

Summary
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein mediates membrane fusion between the virus and the target cells, triggering viral entry into the latter. Here, we describe a SARS-CoV-2 spike-protein-mediated membrane fusion assay using a dual functional split reporter protein to quantitatively monitor the fusion kinetics of the viral and target cell membranes in living cells. This approach can be applied in various cell types, potentially predicting the pathogenicity of newly emerging variants.
For complete details on the use and execution of this protocol, please refer to Kimura et al. (2022b), Kimura et al. (2022c), Motozono et al. (2021), Saito et al. (2022a), Saito et al. (2022b), Suzuki et al. (2022), and Yamasoba et al. (2022).
Subject areas: Cell-based Assays, Microbiology
Graphical abstract
Highlights
-
•
Quantitatively monitor SARS-CoV-2 spike (S) fusion activity using a dual split protein
-
•
Normalize SARS-CoV-2 S fusion activity by the surface expression levels
-
•
Monitor SARS-CoV-2 S-mediated membrane fusion kinetics in live cells
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike-protein mediates membrane fusion between the virus and the target cells, triggering viral entry into the latter. Here, we describe a SARS-CoV-2 spike-protein-mediated membrane fusion assay using a dual functional split reporter protein to quantitatively monitor the fusion kinetics of the viral and target cell membranes in living cells. This approach can be applied in various cell types, potentially predicting the pathogenicity of newly emerging variants.
Before you begin
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is continually evolving, with mutations appearing in its RNA genome. Mutations accumulating in the spike (S) gene influence transmissibility, pathogenicity, and resistance to immunity induced by viral infection and vaccines (Kimura et al., 2022b, 2022c; Meng et al., 2022; Motozono et al., 2021; Saito et al., 2022a, 2022b; Suzuki et al., 2022; Yamasoba et al., 2022). The S protein plays a pivotal role in the fusion of the viral membrane with the target cell membrane, allowing penetration and transfer of the viral genetic material into the host cell (Jackson et al., 2021; Li, 2016). We previously reported that the fusogenicity of the S protein in newly emerged SARS-CoV-2 variants is closely associated with the pathogenicity of the virus (Kimura et al., 2022c; Saito et al., 2022a, 2022b; Suzuki et al., 2022; Yamasoba et al., 2022). For example, the Delta variant contains relatively more fusogenic S protein than the Omicron BA.1 variant (Meng et al., 2022; Saito et al., 2022a; Suzuki et al., 2022) and also has greater pathogenicity (Saito et al., 2022a; Suzuki et al., 2022). Therefore, monitoring the fusogenicity of the S protein may be a potential marker to predict the pathogenicity of newly emerging viruses.
We established a SARS-CoV-2 S protein-mediated membrane fusion assay using a dual split protein (DSP) to monitor viral fusogenicity (Figure 1). This system monitors the signal from the DSP, which indicates successful cell–cell fusion after mixing cells expressing SARS-CoV-2 S and ACE2 (SARS-CoV-2 receptor) as well as the serine protease, TMPRSS2. DSP is a hybrid protein of split Renilla luciferase (RL) and split green fluorescence protein (GFP) and is composed of DSP1–7 and DSP8–11 (Kondo et al., 2010, 2011). The assembly of DSP1–7 and DSP8–11 after fusion produces luminescence and fluorescence by the reconstituted split proteins (Figure 1). This cell-based membrane fusion assay allows the quantitative monitoring of fusion kinetics in live cells.
Figure 1.

SARS-CoV-2 S-protein-mediated membrane fusion assay
Schematic showing that luminescence (RL) and fluorescence (GFP) only occur when the fragments of the split RL and GFP protein are reconstituted in the same cytosol after fusion.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-SARS-CoV-2 S S1/S2 polyclonal antibody (1:100 for immunoblotting) | Thermo Fisher Scientific | Cat# PA5-112048; RRID: AB_2866784 |
| Rabbit anti-SARS-CoV-2 S S1 monoclonal antibody (clone HL6) (1:100 for FACS analysis) | GeneTex | Cat# GTX635654; RRID: AB_2888548 |
| Mouse anti-SARS-CoV-2 S monoclonal antibody (clone 1A9) (1:100 for FACS analysis; 1:10,000 for immunoblotting) | GeneTex | Cat# GTX632604; RRID: AB_2864418 |
| Normal rabbit IgG (1:100 for FACS analysis) | SouthernBiotech | Cat# 0111-01; RRID: AB_2732899 |
| Normal mouse IgG (clone MG1-45) (1:100 for FACS analysis) | BioLegend | Cat# 401401; RRID: AB_2801452 |
| APC-conjugated goat anti-rabbit IgG polyclonal antibody (1:50 for FACS analysis) | Jackson ImmunoResearch | Cat# 111-136-144; RRID: AB_2337987 |
| APC-conjugated goat anti-mouse IgG polyclonal antibody (1:50 for FACS analysis) | Jackson ImmunoResearch | Cat# 115-136-146; RRID: AB_2338651 |
| Rabbit anti-beta actin (ACTB) monoclonal antibody (clone 13E5) (1:5,000 for immunoblotting) | Cell Signaling Technology | Cat# 4970; RRID: AB_2223172 |
| HRP-conjugated donkey anti-rabbit IgG polyclonal antibody (1:10,000 for immunoblotting) | Jackson ImmunoResearch | Cat# 711-035-152; RRID: AB_10015282 |
| HRP-conjugated donkey anti-mouse IgG polyclonal antibody (1:10,000 for immunoblotting) | Jackson ImmunoResearch | Cat# 715-035-150; RRID: AB_2340770 |
| Chemicals, peptides, and recombinant proteins | ||
| Fetal bovine serum | Nichirei | Cat# 175012 |
| Penicillin-streptomycin | Wako | Cat# 168-23191 |
| DMEM (high glucose) | Wako | Cat# 044-29765 |
| DMEM (low glucose) | Wako | Cat# 041-29775 |
