

Summary
Neuroblastoma is a leading cause of cancer-related death in children. Accumulated data suggest that differentiation arrest of the neural crest derived sympathoadrenal lineage contributes to neuroblastoma formation. The developmental arrest of these cell types explains many biological features of the disease, including its cellular heterogeneity, mutational spectrum, spontaneous regression, and response to drugs that induce tumor cell differentiation. In this review, we provide evidence that supports the notion that arrested neural crest derived progenitor cells give rise to neuroblastoma and discuss how this concept could be exploited for clinical management of the disease.
Introduction
The link between cancer and arrested differentiation is a concept dating back more than a century, when pathologists first noticed that tumor cells resemble immature cells from developing tissues (Telloni, 2017). For many types of cancer, the inverse relationship between tumor cell differentiation and prognosis has been well established and led to the development of treatment regimens that induce tumor cell differentiation (Sartorelli, 1985). Extensive cancer genomic studies from the past decade have further bolstered the concept that some cancers are a disease of arrested development. For example, somatic mutations that perturb transcription factors or signaling pathways important in development are often found in combination with mutations in oncogenes and tumor-suppressor genes. Similarly, mutations in epigenetic regulators are highly prevalent and thought to further contribute to changes in the epigenetic landscape that block differentiation and promote tumorigenesis.
The connection between arrested differentiation and oncogenesis is particularly prevalent in pediatric cancer (Chen et al., 2015). Many childhood cancers are not found in adults because they arise during a particular stage of development. For example, retinoblastoma is found only in children younger than 5 years of age because it begins during retinal development in utero (Benavente and Dyer, 2015). Also, the genomic landscape of pediatric cancer is less complex than that of adult cancers (Gröbner et al., 2018), suggesting that differentiation arrest combined with oncogenic mutations in the appropriate developmental milieu underlies tumorigenesis in infants, children, adolescents, and young adults.
One of the best examples of this concept is neuroblastoma, a tumor of the developing sympathoadrenal lineage. Neuroblastoma is the most common extracranial solid tumor of childhood, and most patients are diagnosed under the age of 10 years (Cheung and Dyer, 2013; Matthay et al., 2016). Age at diagnosis inversely correlates with outcome. Infants (< 18 months) have an 88% overall survival rate, and some have spontaneous regression (4S neuroblastoma) without therapeutic intervention (Brodeur, 2018; Kawano et al., 2021). Children between 18 months and 12 years have a 49% overall survival which drops below 10% for adolescents and young adults (>12 years). To date, mutations in ATRX, which encodes a chromatin remodeling protein, are the only genetic lesions associated with age at diagnosis in neuroblastoma (Cheung et al., 2012). Other genetic lesions associated with high-risk disease, such as MYCN amplification, oncogenic ALK and RAS pathway mutations and TRKA tumor suppressor mutations, are distributed across age groups (Brodeur et al., 2009; Eleveld et al., 2015; Ho et al., 2011; Matthay et al., 2016; Mosse et al., 2008). Though many of these genes play a role in sympathoadrenal development, the biological mechanism underlying highly variable survival outcomes for children with neuroblastoma remains an unanswered question.
In this review, we discuss the molecular, cellular, genetic, and epigenetic evidence in support of neuroblastoma as a disease of arrested differentiation. We review findings from recent single-cell transcriptional profiling of neuroblastoma, which resemble the developmental heterogeneity of neural crest-derived lineages. We also highlight the importance of cellular heterogeneity and arrested differentiation in neuroblastoma biology and treatment. The differentiation arrest of neuroblastoma is relevant for treatment and outcome as shown with retinoic acid (RA) therapy. However, RA is not curative and rare clones of undifferentiated cells may continue to grow and disseminate, emphasizing the importance of further research in this area.
Neuroblastoma arises from neural crest–derived lineages
Neuroblastoma was first reported as a pediatric cancer in the mid-nineteenth century (Dalton, 1885). By 1910, the neuronal origin of the disease was established based on the presence of fibrils resembling those of sympathetic ganglionic neurons (Wright, 1910). The ability of neuroblastoma cells to differentiate into mature neurons was also recognized early (Bailey, 1926). By comparing the neuronal features of neuroblastomas at diagnosis and recurrence in the same patients, Cushing and Wolbach postulated that environmental factors influence the differentiation of tumor cells (Cushing and Wolbach, 1927). The direct connection between neuroblastoma and sympathetic ganglionic neuronal differentiation was then made by Potter and Parrish, who reported distinct stages of differentiation in a widely disseminated neuroblastoma from a stillborn infant (Potter and Parrish, 1942). These early observations are particularly impressive given our current understanding of neuroblastoma as a cancer of the developing sympathoadrenal lineage.
More than a century and a half of research on the molecular and cellular mechanisms of embryogenesis have provided a strong foundation for contextualizing the developmental origins of neuroblastoma (Bechmann et al., 2021). The sympathoadrenal lineage is derived from neural crest cells that emigrate from the dorsal neural tube and migrate to distant sites during the early stages of embryogenesis (Kerosuo and Bronner-Fraser, 2012). Neural crest cells are proliferating pluripotent progenitor cells whose repertoire of cell fates are determined by their location along the rostro-caudal axis of the developing embryo (Fig. 1A). For example, cranial neural crest gives rise to cartilage and bone whereas sacral neural crest gives rise to enteric neurons. While intrinsic programs may contribute to differentiation of neural crest derived lineages, cell fate specification is regulated primarily by non-cell autonomous cues (Graham, 2003). For example, trunk neural crest will produce mesenchymal cell types (e.g., bone, cartilage) when transplanted into the cranial region even though they do not normally produce those cell types during development (Dupin et al., 2018). Conversely, cranial neural crest cells that never make neurons can produce the entire sympathoadrenal lineage (sympathetic neurons, chromaffin cells, Schwann cells and melanocytes; Box 1) when transplanted into the trunk (Bronner-Fraser and Fraser, 1988). The specification of neural crest-derived neurons into cholinergic (produce acetylcholine) versus adrenergic (produce noradrenaline) can also be regulated by extrinsic cues (Le Douarin, 1975). As mentioned above, cells of the sympathoadrenal lineage are produced by trunk neural crest and the route of migration correlates with cell fate specification (Fig. 1B), further emphasizing the importance of extrinsic cues (Kulesa and Gammill, 2010). For example, the bone morphogenetic protein (BMP) pathway induces the specification of the sympathoadrenal lineage by upregulating the expression of SOX10, ASCL1, HAND2, GATA3, PHOX2B and other transcription factors (Huber, 2006). In addition to the dormant mesenchymal cell fate potential described above from transplantation studies, recent studies have shown that mesenchymal stem cells (MSCs) in the bone marrow niche are derived from trunk neural crest cells (Isern et al., 2014). Taken together, these data suggest that sympathoblasts (Box 1) from the trunk neural crest are bipotential and can generate both neuronal and mesenchymal cell types (Fig. 2).
Figure 1. Extrinsic cues regulate neural crest lineage specification.
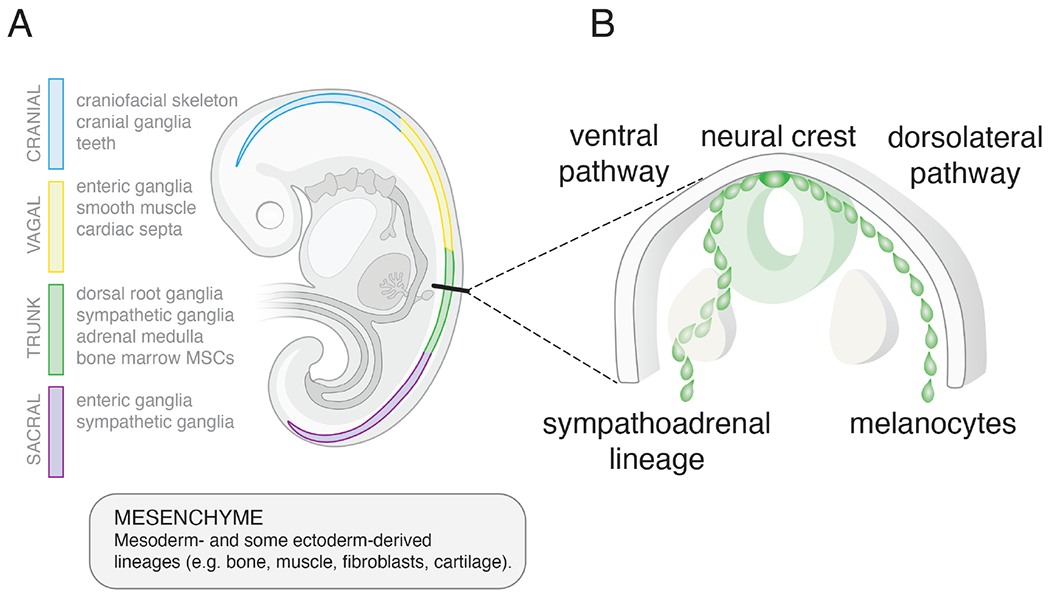
A) Drawing of a human embryo showing the neural crest (4-color). The cellular lineages derived from the neural crest vary based on their rostral-caudal location as shown (cranial (blue), vagal (yellow), trunk (green), sacral (purple)). Transplantation studies have shown that lineage specification is regulated primarily by the extrinsic cues rather than cell-autonomous factors. Trunk neural crest cells transplanted into the cranial region will produce cranial lineages and vice versa. Neural crest cells are multipotent and can give rise to both neuronal and non-neuronal (mesenchymal) cell types. This is particularly relevant for neuroblastomas that arise from the sympathoadrenal lineage of the trunk neural crest because they have both neuronal (N-type) and mesenchymal (S-type) cell populations. B) Drawing of a cross section of the trunk neural crest showing the two paths of migration. Both paths occur bilaterally but for simplicity the ventral pathway is shown on the left side of the embryo and the dorsolateral pathway is shown on the right. Extrinsic cues in these different regions of the embryo contribute to specification of the sympathoadrenal lineage and melanocytes. Thus, not only is the position along the rostral caudal axis important for neural crest derived cell lineage specification but the path of migration (ventral versus dorsolateral) is also important.
BOX 1.
