

Abstract
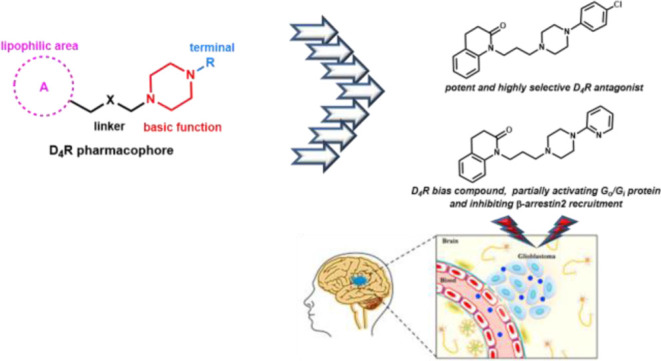
To better understand the role of dopamine D4 receptor (D4R) in glioblastoma (GBM), in the present paper, new ligands endowed with high affinity and selectivity for D4R were discovered starting from the brain penetrant and D4R selective lead compound 1-(3-(4-phenylpiperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (6). In particular, the D4R antagonist 24, showing the highest affinity and selectivity over D2R and D3R within the series (D2/D4 = 8318, D3/D4 = 3715), and the biased ligand 29, partially activating D4R Gi-/Go-protein and blocking β-arrestin recruitment, emerged as the most interesting compounds. These compounds, evaluated for their GBM antitumor activity, induced a decreased viability of GBM cell lines and primary GBM stem cells (GSC#83), with the maximal efficacy being reached at a concentration of 10 μM. Interestingly, the treatment with both compounds 24 and 29 induced an increased effect in reducing the cell viability with respect to temozolomide, which is the first-choice chemotherapeutic drug in GBM.
Introduction
Dopamine (DA) is a catecholamine neurotransmitter that mediates a wide variety of functions via binding with five dopamine receptor subtypes (DRs), belonging to class-A G protein-coupled receptor (GPCR) family. The binding site of DA is located in the extracellular region of DRs between the transmembrane (TM) helices. Based on structural characteristics, DRs are divided into two subfamilies, namely, D1-like receptors, comprising D1R and D5R, and D2-like receptors, including D2R, D3R, and D4R.1−4 After DA binding, D1-like receptors activate stimulatory G-proteins (Gαs/olf) and upregulate intracellular levels of adenosine 3′,5′-cyclic monophosphate (cAMP) by stimulating adenylyl cyclase (AC). Differently, D2-like receptors activate inhibitory G-proteins (Gαi/o) and downregulate the AC activity.5,6 Moreover, DRs have demonstrated to modulate other G-protein-dependent or -independent pathways, involving protein kinases, ion channels, phospholipases, and β-arrestins.4,7
Within the D2-like subfamily, D4R has recently emerged as an attractive target for the management of widespread diseases, including cancer, alcohol/substance use disorders, attention deficit hyperactive disorder, and eating disorders.8−10 This subtype is characterized by high polymorphism in the human genome2 and in particular, in the gene region codifying for the third intracellular loop (ICL3) of the receptor. Indeed, the ICL3 of D4R contains from 2- to 11-repeat forms of a 16-amino acid polypeptide, with the most common versions being 4-repeat (64%) followed by 7- and 2-repeat (21 and 8%, respectively). This polymorphism can influence the coupling of D4R to AC.4,9,11,12
D4R subtype is predominantly expressed in the central nervous system (CNS), especially in the frontal cortex, medulla, hippocampus, hypothalamus, pituitary gland, and amygdala.13,14 D4R expression is weak when compared to that of the other dopamine receptors,15 but its anatomical localization in the prefrontal cortex strongly indicates the role of this subtype in cognition and emotions. Moreover, neurobiological evidence suggest a possible relationship between D4R and glioblastoma (GBM)16,17 and particularly, D4R antagonists have proved to selectively inhibit GBM growth with a lower effect on the cell viability of normal neural stem cells. The D4R antagonists PNU 96415E (1) and L-741,742 (2) (Figure 1) have been demonstrated to disrupt the autophagy-lysosomal pathway specifically in GBM neural stem cells, inhibiting their survival and proliferation.17
Figure 1.
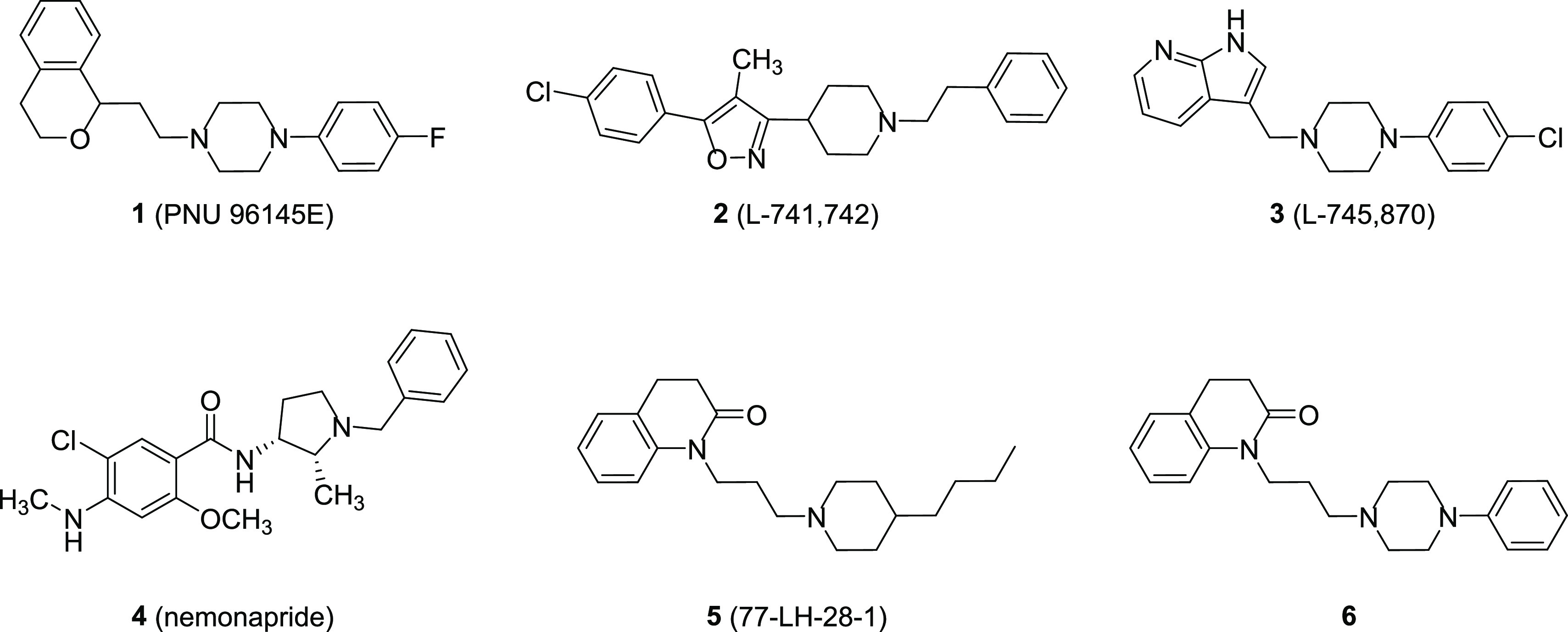
Chemical structures of compounds 1–6.
The resolved crystal structures of the complexes between D4R and the potent antagonist L-745,870 (3) (PDB ID = 6IQL)18 or the antipsychotic drug nemonapride (4) (PDB ID = 5WIU)19 (Figure 1) have greatly ameliorated the knowledge of the molecular mechanisms related to the D4R modulation.
We have recently demonstrated that the known M1 muscarinic bitopic agonist 77-LH-28-1 (5, Figure 1)20 also behaved as a potent D4R antagonist and showed an unexpected D4R selectivity with respect to D2R and D3R (pKi D2R = 6.17; D3R = 6.21; and D4R = 9.01).21 Compound 5 was taken as a starting point for a structure–activity relationship (SAR) study, which led to the discovery of its analogue 6 (Figure 1) characterized by a 4-phenylpiperazine group instead of the 4-butylpiperidine moiety of 5. Compound 6 maintained high affinity for D4R (pKi = 8.54) and showed high selectivity not only over D2R and D3R (selectivity ratio D2/D4 = 380 and D3/D4 = 457) but also over other receptors and transporters. In functional assays, it showed a biased profile behaving as a partial agonist for D4R-Gi protein activation and as an antagonist for β-arrestin recruitment. Moreover, it demonstrated to be highly brain penetrant in mice.
Therefore, due to its promising profile, in the present study, 6 has been chosen as a lead compound for the discovery of new potent and selective D4R ligands useful as pharmacological tools to better understand the role of D4R in GBM. In particular, maintaining the N-arylpiperazine moiety, a well-known scaffold of potent D4R ligands,8,10 including 1 and 3, the following modifications were designed: (i) replacement of the quinolinone portion with other bioisosteric nuclei (compounds 7–12, Figure 2), whose choice was inspired by D4-selective ligands known in the literature;8 (ii) replacement of the propyl linker with chains of different lengths (compounds 13–15, Figure 2), to evaluate the role of the distance between the basic function and the tetrahydroquinolinone nucleus; (iii) introduction of substituents with different electronic and lipophilic contributions in all combinations, such as CH3(+π, −σ), OCH3(−π, −σ), Cl(+π, +σ), and NO2(−π, +σ), in ortho-, meta-, and para-positions of the N-phenyl ring (compounds 16–27, Figure 2).22
Figure 2.
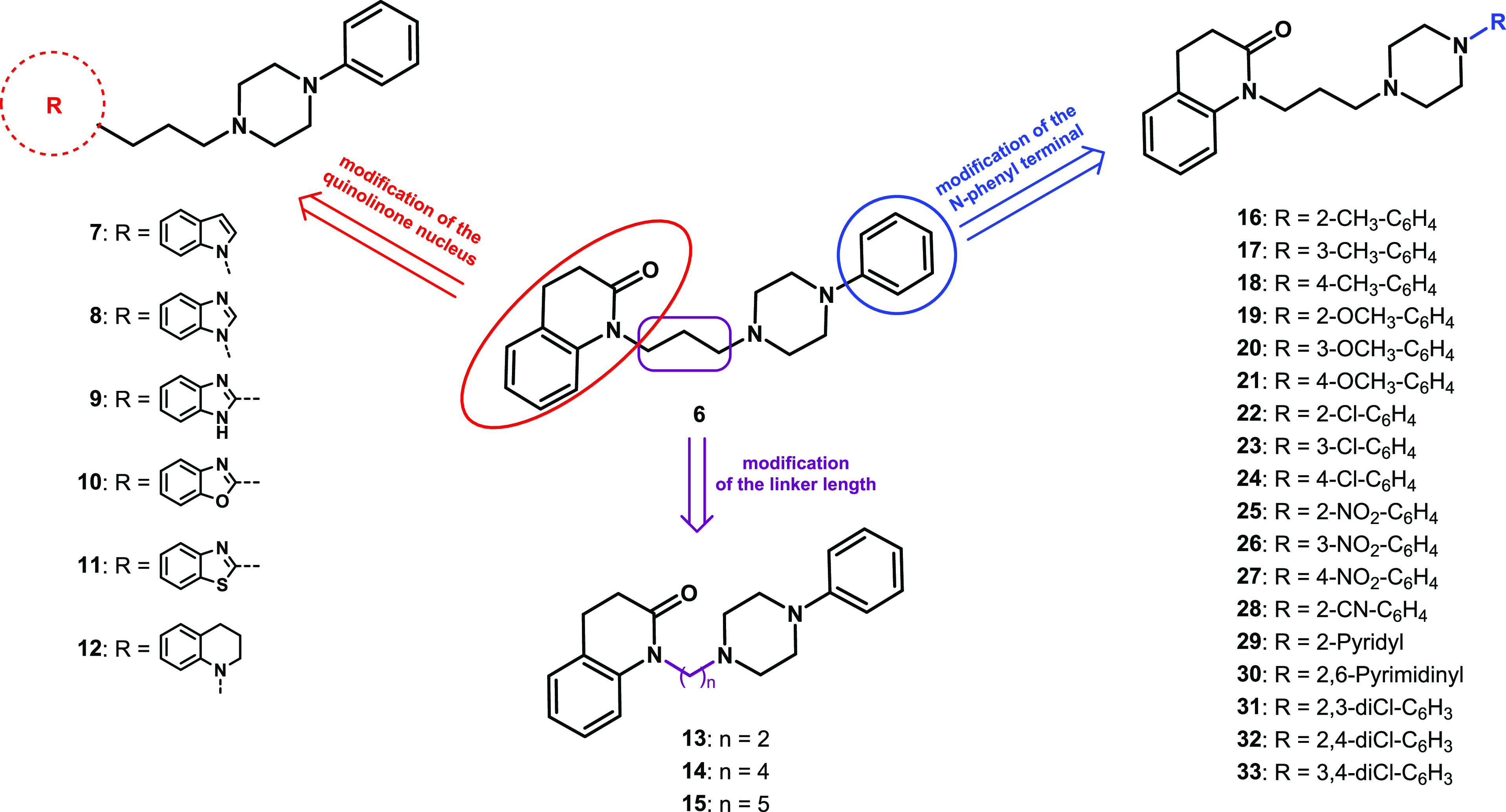
Modifications of the chemical structure of the lead compound 6 yielding derivatives 7–33.
