

Abstract
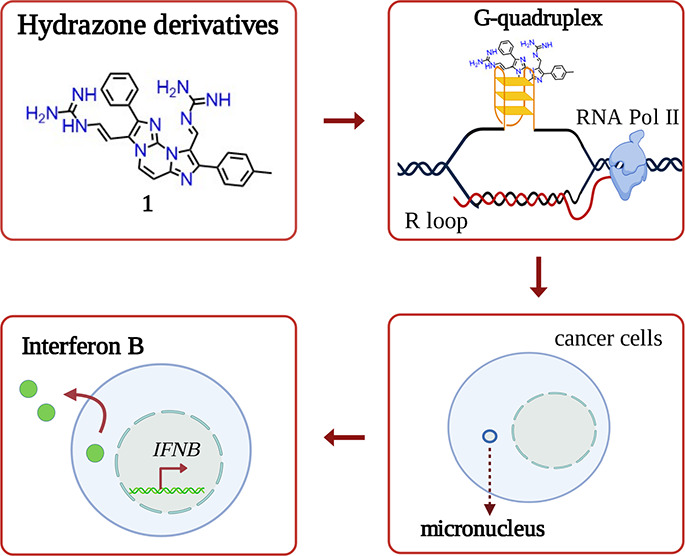
G-quadruplex (G4) ligands are investigated to discover new anticancer drugs with increased cell-killing potency. These ligands can induce genome instability and activate innate immune genes at non-cytotoxic doses, opening the discovery of cytostatic immune-stimulating ligands. However, the interplay of G4 affinity/selectivity with cytotoxicity and immune gene activation is not well-understood. We investigated a series of closely related hydrazone derivatives to define the molecular bases of immune-stimulation activity. Although they are closely related to each other, such derivatives differ in G4 affinity, cytotoxicity, genome instability, and immune gene activation. Our findings show that G4 affinity of ligands is a critical feature for immune gene activation, whereas a high cytotoxic potency interferes with it. The balance of G4 stabilization versus cytotoxicity can determine the level of immune gene activation in cancer cells. Thus, we propose a new rationale based on low cell-killing potency and high immune stimulation to discover effective anticancer G4 ligands.
Introduction
G-quadruplex (G4) ligands are actively investigated to discover new effective anticancer drugs as G4s, non-canonical DNA structures, are considered promising targets.1−4 Despite the large number of specific ligands developed, none has however shown efficacy in cancer patients and very few have reached early phases of clinical trials.5,6 In line with the standard drug discovery rationale, several laboratories have previously searched for G4 binders with a high cell-killing potency.1−5 Interestingly, we have recently demonstrated that the G4 binders pyridostatin (PDS) and PhenDC3 can effectively elicit an innate immune gene response [activation of interferon β (IFN-B) gene and IFN-B-dependent pathways] in human cancer cells, mediated by micronuclei accumulation at non-cytotoxic concentrations.7 As recent advances clearly point to the potential of harnessing innate immunity for cancer immunotherapy,8−12 non-cytotoxic immune-modulators may optimize immunotherapy in unresponsive cancers while having a marginal toxicity against proliferating normal cells. Thus, our recent findings7 indicate that G4 ligands may be exploited as cytostatic immune-stimulating agents for anticancer immune-therapeutic combinations.5 In particular, G4 binders can increase micronuclei,7,13,14 which can be a source of cytoplasmic DNA that is able to induce the cGAS–STING pathway and activate innate immune genes.7,15,16 However, the relationships among G4 affinity/selectivity, cell-killing potency, and genome instability determining a high level of immune gene activation by G4 ligands remains to be established.
Here, to answer this question, we have focused on a highly homogenous series of new compounds able to selectively target G4s. In 2010, some of us identified FG (Chart 1), a bis-guanylhydrazone derivative of diimidazo[1,2-a:1,2-c]pyrimidine, as a potent and selective G4 stabilizer.17 Then, we identified highly selective analogues with a preference for parallel G4 topology and ability to stabilize G4s in living cancer cells,18 including FG and FIM (compounds 1 and 3 by Amato et al.,18 respectively, Chart 1). The results established the diimidazo[1,2-a:1,2-c]pyrimidine core as a scaffold of selective G4 ligands and showed that both the iminoguanidine (Gua) and hydrazinoimidazoline (Imidaz) nitrogen chains are effective in achieving G4 binding properties. Then, FG was shown to induce DNA damage and micronuclei in human osteosarcoma U2OS cells in an R loop-dependent manner.13
Chart 1. Chemical Structures of the Lead Compounds FG and FIM (1 and 3 by Amato et al.,18 respectively).

As these agents are specific and effective G4 binders, we have now synthesized new close derivatives of FG and FIM to investigate the structural features eliciting a high immune gene activation relative to the cell-killing potency. The findings show that a proper balance between G4 affinity/selectivity and cytotoxicity is critical for immune gene activation in cancer cells.
Results
Design of New Hydrazone-Based Compounds
In order to improve affinity and selectivity toward G4 structures and finely tune the biological effects of close FG and FIM analogues, we designed and synthesized a new series of molecules having different electron distribution and similar steric hindrance. For this purpose, the diimidazo[1,2-a:1,2-c]pyrimidine core was maintained unaltered and a chlorine or a methyl group was inserted at the para position of one or both the pending phenyl rings. In fact, chlorine and methyl have almost the same steric hindrance but opposite inductive effects, methyl being an electron donor group while chlorine has an electron withdrawing inductive effect. Both Gua and Imidaz moieties were considered as positively charged chains, either to obtain FG analogues (compounds 1–12, Chart 2) or FIM analogues (compounds 14–18, Chart 2). In addition, since an FG analogue bearing thiophenes instead of phenyl groups proved to be a good G4 binder,18 we also considered this kind of modification along with the replacement of the Gua chains with the Imidaz ones (compound 13, Chart 2). Finally, the formyl group of FIM was replaced with a primary alcohol group, which is able to either accept or donate hydrogen bonds (compounds 19 and 20, Chart 2). The complete synthesis of the derivatives is described in Supporting Information (Scheme S1 and Table S1).
Chart 2. Chemical Structures of New FG and FIM Derivatives Synthesized in This Study.

Circular Dichroism Experiments
The stabilizing effects of compounds 1–20 on G4 structures formed by the G-rich DNA sequences from the nuclease hypersensitive region of the c-KIT (c-kit1 and c-kit2) and c-MYC (c-myc) gene promoters as well as from the human telomeric sequence (tel26) were analyzed by circular dichroism (CD) melting experiments. These DNA sequences were chosen for their ability to adopt different G4 topologies, characterized by parallel (c-kit1, c-kit2, and c-myc) or hybrid (tel26) arrangements.19−21 Consistently, CD spectra of c-kit1, c-kit2, and c-myc displayed a positive band at 264 nm and a negative one around 240 nm (Figure S1), which are characteristics of parallel-stranded G4 topologies.22 On the other hand, tel26 showed a positive band at 290 with a shoulder at ca. 268 nm and a weak negative band at around 240 nm (Figure S1), confirming the presence of a hybrid structure as the main conformation. A 27 residue-long hairpin-forming oligonucleotide (hairpin) was also used to evaluate the selectivity of the new analogues for G4s over a duplex. CD spectra of hairpin showed a positive band at around 280 nm and a negative one at ∼250 nm, confirming the formation of a duplex (Figure S1). Additional CD spectra were recorded to examine the potential of compounds 1–20 to modify the native folding topology of these G4s. DNA/ligand mixtures were prepared by adding each ligand (2 molar equiv) to folded G4 or hairpin structures. No significant variations in the CD signal were observed for any of the analyzed DNA structures (Figures S2–S6), suggesting no G4 topology changes upon addition of compounds. Then, their ability to bind and stabilize the DNA structures was evaluated by CD melting experiments measuring the compound-induced change in the apparent melting temperature (ΔT1/2) of G4 and duplex structures. CD melting curves of DNA with and without each ligand were obtained by following the variations of the intensity of the CD signals at 264, 290, and 252 nm for parallel G4s, hybrid G4, and duplex, respectively (Figures S7–S11). The results show that all compounds are good G4 stabilizers (with one exception, 14) showing a higher preference for parallel than hybrid G4s (Table 1). In addition, as expected for ligands having the same core but different numbers of positive charges, the greater the charge number, the stronger the stabilizing effect on G4s (Table 1). However, compound interactions with dsDNA (hairpin) were also slightly increased by positive charges (Table 1). Thus, to assess the selectivity for G4 structures of this series of compounds, we selected the analogues showing a strong stabilizing effect on at least two G4s and a negligible effect (ΔT1/2 < 2.0 °C) on the hairpin, that is, compounds 1, 2, 8, and 10 among those with two positively charged side chains and 15, 19, and 20 among those with a positive charge only.
