

Abstract
Defects in cellular functions related to altered protein homeostasis and associated progressive accumulation of pathological intracellular material is a critical process involved in the pathogenesis of many neurodegenerative disorders, including Alzheimer’s disease and Parkinson’s disease. Autophagy is an essential mechanism that ensures neuronal health by removing long-lived proteins or defective organelles and by doing so prevents cell toxicity and death within the central nervous system. Abundant evidence has shown that neuronal autophagy pathways are altered in Alzheimer’s disease, Parkinson’s disease and traumas of the central nervous system including Spinal Cord Injury and Traumatic Brain Injury. In this review, we aimed to summarize the latest studies on the role that altered neuronal autophagy plays in brain health and these pathological conditions, and how this knowledge can be leveraged for the development of novel therapeutics against them.
Keywords: Autophagy, Neurons, Central nervous system, Neurodegeneration, Alzheimer’s disease, Parkinson’s disease, Traumatic brain injury, Spinal cord injury
1. Introduction
The physiological control of different cellular processes and activities is defined as cellular homeostasis, a fundamental condition that guarantees normal function and balance of different components and structures of a living organism in terms of cellular health, resilience, and survival. A deficit in maintaining this balance efficient can quickly results in cellular dysfunction and eventually a condition of disease for the organism. Depending on the different cell types, cellular homeostasis is contingent upon multiple factors and among them pH, cell metabolism, metabolites production and excretion, and proteins homeostasis, all of which must be kept under tight control, despite constant environmental changes, challenges and variability in different biochemical processes (Hancock et al., 2017). Defects in protein homeostasis (also known as proteostasis) as well as progressive accumulation of intracellular unwanted materials and organelles is a process that has been critically involved in the onset and progression of neurodegenerative pathologies, including Alzheimer’s disease (AD) and Parkinson’s disease (PD). The major pathways for the degradation of abnormal cellular components are autophagy and cytosolic turnover by the proteasome systems. In mammals, both pathways recruit multiple proteins and enzymes that correctly recognize the “to be degraded” signal of unwanted material and drive the degradation process with the ultimate goal of maintaining cellular homeostasis. However, under particular conditions such as stress, starvation, aging, or disease, the steady state level of most unwanted proteins dramatically changes resulting in the engulfment of the endocytic transport system, protein traffic jam, altered autophagy efficiency and ultimately irreversible loss of cellular homeostasis.
Growing evidence demonstrates how abnormalities of neuronal autophagy could serve as a pro-degenerative triggering mechanism in AD. Defects in this process will lead to an abnormal and progressive build-up of intracellular and extracellular amyloid-beta (Aβ) peptides in AD individuals (Watanabe et al., 2020) facilitating Aβ plaque formation which would trigger microglia activation with secondary pro-inflammatory cytokines release, reactive oxygen species (ROS) production and neuronal cell death (Geng et al., 2018). Besides Aβ, also the accumulation of tau filaments and neurofibrillary tangles (Chen et al., 2020), formed by hyper-phosphorylated tau protein isoforms has been associated with neuronal dysfunction following autophagy impairments (Li et al., 2020). Moreover, genome-wide association studies have identified variants associated with genes involved in the autophagy pathway (Karch & Goate, 2015) with a significant increase the risk of a higher amyloid burden (Caglayan et al., 2014) (Nixon et al., 2007).
Autophagy is essential for the removal of long-lived abnormal proteins or defective organelles to prevent toxicity, cell death, and neurodegeneration also in PD. Thus, accumulation of α-synuclein (α-syn) protein, the major component of Lewy body (LB), was found to be positively associated with autophagy proteins responsible for autophagy machinery activation including microtubule-associated protein 1A/1B-light chain 3 (MAP-LC3 or LC3), autophagy-related proteins (ATGs), sequestrome-1 (SQSTM-1/p62) and lysosome-associated protein 2 (LAMP-2) (Tanida et al., 2008).
Interestingly, the lysosome system, known not only for leading organelles degradation but also as the main regulator of autophagy machinery promoting clearance of pathological aggregates, is also considered as a potential candidate link between Aβ, tau, and α-syn in the design of new treatment against complex neurodegenerative diseases of Central Nervous System (CNS) (Djajadikerta et al., 2019). In recent years, evidence has been accumulating suggesting that both recycling and degradation systems can also be influenced by a master regulator of endosomal protein sorting and trafficking, the retromer complex system. Studies have shown that deficits of this complex can influence autophagy flux as well as lysosome and proteasome pathways (a. Filippone et al., 2021) (b. Filippone et al., 2021).
Moreover, the involvement of both autophagy and lysosomal pathways has been reported in traumatic events of CNS where the primary injury (i.e., mechanical impact) and the secondary injury (i.e., activation of biochemical and molecular processes) affect neuronal tissue in the brain or spinal cord leading to the destruction of the neurovascular unit and neurological function deficits (Wu et al., 2019). After moderate or severe traumas, autophagy dysfunction can influence neuronal tissue repair and modulate the inflammatory cascade through the release of pro-inflammatory cytokines and chemokines (i.e., interferons [IFNs] and interleukins [ILs]) secondary to the activation of CNS resident immune cells such as microglia and astrocytes. In this review, first we will describe the major players controlling neuronal autophagy, then we will focus on the novel concept of modulating neuronal autophagy pathway as a viable therapeutic approach against AD and PD and CNS traumas.
2. Neuronal autophagy overview: machinery and players
Neuronal autophagy is a cellular degradation and recycling process which by sequestering unwanted cytosolic contents and delivering them to the degradative organelles, such as lysosomes and vacuoles, controls their final digestion and elimination. In mammalian cells there are three types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). Among them, macroautophagy, often simply referred as autophagy, is the best and most investigated process (Parzych & Klionsky, 2014) (Fig. 1). Macroautophagy occurs under normal growing condition and cellular differentiation during which one of its major roles is to maintain a proper cellular homeostasis. Moreover, it could be induced under stress condition such as starvation or energy deficit. The process of macroautophagy can be divided into 4 steps (Fig.1A): (1) induction, (2) formation of the autophagosome, (3) fusion with the lysosome or vacuole to form autophagolysosome, (4) breakdown of the encapsulated autophagic body and recycling (Wen & Klionsky, 2016). Initially, portions of cytosol containing dysfunctional organelles and/or unwanted materials are isolated by a phagophore which extends to both sides forming a double membrane vesicle named autophagosome. Subsequently, the autophagosome fuses with the lysosome to form the final autolysosome or autophagic vacuole which is responsible for degrading the cytosolic cargo. Finally, the degraded luminal content is returned to the cytosol for recycling via different metabolic reactions. Microautophagy (Fig. 1B) allows to eliminate small portions of cytoplasm that are directly enveloped by the lysosome. CMA (Fig. 1C) selectively degrades proteins containing the amino acid sequence KFERQ which are transported to the lysosome thanks to the combined actions of the LAMP2A and chaperone heat shock cognate 70 protein (HSC70) (Feng et al., 2014).
Figure 1.
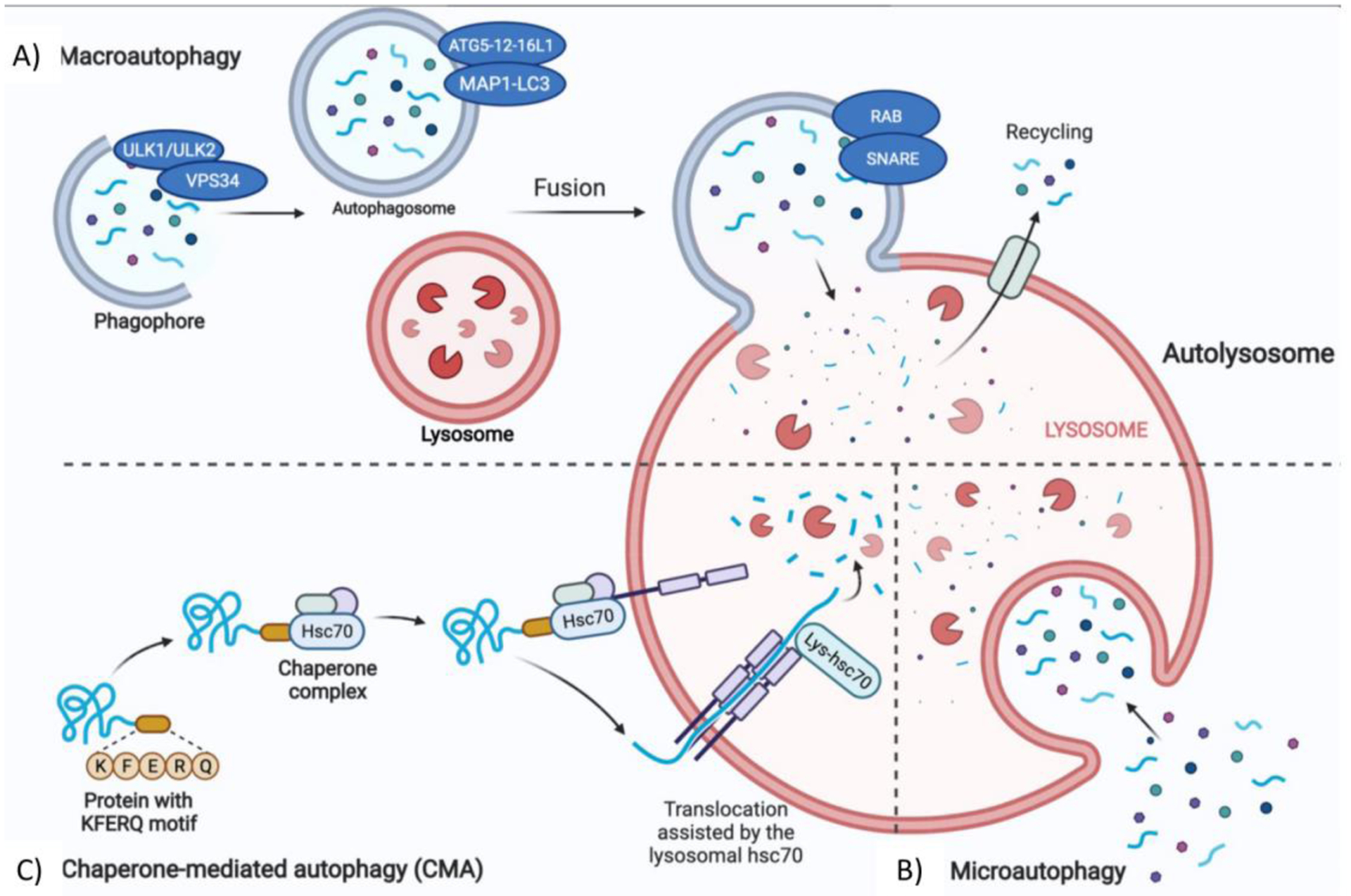
Cellular autophagic pathways. A) Macroautophagy, the process in which cytoplasmic portions such as proteins or organelles are included in a double membrane vesicle called phagophore to form the autophagosome. The external membrane of the autophagosome fuses with the lysosome to form the autolysosome where degradation of the cytoplasmic cargo occurs. B) Microautophagy, the process in which small portions of cytosol are swallowed directly by a lysosomal membrane invagination. C) Chaperone-mediated autophagy (CMA), is the process during which proteins containing a specific pentapeptide sequence (i.e., KFERQ motif) are recognized by cytosolic Hsc70 and cochaperones and subsequently translocated into the lysosomal lumen to be degraded. The figure was created with the help of Biorender.com.