| EMEM | Wako | Cat# 055-08975 |
| G418 | Wako | Cat# 070-06803 |
| Trypsin/EDTA | Wako | Cat# 209-16941 |
| EnduRen live cell substrate | Promega | Cat# E6481 |
| Nonidet P40 substitute | Nacalai Tesque | Cat# 18558-54 |
| Protease inhibitor cocktail | Nacalai Tesque | Cat# 03969-21 |
| SuperSignal west femto maximum sensitivity substrate | Thermo Fisher Scientific | Cat# 34095 |
| SuperSignal west atto ultimate sensitivity substrate | Thermo Fisher Scientific | Cat# A38554 |
| TransIT-LT1 | Takara | Cat# MIR2300 |
| Cell Dissociation Buffer, enzyme-free, Hanks’ Balanced Salt Solution | Gibco | Cat# 13150-016 |
| 7AAD Viability Staining Solution | BioLegend | Cat# 420403 |
| Protein assay dye | Bio-Rad | Cat# 5000006 |
| Experimental models: Cell lines | ||
| Human: HEK293 cells | ATCC | CRL-1573 |
| African green monkey (Chlorocebus sabaeus): VeroE6/TMPRSS2 cells | JCRB Cell Bank (Matsuyama et al., 2020) | JCRB1819 |
| Human: Calu-3/DSP1–7 cells | (Yamamoto et al., 2020) | N/A |
| Human: HeLa-ACE2/TMPRSS2 | (Suzuki et al., 2022) | N/A |
| Human: HOS-ACE2/TMPRSS2 | (Ozono et al., 2021) | N/A |
| Recombinant DNA | ||
| Plasmid: pDSP1–7 | (Kondo et al., 2010, 2011) | N/A |
| Plasmid: pDSP8–11 | (Kondo et al., 2010, 2011) | N/A |
| Plasmid: pC-ACE2 | (Ozono et al., 2021) | N/A |
| Plasmid: pC-TMPRSS2 | (Ozono et al., 2021) | N/A |
| Plasmid: pCAGGS | (Niwa et al., 1991) | N/A |
| Plasmid: pC-SARS-CoV-2 S-D614G | (Ozono et al., 2021) | N/A |
| Plasmid: pC-SARS-CoV-2 S-Delta | (Saito et al., 2022a) | N/A |
| Plasmid: pC-SARS-CoV-2 S-Omicron BA.1 | (Suzuki et al., 2022) | N/A |
| Software and algorithms | ||
| FlowJo software v10.7.1 | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| Image Studio Lite v5.2 | LI-COR Biosciences | https://www.licor.com/bio/image-studio/ |
| Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| SnapGene | Dotmatics | https://www.snapgene.com |
| Fiji software v2.2.0 | ImageJ | https://fiji.sc |
| R v4.1.2 | The R Foundation | https://www.r-project.org/ |
| Other | ||
| 96-well black view plate | PerkinElmer | Cat# 6005225 |
| 96-well flat bottom plate | Thermo Fisher Scientific | Cat# 137103 |
| PVDF membrane | Millipore | Cat# IPVH00010 |
| Centro XS3 LB960 | Berthold Technologies | N/A |
| FACS Canto II | BD Biosciences | N/A |
| SpectraMax ABS | Molecular Devices | https://www.moleculardevices.co.jp/systems/absorbance-readers |
| Amersham Imager 600 | GE Healthcare | N/A |
Materials and equipment
Lysis buffer
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| HEPES (pH 7.2) | 1 M | 25 mM | 1.25 mL |
| Glycerol | 50% | 10% | 10 mL |
| NaCl | 4 M | 125 mM | 1.56 mL |
| Nonidet P40 substitute | 100% | 1% | 0.5 mL |
| Water | N/A | N/A | up to 50 mL |
| Total | 50 mL |
Note: Add 1/100 volume of protease inhibitor cocktail before use. Do not store after adding protease inhibitor cocktail. Store at 4°C for up to 3 months before adding protease inhibitor cocktail.
2 × SDS sample buffer
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| Tris-HCl (pH 6.8) | 1 M | 100 mM | 5 mL |
| SDS | 20% | 4% | 10 mL |
| Glycerol | 100% | 20% | 10 mL |
| Bromophenol blue | 100% | 0.05% | 0.025 g |
| Water | N/A | N/A | up to 50 mL |
| Total | 50 mL |
Note: Add β-mercaptoethanol as the final concentration is 12% before use. Do not store after adding β-mercaptoethanol. Store at 20°C–25°C for up to 6 months before adding β-mercaptoethanol.
10 × Tris-buffered saline (TBS)
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| Tris | N/A | 250 mM | 30 g |
| NaCl | N/A | 1.37 M | 80 g |
| KCl | N/A | 26.8 mM | 2 g |
| Water | N/A | N/A | up to 1 L |
| Total | 1 L |
Note: Adjust to pH 7.4 using 4 N HCl. Store at 20°C–25°C for up to 3 months.
Step-by-step method details
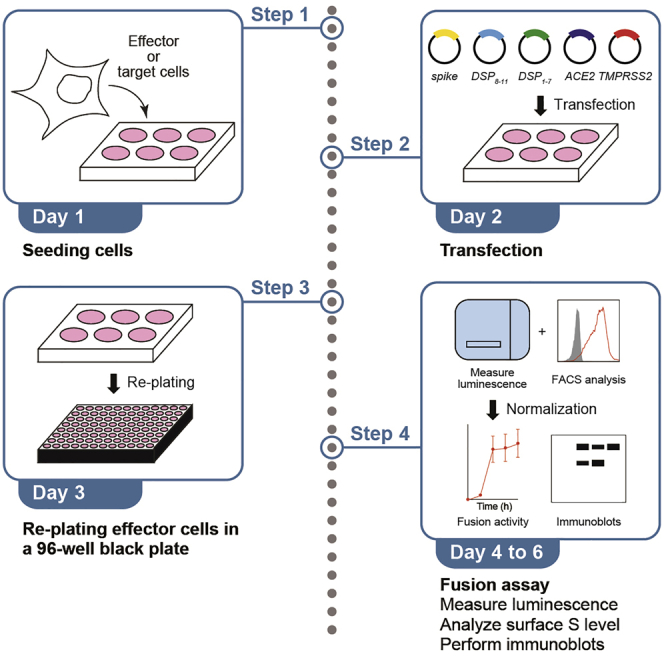
This protocol describes a 5-day SARS-CoV-2 S protein-mediated membrane fusion assay based on the HIV-1 Env protein-based fusion assay (Ikeda et al., 2018; Kondo et al., 2011) with some modifications. Briefly, cells are seeded (day 1) and then transfected with plasmids encoding genes for SARS-CoV-2 S, DSP, and a SARS-CoV-2 receptor, such as ACE2 (and the serine protease, TMPRSS2, if necessary) (day 2). SARS-CoV-2 S protein-expressing cells are then re-plated in a 96-well plate (day 3), RL substrate is incorporated into the target cells, which are then mixed with cells expressing SARS-CoV-2 S (day 4), and RL activity is monitored over 24 h in the living cells (days 4–5).
Cells
HEK293 cells (a human embryonic kidney cell line; ATCC, CRL-1573) and HOS-ACE2/TMPRSS2 cells (HOS cells stably expressing human ACE2 and TMPRSS2) (Ozono et al., 2021) were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). Calu-3/DSP1–7 cells (Calu-3 cells stably expressing DSP1-7) (Yamamoto et al., 2020) were cultured in Eagle’s minimum essential medium (EMEM) supplemented with 20% FBS and 1% P/S. HeLa-ACE2/TMPRSS2 (HeLa cells stably expressing human ACE2 and TMPRSS2; JCRB Cell Bank, JCRB1835) (Suzuki et al., 2022) and VeroE6/TMPRSS2 cells (VeroE6 cells stably expressing human TMPRSS2; JCRB Cell Bank, JCRB1819) (Matsuyama et al., 2020) were cultured in low glucose DMEM containing 10% FBS, 1% P/S, and G418 (1 mg/mL). All cells were maintained at 37°C with 5% CO2. Cells were split in the following ratio for passage; HEK293 cells and HOS-ACE2/TMPRSS2 cells were 1:5 or 1:10. Calu-3/DSP1–7 cells were 1:2 or 1:3. HeLa-ACE2/TMPRSS2 cells were 3:10 or 1:5. VeroE6/TMPRSS2 cells were 4:10 or 3:10. Importantly, Calu-3/DSP1–7 cells needed daily medium change to maintain a healthy condition.
