

Abstract
Prokaryotic adaptive immune systems use Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) and CRISPR Associated (Cas) proteins to target and cleave foreign genetic elements in an RNA-guided manner[1–3]. Type VI CRISPR-Cas systems contain a single effector ribonuclease, Cas13, that binds and processes a CRISPR-RNA (crRNA; also known as a guide-RNA), forming an RNA-guided RNA-targeting effector complex[4, 5]. Previous studies have shown that Cas13 can be engineered to target and modulate RNA processes in human cells, illustrating the versatility and specificity of Cas13 as an RNA knockdown (KD), splicing, editing, or imaging tool[6–8]. While Cas13 has been successfully used by several groups, our lab has observed significant variability in Cas13 KD ability depending which protocol is being followed[9–12]. To further understand this variability and generate a robust Cas13 KD protocol we thoroughly tested which Cas13 ortholog to use, the duration of KD experiments, the amount of plasmid DNA transfected, methods for analyzing KD efficiency, and report an optimized method for carrying out and analyzing Cas13 mediated RNA KD experiments. The method outlined in this paper illustrates a faster and more reliable protocol to iteratively test gRNA performance and target gene KD.
Keywords: CRISPR, Cas13, RNA-targeting, RNA knockdown, Type VI CRISPR-Cas systems, RfxCas13d
Introduction
The implementation of CRISPR-Cas (Clustered Regular Interspersed Short Palindromic Repeats and CRISPR associated proteins) systems as nucleic-acid targeting reagents has quickly become an integral part of the molecular biology research toolkit. Unique to bacteria and archaea, CRISPR-Cas systems function to provide their hosts with adaptive immunity against foreign genetic elements, such as plasmids and bacteriophage[1–3]. Upon initial infection, specific Cas proteins recognize, degrade, and in some cases incorporate foreign RNA/DNA into the host genome at CRISPR arrays[1]. These arrays, composed of spacer and direct repeat sequences are transcribed to produce pre-crRNAs, which are processed into mature crRNAs and incorporated into effector protein complexes to recognize and degrade foreign RNA/DNA upon subsequent infection[1–3]. Divided into two general classes, CRISPR-Cas systems either utilize multiple proteins to process pre-crRNA and degrade target RNA/DNA in an effector complex (Class I) or a single effector protein with the ability to process pre-crRNA (in some cases requiring host factors) and degrade target RNA/DNA (Class II)[13]. These classes are then further divided into types based on the Cas proteins utilized in each system: Class I comprises Type I, III, and IV CRISPR-Cas systems while Class II comprises Type II, V, and VI systems[2, 13].
Several methods are available to target and modulate RNA expression, such as Antisense Oligonucleotides (ASOs), RNAi, and CRISPRi. Optimized ASO designs have high on-target KD efficiency, and are effective in treating diseases caused by dysregulation of splicing events, including Spinal Muscular Atrophy[14]. However, ASOs are very expensive due to the significant chemical modifications required and aren’t amenable to pooled screening approaches. RNAi technologies including miRNA and siRNA targeting have high KD efficiency and can be delivered via lentiviral vectors allowing for large-scale pooled screens, but are prone to significant off-target effects[15–18]. CRISPRi utilizes dCas9 fused to transcriptional activators or repressors to modulate gene expression. Although CRISPRi has high on target activity, regulating RNA at the DNA level does not provide a way to study transcripts produced from alternative splicing events, thereby limiting the applications of CRISPRi in RNA studies[19]. As a result, there is a continued need for more robust and highly specific RNA targeting tools.
Development of new RNA-targeting tools has become possible with the discovery and characterization of RNA-targeting CRISPR-Cas systems. Effector proteins in Type III, VI, and some Type II CRISPR systems naturally target RNA, and other Type II effectors have been engineered to target RNA[13, 20, 21]. Engineering the Type VI effector, Cas13, as an RNA targeting tool has become a particularly attractive option for many groups due to its versatile functions built into a single polypeptide. In native Type VI systems, hosts use Cas13, which has two HEPN domains that form a composite active site with crRNA:target RNA duplex gated RNase activity, to target and degrade foreign single stranded RNA (ssRNA)[2, 4–6]. Type VI systems can be further categorized into Subtypes A-D based on Cas13 phylogeny and contain Cas13a, Cas13b, Cas13c, and Cas13d, respectively. In addition to possessing crRNA-guided RNase activity, Cas13 contains an additional RNase active site to process pre-crRNAs into mature crRNAs[5–8, 22, 23].
