

Abstract
Objective
Describe the clinical course and diagnostic and genetic findings in a cat with X‐linked myotubular myopathy.
Case Summary
A 7‐month‐old male Maine coon was evaluated for progressively worsening gait abnormalities and generalized weakness. Neurolocalization was to the neuromuscular system. Genetic testing for spinal muscular atrophy (LIX1) was negative. Given the progressive nature and suspected poor long‐term prognosis, the owners elected euthanasia. Histopathology of skeletal muscle obtained post‐mortem disclosed numerous rounded atrophic or hypotrophic fibers with internal nuclei or central basophilic staining. Using oxidative reactions mediated by cytochrome C oxidase and succinic dehydrogenase, scattered myofibers were observed to have central dark staining structures and a “ring‐like” appearance. Given the cat's age and clinical history, a congenital myopathy was considered most likely, with the central nuclei and “ring‐like” changes consistent with either centronuclear or myotubular myopathy. Whole genome sequencing identified an underlying missense variant in myotubularin 1 (MTM1), a known candidate gene for X‐linked myotubular myopathy.
New or Unique Information Provided
This case is the first report of X‐linked myotubular myopathy in a cat with an MTM1 missense mutation. Maine coon cat breeders may consider screening for this variant to prevent production of affected cats and to eradicate the variant from the breeding population.
Keywords: CNM, congenital, feline, immunohistochemistry, skeletal muscle, XLMTM
Abbreviations
- BIN1
amphiphysin 2
- CNM
centronuclear myopathy
- CK
creatine kinase
- COX
cytochrome C oxidase
- DNM2
dynamin 2
- EMG
electromyography
- LIX1
limb and CNS expressed 1
- MD
muscular dystrophy
- MTM1
myotubularin 1
- PTPLA
protein tyrosine phosphatase‐like A
- RI
reference interval
- RYR1
ryanodine receptor gene
- SDH
succinic dehydrogenase
- SMA
spinal muscular atrophy
- WGS
whole genome sequencing
- XLMTM
X‐linked myotubular myopathy
1. INTRODUCTION
Congenital myopathies are a genetically and pathologically heterogeneous group of non‐dystrophic muscle diseases that initially are classified based on histological features in muscle biopsy specimens. The histological features include presence of typical rod bodies in nemaline myopathy, cores in central core and multiminicore disease, excessive central nuclei and dark centers with sub‐sarcolemmal peripheral halos with oxidative reactions in centronuclear/myotubular myopathy, and selective type 1 fiber atrophy in congenital fiber type disproportion. 1 Serum creatine kinase (CK) activity is usually normal or only mildly increased. If persistently and markedly increased, a diagnosis of muscular dystrophy (MD) should be considered. 1 Electromyography (EMG) is typically normal or only indicates mild myopathic changes. 1 In recent years, genetic analyses, including whole genome sequencing (WGS), have played a role in identifying gene variants associated with congenital myopathies. 1
The centronuclear/myotubular myopathies are further defined by specific gene variants. These myopathies are classified as either autosomal or X‐linked. Autosomal forms have variants in the genes encoding dynamin 2 (DNM2), amphiphysin 2 (BIN1), and the ryanodine receptor (RYR1). The X‐linked (X‐linked myotubular myopathy) forms have variants in the gene encoding myotubularin (MTM1). In dogs, autosomal forms of centronuclear myopathy are associated with a mutation in the protein tyrosine phosphatase‐like A (PTPLA) gene as described in Labrador retrievers, 2 in BIN1 in Great Danes, 3 and in DNM2 in a Border collie. 4 , 5 The X‐linked form of myotubular myopathy is associated with variants in the gene encoding MTM1 in Labrador retrievers, 6 Rottweilers 7 and recently in Boykin spaniels. 8
To date, variants in genes resulting in autosomal forms of centronuclear myopathy (CNM) or X‐linked myotubular myopathy (XLMTM) have not been described in cats. Here, we describe a missense variant in MTM1 in a young male Maine coon cat.