3. Neuronal autophagy: physiological role ensuring brain health
Neurons together with glia cells are the building blocks of the CNS. Neurons constitute the functional unit of this system since they are specialized cellular structures capable of carrying out its main functions: receive, process, and transmit both excitatory and inhibitory signals (Harris & Attwel, 2012). These functions are supported by the fact that neurons are made up of highly polarized structures that include both dendrites and axons. Furthermore, neurons are among the few post-mitotic cells, which do not undergo cell division but are destined only to differentiation. For these reasons, maintaining the cellular morphology and normal physiological activities of individual neurons is a crucial task to ensure the correct functioning of the entire CNS activity. To do this, neurons need metabolically active biochemical structures that allow the recycling of damaged or aged organelles such as mitochondria, endoplasmic reticulum, and aggregated proteins to maintain cellular homeostasis and preserve their health and survival. The main mechanism of recycling of dysfunctional organelles and degradation of harmful substances is autophagy, in particular macroautophagy. Neuronal macroautophagy prevents cell toxicity by allowing the elimination of aggregated proteins that would otherwise accumulate since they cannot be disposed of due to the absence of mitosis (Kulkarni et al., 2018). Furthermore, it permits to remove extraneous bodies inside the cell, protecting for example against bacterial and viral infections. In addition to mediating these functions, autophagy in neurons also plays an important role in synaptic transmission. A study showed that reducing the autophagic process using ATG5 knockout (ATG5 KO) mice results in a selective accumulation of the tubular endoplasmic reticulum in the axons resulting in an elevation of intracellular calcium, increase in excitatory neurotransmission and an overall impairment of neuronal activity (Kuijpers et al., 2021).
The mechanism of neuronal macroautophagy occurs in several stages. Cytoplasmic cargo is initially sequestered by a phagosome which elongates to form the autophagosome. In this phase, the ATGs that initiate autophagosome formation are the unc-51 like autophagy activating kinase 1 and 2 (ULK1/ULK2) protein complex regulated by subsequent phosphorylation reactions (Fig. 2A). The activated ULK1 complex phosphorylates Beclin1 and Ambra1 proteins to form the active vacuolar protein sorting 34 (Vps34) complex (Sun 2016). Vps34 catalyzes the transformation of phosphatidylinositol into phosphatidylinositol 3-phosphate (PI3P), which recruits the specific autophagic proteins necessary for the formation of the phagophore. Another protein necessary for the formation of the autophagosome is the ATG9 protein, which is recycled in a ULK-dependent manner by the endosomes and Golgi apparatus (Stanley et al., 2014). In the next phase of phagophore elongation and formation of the mature phagolysosome, the proteins most involved are the proteins ATG12 and ATG5 which constitute an active complex ATG5-12-16L1 containing a binding site for MAP1-LC3 which when associated with phosphatidyl ethanolamine forms the lipidated LC3, called LC3II, and capable of crossing the phagolysosome membrane (Runwal et al., 2019). LC3II allows the autophagosome to bind autophagic substrates and / or proteins mediating the selectivity of the cargo (Galluzzi et al., 2017). LC3 is considered as the main marker of autophagosomes and has been used to study the movement of autophagosomes within neurons. Some studies showed that in these cells autophagic vesicles are formed in the axons and travel bidirectionally towards the cell body where the lysosomes are located. The correct movement of autophagosomes along the axon is of fundamental importance for the entire autophagy process and therefore for the maintenance of neuronal homeostasis. This transport is mediated by two microtubule motor proteins: the dynein which allows the autophagosome to move from the axon to the cellular soma and the kinesin which allows the same movement but in the opposite direction (Maday et al., 2012). Once the autophagosome proteins such as the ras-like small GTPase (RAB) and the soluble N-ethylmaleimide sensitive factor attachment protein receptor (SNARE) is synthesized, they initiate the process of fusion of the autophagosome with the lysosome to form the autolysosome. During this process V-ATPase vacuolar proton pump, present in the lysosomal membrane, acidifies the content of the autophagosome and activates the lysosomal hydrolases responsible for the degradation of the cargo (Bento et al., 2013). From the degradation products cells can derive energy and initiate new biosynthetic reactions. For these reasons, malfunction of lysosomal proteolysis due, for example, to mutations of genes encoding for proteins involved in autophagy can lead to the accumulation of protein aggregates, compromising neuronal health. As already mentioned, autophagy allows for the recycling of macromolecular constituents and generates energy in conditions of stress and above all permits the cell to increase its metabolic efficiency in conditions of reduced supply of nutrients. Under physiological conditions, the start of the macroautophagy process is controlled by a particular amino acid detection system. In fact, upstream of the autophagic process, one of the proteins with a central regulatory activity is the mammalian target of the rapamycin 1 complex (mTORC1) which can detect amino acids and intra-cellular nutrients levels. In neurons, active mTORC1 constitutes a powerful repressor of autophagy through a direct link with the ULK1 and ULK2 protein complexes. Especially, mTORC1 phosphorylates the two protein complexes responsible for the activation of the autophagic process by inactivating them, thus blocking the first phases of this process (Kim et al., 2011) (Shang et al., 2011). Under nutrient deficiency conditions ULK1 / ULK2 phosphorylation by mTOR is reduced and adenosine monophosphate (AMP) activated protein kinase (AMPK) phosphorylates, in a site different from that of mTORC1, ULK1/ULK2 protein complex which can then phosphorylate other ATG proteins and induce autophagy thereby providing nutrients. Differently, in the presence of high levels of nutrients mTORC1 is active and inhibits autophagy. A correct balance of the mTOR signaling in physiological conditions is important to ensure normal neuronal health (Dibble & Cantley, 2015) (Fig. 2A). In this regard, studies showed an alteration of the mTOR signaling in PD and AD where there is an mTOR over-activation which in turn reduces neuronal autophagy and promotes accumulation of α-syn, Aβ and tau, respectively (Gao et al., 2015) (Recasens et al., 2014) (Wong & Krainc, 2017) (Li et al., 2017) (Ghavami et al., 2014).
Figure 2.
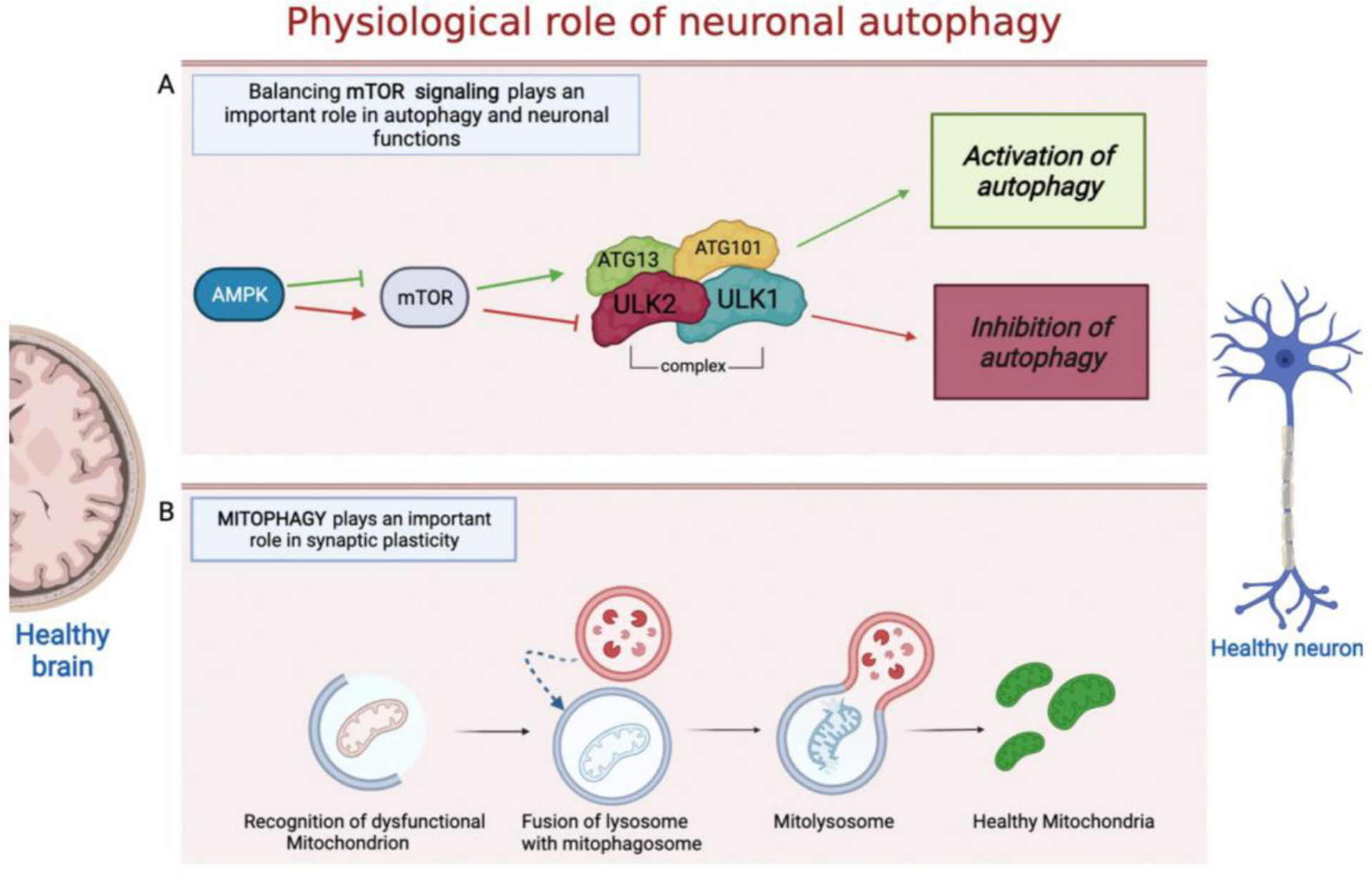
A simplified overview of mTOR signaling pathway and mitophagy. A) mTOR is one of the main regulators of autophagy by controlling the activity of the ULK1/ULK2 complex. Under various cellular stresses active AMPK inhibits the activity of mTORC. Inhibited mTOR promotes the activity of the ULK1/ULK2 complex and induces autophagy. If mTOR is activated, autophagy is inhibited due to inhibitory phosphorylation of ULK1 mediated by mTOR. B) A damaged mitochondrion is recognized by the autophagosome machinery and engulfed by the autophagosome, subsequently the autophagosome lysosome fusion allows the degradation of the mitochondrion and the recycling of healthy mitochondria. The figure was created with the help of Biorender.com.
