

Abstract
Identifying chemical regulators of biological pathways is a time-consuming bottleneck in developing therapeutics and research compounds. Typically, thousands to millions of candidate small molecules are tested in target-based biochemical screens or phenotypic cell-based screens, both expensive experiments customized to each disease. Here, our uncustomized, virtual profile-based screening approach instead identifies compounds that match to pathways based on phenotypic information in public cell image data, created using the Cell Painting assay. Our straightforward correlation-based computational strategy retrospectively uncovered the expected, known small molecule regulators for 32% of positive-control gene queries. In prospective, discovery mode, we efficiently identified new compounds related to three query genes, and validated them in subsequent gene-relevant assays, including compounds that phenocopy or pheno-oppose YAP1 overexpression and kill a Yap1-dependent sarcoma cell line. This image profile-based approach could replace many customized labor- and resource-intensive screens and accelerate the discovery of biologically and therapeutically useful compounds.
One sentence summary:
If a genetic perturbation impacts cell morphology, a computational query can reveal compounds whose morphology “matches”.
eTOC
If a chemical compound and a gene overexpression yield the same cell morphology in the microscopy-based assay Cell Painting, then they are likely to impact the same functions. This principle is exploited to retrieve useful compounds for particular query genes in public Cell Painting datasets.
Graphical Abstract

Introduction
The pace of defining new diseases is rapidly accelerating (Roessler et al., 2021), as is the cost and time required to develop novel therapeutics (Wouters, McKee and Luyten, 2020), creating huge unmet need. The dominant drug-discovery strategies in the pharmaceutical industry and academia are target-based (biochemical) and phenotypic (cell-based) screening. Both require significant setup time, are tailored to a specific target, pathway, or phenotype, and involve physically screening thousands to millions of candidate compounds at great expense (Moffat et al., 2017). Computational approaches that allow virtual screening of small molecule modulators of pathways using the published literature or existing experimental data are beginning to emerge to accelerate drug discovery (Vamathevan et al., 2019; Schneider et al., 2020), but their predictive power is rarely evaluated systematically and prospectively.
Here we test a straightforward, image-based computational matching strategy; querying a compound library using image-based profiles of genes has not to our knowledge been previously attempted systematically. One would not expect the strategy to yield a high-fidelity predictive model for all genes/pathways, but for some subset, the method could readily identify a small set of candidate compounds that could be physically tested to confirm activity, as a sort of pre-screen. We use existing public data capturing the complex morphological responses of cells to a genetic perturbation (in the microscopy assay, Cell Painting (Bray et al., 2016)), then identify small molecules (i.e., chemical compounds) that produce the same (or opposite) response. In this assay, more than a thousand quantitative morphology features (including size, shape, intensity, texture, correlation, and neighbor relationships) are extracted from five-color images of cells stained with six fluorescent stains that label eight cellular components or organelles (Figure 1b), to create an image-based profile of the sample. Conceptually similar to transcriptional profiling (Subramanian et al., 2017), image-based profiling assays like Cell Painting are cheaper and already proven in many applications (Lapins and Spjuth, 2019; Chandrasekaran et al., 2020).
Figure 1: Image profile-based drug discovery offers efficient, virtual screening for pathway modulators.
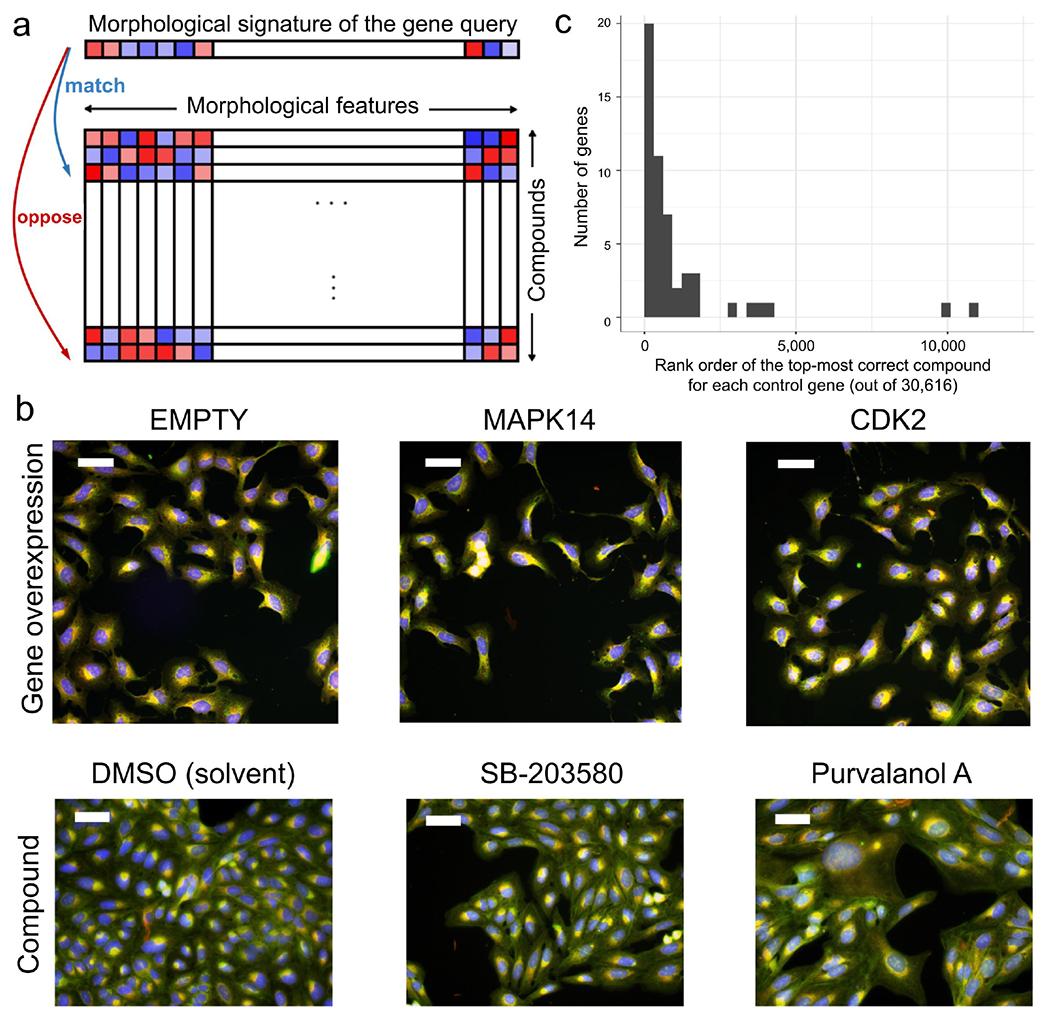
a) If an overexpressed gene changes the morphology of cells, its image-based profile can be used as a query in a database of small molecule profiles, looking for those that match (positively correlate) or oppose (negatively correlate). b) Cell Painting images for two positive control gene-compound matches that yield morphological phenotypes observable by eye (not all are expected to). EMPTY and DMSO are the negative controls in the gene overexpression and compound experiments, respectively; they differ in confluency and image acquisition conditions. The phenotype of p38α (MAPK14) overexpression matches (correlates to) that of SB-203580, a known p38 inhibitor; in both, elongated cells are over-represented. The phenotype of CDK2 overexpression (small cells) negatively correlates to that of purvalanol-a, a known CDK inhibitor, which induces an opposite phenotype (huge cells). Scale bars= 60 μm. c) The rank of the top correct compound for the control genes. Out of 63 genes with known compound matches in the set, 20 genes (32%) had a correct compound match in the top 1% of the 30,616 possible compounds (13 had a correct compound match in the top 0.5% (21%) and 36 genes (57%) had a correct compound match in the top 2.5%). Breakpoints in the histogram are set to [0, 306, 612, …] to correspond with 1% increments. The x-axis does not extend to all 30,616 compounds because all data points are in the top ~12,000.
Using image-based data to identify compounds that match a gene query has never been tested across a set of genes, making it difficult to estimate its potential. It is unknown whether gene perturbations yield profiles specific enough such that correlating compounds are likely to impact the function of a gene. It is also unknown whether anti-correlations of image-based profiles could reveal biological relationships. Evidence in favor of the strategy include anecdotal cases where perturbations of genes phenocopy the compounds targeting them (Mayer et al., 1999), though these usually were done in the context of having a compound of interest and looking for the target instead of vice versa. Almost two decades of research indicates that image profiles can identify compounds inducing phenotypes that match other compounds (Perlman et al., 2004) (reviewed in (Chandrasekaran et al., 2020)); this can be helpful in determining a compound’s mechanism of action (if the matching compounds are annotated)(Futamura et al., 2012; Schulze et al., 2013; Woehrmann et al., 2013; Foley et al., 2020; Schneidewind et al., 2020) and in identifying novel chemical structures with behavior similar to known compounds with desired bioactivity. This latter case can be useful for finding compounds with better physical-chemical or physiological properties, or working around intellectual property concerns, but it requires having a compound with the desired biological impact already. Finding compounds that impact a gene/pathway of interest where existing compounds are not already known is a much harder task, and much more likely to yield groundbreaking medicines.
Recent decades have given rise to an appealing, reductive ideal in the pharmaceutical industry: one drug that targets one protein to target one disease (Hughes et al., 2021). However, diseases often involve many interacting proteins and successful drugs often impact multiple targets (Bunnage, Chekler and Jones, 2013; Lin et al., 2019; Proschak, Stark and Merk, 2019). There is therefore a renewed appreciation for identifying small molecules that can modulate pathways or networks in living cell systems to yield a desired phenotypic effect (Hughes et al., 2021). Because genes in a pathway often show similar morphology (Rohban et al., 2017) and compounds often show similar morphology based on their mechanism of action as summarized above (Chandrasekaran et al., 2020), we examined image profile matching as a promising but untested route to capturing perturbations at the pathway level and accelerating the screening step prior to identifying useful therapeutics and research tool compounds.
Results
Image-based gene-compound matching: validation
We began with 69 unique genes whose overexpression yields a distinctive morphological phenotype by Cell Painting, from our prior study in U2OS cells (Rohban et al., 2017); roughly 50% of overexpression reagents in that study passed this criterion as did 45-54% in a more recent test of 160 overexpressed genes (Chandrasekaran et al., no date). We matched their image-based profiles to our public Cell Painting profiles of 30,616 small molecules (Bray et al., 2017) (Figure 1a), using simple Pearson correlation of population-averaged profiles (see “Scoring gene-compound and compound-compound connections” in Methods), avoiding machine learning methods due to their potential for overfitting and potential heightened sensitivity to experimental batch effects. We restricted matching to the 15,863 tested compounds (52%) whose profiles are distinguishable from negative controls, and confirmed that the profiles show variety rather than a single uniform toxic phenotype (Supplementary Figures S1 and S2).
We first verified that image-based profiles allow compounds to be matched with other compounds that share the same mechanism of action (for the subset that is annotated with this information). Consistent with past work (Chandrasekaran et al., 2020), top-matching compound pairs share a common annotated mechanism-of-action four times more often than for the remainder of pairs (p-value < 5×10−5 permutation test on the Odds ratio, Supplementary Figure S3 and “Permutation test for validation of known compound-compound pairs” in Methods).
We next attempted gene-compound matching. We did not expect a given compound to produce a profile that matches that of its annotated gene target in all cases, nor even the majority. Expecting simple gene-compound matching takes a reductionist view that does not reflect the complexity of typical drug action (see Introduction). We therefore included genes annotated as physically interacting with the gene of interest (using BioGRID; see “Compound annotations” in Methods) as a correct match, given our goal of identifying compounds with the same functional impact in the cell and not only directly targeting the protein product of the gene of interest. In addition, existing annotations are imperfect, particularly given the prevalence of under-annotation, mis-annotation, off-target effects, and polypharmacology, where small molecules modulate protein functions beyond the intended primary target (Lin et al., 2019; Proschak, Stark and Merk, 2019). Finally, technical reasons can also confound matching. The genetic and compound experiments were conducted years apart and by different laboratory personnel, yielding batch effects. They were performed in U2OS cells which may not be relevant for observing the annotated gene-compound interaction. In addition, the negative controls in a gene overexpression experiment (untreated cells), and a small molecule experiment (treated with the solvent control DMSO), do not produce identical profiles (left column, Figure 1b), and must therefore be normalized to align the negative controls in the feature space (see “Feature set alignment” in Methods). Despite these concerns, we persisted because even if the strategy worked in only a small fraction of cases, dozens of virtual screens and small validations of shortlisted predicted compounds could be done for less than the cost of a single traditional screening campaign - testing each compound in singlicate typically costs within an order of magnitude of $1 USD, and companies may screen millions of compounds. Even though downstream characterization, optimization, and target deconvolution of compounds will still be necessary, as for any drug discovery campaign, this virtual pre-screening could still translate to substantial resource savings.
Assessing the 63 genes out of 69 that had a compound annotated as targeting that gene in the set, we found that 32% of the genes successfully matched a compound, using an analysis of the highest-matching compounds per gene, described next. Because the method is intended as a virtual pre-screen that would feed a small proportion of predicted compounds to physical testing, our analysis mirrored this strategy by counting that 20 of the 63 genes had a ‘correct’ compound in the top 1% best-matching/opposing compounds in the experiment (Figure 1c; 1% = 306 out of 30,616 compounds in the library, with the 14,753 compounds not expressing a Cell Painting phenotype placed at the end of the rank order; p-value = 0.026 using 844 random permutations; see “Scoring gene-compound and compound-compound connections” and “Permutation test for validation of number of genes with a relevant compound match” in Methods); 1% represents a typical screen hit rate and a practical proportion for a confirmation screen of shortlisted compounds. Here, “correct” matches include cases where the corresponding protein of a gene interacts physically with at least one known annotated target of the compound (via BioGrid, 98.6% physical interactions). While 32% of genes were successful at the 1% shortlist level, the success rate increased to 36 genes (57%) having a correct compound match in the top 2.5% (Figure 1c).