Similar to Cas9 in mammalian cells, Cas13 generally exhibits highly specific binding and cleavage of its target substrates with negligible off-target effects[5–8, 24]. Across these studies, the RNA targeting ability of Cas13a, Cas13b, and Cas13d have been compared to each other and to shRNAs targeting the same transcript target sequence[6–8]. These studies reveal that all three Cas13 proteins are superior at specifically targeting RNA compared to shRNAs[25–27]. Furthermore, RfxCas13d (a Cas13d ortholog from Ruminococcus flavefaciens), was found to be the most potent of the three Cas13 proteins in knocking down target RNAs[8]. Although it is well known that RNA binding by Cas13 stimulates non-specific “collateral cleavage” activity in vitro and in bacteria, it generally exhibits negligible off-target cleavage activity in mammalian cells[28]. However, recently a growing body of evidence has emerged that indicates collateral still occurs under certain circumstances, specifically when targeting highly expressed endogenous genes or transgenes, and the effect appears to be threshold dependent[9–11, 29, 30], and this effect has also now recently been observed in E. coli[31]. Alternative RNA targeting CRISPR effector proteins, such as Cas7-11, have been shown to have less off-target effects due to collateral activity but it remains to be seen how widely adopted this new technology will become[32].
These inherent abilities of Cas13 have enabled Cas13 to be developed as a programmable RNA targeting tool, which has led to several key additions to the CRISPR toolbox. For example, Specific High-sensitivity Enzymatic Reporter unLOCKing (SHERLOCK) employs the use of Cas13 to target viral RNA and resulting in collateral cleavage of a small molecule fluorescent reporter that can be measured by a detection device[5, 33, 34]. SHERLOCK has been further optimized for greater sensitivity at lower concentrations of input material, multiplex detection, and easy lateral flow readout[24]. Multiplex platforms such as Combinatorial Arrayed Reactions for Multiplex Evaluation of Nucleic acids (CARMEN) employed the SHERLOCK technology to simultaneously test for hundreds of unique samples at a fraction of the cost without sacrificing sensitivity[35]. Nucleic acid detection with Cas13 has even been further developed to detect pathogenetic mutations in cell-free tumor DNA[36].
Catalytically inactive versions of the Cas13a-d isoforms have been engineered with fusion proteins to perform specific base editing of full-length transcripts containing pathogenic mutations. They have also been generally applied to a range of RNA binding protein applications, such as localization studies, transcription and translation modulation, and manipulating splicing events (for more extensive reviews see[28, 37–39]). Even smaller, more compact Cas13 orthologs, including Cas13bt, Cas13X, Cas13Y have been recently discovered and developed for RNA targeting applications, and although they appear to have diminished catalytic activity compared to that of the larger Cas13 proteins, further optimization is being performed to improve their performance[40, 41]. Other orthologs are being tested for clinical applications, for example, RfxCas13d (an ortholog of Cas13d from Rumminococcus flavefaciens) has been injected in mouse models to target disease-associated mRNAs and has shown promise in the treatment of neurological disease, cancer, and macular degeneration[42, 43].
Relatedly, there is increasing evidence that different Cas13 orthologs have different levels of RNA knockdown (KD) activity. For example, when using guide-RNAs (gRNAs) that target the same sequence, Cox et al. found that RanCas13b and PspCas13b exhibited superior KD efficiency compared to LbuCas13a and LwaCas13a, with RanCas13b eliciting the greatest level of target RNA KD[7]. Konermann et al. reported that RfxCas13d had the greatest level of target RNA KD, and greater than that previously reported in the literature for either Cas13a and Cas13b[8]. These same studies have shown that knocking out cleavage activity does not affect binding, showing Cas13 binding is independent of cleavage and that Cas13 binding activities can be harnessed and adapted as programmable RNA-binding tools.
But not all Cas13 proteins are built the same: some orthologs appear to perform better at RNA-binding applications, while others perform better at RNA KD and diagnostics. For example, Yang et al. and Wang et al. showed through fluorescent localization studies targeting the paraspeckle-localized RNA NEAT1 that PspCas13b performs better than RfxCas13d. Specifically, RfxCas13d seems to bind NEAT1 regardless of whether a targeting or non-targeting gRNA were used, whereas PspCas13b does not exhibit this same behavior and can be clearly localized to paraspeckles in specific gRNA-dependent manner[44, 45]. As an RNA targeting tool in other cell types, RfxCas13d has been found to be less toxic at similar concentrations to other Cas13 proteins while achieving greater KD activity, supporting the choice of RfxCas13d in RNA KD experiments [46].