ADR-type neuroblastoma (neuronal)
N-type neuroblastoma cells appear neuroblastic and can undergo partial neuronal differentiation in the presence of retinoic acid. In classic studies, ADR cells express neurofilaments and do not express vimentin. They have also been shown to express neurotransmitter biosynthetic enzymes. Van Groningen et al. showed ADR are PROM1 (CD133)-negative and had PHOX2A, PHOX2B, DBH, TH and 369 other differentially expressed genes relative to MES cells (van Groningen et al., 2017). Additional genes used to identify ADR cells include CHGA, DLK1 and 18 transcription factors including ASCL1, EYA1, GATA3, HAND1, SIX3. Boeva et al. called N-type cells GROUP I and showed that they express many of the same genes as those defined by Goningen et al. with addition of HAND2 (Boeva et al., 2017). Bedoya-Reina et al. used ISL1, PHOX2B, CHGB, CHGA, TH, CARTPT, NTRK1 for this cell population (Bedoya-Reina et al., 2021).
MES-type neuroblastoma (substrate adherent)
MES neuroblastoma cells appear flat and adhere to substrate in culture. They do not express neurofilament and they express vimentin and produce collagen and fibronectin. Alpha smooth muscle actin was found only in MES-cells (Sugimoto et al., 1991). Groningen et al. showed that MES cells expressed PROM1 (CD133) along with VIM, FN1, SNAI2 and 485 other differentially expressed genes relative to N-type cells (van Groningen et al., 2017). Additional genes include WNT5A, IGFBP2, IL13RAI and 20 transcription factors including MEOX1, MEOX2, SIX1, SIX4, SOX9, SMAD3, WWTR1. The Boeva et al. found that they also express FOSL1, FOSL2, JUN (Boeva et al., 2017). Bedoya-Reina et al. used PRRX1, COL12A1, COL6A3, COL12A1, PDGFRB, PDGFRA and YAP1 (Bedoya-Reina et al., 2021). In the Kameneva et al. analysis of normal cells, VIM is most highly expressed in SCPs, subepicardial mesenchyme and melanocytes (Kameneva et al., 2021). This is important because in the historical literature, these MES-cells have been attributed to smooth muscle, Schwann cells and melanocytes.
Schwann Cell Precursor (SCP)
SCPs are derived from the developing neural crest and sympathoblasts and migrate along peripheral nerves during development to give rise to glial cells. More recent studies suggest that SCPs may represent a multipotent progenitor cell population that can respond to stress, injury, or disease in a variety of organ sites. There is now evidence that SCPs can produce chromaffin cells of the adrenal medulla and possibly adrenergic post-ganglionic sympathetic neurons. Kildisiute et al. used SOX10, ERBB3, MPZ, and PLP1 as SCP markers (Kildisiute et al., 2021a). Bedoya-Reina et al. also used S100B, FABP7, FOXD3 (Bedoya-Reina et al., 2021). Jansky et al. used many of the same markers and included CDH19 (Jansky et al., 2021). In Kameneva et al., they include EDNRB and SPARC (Kameneva et al., 2021).
Chromaffin cells
Chromaffin cells are found in the medulla of the adrenal gland and secrete epinephrine and norepinephrine. They are essentially adrenergic post-ganglionic neurons that failed to undergo morphological differentiation and form neurites, axons, and dendrites. Bedoya-Reina et al. reported chromaffin cells have CHGA, CHGB, TH, PHOS2A, PNMT, CARTPT, STMN2 in mature postnatal adrenal glands (Bedoya-Reina et al., 2021). Jansky also used DDC for this cell population and PNMT as a later marker (Jansky et al., 2021). Kameneva added PENK, GNAS, ADM and DLK1 (Kameneva et al., 2021).
Sympathoblasts
Sympathoblasts are proliferating cells derived from the neural crest that can produce postganglionic sympathetic neurons, chromaffin cells, SCPs, and mesenchymal stem cells of the bone marrow. Jansky et al. used NEFM, GAP43, STMN2, ISL1, ALK at early stages to identify sympathoblasts (Jansky et al., 2021). SYN3 and IL7 were used for later stage sympathoblasts. Kameneva et al. also added PRPH, ELAVL3, ELAVL4, PHOX2B, HAND2 (Kameneva et al., 2021).
Figure 2. Sympathoblast cell fate specification.
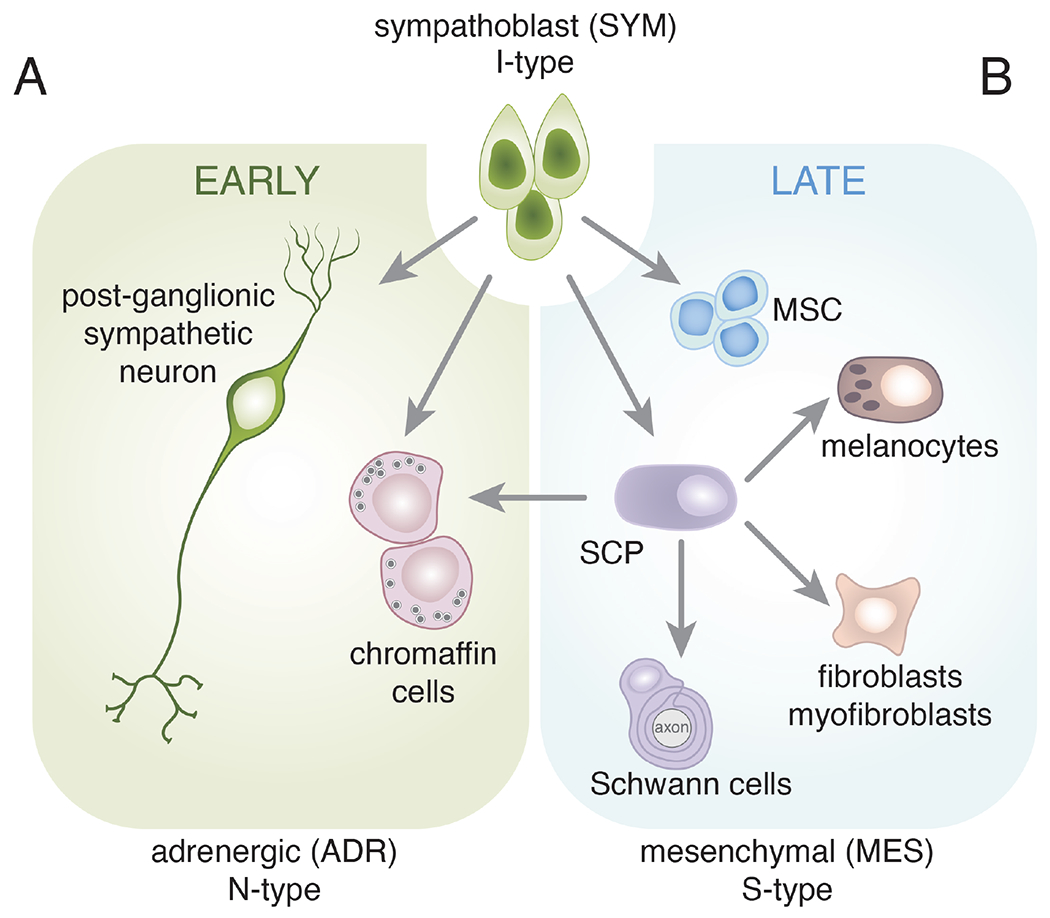
Schematic drawing of major cell fate specification events in the sympathoadrenal lineage and their derivatives. This is not intended to be comprehensive but rather to highlight the multipotency of the sympathoblasts. The neural crest is a transient population and after cells have migrated out of the trunk neural crest along the ventral pathway, a subset of cells commits to become sympathoblasts (I-type). Shortly after the neural crest cells have dispersed and the sympathoblasts have been specified, they form two cell types with neuronal (N-type) features. The post-ganglionic sympathetic neurons may be cholinergic or adrenergic. The chromaffin cells lack traditional dendrites and axons but synthesize catecholamines (epinephrine and norepinephrine) and release those neurotransmitters into the circulation from the adrenal medulla. These are some of the first developmental events of the sympathoadrenal lineage and help to establish the patterning of the sympathetic ganglia and developing adrenal gland. B) Later during development, the sympathoblasts produce Schwann cell precursors (SCPs) and mesenchymal stem cells (MSCs). The SCPs can produce mature Schwann cells that associate with the axons of the sympathetic neurons. They can also produce mesenchymal cell types such as fibroblasts and myofibroblasts. The MSCs migrate to the bone marrow where they contribute to the hematopoietic stem cell niche. Importantly, SCPs have been shown to be the major source of chromaffin cells in the late stages of adrenal development. This resembles the interconversion of N-type and S-type neuroblastoma cells. Therefore, while individual cell types (e.g., SCPs) are multipotent, the sympathoblasts are the most immature cell population in this lineage and may resemble the I-type neuroblastoma cells. This is not intended to be comprehensive, and each arrow may include several distinct stages of cell fate specification and differentiation. For example, bridge cells are thought to be an intermediate state from SCP to chromaffin cells during the later stages of adrenal medulla development.
The migration of the neural crest is completed by E10.5 in mice and post conception day (PCD) 40 in humans (https://www.translatingtime.org/). In the trunk neural crest, there are two overlapping waves of neural crest migration and the sympathoadrenal lineage originates during the first wave (E8.5-E9.5 in mice and PCD 27-30 in humans)(Serbedzija et al., 1990). This is important, because most studies on the specification and differentiation of the sympathoadrenal lineage have focused on the stages of development after sympathoblasts migrate out of the neural crest (Fig. 2). For example, the study by Furlan et al., which showed that Schwann cell precursors (SCPs, Box 1) give rise to chromaffin cells, was performed at E11.5-E15.5 in mice and an analogous study by Jansky et al. was performed on human fetal adrenal from PCD 50-120 (Furlan et al., 2017; Jansky et al., 2021). While it is tempting to speculate that cellular heterogeneity within neuroblastomas reflects the pluripotency of trunk neural crest cells it is more likely to reflect the that of the post migratory cells of the sympathoadrenal lineage.
The autonomic nervous system consists of central nervous system (CNS)-derived cholinergic preganglionic neurons and neural crest derive adrenergic or cholinergic postganglionic neurons (Fig. 3A). The chromaffin cells of the adrenal medulla are also neural crest derived adrenergic cells, but they do not extend dendrites or axons like other autonomic neurons. Rather, they directly secrete neurotransmitters (adrenaline and noradrenaline) into the circulation (Fig. 3B). Chromaffin cells are stimulated by acute exposure to acetylcholine or chronic exposure to pituitary adenylate-cyclase-activating polypeptide PACAP encoded by the ADCYAP1 gene. While many neuroblastomas express high levels of catecholamine biosynthetic enzymes, this is not universally true and a subset of neuroblastomas express PACAP neuropeptide and acetylcholine biosynthetic enzymes (Fig. 3B). As such, it remains an open question whether this variability in neurotransmitter secretion within neuroblastomas is reflective of a sympathoadrenal or chromaffin origin.
