

Abstract
With the growing prevalence of cardiovascular diseases (CVD), there is an urgent need to explore non-conventional therapeutic measures to alleviate the burden of CVD on global healthcare. Among various stakeholders for CVD, mitochondrial injury plays a cardinal role in the pathogenesis of CVD. Mitochondrial dynamics and mitophagy are essential machineries to govern mitochondrial health in cardiomyocytes in physiological and pathophysiological settings. However, with the onset and progression of CVD, homeostasis of mitophagy is disturbed through largely unknown pathological mechanisms, causing mitochondrial damage, and ultimately cardiomyocyte death. In this review, we aim at deciphering the dual regulatory role of mitophagy in CVD pathogenesis, controversies of mitophagy, and highlight recently identified compounds capable of modulating mitophagy. In the end, we will share our perspectives on future research direction for mitophagy in the context of CVD.
Keywords: Mitophagy, Mitophagy Inducers, Cardiovascular diseases, Heart failure, Myocardial infarction, Therapeutics
PINK1 and PRKN, Key Components of Mitophagy
Mitophagy is a type of autophagy “(see the Glossary)” that regulates mitochondrial quality and quantity through the removal and degradation of long-lived/depolarized/damaged/superfluous mitochondria, thereby, perceiving mitochondrial and cellular homeostasis [1–3]. Under physiological conditions, mitophagy helps to maintain the energy supply-demand of cardiomyocytes by removing and recycling impaired mitochondria from a healthy network and fending off mitochondrial apoptosis [4]. Mechanistically, mitochondrial impairment/depolarization induces mitochondrial fission through limited proteolysis of the fusion protein OPA1, mitochondrial dynamin like GTPase (OPA1), combined with altered phosphorylation, acetylation, O-GlcNAcylation, S-nitrosylation or SUMOylation of the fission protein dynamin 1 like (DNM1L/DRP1) [5–10]. Small mitochondria that are unable to restore mitochondrial membrane potential, recruit a set of molecules that mediate mitochondrial entrapment within a double-membrane transient structure named “autophagosome” (Fig 1). Ultimately, the mitochondria-loaded autophagosomes fuse with the lysosomes, resulting in the degradation and recycling of mitochondrial components into their building blocks such as amino acids, lipids, and carbohydrates [11–14]. It is well known that PTEN induced kinase 1 (PINK1) and parkin RBR E3 ubiquitin protein ligase (PRKN) participate in the maintenance of mitochondrial quality through mitophagy activation [15, 16]. Once the serine/threonine kinase PINK1 senses mitochondrial damage, it recruits PRKN to the mitochondria, leading to its activation [17–19]. Mechanistically, PRKN with its E3 ubiquitin ligase capacity, evokes ubiquitination of mitochondrial outer membrane (OMM) proteins, thus, amplifying a complex signaling cascade, leading to the removal and degradation of injured mitochondria (Fig 1) [20]. Also, PINK1 phosphorylates DNM1L in Ser616, inducing mitochondrial fission, an indispensable event prior to mitophagy [21]. Structurally, PRKN is a ubiquitin ligase containing ubiquitin-like (UBL), ring finger protein 1 (RING1), ring finger protein 2 (RING2), three zinc-coordinating in between ring fingers (IBR) domains, and a ring finger protein 0 (RING0) domain. Upon ubiquitination process, binding between E2 enzymes (ubiquitinconjugating enzymes) and RING1 facilitates the transfer of ubiquitin onto the PRKN RING2-resident catalytic cys431, resulting in the formation of the thioester intermediate [22–24]. However, under normal conditions, the UBL domain and repressor element of PRKN (REP) inhibit RING1’s E2-binding site, and RING0 blocks the catalytic cys431 in RING2, leading to the autoinhibited conformation of PRKN [25–27]. In addition, the ubiquitin ligase property of PRKN promotes its translocation to injured mitochondria [28, 29]. For mitochondrial recruitment and activation of PRKN, PINK1 phosphorylates UBL domain of PRKN at ser65 [30, 31]. In the current review, we aim at elucidating the role and molecular mechanisms of mitophagy in the pathogenesis of CVD along with recently identified therapeutic compounds capable of manipulating mitophagy as a therapeutic approach in CVD. In the end, we will share our perspectives on the challenges, gaps, and opportunities in mitophagy research in the context of CVD. In sections below, mitophagy mechanisms will be elucidated in details.
Fig. 1. PINK1-PRKN-dependent Mitophagy.
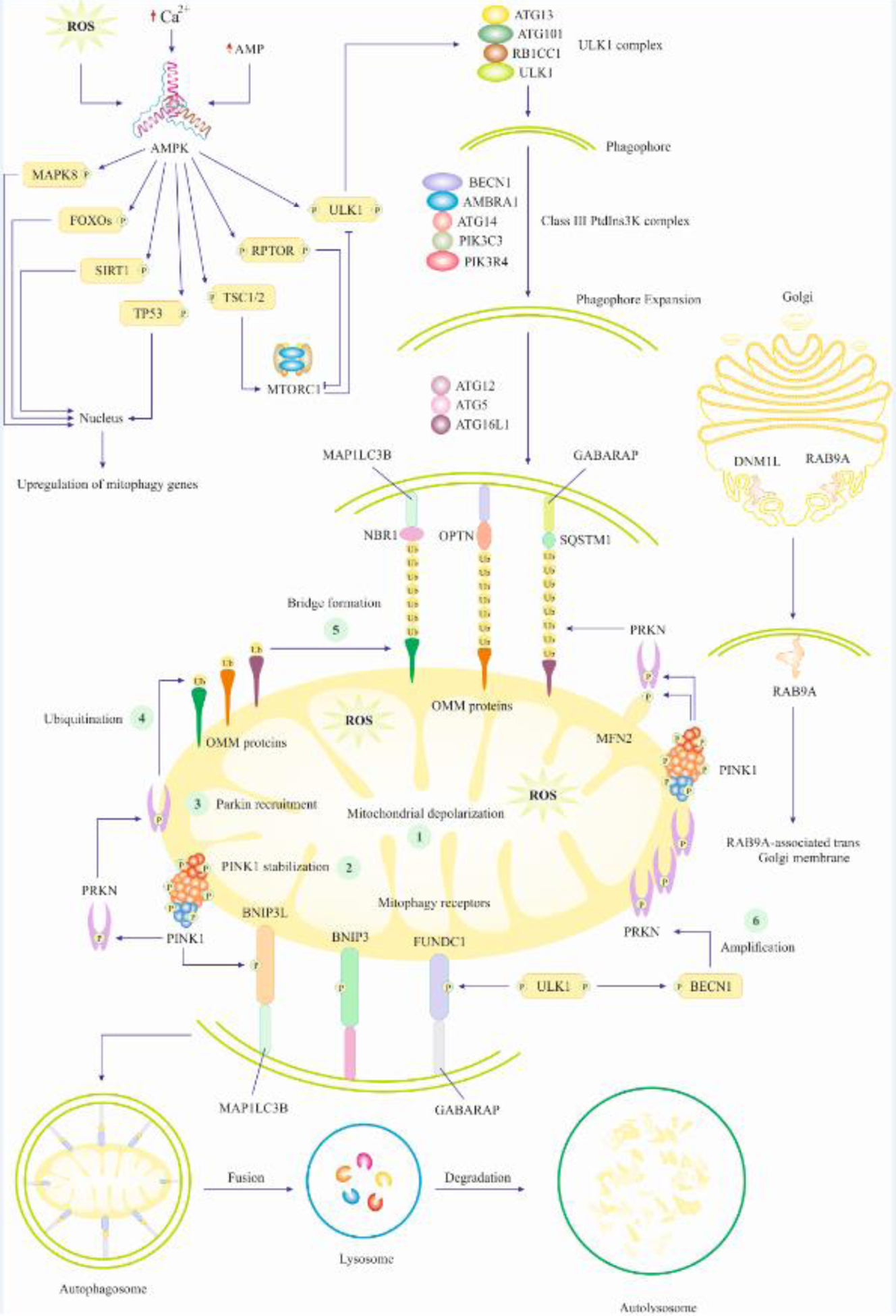
Mitochondrial damage/depolarization instigates PINK1 recruitment and stabilization on the OMM. Subsequently, PINK1 phosphorylates and recruits PRKN to the OMM. PRKN mediates ubiquitination and polyubiquitination of OMM proteins, leading to a bridge formation between targeted mitochondria and the autophagosome. ULK1 and BECN1 also participate in further recruitment of PRKN, a step that is referred to as amplification of PINK1/PRKN mitophagy. Ultimately, damaged/depolarized mitochondria engulfment within the autophagosome will end up with lysosomal fusion and degradation, leaving the cardiomyocytes free of damaged mitochondria. Initial signaling for mitophagy activation also includes AMP-, ROS-, and Ca2+-mediated activation of AMPK, which turns on transcription factors and autophagy regulators. The transcription factors relocate to the nucleus and transactivate mitophagy/autophagy genes that participate in the formation of autophagosomes. However, autophagy regulators inhibit MTORC1, resulting in the activation of the autophagy initiation complex (ULK1 complex), thus, forming phagophore (the initial membrane that ultimately matures into autophagosome). Of note, mitochondria damage is the initial inducer of AMPK activation as it causes cytosolic accumulation of Ca2+, ROS, and AMP.