Table 1. Compound-Induced Thermal Stabilization of G4 and haipin structures Measured by CD Melting Experiments.
| ΔT1/2 (°C)a |
||||||
|---|---|---|---|---|---|---|
| Comp | tel26 | c-kit1 | c-kit2 | c-myc | hairpin | no. of positive charges |
| FGb | –4.5 | >15.0 | >20 | >20 | 0.1c | 2 |
| 1 | 4.8 (±0.2) | 18.2 (±0.2) | >30d | 16.9 (±0.2) | 1.3 (±0.3) | 2 |
| 2 | 5.3 (±0.2) | 15.2 (±0.2) | >30d | 9.1 (±0.3) | 1.1 (±0.4) | 2 |
| 3 | 6.3 (±0.4) | 25.2 (±0.5) | >30d | 16.9 (±0.4) | 3.2 (±0.3) | 2 |
| 4 | 6.2 (±0.2) | 21.4 (±0.4) | >30d | 13.1 (±0.6) | 2.2 (±0.2) | 2 |
| 5 | 5.5 (±0.2) | 18.5 (±0.3) | >30d | 15.6 (±0.2) | 4.1 (±0.3) | 2 |
| 6 | 4.5 (±0.3) | 17.1 (±0.2) | 26.5 (±0.4) | 8.5 (±0.2) | 2.4 (±0.2) | 2 |
| 7 | 5.0 (±0.2) | 21.4 (±0.4) | >30d | 17.6 (±0.4) | 2.3 (±0.2) | 2 |
| 8 | 6.3 (±0.2) | 15.5 (±0.3) | 22.0 (±0.3) | 7.4 (±0.3) | 1.2 (±0.4) | 2 |
| 9 | 8.3 (±0.2) | 20.1 (±0.3) | >30d | 14.3 (±0.2) | 2.6 (±0.2) | 2 |
| 10 | 6.6 (±0.2) | 17.7 (±0.3) | 19.2 (±0.3) | 10.6 (±0.3) | 1.2 (±0.3) | 2 |
| 11 | 4.1 (±0.4) | 11.1 (±0.2) | >30d | 11.4 (±0.2) | 2.9 (±0.2) | 2 |
| 12 | 3.3 (±0.2) | 9.6 (±0.2) | 13.7 (±0.3) | 5.5 (±0.2) | 3.5 (±0.2) | 2 |
| 13 | 3.8 (±0.3) | 10.3 (±0.2) | 14.9 (±0.3) | 4.7 (±0.2) | 2.0 (±0.2) | 2 |
| FIMb | –3.0 | 2.7 | 9.5 | >20 | –0.8c | 1 |
| 14 | 1.3 (±0.2) | 3.8 (±0.2) | 6.8 (±0.2) | 1.5 (±0.2) | 0.0 (±0.3) | 1 |
| 15 | 4.3 (±0.2) | 11.2 (±0.2) | 24.2 (±0.4) | 5.8 (±0.3) | 1.7 (±0.2) | 1 |
| 16 | 4.8 (±0.3) | 16.1 (±0.2) | 25.2 (±0.3) | 9.1 (±0.3) | 2.7 (±0.2) | 1 |
| 17 | 3.8 (±0.2) | 6.6 (±0.3) | 20.8 (±0.5) | 3.1 (±0.2) | 1.2 (±0.2) | 1 |
| 18 | 2.1 (±0.2) | 6.1 (±0.2) | 16.2 (±0.2) | 4.6 (±0.2) | 2.3 (±0.3) | 1 |
| 19 | 2.0 (±0.2) | 7.1 (±0.2) | 14.7 (±0.3) | 1.7 (±0.2) | 1.3 (±0.2) | 1 |
| 20 | 1.8 (±0.3) | 9.7 (±0.2) | 18.6 (±0.4) | 2.8 (±0.2) | 2.3 (±0.3) | 1 |
ΔT1/2 represents the difference in melting temperature [ΔT1/2 = T1/2 (DNA + 2 ligand equiv) – T1/2 (DNA)]. The T1/2 values of DNA alone are: c-kit1 = 54.0 ± 0.5 °C, c-kit2 = 61.5 ± 0.5 °C, c-myc = 72.0 ± 0.5 °C, tel26 = 47.9 ± 0.5 °C, and hairpin = 75.4 ± 0.2 °C. All experiments were performed in duplicate, and the reported values are the average of two measurements.
Data from ref (18).
A self-complementary 12-mer duplex-forming sequence was used as a duplex model.
ΔT1/2 could not be accurately determined as the compound increases hugely the thermal stability of c-kit2.
FRET Melting Experiments
The Förster resonance energy transfer (FRET) methodology23 was used to further evaluate G4-stabilizing properties and G4 versus duplex selectivity of 1, 2, 8, 10, 15, 19, and 20. In this assay, the G4-forming c-kit1 oligonucleotide labeled with FAM (F) and TAMRA (T) at the 5′ and 3′ ends, respectively, was employed (F-c-kit1-T) since, among the G4s more stabilized by these ligands, c-kit1 is the one that has the lowest T1/2 value, thus allowing to better estimate the stabilizing properties of different ligands and evaluate their ability to discriminate between G4 and duplex structures. Indeed, as the target G4 was the only labeled molecule, it was possible to evaluate the ligand selectivity by adding a large excess of the hairpin oligonucleotide (unlabeled competitor). Therefore, the ability of the investigated compounds to selectively stabilize the G4 was evaluated by measuring the effect of the presence of various concentrations of the competitor on the ΔT1/2 of the G4 in the presence of 2 molar equiv of each ligand. Results of these experiments (Figure S12 and Table 2) confirm that the selected compounds are efficient G4 stabilizers. However, in the case of compounds 1, 2, 8, and 10 (carrying two positively charged side chains), G4/ligand interaction turned out to be somewhat challenged by the hairpin sequence being added in excess. This does not happen for 15, 19, and 20, meaning that these compounds are more selective for G4 than the former.
Table 2. G4 Selectivity of the Selected Compoundsa.
| ΔT1/2 (°C)b |
|||
|---|---|---|---|
| Comp | F-c-kit1-T | F-c-kit1-T + hairpin (1:15) | F-ckit1-T + hairpin (1:50) |
| 1 | 25.4 (±0.5) | 22.2 (±0.5) | 19.2 (±0.5) |
| 2 | 22.4 (±0.5) | 21.4 (±0.5) | 20.2 (±0.5) |
| 8 | 24.6 (±0.5) | 21.4 (±0.5) | 19.6 (±0.5) |
| 10 | 26.6 (±0.5) | 24.8 (±0.5) | 23.8 (±0.5) |
| 15 | 22.0 (±1.0) | 21.6 (±1.0) | 22.2 (±1.0) |
| 19 | 8.0 (±0.4) | 7.8 (±0.4) | 8.4 (±0.4) |
| 20 | 10.0 (±0.4) | 11.1 (±0.5) | 12.1 (±0.5) |
G4/dsDNA competition determined by ligand-induced thermal stabilization of F-c-kit1-T G4 measured by FRET. ΔT1/2 values are the differences between the T1/2 of F-c-kit1-T in the presence (2 molar equiv) and absence of the ligands, without or with large excess of unlabeled hairpin (15 and 50 molar equiv with respect to G4).
The T1/2 of F-ckit1-T is 57.4 (±0.2) °C. All experiments were performed at least in duplicate, and the reported values are the average of the measurements. The differences between results of CD and FRET melting experiments could be explained with different DNA sequences and/or experimental conditions.
