. Right panel ranks cytokines in the second component. (B) Ranking of top 5 cytokines that differentiate diabetes from ND+ (a combination of outcomes from non-diabetes and pre-diabetes) samples identified by mRMR. (C) Two-dimensional representation of profiles derived from mathematical combination of the top five conditions identified in panel B. Blue and red dots indicate “inflammation” from ND+ or diabetes subjects, respectively. (D) Quantification of mRMR model quality. A perfect model would have values of 1.000 for all parameters, which are defined under the table.
4. Discussion
Our kinetic analysis defined inflammatory profiles produced by PBMCs from obese non-diabetes, prediabetes, and diabetes subjects and showed that primary stimulation of T cells, but not myeloid cells, produced a profile that changed over time. Temporal changes indicated that multiple analyses were needed to test the relative contributions of myeloid and T cells to peripheral inflammation in obesity-associated metabolic decline. T cells impact peripheral inflammation over longer time points, suggesting focus on TNFα, would test the possibility that T cells significantly contribute to peripheral metaflammation. Our data show T cells contribute at least as much TNF-α as myeloid cells to systemic metaflammation. More comprehensive analyses supported the same conclusion, that T cells dominate systemic inflammation in obesity-associated diabetes. Our work translates the relative importance of myeloid and T cells in metabolic disease first identified in animal models, to a conceptually new demonstration of the relative role of T cells in peripheral metaflammation, and thus obesity sequelae (37, 38).
Responses to the T cell-targeted CD3/CD28 underscored the distinctiveness of prediabetes inflammation first indicated by our analysis of cytokine competency of purified CD4+ T cells (30), and significantly extends existing analyses of cytokines in prediabetes plasma/serum (39, 40). The acute myeloid response, combined with the more sustained response to CD3/CD28 that dominates overall “inflammation” at 72 hrs., is consistent with a canonical immune response in which innate immune cells respond quickly to prime a sustained adaptive immune response. Whether this temporal pattern is mimicked by shifts in the undefined ligands that regulate chronic low-level inflammation in obesity-associated metabolic decline, or explains the suboptimal immune responses to, for example, vaccines and viruses in obesity/diabetes (41, 42), remains unclear.
The prominence of IFN-γ as an indicator of diabetes with likely co-dominance of Th17 cytokines (IL-17A/F, -21, and -22) is consistent with our demonstrations of mixed peripheral Th1/Th17 profiles in human T2D, although mechanistic support of these two CD4+ subsets differs (14, 19). Our findings are consistent with more IFNγ-secreting T cells/mRNA expression in AT from mice and humans with obesity (43, 44), and demonstrations that IFN-γ−/− mice on a high-fat diet are metabolically more healthy than wild-types (45). The similar profile generated by Kentucky compared to Boston-metro samples is surprising given lower concentrations of Th17 cytokines produced by CD3/CD28 stimulation of PBMCs from Kentuckians. The underlying cause of this geographical difference remains unclear, and is a limitation of the study, as variables like stress, diet, and activity almost certainly contribute although all subjects were generally urban and in areas known for an educated population. The Kaiser Commission on Medicaid and the Uninsured (2016) summarized data indicating northeastern U.S. residents are healthier than those residing further south, but this conclusion is counterintuitive to our findings of higher amounts of selected inflammatory cytokines produced by samples from northeasterners. Number of subjects in each group, though appropriate for bioinformatic analysis, is not appropriate to analyze data by gender or race. Given that both CD4+Th1 and CD8+ T cells produce IFN-γ in response to CD3/CD28 stimulation (amidst low myeloid IFN-γ competency), it is possible that population differences in T cell subsets, including a lower frequency of CD8+ cells in diabetes samples in Boston (19) but not Kentucky (Fig. S5B), along with differences in the model assumptions, contribute to the prominence of IFN-γ in the mRMR model. These outcomes stress the benefit of using multiple analytical tools and comparisons to best understand systemic disorders through the lens of inflammation, and thereby target treatments that take advantage of existing FDA approved pharmaceuticals.
Supplementary Material
Study Importance.
What is known?
Focus on myeloid cells as a key source of the obesity-associated inflammation that supports metabolic decline has not impacted clinical treatment of obesity sequelae like type 2 diabetes.
T cells are a second demonstrated source of peripheral inflammation in obesity-associated prediabetes and type 2 diabetes
What are the new findings?