Molecular Mechanisms of PINK1-PRKN Mitophagy
PINK1 Mediates PRKN’s Recruitment and Activation
Once PINK1 is translocated to healthy mitochondria, it is degraded by mitochondrial presenilin associated rhomboid like (PARL) serine protease in the mitochondrial inner membrane (MIM). PARL also degrades PGAM family member 5, mitochondrial serine/threonine protein phosphatase (PGAM5) to disturb PINK1 stabilization in mitochondria. In contrast, upon mitochondrial damage/depolarization prohibitin 2 (PHB2) deactivates PARL, leading to PINK1 stabilization and subsequent accumulation on the OMM [32]. Subsequently, OMM-accumulated PINK1 phosphorylates ubiquitin (basically linked to OMM proteins) at ser65 to enhance the affinity of PRKN to phosphorylated ubiquitin, leading to PRKN recruitment onto the OMM [33–39]. To activate PRKN, ser65-phosphorylated ubiquitin displaces UBL (an inhibitory domain) and stretches the REP, thus, perturbing autoinhibitory interactions in PRKN, leading to its opening and activation [36, 37, 40]. Then, in the active configuration of PRKN, E2 enzymes bind the RING1 domain and the RING2 catalytic cys431 is unmasked to participate in the formation of ubiquitin thioester intermediate. Subsequently, PRKN conjoins ubiquitin chains to OMM proteins, leading to a subsequent succession of PRKN recruitment to damaged mitochondria, creating a positive feedback loop for PRKN activation (Fig 1) [15, 35]. Ultimately, the consortium between PINK1 and PRKN leads to mitochondrial coating with ubiquitin chains, which act as molecular signals to promote autophagy machinery recruitment to the damaged mitochondria, thus, instigating mitophagy.
Known Mitophagy Receptors and Their Working Mechanisms
Mitophagy receptors are proteins responsible for molecular recognition of long-lived/damaged/depolarized mitochondria. There are different types of receptors in terms of their topology/localization, mechanism of action, and stress induction. Mitophagy receptors participating in PINK1-PRKN mitophagy encompass but are not limited to tax1 binding protein 1 (TAX1BP1), calcium binding and coiled-coil domain 2 (CALCOCO2), optineurin (OPTN), sequestosome 1 (SQSTM1), and NBR1 autophagy cargo receptor (NBR1), all of which capable of being relocated to damaged mitochondria in mitophagy induction [41, 42]. For mitophagy receptors to be recruited onto the OMM, PRKN should be activated (Fig 1) [43]. Besides, lys63-linked ubiquitin chains on mitochondria can further instigate the recruitment of mitophagy receptors [43]. Structurally, mitophagy receptors possess UBD (ubiquitin-binding domains) and LIR (LC3-interacting region), through which, they bind to ubiquitin on injured mitochondria and microtubule associated protein 1 light chain 3 beta (MAP1LC3B) or GABA type A receptor-associated protein (GABARAP) within the autophagosomal membrane, respectively [43]. This results in the formation of a bridge between damaged mitochondria and autophagosome, thus, promoting the engulfment of long-lived/damaged mitochondria for ultimate degradation and removal (Fig 1) [43]. The second type of mitophagy receptor corresponds to OMM proteins that are integrated into the OMM via their C-terminal domain or and thus are a constitutive part of the mitochondria rather than being recruited to the organelle. These receptors also bind to autophagy related 8 (ATG8)-family proteins on the phagophore via the LIR motif [3]. In consequence, mitophagy receptors build a bridge between mitophagy-destined mitochondrial components and autophagosome, thus, facilitating engulfment of mitochondria (Fig 1) [3]. One of these mitophagy receptors is FUN14 domain containing 1 (FUNDC1), a mitochondrial-associated-membrane (MAM) protein, plays a predominant role in hypoxia-induced mitophagy [44]. In FUNDC1-mediated mitophagy, PGAM5 phosphatase dephosphorylates FUNDC1 at ser13, which facilitates its interaction with MAP1LC3B to foster mitophagy and mitochondrial engulfment [45–47]. Moreover, mammalian cells contain two additional OMM proteins, FKBP prolyl isomerase 8 (FKBP8), and BCL2 like 13 (BCL2L13), serving as mitophagy receptors in PINK1-PRKN-independent mitophagy (BOX 1) [48, 49]. The third class of receptors involves proteins that are recruited to mitochondria independent of PINK1-PRKN. For instance, BCL2 interacting protein 3 (BNIP3) is a mitophagy receptor that participates in mitochondrial turnover, particularly, in hypoxic conditions [50]. Also, BNIP3L/NIX, with homology to BNIP3, also serves as a mitophagy receptor [51]. LIR motif on BNIP3L binds to GABA type A receptor associated protein like 1 (GABARAPL1) and 2, GABARAP, and MAP1LC3B receptors (Fig 1) [52]. BOX 1, 2, and 3 further illustrate mitophagy mechanisms.
BOX 1. PRKN-Independent Mitophagy.
Certain mitophagy receptors can trigger mitophagy through PRKN-independent mechanisms. This activation relies on the direct interaction of mitophagy receptors with distinct MAP1LC3B and GABARAP proteins [95]. It was reported that upon high mitochondrial oxidative phosphorylation activity, the small GTPase Rheb is recruited to the OMM and promotes mitophagy independent of PRKN and through physical interaction with BNIP3L/NIX and MAP1LC3B [96, 97]. In addition, the participation of FUNDC1 in PRKN-independent mitophagy was first described in hypoxia-stimulated mitophagy [98]. Under hypoxia, unc-51 like autophagy activating kinase 1 (ULK1) is recruited to fragmented mitochondria where it phosphorylates FUNDC1 at Ser17, enabling its interaction with MAP1LC3B and linking mitochondria to autophagosomes [99]. Also, autophagy and beclin 1 regulator 1 (AMBRA1) was initially identified as a pro-autophagic protein involved in phagophore formation, however, AMBRA1 can induce mitophagy in PRKN deficient mouse embryonic fibroblasts (MEFs) [100, 101]. Mechanistically, AMBRA1 binds to depolarized mitochondria and induces mitophagy by direct recruitment of MAP1LC3B [100]. Moreover, it has been evinced that OPTN is recruited to damaged mitochondria in the absence of PRKN and forms a complex with autophagy related 9A (ATG9A), leading to activation of mitophagy independent of PRKN [43, 102]. Likewise, PINK1 recruitment to the MOM triggers OPTN and CALCOCO2 recruitment under PRKN deficiency [103, 104]. Moreover, the E3 ubiquitin ligases including ariadne RBR E3 ubiquitin protein ligase 1 (ARIH1), mitochondrial E3 ubiquitin protein ligase 1 (MUL1), siah E3 ubiquitin protein ligase 1 (SIAH1), SMAD specific E3 ubiquitin protein ligase 1 (SMURF1), and autocrine motility factor receptor (AMFR) mediate mitophagy independent of PRKN [105–108]. Mechanistically, upon mitochondrial impairment/depolarization, these E3 ubiquitin ligases translocate onto mitochondria and produce ubiquitin chains, thus, recruiting OPTN, calcium binding and coiled-coil domain 2 (CALCOCO2/NDP52), and SQSTM1 [104]. Subsequently, autophagy machinery including ULK1, zinc finger FYVE-type containing 1 (ZFYVE1), and WD repeat domain, phosphoinositide interacting 1 (WIPI1) enter the scene to generate phagophore and expand the autophagosome [104]. Moreover, MEFs having a double knockdown of DNM1L and PRKN showed increased mitochondria accumulation, compared with a single loss, suggesting that DNM1L can also mediate a PRKN-independent mitophagy [109]. It has been proposed that, while Parkin stimulates the degradation of mitochondria by ubiquitination of mitochondrial proteins, DNM1L-mediated mitochondrial fission helps autophagosomes engulf mitochondria [109, 110]. Nonetheless, it should not be ruled out that DNM1L may directly interact with the aforementioned E3 ubiquitin ligases triggering PRKN-independent mitophagy. Of note, a better understanding of mitophagy regulatory mechanisms in pathophysiology of CVD should help to reveal novel maneuverable targets in the management of CVD.