Compounds 28–30 (Figure 2), bearing 2-cyanophenyl, 2-pyridyl, and 2-pyrimidinyl terminals, that are present in known potent and selective D4R ligands,8 as well as the 2,3-, 2,4-, and 3,4-dichlorophenyl derivatives 31–33 (Figure 2) were also prepared. All the compounds were evaluated for their affinity at D2R, D3R, and D4R by radioligand binding assays. Although compounds 7,238,2414,2519,2621,2623,2729,2630,26 and 31(26) had previously been reported in the literature, they had never been studied at D4R. The most selective D4R ligands were also tested for their functional activities by bioluminescence resonance energy transfer (BRET) assays to detect D4R G-protein activation and β-arrestin recruitment. The resolved crystal structure of the human D4R complexed with nemonapride (PDB Id: 5WIU)19 allowed to clarify the binding mode of the proposed derivatives and to support the SAR studies. Finally, the most interesting compounds were evaluated for their potential in affecting the viability of GBM cell lines and primary GBM stem cells (GSC#83).
Results and Discussion
Chemistry
Compounds 7–12 were prepared following the procedure reported in Scheme 1. The N-alkylation of the commercially available 1-phenylpiperazine 34 with 1,3-dibromopropane in the presence of potassium hydroxide afforded intermediate 35,28 which was treated with indole (36) or benzimidazole (37) in the presence of sodium hydride to give 7 and 8, respectively. The reaction of 34 with ethyl 4-bromobutanoate in the presence of sodium bicarbonate yielded intermediate 38,29 whose treatment with benzene-1,2-diamine led to derivative 9. The reaction between 34 and alkyl chlorides 39(30) or 40(31) in the presence of potassium carbonate and potassium iodide gave compounds 10 and 11, respectively. Amine 12 was prepared by reduction of the lead compound 6 with borane dimethyl sulfide complex.
Scheme 1. Synthesis of 7−12.

Reagents: (a) 1,3-dibromopropane, KOH, DMSO; (b) NaH, DMF; (c) ethyl 4-bromobutanoate, NaHCO3, EtOH; (d) benzene-1,2-diamine, 4M HCl in dioxane; (e) K2CO3, KI, DME; (f) BH3·S(CH3)2, THF.
Compounds 13–15 were prepared following the procedure reported in Scheme 2. The N-alkylation of 34 with 1-bromo-2-chloroethane in the presence of potassium carbonate afforded intermediate 42,32 which was reacted with the commercially available 3,4-dihydro-2(1H)-quinolinone 41 to give compound 13. The reaction of 41 with 1,4-dibromobutane or 1,5-dibromopentane in the presence of sodium hydride yielded intermediates 43(33) and 44,34 whose treatment with 34 in the presence of potassium carbonate led to derivatives 14 and 15, respectively.
Scheme 2. Synthesis of 13−15.

Reagents: (a) 1-bromo-2-chloroethane, K2CO3, acetone; (b) NaH, xylene; (c) 1,4-dibromobutane for 43 or 1,5-dibromopentane for 44, NaH, DMF; (d) 34, K2CO3, DMF.
The reaction of 41 with 1,3-dibromopropane in the presence of sodium hydride yielded intermediate 45,35 which was treated with suitable amines 46–63 in the presence of potassium carbonate to give derivatives 16–33, respectively (Scheme 3).
Scheme 3. Synthesis of 16−33.

Reagents: (a) 1,3-dibromopropane, NaH, DMF; (b) K2CO3, DMF.
Binding Studies
The pharmacological profile of compounds 7–33 as oxalate salts was evaluated by radioligand binding assays with human recombinant D2-like receptor subtypes stably expressed in HEK293T cells using the [3H]N-methylspiperone, a high-affinity D2-like antagonist, as radioligand to label DRs, following previously described protocols.36,37
D2R, D3R, and D4R affinity values, expressed as pKi, for ligands obtained by modifying the quinolinone nucleus (compounds 7–12), the linker (compounds 13–15), and the aromatic terminal (compounds 16–33) of the lead compound 6 are reported in Table 1 together with those of compounds 3, 5, and 6, included for useful comparison.
Table 1. Affinity Constants, Expressed as pKi,a of Compounds 3, 5–33 for Human Cloned D2LR, D3R, and D4.4R Expressed in HEK293T Cells.


pKi calculated from Ki values determined by competitive inhibition of [3H]N-methylspiperone binding in membranes harvested from HEK293 cells stably expressing hD2LR, hD3R, or hD4.4R. All values are presented as arithmetic mean ± SEM.
Calculated as a ratio between Ki values at D2R and D4R.
Calculated as a ratio between Ki values at D3R and D4R.
The analysis of the results highlights that, concerning the bioisosteric replacement of the tetrahydroquinolinone nucleus of 6, all the compounds show a slightly decreased D4R affinity, except for the N-indole 7 and N-tetrahydroquinoline 12, which maintain the high D4R affinity and selectivity of the lead. Moreover, although the N-benzimidazole derivative 8 binds D4R with lower affinity with respect to the lead compound 6, it shows higher D2/D4 and D3/D4 selectivity ratios (D2/D4 = 380 and D3/D4 = 457 for 6; D2/D4 = 977 and D3/D4 = 719 for 8).
The reduction of the linker length of compound 6, obtaining 13, causes a marked decrease in the binding affinity only at D4R (pKi D4R = 8.54 for 6 and pKi D4R = 7.20 for 13) and D2R (pKi D2R = 5.96 for 6 and pKi D4R = 4.89 for 13), with a consequent decrease in a D3/D4 selectivity ratio (D3/D4 = 457 for 6 and D3/D4 = 17 for 13). Differently, compound 14, the higher homologue of 6 obtained by inserting a methylene unit in the linker, maintains similar D4R (pKi D4R = 8.54 for 6 and pKi D4R = 8.37 for 14) and D2R (pKi D2R = 5.96 for 6 and pKi D2R = 5.95 for 14) affinity values but shows an increase in D3R affinity (pKi D3R = 5.88 for 6 and pKi D3R = 7.09 for 14). Therefore, also in this case, the D3/D4 selectivity ratio is reduced. Although compounds 13 and 14 show different affinities for D2R, D3R, and D4R subtypes, the D2/D4 and D3/D4 selectivity ratios are similar (D2/D4 = 204 and D3/D4 = 17, for 13 and D2/D4 = 263 and D3/D4 = 19 for 14). Further elongation of the linker, yielding 15, induces lower D4R affinity, with a consequent decrease of D2/D4 and D3/D4 selectivity ratios (12 and 8, respectively). Taken together, these results highlight that the propyl chain represents the optimal distance between the quinolinone nucleus and the basic function.
The presence of a substituent on the terminal phenyl ring of 6 markedly affects the D2-like affinity and selectivity profiles of the ligands. All the ortho-, meta-, and para-substituted derivatives show high D4R affinity. However, the derivatives 16–18 and 22–24, bearing substituents with +π values (CH3 and Cl), display similar pKi values at D4R regardless of their position on the phenyl ring, while substituents with -π values (OCH3 and NO2) confer to the ligands the highest affinity when they are in ortho positions (19 vs 20 and 21 and 25 vs 26 and 27). Interestingly, whatever the nature of the substituent is, the most D4R selective compounds are the para-substituted ones (18, 21, 24, and 27) (D2/D4 = 5248 and D3/D4 = 1738 for 18; D2/D4 = 3020 and D3/D4 = 1202 for 21; D2/D4 = 8318 and D3/D4 = 3715 for 24; and D2/D4 = 3631 and D3/D4 = 1660 for 27). The improved selectivity is due to the decrease in D2R and D3R affinity when the substituent is shifted from the ortho to meta- and, especially, to para-positions.
Considering that, among the para-substituted compounds, the best selectivity profile is shown by 4-chloro derivative 24, the influence of the dichlorophenyl disubstitution was probed by the synthesis and study of derivatives 31–33. The results confirm that the presence of a substituent in the para-position of the phenyl ring is detrimental for D2R and D3R binding affinity. Indeed, the ortho/para- and meta/para-disubstituted compounds 32 and 33 show D2/D4 and D3/D4 selectivity ratios significantly higher than those of the ortho/meta-disubstituted compound 31.
To extend the SARs concerning the aromatic terminal, the phenyl ring was replaced by other aromatic pendants, such as 2-cianophenyl (28), 2-pyridyl (29), and 2,6-pyrimidinyl (30) rings, which are also present in known potent and selective D4R ligands. Compounds 28–30 show D4R affinity values similar to that of the lead compound 6. Moreover, 29 and 30 exhibit a slight reduction in affinity for D2R and D3R subtypes and, consequently, are more selective for D4R with respect to 6. In particular, the 2-pyridyl derivative 29 shows the best selectivity profile (D2/D4 = 1230, D3/D4 = 1148).
It has been observed that ortho-, meta-, and para-regiosubstitutions on the terminal aryl ring might modulate efficacy at D4R of arylpiperazines.38,39 However, previously reported D4R partial or highly efficacious agonists demonstrated to bind more readily when in competition against an agonist radioligand (i.e., [3H]-7-OH-DPAT) instead of the classic antagonist [3H]N-methylspiperone. On the other hand, antagonists showed <10-fold difference in binding Ki, or almost no difference at all, independently from the radioligand used.39 Based on these observations, the D4R affinity of the ortho-, meta-, and para-chlorophenylpiperazines 22–24 has also been assessed using the agonist radioligand [3H]-7-OH-DPAT. All the compounds did not show any major shift in their pKi values when tested in the agonist-radioligand mode(22: pKi = 9.29 ± 0.06; 23: pKi = 9.11 ± 0.12; and 24: pKi = 8.83 ± 0.11) compared to the already reported affinity obtained with [3H]N-methylspiperone (22: pKi = 9.22 ± 0.04; 23: pKi = 8.98 ± 0.13; and 24: pKi = 9.18 ± 0.06), suggesting that they might behave as D4R antagonists.
Functional Assays
Based on their remarkable D4R affinity/selectivity profiles, compounds 18, 21, 24, 27, and 29 were selected to be evaluated for their functional activities in BRET-based assays at D4R. Unfortunately, 27 seemed to have an intrinsic light absorption property that interfered with BRET and, therefore, it was not possible to determine its functional profile. The potencies and efficacies, expressed as pEC50 (−log EC50) and Emax (maximum efficacy), respectively, of 18, 21, 24, and 29 are reported in Table 2 along with those of DA (D4R full agonist) and 3 (L745,870, D4R antagonist) as reference compounds.