We performed an alternate analysis, focusing not on one compound list per gene as in the two analyses above, but rather on the single ranked list of all 69 x 1177 gene-compound pairs together (69 genes with a phenotype and 1177 compounds that target any of them). Top-matching gene-compound pairs are correct matches 2.5 times more often than for the remainder of pairs (p-value = 0.002 one-sided Fisher test; Supplementary Figure S4 and Supplementary Table S1).
For some matches, we visually confirmed that gene overexpression phenocopies or pheno-opposes the matching/opposing compound (Figure 1b), although we emphasize that computationally-discovered phenotypes are not always visible to the human eye, particularly given cell heterogeneity. Examining the potential impact of polypharmacology, we found no relationship between the strength of correct gene-compound matches and the number of annotated targets a compound has (Supplementary Figure S5 and S6).
Throughout this study, we looked for compounds that both match (positively correlate) and oppose (negatively correlate) each overexpressed gene profile for several reasons. First, inhibitors and activators of a given pathway may both be of interest. Second, it is known that negative correlations among profiles can be biologically meaningful (Rohban et al., 2017). Third, overexpression may not increase activity of a given gene product in the cell; it could be neutral or even decrease it via a dominant-negative or feedback loop/compensatory effect. Fourth, the impact of a gene or compound perturbation could be cell-type specific; in our evaluation above, the existing compound annotations deemed as ground truth were determined in particular cell contexts; their behavior might differ in the U2OS cells used here. Supporting these theoretical arguments, we found that, empirically, both positively- and negatively-correlating matches were seen in our validation set (Supplementary Figure S4e). Furthermore, among the top 12 known gene-compound matches in our validation set, six showed correlation of the opposite directionality than expected (where expected is that an inhibitor’s profile would have the opposite correlation to its overexpressed target gene, Supplementary Figure S7).
Framing our approach as a virtual pre-screen, success even for 32% of Cell Painting-compatible genes would eliminate the need to carry out many dozens of large-scale customized screens in the pharmaceutical setting, instead advancing a few hundred compounds immediately to disease-relevant assays and saving hundreds of millions of dollars (see also Discussion for elaboration on success rates).
Image-based gene-compound matching: discovery
We next searched virtually for novel small molecule regulators of pathways (defined loosely here as groups of genes whose disruption has a similar impact on biological outcomes). For each of the 69 genes, we created a rank-ordered list of compounds (from the 15,863 impactful compounds of the 30,616 set) based on the absolute value of correlation to that gene, enforcing a minimum of 0.35 (https://github.com/carpenterlab/2021_Rohban_submitted/blob/1de4fe928c30b8118a19be0736d15c3adae0d7d9/corr_mat.csv.zip). Because there is no systematic experiment to validate compounds impacting diverse pathways, we took a customized expert-guided approach to ensure the results are biologically meaningful rather than just statistically significant. We found seven experts studying pathways with strong hits who were willing to conduct experiments; they chose the most relevant biological systems and readouts, rather than simply attempting to validate the original image-based finding. The experts decided on the number of compounds reasonable for them to test in their experimental assay setup, and we provided them with the top matches from our list (subject to availability of sufficient compound). In a pharmaceutical context, we would recommend choosing the top 1% of compounds to test (n=306 in this case); due to throughput limitations here, we tested < the top 0.1% of the compounds (n = 9-33, except for RAS where n = 236).
Two cases yielded no confirmation (data not shown): SMAD3 and RAS. Nine compounds matching or opposing the SMAD3 overexpression profile failed to yield activity in a transcription reporter assay in A549 lung carcinoma cells involving tandem Smad binding elements, with and without Transforming growth factor beta 1 (TGF-β1). 236 compounds with positive or negative correlations to the wild-type RAS or oncogenic HRAS G12V differential profile (see Methods) failed to elicit a RAS-specific response in a 72-hour proliferation assay using isogenic mouse embryonic fibroblast (MEF) cell lines driven by human KRAS4b G12D, HRAS WT, or BRAF V600E alleles but otherwise devoid of RAS isoforms (Drosten et al., 2010). We cannot distinguish whether the compounds were inactive due to major differences in the cell types or readouts, or whether these represent a failure of morphological profiling to accurately identify modulators of the pathway of interest.
A third case affirmed the approach but the novel compound identified was not very potent. We tested 17 compounds that negatively correlated with CSNK1E overexpression in a biochemical assay for the closely related kinase CSNK1A1. Three (SB 203580, SB 239063, and SKF-86002) had inhibitory IC50 concentrations in the nanomolar range at Km ATP. Inhibition of CSNK1 family members by these compounds is supported by published kinase profiling studies (Shanware et al., 2009; Davis et al., 2011; Klaeger et al., 2017). A fourth compound, BRD-K65952656, failed to bind any native kinases in a full KINOMEscan panel, suggesting it mimics CSNK1A1 inhibition via another molecular target. We chose not to pursue the expensive step of target deconvolution given its weak inhibition of CSNK1A1 (IC50 12 μM).
A fourth case affirmed the approach but the novel compound failed to replicate following compound resynthesis, suggesting the desired activity, although validated, was not due to the expected structure, perhaps due to breakdown. We tested 16 compounds that positively correlated and 17 compounds that negatively correlated to GSK3B overexpression, for impact on GSK3α and GSK3β (which generally overlap in function) in a non-cell-based, biochemical assay. This yielded four hits with GSK3α IC50s ≤ 10 μM; the two most potent failed to show activity following resynthesis and hit expansion (testing of similarly-structured compounds) (Supplementary Table S2).
We did not pursue these cases further in light of the success for the three other cases, described next.
Discovery of hits modulating the p38α (MAPK14) pathway
p38α (MAPK14) inhibitors are sought for a variety of disorders, including cancers, dementia, asthma, and COVID-19 (MAPK14 - ClinicalTrials.Gov, no date; Martínez-Limón et al., 2020). We chose 20 compounds whose Cell Painting profile matched (n=9) or opposed (n=11) that of p38α overexpression in U2OS cells. In a single-cell p38 activity reporter assay in retinal pigment epithelial (RPE1) cells (Regot et al., 2014; Liu et al., 2018), we identified many activating compounds; these are less interesting given that the p38 pathway is activated by many stressors but rarely inhibited. We also found several inhibiting compounds and confirmed their activity (Figure 2, Supplementary Figure S8), including a known p38 MAPK inhibitor; most had diverse chemical structures (Supplementary Figure S9a). Although the novel compounds are relatively weak, they nevertheless prove the principle that p38 pathway modulators can be found by image-profile matching, without a specific assay for the gene’s function.
Figure 2: Cell Painting profiles identify compounds impacting the p38 pathway.
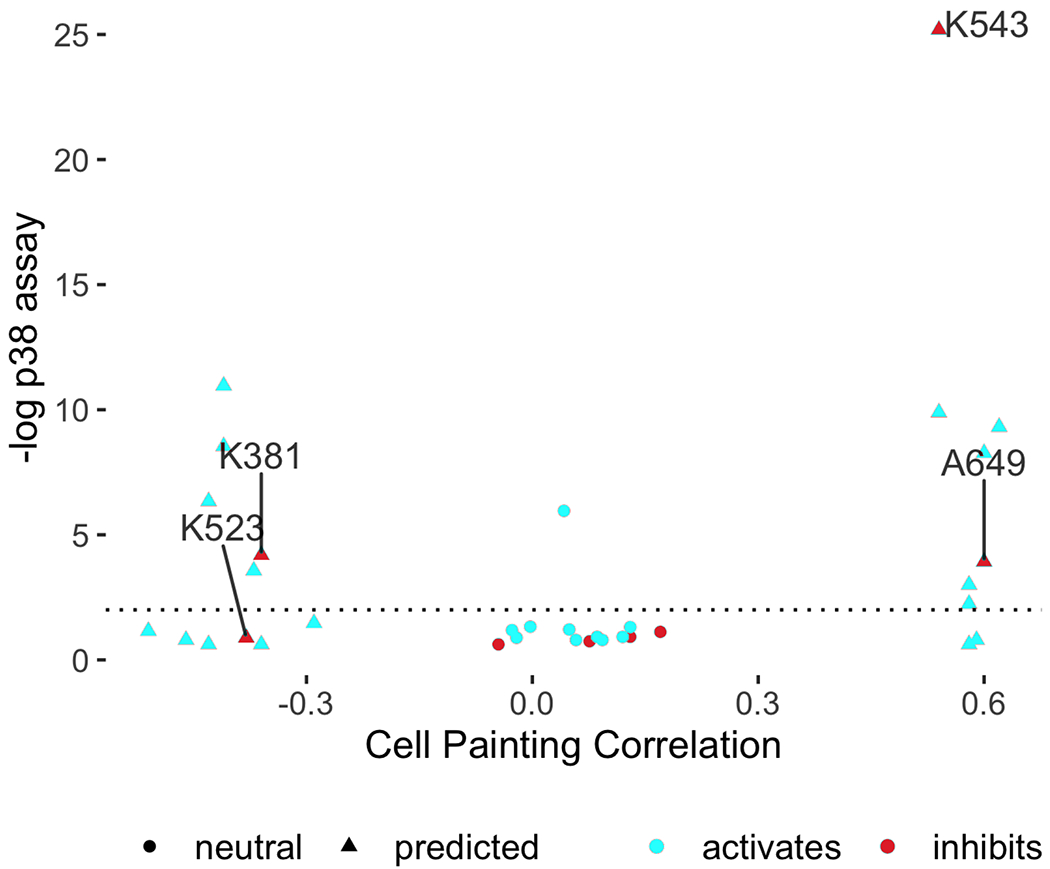
Compounds predicted to perturb p38 activity (triangles) and a set of 14 neutral compounds (Cell Painting profile correlations to p38α between −0.2 to 0.2; circles) were tested for their influence on p38 activity at 1 μM using a two-sided t-test on the single cell distributions of a p38 activity reporter (Kaufman et al., 2020) (FDR-adjusted −log10 p-values shown). Two potential inhibitors were found (BRD-K38197229 <K381> and BRD-A64933752 <A649>); an additional one (BRD-K52394958 <K523>) was identified via an alternative statistical test (Supplementary Figure S8a, h–i). K543 (BRD-K54330070) denotes SB-202190, a known p38 inhibitor found as a match (other known inhibitors such as SB-203580 from Figure 1 were strong matches but excluded from this experiment because they were already known). The x-axis reports the Pearson correlation between the average Cell Painting profile of the compound with the average Cell Painting profile of MAPK14 overexpression, where the averages are computed across five replicates for MAPK14, eight replicates for bioactive compounds, and four replicates for diversity-oriented synthesis (DOS) compounds. The y-axis reports the average FDR-adjusted −log10 p-values across four replicates.
Discovery of hits impacting PPARGC1A (PGC-1α) overexpression phenotypes
We next identified compounds with strong morphological correlation to overexpression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α, encoded by the PPARGC1A gene). We found that these compounds tend to be hits in a published, targeted screen for PGC1α activity (p=7.7e-06, Fisher’s exact test) (National Center for Biotechnology Information, no date), validating our image profile-based matching approach. The dominant matching phenotype is mitochondrial blobbiness, which can be quantified as the high standard deviation of the MitoTracker staining at the edge of the cell (Figure 3a,b) without major changes to cell proliferation, size, or overall protein content. Cell subpopulations that are large, multi-nucleate, and contain fragmented mitochondria are over-represented when PGC-1α is overexpressed while subpopulations whose organelles are asymmetric are under-represented (Supplementary Figure S10). More symmetric organelle morphology has been associated with reduced motility and PGC-1α overexpression (Lee et al., 2009). The role of PGC-1α in mitochondrial biogenesis is well-appreciated (Luo, Widlund and Puigserver, 2016). The phenotype uncovered here using image profile matching is consistent with other recently discovered mitochondrial phenotypes associated with this gene (Halling and Pilegaard, 2020).
Figure 3: Cell Painting profiles identify compounds impacting PPARGC1A (PGC-1α) overexpression phenotypes.
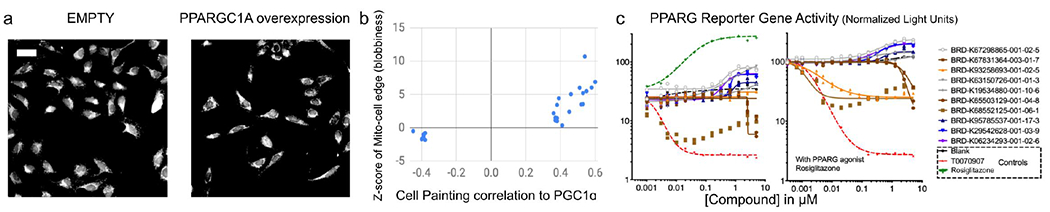
a) Cell Painting images for PPARGC1A (PGC-1α) overexpression compared to negative control (EMPTY, same field as in Figure 1a). Scale bar = 60 ϋm. b) Compounds with high or low correlations of their Cell Painting profiles to PGC-1α overexpression were chosen for further study (hence all samples are below ~−0.35 or above ~0.35 on the X axis, which shows the Pearson correlation between the single, consensus gene profile and the single, consensus compound profile. The consensus profiles are created using median across 5 replicates for gene overexpressions, eight replicates for bioactive compounds, and four replicates for diversity-oriented synthesis (DOS) compounds). Correlation to PGC-1α overexpression is dominated by one feature, the standard deviation of the MitoTracker staining intensity at the edge of the cell, which we term blobbiness (displayed on the Y axis as a z-score with respect to the negative controls and is calculated based on eight replicates for bioactive compounds, and four replicates for DOS compounds). c) PPARG reporter gene assay dose-response curves in the absence (left) or presence (right) of added PPARG agonist, Rosiglitazone. The ten most active compounds are shown and reported as normalized light units; data representative of two experiments; two replicate data points are shown for each BRD compound but sometimes overlap. Line of best fit generated using GraphPad PRISM using variable slope four parameters fit of log(inhibitor) vs. response. Compounds highlighted in blue/purple are structurally related pyrazolo-pyrimidines.