Fluorescence Intercalator Displacement Assay
To gain insight into the affinity of the selected compounds for G4s, fluorescence intercalator displacement (G4-FID) experiments were performed by using the light-up fluorescent probe thiazole orange (TO), which binds to the DNA structure of interest.24 The competitive displacement of TO from DNA by candidate ligands was monitored, thus enabling the determination of their relative binding affinity to the structures under examination, namely, c-kit1, c-kit2, and c-myc G4s, which were selected as they turned out to be those most stabilized by the ligands. Dose–response curves were obtained by plotting the percentage of TO displacement versus the concentration of each compound (Figure S13), and the concentrations at which 50% displacement was achieved (DC50) were calculated. The lower the DC50 value, the higher should be the affinity of the compound for the DNA structure. Results of G4-FID assay (Table 3) indicate a good TO displacement ability for compounds 1, 2, 8, and 10. These ligands exhibited almost similar results for the investigated parallel G4s, suggesting that ligand/G4 interaction is not sequence-specific. On the other hand, compounds 15, 19, and 20 were not able to reach 50% displacement in any case, suggesting that FIM derivatives have a lower affinity for G4s than FG ones. Therefore, the TO-displacing ability seems to be in direct correlation with the number of positive charges on the ligands: the highly cationic molecules are the most efficient TO displacers. As for 15, the apparent discrepancy between the results of G4-FID assay and melting experiments (CD and FRET) could also be explained considering that this ligand may bind to G4s without strictly competing with the TO.25
Table 3. Ligand DC50 Values for c-kit1, c-kit2, and c-myc G4s Determined with G4-FID Assay.
| DC50 (μM)a |
|||
|---|---|---|---|
| Comp | c-kit1 | c-kit2 | c-myc |
| 1 | 1.4 | 1.4 | 1.6 |
| 2 | 1.3 | 1.2 | 1.3 |
| 8 | 2.7 | 1.7 | 1.8 |
| 10 | 1.2 | 1.4 | 1.3 |
The error in DC50 values is ±5%.
Microscale Thermophoresis Assay
Quantitative data on the binding affinity of 1, 2, 8, 10, 15, 19, and 20 for the selected G4s were obtained by microscale thermophoresis (MST), which is a powerful method for the quantitative analysis of the interactions between small molecules and nucleic acids in solution.26 To perform MST experiments, one of the binding partners must be fluorescent (either intrinsically fluorescent or conjugated to a given fluorophore). Therefore, serial dilutions of ligands were prepared, mixed with a constant concentration of Cy5.5-labeled G4s (c-kit1, c-kit2, and c-myc), and analyzed by MST. Results of the binding curves showed that the compounds bind to G4s with different affinity (Table 4 and Figures S14–S16). In particular, compounds showed higher affinity values for c-kit1 and c-kit2 than for c-myc, and a slight preference for c-kit1 over c-kit2, except for 19 and 20. Noteworthy, compound 1 turned out to be the strongest G4 binder of the series, showing Kd values in the nanomolar range for the interaction with c-kit1 and c-kit2 [Kd = 0.03 and 0.04 μM, respectively], while 19 and 20 turned out to be the worst of the series in terms of affinity for G4s.
Table 4. Equilibrium Dissociation Constants for the Binding of the Ligands to c-kit1, c-kit2, and c-myc G4s Obtained by MST Experimentsa.
|
Kd (μM) |
|||
|---|---|---|---|
| Comp | c-kit1 | c-kit2 | c-myc |
| 1 | 0.03 ± 0.01 | 0.04 ± 0.01 | 1.5 ± 0.4 |
| 2 | 0.12 ± 0.02 | 0.13 ± 0.02 | n.d. |
| 8 | 0.38 ± 0.09 | 0.49 ± 0.09 | 3.0 ± 1.0 |
| 10 | 0.07 ± 0.02 | 0.50 ± 0.04 | 1.2 ± 0.4 |
| 15 | 0.30 ± 0.04 | 0.37 ± 0.03 | 7.0 ± 1.0 |
| 19 | 8.0 ± 2.0 | 2.5 ± 0.3 | 37 ± 1 |
| 20 | 2.0 ± 0.2 | 1.3 ± 0.2 | 8.0 ± 3.1 |
Dissociation constant values were obtained with MST experiments. Comp, compound. n.d., not determined.
Cytotoxicity of Selected Hydrazone Derivatives
Next, we determined the cytotoxic potencies of compounds with two (FG, 1, 2, and 8) or one (FIM, 15, 19, and 20) positively charged chain in human osteosarcoma U2OS and murine fibrosarcoma MNMCA1 cells following 24 h of treatments by using PDS as a reference compound (Table 5). We selected these two lines as the former has been used in several G4 studies, included ours,13,14,18 and the latter murine line is known to produce high levels of IFN-B.27 The results show that FIM and 15, both bearing an aldehyde moiety, are the most cytotoxic compounds among those analyzed (Table 5). In particular, they exhibited IC50 values around 6-fold and 5–18-fold higher than those of 19 and 20, respectively, indicating that an aldehyde moiety confers a greater cytotoxicity than an alcohol group. Among the FG analogues, 8 is more cytotoxic than 1 and 2. The compounds have similar IC50 in both the two lines; however, imino-guanidine chains confer around twofold higher cytotoxic activity than 2-hydrazino-2-imidazoline chains in humans but not in murine cells (compare 1vs2, and 20vs19, Table 5). To better define the interplay among G4 affinity/selectivity, G4 stabilization in cells, induction of genome instability and activation of IFN-B, FG and FIM analogues were discussed separately. The tested analogues show similar cytotoxic potency in normal human MRC5 fibroblasts (Table S2) as expected for a cell assay which measures cell-killing effects against proliferative cells.
Table 5. Cytotoxic Potency of Selected Hydrazone Derivativesa.
| Comp | human U2OS | murine MNMCA1 |
|---|---|---|
| FG | 15.9 ± 1.2b | n.d. |
| 1 | 46.8 ± 12.7 | 38.5 ± 5.8 |
| 2 | 108.3 ± 35.4 | 28.6 ± 8.4 |
| 8 | 20.2 ± 1.0 | 23.5 ± 5.1 |
| FIM | 4.0 ± 0.33 | 2.3 ± 0.92 |
| 15 | 2.6 ± 0.87 | 1.9 ± 0.33 |
| 19 | 24.5 ± 1.2 | 12.1 ± 2.5 |
| 20 | 14.3 ± 9.3 | 35.7 ± 0.67 |
| PDS | >50b | 27.0 ± 16.0 |
Data are IC50 (μM, concentration inhibiting 50% of cell growth) of each compound in human osteosarcoma U2OS cells and murine fibrosarcoma MNMCA1 cells. Treatments were for 24 h in exponentially growing cells. Cell survival was evaluated with the MTT test after 48 h of cell recovery in drug-free medium. IC50 values are means ± SEM of two independent experiments performed in triplicate.
See ref (13). n.d., not determined.
G4 Stabilization and DNA Damage by Hydrazone Derivatives with Two Side Chains
Next, we have evaluated cellular effects of closely related analogues starting with compounds 1, 2, and 8, which have two positively charged side chains (Figure 1). We used an immunofluorescence (IF) assay to determine their ability to stabilize G4 structures in U2OS cells using the BG4 antibody, which specifically binds to G4 structures,13,28 and PDS as the positive control.13 The results (Figure 1A) show that 1 and 2 can stabilize G4 structures (around 2.4-fold increase) in living cells whereas compound 8 was ineffective (0.77-fold change). Analogues 1 and 2 showed a G4 stabilization in vivo similar to that of PDS (Figure 1A) and much higher than that of 8, suggesting that the latter has other or additional cytotoxic mechanisms. Overall, these results agree with a higher ligand-induced G4 thermal stabilization observed for analogues 1 and 2 than that for 8, particularly for c-kit2 and c-myc G4s (Tables 1 and 3). The complete lack of G4 stabilization with 8 in nuclei suggests that G4 binding in living cells may be affected by interactions with other cellular components.
Figure 1.