2. CASE SUMMARY
A 7‐month‐old male intact Maine coon cat was referred for further investigation of progressively worsening gait abnormalities and generalized weakness. The owner described the cat as showing apparent pain or difficulty while eating. At 4 months of age, the cat was tentatively diagnosed with nutritional secondary hyperparathyroidism by the referring veterinarian. This diagnosis was based on a history of the cat being fed solely a raw meat diet since weaned at 8 weeks of age, mild diffuse osteopenia and cortical thinning on initial radiographic studies of the skull, thorax, abdomen, pelvis, and selected dental structures. A focal deviation of the medial cortex of the right femur, suggestive of a healing pathologic fracture, also was noted. The cat subsequently was fed a complete and balanced, Association of American Feed Control Officials—formulated commercial diet for the next 3 months until the time of referral. Repeat radiographs performed by the referring veterinarian immediately before referral indicated resolution of the previously identified osteopenia and femoral cortical irregularity.
On physical examination, inability to fully open the jaw, generalized decreased muscle mass, and a body condition score of 1 of 9 were the most clinically relevant findings. No abnormalities were identified on orthopedic examination. No cranial nerve deficits or abnormal spinal reflexes were present on neurological examination. However, weakness of neuromuscular origin was evident.
No abnormalities were noted on hematology. Serum biochemistry identified mild hypoproteinemia (62 g/L; reference interval [RI], 63‐83 g/L), composed of normal albumin (34 g/L; RI, 26‐40 g/L) and low normal globulin (28 g/L; RI, 27‐49 g/L) concentrations, along with mild hyperkalemia (5.2 mmol/L; RI, 3.5‐5.0 mmol/L), and mildly decreased serum creatinine concentration (42 μmol/L; RI, 70‐159 μmol/L). Creatine kinase activity was within normal limits (266 IU/L; RI, 0‐344 IU/L). Serum total thyroxine concentration was also within the reference range (26 nmol/L; RI, 20‐40 nmol/L). The cat was negative for retroviral disease (feline immunodeficiency virus [antibody], feline leukemia virus [antigen]; IDEXX SNAP FIV/FeLV Combo Test, IDEXX Laboratories, Inc., Pukete, Hamilton, New Zealand). The urine was adequately concentrated (specific gravity, 1.058), with 30 mg/dL protein and 3+ blood identified on dipstick analysis, and <5 epithelial cells per high power field (hpf) present on sediment examination.
An edrophonium chloride (Enlon, 150 mg/15 mL; Mylan Institutional LLC, Rockford, Illinois, USA) challenge was performed to rule out congenital myasthenic syndrome. No appreciable improvement in muscle strength was noted after IV administration of 0.25 mg edrophonium chloride. Survey thoracic radiographs indicated mild cardiomegaly without megaesophagus.
Biopsy specimens obtained under general inhalational anesthesia from the right frontalis, triceps, and lateral gastrocnemius muscles were submitted as formalin‐fixed specimens to a commercial veterinary pathology laboratory for analysis by an anatomic pathologist. Histology indicated a mild to moderate, multifocal polyphasic myopathy, suggestive of MD. No abnormal inclusions or aggregates were present within the skeletal muscle after periodic acid‐Schiff staining.
A buccal swab sample was submitted for genetic testing for spinal muscular atrophy (SMA). The cat tested negative for the 140 bp deletion in LIX1 previously identified in Maine coon cats 9 (Orivet Genetic Pet Care, St Kilda, Victoria, Australia).
Given the progressive nature of the disease and suspected poor long‐term prognosis, the owners elected euthanasia. Consent was given for immediate post‐mortem collection of muscle samples at the referring veterinary clinic. Samples were obtained from the triceps, superficial digital flexor, and heart muscle and kept fresh‐frozen at −20°C until transport for further testing.
Specimens from the triceps, superficial digital flexor, and heart muscles were shipped by courier service under refrigeration for evaluation of cryosections by histochemical and immunohistochemical staining. Numerous rounded atrophic or hypotrophic fibers were noted in both limb muscles (Figure 1A) with a type 1 fiber predominance (Figure 1B, NCL‐MHCs 1:10 and NCL‐MHCf 1:10 antibodies used to detect slow and fast muscle fibers, respectively; both from Novocastra Laboratories). Several myofibers contained internal nuclei or central basophilic staining with hematoxylin and eosin staining (Figure 1A). Intramuscular nerve branches were normal in appearance. Using the mitochondrial specific reactions of cytochrome C oxidase (COX, Figure 1C) and succinic dehydrogenase (SDH, Figure 1D), central nuclei were highlighted and scattered myofibers had a “ring‐like” appearance that was dark brown with the COX reaction (Figure 1C) and dark blue with the SDH reaction (Figure 1D). No inflammation, necrosis, fibrosis, fiber loss, organisms, or other specific cytoarchitectural abnormalities were observed. No structural abnormalities were identified in the heart.