Figure 3. Expression of sympathoadrenal neurotransmitters and neuropeptides in neuroblastoma.
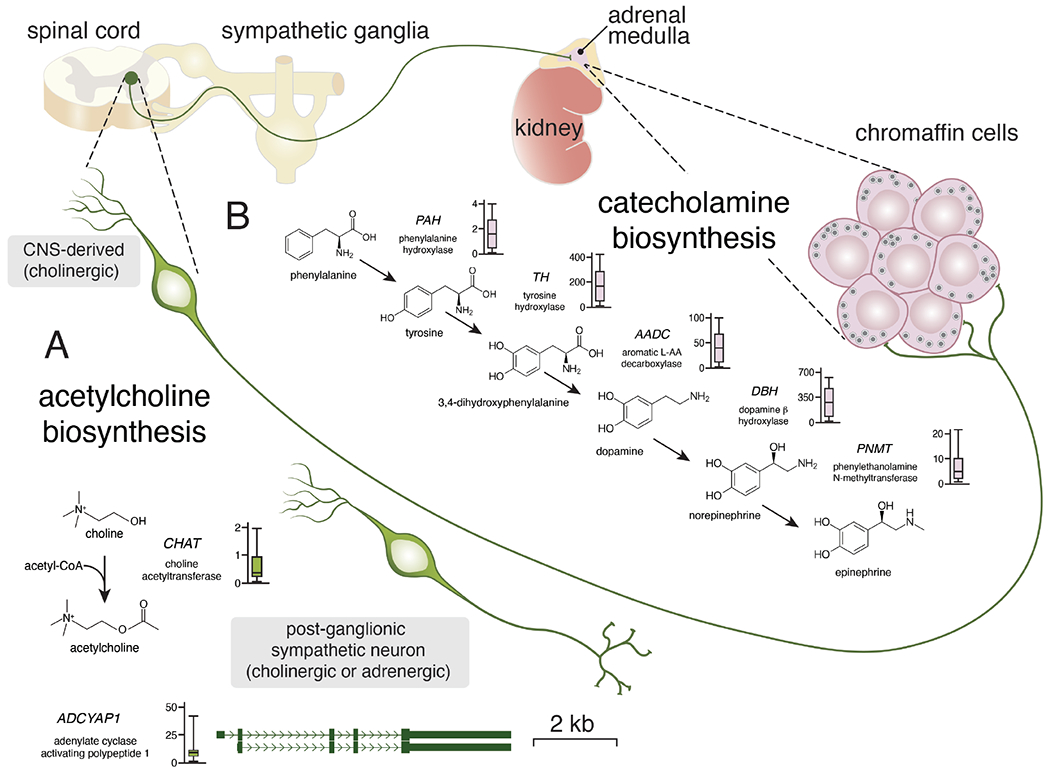
Simplified drawing of the neuronal connections in the sympathoadrenal lineage. Central nervous system (CNS) derived pre-ganglionic neurons synapse directly with chromaffin cells of the adrenal medulla where they release acetylcholine. Once stimulated, the chromaffin cells release epinephrine or norepinephrine into the circulation. Some chromaffin cells release epinephrine and some release norepinephrine from their dense core vesicles. A) Acetylcholine biosynthesis is regulated by choline acetyltransferase and the expression is relatively low (box plot) in the 191 representative neuroblastoma tumors. Acetylcholine is used for acute stimulation of the chromaffin cells. The neuropeptide encoded by ADCYAP1 is used for more chronic stimulation. While expression of ADCYAP1 is low in most neuroblastomas, a subset has very high levels of expression suggesting inter-tumor heterogeneity.
B) Drawing of the biosynthetic pathway for catecholamines with relevant gene and abbreviation for each enzyme. Next to each gene name is a boxplot of RNA expression (FPKM) from 191 neuroblastomas tumors (www.stjude.org/pecan). The chromaffin cells of the adrenal medulla secrete catecholamines as do a subset of post-ganglionic sympathetic neurons that are derived from sympathoblasts.
In addition to these peripheral autonomic neurons, neural crest-derived sympathoblasts also produce SCPs (Fig. 2) (Bechmann et al., 2021; Furlan et al., 2017). SCPs are multipotent progenitor cells that can differentiate into Schwann cells, as well as melanocytes and mesenchymal cells (fibroblasts and myofibroblasts) (Fig. 2B) (Solovieva and Bronner, 2021). Thus, sympathoblasts have the potential to produce both neuronal and mesenchymal cell types during normal development, and recent studies suggest that SCPs can cross the mesenchymal-neuronal developmental boundary (Furlan et al., 2017). Genetic lineage tracing studies in mice have shown that the major source of chromaffin cells in the adrenal medulla is from SCPs, and that this process occurs late in development (E11.5-E14.5). Single cell RNA-seq analysis of human fetal adrenal at similar stages of development was consistent with that model (Jansky et al., 2021; Kildisiute et al., 2021a). Thus, there is an early source of chromaffin cells from the sympathoblast and a later source of chromaffin cells from SCPs (Bechmann et al., 2021; Furlan et al., 2017). For the remainder of this review, we will refer to the specification and differentiation of chromaffin cells and sympathetic neurons from sympathoblasts as “early” sympathoadrenal development (Fig. 2). The specification of SCPs from sympathoblasts and subsequent differentiation into chromaffin cells will be referred to as the “late” sympathoadrenal development (Fig. 2). Differentiation of chromaffin cells from SCPs is accompanied by progressive downregulation of SCP genes (SOX10, FOXD3) and upregulation of chromaffin cell genes (PHOX2B, ASCL1, TH) in a “bridge” cell state (Furlan et al., 2017). While the process of sympathoadrenal development started with epithelial to mesenchymal transition (EMT), it ends with a mesenchymal to epithelial transition (MET) as cells re-establish their polarity and cellular contacts (Kerosuo and Bronner-Fraser, 2012). This is particularly relevant for neuroblastoma, as EMT and MET associated with the normal development of the sympathoadrenal lineage may play a role in intra- and inter-tumor heterogeneity, metastasis, and prognosis. Collectively, decades of intensive study dissecting neural crest differentiation have combined multiple lines of investigation to discern the developmental hierarchy of neural crest differentiation. These findings have invigorated efforts to map and compare intratumoral heterogeneity within neuroblastoma to normal neural crest developmental states.
Neuronal and mesenchymal features of neuroblastoma
The first indication that neuroblastoma tumors exhibited cellular heterogeneity reflecting sympathoadrenal developmental heterogeneity came from studies in neuroblastoma cell lines. In the early 1980s, methods were developed to grow patient tumor samples as primary cultures and to generate immortalized cell lines (Thiele, 1998). Even with the limited availability of molecular probes at that time, it was quickly established that there were at least 2 types of cellular phenotypes in neuroblastoma cultures. The neuronal (N-type) neuroblastoma cells have features of neurons of the sympathoadrenal lineage ranging from expression of neuron specific neurofilaments to morphological differentiation and secretion of neurotransmitters (Fig. 2,3) (Thiele, 1998). The substrate adhesive (S-type) neuroblastoma cells were originally defined based on their flat, adherent cellular morphology as they spread out over the substrate in culture. These cells were later found to express genes and proteins (e.g., vimentin) found in non-neuronal (mesenchymal) neural crest derivatives (Thiele, 1998).
Over subsequent decades of research on neuroblastoma cell lines, it has been reported that S-type cells have molecular features of Schwann cells, melanocytes, ectomesenchymal derivatives and smooth muscle (Fig. 2) (Ciccarone et al., 1989; Rettig et al., 1987; Sadée et al., 1987; Sugimoto et al., 1991). It was not known if those different cellular phenotypes reflected inter- or intra-tumor developmental heterogeneity, or if it was an artifact of changes in the extrinsic cues in culture relative to the tumor microenvironment in patients. While there was evidence for S-type cells in patient tumors, it has been difficult to definitively demonstrate that S-type cells within patient tumors were malignant. Specifically, NB tumors often have non-malignant Schwann cells and mesenchymal cells in the tumor microenvironment and discriminating between cells of the tumor microenvironment from malignant cells with mesenchymal phenotypes remains a challenge.
Advances in molecular biology techniques have enabled more precise exploration of neuroblastoma heterogeneity. Boeva et al. identified super-enhancers (SEs) with neuroblastoma cell lines using H3K27Ac ChIP-seq data (Boeva et al., 2017). This in-silico analysis identified two major core regulatory circuits (CRCs) and provided a transcriptional signature of the neuronal and mesenchymal neuroblastoma cells in patient tumors. MYCN amplified tumors had stronger neuronal signatures, but there was no absolute correlation between any known genetic or clinical feature to the cell state-specific gene expression signatures within patient tumors (Boeva et al., 2017).
In another study, van Groningen et al. analyzed super-enhancers in primary cultures of 3 different isogenic patient neuroblastoma tumors that had gene expression signatures of both neuronal- and mesenchymal- cell types which they referred to as adrenergic (ADR) and mesenchymal (MES), respectively (van Groningen et al., 2017). Using CD133 cell surface expression as a marker for MES-type cells, they separated ADR-type and MES-type cells from mixed primary patient samples. Purified MES-type and ADR-type cells were able to interconvert between cell states, and the authors were able to classify ADR and MES neuroblastoma cells using 369 gene and 485 gene signatures respectively. As shown in Boeva et al., patient tumors had mixed gene expression signatures and the MES-type cells were more resistant to chemotherapy.