A typical feature of neuronal cells is the fact that to produce energy they use exclusively oxidative metabolism, so a correct mitochondrial function in neurons is of fundamental importance to ensure their health. The recycling of mitochondria or the degradation of damaged mitochondria and the restoration of healthy ones is called mitophagy (Yoo & Jung, 2018). Unlike autophagy which degrades dysfunctional organelles in a non-selectively manner, mitophagy allows the selective degradation of damaged mitochondria (Fig. 2B) (Youle & Narendra, 2011). The process of mitophagy begins when the PTEN-induced protein kinase 1 (PINK1) present on the external mitochondrial membrane stabilizes itself because the depolarization of the mitochondrial membrane that occurs when the mitochondria are damaged. PINK1 phosphorylates mitofusin 2 (Mfn2) and ubiquitin which allow the recruitment of the Parkin protein. Parkin can in turn activate several ubiquitin binding proteins such as optineurin (OPTN) and sequestrosome-1 (SQSTM/p62), that initiate mitochondria to the pathway of mitophagy (Lazarou et al., 2015). Some in vivo studies have shown that neuronal mitophagy occurs predominantly in the cell body (Stavoe & Holzbaur, 2019). Together with macroautophagy, mitophagy plays a central role in ensuring the well-being and homeostasis of neuronal cells. This concept is very important considering that neurons are particularly vulnerable to damaged mitochondria which can act as source of ROS, resulting in chronic oxidative stress leading to cell death and neurodegeneration.
4. Neuronal autophagy dysfunction in Alzheimer’s and Parkinson’s diseases
Abnormal protein aggregation and deficits of the degradation processes of misfolded proteins are the most common pathological hallmarks of neurodegenerative diseases such as AD and PD. Once the autophagy pathway is compromised, the clearance of proteins such as α-syn (major component of LB), Aβ and tau (major components of amyloid plaques and neurofibrillary tangles) fails (Guo et al., 2018). The accumulation of α-syn in LB has been reported in the dopaminergic neurons of PD patients as well in MPTP-treated animals and neuronal cells (Zhang et al, 2017). Neuronal cells infected with lentivirus for α-syn accumulated autophagic vesicles positive for lysosomal/autophagy markers such as cathepsin D and LC3 and had an increase in Beclin1 expression suggesting that the accumulation of α-syn alters the autophagic degradation pathway in these cells. Under the same experimental condition, the use of rapamycin, an inhibitor of mTOR, was beneficial via the activation of autophagy (Spencer et al., 2009). The vicious cycle between α-syn accumulation, autophagy-lysosome dysfunctions and mitophagy engulfment can be initiated by mutations of several genes ranging from highly rare familial mutations (A30P, A53T, PINK1/Parkin, and LRRK2) to other genetic risk genes (cathepsin B and D) (CTSD and CTSB) considered relevant to PD pathophysiology (Bellomo et al., 2020) (Fig. 3A). Thus, α-syn mutations A30P and A53T accelerate the formation of protofibrils, which accumulate in the lysosome membranes leading to lysosomal dysfunctions and failure of autophagy pathway. Moreover, α-syn overexpression in neuroblastoma cells SHSY5Y results in changes of Ca2+ signaling, decrease of LAMP1 protein expression, alkalinization of lysosomal pH and increase LC3 positive vesicles compared to WT cells (Nascimento et al., 2020). On the other hand, leucine-rich-repeat kinase 2 (LRRK2) mutation at G20195 site, disrupts autophagic vesicles (AV) transport in cortical neurons of G20195 knock in mouse, indicating defective trafficking of neuronal autophagosomes and loss of AV motility (Boecker et al., 2021). Two common gene mutations associated with early onset PD are linked to PINK1 (PTEN-induced kinase 1) and Parkin (encoded by PARK2) genes. PINK1 mutations have been linked to defects in mitochondrial turnover (via mitophagy) and immune cells infiltration in the brain leading to dopaminergic neurons degeneration in PD (Matheoud et al., 2019). Moreover, loss of PINK1 besides inducing accumulation of dysfunctional mitochondria is associated with mitochondrial Ca2+ imbalance. Parkin mutations by causing a deficit in its recruitment to depolarized mitochondria ultimately impair mitophagy as well (Zeng et al., 2018) (Fig. 3A).
Figure 3.
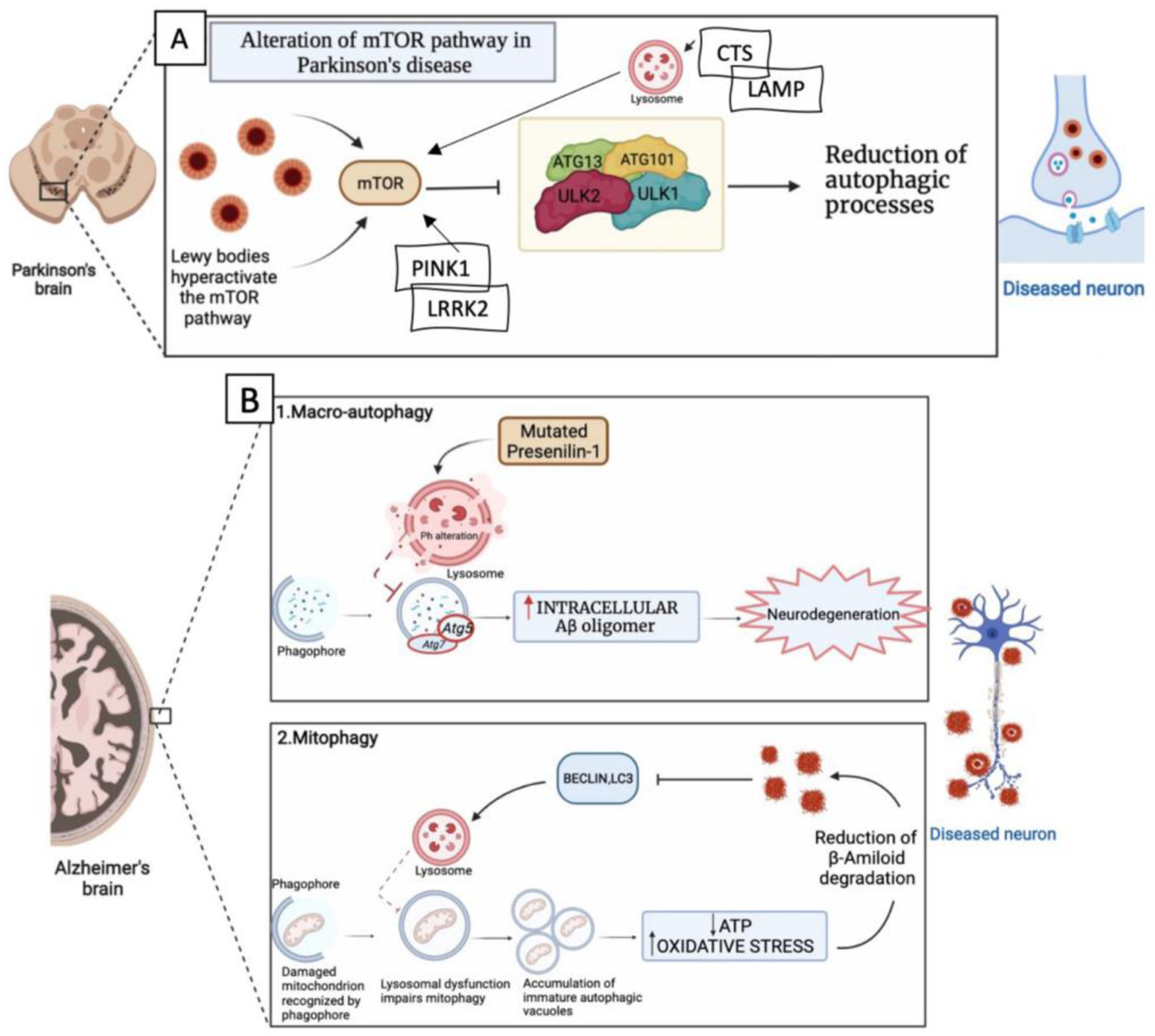
A) In PD, Lewy bodies increase the levels of mTOR which inhibits ULK1 / ULK2 activation with consequent reduction of the autophagic process and subsequent accumulation of protein aggregates that increase neurodegeneration. B) AD and autophagy. B1) In AD defects in the macro-autophagy pathway due to genetic mutations, environmental factors or aging can contribute to the accumulation of abnormal protein aggregates. For example, mutations in presenilin-1 alter the pH of the lysosome, thereby decreasing the activity of autophagy that can increase accumulation of Aβ peptides and neurodegeneration. B2) In AD there is a defect in lysosomal activity which causes a reduction in the mitophagy process with consequent accumulation of autophagic vacuoles. Autophagic vacuoles accumulations increase oxidative stress, while the reduction of healthy mitochondria causes a reduction of ATP. These events lead to a reduction in the degradation of Aβ peptides whose accumulation blocks the proteins activating mitophagy thus establishing a vicious circle that further facilitate neurodegeneration. The figure was created with the help of Biorender.com.