BOX 2. Other Regulators of PINK1-PRKN Mitophagy.
Heat shock protein family A (Hsp70) member (HSPA/HSP70) promotes mitochondrial stabilization of PINK1 to prevent its degradation, thereby, engaging PINK1-PRKN mitophagy [111]. Meanwhile, PRKN tags mitochondrial ras homolog family member T1 (RHOT1) protein for degradation, which fosters segregation of injured mitochondria [112]. Also, deubiquitylating enzymes (DUBs) acting as proteases remove or remodel ubiquitin chains on target proteins, thereby, offering key roles in mitophagy regulation [113, 114]. For instances, the ubiquitin specific peptidase 30 (USP30) is an OMM-localized DUB reported to inhibit mitophagy through specific hydrolysis of lys11- and lys6-linked ubiquitin chains [113, 114]. Furthermore, USP15 negatively regulates mitophagy by removing lys63- and lys48-linked ubiquitin chains on mitochondria [115].
BOX 3. Integration of Upstream Mitophagy with Canonical Mechanisms of Autophagy.
Mammalian cells including cardiomyocytes undergo autophagy under stressful conditions. For instance, cardiomyocytes under nutrient deprivation experience metabolic changes and intracellular events that commence initial triggers of autophagy and mitophagy. Glucose deprivation induces reduction of glycosylation and intracellular elevation of adenosine monophosphate (AMP) levels. Similarly, amino acid deprivation culminates in intracellular elevation of AMP and Ca2+. Subsequently, AMP and Ca2+ activate AMPK, which in turn, phosphorylates/activates positive regulators of autophagy such as beclin 1 (BECN1), ULK1, regulatory associated protein of MTOR complex 1 (RPTOR), and TSC complex subunit 1/2 (TSC1/2). Mechanistically, ULK1 forms autophagy initiation complex with autophagy-related (ATG) proteins, leading to the formation of autophagosomes. To achieve this, RPTOR and TSC1/2 proteins block MTORC1 (a negative regulator of ULK1), resulting in ULK1 activation and autophagy initiation. On the other hand, elevated intracellular levels of Ca2+ causes mitochondrial depolarization, thus, fostering the recruitment of PRKN and initiation of PINK1-PRKN mitophagy. In this stage, autophagy meets mitophagy as the formulated autophagosome and is recruited to the damaged/depolarized mitochondria, where it interacts with mitophagy receptors, thereby, engulfing damaged mitochondria (Fig 1) [11, 116, 117].
Mild/Adaptive vs Excessive/Maladaptive Mitophagy and Merits of Mitophagy Modulation
Despite the lack of consensus and controversies in the field, adaptive mitophagy is defined as a type or a mode of mitophagy conferring protection against cell damage resulting in overall alleviation of pathologies. To the contrary, maladaptive mitophagy is defined as a type of mitophagy or a mode of mitophagy activation that promotes cell injury, leading to cell death and overall exacerbation of a pathological state [53]. Mechanistically, current evidence suggests that the extent, intensity, and duration of mitophagy activation will determine adaptive vs maladaptive mode of mitophagy. In this regard, activation of mitophagy with mild or modest intensity, extent, and duration culminates in adaptive effects such as the removal of injured mitochondria and overall well-being of cells, thereby, adaptive mitophagy is also termed “mild mitophagy”. On the other hand, if the extent, intensity, or duration of mitophagy activation is in excessive or constitutive mode then, adaptive mitophagy shifts to maladaptive mitophagy and exerts detrimental effects on cells such as provoking cell death mechanisms or targeting healthy mitochondrial rather than merely injured mitochondria. Therefore, maladaptive mitophagy is also termed “excessive/constitutive mitophagy”. For instance, mild activation of PINK1-PRKN mitophagy in cardiomyocytes was evinced to be adaptive to alleviate cardiac damage [54]. However, PINK1-PRKN mitophagy exerted not only adaptive but also maladaptive outcomes courtesy of its constitutive/excessive activation in obese hearts [55]. Besides, AMP-activated protein kinase (AMPK) was shown to induce mild activation of PINK1-PRKN mitophagy thereby, AMPK-mediated mitophagy is an adaptive type/mode in cardiomyocytes [56]. Similarly, mild activation of forkhead Box O3 (FOXO3)-dependent mitophagy conferred adaptive effects on cardiomyocytes [57]. Also, mild induction of FUNDC1-mediated mitophagy served as an adaptive form of mitophagy upon myocardial injury [58]. Nonetheless, an alternative notion also exists suggesting that maladaptive mitophagy not only requires excessive/constitutive activation but also specific molecular mechanisms. However, the current literature does not support this notion as different mechanisms of mitophagy were shown to be both adaptive and maladaptive depending on extent, intensity, and duration of activation. Based on this information, therapeutic modulation of mitophagy should be exercised with caution to maintain mild but not unchecked activation.
Mitophagy in Cardiovascular Diseases: Molecular Pathogenesis and Therapeutic Targets
Mitophagy in Heart Failure
Isoform shift from protein kinase AMP-activated catalytic subunit alpha 2 (PRKAA2/AMPKα2) to PRKAA1/AMPKα1 induced mitophagy dysfunction, thereby, exacerbated the progression of chronic heart failure (HF) in human and mouse model of HF [59]. However, forced overexpression of PRKAA2 prevented chronic HF by reactivating mitophagy in mouse hearts [59]. Mechanistically, PRKAA2 interacted with PINK1, leading to activation of the PINK1-PRKN-SQSTM1 mitophagy pathway, removal of reactive oxygen species (ROS), and inhibition of apoptosis in cardiomyocytes [59]. These findings denote that PRKAA2 is a potential therapeutic target the activation of which could be translated into clinics for the treatment/alleviation of HF. However, the challenge is the development of a specific activator of PRKAA2 or gene therapy of PRKAA2 to accommodate clinical applications. Moreover, binding of cytosolic tumor protein p53 (TP53) to PRKN was shown to retard PRKN translocation to mitochondria, resulting in mitophagy dysfunction in murine models of HF and cardiac aging [54]. Consistently, induction of TP53 deficiency or PRKN overexpression reactivated mitophagy, thereby, alleviated HF, cardiac dysfunction, and aging [54]. Hence, cytosolic TP53 acts as mitophagy inhibitor, therefore, restricting its cytosolic activity could be a therapeutic strategy to combat mitophagy dysfunction and HF. Nonetheless, the challenge is that nuclear TP53 as a transcription factor was shown to induce mitophagy in non-cardiovascular setting [60], raising doubt on therapeutic value of targeting TP53. Certain tumor cells may hijack TP53 to induce BNIP3-mediated mitophagy and preserve mitochondrial integrity [61]. However, given the anti-oncogenic role of TP53 in non-malignant cells, targeting TP53 may predispose cardiomyocytes to a pre-oncogenic state [62]. Collectively, due to technological barriers to specific targeting of cytosolic TP53, diverse roles of cytosolic versus nuclear TP53 in mitophagy activation, and the pre-oncogenic perils of TP53, inhibitory targeting TP53 might not be a good choice for mitophagy activation in HF. Nonetheless, these studies suggest that inducible mild activation of PINK1-PRKN mitophagy in cardiomyocytes may prevent HF development and alleviate cardiac damage. On the contrary, BNIP3-mediated mitophagy potentiated HF [63]. Mechanistically, pressure overload (PO) triggered activation of mitogen-activated protein kinase 8 (MAPK8), which in turn, promoted FOXO3, leading to upregulation of BNIP3, resulting in HF and cardiac remodeling both in vitro and in vivo [63]. This study suggests that activation of MAPK8-FOXO3-BNIP3-mediated mitophagy in cardiomyocytes fosters HF development. Also, it is postulated that MAPK8-FOXO3-BNIP3-mediated mitophagy functions as a type of maladaptive mitophagy the constitutive activation of which is maladaptive and detrimental to cardiomyocytes. Hence, therapeutic inhibition of this type of mitophagy could represent a novel approach in the treatment of HF. In this sense, inhibition of MAPK8 signaling using 3-methyladenine was shown to retard cardiac remodeling in HF [63].