FIGURE 1.
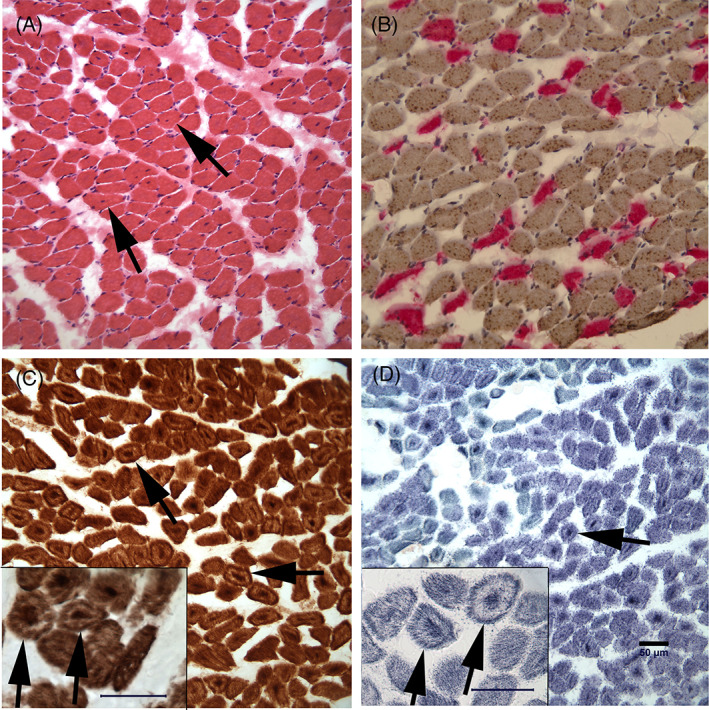
Cryosections from the superficial digital flexor muscle showed a phenotype consistent with centronuclear/myotubular myopathy. Sections were stained with hematoxylin and eosin (A), incubated with monoclonal antibodies against slow (type 1 fibers, brown color) and fast (type 2 fibers, pink color) myosin heavy chain (B), and reacted for cytochrome C oxidase (C) and succinic dehydrogenase (D) activity. Atrophic fibers had a round to polygonal shape with several myofibers containing internal nuclei (arrows in A). Dark brown central deposits and subsarcolemmal rings were present using the cytochrome C oxidase reaction (C, arrows) and dark blue central stained areas were noted with the succinic dehydrogenase reaction (D, arrows). Inserts are included in figures (C) and (D) showing higher power images of ringed fibers. Bar in D = 50 μm for images A to D and individual bars are included in the inserts.
Given the signalment, clinical history and pathological changes in the muscle biopsy samples, a congenital myopathy with central nuclei and subsarcolemmal “ring‐like” structures consistent with CNM or XLMTM was considered most likely. Deoxyribonucleic acid was isolated from 0.25 g of heart muscle using organic extraction methods. 10 Approximately 3 μg was submitted for WGS, which was performed as previously described for the 99 Lives Cat Genome Sequencing Consortium genomic material (http://felinegenetics.missouri.edu/99lives). 11 , 12 The WGS workflow for processing the sequences, alignment to the Felis catus V9.0 reference sequence, 13 and variant calling were conducted as previously described. 12 The DNA variants were viewed, filtered, and annotated using VarSeq (Golden Helix, Bozeman, Montana, USA) with RefSeq release 99 (May 11, 2020). Candidate variants were identified as any variants homozygous in the case cat and absent in 339 cat genomes included in the 99 Lives consortium. Sequencing data are available at NCBI BioProject PRJNA308208.
The 99 Lives dataset of WGS from 340 cats has 3 255 533 exonic variants, including variants within 20 bp of the exon boundaries. Nine variants were unique to the case cat and homozygous, including only 1 missense variant in MTM1 (Table S1A). The MTM1 variant is located on the Xq chromosome at position X:125938001, as a c.455C>T alteration causing a p.ala152val amino acid change in Ensembl transcript ENSFCAT00000024072 (Refseq transcript XM_004000974.4). The read depth at this site was 41× coverage suggesting high accuracy in the variant call. This variant is in exon 6 of the 4 annotated transcripts of MTM1 in the domestic cat. The MTM1 cDNA has 72% identity and 96% homology with the human homolog (Accession no: ENST00000370396.7, Refseq transcript NM_001164191.1), indicating strong conservation of the coding sequence (Figure S1). A cross‐species protein alignment using PRALINE 14 indicated 578 of 603 amino acids are conserved between humans and cats (Figure S2). The missense variant was scrutinized against the cat transcript (MTM1 isoform X1—XP_023105234.1) using protein variation effect analyzer (PROVEAN v1.1.3) web‐based software (http://provean.jcvi.org) and was determined to be deleterious with a PROVEAN score = −3.870. 15 Alternatively, considering the condition as dominant and the case cat to be heterozygous, no other CNM candidate genes were identified (Table S1B).