We chose 24 compounds whose Cell Painting profiles correlated positively or negatively with PGC-1α overexpression in U2OS cells (Supplementary Table S3); they have generally diverse chemical structures (Supplementary Figure S9b) and one is a known direct ligand for PPAR gamma, GW-9662 (BRD-K93258693). PGC-1α is a transcriptional coactivator of several nuclear receptors including PPAR gamma and ERR alpha (Handschin and Spiegelman, 2006). We therefore tested compounds in a reporter assay representing FABP4, a prototypical target gene of the nuclear receptor, PPARG (Goldstein et al., 2017), in a bladder cancer cell line (Figure 3c). Three of the five most active compounds leading to reporter activation (among all the 24 compounds tested) were structurally related and included two annotated SRC inhibitors, PP1 and PP2, which have a known link to PGC1α (Usui, Uno and Nishida, 2016), as well as a novel analog thereof. Inhibitors uncovered were CCT018159 (BRD-K65503129) and Phorbol 12-myristate 13-acetate (BRD-K68552125). Many of the same compounds also showed activity in a ERRalpha reporter assay in 293T cells, albeit with differing effects (Supplementary Figure S11).
Encouraged by these results, we tested the impact of the compounds on mitochondrial motility, given the mitochondrial phenotype we observed and the role of PGC1α in mitochondrial phenotypes and neurodegenerative disorders (Nierenberg et al., 2018). In an automated imaging assay of rat cortical neurons (Shlevkov et al., 2019), we found several compounds decreased mitochondrial motility; none increased motility (Supplementary Figure S12). Although the latter is preferred due to therapeutic potential, this result suggests that the virtual screening strategy, applied to a larger set of compounds, might identify novel motility-promoting compounds. We found 3 of the tested compounds suppress motility but do not decrease mitochondrial membrane potential; this is a much higher hit rate (13.0%) than in our prior screen of 3,280 bioactive compounds, which yielded two such compounds (0.06%)(Shlevkov et al., 2019).
Discovery of small molecules impacting YAP1-related phenotypes
The Hippo pathway affects development, organ size regulation, and tissue regeneration. Small molecule regulators are highly sought for research and as potential therapeutics for cancer and other diseases; the pathway has been deemed relatively undruggable (Dey, Varelas and Guan, 2020; Tang et al., 2021). We tested 30 compounds (Supplementary Table S4) whose Cell Painting profile matched (25 compounds) or opposed (5 compounds) the overexpression of the Hippo pathway effector Yes-associated protein 1 (YAP1), which we previously explored (Rohban et al., 2017) (Supplementary Table S5, images shown in Supplementary Figure S13). The compounds have generally diverse chemical structures (Supplementary Figure S9c). One hit, fipronil, has a known tie to the Hippo pathway: its impact on mRNA profiles matches that of another calcium channel blocker, ivermectin, a potential YAP1 inhibitor (Nishio et al., 2016) (99.9 connectivity score in the Connectivity Map(Subramanian et al., 2017)). After identifying five promising compounds from the 30 compounds tested in a cell proliferation assay in KP230 cells (described later), we focused on the three strongest in various assays and cell contexts, as follows.
N-Benzylquinazolin-4-amine (NB4A, BRD-K43796186) is annotated as an EGFR inhibitor and shares structural similarity with kinase inhibitors. NB4A showed activity in 30 of 606 assays recorded in PubChem, one of which detected inhibitors of TEAD-YAP interaction in HEK-TIYL cells. Its morphological profile positively correlated with that of YAP1 overexpression (0.46) and, consistently, negatively correlated with overexpression of STK3/MST2 (−0.49), a known negative regulator of YAP1.
Because the Hippo pathway can regulate the pluripotency and differentiation of human pluripotent stem cells (hPSCs) (Musah et al., 2014; Zaltsman et al., 2019), we investigated the effect of NB4A in H9 hPSCs. NB4A did not affect YAP1 mRNA expression but increased the expression of YAP1 target genes (CTGF and CYR61) in a dose-dependent manner (Figure 4a), confirming it impacts YAP1 phenotypes. Accordingly, NB4A increased YAP1 nuclear localization (Figure 4b). While decreasing total YAP1 protein levels, NB4A also reduced YAP1 S127 phosphorylation (Figure 4c and Supplementary Figure S14a), which promotes YAP1 cytoplasmic sequestration (Zhao et al., 2007).
Figure 4: Cell Painting profiles identify compounds impacting YAP1 phenotypes.
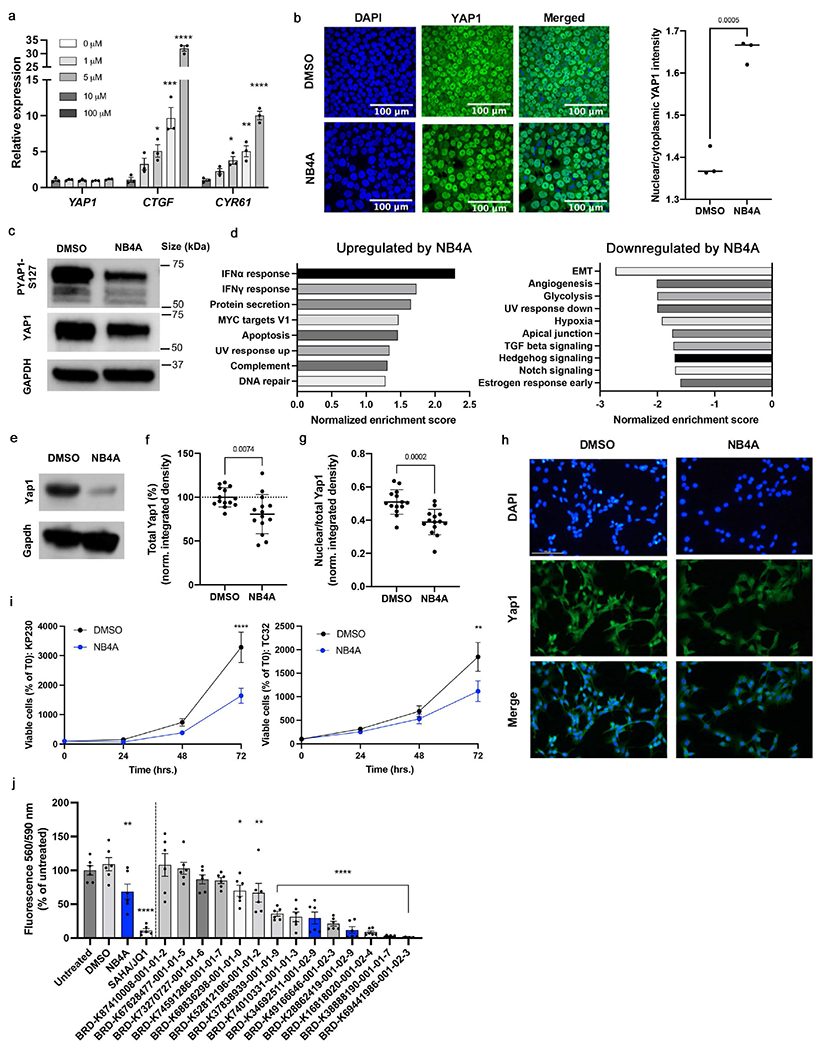
a) Relative transcript levels of YAP1, CTGF, and CYR61 in H9 human pluripotent stem cells treated with NB4A or DMSO control for 24 hrs. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001 (one-way ANOVA with Dunnett’s multiple comparisons test). Mean ± SEM. n = 3 biologically independent experiments. b) Representative images of YAP1 immunofluorescence (left) and quantification of nuclear/cytoplasmic YAP1 mean intensity (right) in H9 cells after treatment with 10 μM NB4A or DMSO control for 24 hours. Two-tailed student’s t-test. n = 3 biologically independent experiments; an average of mean intensities from 3 fields of each biological replicate is calculated. c) Representative blot of n = 3 biologically independent experiments for phospho-YAP1 (S127) and total YAP1 from H9 cells treated with DMSO or 10 μM NB4A for 24 hrs, with GAPDH as loading control (quantified in Supplementary Figure S14a). d) Normalized enrichment scores of GSEA show up to 10 of the most significant Hallmark pathways up- and down-regulated in NB4A-treated vs. control KP230 cells (FDR-adjusted P<0.25). n = 3. IFNα = interferon alpha; IFNγ = interferon gamma; EMT = epithelial-mesenchymal transition. e) Representative western blot for Yap1 in NB4A-treated and control KP230 cells. f) Immunofluorescence-based analysis of total Yap1 in NB4A-treated and control KP230 cells. Two-tailed student’s t-test. Mean ± SEM. n = 3. g) Immunofluorescence-based analysis of nuclear Yap1 in NB4A-treated and control KP230 cells (normalized to total Yap1). Two-tailed student’s t-test. Mean ± SEM. n = 3. For f and g, the Y axis is integrated density normalized to cell number and representative images are shown in (h), out of 5 fields acquired per condition. Scale bar (top left panel) = 100 μM. i) Growth curves of NB4A-treated and control KP230 and TC32 sarcoma cells. **P<0.01; ****P<0.0001 DMSO vs. NB4A (12 hrs.; 2-way ANOVA with Sidak’s multiple comparisons test). Mean ± SEM. n = 3. For panels d-i, cells were treated with 10 μM NB4A daily for 12 hours. j) PrestoBlue screen of KP230 cells treated with various analogs of BRD-K28862419 and BRD-K34692511 (10 μM daily for 48 hours). *P<0.05; **P<0.01; ****P<0.0001 vs. DMSO (one-way ANOVA with Dunnett’s multiple comparisons test). Mean ± SEM. n = 2 biological replicates, each with 3 technical replicates. SAHA (2 μM)/JQ1 (0.5 μM), which inhibits Yap1 expression and transcriptional activity in sarcoma cells (Ye et al., 2018), was used as a positive control. Blue bars indicate the original hits (NB4A, BRD-K28862419 and BRD-K34692511). All test compounds were verified at purity > 75%.
Effects of NB4A on YAP1 mRNA expression were not universal across cell types, consistent with the Hippo pathway’s known context-specific functions. In most cell types represented in the Connectivity Map, YAP1 mRNA is unaffected, but in HT29 cells, YAP1 mRNA is up-regulated after six hours of NB4A treatment (z-score = 3.16; also z-score = 2.04 for TAZ) and in A375 cells, YAP1 mRNA is slightly down-regulated (at 6 and 24 hours; z-score ~ −0.7) (Subramanian et al., 2017). NB4A had no effect in a YAP1-responsive reporter assay following 48h of YAP overexpression in HEK-293 cells (Supplementary Figure S14b).
Compounds influencing the Hippo pathway might be therapeutic for undifferentiated pleomorphic sarcoma (UPS), an aggressive mesenchymal tumor that lacks targeted treatments (Ye et al., 2018). In UPS, YAP1 promotes tumorigenesis and is inversely correlated with patient survival (Ye et al., 2018). In KP230 cells, derived from a mouse model of UPS (Ye et al., 2018), Yap1 protein levels were reduced after 72 hours of NB4A treatment (Figure 4e–f, h). NB4A also significantly attenuated Yap1 nuclear localization (Figure 4g–h), which is known to reduce its ability to impact transcription. Interestingly, NB4A did not directly alter transcription of Yap1, its sarcoma target genes (Foxm1, Ccl2, Hbegf, Birc5, and Rela), nor Yap1’s negative regulator, angiomotin (Amot) (data not shown). Instead, pathways such as interferon alpha and gamma responses were up-regulated, whereas pathways such as the epithelial-mesenchymal transition, angiogenesis, and glycolysis were down-regulated, according to RNA sequencing and gene set enrichment analysis (Figure 4d; Supplementary Table S6). This indicates a potentially useful mechanism distinct from transcriptional regulation of YAP1.
Genetic and pharmacologic inhibition of Yap1 is known to suppress UPS cell proliferation in vitro and tumor initiation and progression in vivo (Ye et al., 2018). Consistent with being a Hippo pathway regulator, NB4A inhibited the proliferation of two YAP1-dependent cell lines: KP230 cells and TC32 human Ewing’s family sarcoma cells (Hsu and Lawlor, 2011) (Figure 4i). NB4A did not affect the proliferation of two other YAP1-dependent lines, STS-109 human UPS cells (Supplementary Figure S15a) and HT-1080 fibrosarcoma cells (Supplementary Figure S15b) (Eisinger-Mathason et al., 2015; Ye et al., 2018), nor YAP1-independent HCT-116 colon cancer cells (Supplementary Figure S15c–e). Interestingly, NB4A treatment did not exhibit overt toxicity by trypan blue staining in any of these (not shown), suggesting it inhibits cell proliferation by a mechanism other than eliciting cell death.
Next, we investigated two structurally similar compounds (BRD-K28862419 and BRD-K34692511, distinct from NB4A’s structure) whose Cell Painting profiles negatively correlated with YAP1’s overexpression profile (−0.43 for BRD-K28862419 and −0.45 for BRD-K34692511) and positively correlated with TRAF2 overexpression (0.41 for BRD-K28862419 and 0.29 for BRD-K34692511) (Supplementary Figure S13). These compounds are not commercially available, limiting our experiments and past literature.
We assessed the compounds’ impact on mesenchymal lineage periosteal cells isolated from 4-day old femoral fracture callus from mice with DOX-inducible YAP-S127A. BRD-K34692511 substantially upregulated mRNA levels of relevant Hippo components including Yap1 and Cyr61 after 48 hours of treatment, but not at 1 and 4 hours (Supplementary Figure S14c–f). By contrast, the compounds had no effect on YAP1 or its target genes in H9 hPSCs (Supplementary Figure S14g), nor in a 48 h YAP-responsive reporter assay following YAP overexpression in HEK-293 cells (Supplementary Figure S14b).