Table 2. Potency (Expressed as pEC50a or pIC50a) and Efficacy Values (%a, Normalized to Dopamine Emax) of Dopamine (DA) and Compounds 3 (L745,870), 18, 21, 24, and 29 for D4R Expressed in HEK293T Cells.
|
Go activation (n ≥ 5) |
Gi activation (n ≥ 5) |
β-arrestin2 recruitment (n ≥ 5) |
||||
|---|---|---|---|---|---|---|
| pEC50 (pIC50) | Emax(Imax) | pEC50(pIC50) | Emax(Imax) | pEC50(pIC50) | Emax(Imax) | |
| DA | 7.83 ± 0.09 | 100 ± 2.8 | 7.68 ± 0.15 | 100 ± 5.1 | 6.57 ± 0.24 | 100 ± 5.7 |
| 3 | (6.84 ± 0.18) | (−75.4 ± 4.7) | (5.71 ± 0.3) | (−83.9 ± 16.3) | (6.79 ± 0.30) | (−89.4 ± 9.3) |
| 18 | ND (6.48 ± 0.20) | 0 (−91.6 ± 8.2) | ND (6.96 ± 0.66) | 0 (−53.2 ± 14.5) | 7.79 ± 1.39 (5.91 ± 0.26) | –30.2 ± 13.9 (−130 ± 15.1) |
| 21 | ND (6.97 ± 0.13) | 0 (−97.2 ± 5.1) | ND (6.57 ± 0.32) | 0 (−104.8 ± 7.3) | ND (6.41 ± 0.41) | 0 (−64.6 ± 9.4) |
| 24 | ND (6.42 ± 0.35) | 0 (−91.9 ± 15.0) | ND (6.60 ± 0.30) | 0 (−88.2 ± 11.7) | 7.29 ± 0.86 (4.98 ± 0.37) | –43.4 ± 13.8 (−144 ± 31.9) |
| 29 | 8.07 ± 0.13 (ND) | 46.2 ± 2.4 (0) | 7.91 ± 0.50 (ND) | 26.6 ± 5.4 (−20.2 ± 14.1) | ND (7.17 ± 0.27) | 0 (−89.4 ± 7.8) |
The values represent the arithmetic mean ± SEM. ND = cannot be determined.
In parallel, the presence of antagonist effects of the tested compounds was studied using a fixed amount of dopamine (1 μM) at D4R (Table 2, pIC50 and Imax). Because functionally selective compounds that exert preferential modulation on the G protein or β-arrestin are deemed to be therapeutically useful approaches, β-arrestin2 recruitment assays at D4R were also performed to characterize the functional properties of the ligands (Table 2, β-arrestin2 recruitment).
From the data analysis, it emerges that all the para-substituted compounds 18, 21, and 24 behave as antagonists toward both Gi-/Go-protein activation and β-arrestin recruitment. On the contrary, 2-pyridyl derivative 29 shows an interestingly biased profile, being a partial agonist with pEC50 values similar to those of dopamine toward D4R Gi-/Go-protein activation and an antagonist toward β-arrestin recruitment with inhibitory potency and maximal inhibition (Imax) similar to 3. These results confirm previous findings reporting that ligands with substituents in the para-position behave as antagonists and those with substituents in the ortho-position or bearing a 2-pyridine ring behave as partial agonists.8 The functional selectivity of 29 might be exploited to improve the knowledge of the biological functions associated with G-protein activation and β-arrestin recruitment pathways.
Molecular Modeling Studies
To better rationalize the reported D4R affinity values, docking simulations involving the resolved D4R structure in complex with nemonapride were performed by using PLANTS. Figure 3A shows the putative complex for 6 and reveals the key ion pair that the protonated piperazine elicits with Asp115 reinforced by the interaction with Tyr389. The quinolinone ring is engaged by a rich set of π–π stacking interactions with the surrounding aromatic residues (e.g., Trp358, Phe361, Phe362, and His365), which can also involve the lactam group. The key role of π–π stacking is confirmed by derivatives 7–11, in which the quinolinone moiety is replaced by bioisosteric heteroaromatic nuclei. The affinity of these bioisosters is indeed in good agreement with the calculated stacking interactions between heterocycles and aromatic residues (pyrrole > imidazole > tiazole > oxazole).40 Quinolinone is also involved in hydrophobic contacts with Leu187 and Val116, and this can explain the good binding affinity of 12. Lastly, the propyl linker elicits apolar contacts with Met112 and Val193, while the N-linked phenyl ring stabilizes π–π stacking with Phe91 and Trp101.
Figure 3.

Main interactions stabilizing the putative complexes of 6 within the binding sites of D4R (PDB id: 5WIU) (A) and D2R (PDB Id: 6CM4) (B). Focus on the interactions engaged by the substituted phenyl ring of 24 (C) and 25 (D) within the D4R.
The residues surrounding the phenyl ring can explain the different roles exerted by the added substituents. Specifically, hydrophobic and small substituents (i.e., methyl and chlorine groups) afford a positive contribution regardless of their position because they can always interact with the surrounding apolar residues without inducing steric constraints. As an example, Figure 3C focuses on the arrangement of the para-chloro derivative 24 in which the chlorine atom reinforces the hydrophobic contacts, which also involve Val87, Leu90, and Leu111, and can be engaged by a halogen bond with Ser94. Similar patterns of interactions are seen when the chlorine atom is in meta- or in ortho-positions. In contrast, polar and large substituents can be properly accommodated only in the ortho-position, where they can interact with the Ser94 without exerting steric clashes as exemplified by the ortho-nitro derivative 25 (Figure 3D). In meta- and in para-positions, the added substituents clash against Trp101, as well as against the backbone atoms of Leu90 and Phe91 which closely surround the ligand’s phenyl ring.
With a view to delve into the factors governing the ligand selectivity, similar docking simulations were performed by using the resolved D2R structure in complex with the high-affinity D2R antagonist risperidone. Figure 3B depicts the putative complex for 6 within the D2R binding site and emphasizes some differences with the corresponding complex with D4R (Figure 3A) that deserve further attention. The quinolinone ring is completely surrounded by aromatic residues and the more flexible alkyl side chains (as seen in D4R) have a marginal impact in this case. On the other side, the N-linked phenyl ring is also accommodated within a narrower subpocket (compared to D4R), which is lined by Trp100, Phe110, and Tyr408. This can explain why substituents on this ring generally have a detrimental role on the D2R affinity unless they can elicit H-bond with Tyr408 or Thr412 (as seen, e.g., with 19). More generally, the orthosteric D2R cavity appears to be smaller and narrower compared to the D4R pocket as clearly evidenced by the comparison of their void volumes as computed by FPocket (void volumes equal to 5694 and 4275 Å3 for D4R and D2R, respectively). This can explain why ligand modifications, that increase the steric hindrance or reduce the flexibility, enhance the D2/D4 selectivity. The flexibility role is noticeable by considering the positive correlation between the linker length and the D2R affinity (as observed for 13, 14, and 15).
Computational analyses were also employed to characterize the ADME/Tox profile of the studied compounds. Thus, Table S2 compiles some relevant physico-chemical descriptors for all the considered compounds. In detail, Table S2 reveals that all compounds show satisfactory physico-chemical profiles (e.g., MW < 500; logP <5; HBA <10; HBD <5; PSA <140 Å2; Rotors <10).41
The in silico ADME profile of compounds 24 and 29 was further investigated by interrogating the swissADME webserver.42 Compound 24 is predicted to be orally bioavailable, brain–blood barrier (BBB) permeant, P-gp substrate with no CYP inhibition apart from CYP2D6. The compound does not violate the most common druglikeness sets of rules (e.g., Lipinski, Ghose, and Veber) without PAINS and Brenk alerts. Its metabolic profile as predicted by the MetaClass method43 indicates that 24 can undergo red-ox reactions on nitrogen and Csp2 aromatic atoms. Compound 29 has an ADME profile almost superimposable to that of 24 except for being predicted BBB non-permeant, reasonably due to its lower lipophilicity.
Biological Studies in GBM Cell Lines
The D4R antagonist 24, showing the highest affinity and selectivity over D2R and D3R, and the ligand 29, showing a distinct biased profile, were selected to be evaluated for their potential in affecting the viability of the temozolomide-resistant T98 and temozolomide-sensitive U251 GBM cell lines,44 and the primary GBM stem cells GSC#83 as well. In particular, GSC and GBM cell lines were treated with the compounds 24 and 29 (from 5 to 50 μM) for 24h (experimental groups). Parallel cultures (control groups) were incubated for 24 h with temozolomide (Tocris), which is the first-choice chemotherapeutic drug in GBM, the known D4R receptor antagonist 1 (Tocris), the D4R agonist A412997 (Tocris) (all used at the concentrations ranging from 5 to 50 μM), or the only vehicle.
Dose–response studies show decreased GBM cell lines and GSC#83’s viability in cultures treated with both compounds 24 and 29, as well as with controls temozolomide and 1, with respect to the only vehicle incubated cultures. Conversely, the selective D4R agonist A412997 does not significantly modulate cell viability (Figure 4). The maximal efficiency of the compounds, both in the experimental and in the control groups, is reached at a concentration of 10 μM and, more importantly, the treatment with both compounds 24 and 29 induces an increased effect in reducing the T98, U251 cell lines, and GSC#83 viability with respect to the control drugs temozolomide and 1 (Figure 4). Moreover, the results confirm the higher sensitivity of T98 cells versus U251 cells to temozolomide treatment. On the contrary, both the GBM cell lines were equally sensitive in vitro to treatment with D4R compounds 24 and 29.
Figure 4.
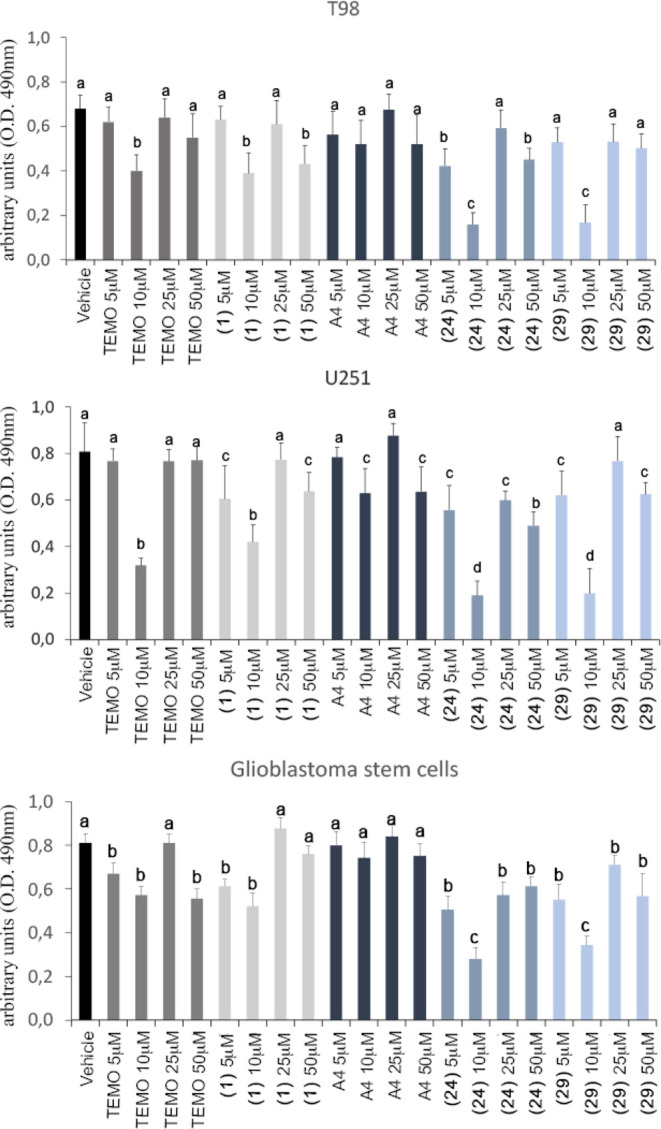
Cell viability assay performed in GBM T98 and U251 cell lines, and in GSC#83. Data were analyzed using two-way analysis of variance. Lowercase letters denote homogeneous subsets (n = 6, data shown are means ± standard error, p < 0.05). Vehicle = DMSO. TEMO = temozolomide. A4 = A412997.
Because all compounds show maximal antiproliferative activity at a dose of 10 μM, it has been considered of interest to evaluate their activity also in the narrower range of concentration from 10 to 20 μM. Moreover, because all the compounds show a similar activity against the three considered cell lines, the experiment was performed only on T98 cell line. Figure 5 shows that the maximal activity of all the tested compounds is further confirmed at a dose of 10 μM.