Myeloid inflammation may mediate earlier stages of obesity-associated inflammation, although T cells become more important over longer time periods of unresolved inflammation.
Peripheral T cells compared to myeloid cells produce more of one critical pro-inflammatory cytokine, TNFα, which has been strongly implicated in obesity sequalae.
Combinatorial analysis indicated that T cell as compared to myeloid cytokines rank higher as predictors of obesity-associated inflammation in the periphery.
How results might change the focus of clinical practice?
This finding suggests that refocus of clinical trials from myeloid to T cell-targeted treatments may leverage the appreciation of obesity/diabetes as inflammatory diseases into more effective treatments.
Acknowledgements
The authors thank Drs. Elizabeth Proctor (Pennsylvania State University) and Doug Lauffenberger (Massachusetts Institute of Technology) for providing valuable insights on data troubleshooting and bioinformatic analysis, respectively. Dr. Jamie Sturgill brainstormed explanations for Kentucky versus Boston-metro differences.
Funding
This work was supported by NIH Training Grants T32 DK007778 and TL1 UL1TR001998 (GHP), R01DK108056 (BSN and PAK), the Shared Resource Facility of the University of Kentucky Markey Cancer Center P30 CA177558, University of Kentucky College of Medicine (BSN), Barnstable Brown Diabetes and Obesity Center (BSN and PAK), The Center for Clinical and Translational Research (UL1TR000117), and NIH National Center for Advancing Translational Sciences UL1TR001998 (PAK).
Footnotes
Duality of Interest
The authors report no potential conflicts of interest relevant to this article.
References
- 1.Maury E, Ehala-Aleksejev K, Guiot Y, Detry R, Vandenhooft A, and Brichard SM. Adipokines oversecreted by omental adipose tissue in human obesity. Am J Physiol Endocrinol Metab. 2007;293(3):E656–65. [DOI] [PubMed] [Google Scholar]
- 2.Pang C, Gao Z, Yin J, Zhang J, Jia W, and Ye J. Macrophage infiltration into adipose tissue may promote angiogenesis for adipose tissue remodeling in obesity. Am J Physiol Endocrinol Metab. 2008;295(2):E313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17(5):610–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fruhbeck G, Catalan V, Rodriguez A, Ramirez B, Becerril S, Salvador J, et al. Adiponectin-leptin Ratio is a Functional Biomarker of Adipose Tissue Inflammation. Nutrients. 2019;11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lumeng CN, Bodzin JL, and Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, et al. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010;59(7):1648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laparra A, Tricot S, Le Van M, Damouche A, Gorwood J, Vaslin B, et al. The Frequencies of Immunosuppressive Cells in Adipose Tissue Differ in Human, Non-human Primate, and Mouse Models. Frontiers in immunology. 2019;10:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol. 2015;16(1):85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ip BC, Hogan AE, and Nikolajczyk BS. Lymphocyte roles in metabolic dysfunction: of men and mice. Trends Endocrinol Metab. 2015;26(2):91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52(3):812–7. [DOI] [PubMed] [Google Scholar]
- 12.Pradhan AD, Manson JE, Rifai N, Buring JE, and Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286(3):327–34. [DOI] [PubMed] [Google Scholar]
- 13.McLaughlin T, Liu LF, Lamendola C, Shen L, Morton J, Rivas H, et al. T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans. Arterioscler Thromb Vasc Biol. 2014;34(12):2637–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ip B, Cilfone NA, Belkina AC, DeFuria J, Jagannathan-Bogdan M, Zhu M, et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFalpha production. Obesity (Silver Spring). 2016;24(1):102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. 2011;186(2):1162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15(8):914–20. [DOI] [PubMed] [Google Scholar]
- 17.Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15(8):921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldfine AB, and Shoelson SE. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J Clin Invest. 2017;127(1):83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicholas DA, Proctor EA, Agrawal M, Belkina AC, Van Nostrand SC, Panneerseelan-Bharath L, et al. Fatty Acid Metabolites Combine with Reduced beta Oxidation to Activate Th17 Inflammation in Human Type 2 Diabetes. Cell Metab. 2019;30(3):447–61 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Defuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U S A. 2013;110(13):5133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholas D, Proctor EA, Raval FM, Ip BC, Habib C, Ritou E, et al. Advances in the quantification of mitochondrial function in primary human immune cells through extracellular flux analysis. PLoS One. 2017;12(2):e0170975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng H, Long F, and Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005;27(8):1226–38. [DOI] [PubMed] [Google Scholar]