G4 stabilization, DNA damage, and IFN-B stimulation induced by FG derivatives. (A) Quantification of fluorescence signals of BG4 foci in U2OS cells being treated for 10 min with PDS or FG derivatives (compounds 1, 2, and 8) at 10 μM concentration. The graph shows the fold increase reported as the mean ± SEM of three biological replicates, and the IF representative images are reported (left). (B) Quantification of fluorescence signals of γH2AX in U2OS cells being treated with PDS (10 μM) and FG derivatives at IC50 concentrations (46, 100, and 20 μM for 1, 2, and 8, respectively) for 24 h of treatment. The graph shows the fold increase reported as the median ± SEM of two biological replicates and the IF representative images are reported (left). (C) Micronuclei quantification by DAPI staining in MNMCA1 cells treated (15 μM) after 24 h of treatment followed by 24 h of drug-free recovery. PDS (10 μM)-treated cells are also shown. The graph shows the mean ± SEM of two biological replicates, and the IF representative images are reported (left). Above the bar chart, the p-value are reported. The scale bar is 10 μm. (D) Quantification of IFN-B produced by MNMCA1 cells treated with FG derivatives at different concentrations (15 and 30 μM). PDS (10 μM)-treated cells are also shown. The IFN-B detection was performed with ELISA assay after 24 h of compounds treatment followed by 48 h of recovery. The bar chart reports the mean ± SEM of three biological replicates. Significance in all the graph was calculated by Mann–Witney test (*p < 0.05, **p > 0.01, ***p > 0.001, and ****p < 0.0001).
Next, we determined DNA damage induced by 1, 2, and 8 by evaluating the levels of S139-phosphorylated histone H2AX (γH2AX) (Figure 1B). We treated U2OS cancer cells with compounds for 24 h, at equal cytotoxic concentrations. PDS (10 μM) was used a reference compound.13 The results show that 2 and 8 increased γH2AX foci levels (1.45- and 1.37-fold increase, respectively) whereas 1 did not. Thus, as 8 can induce DNA damage (Figure 1B) even without stabilizing G4s in cell (Figure 1A), while compound 2 induces G4 stabilization, it is reasonable to speculate that the cytotoxicity mechanism is likely different between the two compounds. On the other hand, G4 stabilization may lead to different levels of DNA damage, likely depending on in vivo G4 targeting. In contrast to 2, compound 1 stabilizes G4 structures in cells (Figure 1A) but does not promote DNA damage (Figure 1B).
Micronuclei and IFN-B Activation by Hydrazone Derivatives with Two Side Chains
As non-cytotoxic doses of G4 binders, PDS and PhenDC3, can activate IFN-B-dependent pathways through micronuclei induction in human cancer cells,7 we next ask if the new analogues can also affect immune gene expression in cancer cells. First, we determined the induction of micronuclei. 1, 2, and 8 at similar cytotoxic concentrations (15 μM, corresponding to 0.4–0.65 of their IC50s) can induce almost the same micronuclei levels in MNMCA1 cells but less than those of PDS (Figure 1C). Then, we measured the amount of IFN-B secreted by murine MNMCA1 cells into the medium with ELISA assay. In agreement with experimental conditions reported for PDS previously,7 murine cells were treated for 24 h with two concentrations (15 and 30 μM) of compounds and then allowed to recover for 2 days in fresh medium (Figure 1D). The results show that PDS induced higher IFN-B levels than the tested analogues, consistently with higher numbers of PDS-stimulated micronuclei. Among the studied analogues, 1 was more effective in the induction of IFN-B than 2, whereas 8 was completely ineffective (Figure 1D). In particular, 1 induced IFN-B production at higher levels at non-cytotoxic concentrations (15 μM) and 8 was ineffective even at concentrations higher (30 μM) than the IC50 (Figure 1D and Table 5). Thus, compound 1, which induces in vivo G4 stabilization without promoting DNA damage, can activate IFN-B gene expression at non-cytotoxic concentrations.
G4 Stabilization and DNA Damage by Hydrazone Derivatives with One Side Chain
Next, we have evaluated cellular effects of closely related analogues with one positively charged side chain, FIM, 15, 19, and 20. Although these analogues showed a markedly decreased affinity for the tested G4 structures with respect to two positively charged analogues (Tables 1–3), their cytotoxic potencies are equal or higher than those of the latter (Table 5). Therefore, we asked whether FIM analogues could trigger G4 stabilization and DNA damage similar to FG analogues. The results show that they are all good G4 stabilizers in living cells (Figure 2A). 15 and 20 induced a somewhat higher stabilization (2.32–2.45-fold change) than FIM and 19 (1.55–1.91-fold change), indicating that the Gua moiety favors G4–ligand interactions better than the 2-hydrazino-2-imidazoline group (see also Table 1) probably due to its higher flexibility. Then, we investigated the ability of these analogues to induce DNA damage under the experimental conditions described above for FG analogues. The results show that these analogues increased γH2AX levels at similar levels in cancer cells, even though 15 was somewhat less effective (Figure 2B). As FIM and 15 were more cytotoxic than 19 and 20, DNA damage features of the former are likely more lethal than those of the latter.
Figure 2.

G4 stabilization, DNA damage, and IFN-B stimulation induced by FIM derivatives. (A) Quantification of fluorescence signals of BG4 foci in U2OS cells treated for 10 min with PDS or FIM derivatives at 10 μM concentration. Graphs show the fold increase reported as the mean ± SEM of three biological replicates. The images are representative of IF assays performed at reported concentrations (left). (B) Quantification of fluorescence signals of γH2AX in U2OS cells treated with PDS (10 μM) and FIM derivatives at IC50 concentrations (4, 2.5, 24, and 14 μM for FIM, 15, 19, and 20, respectively). The graph shows the fold increase reported as the median ± SEM of two biological replicates, and the IF representative images are reported (left). (C) Micronuclei quantification by DAPI staining in MNMCA1 cells treated with 1 μM of compounds FIM and 15 and 5 μM for the analogues 19 and 20. PDS (10 μM)-treated cells are also shown. Left, the graph shows the mean ± SEM of two biological replicates; right, representative cell images. The scale bar is 10 μm. Above the bar chart, the p-value are reported. (D) Quantification of IFN-B stimulated at the reported concentration has been detected after 24 h of treatment followed by 48 h of recovery. PDS (10 μM)-treated cells are also shown. The IFN-B protein levels were detected with ELISA assay. The bar chart reports the mean ± SEM of two biological replicates. Significance in all the graphs was calculated by Mann–Whitney test (*p < 0.05, **p > 0.01, ***p > 0.001, and ****p < 0.0001).
Micronuclei and IFN-B Activation by Hydrazone Derivatives with One Side Chain
Similar to FG analogues (Figure 1), we then tested the FIM analogues for the induction of micronuclei and the activation of IFN-B genes in murine MNMCA1 cells by using sub-cytotoxic concentrations (Figure 2C,D). The results show that FIM, 15, 19, and 20 induced a 2.5–4.0-fold increase in micronuclei levels in comparison to untreated cells with little difference among them (Figure 2C). Overall, the FIM analogues did not affect significantly IFN-B expression, showing a low, if any, with a maximum of less than twofold change for FIM (Figure 2D). No difference was observed between derivatives bearing Gua or 2-hydrazino-2-imidazoline groups as chains. Overall, the results indicate that analogues with one positively charged side chain were less effective in activating the IFN-B gene expression than the two positively charged analogues (Figure 1). The effect on gene expression was thus correlated with G4 affinity of the studied analogues.
Discussion
Hydrazone-based compounds, including FG and FIM (Chart 1), are known to have a high selectivity for G4 structures relative to duplex DNA and to induce DNA damage and genome instability.14,17,18 Here, we provide evidence that these agents can activate IFN-B gene expression in cancer cells at non-cytotoxic doses, therefore pointing to the exploitation of hydrazone-based G4 ligands as immunomodulating agents. In particular, 1 can be considered as a core structure for further analyses aiming at establishing a hit ligand with immune-stimulating anticancer activity.
In vivo G4 selectivity of structurally different G4 ligands is substantially unknown as the number and types of G4 structures in a living cell can be very high.3−5 In addition, a ligand can have more molecular interactions affecting its biological outcome, in particular the cell-killing potency. Thus, our present investigation has been focused on very closely related analogues to minimize putative variations of unpredictable molecular interactions. In particular, a new series of FG and FIM having different electron distribution and similar steric hindrance were designed and synthesized. For this purpose, the diimidazo[1,2-a:1,2-c]pyrimidine core was maintained unaltered and a chlorine or a methyl group was inserted at the para-position of one or both the pending phenyl rings. In fact, chlorine and methyl have almost the same steric hindrance but opposite inductive effects, methyl being an electron donor group while chlorine has an electron-withdrawing inductive effect. Both Gua and Imidaz moieties were considered as positively charged chains, either to obtain FG analogues (compounds 1–12, Chart 2) or FIM analogues (compounds 14–18). In addition, since an FG analogue bearing thiophenes instead of the phenyl groups proved to be a good G4 binder,14 we also considered this kind of modification along with the replacement of the Gua chains with Imidaz ones (compound 13). Finally, the formyl group of FIM was replaced with a primary alcohol group, which is able to either accept or donate hydrogen bonds (compounds 19 and 20).