Taken together, decades of research on neuroblastoma cell lines have shown that there are at least two types of neuroblastoma cells. There are cells with neuronal features referred to as N-type, group I, or ADR-type; hereafter, we will refer to these cells as ADR cells (Box 1). The other major cell type is non-neuronal and has been referred to as S-type, group II, or MES-type. Their precise cellular identity has remained elusive, but they share similarities with a variety of non-neuronal neural crest derivatives including Schwann cells, melanocytes, smooth muscle and ectomesenchyme cells. This cell type will be referred to as MES cells for the remainder of this review (Box 1). Individual neuroblastoma cells may be biased toward the ADR- or MEScellular phenotypes but they can interconvert in culture and in vivo (Ross et al., 1983; van Groningen et al., 2017). The interconversion may reflect the formation of chromaffin cells or post-ganglionic sympathetic neurons from SCPs late in sympathoadrenal development or it may reflect the bipotentiality of the sympathoblast to produce both cell types (Fig. 1C). There are also reports of an intermediate-type cell that may be a precursor to both mesenchymal and neuronal cell fates during normal development (Fig. 2) (Thiele, 1998).
Mapping neuroblastoma cellular heterogeneity to sympathoadrenal development
Despite these important advances in our understanding of cellular heterogeneity in neuroblastoma, it is possible that there are additional neuroblastoma cell populations yet to be identified or that ADR and MES cells can be further subdivided. While bulk RNA analyses of patient tumors suggest that most neuroblastomas have a mixture of cell populations, it is not clear how much of that apparent heterogeneity is from the non-malignant tumor microenvironment. Similarly, we cannot distinguish between separate cell populations with ADR and MES gene expression signatures and a single cell population with a hybrid ADR/MES gene expression signature. That is, while neuronal (ADR) and non-neuronal (MES) cell fate specification programs are mutually exclusive during normal development, that may not be the case in tumors. Finally, it is not known if all the cell populations are intermixed or if there are geometrical partitions within the tumor and how these developmental aspects of intra- and intertumor cellular heterogeneity relates to patient outcome.
To begin to answer some of these questions, multiple groups have carried out single-cell gene expression profiling of neuroblastomas and the developing sympathoadrenal lineages in embryos (Bedoya-Reina et al., 2021; Dong et al., 2020; Jansky et al., 2021; Kameneva et al., 2021; Kildisiute et al., 2021a). To date, 64 tumors have been profiled along with human embryos, fetal adrenal and postnatal adrenal. In the first study by Dong et al., researchers performed single cell RNA-seq (scRNA-seq) on human embryos (4 weeks post-conception), fetal adrenal glands (8-14 weeks post-conception) and neuroblastoma tumors located near the adrenal gland. In the fetal adrenal gland, they manually annotated 3 cell types as SCPs, sympathoblasts and chromaffin cells. The researchers then analyzed 14 neuroblastomas and 2 ganglioneuroblastomas and concluded that most of the tumor cells resembled developing chromaffin cells with fewer sympathoblasts (Dong et al., 2020). However, independent reanalysis of the data showed that the authors had mistakenly switched chromaffin and sympathoblast assignments because of their manual annotation of cell types and cross species differences (Bedoya-Reina and Schlisio, 2021; Kildisiute et al., 2021b; Yang et al., 2021). Specifically, they used CARTPT as a marker of sympathoblasts based on data from murine adrenal development. While Cartpt is expressed in murine sympathoblasts, CARTPT is expressed in chromaffin cells in humans (Kameneva et al., 2021). This experience reinforces the importance of using well-validated and robust species-specific gene signatures to annotate cell types before applying them to tumor datasets.
In a subsequent study comparing scRNA-seq of neuroblastoma to human fetal adrenal medullae, the Behjati lab used multiple unbiased approaches to assign cell-type identity to clusters (Kildisiute et al., 2021a). In human fetal adrenal medullae, the authors identified SCPs, bridge cells, chromaffin cells and sympathoblasts. Tumors were then analyzed using either the 10x Genomics (5 tumors) or CEL-Seq2 (16 tumors) platform. Tumor cells (3,396 cells), identified using inferred copy number analysis, were found to most closely resembled ADR cells. Analysis of bulk RNA-seq from 650 tumors in the TARGET dataset confirmed the presence of their ADR gene expression signature in all tumors and suggested that patients with less adrenergic neuronal differentiation had worse outcomes.
A third study published by the Schlisio lab analyzed 11 neuroblastomas (3,212 cells) using SMART-seq2 (Bedoya-Reina et al., 2021). In their neuroblastoma cohort, they identified two distinct groups of tumor cells. Cells in the first were undifferentiated, expressed PDGFRA, and resembled MES neuroblastoma cell lines; the second group resembled ADR neuroblastoma cell lines. In agreement with prior studies, high-risk neuroblastomas had less of a neuronal differentiation cell signature. Though the authors found cells resembling MES cell lines, they concluded that there were no tumor cells mimicking fetal SCPs. This highlights the challenge in the field regarding the precise identity of the MES cells.
The most recent study from the Westermann lab performed single-cell transcriptomic analysis of normal human fetal adrenal identified SCPs, chromaffin cells, a bridge population, and sympathoblasts (Jansky et al., 2021). The most proliferative cells were either SCPs or sympathoblasts. All 14 neuroblastomas analyzed in this study had malignant cells with the ADR signature and 3 had a subpopulation of cells with the MES signature. The authors proposed that the MES neuroblastoma tumor cells most closely resembled bridge cells (Jansky et al., 2021).
There are several themes that have emerged from these independent scRNA-seq studies of neuroblastoma and developing sympathoadrenal lineage. First, ADR cells were identified in every patient tumor and there is agreement that tumor cells have features of neuronal differentiation. Second, there is general agreement that patients with more differentiated ADR gene expression signatures have better prognoses. Some studies identified tumor cells with the MES signature, but the precise cell-type identity (SCPs, Schwann cells, melanocytes, smooth muscle, MSCs, fibroblasts, myofibroblasts) remains elusive. Third, all studies used inferred copy number analysis to identify tumor cells; this method is imprecise, and it is possible that the presence or absence of MES cells reflect differences in the thresholds used for copy number inference, differences in data quality or variation in computational methods to annotate tumor cells from the microenvironment. Fourth, cell-type assignments can also by imprecise based on whether clusters are manually annotated using a small number of genes or if cell type identities were assigned using unbiased methods. For example, hybrid gene expression signatures of multiple cell fates in a single cell type were not evaluated. Finally, there may be differences in sympathoadrenal development between mice and humans and this must be considered when assigning cell types within scRNA-seq data.
Despite these important advances in our understanding of the cellular heterogeneity of neuroblastoma and the developing sympathoadrenal lineage, there are several challenges to consider when mapping neuroblastoma gene expression signatures to the sympathoadrenal lineage. The first challenge is the role of non-cell-autonomous cues in mesenchymal cell fate specification along the rostral caudal axis of the developing neural tube. We know from cell transplantation studies that trunk neural crest cells that normally give rise to the sympathoadrenal lineage have the dormant potential to produce mesenchymal cell fates if exposed to different extrinsic cues. Moreover, they also produce mesenchymal stem cells of the bone marrow during normal development (Isern et al., 2014). Furthermore, the discovery that SCPs can produce both chromaffin cells and mesenchymal cells further complicates our ability to interpret the heterogeneity within neuroblastoma tumors in relation to developmental heterogeneity. Future studies should focus on discerning the role of extrinsic signals and intrinsic programs in the ADR and MES cellular heterogeneity in neuroblastomas and refining the gene expression signatures of each normal cell population.
Another challenge is the interpretation of the directionality of the developmental transitions as it relates to neuroblastoma interconversion. During normal development, the sympathoblast and SCPs can give rise to both neuronal and mesenchymal fates (Fig. 2). Therefore, it is not known if the interconversion between ADR and MES neuroblastoma cell states reflects that normal developmental potential, or a consequence of different extrinsic cues. In either case, we cannot infer cellular origin from the observation of ADR and MES neuroblastoma cell interconversion. As the study by Wang et al. showed, definitive identification of cell types is challenging for the sympathoadrenal lineage, and this is even more complicated for neuroblastoma tumors (Wang et al., 2019). While there are two major types of tumor cell lines (ADR and MES), and those cell populations are present in patients’ tumors at different ratios, identifying their correlates in normal development is not straightforward. The ADR cells have many similarities with adrenal chromaffin cells in that they produce catecholamines. However, they form neuronal processes in culture even though chromaffin cells in the adrenal medulla lack any neuronal processes. The post-ganglionic sympathetic neurons with dendrites and axons can be cholinergic or adrenergic and there is evidence for acetylcholine production in a subset of neuroblastomas. The MES cells have features of SCPs and their non-neuronal (mesenchymal) derivatives (Furlan et al., 2017), but unambiguous cell-type assignments remain elusive. This may reflect the dormant mesenchymal potential of trunk neural crest, the ability of sympathoblasts to produce MSCs or presence of a hybrid cell state that doesn’t normally exist during development. It is possible that our expectation for tumor cells to accurately recapitulate the normal gene expression programs is misguided. We know in other tumors with neuronal features that the cells can express hybrid signatures that never exist normally during development due to deregulation of the normal process (Norrie et al., 2021). Rather than trying to precisely align developmental and cell heterogeneity, it may be more informative to study the lineage relationships in tumors with molecular barcoding or other methods. This could help us gain a deeper understanding of the clonal dynamics in tumors and how developmental heterogeneity contributes to clonal evolution and selection during treatment. Indeed, emerging evidence suggests that MES cells preferentially survive treatment and contribute to neuroblastoma recurrence (van Groningen et al., 2017).
Mutational spectrum of neuroblastoma
The landscape of genomic mutations provides further evidence for neuroblastoma as a disease of arrested differentiation (Brady et al., 2020). One of the best examples is the homeobox transcription factor, PHOX2B. Sympathoadrenal development requires PHOX2B to activate transcription factors (HAND2, GATA2/3, ASCL1), which are required for cell fate specification within the sympathoadrenal lineage (Tomolonis et al., 2018). For example, ASCL1 activates PHOX2A which in turn activates genes required for catecholamine biosynthesis (Tomolonis et al., 2018). Due to this prominent role in sympathoadrenal development, it is not surprising that reduced expression of PHOX2B in familial neuroblastoma can promote malignant transformation (Raabe et al., 2008).
Another example is the ALK receptor tyrosine kinase, which is the most common familial neuroblastoma predisposition gene and is somatically mutated in sporadic cases (Mosse et al., 2008). Mutations in ALK are oncogenic by promoting ligand-independent activation of the kinase (Bresler et al., 2014). In mouse embryos, Alk is expressed in the developing neural crest (Gonzalez Malagon et al., 2018) and Alk-deficient mice have defects in neuronal differentiation (Weiss et al., 2012). While the precise molecular and cellular function of ALK in sympathoadrenal development has not yet been elucidated, it is possible that ALK plays a role in regulating the proliferation of sympathoblasts and deregulation of ALK signaling through oncogenic mutations leads to hyperplasia and malignant transformation.