Dysfunction of neuronal mitophagy, autophagy and lysosome-endosomal system degradation pathways are reported in AD individuals, induced pluripotent stem cells (iPSC)-derived neurons, and AD transgenic (Tg) mice and implicated in AD pathophysiology since all of them have been associated with defective APP/Aβ clearance and tau turnover (Bera et al., 2020) (Fang et al., 2019) (Fig. 3B1 and B2). Regarding the neuronal endosome system, it has been reported that mutations of the sorting receptor-encoding gene SORL1, which encodes a regulator of protein trafficking between the trans-Golgi network and endosomes and highly expressed in the brain, significantly increase the risk for late-onset of AD. Moreover, complete loss of SORL1 results in neuronal endosome enlargement, increase of Aβ production secondary to a significant increase in the ratio of extracellular soluble APPβ (sAPPβ) to sAPPα, suggesting a shift toward β-secretase-mediated Aβ peptide production and upregulation of autophagosome LC3II, which prevents autophagosome formation (Hung et al., 2021). The autophagy factor Beclin, mainly involved in the initiation phase of autophagy, is also reduced in AD individuals, and its induction promotes Aβ plaque reduction (Pickford et al., 2008).
Recent evidence suggests that Aβ oligomers formed during the early stages of AD pathogenesis by creating an unfavorable environment both intra- and extracellular are responsible for the development of dystrophic neurites (DNs). Once formed, DNs by surrounding Aβ plaques can contribute to altering neuronal processes and exacerbating AD progression (Sharoar et al., 2021). Interestingly, ATG9A, which is an early autophagy protein involved in initiating the formation of pre-autophagosomes, was found accumulated in DNs in proximity to Aβ plaques at their earliest stages of development and also bound their core (Sharoar et al., 2019).
Recently, intracellular protein sorting and trafficking via the endosomal system, which includes early endosomes, recycling endosomes and late endosomes, have been shown to play a major role also in the regulation of autophagy and other degradation pathways in neuronal cells. The main regulator of this system is the retromer complex, a highly conserved multimeric structure composed by a cargo recognition core, which includes the VPS35, VPS29, VPS26 protein trimer, and a dimer consisting of different sorting nexin proteins (Tu et al., 2021). Interestingly, deficits or mutation in one or more components of the retromer complex system have been associated with the onset of AD and PD via the progressive accumulation of pathological protein aggregates (Vagnozzi et al., 2019). Studies in vitro have established a biological link between neuronal VPS35 deficiency and or dysfunction with an impaired activation of autophagy-lysosome and proteasome pathways. Thus, downregulation of VPS35 in neuronal cells carrying the human Swedish mutant of the amyloid precursor protein resulted in a significant alteration of autophagy flux and intracellular accumulation of acidic and ubiquitinated aggregates (a. Filippone et al., 2021). Moreover, dysregulation of the retromer complex system in brain human endothelial cells by impairing autophagy-lysosome degradative pathways resulted in an accumulation of pathologic tau protein (b. Filippone et al., 2021).
5. Dysfunctional neuronal autophagy in Spinal Cord Injury and Traumatic Brain Injury
Tissue damage resulting from traumatic injuries to the CNS is characterized by complex biochemical changes and progressive neurodegeneration. There are different cellular processes that can be initiated as result of a neurotrauma including apoptosis, necrosis and autophagy. Among them, recently an increasing amount of work has been performed on the role that altered neuronal autophagy responses play after this type of injury, with particular attention on how it may contribute to the secondary pathological events following both spinal cord injury (SCI) and traumatic brain injury (TBI) (Liu et al., 2018).
According to the National Spinal Cord Injury Statistical Center [NSCISC, 2018], in the United States around 200,000 people are living with severe motor-sensory deficits secondary to SCI and that may lead to death (Ackery et al., 2004). Initially, SCI consists of the primary injury which occurs immediately after the mechanical trauma and tissue compression which lead to disruption of axons and blood vessels. The second phase is characterized by activation of the inflammatory cascade, microglial activation, vascular changes, ROS formation, apoptosis, and autophagy (Filippone A et al., 2020). So, targeting one of these processes especially the autophagy pathway could be a valid strategy for reducing the secondary phase-related damage typically observed after SCI and promoting clinical recovery (Abbaszadeh et al., 2020). A key set of ATG genes and transcript proteins are influenced after SCI, including ATG5, ATG7 and LC3 which during trauma localize in the damaged axons (called “retraction bulbs”) of the perilesional area. There, levels of the autophagosome marker LC3, which regulates autophagosome development, increase as early as 30 minutes after SCI and remain at a high level up to 6 weeks (Ribas et al., 2015). This change suggests an increase of autophagic flux, a rate of autophagosome degradative activity, which persists chronically following SCI. Alterations in the autophagy flux can also potentiate and induce organelles stress since neurons form ATG5 −/+ mice have an increase in autophagosome proteins such as Beclin, ATG5, lysosome markers LAMP2 and cathepsin-D (CTS-D) (Zhou et al., 2020).
Several complex pathological and biochemical events during SCI are influenced by the autophagic process, which is considered as a cytoprotective one following the trauma almost equally to apoptosis. Interestingly, apoptosis can be influenced by autophagy and vice versa in the contribution to improving functional outcome after SCI, during which there is an upregulation of essential ubiquitin-like conjugation systems (ATG5 - ATG12 - ATG8) that remove damaged organelles and abnormally aggregated proteins (Casili et al., 2020).
Since it has been reported that the efficiency and activity of autophagy players including mTOR, and Beclin changes when SCI occurs, it could be important to consider them as potential targets for modulating autophagy activation (Fig. 4). Some of the neuronal autophagy players are influenced by zinc homeostasis that possesses neuroprotective effects through autophagy stimulation by activating the mTOR signaling pathway after SCI (Lin et al., 2020). Moreover, mTOR inhibition result in functional improvement and structural recovery in acute SCI via the modulation of immune response and release of proinflammatory cytokines including IL-6, IL-1β, and IL-2 and tumor necrosis factor-α (TNF-α) (Vargova et al., 2021) (Fig. 4).
Figure 4.
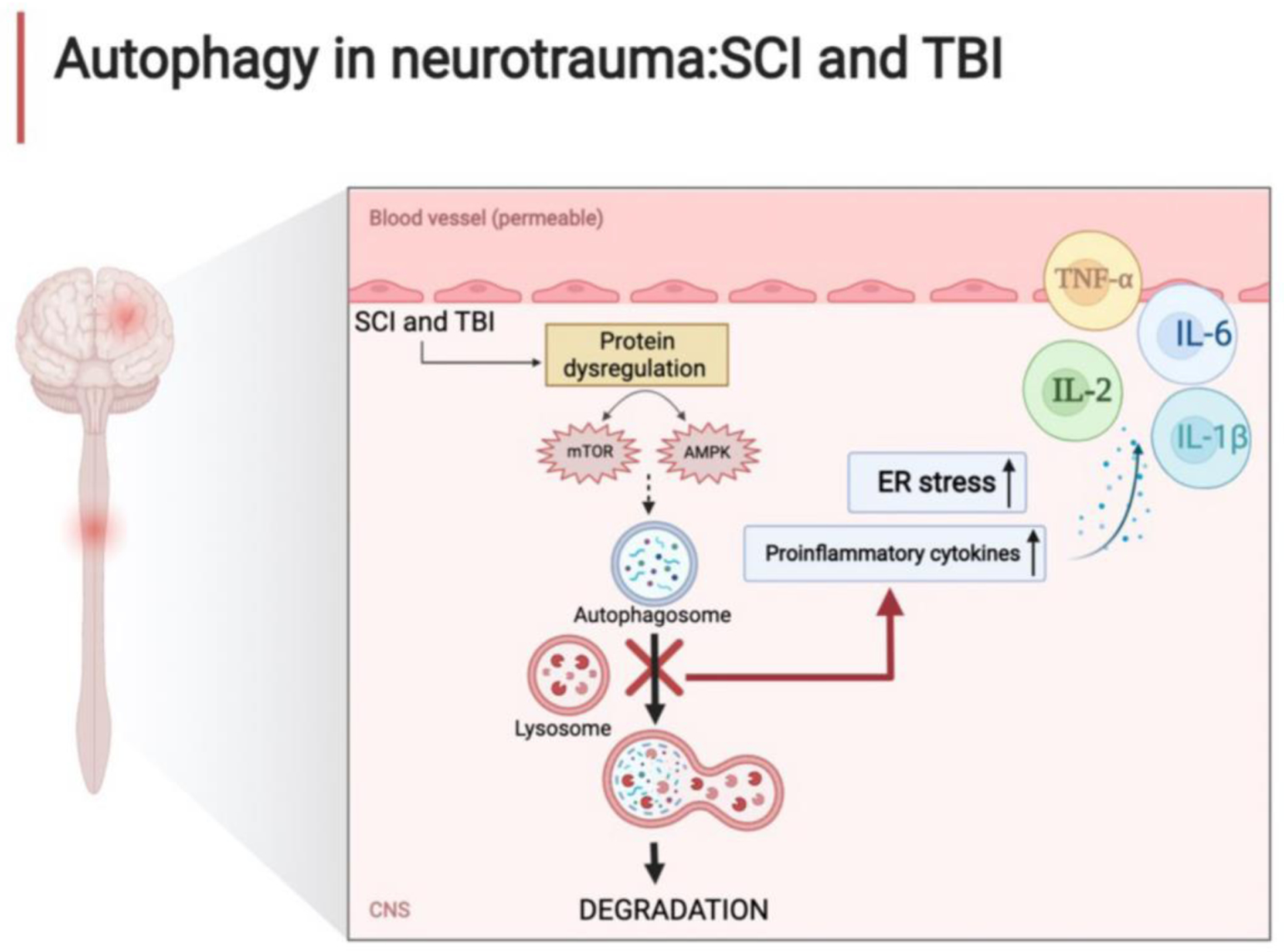
Autophagy in SCI and TBI. In neurotrauma as in SCI and TBI, physical trauma causes an alteration of the cellular microenvironment and modifies the homeostasis of autophagic flux with dysregulation of the mTOR signal and AMPK activity. These events increase ER stress and proinflammatory cytokine release leading to necrosis. The figure was created with the help of Biorender.com.