Mitophagy in Myocardial Infarction and Ischemia-Reperfusion Injury
At the reperfusion stage of myocardial ischemia-reperfusion (I/R) injury solute carrier family 39 member 7 (SLC39A7) was upregulated, leading to suppression of mitophagy and exacerbation of I/R pathology in mice model [64]. However, cardiac-specific SLC39A7 knockout enhanced mitochondrial Zn2+ levels, causing mitochondrial depolarization and mitophagy activation, leading to ROS scavenging and alleviation of myocardial infarction (MI) insult [64]. This study suggests that timely inhibition of SLC39A7 could be considered a therapeutic strategy in the mitigation of reperfusion injury via mitophagy activation. Also, in murine model of I/R and cardiomyocytes under hypoxia-reoxygenation (H/R), Mir302a-3p was remarkably upregulated, thus, inhibiting FOXO3 expression, leading to mitophagy inactivation [57]. Hence, inducing mitophagy via modulation of the Mir302a-3p-FOXO3 axis could be a potential therapeutic strategy in the treatment or alleviation of myocardial I/R injury as inhibition of Mir302a-3p conferred cardioprotection against cardiomyocyte apoptosis and ROS [57]. Also, it can be postulated that FOXO3-dependent mitophagy is a type of adaptive mitophagy the activation of which protects cardiomyocytes against I/R injury. Taken together, inducible mild activation of mitophagy along with scavenging ROS could be a combination therapeutic dogma to cope with the challenge of MI insult. In addition, ROS-mediated activation of the TP53-TIGAR (TP53 induced glycolysis regulatory phosphatase) axis inhibited cardiomyocyte mitophagy resulting in apoptosis and ventricular remodeling upon MI insult [65]. Hence, cytosolic TP53 also acts as mitophagy inhibitor in the context of MI similar to HF as discussed above [54]. Interestingly, one of the underlying mechanisms of TP53-mediated apoptosis is through inhibition of adaptive mitophagy in cardiomyocytes.
In another setting, upregulation of receptor interacting serine/threonine kinase 3 (RIPK3) in murine model of MI and cardiomyocytes under H/R interrupted PINK1-PRKN mitophagy via suppression of AMPK, culminating in mitochondrial transition pore opening, cardiomyocyte necroptosis, and overt cardiac remodeling and dysfunction [56]. Therefore, therapeutic inhibition of RIPK3 protein or gene may reactivate adaptive PINK1-PRKN mitophagy as abrogation of RIPK3 was shown to avert necroptotic cell death and alleviate MI insult [56]. Besides, AMPK could be another therapeutic target the activation of which may reactivate adaptive mitophagy to rescue cardiomyocytes from MI damage. Given that MI is accompanied by metabolic stress and AMPK activation [66], it can be perceived that AMPK leads to activation of PINK1-PRKN mitophagy and removal of dysfunctional mitochondria (responsible for energy deficit), indicating an adaptive machinery for mitophagy to halt energy crisis in cardiomyocytes. In addition, reperfusion triggered RIPK3 upregulation, which promoted FUNDC1 phosphorylation/inactivation, resulting in dysfunction of FUNDC1-mediated mitophagy and prominent apoptosis in a murine model of I/R injury [58]. Therefore, the translational merit of these findings is that the RIPK3-FUNDC1 axis could be considered a novel therapeutic target the inhibition of which may reinvigorate FUNDC1-mediated mitophagy and mitigate myocardial I/R injury. In line with this, RIPK3 deficiency prevented mitochondrial apoptosis and caspase 9 (CASP9) activation, thereby, reduced infarcted area [58]. In other words, FUNDC1-mediated mitophagy serves as an “adaptive form” of mitophagy upon myocardial I/R injury. Collectively, these findings denote that RIPK3 serves as an inhibitor of FUNDC1- and PINK1-PRKN-mediated mitophagy, thereby, fostering MI insult and I/R injury. Developing clinically compatible RIPK3 inhibitors or gene therapy tools targeting RIPK3 gene should help to open a new avenue for rescuing adaptive mitophagy in cardiomyocytes to alleviate myocardial damage. Also, in an alternative therapeutic approach independent of RIPK3, inducible overexpression of polo like kinase 1 (PLK1) activated FUNDC1-mediated mitophagy via upregulation of FUNDC1 and PRKAA (encoding AMPK), leading to suppression of cardiomyocyte apoptosis and alleviation of I/R injury in mice and H9c2 cells [67]. In addition, cardiac-specific genetic ablation of casein kinase 2 alpha 1 (CSNK2A1) promoted FUNDC1-mediated mitophagy, resulting in alleviation of myocardial I/R injury and infarction area in mice [68]. Moreover, given that nuclear receptor subfamily 4 group A member 1 (NR4A1) impaired mitophagy via upregulation of CSNK2A1 expression in myocardial I/R injury [69], pharmacological inhibition or genetic ablation of NR4A1 could be another alternative approach to reactivate FUNDC1-mediated mitophagy and revert myocardial I/R injury. In a controversial report, aldehyde dehydrogenase 2 family member (ALDH2) inhibited PINK1-PRKN mitophagy via downregulation of PINK1 and PRKN expression, leading to alleviation of I/R injury in a rat model and H9C2 cells under H/R [70]. This study raised doubts on significance of activating PINK1-PRKN mitophagy as a therapeutic strategy in the combat against MI and I/R injury. One possible explanation of this study is that the PINK1-PRKN mitophagy is essentially an adaptive mechanism in cardiomyocytes, although its constitutive activation may pose maladaptive rather than beneficial effects on myocardial I/R injury. Thus, for translational purposes the best therapeutic strategy would be to induce mild activation of PINK1-PRKN mitophagy and avoid constitutive activation in cardiomyocytes (see the Clinician’s Corner).
Clinician’s Corner.
Once mitochondrial health is compromised (a common phenomenon in CVD), an array of maladaptive conditions occur including mitochondrial ROS generation, cytosolic Ca2+, and ROS overload, as well as mitochondria-initiated apoptosis, ultimately, leading to cardiomyocyte death and CVD pathogenesis. Regarding the seminal role of mitophagy in restoring mitochondrial health, inducing mitophagy activation in cardiomyocytes could be a promising therapeutic approach in combat against various subset of CVD.
For proper targeting of mitophagy researchers should implement studies to further elaborate on molecular mechanisms instigated by CVD pathology, which induce partial/holistic mitophagy defects. Once underlying mechanisms of mitophagy defects have been quite understood, proper therapeutic interventions could be designed.
To alleviate CVD mitophagy activation should be maintained at mild level. Mainly, PINK1-PRKN mitophagy, FUNDC1-mediated mitophagy, and FOXO3-dependent mitophagy are adaptive types of mitophagy whose mild activation may exert remarkable advances in clinical treatment and management of CVD.
PINK1-PRKN mitophagy is the most common type of adaptive mitophagy which is regulated by key molecules such as AMPK. Therefore, immense effort is warranted to develop clinically meetable drugs or strategies for targeting these molecules and reactivation of mitophagy in CVD patients.
It is not quite understood whether it is the constitutive/excessive activation that is merely responsible for detrimental effects of mitophagy or other mitophagy mediators exist, activation of which, leads to such effects. Thus, therapeutic strategies involving mitophagy activation should be applied with extra caution to avoid excessive activation of mitophagy, which may result in exacerbating effects rather than alleviating effects on CVD.