Approximately 500 bp flanking the variant was obtained from the cat reference sequence from Ensembl ENSFCAG00000004332.6. Polymerase chain reaction primers (MTM1‐F: AGCCATGGAAACTTGAGGTC; MTM1‐R: ATGCGCGTTTGAGATTTACC) were developed in the flanking intronic sequence using the software Primer3Plus 16 and optimized to produce a single amplicon for Sanger sequencing as previously described (Figure 2). 12 Besides the 339 cats and the reference genome cats without the variant, the variant additionally was genotyped by direct Sanger sequencing and not identified in 11 unrelated Maine coon cats, 1 random bred cat, and 1 cat of a different breed.
FIGURE 2.

Electropherogram of MTM1 variant in a Maine coon cat with centronuclear/myotubular myopathy. Presented are the electropherograms for the Sanger sequences of the region of MTM1 with the identified variant of a normal cat (top) and the affected Maine coon (bottom), which demonstrates replacement of a homozygous cytosine (blue peak) with a homozygous thymidine (red peak) within the box. The c.455C>T alteration causes a p.ala152val amino acid change. Image produced by software, Sequencer (GeneCodes Corporation, Ann Arbor, Michigan) V5.1.
3. DISCUSSION
This case represents the first report of a gene variant likely resulting in XLMTM in cats. X‐linked myotubular myopathy and CNM have been extensively described in both humans and dogs, with the Labrador retriever serving as a valuable model for the human disease for decades. 2 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 The condition also has been reported in other dog breeds (including Boykin spaniels, Great Danes, Rottweilers, and a Border collie), an Arabian‐cross foal, and Japanese Black calves. 4 , 7 , 8 , 26 , 27
The main differential diagnosis initially considered given the clinical presentation, age of onset, and signalment was SMA, which has been well‐described in Maine coon cats. 28 Kittens with SMA typically exhibit signs of tremors, proximal muscle weakness, and muscle atrophy beginning at approximately 4 months of age. 28 Initially, loss of function is rapid, but progression tends to slow after 7 to 8 months of age, and affected cats can live for 8 years or more with variable degrees of disability. 28 Genetic testing facilitated the exclusion of this differential diagnosis, highlighting the need for further histopathological and histochemical analysis.
Another disease process that can manifest similarly in a clinical setting is MD. Laminin alpha 2 deficient MD has been described in a Maine coon cat. 29 , 30 , 31 , 32 , 33 , 34 Although initial biopsy findings were suggestive of MD, further analysis on muscle cryosections with histochemical analysis was consistent with a diagnosis of CNM or XLMTM. Whole genome sequencing suggested the MTM1 variant as a potentially causative mutation and supported the diagnosis of XLMTM. In addition, CK activity was within normal limits, which does not support a diagnosis of a form of MD.
The initial formalin‐fixed biopsy specimens in this case were suggestive of MD, in contrast to the fresh‐frozen samples that were later analyzed using both histochemical and immunohistochemical stains and found to be consistent with either a centronuclear or myotubular myopathy. This finding highlights the limitation of routine histological assessment of muscle biopsy specimens in identifying features of congenital myopathies. It also emphasizes the importance of obtaining fresh‐frozen samples for a more comprehensive muscle profile, especially for cases where clinical signs are suggestive of a congenital myopathy and more common (breed‐ and species‐specific) differential diagnoses have been ruled out.