According to the Centers for Disease Control and Prevention, TBI is caused by a bump, blow, or jolt to the head, or a penetrating head injury that alters the normal function of the brain (Centers for Disease Control and Prevention, U.S. Department of Health and Human Services, 2014). Based on the data in the literature male cases greatly outnumbered female cases of total TBI with more than 40% of patients with moderate to severe injuries having long-term disabilities (TBI United States National database, 2017). TBI triggers a complex sequence of events in which damaged neuronal cells rapidly release cytokines and chemokines, these, in turn, recruit immune/inflammatory cells which can assist in limiting the spread of injury and removing cellular debris, or, by contrast, at times also trigger degeneration, cell death and permanent neurological deficits (Alam et al., 2020). New research has shown that after TBI, autophagy organelles including autophagosomes and lysosomes markedly spread in the neurons surrounding damaged areas (Zhang et al., 2014). Dysregulated post-TBI neuronal autophagy activation is also characterized by a decrease in SQSTM-1/p62 and increase in the LC3II/LC3I ratio and associated with an attenuation of IL-13 and changes in the JAK-1-STAT-1 signaling pathway suggesting a link between autophagy and cytokines release (Gao et al., 2020).
In general, activation of autophagy after TBI is considered a double-edged sword via the involvement of the immune system and inflammation. In fact, the ability to maintain appropriate levels of neuronal autophagy can promote the removal of abnormal proteins or damaged organelles, but at the same time the failure to do so results in abnormal neuronal autophagy which will then favor tissue damage.
6. Autophagy: pharmacological targets
As discussed above, the term neuronal autophagy is used to describe a series of catabolic events from the degradation of organelles to the recycling of cytoplasmatic material or pathogens, and it includes three major subtypes, macroautophagy, CMA and microautophagy. Currently more than 30 autophagy-related proteins can potentially be considered as therapeutic targets for modulating this pathway with the goal of restoring it to a physiological level to favor homeostasis and neuronal health with the goal of preventing or halting neurodegeneration, a process shared by AD and PD and CNS traumas. Targeting one (or more) of these players by using molecules that possess abilities to correct altered neuronal autophagy at different steps is an attractive and interesting approach that is becoming more and more the focus of recent pre-clinical research.
As mentioned above, the neuronal autophagy machinery is mainly coordinated at the signaling level by mTOR, a dimeric complex formed by two subunits known as TORC 1 and TORC2 that in turn regulate autophagy flux and drive autophagy initiation through various related kinases and complexes. Among them, the phosphatidylinositide 3-kinase (PI3K) complex consisting of Beclin1, vacuolar proteins sorting 15–34 (VPS15, VPS34) and autophagy-related protein 14L (ATG14L), necessary to the growing of phagophore, ATG8/LC3 family conjugation systems, the endoplasmic reticulum and lysosomes are all involved in neuronal autophagy-mediated degradation (S.N. Suresh 2020). Modulation of autophagy at the mTOR signaling level is possible using mTOR-dependent modulator autophagy enhancer-67 (AUTEN-67), a small molecule identified as a potent candidate possessing antiaging and neuroprotective effects, by significantly increasing autophagic flux in neurons and protecting them from undergoing AD stress-induced cell death (Papp et al., 2016).
A different approach for mTOR modulation could be also via the endogenous inhibitor of cysteine protease activity known as Cystatin C (CysC), ubiquitously expressed by mammalian tissue, and implicated in the response of the CNS to neurodegeneration and oxidative stress (Nagai et al., 2008). CysC exerts its inhibitory effects on several cathepsins (Cat) including CatB, CatC, and CatD that can influence cell proliferation and autophagy. Interestingly, CysC possesses a high affinity for CatB which is fundamental to the regulation of lysosomal proteolysis and activity involved in the main neuronal autophagy pathway (Bonam et al., 2019). Given that the CysC and CatB levels are increased in AD, and that CysC - CatB axis influences lysosomes, CysC can be considered as pharmacological target by enhancing neuronal autophagy pathway activation and inhibiting accumulation of Aβ aggregates that characterized AD pathophysiology (Mathews et al., 2016; Perlenfein et al., 2017).
Tightly linked to autophagy is the lysosome degradative activity which involves the participation of hydrolytic enzymes that within the lumen disrupt various substrates including protein aggregates as Aβ and tau in AD. Lysosome pH gradient is mainly sustained by the vacuolar multi-subunits enzyme (H+) ATPase (or V-ATPase), which pumps protons into the lysosomal lumen by consuming ATP (Song et al., 2020). (H+) ATPase consists of two parts, V0 which induces protons translocation, and V1 that catalyzes ATP hydrolysis. Thus, since ATPase H+ transporting V1 subunit A (ATP6V1A) drives (H+) ATPase to specific compartments (Jansen et al., 2008), it could be pharmacologically targeted to improve neuronal health and reduce the neurodegenerative phenotypes induced by ATP6V1A deficit, which has been reported in AD (Wang et al., 2021). However, ATP6V1A is also involved in the phagosome formation pathway, a degradation process for the lysosome activated after it receives and digests macromolecules via endocytosis, phagocytosis, and autophagy (Saffi et al., 2019). Therefore, the enhancement of ATP6V1A expression could be neuroprotective in AD by targeting this mechanism which is relevant to the disease pathogenesis (Zhou et al., 2021).
Targeting mTOR with rapamycin and trehalose has also neuroprotective effects in PD via the suppression of altered autophagy induction. Thus, blockage of dysregulated autophagy activation in the striatum and substantia nigra, as indicated by LC3II expression, in a mouse model of neurotoxin MPTP-induced PD-like syndrome results in tyrosine hydroxylase restoration (Pupyshev et al., 2019).
Interestingly, inhibition of macroautophagy flux and accumulation of α-syn have been associated with a decrease in glucocerebrosidase (GCase) levels in several PD-relevant brain areas including substantia nigra, putamen, and amygdala (Li et al., 2019). GCase is a lysosome enzyme present in the endoplasmic reticulum, trafficks to the Golgi apparatus, and then is delivered through vesicular fusion to the lysosomal lumen where cleaves glucosylceramide and glucosylsphingomyelin. Recently, mutant GCase (GBA1) has been found to inhibit CMA when at the lysosomal surface leading to an accumulation of substrates including α-syn (Kuo 2022). Based on this knowledge, research effort is being made towards the development of therapies aimed at restoring or increasing brain GCase levels via gene therapy replacement, substrate reduction, small molecule chaperones and small molecules modulators of GCase activity (Chen et al., 2020).
Studies have demonstrated a link between redox changes and autophagy including oxidative stress, formation of oxidation products with subsequent lipidation of important autophagy molecule and substrates such as AGT proteins (Qiu-Qi et al., 2018). To this end, genetic ablation of 12–15 Lipoxygenase, a major source of oxidized lipids, results in enhanced autophagy by increasing the LC3 protein levels in the brain of mice (Jang et al., 2014). In vitro pharmacological inhibition of this enzyme also resulted in increased autophagy and protection against the accumulation of unwanted materials (Jang et al., 2014). We have reported that a transgenic mouse model of AD (3xTg AD mice) chronically receiving a selective pharmacologic inhibitor of 12–15 Lipoxygenase activation had an improvement of the AD-like phenotype (i.e., Aβ, tau tangle and memory deficits) which was secondary to an activation of autophagy as shown by the significant elevation of ATG5–12 and LC3B2/1 ratio (Di Meco et al., 2017). These data would support that targeting this oxidizing enzyme could also be a therapeutic approach against AD.
Consistent epidemiological and clinical data in the literature have shown the beneficial effects of extra virgin olive oil (EVOO) on brain health, cognition, and dementia (Lauretti et al, Chapter, pp.415–422). Additionally, several preclinical studies using rodent models of AD have confirmed these findings showing that indeed chronic administration of EVOO results in a positive effect on memory deficits and synaptic function, a significant reduction of neuroinflammation, and a decrease in both Aβ and tau neuropathology (Qosa et al., 2015; Farr et al., 2012). Interestingly from a mechanistic point of view it was demonstrated that these beneficial effects of EVOO were mediated by a restoration of altered neuronal autophagy (Lauretti et al., 2017).