Plasmids
Plasmids expressing the codon-optimized SARS-CoV-2 S proteins of parent (S gene bearing D614G mutation) (Ozono et al., 2021), Delta (Saito et al., 2022a) and Omicron BA.1 (Suzuki et al., 2022) were prepared in previous studies. Each S gene includes the following amino acid substitution compared to the reference sequence of Wuhan-Hu-1 S gene (GenBank accession number: NC_045512.2); parental S: D614G. Delta S: T19R/G142D/E156G/FR157-158del/L452R/T478K/D614G/P681R/D950N. Omicron BA.1 S: A67V/HV69-70del/T95I/G142D/VYY143-145del/NLVR211-214IVREPE/G339D/S371L/S373P/S375F/K417N/N440K/G446S/S477N/T478K/E484A/Q493R/G496S/Q498R/N501Y/Y505H/T547K/D614G/H655Y/N679K/P681H/N764K/D796Y/N856K/Q954H/N969K/L981F.
The creation and generation of human ACE2 expression vector pC-ACE2 and human TMPRSS2 expression vector pC-TMPRSS2 have been described previously (kindly provided by Kenzo Tokunaga) (Ozono et al., 2021). The function of ACE2 and TMPRSS2 proteins expressed by these vectors in HEK293T cells was also previously reported (Ozono et al., 2021).
The creation and generation of DSP1–7 expression vector pDSP1–7 and DSP8–11 expression vector pDSP8–11 have been described previously (kindly provided by Jin Gohda) (Kondo et al., 2010). It has been reported that the expression of the engineered dual split reporter proteins can monitor the fusion kinetics of HIV-1 Env protein (Ikeda et al., 2018; Kondo et al., 2010, 2011).
Maps of pC-SARS-2 S D614G, pC-ACE2, pC-TMPRSS2, pDSP1–7 and pDSP8–11 expression vectors created by SnapGene are shown in Figure 2.
Figure 2.

Maps of expression vectors used in this study
(A) The map of a vector expressing SARS-CoV-2 B.1.1 S protein harboring a D614G mutation, pC-SARS-2 S D614G. The SARS-CoV-2 Delta and Omicron BA.1 S protein expression vector maps are the same except for the size of each S gene. The B.1.1, Delta, and Omicron BA.1 S genes are 3822 bp, 3816 bp, and 3813 bp, respectively.
(B) The pC-ACE2 vector map.
(C) The pC-TMPRSS2 vector map.
(D) The pDSP1–7 vector map.
(E) The pDSP8–11 vector map. All vector maps were created by SnapGene.
Seeding cells (day 1)
Timing: 20 min
-
1.
Plate HEK293 cells at a density of 0.6–0.8 × 106 cells per well in a 6-well plate.
Note: This step is for preparing effector cells (i.e., SARS-CoV-2 S protein-expressing cells) (step 3).
Note: At least two wells of a 6-well plate containing 0.6 × 106 cells per well are required for each plasmid encoding the SARS-CoV-2 S gene. One well is used for re-seeding (step 7) and cell lysate preparation (step 21) and the other is used for quantifying surface S protein expression by flow cytometry (step 13).
-
2.
Seed 0.6–0.8 × 106 target cells per well in a 6-well plate.
Note: Various target cells can be used, such as HEK293, Calu-3/DSP1–7, HeLa-ACE2/TMPRSS2, HOS-ACE2/TMPRSS2, or VeroE6/TMPRSS2 cells.
Note: In general, 0.75 × 106 target cells per well in 6-well plates are sufficient for mixing with 10–12 SARS-CoV-2 S protein-expressing effector cells if experiments are performed in quadruplicates (i.e., the number of target cells plated on day 1 are comparable to that in 40–48 wells of a 96-well black plate on day 4) (step 11). Calu-3/DSP1–7 cells should be seeded at a density of 0.8 × 106/well and the medium should be changed daily to maintain healthy cells until mixing with effector cells (step 11).
Preparing effector cells and target cells by the transfection of expression vectors containing SARS-CoV-2 S, DSP, and ACE2 (and TMPRSS2) (day 2)
-
3.Transfect the HEK293 cells with pCAGGS vectors expressing the SARS-CoV-2 S gene and pDSP8–11 using TransIT-LT1.
-
a.Dilute the following plasmids in a serum-free medium in an Eppendorf tube.
Plasmid Stock concentration Final concentration Amount SARS-CoV-2 S expression vector or pCAGGS 100 ng/μL 400 ng 4 μL pDSP8–11 100 ng/μL 400 ng 4 μL Serum-free DMEM N/A N/A up to 100 μL Total 100 μL -
b.Add 2.4 μL of TransIT-LT1 reagent to the mixture.
-
c.Mix gently by pipetting, followed by incubation for 15 min at 20°C–25°C.
-
d.Add the transfection mixture dropwise to the corresponding well of a 6-well plate.
-
e.Incubate the plates for 24 h at 5% CO2 and 37°C.Note: This step is for preparing effector cells (SARS-CoV-2 S and DSP8–11-expressing HEK293 cells).Note: A vector control using the pCAGGS vector (Niwa et al., 1991) should be included.Note: In this study, we used the SARS-CoV-2 S genes of the B.1.1 variant bearing a D614G mutation, the Delta variant, and the Omicron BA.1 variant.Note: This is for one well of a 6-well plate. At least two wells of a 6-well plate are required for each SARS-CoV-2 S gene-encoding plasmid. Therefore, the double amount of the aforementioned transfection mixture is needed to prepare one S protein-expressing cells.Note: Many S protein expression plasmids have been used to characterize the virological phenotypes of newly emerged SARS-CoV-2 variants to date (Kimura et al., 2022a, 2022b, 2022c; Motozono et al., 2021; Saito et al., 2022a, 2022b; Suzuki et al., 2022; Yamasoba et al., 2022).
-
a.
-
4.In parallel, prepare the target cells by transfecting HEK293 cells with pC-ACE2 and pDSP1–7.
-
a.Dilute the following plasmids in a serum-free medium in an Eppendorf tube.
Plasmid Stock concentration Final concentration Amount pC-ACE2 100 ng/μL 200 ng 2 μL pDSP1–7 100 ng/μL 400 ng 4 μL Serum-free DMEM N/A N/A up to 100 μL Total 100 μL -
b.Add 1.8 μL of TransIT-LT1 reagent to the mixture.
-
c.Mix gently by pipetting, followed by incubation for 15 min at 20°C–25°C.
-
d.Add the transfection mixture dropwise to the corresponding well of a 6-well plate.
-
e.Incubate the plates for 24 h at 5% CO2 and 37°C.Note: This step is for preparing HEK293 cells transiently expressing ACE2 and DSP1–7 (HEK293-ACE2).Note: The requirement for ACE2 in this assay was previously reported (Saito et al., 2022a).
-
a.