Mitophagy in Cardiomyopathy and Atherosclerosis
In mice under high-fat diet (HFD), mitophagy impairment culminated in lipid accumulation, and mitochondrial dysfunction, and therefore, promoted the progression of diabetic cardiomyopathy [71]. Although this study did not elucidate underpinning mechanisms of mitophagy defect in diabetic cardiomyopathy, inducible mitophagy induction ameliorated diabetic cardiomyopathy [71]. Moreover, in a murine model of diabetic cardiomyopathy, mammalian sterile 20-like kinase 1 (MST1), a key component of the “Hippo” signaling pathway, impeded PINK1-PRKN mitophagy via inhibition of sirtuin 3 (SIRT3) [72]. Hence, MST1/Mst1 could be a therapeutic target, the inhibition of which may reactivate mitophagy and alleviate diabetic cardiomyopathy. In this sense, deletion of Mst1 gene successfully reduced mitochondrial fission, cardiac remodeling, and improved systolic function in diabetic mice [73]. In addition, inducible activation of SIRT3 could be another therapeutic approach as SIRT3 reactivated PINK1-PRKN mitophagy and conferred cardioprotection in a murine model of diabetic cardiomyopathy [74]. Also, in doxorubicin-induced rat model of cardiomyopathy, PINK1-PRKN mitophagy was impaired owing to downregulation of sestrin 2 (SESN2) expression, suggesting a role for SESN2 as a positive regulator of PINK1-PRKN mitophagy [75]. Therefore, modulation of SESN2 could be a therapeutic maneuver as SESN2 upregulation was shown to promote PRKN accumulation on mitochondria and thereby, reactivated PINK1-PRKN mitophagy and mitigated cardiomyopathy [75]. Furthermore, tafazzin, phospholipid-lysophospholipid transacylase (TAFAZZIN) deficiency activated the mechanistic target of rapamycin kinase complex 1 (MTORC1) signaling, leading to inhibition of mitophagy and accumulation of autophagic vacuoles in cardiomyocytes, ultimately cardiomyopathy in a murine model of Barth syndrome [76]. Based on this study, inducing TAFAZZIN expression, as well as pharmacological inhibition of MTORC1 could be promising approaches to reverse dampened mitophagy and rescue cardiomyocytes. In this regard, rapamycin inhibited MTORC1, thus, reactivated mitophagy and ameliorated dilated cardiomyopathy [76]. Overall, the consensus is that PINK1-PRKN mitophagy dysfunction is linked to pathogenesis of cardiomyopathy, therefore, reactivation of mitophagy could be promising in the management or alleviation of cardiomyopathy. Hence, developing gene therapy approaches or therapeutic agents to modulate the expression or activation of SIRT3, MST1/Mst1, SESN2, TAFAZZIN, and MTORC1 may significantly improve adaptive mitophagy in cardiomyocytes. In a murine model of atherosclerosis, apelin-13 induced PINK1-PRKN mitophagy via enhanced phosphorylation/activation of PRKAA in VSMC, resulting in the enhanced proliferation and aggravation of atherosclerotic lesions [77]. Therefore, in contrast to cardiomyopathy, PINK1-PRKN mitophagy in VSMC promotes atherosclerosis. A plausible explanation for this oddity could be that under atherosclerotic conditions, PINK1-PRKN mitophagy is constitutively activated in VSMC, resulting in the maladaptive progression of atherosclerosis. Hence, mild activation of PINK1-PRKN mitophagy in VSMC may confer adaptive responses and alleviate atherosclerosis. Beside VSMC, it was reported that induction of PINK1-PRKN mitophagy in macrophages of atherosclerotic lesions prevented activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome and interleukin 1 beta (IL1B/IL-1β) secretion and thereby, alleviated atherosclerosis [78, 79]. Although the precise role of mitophagy in atherosclerogenesis remains unclear and requires further exploration, the currently available evidence demonstrates a “double-edged sword” role for PINK1-PRKN mitophagy in the pathogenesis of atherosclerosis and cardiomyopathy depending on the mode of activation. While mild mitophagy seems to be benign and protective, excessive or sustained activation of mitophagy might be detrimental to vascular cells and cardiomyocytes in these diseases.
Mitophagy in Metabolic Cardiovascular Disorders
Long non-coding RNA H19 (LncRNA H19) inhibited eukaryotic translation initiation factor 4A2 (EIF4A2) binding to PINK1 mRNA, which suppressed PINK1 and excessive mitophagy, thus, alleviating obesity-induced cardiac disorders in H9c2 cells and in vivo mouse models [55]. In this regard, inducing upregulation of LncRNA H19 could be an effective strategy to halt excessive mitophagy and prevent cardiac dysfunction in obesity. Also, this study indicated that constitutive activation of PINK1-PRKN mitophagy may exert maladaptive effects in obese hearts. In an independent study, FUNDC1 interacted with F-Box and leucine rich repeat protein 2 (FBXL2) to promote degradation of inositol 1,4,5-trisphosphate receptor type 3 (ITPR3), therefore, maintaining mitochondrial Ca2+ homeostasis and alleviating obesity-induced cardiac remodeling in HFD-fed mice [46]. Hence, inducible activation of FUNDC1 could be a proper strategy to prevent cardiac remodeling via mitophagy activation and rescuing mitochondrial Ca2+ homeostasis. Moreover, it was revealed that T2DM exacerbated HF following MI through impaired mitophagy, upregulation of inflammasome signaling, interleukin 18 (IL18) production, and cell death in murine models, suggesting therapeutic importance of activating mitophagy in the alleviation of T2DM-associated HF and cardiomyopathy [80]. Collectively, these studies consolidated presence of mitochondrial injury and mitophagy inactivation in both obesity and diabetic cardiomyopathy. Therefore, induction of mitophagy (or more precisely mild activation of mitophagy while mitigating excessive mitophagy) along with suppression of inflammasome activity may be a possible therapeutic approach in the alleviation of obesity- and T2DM-associated HF.
Mitophagy in Cardiovascular Aging
Advanced aging was reported to impair mitophagy via downregulation of PRKN, SQSTM1, MAP1LC3B, TANK binding kinase 1 (TBK1), and autophagosomes in old mice [81]. However, PRKN overexpression reversed all of these changes, alleviated myocardial aging and irregularity, and inhibited cellular senescence by interacting with TBK1, leading to TBK1 polyubiquitination (at lys63 site) and mitophagy induction [81]. These observations denote that inducing mitophagy by way of PRKN overexpression effectively reverts cardiac aging. Besides, induction of PINK1-PRKN mitophagy could be an anti-aging maneuver in the combat against cardiovascular aging. Moreover, it was revealed that DNM1L, a mitochondrial fission protein, induced mitophagy, while inhibiting PINK1-PRKN mitophagy in aged cardiomyocytes, and thereby, protected against cardiomyocyte apoptosis in cardiac aging [82]. It seems like DNM1L-mediated mitophagy was independent of PINK1-PRKN mitophagy thus, urging further research to shed more light on such mitophagy regulatory dogma reported in this study. Altogether, these results indicate a cardinal role of mitophagy in the retardation of cardiac aging and the overall improvement of elderly patients with heart diseases. In the section below, potential mitophagy inducers and therapeutics have been outlined in details.
Emerging Mitophagy Inducers in Therapeutic Targeting of CVD
In recent years, much attention has been drawn to the use of natural compounds and hormones in the alleviation of CVD through the modulation of mitophagy. For instance, berberine is a natural plant-derived isoquinoline flavonoid, offering beneficial effects on chronic HF via activation of PINK1-PRKN mitophagy in mouse hearts [83]. Berberine also attenuated cardiomyocyte apoptosis and myocardial infarction by enhancing the binding of hypoxia inducible factor 1 subunit alpha (HIF1A) to BNIP3 promoter, leading to activation of BNIP3-mediated mitophagy [83]. Collectively, berberine activates both PINK1-PRKN- and BNIP3-mediated mitophagy, resulting in alleviation of chronic HF. Also, BNIP3 could be a novel target for developing mitophagy inducers for the treatment of HF. In addition, dexpramipexole is a synthetic compound which induced PINK1-PRKN mitophagy, thereby, attenuated tissue damage and myocardial infarction in both in vivo and in vitro models of myocardial I/R injury [84]. Donepezil is another chemical compound which scavenged ROS, inhibited apoptosis, and enhanced mitophagy in male Wistar rats with cardiac I/R injury [85]. Also, chemically reduced donepezil attenuated arrhythmia and infarct size, and alleviated left ventricular dysfunction mainly through activation of mitophagy [85]. Collectively, these observations indicate that both donepezil and dexpramipexole are two potential mitophagy inducers that may rescue cardiomyocytes under cardiac I/R injury. However, their clinical application requires further preclinical studies and validations. Also, quercetin is a natural flavonoid which activated sirtuin 1 (SIRT1)-transmembrane BAX inhibitor motif containing 6 (TMBIM6) axis, leading to activation of mitophagy and ROS scavenging in cardiomyocytes under H/R [86], suggesting that quercetin exhibits a promising capacity in the treatment/management of hypoxia-induced CVD.
In addition, acacetin is a natural flavone that remarkably upregulated PINK1 and PRKN, and induced SIRT1 activation, leading to activation of the sirtuin 6 (SIRT6)-AMPK signaling, which promoted PINK1-PRKN mitophagy and thereby, prevented cellular senescence in a concentration-depending manner in rat H9C2 cardiomyocytes and C57/BL6 mice [87]. Hence, acacetin is a natural mitophagy inducer, deserving further examination for clinical applications. Likewise, kanglexin is a natural compound derived from Chinese herbs such as rhubarb which effectively mitigated aging-induced cardiac remodeling and diastolic dysfunction via activation of PINK1-PRKN mitophagy by interacting with UBD on PRKN to engage its stability on mitochondria in mice and neonatal murine cardiomyocytes [88]. Taken together, these findings denote that kanglexin and acacetin are natural mitophagy inducers that may be considered in the alleviation of cardiovascular aging.