The G4 binding properties of 1–20 in terms of either G4 stabilization, affinity, and selectivity over the duplex structure were measured by means of several biophysical techniques, including CD, G4-FID, MST, and competition FRET-melting. We used the results of CD melting experiments to select the best binders from FG and FIM series. In particular, the ligands were chosen on the basis of their selectivity for G4 over the duplex, that is, those compounds showing the most negligible effects on the hairpin–duplex model (compounds 1, 2, 8, and 10 belonging to the FG series; 15 and 17 belonging to the FIM series and exhibiting the formyl group; and 19 and 20 in which the formyl group of FIM is reduced to the corresponding hydroxyl group). Next, among these compounds, we selected, within each series, those that showed the greatest stabilizing effects on at least two G4s and differed in the presence of Gua or Imidaz pendant groups, that is, compounds 1 and 2 as FG analogues (with Gua and Imidaz substituents, respectively), compound 15 among the FIM derivatives (with a formyl group in R2 and a Gua substituent in R4), and compounds 19 and 20 among the hydroxyl group-containing FIM derivatives (carrying a Gua and an Imidaz pendant group in R4, respectively). Since compounds 8 and 10 belonging to the FG series also showed good stabilizing properties on the investigated G4s, we decided to include them in further biophysical assays aimed at assessing the selectivity of the ligands (FRET) and their affinity for G4s (G4-FID assay).
The results of these studies confirmed that compounds 1, 2, 8, 10, and 15 are stronger G4 stabilizers than 19 and 20, and revealed that compounds with one positively charged side chain (i.e., 15, 19, and 20) have less affinity for G4s but are more selective binders compared to those having two positively charged side chains (1, 2, 8, and 10), with compounds 1, 2, and 10 being the most efficient TO displacers. Despite the high chemical similarity between 8 and 10 (they differ in the inversion of the Phe and ClPhe substituents in R1 and R3), only compound 10 performed similarly to 1 and 2, while compound 8 showed a slightly lower G4 affinity. These results were also confirmed by MST experiments, which allowed to evaluate the affinity of the ligands for the G4s. Indeed, compound 1 turned out to be the strongest G4 binder, followed by 2 and 10, while 19 and 20 were the worst of the series.
Based on the whole set of biophysical data, the compounds were classified according to their affinity for G4s: strong binders in the case of 1, 2, and 10; moderate binders for 8 and 15; and modest binders for 19 and 20.
Therefore, aimed at defining the interplay between G4 affinity, stabilization in cells, cytotoxicity, and immune-stimulation activity of these hydrazone-based compounds, derivatives 1, 2, 8, 15, 19, and 20, having different affinity for G4s, were selected for the biological investigations.
Interestingly, despite the minimal structural differences among the FG analogues 1, 2 and 8, they showed interesting differences in cytotoxic potency, in-cell G4 stabilization, and IFN-B gene activation. Compound 8 is more cytotoxic than 1 and 2 (Table 5); however, it minimally stabilizes G4 in nuclei (Figure 1A) and it does not trigger IFN-B production (Figure 1D). On the contrary, 1 shows a high G4 stabilization in vivo (Figure 1A) and the least cytotoxic potency in murine cells (Table 5), where it triggers a good activation of IFN-B genes (Figure 1D).
Conversely, FIM analogues bearing the chemically reactive aldehyde group (FIM and 15) or the hydroxymethyl group (19 and 20) exhibit a greater cytotoxic potency than FG derivatives (Table 5), with compounds FIM and 15 being the most cytotoxic compounds of the series. In addition, they are able to stabilize G4 in cells (Figure 2A) but not able to trigger IFN-B activation. These data clearly show that a high cytotoxic ability interferes with the ability of a G4 ligand to activate the expression of IFN-B genes. Overall, FG analogues, characterized by two side chains, exhibit a markedly higher G4 affinity than that of FIM analogues (Tables 1 and 3), albeit with a reduction of G4 selectivity (Table 2). As FIM analogues overall do not activate IFN-B genes (Figure 2D), whereas the FG analogue 1 does (Figure 1D), we speculate that a high ligand affinity for G4 may be required for immune gene activation. FG and FIM analogues can stabilize G4s at similar levels in nuclear chromatin; however, we do not know whether G4 structures stabilized by each analogue are the same or not. Our data indicate that the specific pattern of stabilized G4s and, likely, the specific time and location may affect the molecular response to G4 ligand activity.
An important observation was that IFN-B activation was independent of the level of induced micronuclei (Figures 1 and 2, panels C and D), suggesting that cytosolic DNA from micronuclei was necessary but not sufficient for immune gene expression.29,30 Even though the definition of the mechanism likely needs future investigations, however, the activation of other cytoplasmic signaling pathways may affect the recognition of micronuclei and activation of the STING pathway.5,29,30 Autophagic processes are known to be activated by G4 binders3,31,32 and can regulate the STING pathway through recycling micronuclei and DNA by forming autophagosomes.33,34 Interestingly, autophagic gene pathways were not activated at high levels in MCF-7 cells treated with PDS, which can activate at very high levels the IFN-B gene and other genes stimulated by IFN-B.7 Therefore, differences in autophagic pathway activation might explain differences in IFN-B production between analogue 1 and other studied derivatives.
Conclusions
Comparing very closely related G4 binders has allowed us to demonstrate that a proper balance between G4 affinity/selectivity and cytotoxicity is critical for immune gene activation, in particular a high G4 affinity and a relatively low cytotoxic potency are necessary for a G4 ligand to activate IFN-B genes in cancer cells (Figure 3). Thus, we propose a new rationale, based on low cell-killing potency and high G4 affinity, to discover effective anticancer G4 ligands with immune-stimulation activity.
Figure 3.
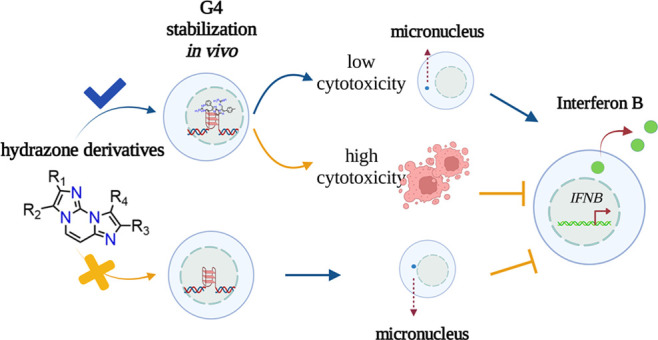
Schematic representation of cellular effects of hydrazone derivatives.
Experimental Section
Compound Synthesis and Materials
The synthesis and NMR spectra of FG and FIM analogues are reported in Supporting Information. All compounds are >95% pure by elemental analysis (see Supporting Information). Controlled pore glass supports, DNA phosphoramidites, all reagents for oligonucleotide synthesis and purification, and all other reagents and solvents were purchased from Merck KGaA (Darmstadt, Germany) and used without further purification. Dual-labeled FAM/TAMRA oligonucleotides and Cy5.5-labeled oligonucleotides were purchased from Biomers (Ulm, Germany).