The clinical picture in humans with the various genetic forms of CNM can be quite variable and is dependent on the causative mutation. 35 In infants with XLMTM, a severe phenotype is present in males at birth with marked weakness, hypotonia, external ophthalmoplegia, and respiratory failure. 35 The disease course is fatal in most cases within the first months of life, but a proportion of affected males may survive into their teenage years or even beyond. 35 In dogs, similar clinical signs and progression have been reported. Labrador retrievers with a missense variant in MTM1 showed generalized weakness and muscle atrophy from as early as 7 weeks of age with signs evident to most owners by 14 weeks of age. 25 Clinical signs progressed rapidly in this cohort, with the majority of affected puppies unable to walk within 3 to 4 weeks after clinical signs were first noted. 25 Progressive weakness also has been documented in a Boykin spaniel with a premature stop codon in exon 13 of the MTM1 gene, with progressive, generalized neuromuscular weakness, and tetraparesis. 8 Severe muscle weakness and atrophy resulting in either early euthanasia or death also was described in Rottweiler puppies with a missense mutation in MTM1. 7 In contrast, Labrador retrievers and other breeds with autosomal CNM have a more variable course with mild to moderately severe weakness in the PTPLA mutant dogs.
Interestingly, feeding difficulties usually occur in human infants with XLMTM, as was seen in our case with the owner describing the cat as showing apparent pain or difficulty while eating. 35 Also, in Labrador retrievers with XLMTM surviving beyond 4 months of age, laryngeal and esophageal dysfunction and weakness of the masticatory muscles have been reported. 25 Muscle contracture (secondary to both disease progression and a lack of appropriate physiotherapy to preserve muscle power and function) may play a role in the development of feeding difficulties and even dysarthria in affected human infants. In the cat described here, muscle contracture may have contributed to the clinical signs reported, but no morphological changes were identified to support this mechanism. In addition, given the history of suspected nutritional secondary hyperparathyroidism, underlying orthopedic disease may have contributed. Survey radiographs of the skull did not identify clinically relevant temporomandibular joint (TMJ) pathology that would explain inability to fully open the jaw. Ideally, advanced imaging such as computed tomography of the skull could have been performed to better evaluate any degenerative changes of the TMJ, secondary to the previously identified osteopenia, and also to identify how any underlying bone pathology may have contributed to the cat's clinical signs. In humans, muscle magnetic resonance imaging has proven useful in identifying muscle pathology in equivocal cases of congenital myopathy and can even guide genetic testing, 35 but the utility of this imaging modality for such pathology in veterinary medicine is currently unknown.
Currently, no curative treatment is available for any form of XLMTM. Management is therefore supportive and requires a multidisciplinary approach. 35 Regular physiotherapy plays an important role in preserving muscle strength and function, and preventing contractures. 35 Gene therapy appears to be a promising treatment modality that has been shown to prolong survival and restore function in both murine and canine models of XLMTM. 36 In particular, systemic gene therapy not only prolonged lifespan in Labrador retrievers with XLMTM but importantly corrected the skeletal musculature throughout the body in a dose‐dependent manner, with normal muscular performance and neurological function being reported. 37 , 38 Such research has established the foundation for clinical trials in human patients.
In humans, the prognosis appears to be related to the mode of inheritance, with the X‐linked form generally considered more severe than the dominant or recessive forms. 35 Labrador retrievers show similar variability in prognosis that also is related to the mode of inheritance. 25 In a recent study comparing approximately 100 000 mixed and purebred dogs, the genetic variants associated with CNM were identified in 0.121% of mixed breed dogs (n = 83 022; 201 alleles, including 3 homozygotes) and in 0.117% of a combination of purebred dogs (n = 26 557; 31 alleles, including 6 homozygotes). 39 The CNM variant described in the Great Dane (n = 92) was not observed, and the CNM variant from the Labrador retriever (n = 476) was identified in both mixed bred and purebred dogs. 39 Given the rarity of this disorder in other affected breeds, coupled with often small case numbers, limited information is available regarding prognosis beyond the Labrador retriever breed.
One limitation of our study was the lack of EMG examination, which was not available at the time. Although EMG findings may not have changed the management of the case, the EMG results could have proved useful in establishing data and criteria that may differentiate this condition from other inherited myopathies in cats. The normal CK activity and absence of degeneration and regeneration in muscle biopsy specimens in this case made a myopathy associated with MD unlikely. Evaluation of muscle biopsy specimens in cryosections using histochemical staining provided a diagnosis of a congenital myopathy of the CNM/XLMTM group. The pathological diagnosis identified specific candidate genes for sequencing.
Another limitation was the lack of genetic testing of parents, littermates, and related cats. Such testing would have helped elucidate the Mendelian characteristics and may have served to identify a previous lineage or lineages from which this mutation originated. Unfortunately, the breeder was not cooperative and additional testing was not possible. Because XLMTM is passed through the female with the female clinically unaffected, genotyping for detection of carrier females would be valuable for the breeding program.