Oncogenic pathways and developmental pathways are often integrated because proliferation and differentiation must be precisely coordinated during development. Indeed, some genetic lesions in neuroblastoma play a dual role by blocking differentiating and promoting tumorigenesis. For example, MYCN amplification leads to persistent high levels of expression and is a strong indicator of poor prognosis in neuroblastoma patients (Cheung and Dyer, 2013). During normal development, transient low-level expression of MYCN is important for promoting neural crest cell migration and neuronal cell fate specification (Wakamatsu et al., 1997). However, when MYCN is persistently expressed at high levels in the sympathoadrenal lineage, this leads to sympathoblast hyperplasia and neuroblastoma formation (Cohen et al., 2020; Weiss et al., 1997). Thus, MYCN amplification blocks differentiation and promotes deregulated proliferation which can ultimately leads to tumor formation in patients.
Disruption of the normal epigenetic mechanisms during neural crest development may result in differentiation arrest and neuroblastoma formation. Somatic mutations in chromatin remodelers, including CHD5 (Koyama et al., 2012), ARID1A/ARID1B (Sausen et al., 2013), BRG1 (Jubierre et al., 2016), and ATRX (Cheung et al., 2012) have been found in neuroblastoma. As for transcription factors like PHOX2B and MYCN, a major question in the field is whether these mutations block sympathoadrenal cell differentiation, promote tumorigenesis or both.
Among the recurrent somatic mutations in chromatin remodelers, ATRX mutations are the most common and are associated with high-risk disease, older age at diagnosis, and poor prognosis (Cheung et al., 2012; Zeineldin et al., 2020). ATRX is crucial for the development of the nervous system, and germline mutations of ATRX cause developmental defects and neuronal cell death (Berube et al., 2005). Unlike PHOX2B, germline ATRX mutations do not increase neuroblastoma susceptibility, suggesting that additional mutations are required for tumorigenesis (Cheung and Dyer, 2013).
ATRX is involved in the deposition of histone H3.3 at repetitive regions across the genome, including telomeres (Wong et al., 2010). Altered H3.3 deposition at DNA repeats can cause accumulation of secondary DNA structures, particularly at G-rich regions (G-quadruplex structures) that can interfere with DNA replication and transcription (Law et al., 2010). As a result of defects in H3.3 deposition at telomeres, ATRX mutant neuroblastomas undergo homologous recombination and have the alternative lengthening of telomeres (ALT) phenotype (Clynes et al., 2015). This allows ATRX mutant neuroblastomas to maintain long telomeres and prevent replication-induced telomere attrition and crisis (Cheung et al., 2012). The defects in H3.3 deposition also disrupt expression of genes important for neuronal differentiation including the retinoic acid responsive genes (Zeineldin et al., 2020). Together, these findings suggest that ATRX mutations promote tumorigenesis through the ALT telomere maintenance program and block neuronal differentiation. Interestingly, ATRX mutations are mutually exclusive with MYCN amplification (Zeineldin et al., 2020). This is unusual because in most tumors, potent oncogenic lesions (e.g., MYCN) and tumor suppressor mutations (e.g., ATRX) cooperate in the aggressive forms of disease. Our work showed that MYCN amplification and ATRX mutations are incompatible in neuroblastoma because each lead to DNA replicative stress and the combination is synthetic lethal (Zeineldin et al., 2020). Ongoing efforts are focused on exploiting this synthetic lethality with drug combinations that induce DNA replicative stress to improve outcomes for patients.
Spontaneous regression of neuroblastoma
Despite an overall poor outcome, neuroblastoma has one of the highest rates of spontaneous regression without therapeutic intervention (Kawano et al., 2021; Matthay et al., 2016). Most cases of spontaneous regression are seen in patients younger than 18 months at diagnosis. Brodeur has proposed candidate mechanisms for the spontaneous regression including loss of paracrine signaling, failure to maintain telomeres, immune rejection, and epigenetic activation of the sympathoadrenal-differentiation program (Brodeur, 2018). Signaling through the neurotrophin receptors (NTRK1,2,3) and their ligands (NGF, BDNF, NTF3) is important for neural crest and sympathoadrenal development and in neuroblastoma cells (Anderson, 1993). For example, neuroblastoma cells with high NTRK1 expression can undergo apoptosis in the absence of its ligand (NGF) and when NTRK1 is stimulated by its ligand, they differentiate (Nakagawara et al., 1993). Thus, a 4S neuroblastoma tumor with high NTRK1 in an infant may grow for a period when NGF is present but spontaneously regress when the ligand dissipates as part of normal development. In contrast, BDNF signaling through NTRK2 in neuroblastoma is often found in MYCN amplified neuroblastomas with poor prognosis (Nakagawara et al., 1994). Thus, developmental signaling through the NTRK family of receptors is perturbed in neuroblastoma and may contribute to the heterogeneity of patient response based on age of diagnosis.
The rate of spontaneous regression of neuroblastoma is also thought to be higher than that reported for clinical cases, because some tumors probably regress without ever being diagnosed. Large-scale screening of young children for elevated catecholamine levels in urine led to a higher rate of neuroblastoma diagnoses but no improvement in overall survival, because most of those additional diagnoses spontaneously regress without intervention (Yamamoto et al., 1998). In addition, hyperplastic foci that histologically resemble neuroblastoma have been identified at autopsy in 1 of every 40 infants younger than 3 months who died of non-neoplastic causes (Beckwith and Perrin, 1963). The authors called these foci “in situ neuroblastoma” and speculated that they are like neuroblastoma that undergoes spontaneous regression. They reasoned, that as the incidence of in situ neuroblastoma is 200 times greater than that of malignant neuroblastoma, most neuroblastomas undergo spontaneous regression. Together, these data suggest that in situ neuroblastoma and rests represent a form of normal hyperplasia involving progenitors of sympathoadrenal lineage during development. When those hyperplastic cells sustain an oncogenic mutation, they may progress to neuroblastoma.
It is possible that the switch between “early” and “late” modes of sympathoadrenal development may play a role in the spontaneous regression of 4S NB in infants (Kastriti et al., 2020). That is, early in development when SCPs do not contribute to the sympathoadrenal lineage, the rests may lack SCPs or SCP-derived cells and then spontaneously regress. However, at later stages of development when SCPs contribute to sympathoadrenal development, the SCPs may interfere with this spontaneous regression. Thus, further studies on the signaling between SCPs and their derivatives and the different populations of tumor cells, immune cells and the remainder of the tumor microenvironment are needed to determine if any aspect of the “early” and “late” mechanisms of development contribute to spontaneous regression of 4S NB in infants.
Inducing neuronal differentiation as a therapeutic approach in neuroblastoma
To date, the most effective agent for inducing neuroblastoma differentiation in culture and in vivo is retinoic acid (RA). Sidel treated a neuroblastoma cell line with RA and showed an irreversible reduction in proliferation and an increase in neurite formation across several neuroblastoma cell lines (Seeger et al., 1982). The RA derivative, 13-cis RA, moved quickly into clinical testing for neuroblastoma and is now part of the standard of care (Matthay et al., 1999). The rationale is that 13-cis RA induces differentiation of residual tumor cells after patients receive chemotherapy, surgical resection, and bone marrow transplantation.
Another strategy for exploiting the differentiated features of neuroblastoma cells for therapeutic benefit incorporates immunotherapy. Two groups identified the GD2 ganglioside on the surface of neuroblastomas and other tumors of neuroectodermal origin (Cahan et al., 1982; Schulz et al., 1984). Research on myelin sheath composition in relation to multiple sclerosis showed that GD2 is a minor proportion (approximately 2%) of the gangliosides expressed on cells in the brain and spinal cord but makes up a higher proportion (5%-8%) of those expressed on peripheral nerves (Svennerholm et al., 1994). Therefore, researchers incorporated anti-GD2 immunotherapy in the treatment of neuroblastoma and demonstrated efficacy in preclinical models and patients (Nguyen et al., 2018; Yu et al., 2010). Anti-GD2 immunotherapy is now standard of care for patients with high-risk neuroblastoma, and efforts are ongoing to reduce the side effects and improve efficacy of anti-GD2 immunotherapy (Furman et al., 2019; Furman et al., 2022).
The ability of neuroblastoma cells to reuptake catecholamines and catecholamine metabolites also has been exploited clinically for neuroblastoma diagnosis and treatment. Metaiodobenzylguanidine (MIBG) is a catecholamine analog that is taken up by catecholaminergic cells. There are two forms of MIBG based on the half-life of their iodine radiolabel: 131I-MIBG has a half-life of 8 days and is being tested for treatment, and 123I-MIBG has a half-life of 13 hours and is used for diagnostic imaging (Matthay et al., 2007). MIBG was first used to treat pheochromocytoma, another tumor that arises from adrenal medullary cells (Wieland et al., 1980). Currently, MIBG is used for recurrent neuroblastoma and is being tested in newly diagnosed patients with high-risk disease.
While RA, anti-GD2 immunotherapy, and MIBG are three examples of basic developmental neurobiology research being integrated with cancer biology to improve outcomes, it is important to emphasize that current chemotherapeutic agents can eliminate neuroblastoma in a significant fraction of patients. However, there are limitations due to the cellular heterogeneity of neuroblastoma. Specifically, anti-GD2 immunotherapy and MIBG approaches are focused on targeting the tumor cell population(s) in neuroblastoma with the ADR phenotype while RA is used to induce differentiation in neuroblastoma precursors that underwent differentiation arrest. While data are still emerging regarding the lineage relationships between the MES and ADR cells, our current understanding is that the MES cells are not responsive to RA, do not display GD2 on their cell surface and are not likely to take up MIBG because they are not catecholaminergic. Thus, future efforts should focus on understanding the implications of neuroblastoma heterogeneity in responding to different therapeutic strategies.