Initial studies exploring gRNA design have set the foundation for optimization of RNA targeting with Cas13. The first algorithm for Cas13 gRNA design published used a random forest model and a set of 22 features to accurately predict highly active gRNAs [47]. Although several predicted secondary structure parameters were included in this algorithm and appeared to help the model select efficient gRNAs, experimental data on the effect of secondary structure on Cas13 targeting efficiency still needed to be explored. To this end, Tatsuo et al. experimentally tested the impact of secondary structure on Cas13 mediated RNA cleavage. They found that optimal cleavage was observed when targeting single stranded regions of the target RNA[48]. Certain chemical modifications to Cas13 gRNAs have also been found to increase targeting and transcript KD in human cells [49]. Since these initial studies, additional gRNA design studies have been conducted, providing the scientific community with additional tools that can predict Cas13 RNA KD performance across many different targets. For example, Guo et al. built upon the initial work described above, performing expanded screens in six model organisms and four viral families, targeting all types of RNAs to enhance the ability of their web application predicting highly efficient gRNAs [50]. More recently, Wessels et al. combined Cas13d KD and the ability of Cas13 to process its own gRNA with single cell pooled gRNA sequencing approaches to make combinatorial perturbations at the RNA level[51].
Beyond this work, several other gRNA selection tools have been developed. Recently, Wei et al. developed a comprehensive gRNA selection model incorporating sequence and structural information to determine optimal gRNA design. They employed a positive selection screen, tiling 55 known essential genes with a total data set of over 127,000 gRNAs. They then trained a convolutional neural network that can predict gRNA performance using the gRNA sequence alone. This study also incorporated secondary structure, free energy, target site position and isoform abundance. They also evaluated feature contributions so the user knows the contribution of each feature in impacting overall gRNA KD performance [52]. Similarly, Krohannon et al. developed CASowary, a gRNA prediction tool that harnessed a screen of gRNAs targeting RNAs of 5000 random genes. They considered features including k-mer composition, gRNA and target sequence composition, gRNA position, target RNA position, and secondary structure of the target RNA [53]. Other groups have been working towards optimizing high fidelity Cas13 variants for RNA targeting, reducing trans or collateral cleavage while maintaining high levels of on-target activity compared to wild type Cas13s[54]. This is critical in recognizing that elements of both components of the effector binary complex can be improved to achieve efficient and specific target RNA KD.
Despite all this exciting progress in Cas13 applications development, there is still much that we do not understand about how to consistently and successfully employ Cas13 in mammalian RNA targeting experiments. This study explores a range of experimental conditions for performing Cas13 RNA KD experiments with the aim of providing a clear protocol for scientists to obtain consistent RNA KD in their experiments. First, we validate which Cas13 ortholog performs the most efficient target RNA KD. Second, in transient transfection experiments, we show that by reducing the amount of plasmid DNA by nearly half of what is commonly used in published experiments, we are still able to achieve optimal KD efficiency without affecting cell morphology and growth. Further, by transfecting the cells for twice the amount of time (compared to previous reports) using a mild transfection reagent, we improve Cas13’s KD performance. We validated the gRNA design algorithm designed by Wessels et al. and determined it was sufficient to design highly efficient gRNAs [47]. Lastly, we confirm that measuring protein expression by flow cytometry is a fast and easy way to scale up experiments to multiplex gRNAs and/or targets when targeting mRNAs that encode for a protein whose expression can be measured by flow cytometry. Together with all these observations, we provide an updated, easy-to-follow Cas13 KD protocol that can be used by scientists wanting to optimize Cas13 KD experiments in their lab.