-
5.Co-transfect the target HEK293 cells in selected wells using pC-TMPRSS2 in addition to pC-ACE2 and pDSP1–7.
-
a.Dilute the following plasmids in a serum-free medium in an Eppendorf tube.Note: This is for preparing HEK293 cells transiently expressing ACE2, TMPRSS2, and DSP1–7 (HEK293-ACE2/TMPRSS2).
Plasmid Stock concentration Final concentration Amount pC-ACE2 100 ng/μL 200 ng 2 μL pC-TMPRSS2 10 ng/μL 40 ng 4 μL pDSP1–7 100 ng/μL 400 ng 4 μL Serum-free DMEM N/A N/A up to 100 μL Total 100 μL -
b.Add 1.92 μL of TransIT-LT1 reagent to the mixture.
-
c.Mix gently by pipetting, followed by incubation for 15 min at 20°C–25°C.
-
d.Add the transfection mixture dropwise to the corresponding well of a 6-well plate.
-
e.Incubate the plates for 24 h at 5% CO2 and 37°C.Note: This step is needed to test how TMPRSS2 affects the SARS-S-CoV-2 S protein-mediated cell entry.Note: The enhancement by TMPRSS2 expression in this assay was previously reported (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022a; Suzuki et al., 2022).
-
a.
-
6.To prepare other target cells beyond HEK293 cells, transfect HeLa-ACE2/TMPRSS2, VeroE6/TMPRSS2, and HOS-ACE2/TMPRSS2 cells with pDSP1–7.
-
a.Dilute pDSP1–7 vector in a serum-free medium in an Eppendorf tube to prepare addition al target cells, HeLa-ACE2/TMPRSS2, VeroE6/TMPRSS2, and HOS- ACE2/TMPRSS2.
Plasmid Stock concentration Final concentration Amount pDSP1–7 100 ng/μL 400 ng 4 μL Serum-free DMEM N/A N/A up to 100 μL Total 100 μL -
b.Add 1.2 μL of TransIT-LT1 reagent to the mixture.
-
c.Mix gently by pipetting, followed by incubation for 15 min at 20°C–25°C.
-
d.Add the transfection mixture dropwise to the corresponding well of a 6-well plate.
-
e.Incubate the plates for 24 h at 5% CO2 and 37°C.Note: We used various cell lines as target cells in the SARS-CoV-2 S protein-mediated membrane fusion assay. For example, HEK293-ACE2 (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022a; Suzuki et al., 2022), HEK293-ACE2/TMPRSS2 (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022a; Suzuki et al., 2022), Calu-3/DSP1–7 (Kimura et al., 2022c; Saito et al., 2022b; Suzuki et al., 2022; Yamasoba et al., 2022), and VeroE6/TMPRSS2 (Kimura et al., 2022c; Suzuki et al., 2022; Yamasoba et al., 2022) cells have been used as target cells in previous studies. HEK293, HeLa-ACE2/TMPRSS2, VeroE6/TMPRSS2, and HOS-ACE2/TMPRSS2 cells require supplementation with pDSP1–7, whereas Calu-3/DSP1–7 cells do not; therefore, the target cells used may require supplements (DSP, ACE2, and TMPRSS2) for successful SARS-CoV-2 S protein-mediated membrane fusion assay.
-
a.
Re-plating the SARS-CoV-2 S-protein-expressing cells (day 3)
-
7.
At 24 h posttransfection, collect and re-seed 16,000 effector cells into each well of a 96-well black view plate (Key resources table).
Alternatives: The 96-well flat bottom plate listed in the Key resources table can be used instead of a 96-well black view plate.
Note: Occasionally, the RL signal indicating successful cell–cell fusion varies between the wells. Experiments should be performed in quadruplicate at least to obtain sufficient data for statistical analysis.
Note: Detach the effector cells by adequate pipetting without using trypsin or any cell dissociation buffer. To avoid cell clumps, we repeated pipetting at least 20–30 times and verified the cells under a microscope. Adequate pipetting always detaches more than 95% of cells; unrepresentative cells, if any, are usually under 1%.
-
8.
Re-seed the remaining effector cells at a density of 0.6–0.8 × 106 cells per well in at least two wells of a 6-well plate.
Note: one well is used for cell lysate preparation of immunoblotting and the other well is for flow cytometry in triplicate.
Note: Approximately 64,000 effector cells are required to perform fusion assays in quadruplicate for each target cell type. A total of 1.5 × 106 effector cells is sufficient for a fusion assay, including cell mixing with one target cell type (step 7), flow cytometry (step 13), and immunoblotting (step 21).
-
9.
Re-seed the target cells at a density of 1 × 106 cells/2 mL/well in 6-well plates to prepare for the incorporation of EnduRen live cell substrate (step 10).
Note: HEK293-ACE2, HEK293-ACE2/TMPRSS2, Calu-3/DSP1–7, HeLa-ACE2/TMPRSS2, HOS-ACE2/TMPRSS2 and VeroE6/TMPRSS2 cells were used as target cells.
Note: The detachment of HEK293-ACE2 and HEK293-ACE2/TMPRSS2 cells is usually performed by adequate pipetting (step 7). However, the detachment of firmly adherent cells, such as that of Calu-3/DSP1–7 and VeroE6/TMPRSS2 cells, requires the use of an enzyme-free cell dissociation buffer (Gibco, Cat# 13150-016; see https://www.thermofisher.com/order/catalog/product/13150016). In this case, cells are washed with phosphate-buffered saline (PBS) and treated with cell dissociation buffer for 10 min at 37°C. Next, the cells are harvested from the plates by gentle pipetting, transferred to an Eppendorf tube and washed with PBS. Finally, an appropriate number of cells is resuspended in a fresh medium to be added to 6-well plates.
Note: One well of the 6-well plate contains sufficient cells on day 1 (step 1) to re-plate into two wells of the 6-well plate, which is sufficient for 10–12 effector cells if experiments are performed in quadruplicate (i.e., they are comparable to 40–48 wells of a 96-well black plate on day 4).
Note: Step 9 is not necessary for Calu-3/DSP1–7 cells.
Incorporation of RL substrate into target cells and mixing of target cells with SARS-CoV-2 S-protein-expressing cells (day 4–5)
-
10.At 48 h posttransfection, incubate the target cells with EnduRen live cell substrate for 3 h at 5% CO2 and 37°C.
-
a.Dissolve 60 mM EnduRen solution in dimethyl sulfoxide.
-
b.Dilute to a final concentration of 60 μM with a prewarmed medium (1.25 mL of medium per well of target cells).
-
c.Replace the cell culture medium with 60 μM EnduRen-containing working solution.
-
d.Incubate the target cells with EnduRen for 3 h at 37°C.
-
a.
Note: Complete resuspension of EnduRen in a prewarmed medium is mandatory and is performed by adequate pipetting immediately prior to adding the medium to cells.
-
11.
After detaching the cells, add 32,000 target cells to each well of the 96-well view plate containing the effector cells.