Figure 5.
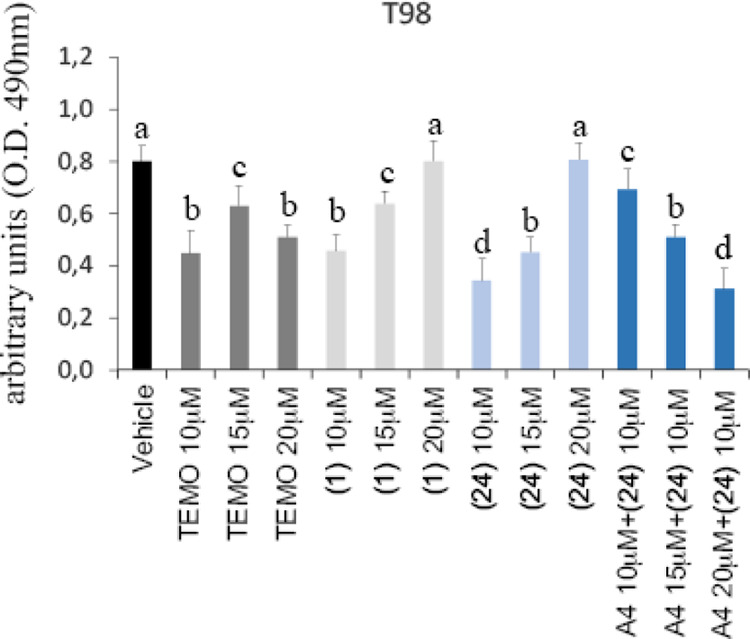
Cell viability assay performed in the T98 cell line. Data were analyzed using two-way analysis of variance. Lowercase letters denote homogeneous subsets (n = 6, data shown are means ± standard error, p < 0.05). Vehicle = DMSO. TEMO = temozolomide. A4 = A412997.
The fact that the maximal efficacy was found by using a medium-low dose of effectors tends to exclude the possibility of a non-specific/toxic effect, which usually occurs when high doses of stimulators are used.
Compound 24 was also tested at 10 μM concentration in the presence of increasing concentrations of the D4R agonist A412997 (Figure 5). The observation that the agonist at 10 μM contrasts the effect of 24 supports the hypothesis that D4R is involved in the antitumor activity of this compound. Analogous to what was observed with 24 and 29, the effect of A412997 decreases at higher doses (15 and 20 μM).
Conclusions
Starting from the brain penetrant and D4R selective lead compound 6, in the present study, new ligands endowed with high affinity and selectivity for D4R were discovered. In particular, maintaining the N-arylpiperazine moiety, the quinolinone portion was replaced by bioisosteric nuclei and the propyl linker by chains of different lengths. Moreover, substituents with different electronic and lipophilic contributions were inserted in ortho-, meta-, and para-positions of the N-aryl terminal. SAR studies, supported by molecular modeling simulations, highlighted that the tetrahydroquinolinone nucleus of 6 can be replaced by an N-indole or an N-tetrahydroquinoline moiety and the propyl linker represents the optimal distance between the lipophilic portion and the basic function. Interestingly, concerning the substitution in the aromatic terminal, the most D4R selective compounds were the para-substituted ones, due to the decrease in D2R and D3R affinity when the substituent is shifted from ortho-to meta- and especially to para-position. From functional studies, while the para-substituted compounds 18, 21, and 24 behaved as D4R antagonists, the 2-pyridyl derivative 29 showed an interestingly biased profile, being a partial agonist toward D4R Gi-/Go-protein activation and an antagonist toward β-arrestin recruitment. In particular, the antagonist 24, showing the highest affinity and selectivity for D4R over D2R and D3R, and the biased ligand 29 were evaluated for their GBM antitumor activity. They both induced a decreased viability of GBM cell lines and GSC#83, with the maximal efficacy being reached at a concentration of 10 μM. Interestingly, the treatment with both compounds 24 and 29 induces an increased effect in reducing the cell viability with respect to temozolomide, which is the first-choice chemotherapeutic drug in GBM. The observation that the effect of 24 is contrasted by the D4R agonist A412997 (10 μM) supports that D4R is involved in the antitumor activity of this compound.
Therefore, the new selective D4R ligands of the present paper might further shed light on the role played by this subtype in GBM and, especially, become lead compounds for the discovery of new alternatives to the standard treatments such as surgery and radiotherapy, that cannot always be applied, and pharmacological treatments, that are still very limited because of drug resistance.
Experimental Section
Chemistry
General
Melting points were taken in glass capillary tubes on a Büchi SMP-20 apparatus and are uncorrected. 1H NMR spectra were recorded either with a Bruker 500 Ascend (Bruker BioSpin Corporation, Billerica, MA, USA) and Varian Mercury AS400 instruments, and chemical shifts (ppm) are reported relative to tetramethylsilane. Spin multiplicities are given as s (singlet), d (doublet), dd (double doublet), t (triplet), or m (multiplet). IR spectra were recorded on a PerkinElmer 297 instrument and spectral data (not shown because of the lack of unusual features) were obtained for all compounds reported and are consistent with the assigned structures. The microanalyses were recorded on a FLASH 2000 instrument (ThermoFisher Scientific). The elemental composition of the compounds agreed to within ±0.4% of the calculated value. All reactions were monitored by thin-layer chromatography using silica gel plates (60 F254; Merck), visualizing with ultraviolet light. Chromatographic separations were performed on silica gel columns (Kieselgel 40, 0.040–0.063 mm, Merck) by flash chromatography. Compounds were named following IUPAC rules as applied by ChemBioDraw Ultra (version 12.0) software for systematically naming organic chemicals. The purity of the novel compounds was determined by combustion analysis and was ≥95%.
1-(3-(4-Phenylpiperazin-1-yl)propyl)-1H-indole (7)
A solution of 36 (1 mmol) in dimethyl formamide (5 mL) was added dropwise to a suspension of sodium hydride (0.04 g, 60% in mineral oil) and dimethyl formamide (5 mL). The resulting mixture was stirred at room temperature for 10 min, followed by the addition of a solution of 35 (1 mmol) in dimethyl formamide (5 mL). The resulting mixture was stirred at 60 °C for 20 h. Then, it was poured onto ice, and the aqueous phase was extracted with EtOAc (2 × 30 mL). The combined organic phases were washed with brine (5 × 30mL) and dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with cyclohexane/EtOAc (75:25). Oil was obtained (70% yield). 1H NMR (CDCl3, 500 MHz): δ 7.62 (d, 1H, J = 7.9 Hz), 7.39 (d, 1H, J = 8.2 Hz), 7.24–6.87 (m, 8H), 6.50 (d, 1H, J = 3.1 Hz), 4.26 (t, 2H, J = 6.6 Hz), 3.33–3.20 (m, 4H), 2.74–2.34 (m, 6H), 2.17–2.05 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a white solid: mp 99–100 °C, ESI/MS m/z: 320 [M + H]+, 342 [M + Na]+. Anal. Calcd (C21H25N3.C2H2O4) C, H, N.
1-(3-(4-Phenylpiperazin-1-yl)propyl)-1H-benzo[d]imidazole (8)
This compound was prepared starting from 37 and 35 following the procedure described for 7: oil was obtained (42% yield). 1H NMR (CDCl3, 500 MHz): δ 8.98 (s, 1H), 7.86 (d, 1H, J = 7.9 Hz), 7.64 (d, 1H, J = 8.0 Hz), 7.47–7.27 (m, 4H), 6.95 (t, 1H, J = 7.3 Hz), 6.89 (d, 2H, J = 8.1 Hz), 4.64 (t, 2H, J = 6.8 Hz), 3.60–3.07 (m, 8H), 2.73–2.64 (m, 2H), 2.25–2.06 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a white solid: mp 195–196 °C, ESI/MS m/z: 321 [M + H]+, 343 [M + Na]+. Anal. Calcd (C20H24N4.C2H2O4) C, H, N.
2-(3-(4-Phenylpiperazin-1-yl)propyl)-1H-benzo[d]imidazole (9)
Benzene-1,2-diamine (1 mmol) was added to a solution of 38 (1.2 mmol) in 4N HCl in dioxane (10 mL) and the mixture was stirred at reflux for 24 h. The reaction mixture was cooled to room temperature, poured over ice-cold H2O (20 mL), neutralized to pH = 7 with NaOH, and extracted with CHCl3 (3 × 20 mL). The organic phase was dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with EtOAc/MeOH (8:2). A yellow solid was obtained (25% yield). 1H NMR (CDCl3, 500 MHz): δ 7.59–7.15 (m, 6H), 6.99 (d, 2H, J = 7.8 Hz), 6.93 (m, 1H), 3.38–3.32 (m, 4H), 3.17–3.12 (m, 2H), 2.82–2.67 (m, 6H), 2.09–2.04 (m, 3H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a white solid: mp 216–219 °C, ESI/MS m/z: 321 [M + H]+, 343 [M + Na]+. Anal. Calcd (C20H24N4.C2H2O4) C, H, N.
2-(3-(4-Phenylpiperazin-1-yl)propyl)benzo[d]oxazole (10)
K2CO3 (5 mmol) and KI (0.2 mmol) were added to a solution of 34 (1 mmol) in DME (10 mL) and the mixture was stirred at room temperature for 10 min, followed by the addition of a solution of 39 (5 mmol) in DME (5 mL). The resulting mixture was stirred at reflux for 15 h. Then, after cooling, EtOAc (20 mL) was added, and the mixture was extracted with brine (3 × 20 mL). The organic phase was dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with cyclohexane/EtOAc (7:3). A white solid was obtained (59% yield). 1H NMR (CDCl3, 500 MHz): δ 7.72–7.26 (m, 6H), 6.96–6.85 (m, 3H), 3.21–3.15 (m, 4H), 3.04 (t, 2H, J = 7.5 Hz), 2.66–2.63 (m, 4H), 2.56 (t, 2H, J = 7.1 Hz), 2.18–2.12 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from MeOH to give a white solid: mp 215–216 °C, ESI/MS m/z: 322 [M + H]+, 344 [M + Na]+. Anal. Calcd (C20H23N3O.C2H2O4) C, H, N, O.
2-(3-(4-Phenylpiperazin-1-yl)propyl)benzo[d]thiazole (11)
This compound was prepared starting from 34 and 40 following the procedure described for 10: oil was obtained (11% yield). 1H NMR (CDCl3, 500 MHz): δ 7.97 (d, 1H, J = 8.1 Hz), 7.86–7.24 (m, 4H), 6.95–6.85 (m, 4H), 3.28–3.18 (m, 6H), 2.75–2.59 (m, 8H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a white solid: mp 184–186 °C, ESI/MS m/z: 338 [M + H]+, 360 [M + Na]+. Anal. Calcd (C20H23N3S.C2H2O4) C, H, N, S.
1-(3-(4-Phenylpiperazin-1-yl)propyl)-1,2,3,4-tetrahydroquinoline (12)
BH3·S(CH3)2 (0.34 mL) was added to a ice-cooled solution of 6 (1 mmol) in THF (10 mL) at 0 °C under nitrogen, and the mixture was stirred at reflux for 3 h. Then, after cooling to 0 °C, MeOH (10 mL) was added. The mixture was acidified with 2N HCl (5 mL) and stirred at reflux for 1 h. Then, it was cooled to room temperature, basified with 2N NaOH and extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were dried over anhydrous Na2SO4.The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with cyclohexane/EtOAc (7:3). Yellow oil was obtained (59% yield). 1H NMR (CDCl3, 500 MHz): δ 7.29–6.92 (m, 6H), 6.87 (t, 1H, J = 7.3 Hz), 6.62 (d, 1H, J = 8.2 Hz), 6.56 (t, 1H, J = 7.3 Hz), 3.36–3.22 (m, 8H), 2.79–2.47 (m, 8H), 1.98–1.84 (m, 4H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a white solid: m.p. 292–294 °C, ESI/MS m/z: 336 [M + H]+, 358 [M + Na]+. Anal. Calcd (C22H29N3.C2H2O4) C, H, N.