Like NB4A, the effects of these compounds on proliferation varied across cell types. In the U2OS Cell Painting images, BRD-K28862419 reduced proliferation (−2.0 st dev). Per PubChem, it inhibits cell proliferation in HEK293, HepG2, A549 cells (AC50 5-18 μM) and it inhibits PAX8, which is known to influence TEAD/YAP signaling(Elias et al., 2016). BRD-K34692511 had none of these impacts.
Both compounds had the desired effect of inhibiting KP230 cell proliferation (Supplementary Figure S15f). Also noteworthy, BRD-K28862419 modestly yet significantly reduced KP230 cell viability (Supplementary Figure S15g), indicating its mechanism of action and/or therapeutic index may differ from that of NB4A and BRD-K34692511.
Finally, we resynthesized both of these non-commercially available compounds to ensure their integrity and identity, and selected a set of close analogs of such compounds to investigate structure-activity relationship. Using a high-throughput resazurin-based screen (Presto Blue assay), we confirmed the activity of the two resynthesized compounds to reduce proliferation of KP230 cells, and furthermore found eight additional analogs with activity (Figure 4J). In fact, four of the analogs were more potent than one or both of the original compounds, reducing KP230 cell growth by 78.70-98.95% vs. untreated cells (Figure 4J). Structural analysis reveals that the macrocycle is quite robust to changes in stereochemistry, and that compounds with an amide linker tend to outperform those with a urea linker (Supplementary Figure S16).
In summary, although deconvoluting the targets and behaviors of these compounds in various cell contexts remains to be further ascertained, we conclude that the strategy identified compounds that modulate YAP1-related phenotypes, in particular an unusual ability to reduce growth of certain aggressive sarcoma lines. This demonstrates that, although the directionality and cell specificity will typically require further study, image-based pathway profiling can identify modulators of a given pathway.
Discussion
We found that hit-stage small molecule regulators of pathways of interest can be discovered by virtual matching of genes and compounds using Cell Painting profiles, which we term image profile-based compound screening. The approach provides a simple, minimal-resource approach to shortlist top candidates to target the pathway of a gene of interest, so long as the gene produces a detectable image-based profile. It can reduce the cost of compound screening by orders of magnitude by enriching the signal to noise ratio from compound libraries. We do not claim the particular compounds we uncovered in this study are sufficiently potent, specific, and non-toxic for human therapeutics. As with all screening approaches, significant further work is necessary to develop hits into useful therapeutics; this includes confirming activity and directionality of hits in a relevant cell type or model system (as we did successfully for three cases here), improving potency and specificity, and identifying the molecular target(s) (including so-called off-target effects that may or may not be useful to achieve the desired phenotypic effect); these latter are significant steps. Further, like all drug discovery, the eventual clinical success relies on the therapeutic hypothesis for the gene, pathway, and/or phenotype being correct, which is also challenging.
Even so, virtualizing a large-scale screen by computationally matching the phenotypic effect of compounds to that of gene manipulation will in many cases enable rapid and inexpensive identification of compounds with desired phenotypic impacts, and avoid an expensive large-scale physical screen. Our approach yielded hits with substantial chemical structure diversity, which is another advantage.
Other virtual drug screening methods have been proposed; to our knowledge, none have been systematically evaluated that take a gene name as input. For those using alternative strategies (e.g. predicting outcomes in a particular assay that is used to train the model), their success rates across a broad swath of biology are unavailable or difficult to compare to this study. Many report retrospective rates on known compound hits (which can unfortunately be prone to overfitting) but do not prospectively identify novel hits; others report success in identifying a novel compound prospectively but do not mention the failures, making it impossible to compute the success rate across multiple diverse genes. To our knowledge, ours is the only study to test a sufficient number of genes to report success rates at the gene level, both retrospectively (63 genes) and prospectively (7 genes). Other methods report predictions with ~8-70% success rates, but at the assay level (Simm et al., 2018; Martin et al., 2019; Trapotsi et al., 2021) (and with varying degrees of stringency in how success is defined). In other words, they predict compounds’ assay activity rather than compounds impacting a given gene function. To study a new area of biology, these methods require (a) developing an assay to target the biological phenotype of interest (this criterion means these methods’ reported success rates have come from testing on only a subset of biological “space”, which is likely better-studied), and (b) testing a pilot set of compounds in the assay to train a machine learning model (typically thousands of compounds, which is resource intensive). This is much more intensive effort than what is needed for the strategy presented here: a morphological profile of a single gene perturbation (which will soon be publicly available at the genome-scale, see below, but can be done in any laboratory with a suitable microscope within a few days), and profiles for compounds (which are already publicly available in the tens of thousands). No other data is required (e.g. to pre-train models), nor special equipment, nor extensive computational expertise nor compute time.
As mentioned, most virtual compound prediction studies report only retrospective assay-level success rates - these are certainly valuable, but a lower bar that, unfortunately, can be inflated by unexpected sources of overfitting. For example, if too-similar assays or too-similar compounds are included in both training and test sets, success rates will be inflated; only some studies control for this well. To clarify success rates for this study in particular, using any gene as an input, roughly 50% will pass at the first step (having a distinguishable morphological phenotype under the assay conditions, though other cell types or stains could be attempted), and the remainder are estimated to succeed at a rate of 32-71% as follows: our study showed a 32% retrospective success rate for known gene-compound connections (for a 1% shortlist); given these known pairs include highly optimized chemical matter, we wondered whether we would see a lower success rate in actual prospective validation. Nevertheless, we discovered a 43% success rate (albeit with low n: 3 out of 7 genes tested); we also note the rate might even be considered 71% (5 of 7) if we consider the other two genes as successes whose hits were abandoned due to chemistry and potency. Due to resource limits, we tested ~0.1% of the best-matching compounds rather than the recommended 1%, which would likely also improve results (we tested for SMAD3: 9 compounds, RAS: 236, CSNK1A1: 17, GSK3B: 33, p38 MAPK14: 20, PPARGC1A: 24, YAP1: 30). Overfitting is not a concern in our study; we used simple metrics of correlation rather than machine learning that runs that risk; avoiding machine learning also may make matching of profiles across datasets created under very different conditions more successful. As a side note, we do not emphasize success rates in terms of how many of the selected compounds for each gene were validated in the followup assays, because in practice, hundreds of compounds should be shortlisted for testing and the major metric of interest to the screener is whether good chemical matter is in that set, not whether the rate within the set is 1% or 100%.
We expect future iterations of this strategy to be more successful. First, we would expect better-quality chemical matter from larger libraries; only 30,000 were screened in this work whereas a pharmaceutical screening campaign can test millions (Mullard, 2019). Large-scale data production efforts are underway that will increase the potential for matching profiles against public data: the JUMP-Cell Painting Consortium is producing a public dataset of 140,000 chemical and genetic perturbations. It is remarkable that the image-based profile matching strategy worked for two datasets created years apart by different personnel and equipment; we expect improvements if the query gene and compound library were created more consistently. The limits of the approach remain to be tested: how different could image sets be, in terms of resolution, confluency, imaging modality, and even cell type? Creating data using other staining sets, cell contexts, or more complex biological models, such as co-cultures, primary cells, or organoids could increase the probability of success for some pathways, as could assessing whether gene knockdown profiles (e.g. by CRISPR) yield better results in practice than gene overexpression. Pathways where overexpression and knockdown give opposite profiles may be even better starting points for virtual screening, as might merged profiles based on several pathway members of interest rather than a single gene. In general, further study is needed to understand the frequency and general principles governing the directionality of matching between compounds and gene overexpression and knockdown. The JUMP dataset should provide sufficient scale and annotation to assess the hypothesized mechanisms described in the validation section above.
Another major direction for the future is making better predictions by integrating morphology profiles with other data sources when available at scale, such as transcriptomic(Haghighi et al., 2021), proteomic, and metabolomic(Holbrook-Smith, Durot and Sauer, 2022) profiles, or historical assay data(Martin et al., 2019). Chemical structure information might also be useful(Trapotsi et al., 2021), though this would require significant adaptation to incorporate because it is not a property one can obtain for the genes used as queries in our matching approach, and the goal is not to identify compounds of similar structure (diversity is usually preferred). More advanced computational methods are also on the horizon, from feature extraction (Pratapa, Doron and Caicedo, 2021) to machine learning on new benchmark datasets of gene-compound pairs (Chandrasekaran et al., 2021); we would expect supervised machine learning to work better than our unsupervised correlation-based approach (Chandrasekaran et al., 2020).
We hope that image profile-based virtual screening will be a new accelerant toward meeting the pressing need for novel therapeutics. This study adds to the growing evidence that image-based profile matching might accelerate drug discovery in multiple ways (Figure 5). Here we used, for the first time, a gene-based query to identify matching compounds in a virtual pre-screening strategy. Other applications have been demonstrated already, such as identifying a compound’s mechanism of action by matching to other compounds, another significant bottleneck in the drug discovery process (Ha et al., 2021). The release of large public datasets offers opportunities for laboratories to carry out these strategies using public data as input queries, or profiles generated via their own small-scale laboratory experiments.
Figure 5: Multiple potential strategies to accelerate drug discovery by image-profile matching.

(A) Many perturbations can yield distinctive image-based profiles to be used as queries against available databases of image-based profiles. In the study, exogenous overexpression of a gene was used as the query (blue, left) to interrogate a public library of small molecule profiles (blue, right) to identify candidate regulators of that gene’s function, to achieve virtual compound screening. (B) Many public and private sources of profiles could be used to achieve many goals, reviewed in detail in (Chandrasekaran et al., 2020). *JUMP-CP data is scheduled for public release November 1, 2022.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anne Carpenter (anne@broadinstitute.org).
Materials Availability
Cell line and DNA construct availability
Cell lines and DNA constructs are available from the laboratories that performed the experiments using them, or where restricted by licensing, from commercial sources.
Data and Code Availability:
Source data statement
The large-scale Cell Painting datasets used in this paper are publicly available and their details and locations are described in publications (gene overexpression dataset(Rohban et al., 2017) and compound dataset(Bray et al., 2017)). RNA-sequencing data have been deposited into the NCBI Gene Expression Omnibus (GEO; accession number GSE198909). The accession numbers are also listed in the key resources table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HRP-conjugated anti-rabbit IgG antibodies at 1:10000 | Kiessling lab; Jackson ImmunoResearch Laboratories | #111-035-003 |
| HRP-conjugated anti-rabbit IgG antibodies at 1:2500 | Eisinger lab; Cell Signaling Technology [CST] | #7074 |
| anti-Yap1 primary antibodies | Cell Signaling Technology [CST] | #14074, #4912 |
| Alexa Fluor 488-conjugated secondary antibodies | Thermo Fisher Scientific | #A-11008 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| BRD-K43796186 (NB4A) | MuseChem | cat. #M189943 |
| BRD-K43796186 (NB4A) | Ambinter | cat # Amb2554311 |
| 8xGTIIC-luciferase | Gift from Stefano Piccolo, Addgene | plasmid # 34615 |
| Critical commercial assays | ||
| Deposited data | ||
| RNA-sequencing data | this paper | GSE198909 |
| Compound Dataset | Bray et al., 2017 | DOI: 10.1093/gigascience/giw014 |
| Gene Overexpression Dataset | Rohban et al., 2017 | DOI:10.7554/eLife.24060 |
| Experimental models: Cell lines | ||
| RT112/84 | Cancer Cell Line Encyclopedia (Broad Institute, Cambridge, MA) | |
| HEK293T | ATCC | |
| Murine KP230 | Eisinger lab | |
| STS-109 UPS | Rebecca Gladdy, MD (University of Toronto) | |
| TC32 | Patrick Grohar, MD, PhD (Children’s Hospital of Philadelphia) | |
| HT-1080 | ATCC | |
| HCT-116 | ATCC | |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Knockdown of YAP1 in HCT-116 cells | Dharmacon | TRCN0000107266 and TRCN0000107267 |
| Software and algorithms | ||
| GitHub Repository to reproduce the results | this paper | DOI 10.5281/zenodo.6970733 |
| Other | ||
Code statement
The code used in this study is available at https://github.com/broadinstitute/GeneCompoundImaging. It is available for use under the BSD 3-clause license, a permissive open-source license. The DOI is listed in the key resources table.
• Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Research animals
Boerckel lab: Mouse experiments were conducted in compliance with all relevant regulations. All animal experiments were performed at the University of Pennsylvania under IACUC review and in compliance with IACUC protocol #806482.
Cell lines
Cell line details and culture conditions are described within the method details sections describing the experiments for which each cell line was used.
Method details
Feature set alignment
As the two experiments (gene overexpression and small molecule treatmen6=t) were analyzed by a slightly different CellProfiler pipeline, and also the phenotype of the negative controls are quite different (Figure 1b), an extra data preprocessing step is needed to make the feature sets comparable. To achieve this, we first took the intersection of features in the two datasets, which resulted in 605 features (1399 features in the genetic screen, without any feature selection; and 729 features in the compound screen, obtained using the findCorrelation with threshold of 0.90 on the original 1,783 dimensional feature set). In order to compare values of the corresponding features across experiments, each feature is standardized (mean-centered and scaled by standard deviation) with respect to the negative control. This was done platewise based on the mean and standard deviation of the controls at profile level for the compound dataset. The normalization parameters are slightly different for the genetic screen, where median and median absolute deviation (MAD) are used instead, to remove outlier effects(Rohban et al., 2017). The code repository for all the analyses are publicly available as described in Code Availability.