Multiple groups have started to explore the biological basis underlying the differential response of ADR versus MES neuroblastoma cells to therapy. In one study, knocking down ASCL1, an ADR TF, disrupted the neuroblastoma CRC and made these cells more responsive to RA alone or in combination with JQ1, an epigenetic agent (Alleboina et al., 2021). Disrupting MES CRCs by targeting SNAI2 was also shown to induce RA-sensitivity in neuroblastoma cells (Vrenken et al., 2020). Zimmerman et al. treated two MYCN-amplified neuroblastoma cells lines with all-trans retinoic acid (ATRA), the active metabolite of RA, and assessed changes in CRCs (Zimmerman et al., 2021). ATRA reduced the expression level of several ADR CRC-associated TFs including MYCN, GATA3, PHOX2B and ASCL1. Moreover, ATRA induced the emergence of a novel CRC characterized by the binding of the transcription factors MEIS1, SOX4, and RARA to novel super-enhancers. The overall result was a mixed picture, with preservation of a fraction of the classical ADR CRC-associated TFs, suggesting that some ADR-associated CRCs are important in RA-inducing differentiation (retino-sympatho CRCs). This is intriguing because the absence of these retino-sympatho CRCs in MES neuroblastoma cells may explain the lack of MES neuroblastoma response to RA-inducing differentiation. This discrepancy in RA response between MES and ADR cells highlights the importance of understanding neuroblastoma transcriptional and cellular heterogeneity for better treatment of the disease.
The differential response of MES and ADR cells is not limited to RA. Recently, Westerhout et al. tested the sensitivity of two pairs of isogenic MES/ADR cell lines with activating ALK mutations to two ALK inhibitors (Westerhout et al., 2022). They found, that unlike ADR cells, MES cells with ALK mutations were resistant to ALK inhibition. The authors showed that ADR cells had an enrichment for H3K27Ac near the ALK locus with a possible physical interaction between the site of the peak of this chromatin mark and the ALK transcription start site, suggesting a functional super-enhancer. This super-enhancer was absent in MES cells with no detectable expression of mutant or wildtype ALK. Using previously published scRNA-seq data, the authors showed that ALK is expressed in committed sympathoblasts but not SCP cells in mouse developing adrenal medullae, supporting the model that MES cells represent more immature forms of cells relative to ADR cells. Taken together, these examples highlight the importance of studying the normal sympathoadrenal differentiation program in neuroblastoma and elucidating the lineage relationships before, during and after treatment.
Developmental heterogeneity and clonal evolution
Extensive genomic studies have demonstrated evidence of parallel clonal evolution of primary and metastatic sites and treatment resistant clones that contribute to clonal selection in refractory/recurrent neuroblastoma (Abbasi et al., 2017; Chicard et al., 2018). While those studies were useful in elucidating the clonal architecture of tumors before and after treatment, they provided little insight into the underlying phenotypes of the cells that survive treatment and contribute to clonal selection and disease recurrence (Fig. 4). However, when combined with sc/sn RNA-seq analyses or other methods, researchers can begin to understand the properties of the cells that survive treatment and how they may change during clonal selection and evolution (Patel et al., 2022). This is particularly relevant for neuroblastoma because of the observation that ADR and MES cells can interconvert and have differential sensitivity to chemotherapy (Fig. 4A,B). For example, it is possible that MES cells represent a minor population in a patient’s tumor at diagnosis because they are dividing slowly or are quiescent. During treatment, the more rapidly dividing ADR cells may be eliminated, and a subset of the MES cells survive. This would be consistent with the clonal selection from genomic studies (Fig. 4C,D). Those residual MES cells may then become re-activated and interconvert to ADR cells to re-establish the cellular heterogeneity of the original tumor. Indeed, it was shown that clonal expansion of neuroblastoma cells in vitro usually results in a mixed population of cells with both adrenergic and mesenchymal phenotypes (Ross et al., 1983). Moreover, patient-derived neuroblastoma cells sorted into ALDH1A3-positive (MES), or ALDH1A3-negative (ADR) cells grew in mouse adrenals to form heterogenous population that contains both ALDH1A3 positive and negative cells (van Groningen et al., 2021). Recently, NOTCH signaling was shown to switch the transcriptional profile of ADR cells to that of MES cells emphasizing the complex intersection of cell intrinsic and extrinsic signaling (van Groningen et al., 2019). While this is just one possible model (Fig. 4B–D), it highlights the complexity of developmental heterogeneity and clonal selection in the context of treatment and recurrence (Fig. 4).
Figure 4. Neuroblastoma differentiation.
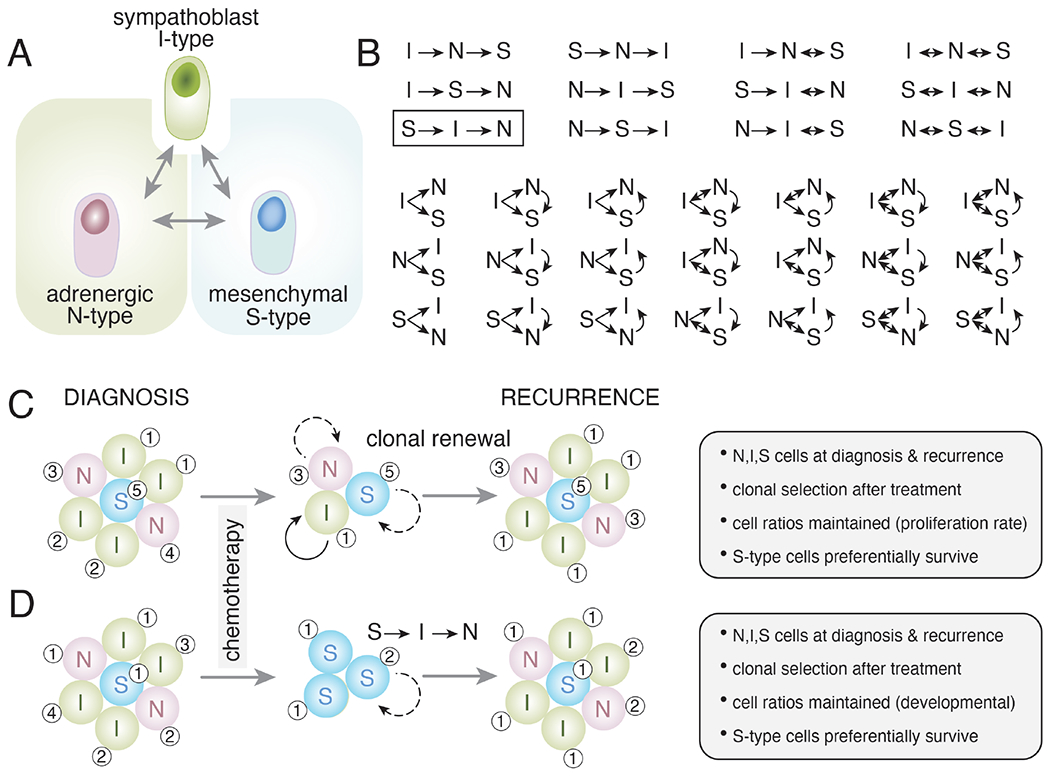
A) Drawing of one hypothetical simplified relationship between immature (I-type) neuronal (N-type) and mesenchymal (S-type) neuroblastoma cells. B) With just these 3 cell types, there are 34 possible relationships in terms of the ability of cells to produce other cell phenotypes and to interconvert. C,D) These 34 different possible relationships are relevant when considering clonal selection and clonal evolution after treatment with chemotherapy. Broad spectrum systemic chemotherapy primarily kills the proliferating cells. In both examples (C,D), the tumor composition is equal with primarily I-type (sympathoblast-like) cells and some with more differentiated neuronal features (N-type) and a small subset of mesenchymal cells (S-type). Chemotherapy eliminates most dividing cells, and it has been shown previously that S-type cells are more likely to survive treatment. This may be due to intrinsic drug resistance or a lower rate of proliferation. In the model in (C), each cell population is clonal and repopulates the same cell phenotype. Some cells may divide more rapidly (solid arrows) than others (dashed arrows). The numbers (1-5) indicate hypothetical genetic clonal relationships. In the model in (D), the S-type cells that survive treatment can produce cells with the other phenotypes (I-type, N-type) according to one of the 34 possible models (box in (B)). The S-type cells produce I-type cells, and they produce N-type cells thereby reconstituting the cellular heterogeneity of the original tumor. Importantly, the two models give very similar results (gray boxes to right), and the major difference is in the cellular mechanism. Specifically, in both models, N-type, I-type, and S-type cells are present at diagnosis and recurrence. Clonal selection occurs in both models as there are fewer clones in the recurrent tumor than the tumor at diagnosis. The approximate cell rations are maintained in both models either by their proliferation rate, developmental trajectory, or both. In both models, the S-type cells preferentially survive and therapeutic intervention that promotes the differentiation of I-type cells to N-type cells would slow recurrence but may not be curative. Therefore, these two models are consistent with our current understanding of neuroblastoma and the major difference is in the cellular mechanisms that lead to recurrence. These examples are illustrative only and future research should focus on detailed in vivo clonal analysis to discern which of the 34 possible combinations of lineage relationships are relevant in neuroblastoma.
There are now tools available to unambiguously label individual clones of cells with unique barcodes and profile their cellular identity at single cell resolution to gain a deeper understanding of developmental heterogeneity and clonal evolution during treatment (Patel et al., 2022). This type of research has the potential to transform our understanding of disease recurrence and develop biomarkers that could predict tumor behavior and/or therapeutic response. It can also help adopting better therapeutic combinations that eliminate all the clones (major and minor) during initial treatment to prevent disease recurrence in pediatric solid tumor. Indeed, this is particularly important for neuroblastoma, because patients with recurrent neuroblastoma have survival rates below 10%.
Conclusions and future directions
Neuroblastomas arise because of differentiation arrest of the developing sympathoadrenal lineage and includes adrenergic (ADR) and non-neuronal mesenchymal (MES) cell populations. Studies on neuroblastoma cell lines dating back more than 3 decades suggested that tumor cells are heterogeneous with cellular phenotypes that may resemble some aspects of normal developmental heterogeneity. Recent sc/sn-RNA-seq studies have further refined this developmental heterogeneity and provide a foundation for understanding their lineage relationships and their ability to interconvert. A detailed understanding of those relationships is essential to understand clonal evolution and selection with treatment, with the goal of preventing recurrence by effectively targeting residual tumor cells. The efficacy of targeting neuroblastoma with chemotherapeutic agents have improved by targeting the neuronal cell types (anti-GD2 and MIBG) and by inducing differentiation in precursor cells (RA). However, this regimen is not effective against the mesenchymal (MES) cells and these tumor cell population might contribute to disease recurrence so new agents to target the mesenchymal population are needed. This will help researchers design more effective treatment regimens that achieve the goal of total clonal elimination to achieve cure.