Although the exact role of modulating autophagy in TBI is not yet completely elucidated, the use of selective inhibitors that target VPS34 (also known as PIK3C3, which stands for PI3K class III) which is involved in autophagy initiation complex, might provide some evidence on autophagy machinery regulation and vesical trafficking. Recently, a new molecule called SAR405, an inhibitor low-molecular-mass that specifically target VPS34 by influencing various PI3 kinases, has been characterized (Ronan et al., 2014). Thus, administration of this compound reduced brain edema and the associated TBI-induced neuronal deficits by decreasing LC3II/LC3I ratio and increasing the SQSTM1 levels (Quan et al., 2021). Another study showed that treatment with valproic acid, a stabilizer for mood disorders, improved animal behavior deficits, reduced brain cell apoptosis and microglial activation after TBI by enhancing autophagic flux (Zheng et al., 2019).
SCI is also characterized by changes in autophagy that can reduce cell apoptosis and at the same time jeopardize neural functional recovery after trauma (James et al., 2018). Previous studies reported that dysregulation of Zinc, a structural element and cofactor for different enzymes, is involved in CNS disorders because of its role in neurotransmission (Mlyniec et al., 2015) and the ability to inhibit microglial activation after lipopolysaccharide-induced inflammation (Hongxia et al., 2019). Recently, a paper proposed that this element could promote autophagy initiation in mice after SCI through the AMPK/mTOR signaling pathway by influencing Beclin, LC3B expression levels (Lin et al., 2020). Other researchers have compared two mTOR inhibitors: rapamycin (RAPA) an inhibitor of mTORC1, and pp242, a second-generation inhibitor, that targets both mTORC1 and mTORC2. The study conducted on rats subjected to SCI demonstrates that pp242 is less effective than RAPA treatment after SCI by showing that selective inhibition of mTORC1 with RAPA provides a better recovery for the altered autophagy processes and reduction of inflammation (Vargova et al., 2021).
Another study showed that an additional potential drug for SCI could be the transcription factor E3 (TFE3), which belongs to the microphthalmic family of transcription factors bHLH-LZ (MiT / TFE). TFE3 improves autophagic flow by promoting autophagosome-lysosome activity, reduces intracellular ROS formation, and decreases ER stress-induced neuronal apoptosis and ultimately ameliorates functional recovery after SCI (Stavoe et al., 2020). Another group of researchers investigated how resveratrol upregulation of the Sirtuin 1(SIRT1)/Adenosine 5′ monophosphate-activated protein kinase (AMPK) signal could restore altered neuronal autophagy after SCI. Resveratrol is a natural polyphenolic compound that exhibits several beneficial health properties including antioxidant, anti-inflammatory and anticancer effects. Intraperitoneal administration of resveratrol in rats undergoing SCI significantly increased the expression of SIRT1, p-AMPK, Beclin-1 and LC3-B thereby improving neuronal autophagy and inhibiting expression of apoptosis proteins. Taken together these effects further demonstrate that restoration of altered neuronal autophagy after SCI reduces tissue injury and significantly promote the recovery of motor function (Zhao et al., 2017).
In search for additional compounds targeting autophagy, a recent approach has been to screen drugs already approved to treat other human diseases for their potential pro-autophagy action with the goal of repurposing them for neurodegenerative diseases such as AD and PD. Besides rapamycin and trehalose, which we have discussed earlier, other compounds with different known activities have been identified (Sarkar 2008). Among them, ATP analogs such as dactolisib, phosphoinositide cycle inhibitors such as valproic acid and carbamazepine, AMPK activators such as metformin, and calcium antagonists such as verapamil, have all been shown to have neuroprotective action via a pro-autophagic mechanism and for this reason they are currently considered as potential ideal candidates to be repositioned against dysregulated proteostasis (Zhang et al 2007; Thellung 2019).
7. Conclusions and future perspectives
Today there is a consensus on the fact that altered neuronal autophagy not only is a constant feature of AD, PD and CNS traumas, but it could also play a functional role in the onset and progression of their pathogenesis. In light of this evidence, the idea that targeting dysregulated neuronal autophagy with the goal of restoring its proper function would be beneficial for neuronal health and homeostasis could be envisioned as a novel and viable therapeutic approach against AD, PD and CNS traumas. However, despite the impressive recent progress in this field which has significantly widened our understanding of the complex molecular and cellular mechanisms regulating neuronal autophagy through different signaling pathways in health and pathology, there is much work to be done. In particular, we believe that future areas of emphasis should be considered the pharmacology of neuronal autophagy, which can be achieved by focusing on drug discovery and development with the goal of identifying novel and promising therapeutic tools useful in preclinical models and ultimately in clinical settings.
In fact, although promising and exciting data are available in the literature, overall there is a general deficit in translating the few pre-clinical studies with small molecules targeting altered neuronal autophagy into clinically relevant conditions aiming at stopping or slowing the progress of AD and PD, or preventing the primary and secondary effects of SCI and TBI. To this end, the study of old and newly identified pharmacological targets, the elucidation of the molecular mechanisms through which neuronal autophagy maintains cellular homeostasis and proteostasis, and the precise role whereby altered autophagy influence these neurological diseases are some of the future strategies that need to be implemented in this area with tremendous implication for human health.
Acknowledgements
Domenico Praticò is the Scott Richards North Star Charitable Foundation chair for Alzheimer’s research. Some of the discussed work performed in the author’s lab was in part supported by grants from the National Institute of Health (AG055707, and AG056689).
Abbreviations:
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- Aβ
amyloid-beta
- ROS
reactive oxygen species
- α-syn
α-synuclein
- LB
Lewy body
- MAP-LC3 or LC3
microtubule-associated protein 1A/1B-light chain 3
- ATGs
autophagy-related proteins
- SQSTM-1/p62
sequestrome-1
- LAMP-2
lysosome-associated protein 2
- CNS
Central Nervous System
- IFNs
interferons
- ILs
interleukins
- SCI
spinal cord injury
- TBI
traumatic brain injury
- CMA
chaperone-mediated autophagy
- HSC70
chaperone heat shock cognate 70
- ULK1/ULK2
unc-51 like autophagy activating kinase 1 and 2
- VPS
vacuolar protein sorting
- PI3P
phosphatidylinositol 3-phosphate
- PI3K
phosphatidylinositol 3- kinase
- RAB
ras-like small GTPase
- SNARE
soluble N-ethylmaleimide sensitive factor attachment protein receptor
- mTORC1
mammalian target of the rapamycin 1 complex
- AMPK
AMP activated protein kinase
- PINK1
PTEN-induced protein kinase 1
- Mfn2
mitofusin 2
- LRRK2
leucine-rich-repeat kinase 2
- OPTN
optineurin
- CTSD
cathepsin-D
- CTSB
cathepsin-B
- AV
autophagic vesicles
- TNF-α
tumor necrosis factor-α
- AUTEN-67
mTOR-dependent modulator autophagy enhancer-67
- CysC
Cystatin C
- EVOO
extra virgin olive oil
- RAPA
rapamycin
- SIRT1
Sirtuin 1
- iPSC
induced pluripotent stem cells
- SORL1
sortlin-related receptor 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing interest
All the authors have no competing interest to declare.
References
- Abbaszadeh F, Fakhri S, & Khan H Targeting apoptosis and autophagy following spinal cord injury: Therapeutic approaches to polyphenols and candidate phytochemicals. Pharmacological Research, (2020) 105069. [DOI] [PubMed] [Google Scholar]
- Ackery A; Tator C; Krassioukov A A global perspective on spinal cord injury epidemiology. J. Neurotrauma (2004), 21, 1355–1370. [DOI] [PubMed] [Google Scholar]
- Alam A, Thelin EP, Tajsic T et al. Cellular infiltration in traumatic brain injury. J Neuroinflammation (2020) 17, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauquis J, Pavia P, Pomilio C, Vinuesa A, Podlutskaya N, Galvan V, Saravia F. Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp Neurol (2013) 239:28–37. [DOI] [PubMed] [Google Scholar]
- Bellomo G, Paciotti S, Gatticchi L, and Parnetti L. The Vicious Cycle Between α-Synuclein Aggregation and Autophagic-Lysosomal Dysfunction. Movement Disorders, 1, 2020. Vol. 35. [DOI] [PubMed] [Google Scholar]
- Bento CF; Puri C; Moreau K; Rubinsztein DC The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci (2013), 126, 1059–1069. [DOI] [PubMed] [Google Scholar]
- Bera S, Camblor-Perujo S, Calleja Barca E, Negrete-Hurtado A, Racho J, De Bruyckere E AP-2 reduces amyloidogenesis by promoting BACE1 trafficking and degradation in neurons. EMBO Rep. (2020) 21:e47954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boecker C, Goldsmith Juliet, Dou Dan, Cajka Gregory G., Holzbaur Erika L.F.. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Current Biology. 2021. :2140–2154.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonam SR, Wang F & Muller S Lysosomes as a therapeutic target. Nat Rev Drug Discov 18, (2019) p923–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caglayan S, Takagi-Niidome S, Liao F, Carlo A-S, Schmidt V, Burgert T, Kitago Y, Füchtbauer E-M, Füchtbauer A, Holtzman DM. Lysosomal sorting of amyloid-β by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Sci. Transl. Med, 6 (2014), p. 223ra20. [DOI] [PubMed] [Google Scholar]