Whole genome sequencing as part of Precision/Genomic Medicine is rapidly becoming a feasible standard of care in companion animals and can confirm suspected diagnosis of a heritable condition or identify new disease‐causing variants. The 99 Lives project has sufficient genome sequences that have produced a variant database useful for identifying protein‐coding rare disease variants in cats. 40 , 41 , 42 The identified c.455C>T alteration causing a p.ala152val in MTM1 suggested an X‐linked recessive cause for the XLMTM in this case, particularly because the altered amino acid was highly conserved and changes were predicted to be deleterious. The histopathological phenotype was consistent with this diagnosis. No other DNA samples were available from the breeding lineage to provide segregation support, but this discovery suggests Maine coon cats should be genetically tested for the variant and monitored for the disease. Notably, because X‐linked diseases are passed through the female, all breeding females should be tested for the carrier status of this condition.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Animal ethics approval was not needed for this study. Animal exemption protocol for the acceptance of feline DNA samples for DNA studies is MU IACUC 9178 (Leslie A. Lyons).
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Appendix S1 99 Lives Cat Genome Sequencing Consortium.
Figure S1 Coding sequencing alignment of MTM1. Alignment of domestic cat cDNA (Query: ENSFCAT00000024072.4, Refseq XM_023249466.1) and Homo sapiens cDNA (Subject: ENST00000370396.7, Refseq NM_000252.3) using the Needleman‐Wunsch tool. Position of the cat MTM1 cytosine to thymidine change highlighted
Figure S2 Protein alignment of MTM1 for diverse species. Domestic cat (FCAT, ENSFCAT00000024072.4, Refseq XM_023249466.1), human (HSA, ENST00000370396.7, Refseq NM_000252.3), mouse (MMU, ENMUST00000051970.12, Refseq NM_001164191.1), and zebrafish (DR, ENSDART00000149835.3, Refseq NM_001037684.2). Protein alignment produced using PRALINE14. The highlighted alanine at position 152 is conserved across diverse species indicating function importance and a high conservation score. The c.455C > T alteration causes a p.ala152val amino acid change
Table S1A 2021 99 LIves 340WGS41WES.
Table S1B Dominent.
ACKNOWLEDGMENT
Support has been provided in part from the MU Gilbreath McLorn Endowment, the Winn Feline Foundation and George and Phyllis Miller Trust (MT18‐009, MTW18‐009, MT19‐001) (Leslie A. Lyons), Mars Veterinary (Reuben M. Buckley), and the National Institute of General Medical Science (NIGMS) MARC Fellows Program (T34 GM 136493) (Juan A. Valencia). The authors acknowledge Dr Katherine R. Crosse for performing the surgical muscle biopsies, Dr Susan P. Piripi for her histopathological interpretation of the initial biopsy samples, and Dr Steve Roberts for assistance with obtaining post‐mortem muscle samples. We also appreciate the technical assistance of Thomas R. Juba, MS.
Kopke MA, Diane Shelton G, Lyons LA, et al. X‐linked myotubular myopathy associated with an MTM1 variant in a Maine coon cat. J Vet Intern Med. 2022;36(5):1800‐1805. doi: 10.1111/jvim.16509
Funding information National Institute of General Medical Sciences, Grant/Award Number: T34GM136493; Mars Veterinary; Winn Feline Foundation and George and Phyllis Miller Trust, Grant/Award Numbers: MT19‐001, MTW18‐009, MT18‐009; MU Gilbreath McLorn Endowment
REFERENCES
- 1. North KN, Wang CH, Clarke N, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord. 2014;24(2):97‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maurer M, Mary J, Guillaud L, et al. Centronuclear myopathy in Labrador retrievers: a recent founder mutation in the PTPLA gene has rapidly disseminated worldwide. PLoS One. 2012;7(10):e46408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bohm J, Vasli N, Maurer M, et al. Altered splicing of the BIN1 muscle‐specific exon in humans and dogs with highly progressive centronuclear myopathy. PLoS Genet. 2013;9(6):e1003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eminaga S, Cherubini GB, Shelton GD. Centronuclear myopathy in a Border collie dog. J Small Anim Pract. 2012;53(10):608‐612. [DOI] [PubMed] [Google Scholar]