Compound annotations
Compound MOAs and target annotations were mainly acquired from the “Repurposing hub” (REPURPOSING, no date) and then curated to include missing annotations from other sources, such as DrugBank (DrugBank Online, no date). This results in 747 compounds annotated with the gene(s) that they target. The protein interaction data, which was used to assess relevance of a protein to compound targets, was collected from BioGRID (Lab, no date). We note that 98.6% of BioGrid human gene annotations are based on physical interaction rather than genetic (Oughtred et al., 2021). For the Gene Ontology-based functional similarity, we used the R package ontologylndex and ontologySimilarity (Greene, Richardson and Turro, 2017), which were previously developed to calculate semantic similarity between ontological objects. The similarity score is calculated based on the GO annotations under biological processes through the get_sim_grid function.
Scoring gene-compound and compound-compound connections
We use Pearson correlation on aligned profiles of a gene and compound to score their connection. The profiles are obtained by averaging the replicate profiles feature-wise. We empirically found that for gene-compound connections, an absolute score value greater than 0.35 indicates similar/opposite phenotypes in the two profiles and used this for validation experiments. For the follow-up experiments of a gene, we typically used a more stringent filter of 0.40 and picked a balanced number of strongest-correlating compounds and strongest anti-correlating, subject to availability in the chemical library.
To validate how many genes find a compound with relevant targets among their top matches, we obtain the rank of the first relevant compound match for each gene, and simply count the number of genes for which the rank is within the top 306 compounds (1% of compounds, out of 30,616).
The 69 genes used as queries in the experiment were taken from the 110 reagents scored as expressing a Cell Painting phenotype in (Rohban et al., 2017) by excluding engineered variants of genes, and by selecting a single reference / wild-type construct when multiple were available (e.g. isoforms).
Permutation test for validation of known compound-compound pairs
The odds ratio of how likely compound pairs that are strongly correlated have the same MoA is considered as the test statistic. The compound names are then shuffled and the odds ratio is re-calculated. This yields a sample from the null hypothesis. The re-calculation is repeated n times. The actual odds ratio, without any shuffling of compound names, is ranked against the null distribution samples to produce a p-value estimation.
Permutation test for validation of number of genes with a relevant compound match
We run a permutation test to test the significance of the number of genes (20 out of 63) that have a match in the top 1% of their rank ordered list of compounds (with the rank based on their correlation to the gene). To generate a sample from the null distribution, the compounds are shuffled randomly, and the number of such genes is calculated. The p-value is calculated based on the empirical sampled null distribution. Note that (1) we perform a single test, where the test statistic is the number of genes, and therefore no correction for multiple testing is required here, and (2) that this procedure automatically accounts for the fact that some genes have many associated compounds (many matches) in the set.
Enrichment analysis plots
We follow the same logic as the Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005). The set of gene-compound pairs are sorted based on their profile correlations on the x-axis. On the y-axis, the plot goes up by a certain fixed amount if the corresponding gene-compound is a valid pair. Otherwise, the plot goes down by the same fixed amount. Scanning the x-axis from left to right, early existence of abundant valid pairs results in a rapid jump of the plot, and illustrates higher enrichment of profile correlation for being indicative of biological relevance between the gene-compound pair.
Relationship between strength of gene-compound matching and the number of correct gene annotations
For the analysis shown in Supplementary Figure S5, we calculated the spearman correlation between the absolute correlation of correct gene-compound pairs (including BioGrid-extended genes) and the number of direct protein targets of the compound (including only the direct annotated target genes of each compound, to better capture true polypharmacology).
Chemical structure similarity (calculating Tanimoto coefficients for hit compounds)
Similarity matrices were calculated using the Pipeline Pilot (Biovia, version 18.1.0) “Molecular Similarity NxN” function with ECFP4 fingerprints (Rogers and Hahn, 2010) and Tanimoto similarity coefficients (Bajusz, Rácz and Héberger, 2015). Heatmaps were generated using the Seaborn library (0.11.2) for Python.
Assessing compound purity and identity
Compound purity and identity were determined by ultra performance liquid chromatography coupled with mass spectroscopy (UPLC-MS). Purity was measured by UV absorbance at 210 nm. Identity was determined on a SQ mass spectrometer by positive and/or negative electrospray ionization. Mobile phase A consisted of either 0.1% ammonium hydroxide or 0.05% trifluoroacetic acid in water, while mobile phase B consisted of either 0.1% ammonium hydroxide or 0.06% trifluoroacetic acid in acetonitrile. The gradient ran from 5% to 95% mobile phase B over 2.65 min at 0.9 mL/min. An Acquity BEH C18, 1.7 μm, 2.1x50 mm column was used with column temperature maintained at 65 degrees C. Compounds were dissolved in DMSO at a nominal concentration of 1 mM, and 1.0 μL of this solution was injected.
SMAD3 experiments
For SMAD3 compounds, given a limit of 10 compounds to study, we chose the top five positive matches and the top two negative matches (which were somewhat cytotoxic based on cell count in the Cell Painting assay), along with three additional negative matches (among the top 15) which were less cytotoxic. One was unavailable.
A549 lung carcinoma cells were transfected with the luciferase reporter plasmids 4xSBE-Luc to measure TGF-b/Smad3-activated transcription (Feng et al., 1998) and pRL-TK (low expressing, constitutively active Renilla luciferase under the HSV-thymidine kinase promoter) (Promega cat# E224A) to normalize for the 4xSBE Firefly luciferase values. The transfected cell lysates were processed for luciferase assays as described56 and per manufacturer’s protocol (Promega). In brief, the cells were seeded in 24-well plates at 80% confluency and, after adhering, the media was changed to growth or starvation media (RPMI-1640 with 10% or 2% FBS respectively) for 6 hours. The cells were then transfected with 200 ng 4xSBE-Luc and 100ng Rl-Tk-Luc reporter plasmids per well using Lipofectamine 2000 per manufacturer recommendations (Thermo Fisher cat# 11668019). 12 hours after transfection cells were treated for 24 hours with 5 ng/ml TGF-β1 or 5 μM SB431542 to inhibit TGF-b-induced Smad activation, and either of 9 compounds at 10μM in triplicate. All cells were harvested with 200 μl of passive lysis buffer (Promega). Luciferase assays were performed using a Dual-Luciferase assay kit (Promega), and luciferase activities were quantified with a SpectraMax M5 plate luminometer (Molecular Devices) and normalized to the internal Renilla luciferase control and DMSO control.
Ras experiments
Isogenic RAS-less mouse embryonic fibroblast cell lines driven by human KRAS4b G12D, HRAS WT, or BRAF V600E alleles were plated in 384-well plates and dosed with compound or DMSO 18 hours later using an Echo acoustic liquid handler in a 10 point, 2-fold dilution in 0.2% DMSO, with 10μM as the top concentration. After 72 hours, Promega CellTiter-Glo® reagent was added, and the signal was read using Envision software. Values were normalized using day zero and DMSO control readings. Hits were determined by a one log difference in IC50 values between BRAF V600E and RAS-driven cell line responses.
Casein-kinase 1 alpha experiments
CSNK1A1 enzymatic assays were performed by mobility shift assay using the Labchip EZ Reader II (Perkin Elmer). GST-tagged human CSNK1A1 (Carna Biosciences) protein was incubated with ATP, substrate, and assay buffer (20 mM Hepes - pH 7.5, 5 mM MgCl2, and 0.01% Triton X-100). The assay reaction was initiated with 5 μM ATP, 2 mM DTT, and 1 μM Profiler Pro FL-Peptide 16 substrate (Perkin Elmer). Curve fitting and determination of AC50 values for phosphorylation inhibition were performed using Genedata.
GSK3B experiments
The compounds with a Cell Painting profile matching or opposing GSK3 overexpression were tested against GSK3α and GSK3β as previously reported (Wagner et al., 2018). Purified GSK3β or GSK3α was incubated with tested compounds in the presence of 4.3 μM of ATP (at or just below Km to study competitive inhibitors) and 1.5 μM peptide substrate (Peptide 15, Caliper) for 60 minutes at room temperature in 384-well plates (Seahorse Bioscience) in assay buffer that contained 100 mM HEPES (pH 7.5), 10 mM MgCl2, 2.5 mM DTT, 0.004% Tween-20, and 0.003% Brij-35. Reactions were terminated with the addition of 10 mM ethylenediaminetetraacetic acid (EDTA). Substrate and product were separated electrophoretically, and fluorescence intensity of the substrate and product was determined by Labchip EZ Reader II (Perkin Elmer). The kinase activity was measured as percent conversion to product. The reactions were performed in duplicate for each sample. The positive control, CHIR99021, was included in each plate and used to scale the data in conjunction with “in-plate” DMSO controls. The results were analyzed by Genedata Assay Analyzer. The percent inhibition was plotted against the compound concentration, and the IC50 value was determined from the logistic dose-response curve fitting. Values are the average of at least three experiments. Compounds were tested using a 12-point dose curve with 3-fold serial dilution starting from 33 μM. The two most active compounds were resynthesized for validation and tested along with closely related analogs (Supplementary Methods).
p38 experiments
Cell Painting profiles for two wild-type variants of p38α (MAPK14) were averaged to create a p38α Cell Painting profile. 20 compounds whose Cell Painting profile correlated positively or negatively to that of p38α overexpression were selected; we also chose 14 “non-correlated” compounds (i.e. absolute value of correlation <0.2) as negative/neutral controls. The compounds were tested for their influence on p38 activity using the RPE1-p38 kinase translocation reporter (KTR) line that was previously generated(Liu et al., 2018). This cell line has been tested and confirmed to be negative for mycoplasma contamination, but not authenticated. p38 activity is measured by phosphorylation of its substrate, MEF2C, which is preferentially phosphorylated by p38α, while p38β and p38δ contribute less (Zetser, Gredinger and Bengal, 1999). RPE1-p38KTR cells were cultured in DMEM/F12 medium supplemented with 10% Fetal Bovine Serum at 37C in a humidified atmosphere with 5% CO2. 1000 cells were plated per well in 96-well plates and treated with 1μM and 10μM of each compound (n=4 well per concentration per compound, no replicates) for 48 hours. Only the middle 60 wells were used to prevent potential confounds from the edge effect. Cells were then fixed in 4% paraformaldehyde for 10min, followed by permeabilization in cold methanol at −20C for 5min. Cells were stained with 0.4 μg/mL Alexa Fluor 647 carboxylic acid, succinimidyl ester for 2hr at RT, followed by 1μg/mL DAPI for 10min at RT to facilitate the segmentation of individual cells.
p38 activity in single cells was calculated using the ratio of the median intensity of the p38-KTR in a 5-pixel-wide cytoplasmic ring around the nucleus to the median intensity of the p38-KTR in the nucleus. p38 activity measurements were normalized to DMSO within the same plate and column. The Student’s t-test or Kolomogorov-Smirnov (KS) test was used to assess the significance of changes in the single cell distributions of p38 activity for each compound relative to control; we note that even for the positive control known inhibitor the effect sizes are small. When reporting hits from the assay, KS test and t-test p-values were adjusted to control the false discovery rate using the Benjamini-Hochberg method, using the p.adjust(method=‘BH”) method in R.
PPARGC1A (PGC-1α) experiments
Reporter assays:
To measure PGC-1α activity related to PPARG, RT112/84 cells were obtained from the Cancer Cell Line Encyclopedia (Broad Institute, Cambridge, MA), which obtained them from the original source and performed cell line authentication. The cell line was engineered with the NanoLuc gene cloned into the 3’ UTR of the FABP4 (previously described(Goldstein et al., 2017)) followed by stable expression of nuclear GFP (pTagGFP2-H2B, Evrogen) and tested negative for mycoplasma (MycoAlert, Lonza). Cells were plated in 384-well plates at ~10,000 cells/well and dosed with indicated compounds in the absence or presence of EC50 of PPARG agonist, rosiglitazone, using an HP D300 digital dispenser. The following day nuclei were counted for normalization (IncuCyte S3, Essen Bioscience) and the reporter activity was evaluated using the NanoGlo Luciferase Assay System (Promega). Normalized data is reported as NanoGlo arbitrary light units divided by cell number. PPARG agonist, rosiglitazone, and inhibitor, T0070907, were obtained from Tocris and included as controls.
To measure effects on PGC1a/ERRalpha, HEK293T cells purchased from ATCC were co-transfected with Gal4-ERRalpha, with and without PGC1a (pCDNA3.1-Flag-HA-PGC-1alpha (Rodgers et al., 2005)), kind gifts from Pere Puigserver, in combination with the Gal4 UAS reporter construct, pGL4.35 [luc2P/9XGAL4UAS/Hygro] (Promega) modified by subcloning the HSV-TK promoter into the unique HindIII site that is downstream of the 9xGal4 UAS sites, in addition to a Renilla luciferase expression vector pRL (Promega) for normalization. Cells were dosed with compounds and 24 hours later, plates were analyzed using Dual-Glo Luciferase Assay System (Promega). Normalized light units are reported as Firefly luciferase divided by Renilla luciferase. ERRalpha modulators XCT790, Daidzein, and Biochanin A (Cayman Chemical) were included as controls. 293T cells were not authenticated nor tested for mycoplasma.
High content mitochondrial motility screen:
We used our previously published assay to assess mitochondrial motility(Shlevkov et al., 2019). Briefly, we plated E18 rat cortical neurons in the middle 60 wells of 96 well plates (Greiner) – 40,000 cells per well in 150 μl enriched Neurobasal media. Neurons were transfected with mito-DsRed at DIV7 using Lipofectamine2000 (Life Technologies). Plating and transfection were all done using an Integra VIAFLO 96/384 automated liquid handler. At DIV9, test compounds were added into wells to achieve a final concentration of 10 μM each (4 wells per compound), as well at 10 μM calcimycin for neg. control (Wang and Schwarz, 2009), and DMSO only for mock treatment. Following a 1-2 hour incubation, plates were imaged on a ArrayScan XTI (Thermo Fisher). Mitochondrial motility data was extrapolated from imaging data using a MATLAB and CellProfiler based computational pipeline. Compounds A01-A12 were tested on one plate; B01-B11 were tested separately on another plate on the same day. The experiment was repeated twice in different weeks. In the second week, TMRE was added to all wells after imaging was completed (20min, then 2 washes) and imaged to measure mitochondrial membrane potential in order to determine mitochondrial and cell health.