Acknowledgement
This work was supported by Cancer Center Support (CA21765) and grants to M.A.D. from the National Institutes of Health (EY014867, EY018599, and CA168875). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Research was also supported by American Lebanese Syrian Associated Charities. M.A.D. was also supported by the Howard Hughes Medical Institute, Alex’s Lemonade Stand, and Tully Family and Peterson Foundations. A.G.P. was supported by Alex’s Lemonade Stand, Hyundai Hope on Wheels, and the National Pediatric Cancer Foundations. A.G.P is a Damon Runyon-Sohn Pediatric Cancer Fellow supported by the Damon Runyon Cancer Research Foundation and The Sohn Conference Foundation (DRSG-33P-20).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbasi MR, Rifatbegovic F, Brunner C, Mann G, Ziegler A, Pötschger U, Crazzolara R, Ussowicz M, Benesch M, Ebetsberger-Dachs G, et al. (2017). Impact of Disseminated Neuroblastoma Cells on the Identification of the Relapse-Seeding Clone. Clin Cancer Res 23, 4224–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alleboina S, Aljouda N, Miller M, and Freeman KW (2021). Therapeutically targeting oncogenic CRCs facilitates induced differentiation of NB by RA and the BET bromodomain inhibitor. Mol Ther Oncolytics 23, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ (1993). Molecular control of cell fate in the neural crest: the sympathoadrenal lineage. Annu Rev Neurosci 16, 129–158. [DOI] [PubMed] [Google Scholar]
- Bailey P (1926). A classification of the tumors of the glioma group on a histogenetic basis with a correlated study of prognosis (Philadelphia: J.B. Lippincott Company; ). [Google Scholar]
- Bechmann N, Berger I, Bornstein SR, and Steenblock C (2021). Adrenal medulla development and medullary-cortical interactions. Mol Cell Endocrinol 528, 111258. [DOI] [PubMed] [Google Scholar]
- Beckwith JB, and Perrin EV (1963). IN SITU NEUROBLASTOMAS: A CONTRIBUTION TO THE NATURAL HISTORY OF NEURAL CREST TUMORS. Am J Pathol 43, 1089–1104. [PMC free article] [PubMed] [Google Scholar]
- Bedoya-Reina OC, Li W, Arceo M, Plescher M, Bullova P, Pui H, Kaucka M, Kharchenko P, Martinsson T, Holmberg J, et al. (2021). Single-nuclei transcriptomes from human adrenal gland reveal distinct cellular identities of low and high-risk neuroblastoma tumors. Nat Commun 12, 5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoya-Reina OC, and Schlisio S (2021). Chromaffin Cells with Sympathoblast Signature: Too Similar to Keep Apart? Cancer Cell 39, 134–135. [DOI] [PubMed] [Google Scholar]
- Benavente CA, and Dyer MA (2015). Genetics and epigenetics of human retinoblastoma. Annual review of pathology 10, 547–562. [DOI] [PubMed] [Google Scholar]
- Berube NG, Mangelsdorf M, Jagla M, Vanderluit J, Garrick D, Gibbons RJ, Higgs DR, Slack RS, and Picketts DJ (2005). The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. The Journal of clinical investigation 115, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugène C, Raynal V, Etchevers HC, Thomas S, Lermine A, Daudigeos-Dubus E, et al. (2017). Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nature genetics 49, 1408–1413. [DOI] [PubMed] [Google Scholar]
- Brady SW, Liu Y, Ma X, Gout AM, Hagiwara K, Zhou X, Wang J, Macias M, Chen X, Easton J, et al. (2020). Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat Commun 11, 5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, et al. (2014). ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26, 682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM (2018). Spontaneous regression of neuroblastoma. Cell Tissue Res 372, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM, Minturn JE, Ho R, Simpson AM, Iyer R, Varela CR, Light JE, Kolla V, and Evans AE (2009). Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res 15, 3244–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner-Fraser M, and Fraser SE (1988). Application of new technologies to studies of neural crest migration and differentiation. Am J Med Genet Suppl 4, 23–39. [DOI] [PubMed] [Google Scholar]
- Cahan LD, Irie RF, Singh R, Cassidenti A, and Paulson JC (1982). Identification of a human neuroectodermal tumor antigen (OFA-I-2) as ganglioside GD2. Proc Natl Acad Sci U S A 79, 7629–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Pappo A, and Dyer MA (2015). Pediatric solid tumor genomics and developmental pliancy. Oncogene 34, 5207–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung NK, and Dyer MA (2013). Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, Heguy A, Pappo AS, Federico S, Dalton J, et al. (2012). Association of age at diagnosis and genetic mutations in patients with neuroblastoma. Jama 307, 1062–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicard M, Colmet-Daage L, Clement N, Danzon A, Bohec M, Bernard V, Baulande S, Bellini A, Deveau P, Pierron G, et al. (2018). Whole-Exome Sequencing of Cell-Free DNA Reveals Temporospatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clin Cancer Res 24, 939–949. [DOI] [PubMed] [Google Scholar]
- Ciccarone V, Spengler BA, Meyers MB, Biedler JL, and Ross RA (1989). Phenotypic diversification in human neuroblastoma cells: expression of distinct neural crest lineages. Cancer Res 49, 219–225. [PubMed] [Google Scholar]
- Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, and Gibbons RJ (2015). Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun 6, 7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MA, Zhang S, Sengupta S, Ma H, Bell GW, Horton B, Sharma B, George RE, Spranger S, and Jaenisch R (2020). Formation of Human Neuroblastoma in Mouse-Human Neural Crest Chimeras. Cell Stem Cell 26, 579–592.e576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing H, and Wolbach SB (1927). The Transformation of a Malignant Paravertebral Sympathicoblastoma into a Benign Ganglioneuroma. Am J Pathol 3, 203–216.207. [PMC free article] [PubMed] [Google Scholar]
- Dalton N (1885). Infiltrating growth in liver and suprarenal capsule. . Pathological Society of London. [Google Scholar]
- Dong R, Yang R, Zhan Y, Lai HD, Ye CJ, Yao XY, Luo WQ, Cheng XM, Miao JJ, Wang JF, et al. (2020). Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 38, 716–733.e716. [DOI] [PubMed] [Google Scholar]
- Dupin E, Calloni GW, Coelho-Aguiar JM, and Le Douarin NM (2018). The issue of the multipotency of the neural crest cells. Dev Biol 444 Suppl 1, S47–s59. [DOI] [PubMed] [Google Scholar]
- Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, Schild L, Bentahar NB, Bellini A, Chicard M, et al. (2015). Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nature genetics 47, 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan A, Dyachuk V, Kastriti ME, Calvo-Enrique L, Abdo H, Hadjab S, Chontorotzea T, Akkuratova N, Usoskin D, Kamenev D, et al. (2017). Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman WL, Federico SM, McCarville MB, Shulkin BL, Davidoff AM, Krasin MJ, Sahr N, Sykes A, Wu J, Brennan RC, et al. (2019). A Phase II Trial of Hu14.18K322A in Combination with Induction Chemotherapy in Children with Newly Diagnosed High-Risk Neuroblastoma. Clin Cancer Res 25, 6320–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman WL, McCarville B, Shulkin BL, Davidoff A, Krasin M, Hsu CW, Pan H, Wu J, Brennan R, Bishop MW, et al. (2022). Improved Outcome in Children With Newly Diagnosed High-Risk Neuroblastoma Treated With Chemoimmunotherapy: Updated Results of a Phase II Study Using hu14.18K322A. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 40, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Malagon SG, Lopez Muñoz AM, Doro D, Bolger TG, Poon E, Tucker ER, Adel Al-Lami H, Krause M, Phiel CJ, Chesler L, et al. (2018). Glycogen synthase kinase 3 controls migration of the neural crest lineage in mouse and Xenopus. Nat Commun 9, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham A (2003). The neural crest. Curr Biol 13, R381–384. [DOI] [PubMed] [Google Scholar]
- Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, Johann PD, Balasubramanian GP, Segura-Wang M, Brabetz S, et al. (2018). The landscape of genomic alterations across childhood cancers. Nature 555, 321–327. [DOI] [PubMed] [Google Scholar]
- Ho R, Minturn JE, Simpson AM, Iyer R, Light JE, Evans AE, and Brodeur GM (2011). The effect of P75 on Trk receptors in neuroblastomas. Cancer Lett 305, 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K (2006). The sympathoadrenal cell lineage: specification, diversification, and new perspectives. Dev Biol 298, 335–343. [DOI] [PubMed] [Google Scholar]
- Isern J, García-García A, Martín AM, Arranz L, Martín-Pérez D, Torroja C, Sánchez-Cabo F, and Méndez-Ferrer S (2014). The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. eLife 3, e03696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansky S, Sharma AK, Kӧrber V, Quintero A, Toprak UH, Wecht EM, Gartlgruber M, Greco A, Chomsky E, Grünewald TGP, et al. (2021). Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nature genetics 53, 683–693. [DOI] [PubMed] [Google Scholar]
- Jubierre L, Soriano A, Planells-Ferrer L, París-Coderch L, Tenbaum SP, Romero OA, Moubarak RS, Almazán-Moga A, Molist C, Roma J, et al. (2016). BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene 35, 5179–5190. [DOI] [PubMed] [Google Scholar]
- Kameneva P, Artemov AV, Kastriti ME, Faure L, Olsen TK, Otte J, Erickson A, Semsch B, Andersson ER, Ratz M, et al. (2021). Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nature genetics 53, 694–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastriti ME, Kameneva P, and Adameyko I (2020). Stem cells, evolutionary aspects and pathology of the adrenal medulla: A new developmental paradigm. Mol Cell Endocrinol 518, 110998. [DOI] [PubMed] [Google Scholar]
- Kawano A, Hazard FK, Chiu B, Naranjo A, LaBarre B, London WB, Hogarty MD, Cohn SL, Maris JM, Park JR, et al. (2021). Stage 4S Neuroblastoma: Molecular, Histologic, and Immunohistochemical Characteristics and Presence of 2 Distinct Patterns of MYCN Protein Overexpression-A Report From the Children’s Oncology Group. Am J Surg Pathol 45, 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerosuo L, and Bronner-Fraser M (2012). What is bad in cancer is good in the embryo: importance of EMT in neural crest development. Semin Cell Dev Biol 23, 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildisiute G, Kholosy WM, Young MD, Roberts K, Elmentaite R, van Hooff SR, Pacyna CN, Khabirova E, Piapi A, Thevanesan C, et al. (2021a). Tumor to normal single-cell mRNA comparisons reveal a pan-neuroblastoma cancer cell. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildisiute G, Young MD, and Behjati S (2021b). Pitfalls of Applying Mouse Markers to Human Adrenal Medullary Cells. Cancer Cell 39, 132–133. [DOI] [PubMed] [Google Scholar]