- 5. Eminaga S, Cherubini GB, Shelton GD. Identification of the mutation causing centronuclear myopathy in a border collie. Vet Rec. 2014;175:124. [DOI] [PubMed] [Google Scholar]
- 6. Beggs AH, Bohm J, Snead E, et al. MTM1 mutation associated with X‐linked myotubular myopathy in Labrador retrievers. Proc Natl Acad Sci USA. 2010;107(33):14697‐14702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shelton GD, Rider BE, Child G, et al. X‐linked myotubular myopathy in Rottweiler dogs is caused by a missense mutation in Exon 11 of the MTM1 gene. Skelet Muscle. 2015;5(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olby NJ, Friedenberg S, Meurs K, et al. A mutation in MTM1 causes X‐linked myotubular myopathy in Boykin spaniels. Neuromuscul Disord. 2020;30(5):353‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fyfe JC, Menotti‐Raymond M, David VA, et al. An approximately 140‐kb deletion associated with feline spinal muscular atrophy implies an essential LIX1 function for motor neuron survival. Genome Res. 2006;16(9):1084‐1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Green MR, Sambrook J. Molecular Cloning: A Laboratory Manual. 4th ed. New York: Cold Spring Harbor Laboratory Press; 2012. [Google Scholar]
- 11. Gandolfi B, Grahn RA, Creighton EK, et al. COLQ variant associated with Devon Rex and Sphynx feline hereditary myopathy. Anim Genet. 2015;46(6):711‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buckley RM, Gandolfi B, Creighton EK, et al. Werewolf, there wolf: variants in hairless associated with hypotrichia and roaning in the Lykoi cat breed. Genes (Basel). 2020;11(6):682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buckley RM, Davis BW, Brashear WA, et al. A new domestic cat genome assembly based on long sequence reads empowers feline genomic medicine and identifies a novel gene for dwarfism. PLoS Genet. 2020;16(10):e1008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simossis VA, Heringa J. PRALINE: a multiple sequence alignment toolbox that integrates homology‐extended and secondary structure information. Nucleic Acids Res. 2005;33(Web Server issue):W289‐W294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745‐2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35(Web Server issue):W71‐W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoskins JD, Root CR. Myopathy in a Labrador retriever. Vet Med Small Anim Clin. 1983;78(9):1387‐1390. [Google Scholar]
- 18. McKerrell RE, Braund KG. Hereditary myopathy in Labrador retrievers: a morphologic study. Vet Pathol. 1986;23(4):411‐417. [DOI] [PubMed] [Google Scholar]
- 19. Watson ADJ, Farrow BRH, Middleton DJ, Smyth JBA. Myopathy in a Labrador retriever. Aust Vet J. 1988;65(7):226‐227. [DOI] [PubMed] [Google Scholar]
- 20. Gortel K, Houston DM, Kuiken T, Fries CL, Boisvert B. Inherited myopathy in a litter of Labrador retrievers. Can Vet J. 1996;37(2):108‐110. [PMC free article] [PubMed] [Google Scholar]
- 21. Tiret L, Blot S, Kessler JL, et al. The cnm locus, a canine homologue of human autosomal forms of centronuclear myopathy, maps to chromosome 2. Hum Genet. 2003;113(4):297‐306. [DOI] [PubMed] [Google Scholar]
- 22. Pele M, Tiret L, Kessler JL, et al. SINE exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs. Hum Mol Genet. 2005;14(11):1417‐1427. [DOI] [PubMed] [Google Scholar]
- 23. Gentilini F, Zambon E, Gandini G, et al. Frequency of the allelic variant of the PTPLA gene responsible for centronuclear myopathy in Labrador retriever dogs as assessed in Italy. J Vet Diagn Invest. 2011;23(1):124‐126. [DOI] [PubMed] [Google Scholar]
- 24. Martin‐Flores M, Pare MD, Tomak EA, et al. Neuromuscular blocking effects of vecuronium in dogs with autosomal‐recessive centronuclear myopathy. Am J Vet Res. 2015;76(4):302‐307. [DOI] [PubMed] [Google Scholar]
- 25. Snead ECR, Taylor SM, van der Kooij M, Cosford K, Beggs AH, Shelton GD. Clinical phenotype of X‐linked Myotubular myopathy in Labrador retriever puppies. J Vet Intern Med. 2015;29(1):254‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Polle F, Andrews FM, Gillon T, et al. Suspected congenital centronuclear myopathy in an Arabian‐cross foal. J Vet Intern Med. 2014;28(6):1886‐1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamada M, Takashima H, Takizawa T, et al. Congenital myopathy in Japanese black calves. J Vet Med Sci. 2004;66(8):997‐1001. [DOI] [PubMed] [Google Scholar]