Urolithin A is another natural compound produced by the gut microbiota which activated FUNDC1-mediated mitophagy, resulting in the alleviation of sepsis-caused cardiac dysfunction in mice [89]. Also, urolithin A removed dysfunctional mitochondria, promoted mitochondrial respiratory capacity, and prolonged lifespan in C. elegans and in rodents, and rescued muscle function and exercise capacity in aged mice due to mitophagy reinvigoration [90]. Furthermore, urolithin A, actinonin, and nicotinamide adenine dinucleotide (NAD+) supplementation induced PINK1-mediated mitophagy in mice and elegans models of Alzheimer disease [91]. Similar results were obtained from two natural mitophagy inducers including rhapontigenin and kaempferol in rodent and nematode models of AD [92]. Moreover, tomatidine is a natural mitophagy inducer found in tomatoes which prevented skeletal muscle atrophy in aged mice via induction of PINK1-dependent mitophagy [93]. Given that muscle function is tightly linked to CVD, exploring potential utility of these mitophagy inducers in CVD is intriguing. In addition, deficiency of XPA (XPA, DNA damage recognition and repair factor) in nematodes induced hyperactivation of poly(ADP-Ribose) polymerase 1 (PARP1), which inhibited NAD+-SIRT1-PPARG coactivator 1 alpha (PPARGC1A) axis, resulting in PINK1 cleavage and mitophagy defect [94]. In this sense, application of NAD+ precursor (nicotinamide riboside) activated mitophagy [94]. In sum, mitophagy inducers such as urolithin A, tomatidine, and actinonin, as well as NAD+ supplementation all displayed promising results in activation of adaptive PINK1-/FUNDC1-dependent mitophagy in preclinical settings. Thus, immense efforts should be warranted to corroborate the utility of these compounds in activation of mitophagy for clinical management of CVD (BOX 4).
BOX 4. Targeting Mitophagy in CVD: Natural Hormones.
Melatonin
Melatonin sustained cardiomyocyte viability and improved myocardial function owing to normalization of OPA1-induced mitophagy and mitochondrial fusion, resulting in inhibition of severe mitochondrial fission, augmentation of mitochondrial energy metabolism, and suppression of CASP9-mediated apoptosis in both in vitro and mouse models of myocardial I/R injury [118]. From mechanistic standpoint, melatonin activated AMPK, which in turn, enhanced OPA1 stabilization and upregulation, leading to mitophagy activation. In sum, these findings indicate that melatonin possesses potential therapeutic efficacy on myocardial I/R injury by inducing mild mitophagy via activation of the AMPK-OPA1 axis. Furthermore, melatonin impeded activation of NLRP3 inflammasome and IL1B production by inducing mild activation of mitophagy through the SIRT3-FOXO3-PRKN axis and ROS scavenging in atherosclerotic macrophages in murine models [78, 119]. This study for the first time revealed that melatonin could be used as a therapy for atherosclerosis via induction of cytoprotective mitophagy [78, 119]. In another setting, melatonin treatment mitigated cardiac function/geometry by reactivating mitophagy via activation of the cyclic GMP-AMP synthase (CGAS)-stimulator of interferon genes (STING)-TBK1 pathway in a manner depending on ALDH2 upregulation in Alzheimer disease (AD) patients and murine models [120]. Hence, these findings support the therapeutic effects of melatonin on rescuing cardiac anomalies in AD patients by inducing mitophagy activation. Last but not least, melatonin alleviated systolic dysfunction associated with diabetic cardiomyopathy via inhibition of MST1 and activation of PRKN-dependent mitophagy [121]. Collectively, melatonin is a natural mitophagy inducer with promising effects on alleviation of CVD. Further studies may pave the way for clinical application of this natural hormone in combat against CVD.
Irisin
Investigating the effect of irisin on infarcted hearts and hypoxic cardiomyocytes revealed that OPA1 was upregulated, leading to activation of mitophagy, cardioprotection against hypoxia, and overall alleviation of MI both in vivo and in vitro [122]. Therefore, OPA1-induced mitophagy is a cardioprotective mechanism and irisin could be a potential candidate to turn on this mechanism and ameliorate cardiomyocyte injury caused by MI. Moreover, irisin treatment induced FUNDC1-dependent mitophagy, resulting in the improvement of mitochondrial ATP production and glucose metabolism, reduction of ROS (due to increased levels of antioxidant enzymes e.g., superoxide dismutase (SOD) and glutathione peroxidase (GPX)), and apoptosis inhibition in the H9C2 model of septic cardiomyopathy [123]. Therefore, this study reveals that irisin is a natural compound that may retard septic cardiomyopathy via induction of FUNDC1-mediated mitophagy. Overall, a growing evidence advocates the promising role of irisin as natural mitophagy inducer in a subset of CVD. However, more stringent studies are required to examine its effects on other CVD and possible clinical applications.
Concluding Remarks
In summary, the majority of currently available literature suggests a cardioprotective role for mitophagy and therapeutic interventional promises in the treatment/management of CVD. Yet, a number of observations have cast doubt with regards to the therapeutic benefits of mitophagy in CVD mainly due to excessive/constitutive activation of mitophagy to damage mitochondria and cardiomyocytes. It is thus perceived that induction of mild mitophagy while mitigating excessive mitophagy could be the appropriate approach to the manipulation of mitophagy in CVD. Hence, the identification of more small molecule drugs and natural compounds to balance mitophagy activation and induce mild mitophagy seems to be pertinent for targeted therapy of CVD. The majority of current mitophagy research has been focused on uncapping the role of mitophagy in pathophysiology of CVD, although translation aspects of these findings are still missing. To fulfill this gap, positive/negative regulators of mitophagy should be further explored to boost our knowledge of mitophagy modulation in cardiomyocytes. Subsequently, maneuverable targets such as PRKAA2, RIPK3, cytosolic TP53, or other unidentified molecules in mitophagy may guide development of clinically applicable drugs or gene therapy tools for therapeutic purpose. Also, the efficacy, specificity, and sensitivity of natural mitophagy inducers have not yet been demonstrated in human models of CVD thus, mandating future clinical examinations (see the Outstanding Questions).
Outstanding questions.
Mitochondrial damage is the initial trigger for activation of mitophagy, although cardiovascular pathologies harbor different patterns of mitochondrial damage including ROS generation, membrane depolarization, and reduced ATP production. It is imperative to identify how mitophagy is distinctly activated in response to unique mitochondrial damage. Hence, the key question is how mitophagy machineries differ in compliance with the different types of mitochondrial damages observed in cardiovascular diseases?
There has been a long-standing debate in the field of mitophagy over the explanation of maladaptive/pathological mitophagy. It is not yet clear whether maladaptive/pathological mitophagy is the result of excessive/constitutive activation or unknown mechanisms underscore the pathological nature of mitophagy in cardiovascular diseases. Hence, future studies should focus on how cardiovascular cells host detrimental mitophagy and which matters the most - the extent of mitophagy activation or the existence of unique mitophagy pathways?
Importantly, stressed cardiomyocytes or vascular cells may contain mitochondria with disparate levels of damage, some are more severe while others are less damaged. We hypothesize that mitophagy acts differently in response to highly-damaged mitochondria compared to less-damaged mitochondria. Hence, do other mitophagy mechanisms exist that respond differently to a damaged mitochondrion based on the level of damage? Or is it just the level of mitophagy activation that fills the gap? Is there a scenario that mild mitophagy is activated in response to less-damaged mitochondria and excessive mitophagy is activated in response to highly-damaged mitochondria?
Highlights.
Mitophagy dysfunction underscores the pathogenesis of cardiovascular diseases, leading to mitochondrial damage, ROS generation, and ultimately onset or progression of various subsets of cardiovascular diseases.
Basal induction of mitophagy perserves mitochondrial health in cardiovascular cells and thereby, prevents mitochondrial damage-/mitophagy dysfunction-initiated cardiovascular diseases.
Unlike basal mitophagy, excessive/unchecked activation of mitophagy may exert negative impacts on mitochondrial health and homeostasis in cardiovascular cells, leading to maladaptive responses, apoptosis, and exacerbation of cardiovascular diseases.