Oligonucleotide Synthesis and Sample Preparation
The following deoxyribonucleotide sequences were used in this study: d(AGG GAG GGC GCT GGG AGG AGG G) (c-kit1), d(CGG GCG GGC GCT AGG GAG GGT) (c-kit2), d(TGA GGG TGG GTA GGG TGG GTA A) (c-myc), d(TTA GGG TTA GGG TTA GGG TTA GGG TT) (tel26), and d(CGC GAA TTC GCG TTT CGC GAA TTC GCG) (hairpin). These oligonucleotides were chemically synthesized on the 1 μmol scale on an ABI 394 DNA/RNA synthesizer (Applied Biosystems, CA, USA) by using the standard β-cyanoethyl phosphoramidite solid-phase chemistry, as described elsewhere.35 After synthesis, oligonucleotides were detached from the support and deprotected by treating with an aqueous solution of concentrated ammonia at 55 °C, for 17 h. The filtrates and washings, after being combined and concentrated under reduced pressure, were solubilized in water and purified using a high-performance liquid chromatography system equipped with a Nucleogel SAX column (Macherey-Nagel, 1000-8/46), using a 30 min linear gradient from 100% buffer A to 100% buffer B at a flow rate of 1 mL/min, with buffer A consisting of a 20 mM KH2PO4/K2HPO4 aqueous solution (pH 7.0) and buffer B consisting of 1.0 M KCl and 20 mM KH2PO4/K2HPO4 aqueous solution (pH 7.0). Both buffer A and B also contained 20% (v/v) CH3CN. The purified fractions were then desalted by means of C-18 cartridges (Sep-Pak). The purity of the isolated oligomers was checked by NMR and proved to be higher than 98%. All oligonucleotides were dissolved in a buffer solution consisting of 5 mM KH2PO4/K2HPO4 (pH 7.0) and 20 mM KCl (or LiCl in the case of c-myc because of its high thermal stability). The concentration of each oligonucleotide was verified by measuring the UV absorption at 90 °C, considering the appropriate molar extinction coefficient values ε (λ = 260 nm) calculated using the nearest-neighbor model.36 Finally, to achieve the correct folding of the DNA sequences, oligonucleotide solutions were annealed by heating at 95 °C for 5 min followed by a slow cooling to room temperature and storage overnight at 4 °C.
CD Experiments
CD experiments were performed on a Jasco J-815 spectropolarimeter equipped with a PTC-423S/15 Peltier temperature controller. All the spectra were recorded at 20 and 100 °C in the wavelength range of 230–320 nm and averaged over three scans. A scan rate of 100 nm/min, with a 0.5 s response time and 1 nm bandwidth, was used. The buffer baseline was subtracted from each spectrum. For the CD experiments, 10 μM G4 and 15 μM duplex DNA in the absence or presence of 2 molar equiv of ligand were used. CD spectra were recorded 10 min after ligand addition. Ligand stock solutions were 10 mM in DMSO. CD melting experiments were carried out in the 20–100 °C temperature range at a 1 °C/min heating rate by following the changes in the CD signal at the wavelengths of the maximum CD intensity (263 nm) for c-kit1, c-kit2, c-myc, and (287 nm) tel26 G4s, or minimum CD intensity (252 nm) for the hairpin. CD melting experiments were recorded both in the absence and presence of compounds (2 molar equiv) added to the folded nucleic acid structures. The apparent melting temperatures (T1/2) were determined from a curve fit using OriginPro 2021 software (OriginLab Corp., MA, USA). ΔT1/2 values were determined as the difference in the T1/2 values of the nucleic acid structures in the presence and absence of the compounds. Normalization of melting curves between 0 and 1 was performed to better compare the results. In cases where the melting process was not completed even at 100 °C due to an exceptional ligand-induced G4 thermal stabilization, the relative melting curves were normalized by dividing only by the maximum.
FRET Melting Experiments
Measurements were carried out on a Jasco FP-8300 spectrofluorometer equipped with a Peltier temperature controller system (PCT-818) using a dual-labeled G4-forming sequence FAM-[d(CGG GCG GGC GCT AGG GAG GGT)]-TAMRA (F-c-kit1-T). The oligonucleotide was dissolved in water at 1 mM, diluted at 1 μM using 5 mM KH2PO4/K2HPO4 (pH 7.0) containing 20 mM KCl, and annealed by heating to 90 °C for 5 min, followed by slow cooling to room temperature overnight and storage at 4 °C for 24 h before data acquisition. Experiments were performed in sealed quartz cuvettes with a path length of 1 cm by using 0.2 μM prefolded F-c-kit1-T target, in the absence and presence of 2 molar equiv of the ligand and of the duplex competitor at 3 and 10 μM final concentrations. In addition, an experiment in the absence of compounds and competitors was also performed. Fluorescence spectra were acquired before and after melting assay (15 and 90 °C, respectively). The dual-labeled oligonucleotide was excited at 492 nm, and emission spectra were recorded between 500 and 650 nm by using a 100 nm/s scan speed. Excitation and emission slit widths were both set to 5 nm. FRET melting experiments were performed by monitoring the emission of FAM at 520 nm (upon excitation at 492 nm), using a heating gradient of 0.2 °C/min over the range 15–90 °C. Emission of FAM was normalized between 0 and 1. Final analysis of the data was carried out using OriginPro 2021 software.
Fluorescent Intercalator Displacement (G4-FID) Assay
A solution containing 0.25 μM G4 DNA (c-kit1, c-kit2, or c-myc) and 0.5 μM TO in 5 mM KH2PO4/K2HPO4 buffer (pH 7.0) containing 20 mM KCl (or LiCl in the case of c-myc) was prepared in a 1 cm-path length cell, and the corresponding fluorescence spectrum was acquired in the absence and presence of increasing concentrations of selected compounds (1 mM stock solution in DMSO). Each ligand addition (from 0.5 to 20 molar equiv) was followed by a 3 min equilibration time before spectrum acquisition. Measurements were run at 20 °C on a Jasco FP-8300 spectrofluorometer equipped with a Peltier cell holder (PCT-818), using an excitation wavelength of 485 nm and recording the emission in the 500–650 nm wavelength range. Both excitation and emission slits were set at 5 nm. Final analysis of the data was carried out using OriginPro 2021 software. The percentage of TO displacement was calculated as follows: TO displacement (%) = 100 – [(F/F0) × 100], where F0 is the fluorescence in the absence of a ligand and F is the fluorescence after each ligand addition. The percentage of displacement was then plotted as a function of the ligand concentration, and DC50 was calculated as the required concentration to displace 50% TO. Each titration was performed in duplicate.
Microscale Thermophoresis
MST measurements were performed using a Monolith NT.115 instrument (NanoTemper Technologies). The Cy5.5 fluorescently labeled oligonucleotides (c-kit1, c-kit2, and c-myc) were prepared at 1 μM in 5 mM KH2PO4/K2HPO4 buffer (pH 7.0) containing 20 mM KCl and annealed as described above. DNA samples were then diluted using the same phosphate buffer supplemented with 0.1% Tween. For the MST experiments, the concentration of the labeled oligonucleotides was kept constant at 20 nM, while a serial dilution of the ligand (1:2 from 5.0, 40, 160, or 400 μM ligand stock solution) in the same buffer used for DNAs was prepared and mixed with the oligonucleotide solution with a volume ratio of 1:1. All the samples, containing 20% DMSO as the final concentration, were loaded into standard capillaries (NanoTemper Technologies). Measurements were performed and analyzed as previously reported.37
Cell Lines and Treatments
Human osteosarcoma U2OS and murine fibrosarcoma MNMCA1 cell lines were grown in monolayer cultures in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% l-glutamine (Gibco). Human fibroblast lung MRC5 cells were grown in a monolayer culture in DMEM, supplemented with Ham’s F-10 nutrient mix (1:1), 10% FBS, and Pen/Strep 100 μg/mL. All cell lines were grown in a humidified incubator at 37 °C and 5% of CO2. Cell line identity was routinely checked by genotyping (BMR Genomics). Compounds were dissolved in dimethyl sulfoxide (Sigma-Aldrich #472301) at 10 mM concentration, stored in aliquots at −20 °C, and diluted to final concentrations immediately prior to use.
MTT Cell Proliferation Assay
U2OS, MNMCA1, and MRC5 cells (3 × 104) were seeded in 24 wells. 24 h after seeding, cells were treated with increasing concentrations of compounds for 24 h. Then, compounds were removed, and the cells were grown in complete drug-free medium for 48 h. Then, thiazolyl blue tetrazolium bromide (MTT) (Merck #2128) solution (0.45 μg/mL) was added to each well and incubated for 1 h at 37 °C. After incubation, the medium was removed and 300 μL of dimethyl sulfoxide was added and incubated for 1 h at room temperature. Then, 100 μL of the solution was put in 96 wells, and absorbance at 540 nm was measured using a multiplate reader. The linear regression parameters were determined to calculate the IC50 (GraphPad Prism 4.0, Graph Pad Software Inc.).