Note: The detachment of HEK293 cells is usually performed by adequate pipetting (step 5). However, the detachment of firmly adherent cells, such as that of Calu-3/DSP1–7 and VeroE6/TMPRSS2 cells, requires the use of an enzyme-free cell dissociation buffer (Gibco, Cat# 13150-016; see step 7) after the 3-h incubation. In this case, the EnduRen-containing medium is removed and stored in an Eppendorf tube. The cells are then washed with PBS and treated with an enzyme-free cell dissociation buffer for 10 min at 37°C. After incubation, the cells are harvested from the plates by gentle pipetting, transferred to an Eppendorf tube and washed with PBS. Finally, an appropriate number of cells is resuspended in the stored medium for mixing with the effector cells.
-
12.
Measure the RL activity at 0, 6, 12, 18, and 24 h using a Centro XS3 LB960 microplate luminometer with a counting time of 1 s per well (Troubleshooting Problem 1).
Measurement of S protein expression levels on the surface of transfected cells (day 4)
Expression levels of SARS-CoV-2 S protein on the cell surface impact S protein-mediated membrane fusion activity. Therefore, the surface expression levels of S protein should be measured by flow cytometry and the fusion activity of SARS-CoV-2 S protein is normalized according to the surface expression levels (Kimura et al., 2022b, 2022c; Motozono et al., 2021; Saito et al., 2022a, 2022b; Suzuki et al., 2022; Yamasoba et al., 2022).
Staining cell surface S proteins for flow cytometry
-
13.
Harvest the effector cells by adequate pipetting (prepared in step 8).
-
14.
Incubate the cells with rabbit anti-SARS-CoV-2 S S1/S2 polyclonal antibody (1:100) or normal rabbit IgG (1:100; as an isotype control) at 4°C for 30 min.
Alternatives: Other anti-SARS-CoV-2 S antibodies are available (Key resources table).
Note: It is critical to use an anti-SARS-CoV-2 S antibody to measure the S protein expression levels on the cell surface because S protein expression levels are used to normalize luminescence and indicate successful cell–cell fusion. The normalized value represents the fusion activity of the S protein.
Note: Fluorescence-activated cell sorting (FACS) data using rabbit anti-SARS-CoV-2 S S1 monoclonal antibody (Motozono et al., 2021; Saito et al., 2022a), rabbit anti-SARS-CoV-2 S S1/S2 polyclonal antibody (Kimura et al., 2022b, 2022c; Meng et al., 2022; Saito et al., 2022a, 2022b; Suzuki et al., 2022; Yamasoba et al., 2022), and mouse anti-SARS-CoV-2 S monoclonal antibody (Suzuki et al., 2022) antibodies are predicted to show variable patterns depending on their epitopes and potentially alter the reactivity with S protein with mutations in the epitope. Therefore, an appropriate anti-S antibody should be considered for each experiment.
-
15.
Wash the cells three times with 2% FBS in PBS (2% FBS/PBS).
-
16.
Incubate the effector cells with APC-conjugated goat anti-rabbit IgG polyclonal antibody (see the Key resources table, 1:50) at 4°C for 30 min.
Alternatives: An appropriate secondary antibody may be selected depending on the primary antibody used for FACS analysis. The secondary antibodies used are shown in the Key resources table.
-
17.
Wash the cells three times with 2% FBS/PBS.
-
18.
Resuspend the cells in 2% FBS/PBS for immediate FACS analysis.
Alternatives: To evaluate the expression levels of surface S proteins in live cells, 7AAD viability staining solution (BioLegend, Cat# 420403) can be used before the FACS analysis. After washing, the cells are treated with 5 μL of 7AAD solution at 20°C–25°C for 5–10 min in the dark, and then proceeded to the FACS analysis (step 19).
-
19.
Measure the surface expression level of S proteins using a FACS Canto II clinical flow cytometry system.
-
20.
Analyze the data using FlowJo software v10.7.1. Representative FACS data are shown in Figure 3 (Troubleshooting Problem 2).
Figure 3.

Surface expression levels of S protein
(A) Representative FACS data of surface S proteins in HEK293 cells. S protein-expressing-HEK293 cells were harvested at 48 h posttransfection and stained using rabbit anti-SARS-CoV-2 S S1/S2 polyclonal antibody. Expression levels of the indicated S proteins on the cell surface were measured by flow cytometry and are shown as the MFI in the histogram. Normal rabbit IgG was used as isotype control (gray). pCAGGS was used as the vector. The parent S protein was derived from the B.1.1 variant bearing a D614G mutation.
(B) A summary of S protein expression levels on the HEK293 cell surface. The graph indicates the mean ± standard deviation (SD) MFI (n = 3). pCAGGS was used as a vector. The parent S protein was derived from the B.1.1 variant bearing a D614G mutation. Statistical significance was assessed using two-sided paired t test. ∗ p < 0.05 compared with the parent S protein (blue) and Omicron BA.1 S protein (green).
Immunoblotting
The S1/S2 cleavage of SARS-CoV-2 S protein by cellular proteases, such as Furin, is important for the fusion of the effector cells to the target cells and subsequent pathogenicity. The Delta variant showed significantly higher fusogenicity of its S protein than that of the parental virus, which harbors a D614G mutation (Saito et al., 2022a). This higher fusogenic potential was partially mediated by a P681R mutation in the S protein that facilitated S1/S2 cleavage and was associated with higher pathogenicity (Saito et al., 2022a). In contrast, the S protein of the Omicron BA.1 variant showed less efficient S1/S2 cleavage and less fusogenicity to target cells compared with that of the Delta variant (Meng et al., 2022; Suzuki et al., 2022).
From sample preparation to protein visualization for immunoblotting
-
21.
Detach the HEK293 cells co-transfected with the S expression plasmids and pDSP8–11 (prepared in step 8) by adequate pipetting 20–30 times.
-
22.
Transfer the cells into an Eppendorf tube on ice and pellet down by centrifugation (300 × g at 4°C for 5 min) for washing twice with 1 × PBS.
-
23.
After discarding the supernatant, tap the tube to loosen the cell pellets.
-
24.
Resuspend the pellets in ice-cold lysis buffer containing the protease inhibitor cocktail (see “Materials and equipment” section).
Note: Approximately 1 × 105 cells are resuspended in 100 μL of ice-cold lysis buffer.
-
25.
Incubate on ice for 30 min.
-
26.
Centrifuge at 20,000 × g at 4°C for 15 min.
-
27.
Collect the supernatant in a new Eppendorf tube as total cell lysate.
-
28.
Quantify total protein using a Bio-Rad protein assay dye reagent (Bio-Rad, Cat# 500006) according to the manufacturer’s instructions.
Note: First, the working assay dye is prepared by diluting the concentrated assay dye at a ratio of 1:5 using deionized water. Then, 5 μL of the total cell lysate is added to 995 μL of the working assay dye by mixing well and the reaction mixture is incubated at 20°C–25°C for 5 min. After incubation, 200 μL of each sample is transferred into a well of the 96-well plate in quadruplicates. The absorbance is read at 595 nm by SpectraMax ABS (Molecular Devices). It is noted that the absorbance should be read as soon as possible as the absorbance increases over time and exceeds the detection limit. Moreover, samples should be incubated at 20°C–25°C for not more than 30 min. Finally, the protein concentration of each sample is calculated by plotting the sample’s OD value at 595 nm against the standard curve. The standard curve is established by plotting the OD value of 6 standard samples prepared by making serial dilutions out of 1 mg/mL bovine serum albumin solution; 5 μL of each standard is mixed with 995 μL of working assay dye and the OD value at 595 nm is recorded after 5 min incubation.