YAP1-related compounds
For the initial experiments, quality control of the compounds revealed that purity was 88% for A15 (BRD-K34692511-001-01-9), 81% for A05 (BRD-K28862419-001-01-9), and > 99% for E07 (BRD-K43796186-001-01-1). For subsequent experiments in the Eisinger lab, BRD-K43796186 (NB4A) was ordered from MuseChem (cat. #M189943) and for the Kiessling lab, from Ambinter (Cat# Amb2554311).
YAP1 cell culture and treatments
Eisinger lab:
Murine KP230 cells, a Yap1-dependent cancer cell line, were derived from a tumor from the KP mouse model (KrasG12/D; Trp53Pfl/fl), as described in(Eisinger-Mathason et al., 2015). STS-109 UPS cells were derived from a human UPS tumor and validated by Rebecca Gladdy, MD (Sinai Health System, Toronto, Ontario, Canada). TC32 cells were a gift from Patrick Grohar, MD, PhD (Children’s Hospital of Philadelphia). HT-1080, HCT-116, and HEK293T cells were purchased from ATCC. KP230, HT-1080, and HEK-293T cells were grown in DMEM with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin (P/S). STS-109 cells were cultured in DMEM with 20% FBS, 1% L-glutamine, and 1% P/S. TC32 cells were grown in RPMI with 10% FBS, 1% L-glutamine, and 1% P/S. HCT-116 cells were cultured in McCoy’s 5A medium with 10% FBS and 1% P/S. All cells were confirmed to be negative for mycoplasma contamination and were maintained in an incubator at 37C with 5% CO2. For experimental purposes, cells were cultured for up to 20 passages before being discarded, and were grown to approximately 50% confluence to circumvent the effects of high cell density on Yap1 expression and activity. All cell lines in the Eisinger laboratory were treated with 10 μM of each inhibitor or an equivalent volume of DMSO every 24 hours for 3 days, except for STS-109 cells, which were treated daily for 8 days.
Kiessling lab:
H9 hPSCs (WiCell) were maintained on vitronectin (Thermo Fisher)-coated plates in Essential 8 (E8) medium. The cells were routinely passaged using 0.5mM EDTA and treated with 5μM Y-27632 dihydrochloride (Tocris) on the first day. For testing the effects of the small molecules, H9 hPSCs were seeded at 50K cells/cm2 on vitronectin-coated plates in E8 medium supplemented with 5μM Y-27632 dihydrochloride (day 0). On day 1, the medium was switched to E8 medium. On day 2, the medium was switched to E8 medium supplemented with the small molecules. Following overnight incubation, the cells were collected for subsequent analysis on day 3. The cells were regularly checked for Mycoplasma contaminations (Sigma Aldrich - Lookout Mycoplasma PCR Detection Kit) but were not authenticated.
Boerckel lab:
Murine periosteal cells were isolated from a transgenic mouse model (CMV-Cre;R26R-rtTAfl; tetO-YAPS127A; C57BI/6 strain/background) in which YAP1 can be activated in a doxycycline inducible manner (Camargo 2011). This mouse model expresses a mutant form of YAP1 (YAPS127A) that escapes degradation. Cells were isolated from 3 female mice (age 15 weeks) from a 4-day old femoral fracture callus. Cells were cultured in a-MEM with 15% Fetal Bovine serum (S11550, R&D Systems), 1% GlutaMAX-I (Gibco, 35050-061) and 1% Penicillin/Streptomycin (Gibco, 15140-122).
YAP1-related lentiviral production
Knockdown of YAP1 in HCT-116 cells was performed with shRNAs (TRC clone IDs: TRCN0000107266 and TRCN0000107267); a scrambled shRNA was used as a negative control. shRNA plasmids (Dharmacon) were packaged using the third-generation lentiviral vector system (pVSV-G, pMDLG, and pRSV-REV; Addgene) and expressed in HEK-293T cells using Fugene 6 transfection reagent (Promega). Virus-containing supernatants were collected 24 and 48 hours after transfection and concentrated 40-fold by centrifugation with polyethylene glycol 8000.
YAP1-related proliferation assays
NB4A treatment:
Cells were treated with 10 μM of each inhibitor or an equivalent volume of DMSO every 24 hours for 3-8 days, and counted with a hemocytometer with trypan blue exclusion daily (KP230, HT-1080, TC32, HCT-116), or every 2 days (STS-109).
BRD-K28862419 and BRD-K34692511 analog screen:
KP230 cells were treated with 10 μM of each compound or an equivalent volume of DMSO every 24 hours for 2 days. Treated cells were then incubated with PrestoBlue viability reagent (ThermoFisher Scientific) for 2 hours according to the manufacturer’s recommendations. Fluorescence (560/590 nm) was read on a Spectramax M2e plate reader (Molecular Devices).
shRNA-mediated YAP1 knockdown:
HCT-116 cells were infected with YAP1 shRNA-encoding lentiviruses in the presence of 8 μg/mL polybrene (Sigma). Antibiotic selection (3 μg/mL puromycin) was performed after 48 hours, after which cells were cultured for an additional 48 hours. Cells were then trypsinized, seeded under puromycin-selection conditions, and counted with a hemocytometer with trypan blue exclusion on days 7, 8, and 9 post-infection.
YAP1-related qRT-PCR
For the Eisinger lab, total RNA from cultured cells was isolated with the QIAGEN RNeasy mini kit, and cDNA was synthesized with the High-Capacity RNA-to-cDNA kit (Life Technologies). qRT-PCR analysis was performed with TaqMan “best coverage” probes on a ViiA7 instrument. Hypoxanthine phosphoribosyltransferase (HPRT) and succinate dehydrogenase subunit A (SDHA) were used as endogenous controls. Relative expression was calculated using the ddCt method.
For the Kiessling lab, the RNA was extracted using TRIzol (Life Technologies) and Direct-zol™ RNA MiniPrep kit (Zymo Research) as per manufacturer instructions. The RNA was reverse transcribed using iScript cDNA synthesis kit (Bio-Rad). The qPCR was performed on CFX Connect (Bio-Rad) using iTaq Universal SYBR Green Supermix (Bio-Rad). GAPDH was used as a reference gene for normalization. The relative gene expression levels were determined using the ddCt method. The primer sequences used are listed in Supplementary Table S7.
For the Boerckel lab, to induce YAPS127A, 1μM doxycycline was added to the cell culture medium for 48 hours. This was used as a positive control to compare YAP1 mRNA expression. Cells were also treated with BRD-K34692511-001-01-9 at 5μM. mRNA was isolated from cells (n=3/group/time point) at 1, 4 or 48 hours after treatment using Qiagen RNeasy Mini kit (Qiagen, 74106). cDNA was prepared as per the manufacturer’s protocol using the High-Capacity Reverse Transcription kit (Thermofisher scientific, 4368814). qPCR analysis was performed using the QuantStudio 6 Pro Real-Time PCR System.
YAP1-related reporter assay
Varelas lab:
HEK293T cells purchased from ATCC were co-transfected using Lipofectamine 3000 (Thermo Fisher) with a TEAD luciferase reporter construct, 8xGTIIC-luciferase (gift from Stefano Piccolo, Addgene plasmid # 34615), a plasmid expressing Renilla Luciferase from a CMV promoter as a transfection control, along with a plasmid expressing 3xFlag-tagged wild-type YAP1 from a CMV promoter (pCMV5 backbone). Following transfection the cells were immediately treated with 0.2% DMSO, 10μM NB4A, BRD-K34692511 or BRD-K28862419 and then lysed 48 hours later. Lysates were examined using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol and measured using a SpectraMax iD3 plate reader (Molecular Devices). Firefly Luciferase activity from the TEAD reporter was normalized to Renilla Luciferase activity and then plotted as relative values. Mycoplasma tests are routinely performed, but cells were not recently authenticated.
YAP1-related RNA-sequencing and data analysis
Total RNA from cultured cells was isolated with the QIAGEN RNeasy Mini Kit with on-column DNase digestion. RNA quality checks were performed with an Agilent 2100 Bioanalyzer (Eukaryotic Total RNA Nano kit). Library preparation (500 ng input RNA) was performed with the NEBNext Poly(A) mRNA Magnetic Isolation Module (#E7490) with SPRIselect Beads (Beckman Coulter), the NEBNext Ultra II Single-End RNA Library Prep kit (#7775S), and the NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) according to the manufacturer’s instructions. Library size was confirmed with an Agilent 2100 Bioanalyzer (DNA1000 chip). Pooled libraries were diluted to 1.8 pM (concentrations checked with the Qubit Fluorometer high-sensitivity assay, Thermo Fisher), and sequenced on an Illumina NexSeq 500 instrument with the NexSeq 500 75-cycle high-output kit.
For data analysis, FASTQ files were generated with the bcl2fastq command line program (Illumina). Transcript alignment was performed with Salmon (Patro et al., 2017). Differential expression analysis (NB4A- vs. DMSO-treated cells) was performed with the DESeq2 R package. DESeq2 “stat” values for each gene were used as inputs to pre-ranked GSEA, where enrichment was tested against the Hallmark gene sets from the Molecular Signatures Database (MSigDB). Access to sequencing data is discussed in the data availability section.
YAP1-related Western blotting
For the Kiessling lab, the cells were lysed in RIPA buffer (Pierce) supplemented with Halt Protease inhibitor cocktail and Halt Phosphatase inhibitor cocktail (Thermo Fisher). The Eisinger lab lysed cells in hot Tris-SDS buffer (pH 7.6) and boiled for 5 minutes at 95°C. The protein concentration of each sample was quantified using the Pierce BCA protein assay (Thermo Fisher). The proteins were resolved by SDS-PAGE and transferred to PVDF membranes using the Trans-Blot Turbo Transfer system (Bio-Rad). The membranes were blocked in 5% non-fat milk in TBS-T for up to 1 hour at room temperature and incubated with primary antibodies in 5% bovine serum albumin in TBS-T overnight at 4°C. Then, the membranes were incubated with HRP-conjugated anti-rabbit IgG secondary antibodies at 1:10000 (Kiessling lab; Jackson ImmunoResearch Laboratories, #111-035-003) or 1:2500 (Eisinger lab; Cell Signaling Technology [CST] #7074) for 1 hour at RT and developed in the ChemiDoc MP Imaging system (Kiessling lab) or on autoradiography film (Eisinger lab) using ECL Prime reagent (Amersham). The band intensities in immunoblots were quantified with Image Lab software. The primary antibodies and dilutions used are: anti-YAP1 (CST 4912S and CST 14074 [clone D8H1X]) at 1:1000, anti-phospho-YAP1-S127 (CST 4911S) at 1:1000, and anti-GAPDH (CST 5174 and CST 2118 [clone 14C10]) at 1:15000 and 1:1000, respectively. Primary antibodies were validated commercially in cells both wild-type and deficient (e.g., knockout) for the gene/protein of interest. YAP1-related immunofluorescence and image analysis
For the Eisinger lab, cells grown on poly-L-lysine-coated chamber slides were fixed in 4% PFA (15 minutes at room temperature), permeabilized with 0.5% Triton-X100/PBS (15 minutes at room temperature), and blocked with 5% goat serum (Vector Laboratories S-1000; 1 hour at room temperature). Cells were then incubated with anti-Yap1 primary antibodies (CST #14074 [clone D8H1X]; 1:1000) diluted in blocking buffer overnight at 4°C. Subsequently, cells were incubated with Alexa Fluor 488-conjugated secondary antibodies (4 ug/mL in blocking buffer; Thermo Fisher Scientific #A-11008) for 1 hour at room temperature. Coverslip mounting was performed with ProLong Gold Antifade reagent with DAPI. Images (5 fields per condition for each of 3 independent experiments) were acquired with a Nikon Eclipse Ni microscope and Nikon NES Elements software. Image analysis was performed with Fiji as follows: For nuclear staining intensity, watershed analysis of DAPI channel images (8-bit) was performed to “separate” nuclei that appeared to be touching. Nuclei were then converted to regions of interest (ROIs) that were “applied” to the corresponding GFP channel image (8-bit format). Analysis of staining intensity in these nuclear ROIs was then performed, excluding objects smaller than 100 pixels2 (integrated density normalized to number of nuclei). A similar process was followed to determine whole-cell staining intensity: using 8-bit GFP channel images, cells were distinguished from background via thresholding, and converted to ROIs that were applied back to the 8-bit GFP channel images. Analysis of staining intensity (integrated density normalized to number of nuclei) was then performed in these ROIs, excluding objects smaller than 500 pixels2. The ratio of nuclear to total Yap1 expression was determined after subtracting out background GFP signal from no-primary antibody controls.
For the Kiessling lab, the cells were fixed with 4% formaldehyde for 15 mins at room temperature. The cells were permeabilized and blocked with PBS containing 2% BSA and 0.1% Triton-X100. The cells were incubated with a primary antibody against YAP1 (Santa Cruz Biotechnology, sc-101199) at 1:200 dilution in a blocking buffer overnight at 4°C. Then, the cells were incubated with a goat anti-mouse Alexa Fluor 488 conjugated secondary antibody (Thermo Fisher, #A11001) at 1:1000 dilution for 1 hour at room temperature. The nuclei were counterstained with DAPI dilactate (Molecular Probes). Images were collected with Olympus FV1200 microscope and analyzed with CellProfiler. Briefly, nuclei and cell bodies were segmented using DAPI and YAP channels respectively. The cell cytoplasm was determined as the region outside nuclei but within the cell bodies. Then, the ratio of mean intensity of YAP in the nucleus to cytoplasm was calculated to determine YAP translocation.