Acknowledgments
We will express our sincere apology to those authors whose important work cannot be included due to space limitations. This work was supported by NIH grant GM131919 to DJK. Also, this work was supported by grants from the Comision Nacional de Investigacion Cientifica y Tecnologica de Chile (FONDAP 15130011 to S.L., M.C.; FONDECYT 1200490 to S.L. and 1220392 to M.C.).
Glossary
- Autophagosome
A double-membraned organelle that engulfs cellular components and organelles during autophagy with a largely unknown origin of formation.
- Autophagy
A natural intracellular process in eukaryotes that removes unwanted cellular components, organelles, and protein aggregates via autophagosomal engulfment and lysosomal degradation.
- Barth syndrome
A rare anomaly manifested by weakened and enlarged cardiac, skeletal muscles weakness, and lowered white blood cells in the body.
- Deubiquitinating enzymes
These enzymes play key roles in multiple cellular processes by posttranslationally detaching ubiquitin chains from certain target proteins.
- Ischemia-reperfusion
Refers to a tissue damage instigated by the return or reperfusion of blood to the damaged tissue after ischemia (a period of blood/oxygen lacking).
- O-GlcNAcylation
A glycosylation process by which O-GlcNAc (a monosaccharide) is transferred to threonine/serine residues of proteins.
- SUMOylation
A type of post-translational modification through which the SUMO protein covalently binds to and regulates other proteins thereby, playing a role in cellular functions and growth.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
None of the authors declare any potential conflict of interest.
REFERENCES:
- 1.Wu NN et al. (2019) Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxidative Medicine and Cellular Longevity 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang M et al. (2019) Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1865 (9), 2293–2302. [DOI] [PubMed] [Google Scholar]
- 3.Ajoolabady A et al. (2020) Mitophagy receptors and mediators: therapeutic targets in the management of cardiovascular ageing. Ageing Research Reviews, 101129. [DOI] [PubMed] [Google Scholar]
- 4.Morales PE et al. (2020) Emerging role of mitophagy in cardiovascular physiology and pathology. Molecular aspects of medicine 71, 100822. [DOI] [PubMed] [Google Scholar]
- 5.Figueroa-Romero C et al. (2009) SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J 23 (11), 3917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu Q et al. (2020) Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ Res 126 (4), 456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gawlowski T et al. (2012) Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 287 (35), 30024–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DS and Kim JE (2018) PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis 9 (9), 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cribbs JT and Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8 (10), 939–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuzmicic J et al. (2011) [Mitochondrial dynamics: a potential new therapeutic target for heart failure]. Rev Esp Cardiol 64 (10), 916–23. [DOI] [PubMed] [Google Scholar]
- 11.Ajoolabady A et al. (2020) Enzyme-based autophagy in anti-neoplastic management: from molecular mechanisms to clinical therapeutics. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 1874 (1), 188366. [DOI] [PubMed] [Google Scholar]
- 12.Ajoolabady A et al. (2021) Targeting autophagy in ischemic stroke: From molecular mechanisms to clinical therapeutics. Pharmacology & Therapeutics, 107848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ajoolabady A et al. (2021) Targeting autophagy in neurodegenerative diseases: from molecular mechanisms to clinical therapeutics. Clinical and Experimental Pharmacology and Physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ajoolabady A et al. (2021) Deciphering the role of autophagy in heart failure. Cardiology Plus 6 (2), 92. [Google Scholar]
- 15.Pickrell AM and Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85 (2), 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deas E et al. (2011) Mitophagy and Parkinson’s disease: the PINK1–parkin link. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1813 (4), 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tolkovsky AM (2009) Mitophagy. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1793 (9), 1508–1515. [DOI] [PubMed] [Google Scholar]
- 18.Ding W-X and Yin X-M (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis. Biological chemistry 393 (7), 547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Twig G and Shirihai OS (2011) The interplay between mitochondrial dynamics and mitophagy. Antioxidants & redox signaling 14 (10), 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onishi M et al. (2021) Molecular mechanisms and physiological functions of mitophagy. The EMBO Journal 40 (3), e104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han H et al. (2020) PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep 21 (8), e48686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wenzel DM et al. (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474 (7349), 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iguchi M et al. (2013) Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. Journal of Biological Chemistry 288 (30), 22019–22032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazarou M et al. (2013) PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. Journal of Cell Biology 200 (2), 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wauer T and Komander D (2013) Structure of the human Parkin ligase domain in an autoinhibited state. The EMBO journal 32 (15), 2099–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trempe J-F et al. (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340 (6139), 1451–1455. [DOI] [PubMed] [Google Scholar]
- 27.Chaugule VK et al. (2011) Autoregulation of Parkin activity through its ubiquitin-like domain. The EMBO journal 30 (14), 2853–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuda N et al. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. Journal of Cell Biology 189 (2), 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng X and Hunter T (2013) Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell research 23 (7), 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kondapalli C et al. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open biology 2 (5), 120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiba-Fukushima K et al. (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Scientific reports 2 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan C et al. (2020) PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 16 (3), 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okatsu K et al. (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. Journal of Cell Biology 209 (1), 111–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiba-Fukushima K et al. (2014) Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS genetics 10 (12), e1004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ordureau A et al. (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Molecular cell 56 (3), 360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wauer T et al. (2015) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524 (7565), 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sauvé V et al. (2015) A Ubl/ubiquitin switch in the activation of Parkin. The EMBO journal 34 (20), 2492–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamano K et al. (2015) Site-specific Interaction Mapping of Phosphorylated Ubiquitin to Uncover Parkin Activation*♦. Journal of Biological Chemistry 290 (42), 25199–25211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kazlauskaite A et al. (2015) Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1-dependent phosphorylation and activation. EMBO reports 16 (8), 939–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar A et al. (2015) Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. The EMBO journal 34 (20), 2506–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarraf SA et al. (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496 (7445), 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan NC et al. (2011) Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Human molecular genetics 20 (9), 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stolz A et al. (2014) Cargo recognition and trafficking in selective autophagy. Nature cell biology 16 (6), 495–501. [DOI] [PubMed] [Google Scholar]
- 44.Wu W et al. (2016) FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 12 (9), 1675–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pei Z et al. (2021) FUNDC1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through ACSL4-mediated ferroptosis. Metabolism 122, 154840. [DOI] [PubMed] [Google Scholar]
- 46.Ren J et al. (2020) FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Science advances 6 (38), eabc8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma K et al. (2020) Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death & Differentiation 27 (3), 1036–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujiwara M et al. (2019) The mitophagy receptor Bcl-2–like protein 13 stimulates adipogenesis by regulating mitochondrial oxidative phosphorylation and apoptosis in mice. Journal of Biological Chemistry 294 (34), 12683–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhujabal Z et al. (2017) FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO reports 18 (6), 947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J and Ney PA (2009) Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death & Differentiation 16 (7), 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsushima M et al. (1998) Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes, Chromosomes and Cancer 21 (3), 230–235. [PubMed] [Google Scholar]
- 52.Marinković M et al. (2021) Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 17 (5), 1232–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ajoolabady A et al. (2020) Mitophagy receptors and mediators: therapeutic targets in the management of cardiovascular ageing. Ageing Research Reviews 62, 101129. [DOI] [PubMed] [Google Scholar]
- 54.Hoshino A et al. (2013) Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nature communications 4 (1), 1–12. [DOI] [PubMed] [Google Scholar]
- 55.Wang S-H et al. (2021) LncRNA H19 governs mitophagy and restores mitochondrial respiration in the heart through Pink1/Parkin signaling during obesity. Cell death & disease 12 (6), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu P et al. (2021) RIPK3 Induces Cardiomyocyte Necroptosis via Inhibition of AMPK-Parkin-Mitophagy in Cardiac Remodelling after Myocardial Infarction. Oxidative medicine and cellular longevity 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lv W et al. (2020) MiR-302a-3p aggravates myocardial ischemia-reperfusion injury by suppressing mitophagy via targeting FOXO3. Experimental and Molecular Pathology 117, 104522. [DOI] [PubMed] [Google Scholar]