IF Microscopy
U2OS cells (3.5 × 105) were seeded in 35 mm dish on coverslips. The BG4 fluorescence signal was determined after 10 min of treatment at the reported concentrations. The BG4 antibody was purified as described.13 Briefly, BG4 was isolated from Escherichia coli extracts by using silica-based resin (Thermo #89964) precharged with Co2+ ions and eluted with 250 μM imidazole/PBS pH 8.0. The eluted antibody was concentrated in Pierce 30k MWCO tubes (Pierce #88529), and imidazole was finally removed by buffer exchange with intracellular cell salt buffer in Pierce 30k MWCO tubes. For BG4 staining, cells were pre-fixed with cell culture medium and fix solution (1:1) and then incubated with the fix solution composed of methanol and acetic acid (3:1) for 10 min at RT. The cells were permeabilized with 0.1% of Triton X-100 in PBS and blocked in 2% non-fat milk for 1 h at RT under gentle shaking. Next, cells were stained with 0.5 μg of BG4 for 2 h at room temperature. Next, cells were incubated with the anti-FLAG antibody (dilution 1/800) (Cell Signaling Technology #2368) for 1 h and then stained with the Alexa Fluor 488 anti-rabbit IgG (Life technologies #A11008). For S139-phosphorylated histone H2AX, γH2AX cells were treated with compounds at the reported concentrations for 24 h. Then, cells were fixed with 4% formaldehyde for 10 min, permeabilized with 0.5% Triton X-100 in PBS for 15 , and then incubated with 8% BSA in PBS for 30 min at RT. Next, cells were stained with anti-γH2AX antibodies (#05-636, Millipore) diluted to 1:500 and next incubated for 1 h with Alexa Fluor 594 anti-mouse IgG (#A11032, Life Technologies). For DNA staining, cells were incubated with 2 μg/μL DAPI for 20 min. The cover glasses were mounted with Mowiol 488. The slides were visualized at room temperature by using a fluorescence microscope (Eclipse TE 2000-S, Nikon) equipped with an AxioCam MRm (Zeiss) digital camera. The fluorescence signal was quantified by using ImageJ software and reported as a fold increase of the non-treated sample. Graphs were prepared with GraphPad Prism 8.
IFN-B ELISA Assay
MNMCA1 cells (8 × 105) were seeded in a 10 mm dish. IFN-B protein levels were measured in cell medium supernatants. Culture medium of untreated and treated MNMCA1 cells was collected after 24 h of treatment followed by 48 h of drug recovery. Supernatants were added with protease inhibitors (1 mg/mL pepstatin, leupeptin, and aprotinin, 2 mM DTT, and 0.5 mM PMSF) and then concentrated around 25-fold by using a Pierce Protein Concentrator PES, 3k MWCO, 5–20 mL (#88525, Thermo Fisher). IFN-B protein levels were quantified with a human IFN-B Quantikine ELISA kit (MIFNB0, R&D Systems) following manufacturer’s instructions. IFN-B levels were normalized over the cell number.
Acknowledgments
We thank all laboratory members for helpful discussion. The research leading to these results had received funding from AIRC (Associazione Italiana per la Ricerca sul Cancro), IG 2019—ID 23032 project—PI G.C., IG 2020—ID 24590 project—PI B.P and IG 2021—ID 26313 project-PI A.R. G.M. is a recipient of a FIRC-AIRC postdoc fellowship from Italy [23953].
Glossary
Abbreviations
- BSA
bovine serum albumin
- CBR
condensed benzene ring
- CD
circular dichroism
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle medium
- DMF
dimethylformamide
- DSB
double-strand break
- FAM
fluorescein
- FBS
fetal bovine serum
- FID
fluorescence intercalator displacement
- FRET
Förster resonance energy transfer
- G4
G-quadruplex
- γH2AX
S139-phosphorylated histone H2AX
- IF
immunofluorescence
- IFN-B
interferon β
- MST
microscale thermophoresis
- NOE
nuclear overhauser effect
- PBS
phosphate-buffered saline
- PDS
pyridostatin
- PIPES
1,4-piperazinediethanesulfonic acid
- TAMRA
carboxytetramethylrhodamine
- TO
thiazole orange
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00772.
Author Contributions
S.M., G.M., and R.M. contributed equally. R.M., A. Locatelli, and A. Leoni synthesized derivatives. S.M., B.P., and J.A. performed oligonucleotide synthesis and biophysical experiments. G.M., A.A., J.M., and M.P. performed the biological experiments. A.R., J.A., and G.C. designed, planned, and supervised experimental work and interpreted the results with all authors. The manuscript was written by G.C., J.A., G.M., and R.M. and with contributions from other authors. All authors approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Balasubramanian S.; Hurley L. H.; Neidle S. Targeting G-Quadruplexes in Gene Promoters: A Novel Anticancer Strategy?. Nat. Rev. Drug Discovery 2011, 10, 261–275. 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidle S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. 10.1021/acs.jmedchem.5b01835. [DOI] [PubMed] [Google Scholar]
- Carvalho J.; Mergny J.-L.; Salgado G. F.; Queiroz J. A.; Cruz C. G-Quadruplex, Friend or Foe: The Role of the G-Quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. 10.1016/j.molmed.2020.05.002. [DOI] [PubMed] [Google Scholar]
- Miglietta G.; Russo M.; Capranico G. G-Quadruplex–R-Loop Interactions and the Mechanism of Anticancer G-Quadruplex Binders. Nucleic Acids Res. 2020, 48, 11942–11957. 10.1093/nar/gkaa944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miglietta G.; Marinello J.; Russo M.; Capranico G.. Ligands Stimulating Antitumour Immunity as the next G-Quadruplex Challenge. Mol. Cancer 2022, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton J.; Gelmon K.; Bedard P. L.; Tu D.; Xu H.; Tinker A. V.; Goodwin R.; Laurie S. A.; Jonker D.; Hansen A. R.; Veitch Z. W.; Renouf D. J.; Hagerman L.; Lui H.; Chen B.; Kellar D.; Li I.; Lee S.-E.; Kono T.; Cheng B. Y. C.; Yap D.; Lai D.; Beatty S.; Soong J.; Pritchard K. I.; Soria-Bretones I.; Chen E.; Feilotter H.; Rushton M.; Seymour L.; Aparicio S.; Cescon D. W. Results of the Phase I CCTG IND.231 Trial of CX-5461 in Patients with Advanced Solid Tumors Enriched for DNA-Repair Deficiencies. Nat. Commun. 2022, 13, 3607. 10.1038/s41467-022-31199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miglietta G.; Russo M.; Duardo R. C.; Capranico G. G-Quadruplex Binders as Cytostatic Modulators of Innate Immune Genes in Cancer Cells. Nucleic Acids Res. 2021, 49, 6673–6686. 10.1093/nar/gkab500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo S.-R.; Corrales L.; Gajewski T. F. Innate Immune Recognition of Cancer. Annu. Rev. Immunol. 2015, 33, 445. 10.1146/annurev-immunol-032414-112043. [DOI] [PubMed] [Google Scholar]
- Poh A. STINGing Antitumor Immunity into Action. Cancer Discovery 2018, 8, 259–260. 10.1158/2159-8290.CD-ND2018-002. [DOI] [PubMed] [Google Scholar]