- Casili G, Campolo M, Lanza M et al. Role of ABT888, a Novel Poly(ADP-Ribose) Polymerase (PARP) Inhibitor in Countering Autophagy and Apoptotic Processes Associated to Spinal Cord Injury. Mol Neurobiol 57, (2020) p4394–4407. [DOI] [PubMed] [Google Scholar]
- Chen X, Li Y, Wang C et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol Neurodegeneration (2020). p15, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Yu, Sam Richard, Sharma Pankaj, Chen Lu, Do Jenny & Sidransky Ellen. Glucocerebrosidase as a therapeutic target for Parkinson’s disease, Expert Opinion on Therapeutic Targets (2020), 24:4, 287–294, DOI: 10.1080/14728222.2020.1733970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meco A, Li JG, Blass BE, Abou-Gharbia M, Lauretti E, Pratico D. 12/15-Lipoxygenase inhibition reverses cognitive impairment, brain amyloidosis, and tau pathology by stimulating autophagy in aged triple transgenic mice. Biol Psychiatry. (2017);81:92–100. [DOI] [PubMed] [Google Scholar]
- Djajadikerta A, Keshri S, Pavel M, Prestil R, Ryan L, Rubinsztein DC. Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases. J Mol Biol. 2020. Apr 3;432(8):2799–2821. [DOI] [PubMed] [Google Scholar]
- Dibble CC & Cantley LC Regulation of mTORC1 by PI3K signaling. Trends Cell Biol (2015), 25, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci (2019). p22 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr SA, Price O, Dominguez LJ, et al. Extra virgin olive oil improves learning and memory in SAMP8 mice. J. Alz. Dis (2012); p28:81–92 [DOI] [PubMed] [Google Scholar]
- Feng Y; He D; Yao Z; Klionsky DJ The machinery of macroautophagy. Cell Res (2014), 24, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippone A, Lanza M, Campolo M, Casili G, Paterniti I, Cuzzocrea S, Esposito E. Protective effect of sodium propionate in Aβ1–42 -induced neurotoxicity and spinal cord trauma. Neuropharmacology. (2020) Apr;166:107977. [DOI] [PubMed] [Google Scholar]
- a. Filippone A, Li JG, Praticò D. VPS35 Downregulation Alters Degradation Pathways in Neuronal Cells. J Alzheimers Dis. 84(3) (2021) p1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- b. Filippone A, Smith T, Pratico D. Dysregulation of the Retromer Complex in Brain Endothelial Cells Results in Accumulation of Phosphorylated Tau. J Inflamm Res. (2021) 29;14: 7455–7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L; Baehrecke EH; Ballabio A; Boya P; Bravo-San Pedro JM; Cecconi F; Choi AM; Chu CT; Codogno P; Colombo MI; Molecular definitions of autophagy and related processes. EMBO J 2017, 36, p 1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Li J, Wu L, et al. Tetrahydrocurcumin provides neuroprotection in rats after traumatic brain injury: autophagy and the PI3K/AKT pathways as a potential mechanism. J Surg Res (2016); 206(1): p67–76. [DOI] [PubMed] [Google Scholar]
- Gao C, Yan Y, Chen Guang, Wang T, Luo C, Zhang M, Chen X, and Tao L. Autophagy Activation Represses Pyroptosis through the IL-13 and JAK1/STAT1 Pathways in a Mouse Model of Moderate Traumatic Brain Injury. ACS Chem. Neurosci (2020), 11, 24, 4231–4239. [DOI] [PubMed] [Google Scholar]
- Gao S; Duan C; Gao G; Wang X; Yang H Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling. Int J Biochem Cell Biol 2015, 64, p25–33. [DOI] [PubMed] [Google Scholar]
- Geng P, Zhang J, Dai W, et al. Autophagic degradation deficit involved in sevoflurane-induced amyloid pathology and spatial learning impairment in APP/PS1 transgenic mice. Front Cell Neurosci. 2018, 12:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, Łos MJ. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014. Jan; 112:24–49. [DOI] [PubMed] [Google Scholar]
- Guo F, Liu X, Cai H, Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. (2018); 28(1):3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock EJ, Ang Jordan, Papachristodoulou Antonis, Stan Guy-Bart. The Interplay between Feedback and Buffering in Cellular Homeostasis. Cell Systems, 2017, 5(5):498–508.e23. [DOI] [PubMed] [Google Scholar]
- Harris JJ; Attwell D The energetics of CNS white matter. J Neurosci 2012, 32, 356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongxia L, Tang Yuxiao, Shen Zhilei, Sun Yan, Qu Yicui, Shen Jiamin, Xu Xin, Yang Jianxin, Mo Fengfeng, Shen Hui. Zinc inhibited LPS-induced inflammatory responses by upregulating A20 expression in microglia BV2 cells. Journal of Affective Disorders. 249:136–142. [DOI] [PubMed] [Google Scholar]
- Hung C, Tuck Eleanor, Stubbs Victoria, van der Lee Sven J., Aalfs Cora, van Spaendonk Resie, Scheltens Philip, Hardy John, Holstege Henne, Livesey Frederick J.. SORL1 deficiency in human excitatory neurons causes APP-dependent defects in the endolysosome-autophagy network. Cell Reports. 2021. 35(11):109259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang I, Park S, Cho JW, et al. Genetic ablation and short-duration inhibition of lipoxygenase results in increased autophagy. Exp. Cell Res (2014);321;276–287. [DOI] [PubMed] [Google Scholar]
- Jansen EJ, Scheenen WJ, Hafmans TG, Martens GJ. Accessory subunit Ac45 controls the V-ATPase in the regulated secretory pathway. Biochim Biophys Acta. Dec;1783(12):2301–10. doi: 10.1016/j.bbamcr.2008.06.020. [DOI] [PubMed] [Google Scholar]
- James ND, McMahon SB, Field-Fote EC, Bradbury EJ. Neuromodulation in the restoration of function after spinal cord injury. Lancet Neurol, 17 (2018), pp. 905–917. [DOI] [PubMed] [Google Scholar]
- Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry, 77 (2015), pp. 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J; Kundu M; Viollet B; Guan KL AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011), 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A; Chen J; Maday S Neuronal autophagy and intercellular regulation of homeostasis in the brain. Curr Opin Neurobiol (2018), 51, p29–36. [DOI] [PubMed] [Google Scholar]
- Kuo SH, Tasset I, Cheng MM, Diaz A, Pan MK, Lieberman OJ, Hutten SJ, Alcalay RN, Kim S, Ximénez-Embún P, Fan L, Kim D, Ko HS, Yacoubian T, Kanter E, Liu L, Tang G, Muñoz J, Sardi SP, Li A, Gan L, Cuervo AM, Sulzer D. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv (2022). 11;8(6):eabm6393. doi: 10.1126/sciadv.abm6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers M; Kochlamazashvili G; Stumpf A; Puchkov D; Swaminathan A; Lucht MT; Krause E; Maritzen T; Schmitz D; Haucke V Neuronal Autophagy Regulates Presynaptic Neurotransmission by Controlling the Axonal Endoplasmic Reticulum. Neuron (2021), 109, p299–313 e299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M; Sliter DA; Kane LA; Sarraf SA; Wang C; Burman JL; Sideris DP; Fogel AI; Youle RJ The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature (2015), 524, p309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauretti E, Iuliano L, Pratico D. Extra virgin olive oil ameliorates cognition and neuropathology if the 3xTg mice: role of autophagy. Ann Clin Transl. Neurol (2017);4(8):p564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauretti E, Iuliano L, Pratico D. Extra-virgin olive oil, cognition and brain health. In: Olives and Olive Oil in health and disease prevention. Editor: Preedy Victor R.; Second edition. Chapter, pp.415–422. 10.1016/B978-0-12-819528-4.00018-3. [DOI] [Google Scholar]
- Lin S, Tian He, Lin Jiaquan, Xu Chang, Yuan Yajiang, Gao Shuang, Song Changwei, Lv Pengfei, Mei Xifan. Zinc promotes autophagy and inhibits apoptosis through AMPK/mTOR signaling pathway after spinal cord injury. Neuroscience Letters. (2020) 25;736: p135263. [DOI] [PubMed] [Google Scholar]
- Liu S; Li Y; Choi HMC; Sarkar C; Koh EY; Wu J; Lipinski MM Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. (2018), 9, p476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li Kai, Chu Fangxuan, Huang Jie, Yang Zhuo. Graphene oxide enhances β-amyloid clearance by inducing autophagy of microglia and neurons. Chemico-Biological Interactions, (2020), 325: p109126. [DOI] [PubMed] [Google Scholar]
- Li Q, Liu Y & Sun M Autophagy and Alzheimer’s Disease. Cell Mol Neurobiol (2017) p37, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QQ, Li Q, Jia JN, Liu ZQ, Zhou HH, Mao XY. 12/15 lipoxygenase: A crucial enzyme in diverse types of cell death. Neurochem Int. (2018) Sep;118: p34–41. [DOI] [PubMed] [Google Scholar]
- Li H, Ham A, Ma TC, Kuo SH, Kanter E, Kim D, Ko HS, Quan Y, Sardi SP, Li A, Arancio O, Kang UJ, Sulzer D, Tang G. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy (2019), 15, pp. 113–130, 10.1080/15548627.2018.1509818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, He Tian, Jiaquan Lin, Chang Xu, Yajiang Yuan, Shuang Gao, Changwei Song, Pengfei Lv, Xifan Mei. Zinc promotes autophagy and inhibits apoptosis through AMPK/mTOR signaling pathway after spinal cord injury. Neuroscience Letters. (2020) 25;736: p135263. [DOI] [PubMed] [Google Scholar]
- Maday S; Wallace KE; Holzbaur EL Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 2012, 196, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheoud D, Cannon T, Voisin A, Penttinen A-M, Ramet L, Fahmy AM Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature (2019) 571, p565–569. [DOI] [PubMed] [Google Scholar]
- Mathews PM, Levy E. Cystatin C in aging and in Alzheimer’s disease. Ageing Res. Rev, 32 (2016), pp. 38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlyniec K, Singewald N, Holst B, Nowak G. GPR39 Zn(2+)-sensing receptor: a new target in antidepressant development?. J. Affect. Disord, 174 (2015), pp. 89–100. [DOI] [PubMed] [Google Scholar]
- Nagai A, Terashima M, Sheikh AM, Notsu Y, Shimode K, Yamaguchi S, Kobayashi S, Kim SU, Masuda J. Involvement of cystatin C in pathophysiology of CNS diseases. Front Biosci. (2008) May 1; 13:p3470–9. [DOI] [PubMed] [Google Scholar]
- Nascimento AC, Erustes AG, Reckziegel P et al. α-Synuclein Overexpression Induces Lysosomal Dysfunction and Autophagy Impairment in Human Neuroblastoma SH-SY5Y. Neurochem Res 45, (2020) 2749–2761. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease J. Cell Sci, 120 (2007), pp. 4081–4091. [DOI] [PubMed] [Google Scholar]