1-(2-(4-Phenylpiperazin-1-yl)ethyl)-3,4-dihydroquinolin-2(1H)-one (13)
Sodium hydride (0.12 g, 60% in mineral oil) was added to a solution of 41 (10 mmol) in xylene (5 mL), and the mixture was stirred at room temperature for 20 min, followed by the addition of a solution of 42 (5 mmol) in xylene (5 mL). The resulting mixture was stirred at reflux for 4 h. Then, after cooling, it was poured onto ice, and the organic phase was extracted with 5% HCl (3 × 20 mL). The aqueous phase was basified with 2N NaOH and extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were washed with brine (2 × 20 mL) and dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with EtOAc/CH3OH (99:1). Oil was obtained (76% yield). 1H NMR (CDCl3, 400 MHz): δ 7.60–6.95 (m, 9H), 4.04 (m, 2H), 3.74–3.62 (m, 6H), 3.32–3.10 (m, 4H), 2.96 (m, 2H), 2.68 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 210–211 °C, ESI/MS m/z: 336 [M + H]+, 358 [M + Na]+. Anal. Calcd (C21H25N3O.C2H2O4) C, H, N.
1-(4-(4-Phenylpiperazin-1-yl)butyl)-3,4-dihydroquinolin-2(1H)-one (14)
A solution of 43 (1 mmol) in DMF (5 mL) was added dropwise to a solution of 34 (1 mmol) and K2CO3 (1.2 mmol) in DMF (10 mL). The reaction mixture was stirred at 70 °C for 4 h; then, it was diluted with water (20 mL) and extracted with EtOAc (2 × 30 mL). The organic layer was washed with brine (5 × 20 mL) and dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with EtOAc/CH3OH (99:1). Oil was obtained (72% yield). 1H NMR (CDCl3, 400 MHz): δ 7.39–6.88 (m, 9H), 4.03 (m, 2H), 3.30 (m, 4H), 2.97–2.42 (m. 10H), 1.90–1.55 (m, 4H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 164–165 °C, ESI/MS m/z: 364 [M + H]+, 386 [M + Na]+. Anal. Calcd (C23H29N3O.C2H2O4) C, H, N.
1-(5-(4-Phenylpiperazin-1-yl)pentyl)-3,4-dihydroquinolin-2(1H)-one (15)
This compound was prepared starting from 44 and 34 following the procedure described for 14: oil was obtained (51% yield). 1HNMR (CDCl3, 400 MHz): δ 7.35–6.82 (m, 9H), 3.95 (m, 2H), 3.30 (m, 4H), 2.95 (m, 2H), 2.81–2.45 (m. 8H), 1.85–1.41 (m, 6H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 156–158 °C, ESI/MS m/z: 378 [M + H]+, 400 [M + Na]+. Anal. Calcd (C24H31N3O.C2H2O4) C, H, N.
1-(3-(4-(o-Tolyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (16)
This compound was prepared starting from 45 and 46 following the procedure described for 14: oil was obtained (47% yield). 1H NMR (CDCl3): δ 7.31–6.92 (m, 8H), 4.05 (m, 2H), 3.14 (m, 4H), 2.92 (m, 2H), 2.71–2.49 (m, 8H), 2.38 (s, 3H), 1.95 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 149–150 °C, ESI/MS m/z: 364 [M + H]+. Anal. Calcd (C23H29N3O.C2H2O4) C, H, N.
1-(3-(4-(m-Tolyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (17)
This compound was prepared starting from 45 and 47 following the procedure described for 14: oil was obtained (55% yield). 1H NMR (CDCl3, 500 MHz): δ 7.27–6.89 (m, 8H), 4.04 (m, 2H), 3.23 (m, 4H), 2.92 (m, 2H), 2.67 (m, 6H), 2.49 (m, 2H), 2.33 (s, 3H), 1.92 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 185–186 °C, ESI/MS m/z: 364 [M + H]+. Anal. Calcd (C23H29N3O.C2H2O4) C, H, N.
1-(3-(4-(p-Tolyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (18)
This compound was prepared starting from 45 and 48 following the procedure described for 14: oil was obtained (61% yield). 1H NMR (CDCl3, 500 MHz): δ 7.29–6.85 (m, 8H), 4.04 (m, 2H), 3.18 (m, 4H), 2.93 (dd, 2H, J = 18.4 and 11.5 Hz), 2.67 (m, 6H), 2.56 (t, 2H, J = 7.1 Hz), 2.29 (s, 3H), 1.92 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 198–199 °C, ESI/MS m/z 364 [M + H]+. Anal. Calcd (C23H29N3O.C2H2O4) C, H, N.
1-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (19)
This compound was prepared starting from 45 and 49 following the procedure described for 14: oil was obtained (46% yield). 1H NMR (CDCl3, 500 MHz): δ 7.31–6.88 (m, 8H), 4.07 (m, 2H), 3.79 (s, 3H), 3.22 (m, 4H), 2.94 (m, 2H), 2.76–2.35 (m, 8H), 1.95 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 164–166 °C, ESI/MS m/z: 380 [M + H]+. Anal. Calcd (C23H29N3O2.C2H2O4) C, H, N.
1-(3-(4-(3-Methoxyphenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (20)
This compound was prepared starting from 45 and 50 following the procedure described for 14: oil was obtained (49% yield). 1H NMR (CDCl3, 500 MHz): δ 7.26–6.42 (m, 8H), 4.04 (m, 2H), 3.81 (s, 3H), 3.23 (m, 4H), 2.92 (m, 2H), 2.71–2.61 (m, 6H), 2.50 (m, 2H), 1.91 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 153–154 °C, ESI/MS m/z: 380 [M + H]+. Anal. Calcd (C23H29N3O2.C2H2O4) C, H, N.
1-(3-(4-(4-Methoxyphenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (21)
This compound was prepared starting from 45 and 51 following the procedure described for 14: a white solid was obtained (46% yield): mp 98–99 °C. 1H NMR (CDCl3, 500 MHz): δ 7.25–6.81 (m, 8H), 4.01 (m, 2H), 3.77 (s, 3H), 3.10 (m, 4H), 2.92–2.58 (m, 8H), 2.49 (t, 2H, J = 7.2 Hz), 1.90 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 175–177 °C, ESI/MS m/z: 380 [M + H]+. Anal. Calcd. (C23H29N3O2.C2H2O4) C, H, N.
1-(3-(4-(2-Chlorophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (22)
This compound was prepared starting from 45 and 52 following the procedure described for 14: oil was obtained (58% yield). 1H NMR (CDCl3, 500 MHz): δ 7.41–6.92 (m, 8H), 4.07 (m, 2H), 3.19 (m, 4H), 2.94 (m, 2H), 2.73-2-55 (m, 8H), 1.94 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a white solid: mp 176–178 °C, ESI/MS m/z: 384 [M + H]+, 406 [M + Na]+. Anal. Calcd (C22H26ClN3O.C2H2O4) C, H, N.
1-(3-(4-(3-Chlorophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (23)
This compound was prepared starting from 45 and 53 following the procedure described for 14: oil was obtained (61% yield). 1H NMR (CDCl3, 500 MHz): δ 7.40–6.96 (m, 8H), 4.05 (m, 2H), 3.11 (m, 4H), 2.92 (m, 2H), 2.67 (m, 6H), 2.53 (t, 2H, J = 7.1 Hz), 1.91 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a white solid: mp 172–173 °C, ESI/MS m/z: 384 [M + H]+, 406 [M + Na]+. Anal. Calcd (C22H26ClN3O.C2H2O4) C, H, N.
1-(3-(4-(4-Chlorophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (24)
This compound was prepared starting from 45 and 54 following the procedure described for 14: oil was obtained (61% yield). 1H NMR (CDCl3, 500 MHz): δ 7.31–6.83 (m, 8H), 4.04 (m, 2H), 3.19 (m, 4H), 2.93 (dd, 2H, J = 19.1 and 12.3 Hz), 2.67 (m, 6H), 2.50 (t, 2H, J = 7.0 Hz), 1.90 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a white solid: mp 212–214 °C, ESI/MS m/z: 384 [M + H]+, 406 [M + Na]+. Anal. Calcd (C22H26ClN3O.C2H2O4) C, H, N.
1-(3-(4-(2-Nitrophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (25)
This compound was prepared starting from 55 following the procedure described for 14: oil was obtained (54% yield). 1H NMR (CDCl3, 500 MHz): δ 7.84–7.00 (m, 8H), 4.07 (m, 2H), 3.12 (m, 4H), 2.94 (m, 2H), 2.63 (m, 6H), 2.52 (m, 2H), 1.97 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a yellow solid: mp 172–173 °C, ESI/MS m/z: 395 [M + H]+, 417 [M + Na]+. Anal. Calcd (C22H26N4O3.C2H2O4) C, H, N.
1-(3-(4-(3-Nitrophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (26)
This compound was prepared starting from 45 and 56 following the procedure described for 14: oil was obtained (49% yield). 1H NMR (CDCl3, 500 MHz): δ 7.84–7.00 (m, 8H), 4.10 (t, 2H, J = 7.0 Hz), 3.73 (m, 6H), 3.21 (m, 2H), 2.94 (m, 2H), 2.69 (m, 2H), 2.40 (m, 2H), 1.97 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a yellow solid: mp 192–193 °C, ESI/MS m/z: 395 [M + H]+, 417 [M + Na]+. Anal. Calcd (C22H26N4O3.C2H2O4) C, H, N.
1-(3-(4-(4-Nitrophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (27)
This compound was prepared starting from 45 and 57 following the procedure described for 14: oil was obtained (49% yield). 1H NMR (CDCl3, 500 MHz): δ 8.15 (d, 2H, J = 9.5 Hz), 7.28–6.99 (m, 4H), 6.84 (d, 2H, J = 9.5 Hz), 4.05 (m, 2H), 3.46 (m, 4H), 2.92 (m, 2H), 2.70–2.62 (m, 6H), 2.51 (t, 2H, J = 7.0 Hz), 1.92 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a yellow solid: mp 206–207 °C, ESI/MS m/z: 395 [M + H]+, 417 [M + Na]+. Anal. Calcd (C22H26N4O3.C2H2O4) C, H, N.
2-(4-(3-(2-Oxo-3,4-dihydroquinolin-1(2H)-yl)propyl)piperazin-1-yl)benzonitrile (28)
This compound was prepared starting from 45 and 58 following the procedure described for 14: oil was obtained (67% yield). 1H NMR (CDCl3, 500 MHz): δ 7.58–7.00 (m, 8H), 4.04 (m, 2H), 3.26 (m, 4H), 2.92 (m, 2H), 2.67 (m, 6H), 2.53 (t, 2H, J = 7.1 Hz), 1.90 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from EtOH to give a pale yellow solid: mp 173–174 °C, ESI/MS m/z 375 [M + H]+, 397 [M + Na]+. Anal. Calcd (C23H26N4O.C2H2O4) C, H, N.
1-(3-(4-(Pyridin-2-yl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (29)
This compound was prepared starting from 45 and 59 following the procedure described for 14: oil was obtained (48% yield). 1H NMR (CDCl3, 500 MHz): δ 8.15 (dd, 1H, J = 5.0 and 1.5 Hz), 7.46–6.96 (m, 5H), 6.62–6.55 (m, 2H) 3.98 (m, 2H), 3.50–2.90 (m, 6H), 2.65–2.48 (m, 8H), 1.90 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 180–182 °C, ESI/MS m/z: 351 [M + H]+, 373 [M + Na]+. Anal. Calcd (C21H26N4O.C2H2O4) C, H, N.
1-(3-(4-(Pyrimidin-2-yl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (30)
This compound was prepared starting from 45 and 60 following the procedure described for 14: oil was obtained (62% yield). 1H NMR (CDCl3, 500 MHz): δ 8.32 (d, 2H, J = 4.7 Hz), 7.27–6.99 (m, 4H), 6.50 (t, 1H, J = 4.7 Hz), 4.04 (m, 2H), 3.84 (m, 4H), 2.94 (m, 2H), 2.67 (m, 2H), 2.50 (m, 6H), 1.90 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 181–182 °C, ESI/MS m/z: 352 [M + H]+, 374 [M + Na]+. Anal. Calcd (C20H25N5O.C2H2O4) C, H, N.