Chemical synthesis of BRD4486
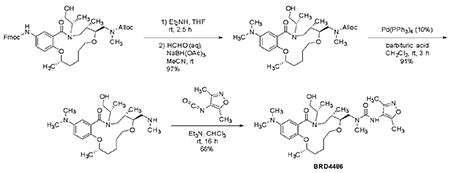
We followed prior work (Marcaurelle et al., 2010). To a solution of the Fmoc-protected intermediate (100 mg, 0.140 mmol, 1 eq) in tetrahydrofuran (1 mL) was added diethylamine (150 μL, 1.44 mmol, 10.3 eq). The mixture was stirred 2.5 h, and the solvent and excess amine were evaporated.
The residue was dissolved in acetonitrile (1 mL), then formaldehyde (37% aqueous, 100 μL, 1.32 mmol, 9.4 eq), catalytic acetic acid (1 drop), and sodium triacetoxyborohydride (100 mg, 0.472 mmol, 3.4 eq) were added. The mixture was stirred 1 h, then quenched with saturated aqueous NaHCO3 and diluted with CH2Cl2. The layers were separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over MgSO4, filtered, and evaporated, and the residue was purified by flash column chromatography (0-10% MeOH/CH2Cl2) to provide the dimethylaniline (70.2 mg, 97% yield).
A yellow solution of the resulting Alloc-protected amine (68 mg, 0.131 mmol, 1 eq), 1,3-dimethyl-barbituric acid (200 mg, 1.28 mmol, 9.8 eq), and tetrakis(triphenylphosphine)palladium(0) (15 mg, 0.013 mmol, 0.1 eq) in dichloromethane (1 mL) was stirred at room temperature for 3 h. The reaction was quenched with 0.1 M HCl and stirred 15 min. The layers were separated, and the organic layer was extracted with 0.1 M HCl. The pH of the combined aqueous layers was adjusted to ~12 with 4 M NaOH, then extracted with CH2O2. The combined organic layers were dried over Na2CO3, filtered, and evaporated to provide the crude free amine (52 mg, 91% yield), which was used directly in the urea formation.
To a solution of the crude secondary amine (52 mg, 0.119 mmol, 1 eq) and 3,5-dimethylisoxazol-4-yl isocyanate (20 mg, 0.145 mmol, 1.2 eq) in chloroform (1 mL) was added triethylamine (16.5 μL, 0.119 mmol, 1 eq). The mixture was stirred at room temperature for 16 h, and the solvent was evaporated. The residue was purified by flash column chromatography (0-10% MeOH/CH2Cl2) to provide BRD4486 (46.2 mg, 68% yield) as a white solid.
MS: 574.5 [M+H]+; 572.3 [M-H]−
Chemical synthesis of BRD4313
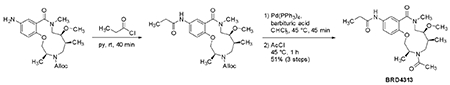
We followed prior work (Fitzgerald et al., 2012). To the aniline intermediate (100 mg, 0.247 mmol, 1 eq) dissolved in pyridine (0.5 mL) was added propionyl chloride (45 μL, 0.514 mmol, 2.1 eq). The resulting mixture was stirred at room temperature for 40 min.
Chloroform (0.5 mL) and 1,3-dimethylbarbituric acid (385 mg, 2.46 mmol, 10 eq) were added, and, after 5 min, tetrakis(triphenylphenylphosphine)palladium(0) (28 mg, 0.024 mmol, 0.1 eq) was added. The yellow-green solution was stirred at 45 °C for 45 min (CO2 evolution), then acetyl chloride (261 μL, 3.69 mmol, 15 eq) was added. After stirring 1 h, the mixture was diluted with CH2Cl2 and poured into 0.1 M HCl. The layers were separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with saturated aqueous NaHCO3, dried over MgSO4, filtered, and evaporated. The residue was purified by flash column chromatograph (2.5-10% MeOH/CH2Cl2) to provide the product (82.4 mg) in 84% purity. The material was further purified by mass-directed prep-HPLC (0-100% MeCN/water + 0.1% TFA) to provide BRD4313 (54.6 mg, 51% yield over 3 steps) as a tan solid.
MS: 420.8 [M+H]+
Analog selection for GSK experiments
Compounds for (stereochemical) structure-activity relationship determination were selected from the Broad compound library in the following order until 24 analogs had been reached for each hit:
All available stereoisomers of the hit compound
Available regioisomers of the hit compound
Analogs with high structural similarity (Tanimoto ≥ 0.75) to the hit compound, selected for chemical diversity
Assay plates for each series (RCM/H2T) were prepared with 4 on-plate controls plus the hit compound from the opposing series. The controls included 3 dual GSK3α/β inhibitors (CHIR-99021, GW8510, and BRD0320) and one GSK3α-specific inhibitor (BRD0705). All compounds tested are shown in Supplementary Figures S17 and S18, with results shown in Supplementary Figure S19.
Mobility shift microfluidics assay protocol
We followed previous protocol (Wagner et al., 2018). Purified GSK3β or GSK3α was incubated with tested compounds in the presence of 4.3 μM of ATP (at or just below Km to study competitive inhibitors) and 1.5 μM peptide substrate (Peptide 15, Caliper) for 60 minutes at room temperature in 384-well plates (Seahorse Bioscience) in assay buffer that contained 100 mM HEPES (pH 7.5), 10 mM MgCl2, 2.5 mM DTT, 0.004% Tween-20, and 0.003% Briji-35. Reactions were terminated by the addition of 10 mM ethylenediaminetetraacetic acid (EDTA). Substrate and product were separated electrophoretically, and fluorescence intensity of the substrate and product was determined by Labchip EZ Reader II (Caliper Life Sciences). The kinase activity was measured as percent conversion. The reactions were performed in duplicate for each sample. The positive control, CHIR99021, was included in each plate and used to scale the data in conjunction with in-plate DMSO controls. The results were analyzed by Genedata Assay Analyzer. The percent inhibition was plotted against the compound concentration, and the IC50 value was determined from the logistic dose-response curve fitting. Values are the average of at least three experiments. Compounds were tested using a 12-point dose curve with 3-fold serial dilution starting from 33 μM.
Chemical synthesis of BRD-K28862419
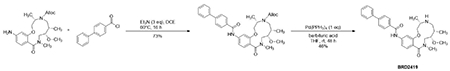
We followed prior work (Fitzgerald et al., 2012). To a solution of the alloc protected aniline intermediate (45 mg, 0.111 mmol, 1.20 eq) and of 4-phenylbenzoyl chloride (20 mg, 0.0923 mmol, 1.00 eq) in CH2Cl2 (0.5 mL) was added triethylamine (0.277 mmol, 3 eq). The mixture was stirred at 60°C for 16 h. The solvent was evaporated, and the residue purified by normal phase chromatography (0-50% ethyl acetate in hexanes) to provide the diphenylamide (39 mg, 73% yield).
A vial of the alloc protected intermediate (51 mg) in tetrahydrofuran (0.5 mL) was sparged with Ag for 10 mins. In a separate vial Pd(PPH3)4 (102 mg, 0.0883 mmol, 1.00 eq) and barbituric acid (207 mg, 1.32 mmol, 15.0 eq) were combined and backfilled with Ag. Then, the THF solution was transferred (by syringe) to the powder. The reaction was stirred under nitrogen for 48 h. The reaction was concentrated then purified by normal phase chromatography (silica, 0 – 40% CH3OH in CH2Cl2) to provide N-((3S,6S,7S)-7-methoxy-3,6,9-trimethyl-10-oxo-3,4,5,6,7,8,9,10-octahydro-2H-benzo[k][1]oxa[4,9]diazacyclododecin-13-yl)-[1,1′-biphenyl]-4-carboxamide (21 mg, 0.0409 mmol, ?, 46% yield). MS: 502.96 [M+H] + 500.62 [M−H]−.
Chemical re-synthesis of and BRD-K34692511
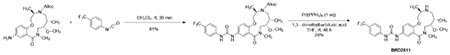
We followed prior work (Fitzgerald et al., 2012). To a vial of the analine intermediate (42 mg, 0.104 mmol, 1.00 eq) in CH2Cl2 (0.5 mL) was added 1-isocyanato-4-(trifluoromethyl)benzene (0.01 mL, 0.104 mmol, 1.00 eq) and stirred at 25 °C for 30 min. The reaction was concentrated and the residue was purified by normal phase chromatography (0-100% ethyl acetate in hexanes) to provide the protected urea intermediate (52 mg, 81% yield).
A vial of the urea intermediate (52 mg, 0.0871 mmol, 1.00 eq) in tetrahydrofuran (0.4 mL) was sparged with Ag for 10 mins. In a separate vial Pd(PPH3)4 (101 mg, 0.0871 mmol, 1.00 eq) and 1,3 - dimethylbarbituric acid (14 mg, 0.0871 mmol, 1.00 eq) were combined and backfilled with Ag. Then the THF solution was transferred (by syringe) to the powder. The reaction was stirred under nitrogen for 48 h. The reaction remained bright yellow. The reaction was concentrated then purifed by normal phase chromatography (0-100% CH3OH in CH2Cl2, product elutes). The collected fractions were concentrated and purified with a focused gradient of 35-45% CH3OH in CH2Cl2 was run to separate remaining impurities. Fractions containing product were collected to provide 1-[(4R,7S,8S)-8-methoxy-4,7,10-trimethyl-11-oxo-2-oxa-5,10-diazabicyclo[10.4.0]hexadeca-1(12),13,15-trien-14-yl]-3-[4-(trifluoromethyl)phenyl]urea, (13 mg, 0.0224 mmol, 26% yield). MS: 510.63 [M+H]+, 553.860 [M+CHOO−]−.
Supplementary Material
Highlights.
Compounds impacting particular genes’ function are highly sought.
Most chemicals and overexpressed genes impact cell morphology in the Cell Painting assay.
Matching these image profiles can find chemicals that impact a particular gene’s function.
This virtual screen using public data found new chemical regulators of several pathways.
Acknowledgements:
The authors thank the researchers who originally helped produce the published data used in this analysis, including the Broad Institute LINCS team, Cancer Program and PRISM team. We appreciate helpful discussions with our colleagues, including Pere Puigserver, Evan Rosen, and Amit Majithia.
Funding:
The Carpenter–Singh lab team was supported by the National Institutes of Health (NIH R35 GM122547 to AEC). R.K. is supported by the Canadian Institutes of Health Research (343437) and the Natural Sciences and Engineering Research Council of Canada (RGPIN-2015-05805). C.T. is supported by a University of Toronto Open Fellowship. The Eisinger lab team was supported by the National Institutes of Health (R01CA229688 to TSKE and T32-HL007971 to AMF) and the American Cancer Society-Roaring Fork Valley Postdoctoral Fellowship (PF-21-111-01MM to AMF), and is grateful to the University of Pennsylvania High-Performance Computing Facility for providing computational capacity and data archiving services. The Kiessling lab team was supported by the National Institutes of Health (U01CA231079 to LK). The Boerckel lab team was supported by the National Institutes of Health (R01AR073809 to JDB) and the National Science Foundation (CMMI: 15-48571 to JDB). S.M.C. is supported by the National Institutes of Health (K08CA230220). Turbyville and Rigby are supported with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E.
Declaration of interests:
The Broad Institute and the University of Pennsylvania have filed a patent on the described compounds related to YAP1 overexpression. AEC reports receiving research funding from pharmaceutical companies in the JUMP-Cell Painting Consortium and Calico Life Sciences. AEC has ownership interest and serves on the scientific advisory board for Recursion, a publicly-traded biotech company using images for drug discovery. JTG reports receiving a commercial research grant from Bayer AG. SMC reports receiving research funding from Bayer and Calico Life Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes:
- Bajusz D, Rácz A and Héberger K (2015) ‘Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?’, Journal of cheminformatics, 7, p. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M-A et al. (2016) ‘Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes’, Nature protocols, 11(9), pp. 1757–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M-A et al. (2017) ‘A dataset of images and morphological profiles of 30 000 small-molecule treatments using the Cell Painting assay’, GigaScience, 6(12), pp. 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnage ME, Chekler ELP and Jones LH (2013) Target validation using chemical probes’, Nature chemical biology, 9(4), pp. 195–199. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran SN et al. (2020) ‘Image-based profiling for drug discovery: due for a machine-learning upgrade?’, Nature reviews. Drug discovery, pp. 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran SN et al. (2021) Three million images and morphological profiles of cells treated with matched chemical and genetic perturbations’. Available at: https://openreview.net/forum?id=rCRyg1-Yovi (Accessed: 12 June 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran SN et al. (no date) Three million images and morphological profiles of cells treated with matched chemical and genetic perturbations’. doi: 10.1101/2022.01.05.475090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI et al. (2011) ‘Comprehensive analysis of kinase inhibitor selectivity’, Nature biotechnology, 29(11), pp. 1046–1051. [DOI] [PubMed] [Google Scholar]
- Dey A, Varelas X and Guan K-L (2020) Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine’, Nature reviews. Drug discovery, 19(7), pp. 480–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten M et al. (2010) ‘Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival’, The EMBO journal, 29(6), pp. 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DrugBank Online (no date). Available at: https://www.drugbank.ca (Accessed: 2 June 2022).