-
29.
Dilute the lysates with 2 × sodium dodecyl sulfate (SDS) sample buffer (see “Materials and equipment” section).
-
30.
Boil the mixture at 98°C on a heat block for 10 min.
-
31.
Load 10 μL of each sample (50 μg of total protein) onto gradient 4%–20% SDS-PAGE gels for separation.
Note: The molecular weight of the full-length SARS-CoV-2 S protein is ∼180 kDa. Therefore, an SDS-PAGE gel that can cover a broad protein size range should be used.
-
32.
Transfer the proteins to methanol-activated PVDF membranes using a Bio-Rad wet/tank blotting system.
Note: A wet tank transfer is recommended to successfully transfer high-molecular weight proteins.
-
33.
Block the membrane with 4% skim milk in Tris-buffered saline containing 0.1% Tween 20 (0.1% TBST) under gentle shaking at 20°C–25°C for 1 h.
-
34.
Incubate the membranes at 20°C–25°C for 1 h or at 4°C for 12–16 h for protein detection with the following primary antibodies (Key resources table).
Note: A mouse anti-SARS-CoV-2 S monoclonal antibody was diluted at a 1:10,000 ratio in 4% skim milk/0.1% TBST. A rabbit anti-beta actin monoclonal antibody was diluted at a 1:5,000 ratio in 4% skim milk/0.1% TBST.
-
35.
Wash the membrane three times with 0.1% TBST under gentle shaking at 20°C–25°C for 10 min.
-
36.
Incubate the membrane with the following secondary antibodies under gentle shaking at 20°C–25°C for 1 h (the secondary antibodies are listed in the Key resources table).
Note: A horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG polyclonal antibody or HRP-conjugated donkey anti-mouse IgG polyclonal antibody was diluted at a 1:10,000 ratio in 1% skim milk/0.1% TBST.
-
37.
Wash the membrane three times with 0.1% TBST under gentle shaking at 20°C–25°C for 10 min.
-
38.
Measure chemiluminescence using SuperSignal west femto maximum sensitivity substrate according to the manufacturer’s instructions.
-
39.
Visualize only the bands that are not overexposed using an Amersham Imager 600 (Troubleshooting Problem 3).
Expected outcomes
The fusion activity of the Delta S protein is expected to be consistently higher than that of the parental S protein, which harbors a D614G mutation, and the Omicron BA.1 S protein in all cell lines used for the S protein-mediated fusion assay (Figure 4). The fusion activity mediated by the parental S protein is expected to be higher than the Omicron BA.1 S protein in all cell lines used (Figure 4). The fusion activity of these S proteins could predict the pathogenicity of new SARS-CoV-2 variants.
Figure 4.

Representative S-protein-mediated membrane fusion assay in various cell lines
S protein-mediated membrane fusion assay was performed in HEK293-ACE2, HEK293-ACE2/TMPRSS2, Calu-3/DSP1-7, HeLa-ACE2/TMPRSS2, HOS-ACE2/TMPRSS2 and VeroE6/TMPRSS2 cells, respectively. Luminescence, indicating successful cell–cell fusion, was monitored at 0, 6, 12, 18, and 24 h after mixing the effector and target cells. The experiments were performed in quadruplicate. The parent S protein was derived from the B.1.1 variant bearing a D614G mutation. Fusion activity (arbitrary unit) was measured as luminescence normalized to MFI and is shown as mean ± SD. Statistical differences between a pair of variants across timepoints were determined by multiple regression and indicated in each graph. The 0 h data were excluded from the analyses. The FWERs calculated using the Holm method are indicated in the figures.
S1/S2 cleavage of the Delta S protein is expected to be higher than that of the parental S protein (Figure 5). On the other hand, the S protein of the Omicron BA.1 variant is expected to show less efficient S1/S2 cleavage (Figure 5), consistent with its lower fusogenicity (Figure 4).
Figure 5.

Cleavage of S protein in HEK293 cells
(A) Representative immunoblots. The cell lysates of HEK293 cells transfected with the S gene were subjected to immunoblotting. The indicated S proteins were detected by mouse anti-SARS-CoV-2 S monoclonal antibody (S2 subunit). pCAGGS was used as the vector. The parent S protein was derived from the B.1.1 variant bearing a D614G mutation. β-actin was used as the loading control.
(B) Summary of the S1/S2 cleavage efficiency of the indicated S proteins in HEK293 cells. Full-length S and S2 band intensities were quantified and the ratio of S2 to the sum of S and S2 [S2/(S + S2)] was calculated from three independent experiments. The parent S protein was derived from the B.1.1 variant bearing a D614G mutation. Data represent the mean S2/(S + S2) ± SD relative to the parent S protein. Statistical significance was performed by a two-sided paired t test. ∗ p < 0.05 compared with the parent S protein (blue), Delta S protein (red), and Omicron BA.1 S protein (green).
Quantification and statistical analysis
-
1.
The level of SARS-CoV-2 S protein expression on the cell surface affects the S protein mediates the membrane fusion activity. Fusion activity is calculated as RL divided by the mean fluorescence intensity (MFI) of the surface S protein. The normalized value (i.e., RL activity per surface S protein MFI) was represented as the fusion activity (Figure 4).
-
2.
The S1/S2 cleavage efficiency of the SARS-CoV-2 S protein is closely associated with its fusion activity to target cells and pathogenicity (Saito et al., 2022a; Suzuki et al., 2022). Therefore, the S1/S2 cleavage of the SARS-CoV-2 S protein was quantified using the band intensities of the full-length S and cleaved S2 using Image Studio Lite v5.2 or Fiji software v2.2.0 (ImageJ).
-
3.
Statistical significance was performed using a two-sided paired t test (Figures 3B and 5B). GraphPad Prism software v8.4.3 was used for these statistical tests.
-
4.
In the time-course experiments (Figure 4), a multiple regression analysis including experimental conditions (i.e., S protein types) as explanatory variables and timepoints as qualitative control variables was performed to evaluate the difference between the experimental conditions thorough all timepoints. The initial time point was removed from the analysis. The P value was calculated by a two-sided Wald test. Subsequently, familywise error rates (FWERs) were calculated using the Holm method. These analyses were performed in R v4.1.2 (https://www.r-project.org/).