1-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (31)
This compound was prepared starting from 45 and 61 following the procedure described for 14: oil was obtained (44% yield). 1H NMR (CDCl3, 500 MHz): δ 7.28–6.93 (m, 7H), 4.02 (m, 2H), 3.09 (m, 4H), 2.89 (dd, 2H, J = 16.4 and 9.6 Hz), 2.66 (m, 6H), 2.52 (t, 2H, J = 7.2 Hz), 1.90 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 190–191 °C, ESI/MS m/z: 419 [M + H]+. Anal. Calcd (C22H25Cl2N3O.C2H2O4) C, H, N.
1-(3-(4-(2,4-Dichlorophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (32)
This compound was prepared starting from 45 and 62 following the procedure described for 14: oil was obtained (41% yield). 1H NMR (CDCl3, 500 MHz): δ 7.39–6.96 (m, 7H), 4.05 (m, 2H), 3.13 (m, 4H), 2.83–2.54 (m, 10H), 1.94 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 209–209 °C, ESI/MS m/z 419 [M + H]+. Anal. Calcd (C22H25Cl2N3O.C2H2O4) C, H, N.
1-(3-(4-(3,4-Dichlorophenyl)piperazin-1-yl)propyl)-3,4-dihydroquinolin-2(1H)-one (33)
This compound was prepared starting from 45 and 63 following the procedure described for 14: oil was obtained (41% yield). 1H NMR (CDCl3, 500 MHz): δ 7.32–6.73 (m, 7H), 4.04 (m, 2H), 3.22 (m, 4H), 2.92 (m, 2H), 2.70–2.44 (m, 8H), 1.91 (m, 2H). The free base was transformed into the oxalate salt, which was crystallized from 2-PrOH to give a white solid: mp 187–189 °C, ESI/MS m/z: 419 [M + H]+. Anal. Calcd (C22H25Cl2N3O.C2H2O4) C, H, N.
1-(3-Bromopropyl)-4-phenylpiperazine (35)
A solution of 34 (10 mmol) in DMSO (10 mL) was added dropwise to a solution of 1,3-dibromopropane (22 mmol) and KOH (0.6 g) in DMSO (30 mL) and the mixture was stirred for 4 h at 70 °C. Then, it was poured into absolute ethanol to precipitate a solid, which was filtered and rinsed with absolute ethanol three times. Evaporation of the solvent gave 35 as a pale yellow hygroscopic solid (71% yield). 1H NMR (CDCl3, 500 MHz): δ 7.26 (t, 2H, J = 8.7 Hz), 6.93 (d, 2H, J = 8.1 Hz), 6.85 (t, 1H, J = 6.9 Hz), 3.50 (t, 2H, J = 6.6 Hz), 3.20 (m, 4H), 2.62 (m, 4H), 2.55 (t, 2H, J = 7.5 Hz), 2.08 (m, 2H).
Ethyl 4-(4-Phenylpiperazin-1-yl)butanoate (38)
Ethyl 4-bromobutanoate (5.0 mmol) was added to the solution of 34 (5.0 mmol) in ethanol (20 mL) at room temperature and the resulting solution was stirred at reflux for 6 h. After the completion of reaction, the mixture was cooled to room temperature. Sat. NaHCO3 (50 mL) was added, and the resulting solution was extracted with CH2Cl2 (3 × 50 mL). The organic layer was dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with CH2Cl2/CH3OH (95:5). Oil was obtained (89% yield). 1H NMR (CDCl3, 500 MHz): δ 7.30–7.26 (m, 2H), 6.97–6.93 (m, 2H), 6.88 (t, 1H, J = 7.3 Hz), 4.18–4.13 (m, 2H), 3.26–3.21 (m, 4H), 2.69–2.63 (m, 4H), 2.49–2.39 (m, 4H), 1.93–1.86 (m, 2H), 1.31–1.26 (m, 3H).
1-(2-Chloroethyl)-4-phenylpiperazine (42)
1-Bromo-2-chloroethane 2 (7.2 mmol) was added dropwise to a solution of 34 (6.15 mmol) and K2CO3 (9.25 mmol) in acetone (10 mL). The reaction mixture was stirred under a nitrogen atmosphere for 15 h. Then, it was filtered, and the filtrate was concentrated under reduced pressure. The residue was diluted with water and extracted with EtOAc (3 × 50 mL). The organic layer was dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with cyclohexane/EtOAc (7:3). Oil was obtained (57% yield). 1H NMR (CDCl3, 400 MHz): δ 7.30–6.80 (m, 5H), 3.63 (t, 2H, J = 8.0 Hz), 3.21 (t, 4H), 2.79 (t, 2H, J = 8.0 Hz), 2.68 (m, 4H).
1-(4-Bromobutyl)-3,4-dihydroquinolin-2(1H)-one (43)
A solution of 41 (13.6 mmol) in DMF (10 mL) was added dropwise to a suspension of sodium hydride (0.54 g, 60% in mineral oil) and DMF (20 mL). The resulting mixture was stirred at room temperature for 20 min, followed by the addition of a solution of 1,4-dibromobutane (13.7 mmol) in DMF (10 mL). The resulting mixture was stirred at room temperature for 20 min. Then, it was poured onto ice, and the aqueous phase was extracted with EtOAc (2 × 30 mL). The combined organic phases were washed with brine (5 × 30mL) and dried over anhydrous Na2SO4. The evaporation of the solvent under reduced pressure afforded a residue, which was purified by flash chromatography, eluting with cyclohexane/EtOAc (7:3). Oil was obtained (84% yield). 1H NMR (CDCl3, 400 MHz): δ 7.23–6.96 (m, 4H), 3.93 (m, 2H), 3.40 (m, 2H), 2.85 (t, 2H, J = 8 Hz), 2.60 (t, 2H, J = 8 Hz), 1.97 (m, 2H), 1.77 (m, 2H).
1-(5-Bromopentyl)-3,4-dihydroquinolin-2(1H)-one (44)
This compound was prepared starting from 41 and 1,5-dibromopentane following the procedure described for 44: oil was obtained (89% yield). 1H NMR (CDCl3, 400 MHz): δ 6.96–7.23 (m, 4H), 3.95 (m, 2H), 3.40 (m, 2H), 2.91 (t, 2H, J = 7.8 Hz), 2.63 (t, 2H, J = 7.8 Hz), 1.94 (m, 2H), 1.68 (m, 2H), 1.52 (m, 2H).
1-(3-Bromopropyl)-3,4-dihydroquinolin-2(1H)-one (45)
This compound was prepared starting from 41 and 1,3-dibromopropane following the procedure described for 44: oil was obtained (56% yield). 1H NMR (CDCl3, 400 MHz): δ 7.30–6.99 (m, 4H), 4.11 (m, 2H), 3.50 (t, 2H, J = 6.5 Hz), 2.93 (m, 2H), 2.67 (dd, 2H, J = 8.5 and 6.6 Hz), 2.26 (m, 2H).
D2-like Radioligand Binding Assays
Membranes were prepared from HEK293 cells stably expressing human D2L, D3, or D4.4, grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM l-glutamine, 0.1 mM non-essential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37 °C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using pre-mixed Earle’s balanced salt solution (EBSS) with 5 mM EDTA (Life Technologies) and centrifuged at 3,000 rpm for 10 min at 21 °C. The supernatant was removed, and the pellet was resuspended in 10 ml hypotonic lysis buffer (5 mM MgCl2, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 20,000 rpm for 30 min at 4 °C. The pellet was then resuspended in the respective fresh binding buffers made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4 for the [3H]N-methylspiperone assay, or 50 mM Tris, 10 mM MgCl2, 1 mM EDTA, pH to 7.4 for the [3H]-(R)-(+)-7-OH-DPAT assay. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration, and membranes were either stored at −80 °C for later use (500 μg/ml for [3H]N-methylspiperone assay), or used fresh {∼500–600 μg/ml for [3H]-(R)-(+)-7-OH-DPAT assay}.
Radioligand competition binding experiments were conducted as previously described.36,37 Test compounds were freshly dissolved in 30% DMSO and 70% H2O to a stock concentration of 10 mM. Each test compound was then diluted into 10 half-log serial dilutions using a 30% DMSO vehicle. For the [3H]N-methylspiperone assay, previously frozen membranes were diluted in fresh EBSS to a 200 μg/mL (for D2 or D3) or 300 μg/mL (D4) stock for binding. Radioligand competition experiments were conducted in 96-well plates containing 300 mL fresh binding buffer, 50 mL of diluted test compound, 100 mL of membranes ([3H]N-methylspiperone: 20 μg/well total protein for D2 or D3, 30 μg/well total protein for D4; [3H]-(R)-(+)-7-OH-DPAT: ∼50–60 μg/well total protein concentration for D4), and 50 mL of [3H]N-methylspiperone (0.4 nM final concentration; Novandi Chemistry, SE) or [3H]-(R)-(+)-7-OH-DPAT (3 nM final concentration, Perkin Elmer), diluted in their respective binding buffer. Nonspecific binding was determined using 10 μM (+)-butaclamol (Sigma-Aldrich, St. Louis, MO) and total binding was determined with 30% DMSO vehicle (3% final concentration in the wells). All compound dilutions were tested in triplicate and the reaction was incubated for 1 h ([3H]N-methylspiperone) or 1.5 h ([3H]-(R)-(+)-7-OH-DPAT), at room temperature. The reaction was terminated by filtration through a PerkinElmer Uni-Filter-96 GF/B or GF/C, presoaked in 0.5% polyethylenimine for all the incubation time, using a Brandel 96-well plates Harvester manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 mL (3 × 1 mL/well) of ice-cold binding buffer. Then, 65 μL of PerkinElmer MicroScint 20 scintillation cocktail was added to each well, and filters were counted using a PerkinElmer MicroBeta microplate counter. The counter efficiency was experimentally determined for each radioligands, and aliquots of the radioligand dilutions were measured to quantify the exact amount of [3H] ligand added in each experiment. IC50 values for each compound were determined from dose–response curves, and Ki values were calculated using the Cheng–Prusoff equation. When a complete inhibition could not be achieved at the highest tested concentrations, Ki values have been extrapolated by constraining the bottom of the dose–response curves (=0% residual specific binding) in the nonlinear regression analysis. Kd values for both radioligands were determined via separate homologous competitive binding experiments. These analyses were performed using GraphPad Prism version 9.00 for Macintosh (GraphPad Software, San Diego, CA). Ki values were determined from at least three independent experiments and are reported as mean ± SEM.
BRET Assays
To perform BRET functional assays, human embryonic kidney cells 293T (HEK-293T) were transfected with constructs that include the donor enzyme RLuc8 (renilla luciferase variant) and the acceptor protein mVenus (yellow fluorescent variant) as a BRET pair fused to the respective proteins under study. In the G protein activation assays, the Gαi1 or GαoA subunit was fused to RLuc8 and the Gγ2 subunit to the mVenus. For the recruitment assays, β-arrestin was fused to mVenus and the D4 receptor was fused to RLuc8, as previously described.45 HEK293T cells were grown on 10 cm dishes in the DMEM culture medium supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine and 1% penicillin–streptomycin and transiently transfected with 15 μg total plasmid cDNA using 30 μg polyethyleneimine (Sigma-Aldrich) as a transfection agent with 6 h incubation terminated by the medium change. After 48 h, the transfected cells were washed, harvested, and resuspended in 1X PBS containing 0.1% glucose and 200 μM Na bisulfite. Approximately, 2 × 105 cells/well were distributed into 96-well plates (White Lumitrac 200, Greiner bio-one, Monroe, NC, USA) and 5 μM of the luciferase substrate, coelenterazine H, was added to each well. After 2min, the ligands were also transferred to each well. Antagonists were preincubated with the cells 10 min prior to the addition of ligands. Luminescence was measured at the RLuc8 wavelength (485 nm) and fluorescence at the m-Venus wavelength window (530nm), 2.5 min after ligands were added, using a PherastarFSX plate reader (BMG Labtech, Cary, NC, USA). The BRET ratio was expressed as the ratio of fluorescence and luminescence and the background determined in cells expressing RLuc8 alone was subtracted to obtain net BRET values. Generation of dose–response curves represented in drug-induced BRET ratios in response to the respective drugs as well as statistical analysis were performed using Prism 9 (GraphPad Software, San Diego, CA, USA).