- 23.Chen T, and Guestrin C. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2016:785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.F P, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel M, et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research. 2011;12(85):2825–30. [Google Scholar]
- 25.Ståhle L, and Wold S. Partial least squares analysis with cross-validation for the two-class problem: A monte carlo study. J Chemometrics 1987;1(3):185–96. [Google Scholar]
- 26.Cao K-AL, González I, and Déjean S. IntegrOmics: an R package to unravel relationships between two omics data sets. Bioinformatics. 2009;25(21):2855–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coquet JM, Schuijs MJ, Smyth MJ, Deswarte K, Beyaert R, Braun H, et al. Interleukin-21-Producing CD4(+) T Cells Promote Type 2 Immunity to House Dust Mites. Immunity. 2015;43(2):318–30. [DOI] [PubMed] [Google Scholar]
- 28.Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15(5):271–82. [DOI] [PubMed] [Google Scholar]
- 29.Gramaglia I, Mauri DN, Miner KT, Ware CF, and Croft M. Lymphotoxin alphabeta is expressed on recently activated naive and Th1-like CD4 cells but is down-regulated by IL-4 during Th2 differentiation. J Immunol. 1999;162(3):1333–8. [PubMed] [Google Scholar]
- 30.Liu R, Pugh GH, Tevonian E, Thompson K, Lauffenburger DA, Kern PA, et al. Regulatory T Cells Control Effector T Cell Inflammation in Human Prediabetes. Diabetes. 2022;71(2):264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tejero JD, Armand NC, Finn CM, Dhume K, Strutt TM, Chai KX, et al. Cigarette smoke extract acts directly on CD4 T cells to enhance Th1 polarization and reduce memory potential. Cell Immunol. 2018;331:121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hotamisligil GS, Shargill NS, and Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. [DOI] [PubMed] [Google Scholar]
- 33.Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, and Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest. 1995;95(5):2111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poitou C, Dalmas E, Renovato M, Benhamo V, Hajduch F, Abdennour M, et al. CD14dimCD16+ and CD14+CD16+ monocytes in obesity and during weight loss: relationships with fat mass and subclinical atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31(10):2322–30. [DOI] [PubMed] [Google Scholar]
- 36.Uysal KT, Wiesbrock SM, Marino MW, and Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389(6651):610–4. [DOI] [PubMed] [Google Scholar]
- 37.Strissel KJ, Denis GV, and Nikolajczyk BS. Immune regulators of inflammation in obesity-associated type 2 diabetes and coronary artery disease. Current opinion in endocrinology, diabetes, and obesity. 2014;21(5):330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu M, and Nikolajczyk BS. Immune cells link obesity-associated type 2 diabetes and periodontitis. J Dent Res. 2014;93(4):346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grossmann V, Schmitt VH, Zeller T, Panova-Noeva M, Schulz A, Laubert-Reh D, et al. Profile of the Immune and Inflammatory Response in Individuals With Prediabetes and Type 2 Diabetes. Diabetes Care. 2015;38(7):1356–64. [DOI] [PubMed] [Google Scholar]
- 40.Brahimaj A, Ligthart S, Ghanbari M, Ikram MA, Hofman A, Franco OH, et al. Novel inflammatory markers for incident pre-diabetes and type 2 diabetes: the Rotterdam Study. Eur J Epidemiol. 2017;32(3):217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiggins KB, Smith MA, and Schultz-Cherry S. The Nature of Immune Responses to Influenza Vaccination in High-Risk Populations. Viruses. 2021;13(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamamoto S, Mizoue T, Tanaka A, Oshiro Y, Inamura N, Konishi M, et al. Sex-associated differences between body mass index and SARS-CoV-2 antibody titers following the BNT 162b2 vaccine among 2,435 healthcare workers in Japan. Obesity. 2022;30:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pacifico L, Di Renzo L, Anania C, Osborn JF, Ippoliti F, Schiavo E, et al. Increased T-helper interferon-gamma-secreting cells in obese children. Eur J Endocrinol. 2006;154(5):691–7. [DOI] [PubMed] [Google Scholar]
- 44.Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103(5):467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Rourke RW, White AE, Metcalf MD, Winters BR, Diggs BS, Zhu X, et al. Systemic inflammation and insulin sensitivity in obese IFN-gamma knockout mice. Metabolism. 2012;61(8):1152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.