Limitations
The SARS-CoV-2 S protein plays a pivotal role in viral infection by mediating the fusion of the viral membrane with the target cell membrane (Jackson et al., 2021; Li, 2016). Incorporating the S protein into a cell-free virion leads to binding with the ACE2 protein expressed in the target cell and facilitates viral entry into the target cells. However, the S protein on the plasma membrane of the infected cell triggers fusion between the infected cell and neighboring cells expressing ACE2 and forms multinucleated cells, known as syncytia. We established a SARS-CoV-2 S protein-mediated membrane fusion assay that allows for the quantification of S protein-mediated fusion activity between S protein-expressing (i.e., effector cell) and ACE2 (and TMPRSS2)-expressing target cells using a DSP (Kimura et al., 2022b, 2022c; Motozono et al., 2021; Saito et al., 2022a, 2022b; Suzuki et al., 2022; Yamasoba et al., 2022). Interestingly, several of our studies have demonstrated that SARS-CoV-2 S protein-mediated fusion activity quantified using this assay is closely associated with viral pathogenicity in an in vivo hamster model (Kimura et al., 2022c; Saito et al., 2022a, 2022b; Suzuki et al., 2022; Yamasoba et al., 2022). However, virion-associated S fusion activity cannot be determined using this assay and an advanced method is required to quantify virion-associated S fusion activity in order to fully understand the S protein function. Furthermore, a system that allows for measuring the fusogenicity of authentic viruses would be required to understand SARS-CoV-2 spread and pathogenesis.
Troubleshooting
Problem 1
Low RL activity (step 12).
Potential solution
Insufficient S protein expression in the effector cells may reduce the luminescence from RL. Thus, the S protein expression levels on the surface of the effector cells should be confirmed by flow cytometry (step 13). If the S protein expression levels are low, fresh HEK293 cells should be used as the effector cells to restore efficient transfection and subsequent luminescence.
In general, the condition of the effector cell (i.e., HEK293 cells) is critical for efficient transfection and appropriate S protein expression levels on the cell surface and subsequent cell–cell fusion. Unsuccessful S protein-mediated membrane fusion can be avoided by going back to an early passage frozen stock of HEK293 cells monthly. In addition, daily cell monitoring and observation of cells throughout the assay are essential to maintain good cell conditions.
Problem 2
Low cell surface S expression levels (step 20).
Potential solution
The MFI of the parental D614G S protein is typically 40- to 60-folds over background (as determined by isotype control staining) under our experimental settings. If the MFI of D614G S protein is less than 20-fold over background, this indicates inappropriate cell conditions and the use of fresh HEK293 cells is recommended.
S protein expression levels are used to normalize luminescence values, which are finally used to represent the S protein fusion activity. Therefore, an anti-SARS-CoV-2 S antibody is essential for FACS analysis. To date, we have seen variable expression levels among S protein variants on the cell surface (Kimura et al., 2022b, 2022c; Motozono et al., 2021; Saito et al., 2022a, 2022b; Suzuki et al., 2022; Yamasoba et al., 2022); however, these differences have not shown more than a twofold difference between the highest and lowest values using a rabbit anti-SARS-CoV-2 S S1/S2 polyclonal antibody. To our knowledge, this polyclonal antibody is one of the best antibodies for the FACS analysis of surface S proteins. However, another anti-S protein antibody should be considered if the parental D614G S protein MFI is less than 20-folds over background.
Problem 3
Unclear S expression levels in effector cells by immunoblotting (step 39).
Potential solution
Fresh HEK293 cells should be used if the RL activity (step 12) and S expression levels on the cell surface (step 20) are low. However, when these parameters are normal, possible reasons for low S expression levels in effector cells by immunoblotting are as follows. (1) Low concentration of total proteins loaded: We recommend using 10 μL of sample, which is equivalent to 50 μg of total protein (step 31). (2) Large S protein >180 kDa: An efficient transfer system should be considered to successfully transfer high-molecular weight proteins (step 32). (3) Unsuitable primary antibody (step 34): The selected primary antibody may not react with the S protein used in the experiments; The target epitope of the selected primary antibody should be verified and another primary antibody should be considered. (4) Low substrate levels (step 38): The sensitivity of substrate used is below the detection limit for the S protein used in the experiments. We used SuperSignal west femto maximum sensitivity substrate. Alternatively, SuperSignal west atto ultimate sensitivity substrate (Thermo Fisher Scientific, Cat# A38554) is available.
Resource availability
Lead contact
Further information for resources and reagents should be directed to and will be fulfilled by the lead contact, Terumasa Ikeda (ikedat@kumamoto-u.ac.jp).
Materials availability
This study did not generate new unique reagents.
Consortia
The Genotype to Phenotype Japan (G2P-Japan) Consortium: Keita Matsuno, Naganori Nao, Hirofumi Sawa, Mai Kishimoto, Shinya Tanaka, Masumi Tsuda, Lei Wang, Yoshikata Oda, Marie Kato, Zannatul Ferdous, Hiromi Mouri, Kenji Shishido, Takasuke Fukuhara, Tomokazu Tamura, Rigel Suzuki, Hayato Ito, Daichi Yamasoba, Izumi Kimura, Naoko Misawa, Keiya Uriu, Yusuke Kosugi, Shigeru Fujita, Mai Suganami, Mika Chiba, Ryo Yoshimura, So Nakagawa, Jiaqi Wu, Akifumi Takaori-Kondo, Kotaro Shirakawa, Kayoko Nagata, Yasuhiro Kazuma, Ryosuke Nomura, Yoshihito Horisawa, Yusuke Tashiro, Yugo Kawai, Takashi Irie, Ryoko Kawabata, MST Monira Begum, Otowa Takahashi, Kimiko Ichihara, Takamasa Ueno, Chihiro Motozono, Mako Toyoda, Yuri L. Tanaka, Erika P. Butlertanaka, Maya Shofa, Kazuo Takayama, Rina Hashimoto, Sayaka Deguchi, Takao Hashiguchi, Tateki Suzuki, Kanako Kimura, Jiei Sasaki, Yukari Nakajima, Kaori Tabata.
Acknowledgments
We would like to thank all members belonging to The Genotype to Phenotype Japan (G2P-Japan) Consortium. We thank Dr. Kenzo Tokunaga (National Institute for Infectious Diseases, Japan) and Dr. Jin Gohda (The University of Tokyo, Japan) for providing reagents. We also thank Ms. Kazuko Kitazato and Haruyo Hasebe for their excellent assistance.
This study was supported in part by AMED Research Program on Emerging and Re-emerging Infectious Diseases (JP21fk0108465 to A.S.; JP21fk0108146 and JP22fk0108146 to K.S.; JP20fk0108413 to K.S. and T.I.; JP20fk0108451 to G2P-Japan Consortium, A.S., K.S., and T.I.; JP21fk0108574 to H.N.; and JP21fk0108494 to G2P-Japan Consortium, K.S., and T.I.); AMED Research Program on HIV/AIDS (JP21fk0410033, JP22fk0410033, and JP22fk0410047 to A.S.; JP21fk0410039 and JP22fk0410039 to K.S.; and JP22fk0410055 to T.I.); AMED CRDF Global Grant (JP21jk0210039 and JP22jk0210039 to A.S.); AMED Japan Program for Infectious Diseases Research and Infrastructure (JP21wm0325009 and JP22wm0325009 to A.S.); JST A-STEP (JPMJTM20SL to T.I.); JST SICORP (e-ASIA) (JPMJSC20U1 to K.S.); JST SICORP (JPMJSC21U5 to K.S.), JST CREST (JPMJCR20H4 to K.S.); JSPS KAKENHI Grant-in-Aid for Scientific Research C (19K06382 to A.S. and 22K07103 to T.I.); JSPS KAKENHI Grant-in-Aid for Scientific Research B (18H02662 and 21H02737 to K.S.); JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (22K16375 to H.N.); JSPS Fund for the Promotion of Joint International Research (Fostering Joint International Research) (18KK0447 to K.S.); JSPS Core-to-Core Program (A. Advanced Research Networks) (JPJSCCA20190008 to K.S.); JSPS Leading Initiative for Excellent Young Researchers (LEADER) (to T.I.); The Tokyo Biochemical Research Foundation (to K.S.); Takeda Science Foundation (to T.I.); Mitsubishi Foundation (to T.I.); Shin-Nihon Foundation of Advanced Medical Research (to T.I.); Waksman Foundation of Japan (to T.I.); a Grant for Joint Research Projects of the Research Institute for Microbial Diseases, Osaka University (to A.S.); an intramural grant from Kumamoto University COVID-19 Research Projects (AMABIE) (to T.I.); and Intercontinental Research and Educational Platform Aiming for Eradication of HIV/AIDS (to T.I.).