Experimental Details of Modeling Studies
Docking simulations involved the resolved D4R structure in complex with nemonapride (PDB Id: 5WIU) as well as the resolved D2R structure in complex with risperidone (PDB Id: 6CM4). The protein structures were prepared as previously described.36,37 The ligands were simulated in their protonated state and their 3D structure was optimized by using the VEGA suite of programs.46 Docking simulations were performed by using PLANTS47 and focusing the searches within a 10 Å radius sphere around the co-crystallized ligand. For each molecule, 10 poses were generated by using the ChemPLP scoring functions and the speed parameter equal to 1. The so computed complexes were finally minimized and analyzed using ReScore+0.48 Authors will release the atomic coordinates upon article publication.
Experimental Details of Biological Studies in GBM Cell Lines
GBM Cell Lines and GSC Cultures
GBM cell lines T98 and U251 (grade IV) were obtained as previously described.49 Cells were grown until 80% of confluence in Eagle’s minimum essential medium (EMEM, Sigma, Merck Life Science S.r.l. Milano, Italy) plus 10% heat-inactivated fetal calf serum (HIFCS, Life Technologies, Monza, Italy), penicillin (100 U/mL), and streptomycin (50 μg/mL) in a humidified atmosphere of 5% CO2 at 37 °C. GSC#83 line previously characterized by Ricci-Vitiani et al.50 was isolated from a surgical sample of adult patients with a primitive brain tumor undergoing partial surgical resection at the Institute of Neurosurgery, Catholic University School of Medicine, in Rome, Italy. Patients were eligible for the study if a diagnosis of glioblastoma multiforme was established histologically according to the WHO classification.51 Informed consent was obtained before surgery according to the Ethical Committee of Catholic University School of Medicine. GSC culture was established from the tumor specimen through mechanical dissociation and culturing in DMEM/F12 serum-free medium containing 2 mM glutamine, 0.6% glucose, 9.6 g/mL putrescine, 6.3 ng/mL progesterone, 5.2 ng/mL sodium selenite, 0.025 mg/mL insulin, and 0.1 mg/mL transferrin sodium salt (Sigma-Aldrich, St. Louis, MO, USA), supplemented with EGF and bFGF. GSC line grown as floating spheres in serum-free medium supplemented with mitogens showed an undifferentiated state, as indicated by their rounded morphology, high nuclear/cytoplasm ratio. Human GSC#83 line was authenticated by short tandem repeat (STR) profiling according to the American National Standards Institute/American Type Culture Collection Standard ASN-0002-2011.12 using the Cell line Integrated Molecular Authentication database (CLIMA),13 and Cellosaurus STR database (CLASTR) of the Cellosaurus database (ExPASy) at the IRCC Ospedale Policlinico San Martino, Interlab Cell Line Collection (ICLC), Biological Resource Center (CRB-HSM), Genova, Italy.52
MTS Assay
T98 and U251 cell lines as well as GSC#83 were plated on 96 well culture plate at a density of 5,000 cells/well and grown as above described until 80% of confluence. Then, cells were treated with the following compounds: 24 and 29 at different concentrations starting from 5 μM to 50 μM diluted in DMSO (Sigma, Milano, Italy) for 24 h. Controls were performed by incubating the cultures for 24 h with different doses (ranging from 5 to 50 μM) of temozolomide, the D4R antagonists 1, the D4R agonist A412997, and with the only vehicle (DMSO). The next steps were performed as previously described.53 Briefly, cultures were incubated with 200 μL/well of CellTiter 96 Aqueous One Solution Reagent (Promega Italia srl, Milano, Italy) and the colored formazan product was measured by reading the absorbance at 490 nm using a 96-well plate reader (Tecan infinite multiplate reader).
Statistical Analysis
All the data were expressed as a mean ± standard error (s.e). Two-way analysis of variance (ANOVA) was used to compare the variables. The Tukey test was used in multiple comparisons among all groups. All the statistical analyses were performed using the GraphPad Prism (v 6.01) on a personal computer O.S. Windows 10. Data were presented as mean ± s.e. Values of P < 0.05 were considered significant.
Acknowledgments
This work was supported by grants from the University of Camerino (Fondo di Ateneo per la Ricerca 2019), and by the Intramural Research Program of the National Institute on Drug Abuse (NIDA-IRP).
Glossary
Abbreviations USED
- AC
adenylyl cyclase
- BBB
brain–blood barrier
- BRET
bioluminescence resonance energy transfer
- cAMP
adenosine 3′,5′-cyclic monophosphate
- CNS
central nervous system
- DA
dopamine
- DR
dopamine receptors
- GBM
glioblastoma
- GPCR
G protein-coupled receptor
- ICL3
third intracellular loop
- TM
transmembrane
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00840.
Author Contributions
○ A.S. and H.Y. equally contributed.
The authors declare no competing financial interest.
Supplementary Material
References
- Missale C.; Nash S. R.; Robinson S. W.; Jaber M.; Caron M. G. Dopamine Receptors: From Structure to Function. Physiol. Rev. 1998, 78, 189–225. 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Beaulieu J. M.; Gainetdinov R. R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Martel J. C.; Gatti McArthur S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. 10.3389/fphar.2020.01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu J. M.; Espinoza S.; Gainetdinov R. R. Dopamine Receptors - Iuphar Review 13. Br. J. Pharmacol. 2015, 172, 1–23. 10.1111/bph.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin J.; Fan T.; Guo P.; Wang J. Identification of Functional Divergence Sites in Dopamine Receptors of Vertebrates. Comput. Biol. Chem. 2019, 83, 107140. 10.1016/j.compbiolchem.2019.107140. [DOI] [PubMed] [Google Scholar]
- Vallone D.; Picetti R.; Borrelli E. Structure and Function of Dopamine Receptors. Neurosci. Biobehav. Rev. 2000, 24, 125–132. 10.1016/s0149-7634(99)00063-9. [DOI] [PubMed] [Google Scholar]
- Huff R. M.; Chio C. L.; Lajiness M. E.; Goodman L. V. Signal Transduction Pathways Modulated by D2-Like Dopamine Receptors. Adv. Pharmacol. 1998, 42, 454. 10.1016/s1054-3589(08)60786-3. [DOI] [PubMed] [Google Scholar]
- Giorgioni G.; Del Bello F.; Pavletic P.; Quaglia W.; Botticelli L.; Cifani C.; Micioni Di Bonaventura E.; Micioni Di Bonaventura M. V.; Piergentili A. Recent Findings Leading to the Discovery of Selective Dopamine D4 Receptor Ligands for the Treatment of Widespread Diseases. Eur. J. Med. Chem. 2021, 212, 113141. 10.1016/j.ejmech.2020.113141. [DOI] [PubMed] [Google Scholar]
- Botticelli L.; Micioni Di Bonaventura E.; Del Bello F.; Giorgioni G.; Piergentili A.; Romano A.; Quaglia W.; Cifani C.; Micioni Di Bonaventura M. V. Underlying Susceptibility to Eating Disorders and Drug Abuse: Genetic and Pharmacological Aspects of Dopamine D4 Receptors. Nutrients 2020, 12, 2288. 10.3390/nu12082288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley C. W.; Hopkins C. R. Return of D4 Dopamine Receptor Antagonists in Drug Discovery. J. Med. Chem. 2017, 60, 7233–7243. 10.1021/acs.jmedchem.7b00151. [DOI] [PubMed] [Google Scholar]
- Tol H. H.; Wu C. M.; Guan H. C.; Ohara K.; Bunzow J. R.; Civelli O.; Kennedy J.; Seeman P.; Niznik H. B.; Jovanovic V. Multiple Dopamine D4 Receptor Variants in the Human Population. Nature 1992, 358, 149–152. 10.1038/358149a0. [DOI] [PubMed] [Google Scholar]
- Asghari V.; Sanyal S.; Buchwaldt S.; Paterson A.; Jovanovic V.; Van Tol H. H. Modulation of Intracellular Cyclic AMP Levels by Different Human Dopamine D4 Receptor Variants. J. Neurochem. 1995, 65, 1157. 10.1046/j.1471-4159.1995.65031157.x. [DOI] [PubMed] [Google Scholar]
- Valerio A.; Belloni M.; Gorno M. L.; Tinti C.; Memo M.; Spano P. Dopamine D2, D3, and D4 Receptor mRNA Levels in Rat Brain and Pituitary During Aging. Neurobiol. Aging 1994, 15, 713–719. 10.1016/0197-4580(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Ariano M. A.; Wang J.; Noblett K. L.; Larson E. R.; Sibley D. R. Cellular Distribution of the Rat D4 Dopamine Receptor Protein in the CNS Using Anti-Receptor Antisera. Brain Res. 1997, 752, 26–34. 10.1016/s0006-8993(96)01422-9. [DOI] [PubMed] [Google Scholar]
- Jaber M.; Robinson S. W.; Missale C.; Caron M. G. Dopamine Receptors and Brain Function. Neuropharmacology 1996, 35, 1503–1519. 10.1016/s0028-3908(96)00100-1. [DOI] [PubMed] [Google Scholar]
- Rosas-Cruz A.; Salinas-Jazmín N.; Velázquez M. A. V.-. Dopamine Receptors in Cancer: Are They Valid Therapeutic Targets?. Technol. Cancer Res. Treat. 2021, 20, 1–13. 10.1177/15330338211027913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S.; Selvadurai H. J.; Lan X.; Lee L.; Kushida M.; Voisin V.; Whetstone H.; So M.; Aviv T.; Park N.; Zhu X.; Xu C.; Head R.; Rowland K. J.; Bernstein M.; Clarke I. D.; Bader G.; Harrington L.; Brumell J. H.; Tyers M.; Dirks P. B. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell 2016, 29, 859–873. 10.1016/j.ccell.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Cao C.; He L.; Wang X.; Zhang X. C. Crystal Structure of Dopamine Receptor D4 Bound to the Subtype Selective Ligand, L745870. ELife 2019, 8, e48822 10.7554/eLife.48822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Wacker D.; Levit A.; Che T.; Betz R. M.; McCorvy J. D.; Venkatakrishnan A. J.; Huang X. P.; Dror R. O.; Shoichet B. K.; Roth B. L. D4 Dopamine Receptor High-Resolution Structures Enable the Discovery of Selective Agonists. Science 2017, 358, 381–386. 10.1126/science.aan5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifazi A.; Yano H.; Del Bello F.; Farande A.; Quaglia W.; Petrelli R.; Matucci R.; Nesi M.; Vistoli G.; Ferre S.; Piergentili A. Synthesis and Biological Evaluation of a Novel Series of Heterobivalent Muscarinic Ligands Based on Xanomeline and 1-[3-(4-Butylpiperidin-1-yl)propyl]-1,2,3,4-tetrahydroquinolin-2-one (77-LH-28-1). J. Med. Chem. 2014, 57, 9065–9077. 10.1021/jm501173q. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Bonifazi A.; Giorgioni G.; Cifani C.; Micioni Di Bonaventura M. V.; Petrelli R.; Piergentili A.; Fontana S.; Mammoli V.; Yano H.; Matucci R.; Vistoli G.; Quaglia W. 1-[3-(4-Butylpiperidin-1-yl)propyl]-1,2,3,4-tetrahydroquinolin-2-one (77-LH-28-1) as a Model for the Rational Design of a Novel Class of Brain Penetrant Ligands with High Affinity and Selectivity for Dopamine D4 Receptor. J. Med. Chem. 2018, 61, 3712–3725. 10.1021/acs.jmedchem.8b00265. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Bonifazi A.; Giannella M.; Giorgioni G.; Piergentili A.; Petrelli R.; Cifani C.; Micioni Di Bonaventura M. V.; Keck T. M.; Mazzolari A.; Vistoli G.; Cilia A.; Poggesi E.; Matucci R.; Quaglia W. The Replacement of the 2-Methoxy Substituent of N-((6,6-diphenyl-1,4-dioxan-2-yl)methyl)-2-(2-methoxyphenoxy)ethan-1-amine Improves the Selectivity for 5-HT1A Receptor over α1-Adrenoceptor and D2-Like Receptor Subtypes. Eur. J. Med. Chem. 2017, 125, 233–244. 10.1016/j.ejmech.2016.09.026. [DOI] [PubMed] [Google Scholar]
- Yarim M.; Koksal M.; Schepmann D.; Wünsch B. Synthesis and in Vitro Evaluation of Novel Indole-Based Sigma Receptors Ligands. Chem. Biol. Drug Des. 2011, 78, 869–875. 10.1111/j.1747-0285.2011.01215.x. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Lirong Z.; Jie Z.; Xin Z.; Peng W.. Benzo Five-Membered Nitrogen Heterocyclic Piperidine or Piperazine Derivatives and Preparation Methods and Pharmaceutical Compositions Thereof. U.S. Patent 9,802,929 B2, 2015.