- Eisinger-Mathason TSK et al. (2015) ‘Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis’, Proceedings of the National Academy of Sciences of the United States of America, 112(26), pp. E3402–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias KM et al. (2016) ‘Epigenetic remodeling regulates transcriptional changes between ovarian cancer and benign precursors’, JCI insight, 1(13). doi: 10.1172/jci.insight.87988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH et al. (1998) The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for smad3 in TGF-beta-induced transcriptional activation’, Genes & development, 12(14), pp. 2153–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald ME et al. (2012) ‘Build/couple/pair strategy for the synthesis of stereochemically diverse macrolactams via head-to-tail cyclization’, ACS combinatorial science, 14(2), pp. 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley DJ et al. (2020) ‘Phenotyping reveals targets of a pseudo-natural-product autophagy inhibitor’, Angewandte Chemie, 59(30), pp. 12470–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futamura Y et al. (2012) ‘Morphobase, an encyclopedic cell morphology database, and its use for drug target identification’, Chemistry & biology, 19(12), pp. 1620–1630. [DOI] [PubMed] [Google Scholar]
- Goldstein JT et al. (2017) ‘Genomic Activation of PPARG Reveals a Candidate Therapeutic Axis in Bladder Cancer’, Cancer research, 77(24), pp. 6987–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene D, Richardson S and Turro E (2017) ‘ontologyX: a suite of R packages for working with ontological data’, Bioinformatics, 33(7), pp. 1104–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi M et al. (2021) ‘High-Dimensional Gene Expression and Morphology Profiles of Cells across 28,000 Genetic and Chemical Perturbations’, bioRxiv. doi: 10.1101/2021.09.08.459417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha J et al. (2021) ‘Recent advances in identifying protein targets in drug discovery’, Cell chemical biology, 28(3), pp. 394–423. [DOI] [PubMed] [Google Scholar]
- Hailing JF and Pilegaard H (2020) ‘PGC-1α-mediated regulation of mitochondrial function and physiological implications’, Applied physiology, nutrition, and metabolism = Physiologie appliquee, nutrition et metabolisme, 45(9), pp. 927–936. [DOI] [PubMed] [Google Scholar]
- Handschin C and Spiegelman BM (2006) ‘Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism’, Endocrine reviews, 27(7), pp. 728–735. [DOI] [PubMed] [Google Scholar]
- Holbrook-Smith D, Durot S and Sauer U (2022) ‘High-throughput metabolomics predicts drug-target relationships for eukaryotic proteins’, Molecular systems biology, 18(2), p. e10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JH and Lawlor ER (2011) BMI-1 suppresses contact inhibition and stabilizes YAP in Ewing sarcoma’, Oncogene, 30(17), pp. 2077–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RE et al. (2021) ‘High-content phenotypic and pathway profiling to advance drug discovery in diseases of unmet need’, Cell chemical biology, 28(3), pp. 338–355. [DOI] [PubMed] [Google Scholar]
- Kaufman T et al. (2020) ‘Visual barcodes for multiplexing live microscopy-based assays’. doi: 10.21203/rs.3.rs-67883/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaeger S et al. (2017) The target landscape of clinical kinase drugs’, Science, 358(6367). doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lab MT (no date) BioGRID interaction database. Available at: https://thebiogrid.org (Accessed: 2 June 2022).
- Lapins M and Spjuth O (2019) ‘Evaluation of Gene Expression and Phenotypic Profiling Data as Quantitative Descriptors for Predicting Drug Targets and Mechanisms of Action’, Cold Spring Harbor Laboratory. doi: 10.1101/580654. [DOI] [Google Scholar]
- Lee H-J et al. (2009) ‘PPAR(gamma)/PGC-1 (alpha) pathway in E-cadherin expression and motility of HepG2 cells’, Anticancer research, 29(12), pp. 5057–5063. [PubMed] [Google Scholar]
- Lin A et al. (2019) Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials’, Science translational medicine, 11(509). doi: 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S et al. (2018) ‘Size uniformity of animal cells is actively maintained by a p38 MAPK-dependent regulation of G1-length’, eLife, 7. doi: 10.7554/eLife.26947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Widlund HR and Puigserver P (2016) ‘PGC-1 Coactivators: Shepherding the Mitochondrial Biogenesis of Tumors’, Trends in cancer research, 2(10), pp. 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAPK14 - ClinicalTrials.Gov (no date). Available at: https://clinicaltrials.gov/ct2/results?term=MAPK14&Search=Apply&age_v=&gndr=&type=&rslt= (Accessed: 1 June 2021).
- Marcaurelle LA et al. (2010) ‘An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: discovery of macrocyclic histone deacetylase inhibitors’, Journal of the American Chemical Society, 132(47), pp. 16962–16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EJ et al. (2019) ‘All-Assay-Max2 pQSAR: Activity Predictions as Accurate as Four-Concentration IC50s for 8558 Novartis Assays’, Journal of chemical information and modeling, 59(10), pp. 4450–4459. [DOI] [PubMed] [Google Scholar]
- Martínez-Limón A et al. (2020) The p38 Pathway: From Biology to Cancer Therapy’, International journal of molecular sciences, 21(6). doi: 10.3390/ijms21061913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer TU et al. (1999) ‘Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen’, Science, 286(5441), pp. 971–974. [DOI] [PubMed] [Google Scholar]
- Moffat JG et al. (2017) Opportunities and challenges in phenotypic drug discovery: an industry perspective’, Nature reviews. Drug discovery, 16(8), pp. 531–543. [DOI] [PubMed] [Google Scholar]
- Mullard A (2019) ‘Machine learning brings cell imaging promises into focus’, Nature reviews. Drug discovery, 18(9), pp. 653–655. [DOI] [PubMed] [Google Scholar]
- Musah S et al. (2014) ‘Substratum-induced differentiation of human pluripotent stem cells reveals the coactivator YAP is a potent regulator of neuronal specification’, Proceedings of the National Academy of Sciences of the United States of America, 111(38), pp. 13805–13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information (no date) PubChem Bioassay Record for AID 651723, Source: Broad Institute. Available at: https://pubchem.ncbi.nlm.nih.gov/bioassay/651723 (Accessed: 30 June 2021). [Google Scholar]
- Nierenberg AA et al. (2018) ‘Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 Alpha as a Novel Target for Bipolar Disorder and Other Neuropsychiatric Disorders’, Biological psychiatry, 83(9), pp. 761–769. [DOI] [PubMed] [Google Scholar]
- Nishio M et al. (2016) ‘Dysregulated YAP1/TAZ and TGF-β signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice’, Proceedings of the National Academy of Sciences of the United States of America, 113(1), pp. E71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oughtred R et al. (2021) The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions’, Protein science: a publication of the Protein Society, 30(1), pp. 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R et al. (2017) ‘Salmon provides fast and bias-aware quantification of transcript expression’, Nature methods, 14(4), pp. 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman ZE et al. (2004) ‘Multidimensional drug profiling by automated microscopy’, Science, 306(5699), pp. 1194–1198. [DOI] [PubMed] [Google Scholar]
- Pratapa A, Doron M and Caicedo JC (2021) ‘Image-based cell phenotyping with deep learning’, Current opinion in chemical biology, 65, pp. 9–17. [DOI] [PubMed] [Google Scholar]
- Proschak E, Stark H and Merk D (2019) ‘Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds’, Journal of medicinal chemistry, 62(2), pp. 420–444. [DOI] [PubMed] [Google Scholar]
- Regot S et al. (2014) ‘High-sensitivity measurements of multiple kinase activities in live single cells’, Cell, 157(7), pp. 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REPURPOSING (no date). Available at: https://clue.io/repurposing-app (Accessed: 2 June 2022).
- Rodgers JT et al. (2005) ‘Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1’, Nature, 434(7029), pp. 113–118. [DOI] [PubMed] [Google Scholar]
- Roessler HI et al. (2021) ‘Drug Repurposing for Rare Diseases’, Trends in pharmacological sciences, 42(4), pp. 255–267. [DOI] [PubMed] [Google Scholar]
- Rogers D and Hahn M (2010) ‘Extended-connectivity fingerprints’, Journal of chemical information and modeling, 50(5), pp. 742–754. [DOI] [PubMed] [Google Scholar]
- Rohban MH et al. (2017) ‘Systematic morphological profiling of human gene and allele function via Cell Painting’, eLife, 6. doi: 10.7554/eLife.24060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider P et al. (2020) ‘Rethinking drug design in the artificial intelligence era’, Nature reviews. Drug discovery, 19(5), pp. 353–364. [DOI] [PubMed] [Google Scholar]
- Schneidewind T et al. (2020) ‘Morphological Profiling Identifies a Common Mode of Action for Small Molecules with Different Targets’, Chembiochem: a European journal of chemical biology, 21(22), pp. 3197–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze CJ et al. (2013) ‘“Function-first” lead discovery: mode of action profiling of natural product libraries using image-based screening’, Chemistry & biology, 20(2), pp. 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanware NP et al. (2009) ‘Non-specific in vivo inhibition of CK1 by the pyridinyl imidazole p38 inhibitors SB 203580 and SB 202190’, BMB reports, 42(3). doi: 10.5483/bmbrep.2009.42.3.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlevkov E et al. (2019) ‘A High-Content Screen Identifies TPP1 and Aurora B as Regulators of Axonal Mitochondrial Transport’, Cell reports, 28(12), pp. 3224–3237.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simm J et al. (2018) ‘Repurposing High-Throughput Image Assays Enables Biological Activity Prediction for Drug Discovery’, Cell chemical biology, 25(5), pp. 611–618.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A et al. (2005) ‘Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles’, Proceedings of the National Academy of Sciences of the United States of America, 102(43), pp. 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A et al. (2017) ‘A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles’, Cell, 171(6), pp. 1437–1452.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TT et al. (2021) ‘Small Molecule Inhibitors of TEAD Auto-palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-deficient Mesothelioma’, Molecular cancer therapeutics, 20(6), pp. 986–998. [DOI] [PubMed] [Google Scholar]
- Trapotsi M-A et al. (2021) ‘Comparison of Chemical Structure and Cell Morphology Information for Multitask Bioactivity Predictions’, Journal of chemical information and modeling, 61(3), pp. 1444–1456. [DOI] [PubMed] [Google Scholar]
- Usui M, Uno M and Nishida E (2016) ‘Src family kinases suppress differentiation of brown adipocytes and browning of white adipocytes’, Genes to cells: devoted to molecular & cellular mechanisms, 21(4), pp. 302–310. [DOI] [PubMed] [Google Scholar]
- Vamathevan J et al. (2019) ‘Applications of machine learning in drug discovery and development’, Nature reviews. Drug discovery, 18(6), pp. 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner FF et al. (2018) ‘Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia’, Science translational medicine, 10(431), p. eaam8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X and Schwarz TL (2009) ‘Chapter 18 Imaging Axonal Transport of Mitochondria’, in Methods in Enzymology. Academic Press, pp. 319–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehrmann MH et al. (2013) ‘Large-scale cytological profiling for functional analysis of bioactive compounds’, Molecular bioSystems, 9(11), pp. 2604–2617. [DOI] [PubMed] [Google Scholar]
- Wouters OJ, McKee M and Luyten J (2020) ‘Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018’, JAMA: the journal of the American Medical Association, 323(9), pp. 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S et al. (2018) ‘YAP1-Mediated Suppression of USP31 Enhances NFkB Activity to Promote Sarcomagenesis’, Cancer research, 78(10), pp. 2705–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaltsman Y et al. (2019) ‘Angiomotin Regulates YAP Localization during Neural Differentiation of Human Pluripotent Stem Cells’, Stem cell reports, 12(5), pp. 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetser A, Gredinger E and Bengal E (1999) ‘p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor’, The Journal of biological chemistry, 274(8), pp. 5193–5200. [DOI] [PubMed] [Google Scholar]
- Zhao B et al. (2007) ‘Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control’, Genes & development, 21(21), pp. 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data statement
The large-scale Cell Painting datasets used in this paper are publicly available and their details and locations are described in publications (gene overexpression dataset(Rohban et al., 2017) and compound dataset(Bray et al., 2017)). RNA-sequencing data have been deposited into the NCBI Gene Expression Omnibus (GEO; accession number GSE198909). The accession numbers are also listed in the key resources table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HRP-conjugated anti-rabbit IgG antibodies at 1:10000 | Kiessling lab; Jackson ImmunoResearch Laboratories | #111-035-003 |
| HRP-conjugated anti-rabbit IgG antibodies at 1:2500 | Eisinger lab; Cell Signaling Technology [CST] | #7074 |
| anti-Yap1 primary antibodies | Cell Signaling Technology [CST] | #14074, #4912 |
| Alexa Fluor 488-conjugated secondary antibodies | Thermo Fisher Scientific | #A-11008 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| BRD-K43796186 (NB4A) | MuseChem | cat. #M189943 |
| BRD-K43796186 (NB4A) | Ambinter | cat # Amb2554311 |
| 8xGTIIC-luciferase | Gift from Stefano Piccolo, Addgene | plasmid # 34615 |
| Critical commercial assays | ||
| Deposited data | ||
| RNA-sequencing data | this paper | GSE198909 |
| Compound Dataset | Bray et al., 2017 | DOI: 10.1093/gigascience/giw014 |
| Gene Overexpression Dataset | Rohban et al., 2017 | DOI:10.7554/eLife.24060 |
| Experimental models: Cell lines | ||
| RT112/84 | Cancer Cell Line Encyclopedia (Broad Institute, Cambridge, MA) | |
| HEK293T | ATCC | |
| Murine KP230 | Eisinger lab | |
| STS-109 UPS | Rebecca Gladdy, MD (University of Toronto) | |
| TC32 | Patrick Grohar, MD, PhD (Children’s Hospital of Philadelphia) | |
| HT-1080 | ATCC | |
| HCT-116 | ATCC | |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Knockdown of YAP1 in HCT-116 cells | Dharmacon | TRCN0000107266 and TRCN0000107267 |
| Software and algorithms | ||
| GitHub Repository to reproduce the results | this paper | DOI 10.5281/zenodo.6970733 |
| Other | ||
Code statement
The code used in this study is available at https://github.com/broadinstitute/GeneCompoundImaging. It is available for use under the BSD 3-clause license, a permissive open-source license. The DOI is listed in the key resources table.
• Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.