Author contributions
H.N., R.S., A.S., K.S., and T.I. performed the experiments. H.N., A.S., K.S., and T.I. designed the experiments and interpreted the results. J.I. and T.I. performed the statistical analyses. H.N. and T.I. wrote the original manuscript. All authors reviewed and proofread the manuscript. The Genotype to Phenotype Japan (G2P-Japan) Consortium contributed to the project administration.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Terumasa Ikeda, Email: ikedat@kumamoto-u.ac.jp.
The Genotype to Phenotype Japan (G2P-Japan) Consortium:
Keita Matsuno, Naganori Nao, Hirofumi Sawa, Mai Kishimoto, Shinya Tanaka, Masumi Tsuda, Lei Wang, Yoshikata Oda, Marie Kato, Zannatul Ferdous, Hiromi Mouri, Kenji Shishido, Takasuke Fukuhara, Tomokazu Tamura, Rigel Suzuki, Hayato Ito, Daichi Yamasoba, Izumi Kimura, Naoko Misawa, Keiya Uriu, Yusuke Kosugi, Shigeru Fujita, Mai Suganami, Mika Chiba, Ryo Yoshimura, So Nakagawa, Jiaqi Wu, Akifumi Takaori-Kondo, Kotaro Shirakawa, Kayoko Nagata, Yasuhiro Kazuma, Ryosuke Nomura, Yoshihito Horisawa, Yusuke Tashiro, Yugo Kawai, Takashi Irie, Ryoko Kawabata, MST Monira Begum, Otowa Takahashi, Kimiko Ichihara, Takamasa Ueno, Chihiro Motozono, Mako Toyoda, Yuri L. Tanaka, Erika P. Butlertanaka, Maya Shofa, Kazuo Takayama, Rina Hashimoto, Sayaka Deguchi, Takao Hashiguchi, Tateki Suzuki, Kanako Kimura, Jiei Sasaki, Yukari Nakajima, and Kaori Tabata
Data and code availability
The raw data supporting the current study are available from the lead contact on request.
References
- Ikeda T., Symeonides M., Albin J.S., Li M., Thali M., Harris R.S. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog. 2018;14:e1007010. doi: 10.1371/journal.ppat.1007010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson C.B., Farzan M., Chen B., Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022;23:3–20. doi: 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I., Kosugi Y., Wu J., Zahradnik J., Yamasoba D., Butlertanaka E.P., Tanaka Y.L., Uriu K., Liu Y., Morizako N., et al. The SARS-CoV-2 Lambda variant exhibits enhanced infectivity and immune resistance. Cell Rep. 2022;38:110218. doi: 10.1016/j.celrep.2021.110218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I., Yamasoba D., Nasser H., Zahradnik J., Kosugi Y., Wu J., Nagata K., Uriu K., Tanaka Y.L., Ito J., et al. SARS-CoV-2 spike S375F mutation characterizes the Omicron BA.1 variant. bioRxiv. 2022 doi: 10.1101/2022.04.03.486864. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I., Yamasoba D., Tamura T., Nao N., Suzuki T., Oda Y., Mitoma S., Ito J., Nasser H., Zahradnik J., Uriu K., et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 subvariants including BA.4 and BA.5. Cell. 2022 doi: 10.1016/j.cell.2022.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo N., Miyauchi K., Matsuda Z. Monitoring viral-mediated membrane fusion using fluorescent reporter methods. Curr. Protoc. Cell Biol. 2011;Chapter 26 doi: 10.1002/0471143030.cb2609s50. Unit 26.9. [DOI] [PubMed] [Google Scholar]
- Kondo N., Miyauchi K., Meng F., Iwamoto A., Matsuda Z. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem. 2010;285:14681–14688. doi: 10.1074/jbc.M109.067090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016;3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Nao N., Shirato K., Kawase M., Saito S., Takayama I., Nagata N., Sekizuka T., Katoh H., Kato F., et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA. 2020;117:7001–7003. doi: 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B., Abdullahi A., Ferreira I.A.T.M., Goonawardane N., Saito A., Kimura I., Yamasoba D., Gerber P.P., Fatihi S., Rathore S., et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature. 2022;603:706–714. doi: 10.1038/s41586-022-04474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motozono C., Toyoda M., Zahradnik J., Saito A., Nasser H., Tan T.S., Ngare I., Kimura I., Uriu K., Kosugi Y., et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. 2021;29:1124–1136.e11. doi: 10.1016/j.chom.2021.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Ozono S., Zhang Y., Ode H., Sano K., Tan T.S., Imai K., Miyoshi K., Kishigami S., Ueno T., Iwatani Y., et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021;12:848. doi: 10.1038/s41467-021-21118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A., Irie T., Suzuki R., Maemura T., Nasser H., Uriu K., Kosugi Y., Shirakawa K., Sadamasu K., Kimura I., et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature. 2022;602:300–306. doi: 10.1038/s41586-021-04266-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A., Tamura T., Zahradnik J., Deguchi S., Tabata K., Kimura I., Ito J., Nasser H., Toyoda M., Nagata K., et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2.75. bioRxiv. 2022 doi: 10.1016/j.chom.2022.10.003. https://www.biorxiv.org/content/10.1101/2022.08.07.503115v1 Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R., Yamasoba D., Kimura I., Wang L., Kishimoto M., Ito J., Morioka Y., Nao N., Nasser H., Uriu K., et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature. 2022;603:700–705. doi: 10.1038/s41586-022-04462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M., Kiso M., Sakai-Tagawa Y., Iwatsuki-Horimoto K., Imai M., Takeda M., Kinoshita N., Ohmagari N., Gohda J., Semba K., et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses. 2020;12:629. doi: 10.3390/v12060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasoba D., Kimura I., Nasser H., Morioka Y., Nao N., Ito J., Uriu K., Tsuda M., Zahradnik J., Shirakawa K., et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell. 2022;185:2103–2115.e19. doi: 10.1016/j.cell.2022.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data supporting the current study are available from the lead contact on request.