- Hartmann G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. 10.1016/bs.ai.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boorn J. G.; Hartmann G. Turning Tumors into Vaccines: Co-Opting the Innate Immune System. Immunity 2013, 39, 27–37. 10.1016/j.immuni.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Fu J.; Kanne D. B.; Leong M.; Glickman L. H.; McWhirter S. M.; Lemmens E.; Mechette K.; Leong J. J.; Lauer P.; Liu W.; Sivick K. E.; Zeng Q.; Soares K. C.; Zheng L.; Portnoy D. A.; Woodward J. J.; Pardoll D. M.; Dubensky T. W.; Kim Y. STING Agonist Formulated Cancer Vaccines Can Cure Established Tumors Resistant to PD-1 Blockade. Sci. Transl. Med. 2015, 7, 283ra52. 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Magis A.; Manzo S. G.; Russo M.; Marinello J.; Morigi R.; Sordet O.; Capranico G. DNA Damage and Genome Instability by G-Quadruplex Ligands Are Mediated by R Loops in Human Cancer Cells. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 816–825. 10.1073/pnas.1810409116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato J.; Miglietta G.; Morigi R.; Iaccarino N.; Locatelli A.; Leoni A.; Novellino E.; Pagano B.; Capranico G.; Randazzo A. Monohydrazone Based G-Quadruplex Selective Ligands Induce DNA Damage and Genome Instability in Human Cancer Cells. J. Med. Chem. 2020, 63, 3090–3103. 10.1021/acs.jmedchem.9b01866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding S. M.; Benci J. L.; Irianto J.; Discher D. E.; Minn A. J.; Greenberg R. A. Mitotic Progression Following DNA Damage Enables Pattern Recognition within Micronuclei. Nature 2017, 548, 466–470. 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie K. J.; Carroll P.; Martin C.-A.; Murina O.; Fluteau A.; Simpson D. J.; Olova N.; Sutcliffe H.; Rainger J. K.; Leitch A.; Osborn R. T.; Wheeler A. P.; Nowotny M.; Gilbert N.; Chandra T.; Reijns M. A. M.; Jackson A. P. CGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465. 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparapani S.; Bellini S.; Gunaratnam M.; Haider S. M.; Andreani A.; Rambaldi M.; Locatelli A.; Morigi R.; Granaiola M.; Varoli L.; Burnelli S.; Leoni A.; Neidle S. Bis-Guanylhydrazone Diimidazo[1,2-a:1,2-c]Pyrimidine as a Novel and Specific G-Quadruplex Binding Motif. Chem. Commun. 2010, 46, 5680. 10.1039/c0cc00020e. [DOI] [PubMed] [Google Scholar]
- Amato J.; Morigi R.; Pagano B.; Pagano A.; Ohnmacht S.; De Magis A.; Tiang Y.-P. Y. P.; Capranico G.; Locatelli A.; Graziadio A.; Leoni A.; Rambaldi M.; Novellino E.; Neidle S.; Randazzo A. Toward the Development of Specific G-Quadruplex Binders: Synthesis, Biophysical, and Biological Studies of New Hydrazone Derivatives. J. Med. Chem. 2016, 59, 5706–5720. 10.1021/acs.jmedchem.6b00129. [DOI] [PubMed] [Google Scholar]
- Hsu S. T. D.; Varnai P.; Bugaut A.; Reszka A. P.; Neidle S.; Balasubramanian S. A G-Rich Sequence within the c-Kit Oncogene Promoter Forms a Parallel G-Quadruplex Having Asymmetric G-Tetrad Dynamics. J. Am. Chem. Soc. 2009, 131, 13399–13409. 10.1021/ja904007p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrus A.; Chen D.; Dai J.; Jones R. A.; Yang D. Solution Structure of the Biologically Relevant G-Quadruplex Element in the Human c-MYC Promoter. Implications for G-Quadruplex Stabilization. Biochemistry 2005, 44, 2048–2058. 10.1021/bi048242p. [DOI] [PubMed] [Google Scholar]
- Dai J.; Carver M.; Punchihewa C.; Jones R. A.; Yang D. Structure of the Hybrid-2 Type Intramolecular Human Telomeric G-Quadruplex in K+ Solution: Insights into Structure Polymorphism of the Human Telomeric Sequence. Nucleic Acids Res. 2007, 35, 4927–4940. 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randazzo A.; Spada G. P.; da Silva M. W. Circular Dichroism of Quadruplex Structures. Top. Curr. Chem. 2013, 330, 67–86. 10.1007/128_2012_331. [DOI] [PubMed] [Google Scholar]
- Decian A.; Guittat L.; Kaiser M.; Sacca B.; Amrane S.; Bourdoncle A.; Alberti P.; Teuladefichou M. P.; Lacroix L.; Mergny J. L. Fluorescence-Based Melting Assays for Studying Quadruplex Ligands. Methods 2007, 42, 183–195. 10.1016/j.ymeth.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Monchaud D.; Allain C.; Teulade-Fichou M. P. Development of a Fluorescent Intercalator Displacement Assay (G4-FID) for Establishing Quadruplex-DNA Affinity and Selectivity of Putative Ligands. Bioorg. Med. Chem. Lett. 2006, 16, 4842–4845. 10.1016/j.bmcl.2006.06.067. [DOI] [PubMed] [Google Scholar]
- Zuffo M.; Guédin A.; Leriche E.-D.; Doria F.; Pirota V.; Gabelica V.; Mergny J.-L.; Freccero M. More Is Not Always Better: Finding the Right Trade-off between Affinity and Selectivity of a G-Quadruplex Ligand. Nucleic Acids Res. 2018, 46, e115 10.1093/nar/gky607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerabek-Willemsen M.; Wienken C. J.; Braun D.; Baaske P.; Duhr S. Molecular Interaction Studies Using Microscale Thermophoresis. Assay Drug Dev. Technol. 2011, 9, 342–353. 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni P.; Landuzzi L.; Nicoletti G.; De Giovanni C.; Giovarelli M.; Lalli E.; Facchini A.; Lollini P. L. Control of H-2 Expression in Transformed Nonhaemopoietic Cells by Autocrine Interferon. Br. J. Cancer 1992, 66, 479–482. 10.1038/bjc.1992.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G.; Di Antonio M.; Tannahill D.; Balasubramanian S. Visualization and Selective Chemical Targeting of RNA G-Quadruplex Structures in the Cytoplasm of Human Cells. Nat. Chem. 2014, 6, 75–80. 10.1038/nchem.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowl J. T.; Gray E. E.; Pestal K.; Volkman H. E.; Stetson D. B. Intracellular Nucleic Acid Detection in Autoimmunity. Annu. Rev. Immunol. 2017, 35, 313–336. 10.1146/annurev-immunol-051116-052331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilger D.; Seymour L. W.; Jackson S. P. Interfaces between Cellular Responses to DNA Damage and Cancer Immunotherapy. Genes Dev. 2021, 35, 602–618. 10.1101/gad.348314.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvarlet J.; Bensadoun P.; Darbo E.; Labrunie G.; Rousseau B.; Richard E.; Draskovic I.; Londono-Vallejo A.; Dupuy J.-W.; Nath Das R.; Guédin A.; Robert G.; Orange F.; Croce S.; Valesco V.; Soubeyran P.; Ryan K. M.; Mergny J.-L.; Djavaheri-Mergny M. Modulation of the ATM/Autophagy Pathway by a G-Quadruplex Ligand Tips the Balance between Senescence and Apoptosis in Cancer Cells. Nucleic Acids Res. 2019, 47, 2739–2756. 10.1093/nar/gkz095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W. J.; Deng R.; Zhang X. Y.; Feng G. K.; Gu L. Q.; Zhu X. F. G-Quadruplex Ligand SYUIQ-5 Induces Autophagy by Telomere Damage and TRF2 Delocalization in Cancer Cells. Mol. Cancer Ther. 2009, 8, 3203–3213. 10.1158/1535-7163.mct-09-0244. [DOI] [PubMed] [Google Scholar]
- Gui X.; Yang H.; Li T.; Tan X.; Shi P.; Li M.; Du F.; Chen Z. J. Autophagy Induction via STING Trafficking Is a Primordial Function of the CGAS Pathway. Nature 2019, 567, 262–266. 10.1038/s41586-019-1006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner K.-P.; Hornung V. Molecular Mechanisms and Cellular Functions of CGAS–STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- Amato J.; Mashima T.; Kamatari Y. O.; Kuwata K.; Novellino E.; Randazzo A.; Giancola C.; Katahira M.; Pagano B. Improved Anti-Prion Nucleic Acid Aptamers by Incorporation of Chemical Modifications. Nucleic Acid Ther. 2020, 30, 414–421. 10.1089/nat.2020.0899. [DOI] [PubMed] [Google Scholar]
- Cantor C. R.; Warshaw M. M.; Shapiro H. Oligonucleotide Interactions. III. Circular Dichroism Studies of the Conformation of Deoxyoligonucleolides. Biopolymers 1970, 9, 1059–1077. 10.1002/bip.1970.360090909. [DOI] [PubMed] [Google Scholar]
- Amato J.; Madanayake T. W.; Iaccarino N.; Novellino E.; Randazzo A.; Hurley L. H.; Pagano B. HMGB1 Binds to the KRAS Promoter G-Quadruplex: A New Player in Oncogene Transcriptional Regulation?. Chem. Commun. 2018, 54, 9442–9445. 10.1039/c8cc03614d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.