- 58.Zhou H et al. (2017) Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox biology 13, 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang B et al. (2018) AMPKα2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circulation research 122 (5), 712–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fu W et al. (2019) Transient p53 inhibition sensitizes aged white adipose tissue for beige adipocyte recruitment by blocking mitophagy. Faseb j 33 (1), 844–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang HW et al. (2019) p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 38 (19), 3729–3742. [DOI] [PubMed] [Google Scholar]
- 62.Efeyan A and Serrano M (2007) p53: guardian of the genome and policeman of the oncogenes. Cell cycle 6 (9), 1006–1010. [DOI] [PubMed] [Google Scholar]
- 63.Chaanine A et al. (2012) JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell death & disease 3 (2), e265–e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang H et al. (2021) The zinc transporter ZIP7 (Slc39a7) controls myocardial reperfusion injury by regulating mitophagy. Basic Research in Cardiology 116 (1), 1–16. [DOI] [PubMed] [Google Scholar]
- 65.Hoshino A et al. (2012) p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. Journal of molecular and cellular cardiology 52 (1), 175–184. [DOI] [PubMed] [Google Scholar]
- 66.Opie LH, Metabolic management of acute myocardial infarction comes to the fore and extends beyond control of hyperglycemia, Am Heart Assoc, 2008, pp. 2172–2177. [DOI] [PubMed] [Google Scholar]
- 67.Mao S et al. (2021) Overexpression of PLK1 relieved the myocardial ischemia-reperfusion injury of rats through inducing the mitophagy and regulating the p-AMPK/FUNDC1 axis. Bioengineered 12 (1), 2676–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou H et al. (2018) Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death & Differentiation 25 (6), 1080–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou H et al. (2018) NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic research in cardiology 113 (4), 1–20. [DOI] [PubMed] [Google Scholar]
- 70.Ji W et al. (2016) Aldehyde dehydrogenase 2 has cardioprotective effects on myocardial ischaemia/reperfusion injury via suppressing mitophagy. Frontiers in pharmacology 7, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tong M et al. (2019) Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circulation research 124 (9), 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang S et al. (2019) Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy. Biochim Biophys Acta Mol Basis Dis 1865 (7), 1905–1914. [DOI] [PubMed] [Google Scholar]
- 73.Feng X et al. (2020) Mst1 Knockout Alleviates Mitochondrial Fission and Mitigates Left Ventricular Remodeling in the Development of Diabetic Cardiomyopathy. Front Cell Dev Biol 8, 628842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu W et al. (2017) Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1863 (8), 1973–1983. [DOI] [PubMed] [Google Scholar]
- 75.Wang P et al. (2019) SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. Journal of molecular and cellular cardiology 133, 125–137. [DOI] [PubMed] [Google Scholar]
- 76.Zhang J et al. (2022) Restoration of mitophagy ameliorates cardiomyopathy in Barth syndrome. Autophagy, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He L et al. (2019) PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKα and exacerbates atherosclerotic lesions. Journal of cellular physiology 234 (6), 8668–8682. [DOI] [PubMed] [Google Scholar]
- 78.Ma S et al. (2018) Melatonin ameliorates the progression of atherosclerosis via mitophagy activation and NLRP3 inflammasome inhibition. Oxidative medicine and cellular longevity 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ajoolabady A et al. (2022) Melatonin-Based Therapeutics for Atherosclerotic Lesions and Beyond: Focusing on Macrophage Mitophagy. Pharmacological Research 176, 106072. [DOI] [PubMed] [Google Scholar]
- 80.Devi TD et al. (2017) Aggravated postinfarct heart failure in type 2 diabetes is associated with impaired mitophagy and exaggerated inflammasome activation. The American journal of pathology 187 (12), 2659–2673. [DOI] [PubMed] [Google Scholar]
- 81.Gao B et al. (2021) Parkin overexpression alleviates cardiac aging through facilitating K63-polyubiquitination of TBK1 to facilitate mitophagy. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1867 (1), 165997. [DOI] [PubMed] [Google Scholar]
- 82.Wu Q et al. (2016) Dynamin-related protein 1 (Drp1) mediating mitophagy contributes to the pathophysiology of nervous system diseases and brain injury. Histology and histopathology 32 (6), 551–559. [DOI] [PubMed] [Google Scholar]
- 83.Abudureyimu M et al. (2020) Berberine promotes cardiac function by upregulating PINK1/Parkin-mediated mitophagy in heart failure. Frontiers in physiology 11, 1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang L et al. (2021) Dexpramipexole attenuates myocardial ischemia/reperfusion injury through upregulation of mitophagy. European Journal of Pharmacology 899, 173962. [DOI] [PubMed] [Google Scholar]
- 85.Khuanjing T et al. (2021) Donepezil attenuated cardiac ischemia/reperfusion injury through balancing mitochondrial dynamics, mitophagy, and autophagy. Translational Research 230, 82–97. [DOI] [PubMed] [Google Scholar]
- 86.Chang X et al. (2021) Quercetin Improves Cardiomyocyte Vulnerability to Hypoxia by Regulating SIRT1/TMBIM6-Related Mitophagy and Endoplasmic Reticulum Stress. Oxidative Medicine and Cellular Longevity 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hong Y-X et al. (2021) Cardiac senescence is alleviated by the natural flavone acacetin via enhancing mitophagy. Aging (Albany NY) 13 (12), 16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H. m. et al. (2021) Kanglexin delays heart aging by promoting mitophagy. Acta Pharmacologica Sinica, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Y et al. (2021) Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biology, 102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ryu D et al. (2016) Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 22 (8), 879–88. [DOI] [PubMed] [Google Scholar]
- 91.Fang EF et al. (2019) Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22 (3), 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xie C et al. (2022) Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat Biomed Eng 6 (1), 76–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fang EF et al. (2017) Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci Rep 7, 46208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fang EF et al. (2014) Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157 (4), 882–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Villa E et al. (2018) No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol 28 (11), 882–895. [DOI] [PubMed] [Google Scholar]
- 96.Melser S et al. (2013) Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab 17 (5), 719–30. [DOI] [PubMed] [Google Scholar]
- 97.Sandoval H et al. (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454 (7201), 232–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu L et al. (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14 (2), 177–85. [DOI] [PubMed] [Google Scholar]
- 99.Wu W et al. (2014) ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15 (5), 566–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Strappazzon F et al. (2015) AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 22 (3), 419–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gatica D et al. (2021) The role of autophagy in cardiovascular pathology. Cardiovasc Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yamano K et al. (2020) Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Biol 219 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wong YC and Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proceedings of the National Academy of Sciences 111 (42), E4439–E4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lazarou M et al. (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524 (7565), 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fu M et al. (2013) Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell 24 (8), 1153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Villa E et al. (2017) Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell reports 20 (12), 2846–2859. [DOI] [PubMed] [Google Scholar]
- 107.Orvedahl A et al. (2011) Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480 (7375), 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Szargel R et al. (2016) The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Human molecular genetics 25 (16), 3476–3490. [DOI] [PubMed] [Google Scholar]
- 109.Kageyama Y et al. (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33 (23), 2798–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Youle RJ and Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12 (1), 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zheng Q et al. (2018) Hsp70 participates in PINK1-mediated mitophagy by regulating the stability of PINK1. Neuroscience letters 662, 264–270. [DOI] [PubMed] [Google Scholar]
- 112.Wang X et al. (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147 (4), 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cunningham CN et al. (2015) USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nature cell biology 17 (2), 160–169. [DOI] [PubMed] [Google Scholar]
- 114.Bingol B et al. (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510 (7505), 370–375. [DOI] [PubMed] [Google Scholar]
- 115.Cornelissen T et al. (2014) The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Human molecular genetics 23 (19), 5227–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ajoolabady A et al. (2021) Targeting autophagy in ischemic stroke: From molecular mechanisms to clinical therapeutics. Pharmacology & Therapeutics 225, 107848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ajoolabady A et al. (2021) Targeting autophagy in neurodegenerative diseases: from molecular mechanisms to clinical therapeutics. Clinical and Experimental Pharmacology and Physiology 48 (7), 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Y et al. (2019) Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. Journal of Pineal Research 66 (2), e12542. [DOI] [PubMed] [Google Scholar]
- 119.Ajoolabady A et al. (2022) Melatonin-Based Therapeutics for Atherosclerotic Lesions and Beyond: Focusing on Macrophage Mitophagy. Pharmacological Research, 106072. [DOI] [PubMed] [Google Scholar]
- 120.Wang S et al. (2020) ALDH2 contributes to melatonin-induced protection against APP/PS1 mutation-prompted cardiac anomalies through cGAS-STING-TBK1-mediated regulation of mitophagy. Signal transduction and targeted therapy 5 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang S et al. (2018) Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition. J Cell Mol Med 22 (10), 5132–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xin T and Lu C (2020) Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging (Albany Ny) 12 (5), 4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jiang X et al. (2021) Irisin attenuates oxidative stress, mitochondrial dysfunction, and apoptosis in the H9C2 cellular model of septic cardiomyopathy through augmenting Fundc1-dependent mitophagy. Oxidative Medicine and Cellular Longevity 2021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]