- Parzych KR and Klionsky DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid Redox Signal. (2014)(3):460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest (2008) Jun; 118(6): p2190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupyshev AB, Tikhonova Maria A., Akopyan Anna A., Tenditnik Michael V., Dubrovina Nina I., Korolenko Tatyana A.. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacology Biochemistry and Behavior. (2018) p177:1–11. [DOI] [PubMed] [Google Scholar]
- Perlenfein TJ and Murphy Regina M.. A mechanistic model to predict effects of cathepsin B and cystatin C on β-amyloid aggregation and degradation. Journal of biological chemistry. (2017) 292(51):21071–21082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parzych KR; Klionsky DJ An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal (2014), 20, 460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp D, Kovács T, Billes V, Varga M, Tarnóci A, Hackler L Jr, Puskás LG, Liliom H, Tárnok K, Schlett K, Borsy A, Pádár Z, Kovács AL, Hegedűs K, Juhász G, Komlós M, Erdős A, Gulyás B, Vellai T. AUTEN-67, an autophagy-enhancing drug candidate with potent antiaging and neuroprotective effects. Autophagy. 2016;12(2): p273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan X, Song Li, Zheng Xiaomei, Liu Shenjie, Ding Huaqiang, Li Sijing, Xu Guanghui, Li Xin, Liu Liang. Reduction of Autophagosome Overload Attenuates Neuronal Cell Death After Traumatic Brain Injury. Neuroscience. (2021). 460: p107–119. [DOI] [PubMed] [Google Scholar]
- Qosa H, Mohamed LA, Bataresh YS et al. Extra virgin olive oil attenuates amyloid0b and tau pathology in the brains of TgSwDI mice. J. Nutr. Biochem (2015);26: p1479–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A; Dehay B; Bove J; Carballo-Carbajal I; Dovero S; Perez-Villalba A; Fernagut PO; Blesa J; Parent A; Perier C; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol (2014), 75, p351–362. [DOI] [PubMed] [Google Scholar]
- Ribas, Vinicius T, and Paul Lingor. “Autophagy in degenerating axons following spinal cord injury: evidence for autophagosome biogenesis in retraction bulbs.” Neural regeneration research vol. 10,2 (2015): p198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan B, Flamand O, Vescovi L et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol (2014) 10, p1013–1019. [DOI] [PubMed] [Google Scholar]
- Runwal G; Stamatakou E; Siddiqi FH; Puri C; Zhu Y; Rubinsztein DC LC3-positive structures are prominent in autophagy-deficient cells. Sci Rep (2019), 9, 10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffi & Botelho. 2019. Lysosome fission: planning for an exit. Trends in Cell Biology (2019).;29(8):635–646. doi: 10.1016/j.tcb.2019.05.003. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Rubinsztein DC Small molecule enhancers of autophagy for neurodegenerative diseases. Mol. Biosyst 2008;4:895–901. [DOI] [PubMed] [Google Scholar]
- Sharoar MG, Hu X, Ma XM. et al. Sequential formation of different layers of dystrophic neurites in Alzheimer’s brains. Mol Psychiatry (2019). 24, 1369–1382. 10.1038/s41380-019-0396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharoar MG, Palko S, Ge Y, Saido TC & Yan R Accumulation of saposin in dystrophic neurites is linked to impaired lysosomal functions in Alzheimer’s disease brains. Mol Neurodegener (2021). 16, 45, doi: 10.1186/s13024-021-00464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L; Chen S; Du F; Li S; Zhao L; Wang X Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A 2011, 108, p4788–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q, Meng B, Xu H et al. The emerging roles of vacuolar-type ATPase-dependent Lysosomal acidification in neurodegenerative diseases. Transl Neurodegener (2020). 9, 17. 10.1186/s40035-020-00196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavoe AKH; Holzbaur ELF Autophagy in Neurons. Annu Rev Cell Dev Biol (2019), 35, p477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. (2009);29: p13578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh SN, Chakravorty Anushka, Giridharan Mridhula, Garimella Lakshmi, Manjithaya Ravi. Pharmacological Tools to Modulate Autophagy in Neurodegenerative Diseases. Journal of Molecular Biology. (2020) 432(8):p2822–2842. [DOI] [PubMed] [Google Scholar]
- Surveillance Report of Traumatic Brain Injury-related Emergency Department Visits, Hospitalizations, and Deaths. In: Centers for Disease Control and Prevention, U.S. Department of Health and Human Services. 2014. Available at: https://www.cdc.gov/traumaticbraininjury/get_the_facts.html. Accessed July 8, (2019). [Google Scholar]
- Sun WL Ambra1 in autophagy and apoptosis: Implications for cell survival and chemotherapy resistance. Oncol Lett (2016), 12, p367–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley RE; Ragusa MJ; Hurley JH The beginning of the end: how scaffolds nucleate autophagosome biogenesis. Trends Cell Biol (2014), 24, p73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. (2008). 445: p77–88. [DOI] [PubMed] [Google Scholar]
- Thellung S, Corsaro A, Nizzari M, Barbieri F, Florio T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. Int J Mol Sci. 2019. Feb; 20(4): 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Yingfeng and Matthew NJ Seaman. “Navigating the Controversies of Retromer-Mediated Endosomal Protein Sorting.” Frontiers in Cell and Developmental Biology 9 (2021): 1469. 17;9:658741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United States (US) National database: 2017 profile of people within the traumatic brain injury model systems. Traumatic brain injury model systems National Data and Statistical Center. Center. Available at: https://msktc.org/lib/docs/Data_Sheets_/2017_TBIMS_National_Database_Update_1.pdf. [Google Scholar]
- Vagnozzi Alana N., and Domenico Praticò. “Endosomal sorting and trafficking, the retromer complex and neurodegeneration.” Molecular psychiatry 24.6 (2019): p 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargova I, Machova Urdzikova L, Karova K, Smejkalova B, Sursal T, Cimermanova V, Turnovcova K, Gandhi CD, Jhanwar-Uniyal M, Jendelova P. Involvement of mTOR Pathways in Recovery from Spinal Cord Injury by Modulation of Autophagy and Immune Response. Biomedicines. (2021); 9(6):593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Li A, Sekiya M, Beckmann ND, Quan X, Schrode N, Fernando MB, Yu A, Zhu L, Cao J, Lyu L, Horgusluoglu E, Wang Q, Guo L, Wang YS, Neff R, Song WM, Wang E, Shen Q, Zhou X, Ming C, Ho SM, Vatansever S, Kaniskan HÜ, Jin J, Zhou MM, Ando K, Ho L, Slesinger PA, Yue Z, Zhu J, Katsel P, Gandy S, Ehrlich ME, Fossati V, Noggle S, Cai D, Haroutunian V, Iijima KM, Schadt E, Brennand KJ, Zhang B. Transformative Network Modeling of Multi-omics Data Reveals Detailed Circuits, Key Regulators, and Potential Therapeutics for Alzheimer’s Disease. Neuron (2020), 10.1016/j.neuron.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y; Taguchi K; Tanaka M Ubiquitin, Autophagy and Neurodegenerative Diseases. Cells (2020). 2;9(9):2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X; Klionsky DJ An overview of macroautophagy in yeast. J Mol Biol (2016), 428, 1681–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC; Krainc D alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med (2017), 23, p1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lipinski MM. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells. (2019) Jul 10;8(7): p693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SM; Jung YK A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol Cells (2018), 41, p18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ; Narendra DP Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011), 12, p9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. USA 2007;104:19023–19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Shan H, Chang P, Wang T, Dong W, Chen X, and Tao L Hydrogen sulfide offers neuroprotection on traumatic brain injury in parallel with reduced apoptosis and autophagy in mice. PLoS One 9, (2014). e87241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Chen S, Gao K, Zhou Z, Wang C, Shen Z, Guo Y, Li Z, Wan Z, Liu C, Mei X. Resveratrol protects against spinal cord injury by activating autophagy and inhibiting apoptosis mediated by the SIRT1/AMPK signaling pathway. Neuroscience. (2017) Apr 21;348: p241–251. [DOI] [PubMed] [Google Scholar]
- Zhang Qiu-shuang, et al. “Reassessment of subacute MPTP-treated mice as animal model of Parkinson’s disease.” Acta Pharmacologica Sinica 38. 10 (2017): p1317–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Xian-Si, Geng Wen-Shuo, Jia Jin-Jing, Chen Lei, Zhang Peng-Peng. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Frontiers in Aging Neuroscience. (2018) 17;10: p109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Yanqing Wu, Zhengmao Li, Luxia Ye, Qi Lu, Yajiao Zhou, Yuan Yuan, Ting Jiang, Ling Xie, Yanlong Liu, Daqing Chen, Junming Ye, Wutigri Nimlamool, Hongyu Zhang, Jian Xiao. Valproic acid affects neuronal fate and microglial function via enhancing autophagic flux in mice after traumatic brain injury. Journal of Neurochemistry (JNC). (2019) 154 (3): p284–300. [DOI] [PubMed] [Google Scholar]
- Zhou K, Zheng Z, Li Y, Han W, Zhang J, Mao Y, Chen H, Zhang W, Liu M, Xie L, Zhang H, Xu H, Xiao J. TFE3, a potential therapeutic target for Spinal Cord Injury via augmenting autophagy flux and alleviating ER stress. Theranostics. (2020) Jul 23;10(20): p9280–9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Bai J, Zhong S, Zhang R, Kang K, Zhang X, Xu Y, Zhao C, Zhao M. Downregulation of ATP6V1A Involved in Alzheimer’s Disease via Synaptic Vesicle Cycle, Phagosome, and Oxidative Phosphorylation. Oxid Med Cell Longev (2021). 19;2021:5555634. doi: 10.1155/2021/5555634. [DOI] [PMC free article] [PubMed] [Google Scholar]