- 28. He QC, Lowrie C, Shelton GD, et al. Inherited motor neuron disease in domestic cats: a model of spinal muscular atrophy. Pediatr Res. 2005;57(3):324‐330. [DOI] [PubMed] [Google Scholar]
- 29. O'Brien DP, Johnson GC, Liu LA, et al. Laminin alpha 2 (merosin)‐deficient muscular dystrophy and demyelinating neuropathy in two cats. J Neurol Sci. 2001;189(1–2):37‐43. [DOI] [PubMed] [Google Scholar]
- 30. Poncelet L, Resibois A, Engvall E, Shelton GD. Laminin alpha 2 deficiency‐associated muscular dystrophy in a Maine coon cat. J Small Anim Pract. 2003;44(12):550‐552. [DOI] [PubMed] [Google Scholar]
- 31. Awamura Y, Uchida K, Arikawa‐Hirasawa E. Long‐term follow‐up of laminin alpha 2 (merosin)‐deficient muscular dystrophy in a cat. J Feline Med Surg. 2008;10(3):274‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin PT, Shelton GD, Dickinson P, et al. Muscular dystrophy associated with alpha‐dystroglycan deficiency in Sphynx and Devon Rex cats. Neuromuscul Disord. 2008;18(12):942‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Salvadori C, Vattemi G, Lombardo R, Marini M, Cantile C, Shelton GD. Muscular dystrophy with reduced beta‐sarcoglycan in a cat. J Comp Pathol. 2009;140(4):278‐282. [DOI] [PubMed] [Google Scholar]
- 34. Salvadori C, Vattemi G, Marini M, et al. Adult‐onset muscular dystrophy in a cat associated with a presumptive alteration in trafficking of caveolin‐3. J Comp Pathol. 2012;147(2‐3):253‐258. [DOI] [PubMed] [Google Scholar]
- 35. Jungbluth H, Wallgren‐Pettersson C, Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. 2008;3:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Childers MK, Joubert R, Poulard K, et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci Transl Med. 2014;6(220):220ra10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mack DL, Poulard K, Goddard MA, et al. Systemic AAV8‐mediated gene therapy drives whole‐body correction of myotubular myopathy in dogs. Mol Ther. 2017;25(4):839‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elverman M, Goddard MA, Mack D, et al. Long‐term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve. 2017;56(5):943‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Donner J, Anderson H, Davison S, et al. Frequency and distribution of 152 genetic disease variants in over 100,000 mixed breed and purebred dogs. PLoS Genet. 2018;14(4):e1007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mauler DA, Gandolfi B, Reinero CR, et al. Precision medicine in cats: novel Niemann‐pick type C1 diagnosed by whole‐genome sequencing. J Vet Intern Med. 2017;31(2):539‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aberdein D, Munday JS, Gandolfi B, et al. A FAS‐ligand variant associated with autoimmune lymphoproliferative syndrome in cats. Mamm Genome. 2017;28(1‐2):47‐55. [DOI] [PubMed] [Google Scholar]
- 42. Jaffey JA, Reading NS, Giger U, et al. Clinical, metabolic, and genetic characterization of hereditary methemoglobinemia caused by cytochrome b(5) reductase deficiency in cats. J Vet Intern Med. 2019;33(6):2725‐2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 99 Lives Cat Genome Sequencing Consortium.
Figure S1 Coding sequencing alignment of MTM1. Alignment of domestic cat cDNA (Query: ENSFCAT00000024072.4, Refseq XM_023249466.1) and Homo sapiens cDNA (Subject: ENST00000370396.7, Refseq NM_000252.3) using the Needleman‐Wunsch tool. Position of the cat MTM1 cytosine to thymidine change highlighted
Figure S2 Protein alignment of MTM1 for diverse species. Domestic cat (FCAT, ENSFCAT00000024072.4, Refseq XM_023249466.1), human (HSA, ENST00000370396.7, Refseq NM_000252.3), mouse (MMU, ENMUST00000051970.12, Refseq NM_001164191.1), and zebrafish (DR, ENSDART00000149835.3, Refseq NM_001037684.2). Protein alignment produced using PRALINE14. The highlighted alanine at position 152 is conserved across diverse species indicating function importance and a high conservation score. The c.455C > T alteration causes a p.ala152val amino acid change
Table S1A 2021 99 LIves 340WGS41WES.
Table S1B Dominent.