- Koyama H, Zhuang T, Light JE, Kolla V, Higashi M, McGrady PW, London WB, and Brodeur GM (2012). Mechanisms of CHD5 Inactivation in neuroblastomas. Clin Cancer Res 18, 1588–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulesa PM, and Gammill LS (2010). Neural crest migration: patterns, phases and signals. Dev Biol 344, 566–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, Mitson M, De Gobbi M, Marra M, Morris A, et al. (2010). ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143, 367–378. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM (1975). The neural crest in the neck and other parts of the body. Birth Defects Orig Artic Ser 11, 19–50. [PubMed] [Google Scholar]
- Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, and Weiss WA (2016). Neuroblastoma. Nat Rev Dis Primers 2, 16078. [DOI] [PubMed] [Google Scholar]
- Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, et al. (1999). Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. The New England journal of medicine 341, 1165–1173. [DOI] [PubMed] [Google Scholar]
- Matthay KK, Yanik G, Messina J, Quach A, Huberty J, Cheng SC, Veatch J, Goldsby R, Brophy P, Kersun LS, et al. (2007). Phase II study on the effect of disease sites, age, and prior therapy on response to iodine-131-metaiodobenzylguanidine therapy in refractory neuroblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 25, 1054–1060. [DOI] [PubMed] [Google Scholar]
- Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. (2008). Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, and Brodeur GM (1993). Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. The New England journal of medicine 328, 847–854. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Azar CG, Scavarda NJ, and Brodeur GM (1994). Expression and function of TRK-B and BDNF in human neuroblastomas. Molecular and cellular biology 14, 759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen R, Houston J, Chan WK, Finkelstein D, and Dyer MA (2018). The role of interleukin-2, all-trans retinoic acid, and natural killer cells: surveillance mechanisms in anti-GD2 antibody therapy in neuroblastoma. Cancer immunology, immunotherapy : CII 67, 615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrie JL, Nityanandam A, Lai K, Chen X, Wilson M, Stewart E, Griffiths L, Jin H, Wu G, Orr B, et al. (2021). Retinoblastoma from human stem cell-derived retinal organoids. Nat Commun 12, 4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Chen X, Huang X, Clay MR, Komorova N, Krasin MJ, Pappo A, Tillman H, Orr BA, McEvoy J, et al. (2022). The myogenesis program drives clonal selection and drug resistance in rhabdomyosarcoma. Developmental cell In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter EL, and Parrish JM (1942). Neuroblastoma, Ganglioneuroma and Fibroneuroma in a Stillborn Fetus. Am J Pathol 18, 141–151. [PMC free article] [PubMed] [Google Scholar]
- Raabe EH, Laudenslager M, Winter C, Wasserman N, Cole K, LaQuaglia M, Maris DJ, Mosse YP, and Maris JM (2008). Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 27, 469–476. [DOI] [PubMed] [Google Scholar]
- Rettig WJ, Spengler BA, Chesa PG, Old LJ, and Biedler JL (1987). Coordinate changes in neuronal phenotype and surface antigen expression in human neuroblastoma cell variants. Cancer Res 47, 1383–1389. [PubMed] [Google Scholar]
- Ross RA, Spengler BA, and Biedler JL (1983). Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J Natl Cancer Inst 71, 741–747. [PubMed] [Google Scholar]
- Sadée W, Yu VC, Richards ML, Preis PN, Schwab MR, Brodsky FM, and Biedler JL (1987). Expression of neurotransmitter receptors and myc protooncogenes in subclones of a human neuroblastoma cell line. Cancer Res 47, 5207–5212. [PubMed] [Google Scholar]
- Sartorelli AC (1985). The 1985 Walter Hubert Lecture. Malignant cell differentiation as a potential therapeutic approach. British journal of cancer 52, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA Jr., Papadopoulos N, et al. (2013). Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nature genetics 45, 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, and Reisfeld RA (1984). Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res 44, 5914–5920. [PubMed] [Google Scholar]
- Seeger RC, Siegel SE, and Sidell N (1982). Neuroblastoma: clinical perspectives, monoclonal antibodies, and retinoic acid. Ann Intern Med 97, 873–884. [DOI] [PubMed] [Google Scholar]
- Serbedzija GN, Fraser SE, and Bronner-Fraser M (1990). Pathways of trunk neural crest cell migration in the mouse embryo as revealed by vital dye labelling. Development 108, 605–612. [DOI] [PubMed] [Google Scholar]
- Solovieva T, and Bronner M (2021). Schwann cell precursors: Where they come from and where they go. Cells Dev 166, 203686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto T, Ueyama H, Hosoi H, Inazawa J, Kato T, Kemshead JT, Reynolds CP, Gown AM, Mine H, and Sawada T (1991). Alpha-smooth-muscle actin and desmin expressions in human neuroblastoma cell lines. Int J Cancer 48, 277–283. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Bostrӧm K, Fredman P, Jungbjer B, Lekman A, Månsson JE, and Rynmark BM (1994). Gangliosides and allied glycosphingolipids in human peripheral nerve and spinal cord. Biochim Biophys Acta 1214, 115–123. [DOI] [PubMed] [Google Scholar]
- Telloni SM (2017). Tumor Staging and Grading: A Primer. Methods in molecular biology (Clifton, NJ) 1606, 1–17. [DOI] [PubMed] [Google Scholar]
- Thiele CJ. (1998). Neuroblastoma Cell Lines. In Human Cell Culture, Masters J, ed. (Lancaster, UK: Kluwer Academic Publishers; ), pp. 21–53. [Google Scholar]
- Tomolonis JA, Agarwal S, and Shohet JM (2018). Neuroblastoma pathogenesis: deregulation of embryonic neural crest development. Cell Tissue Res 372, 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Groningen T, Akogul N, Westerhout EM, Chan A, Hasselt NE, Zwijnenburg DA, Broekmans M, Stroeken P, Haneveld F, Hooijer GKJ, et al. (2019). A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat Commun 10, 1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, Broekmans M, Haneveld F, Nowakowska NE, Bras J, et al. (2017). Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nature genetics 49, 1261–1266. [DOI] [PubMed] [Google Scholar]
- van Groningen T, Niklasson CU, Chan A, Akogul N, Westerhout EM, von Stegingk K, Hamdi M, Valentinjn LJ, and Mohlin S (2021). An immature subset of neuroblastdoma cells synthesizes retinoic acid and depends on this metabolite. bioRxiv 2021.2005.2018.444639. [Google Scholar]
- Vrenken KS, Vervoort BMT, van Ingen Schenau DS, Derks YHW, van Emst L, Grytsenko PG, Middelbeek JAJ, and van Leeuwen FN (2020). The transcriptional repressor SNAI2 impairs neuroblastoma differentiation and inhibits response to retinoic acid therapy. Biochim Biophys Acta Mol Basis Dis 1866, 165644. [DOI] [PubMed] [Google Scholar]
- Wakamatsu Y, Watanabe Y, Nakamura H, and Kondoh H (1997). Regulation of the neural crest cell fate by N-myc: promotion of ventral migration and neuronal differentiation. Development 124, 1953–1962. [DOI] [PubMed] [Google Scholar]
- Wang L, Tan TK, Durbin AD, Zimmerman MW, Abraham BJ, Tan SH, Ngoc PCT, Weichert-Leahey N, Akahane K, Lawton LN, et al. (2019). ASCL1 is a MYCN- and LMO1-dependent member of the adrenergic neuroblastoma core regulatory circuitry. Nat Commun 10, 5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JB, Xue C, Benice T, Xue L, Morris SW, and Raber J (2012). Anaplastic lymphoma kinase and leukocyte tyrosine kinase: functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol Biochem Behav 100, 566–574. [DOI] [PubMed] [Google Scholar]
- Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, and Bishop JM (1997). Targeted expression of MYCN causes neuroblastoma in transgenic mice. The EMBO journal 16, 2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhout EM, Hamdi M, Stroeken P, Nowakowska NE, Lakeman A, van Arkel J, Hasselt NE, Bleijlevens B, Akogul N, Haneveld F, et al. (2022). Mesenchymal-Type Neuroblastoma Cells Escape ALK Inhibitors. Cancer Res 82, 484–496. [DOI] [PubMed] [Google Scholar]
- Wieland DM, Wu J, Brown LE, Mangner TJ, Swanson DP, and Beierwaltes WH (1980). Radiolabeled adrenergi neuron-blocking agents: adrenomedullary imaging with [131I]iodobenzylguanidine. J Nucl Med 21, 349–353. [PubMed] [Google Scholar]
- Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, George AJ, Morgan KA, Mann JR, and Choo KH (2010). ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 20, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JH (1910). Neurocytoma or Neuroblastoma, a Kind of Tumor not Generally Recognized. The Journal of experimental medicine 12, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Hanada R, Kikuchi A, Ichikawa M, Aihara T, Oguma E, Moritani T, Shimanuki Y, Tanimura M, and Hayashi Y (1998). Spontaneous regression of localized neuroblastoma detected by mass screening. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 16, 1265–1269. [DOI] [PubMed] [Google Scholar]
- Yang R, Luo W, Zhan Y, Li K, Wang J, and Dong R (2021). Response to Kildsiute et al. and Bedoya-Reina and Schlisio. Cancer Cell 39, 136–137. [DOI] [PubMed] [Google Scholar]
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. (2010). Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. The New England journal of medicine 363, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeineldin M, Federico S, Chen X, Fan Y, Xu B, Stewart E, Zhou X, Jeon J, Griffiths L, Nguyen R, et al. (2020). MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat Commun 11, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MW, Durbin AD, He S, Oppel F, Shi H, Tao T, Li Z, Berezovskaya A, Liu Y, Zhang J, et al. (2021). Retinoic acid rewires the adrenergic core regulatory circuitry of childhood neuroblastoma. Sci Adv 7, eabe0834. [DOI] [PMC free article] [PubMed] [Google Scholar]