- Mokrosz J. L.; Duszyńska B.; Paluchowska M. H. Structure-Activity Relationship Studies of CNS Agents, XV: N-[omega-(4-aryl-1-piperazinyl)alkyl]-2-oxo-1,2,3,4-tetrahydroquinolines and -4-oxo-1,2,3,4-tetrahydropyrazino[1,2-a]indoles: New, Highly Potent 5-HT1A Ligands. Arch. Pharm. 1994, 327, 529–531. 10.1002/ardp.19943270811. [DOI] [PubMed] [Google Scholar]
- López L.; Selent J.; Ortega R.; Masaguer C. F.; Domínguez E.; Areias F.; Brea J.; Loza M. I.; Sanz F.; Pastor M. Synthesis, 3D-QSAR, and Structural Modeling of Benzolactam Derivatives with Binding Affinity for the D2 and D3 Receptors. ChemMedChem 2010, 5, 1300–1317. 10.1002/cmdc.201000101. [DOI] [PubMed] [Google Scholar]
- Oshiro Y.; Sakurai Y.; Sato S.; Kurahashi N.; Tanaka T.; Kikuchi T.; Tottori K.; Uwahodo Y.; Miwa T.; Nishi T. 3,4-Dihydro-2(1H)-quinolinone as a Novel Antidepressant Drug: Synthesis and Pharmacology of 1-[3-[4-(3-Chlorophenyl)-1-piperazinyl]propyl]-3,4- dihydro-5-methoxy-2(1H)-quinolinone and Its Derivatives. J. Med. Chem. 2000, 43, 177–189. 10.1021/jm980333v. [DOI] [PubMed] [Google Scholar]
- Santos M. A.; Marques S. M.; Tuccinardi T.; Carelli P.; Panelli L.; Rossello A. Design, Synthesis and Molecular Modeling Study of Iminodiacetyl Monohydroxamic Acid Derivatives as MMP Inhibitors. Bioorg. Med. Chem. 2006, 14, 7539. 10.1016/j.bmc.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Na Y. H.; Hong S. H.; Lee J. H.; Park W. K.; Baek D. J.; Koh H. Y.; Cho Y. S.; Choo H.; Pae A. N. Novel Quinazolinone Derivatives as 5-HT7 Receptor Ligands. Bioorg. Med. Chem. 2008, 16, 2570–2578. 10.1016/j.bmc.2007.11.049. [DOI] [PubMed] [Google Scholar]
- Sampson D.; Zhu X. Y.; Eyunni S. V.; Etukala J. R.; Ofori E.; Bricker B.; Lamango N. S.; Setola V.; Roth B. L.; Ablordeppey S. Y. Identification of a New Selective Dopamine D4 Receptor Ligand. Bioorg. Med. Chem. 2014, 22, 3105–3114. 10.1016/j.bmc.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X. Y.; Etukala J. R.; Eyunni S. V. K.; Setola V.; Roth B. L.; Ablordeppey S. Y. Benzothiazoles as Probes for the 5-HT1A Receptor and the Serotonin Transporter (SERT): A Search for New Dual-Acting Agents as Potential Antidepressants. Eur. J. Med. Chem. 2012, 53, 124–132. 10.1016/j.ejmech.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Hong K. H.; Ji M.; Cai J. Design, Synthesis, and Biological Evaluation of Structurally Constrained Hybrid Analogues Containing Ropinirole Moiety as a Novel Class of Potent and Selective Dopamine D3 Receptor Ligands. Chem. Biol. Drug Des. 2018, 92, 1597–1609. 10.1111/cbdd.13324. [DOI] [PubMed] [Google Scholar]
- Zampieri D.; Vio L.; Fermeglia M.; Pricl S.; Wünsch B.; Schepmann D.; Romano M.; Mamolo M. G.; Laurini E. Computer-Assisted Design, Synthesis, Binding and Cytotoxicity Assessments of New 1-(4-(Aryl(methyl)amino)butyl)-heterocyclic Sigma 1 Ligands. Eur. J. Med. Chem. 2016, 121, 712–726. 10.1016/j.ejmech.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Felding J.; Bang-Andersen B.; Smith G.; Paul; Andersen K.. Indole Derivatives Useful for the Treatment of CNS Disorders, ZA,200,209,958 B, 2002.
- Sams A. G.; Hentzer M.; Mikkelsen G. K.; Larsen K.; Bundgaard C.; Plath N.; Christoffersen C. T.; Bang-Andersen B. Discovery of N-{1-[3-(3-oxo-2,3-dihydrobenzo[1,4]oxazin-4-yl)propyl]piperidin-4-yl}-2-phenylacetamide (Lu AE51090): An Allosteric Muscarinic M1 Receptor Agonist with Unprecedented Selectivity and Procognitive Potential. J. Med. Chem. 2010, 53, 6386–6397. 10.1021/jm100697g. [DOI] [PubMed] [Google Scholar]
- Bonifazi A.; Newman A. H.; Keck T. M.; Gervasoni S.; Vistoli G.; Del Bello F.; Giorgioni G.; Pavletic P.; Quaglia W.; Piergentili A. Scaffold Hybridization Strategy Leads to the Discovery of Dopamine D3 Receptor-Selective or Multitarget Bitopic Ligands Potentially Useful for Central Nervous System Disorders. ACS Chem. Neurosci. 2021, 12, 3638–3649. 10.1021/acschemneuro.1c00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bello F.; Ambrosini D.; Bonifazi A.; Newman A. H.; Keck T. M.; Giannella M.; Giorgioni G.; Piergentili A.; Cappellacci L.; Cilia A.; Franchini S.; Quaglia W. Multitarget 1,4-Dioxane Compounds Combining Favorable D2-Like and 5-HT1A Receptor Interactions with Potential for the Treatment of Parkinson’s Disease or Schizophrenia. ACS Chem. Neurosci. 2019, 10, 2222–2228. 10.1021/acschemneuro.8b00677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart A. O.; Cowart M. D.; Moreland R. B.; Latshaw S. P.; Matulenko M. A.; Bhatia P. A.; Wang X.; Daanen J. F.; Nelson S. L.; Terranova M. A.; Namovic M. T.; Donnelly-Roberts D. L.; Miller L. N.; Nakane M.; Sullivan J. P.; Brioni J. D. Dopamine D4 Ligands and Models of Receptor Activation: 2-(4-Pyridin-2-ylpiperazin-1-ylmethyl)-1H-benzimidazole and Related Heteroarylmethylarylpiperazines Exhibit a Substituent Effect Responsible for Additional Efficacy Tuning. J. Med. Chem. 2004, 47, 2348–2355. 10.1021/jm0305669. [DOI] [PubMed] [Google Scholar]
- Keck T. M.; Free R. B.; Day M. M.; Brown S. L.; Maddaluna M. S.; Fountain G.; Cooper C.; Fallon B.; Holmes M.; Stang C. T.; Burkhardt R.; Bonifazi A.; Ellenberger M. P.; Newman A. H.; Sibley D. R.; Wu C.; Boateng C. A. Dopamine D4 Receptor-Selective Compounds Reveal Structure–Activity Relationships That Engender Agonist Efficacy. J. Med. Chem. 2019, 62, 3722–3740. 10.1021/acs.jmedchem.9b00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootsma A. N.; Doney A. C.; Wheeler S. E. Predicting the Strength of Stacking Interactions between Heterocycles and Aromatic Amino Acid Side Chains. J. Am. Chem. Soc. 2019, 141, 11027–11035. 10.1021/jacs.9b00936. [DOI] [PubMed] [Google Scholar]
- Matsson P.; Doak B. C.; Over B.; Kihlberg J. Cell Permeability Beyond the Rule of 5. Adv. Drug Del. Rev. 2016, 101, 42–61. 10.1016/j.addr.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. Swissadme: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzolari A.; Scaccabarozzi A.; Vistoli G.; Pedretti A. MetaClass, a Comprehensive Classification System for Predicting the Occurrence of Metabolic Reactions Based on the MetaQSAR Database. Molecules 2021, 26, 5857. 10.3390/molecules26195857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nifterik K. A.; van den Berg J.; van der Meide W. F.; Ameziane N.; Wedekind L. E.; Steenbergen R. D.; Leenstra S.; Lafleur M. V.; Slotman B. J.; Stalpers L. J.; Sminia P. Absence of the Mgmt Protein as Well as Methylation of the MGMT Promoter Predict the Sensitivity for Temozolomide. Br. J. Cancer 2010, 103, 29–35. 10.1038/sj.bjc.6605712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari P.; Xie B.; Semeano A.; Bonifazi A.; Battiti F. O.; Newman A. H.; Yano H.; Shi L. Chirality of Novel Bitopic Agonists Determines Unique Pharmacology at the Dopamine D3 Receptor. Biomolecules 2021, 11, 11. 10.3390/biom11040570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedretti A.; Mazzolari A.; Gervasoni S.; Fumagalli L.; Vistoli G. The Vega Suite of Programs: An Versatile Platform for Cheminformatics and Drug Design Projects. Bioinformatics 2021, 37, 1174–1175. 10.1093/bioinformatics/btaa774. [DOI] [PubMed] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with Plants. J. Chem. Inf. Model. 2009, 49, 84–96. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Vistoli G.; Mazzolari A.; Testa B.; Pedretti A. Binding Space Concept: A New Approach to Enhance the Reliability of Docking Scores and Its Application to Predicting Butyrylcholinesterase Hydrolytic Activity. J. Chem. Inf. Model. 2017, 57, 1691–1702. 10.1021/acs.jcim.7b00121. [DOI] [PubMed] [Google Scholar]
- Santoni G.; Amantini C.; Nabissi M.; Maggi F.; Arcella A.; Marinelli O.; Eleuteri A. M.; Santoni M.; Morelli M. B. Knock-Down of Mucolipin 1 Channel Promotes Tumor Progression and Invasion in Human Glioblastoma Cell Lines. Front. Oncol. 2021, 11, 578928. 10.3389/fonc.2021.578928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L.; Pallini R.; Biffoni M.; Todaro M.; Invernici G.; Cenci T.; Maira G.; Parati E. A.; Stassi G.; Larocca L. M.; De Maria R. Tumour Vascularization Via Endothelial Differentiation of Glioblastoma Stem-Like Cells. Nature 2010, 468, 824–828. 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- Kleihues P.; Cavenee W. K.. Pathology & Genetics. Tumours of the Nervous System. In World Health Organisation Classification of Tumours; IARC Press: Edinburgh, U.K., 2000, p 314. [Google Scholar]
- Visconti P.; Parodi F.; Parodi B.; Casarino L.; Romano P.; Buccarelli M.; Pallini R.; D’Alessandris Q. G.; Montori A.; Pilozzi E.; Ricci-Vitiani L. Short Tandem Repeat Profiling for the Authentication of Cancer Stem-Like Cells. Int. J. Cancer 2021, 148, 1489–1498. 10.1002/ijc.33370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agas D.; Amaroli A.; Lacava G.; Yanagawa T.; Sabbieti M. G. Loss of P62 Impairs Bone Turnover and Inhibits PTH-Induced Osteogenesis. J. Cell. Physiol. 2020, 235, 7516–7529. 10.1002/jcp.29654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.