Materials
Trypsin-EDTA (0.25%), phenol red (Gibco Cat# 25200056)
Dulbecco’s Modified Eagle’s Medium (Lonza Cat# 12-614F) and Premium Fetal Bovine Serum Volume 500 ml (Biowest Cat# S1620)
JetPrime transfection reagent (Polyplus Cat# 101000027)
TRIzol reagent (Cat# 15596026)/TRIreagent (Molecular Research Center Cat# TR118)
ACS Chloroform (VWR Analytical Product Cat# BDH1109-4LG)
GlycoBlue Coprecipitant (Thermofisher Cat# AM9515)
Quant-iT RNA Assay, Broad Range (Invitrogen by Thermo Fisher Scientific Cat# Q10213)
Hi Capacity cDNA Reverse Transcription Kit (Applied Biosystems by Thermo Fisher Scientific Cat# 4368814)
Omega Bio-Tek E.Z.N.A. Plasmid DNA Mini Kit I (Cat# D6942-02)
PE: anti-human CD46 Clone: TRA-2-10 (Biolegend Cat# 352402)
PBS without Calcium or Magnesium (Lonza Cat # 17-516F)
HEK293T cells (ATCC CRL-3216)
Calf Intestinal Phosphatase (CIP) (NEB Cat# M0290)
Shrimp Alkaline Phosphatase (rSAP) (NEB Cat# M0371S)
T4 DNA Ligase (NEB Cat# M0202S)
T4 Polynucleotide Kinase (PNK, NEB Cat# M0201S)
100 nM Sodium Pyruvate (NaPy) (Gibco Cat#11360070)
200 nM L-Glutamine (L-Glut) (Gibco Cat# 25030081)
100 ml Penicillin-Streptomycin (PenStrep Gibco Cat# 15140-122)
Applied Biosystems TaqMan Fast Advanced Master Mix (Cat# 4444554)
IDT PrimeTime Gene Expression Master Mix (5 ml, Cat# 1055772)
Applied Biosystems MicroAmp Optical 384-well Reaction plate with Barcode (Cat# 4309849)
96-well tissue culture treated plates (flat bottom, round bottom, v-bottom, Fisherbrand)
Equipment
BD Accuri flow cytometer
Quantstudio5 qPCR thermocycler
Nanodrop spectrophotometer
Tecan (or any fluorescence capable) plate reader
Eppendorf (or any) centrifuges
Chemidox imaging system (BioRad)
Eppendorf (or any) thermocycler
Incubator for mammalian tissue culture
Incubator for E. coli culture
2. Methods
2.1. Generation of gRNAs using modified Gateway Cloning procedure
-
Design oligos to clone the gRNA-targeting sequence into gRNA expression backbone (pC0041 for RanCas13b, pC0043 for PspCas13b, and pXR003 for RfxCas13d; see Tables 1 and 2). For example, for RanCas13b or PspCas13b gRNAs the following two oligos were synthesized in the form:
positive strand oligo: 5’–CACCGNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN– 3’
negative strand oligo: 3’–CNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCAAC– 5’- For cloning gRNA for RanCas13b or PspCas13b, CACC flanking sequence is required for directional ligation into the gRNA vector and the G is required for efficient RNA polymerase III transcription. N in the positive strand oligo is the spacer sequence and N in the negative strand oligo is the reverse complement of this sequence (i.e. to form a base-paired dsDNA duplex with the positive strand oligo). The 5′-CAAC-3′ end in the negative stranded oligo is required for directional ligation. The C at the other end is the complement of the G required for RNA polymerase III transcription.
- For cloning gRNAs for RfxCas13d, an AAAC flanking sequence is required for directional ligation into the gRNA vector and the G is required for efficient RNA polymerase III transcription. A 5’-AAAA-3’ flanking sequence in the negative strand oligo is required for directional ligation. The C at the other end is the complement of the G required for RNA polymerase III transcription.
- Make sure to reverse the negative strand above before ordering, as it is depicted in the 3’-5’ direction above and needs to be ordered in the 5’ -3’ direction.
- 30-nt. spacers were used for Cas13bs and 23-nt. spacer for RfxCas13d
- Reconstitute lyophilized oligos to a final concentration of 100 μM.
- Digest at least 1 μg of gRNA vector with BbsI (or other Type IIS restriction enzyme depending on the plasmid being used) for at least 1hr at 37°C. One can omit this step if already have stock of cut vector.
- >1 ug of vector
- 1 μl BbsI (NEB) (or less ;~2-4 units of enzyme is required for an efficient reaction)
- 2 μl 10X Cutsmart buffer (for final 1x concentration)
- X μl MilliQ water (up to 20 μl)
- 20 μl total
- Add 1 μl of Calf Intestinal Phosphatase (CIP) or Shrimp Alkaline Phosphatase (rSAP) for last 1 hour of digestion reaction
- This will dephosphorylate the 5’-phosphates of the digested vector to prevent re-ligating of incompletely cleaved single cut empty vector during the initial ligation step. This step is optional but can help improve the efficiency in the overall ligation reaction if you’re having background re-ligation issues. Also see below in Step 5 regarding phosphorylation of oligos.
Purify the cut vector using a PCR purification kit (e.g. Omega E.Z.N.A Cycle Pure Kit #D6492-01) or other methods used in your laboratory (e.g. magnetic beads etc.)
- Phosphorylate and anneal each pair of oligos (each pair of oligos = one unique spacer sequence)
- 1 μl oligo_positive (100 μM stock concentration)
- 1 μl oligo_negative (100 μM stock concentration)
- 1 μl 10X T4 ligation buffer (NEB, use this buffer instead of PNK buffer because it contains ATP)
- 6.5 μl MilliQ water
- 0.5 μl T4 Polynucleotide Kinase (PNK) (NEB)
- 10 μl total
Incubate at 37°C for 30 mins
- Incubate at 95°C for 5 min, remove metal block from block heater and leave to cool to approximately RT for 30 mins. Alternatively, set up steps 4 and 5 in a thermocycler and for step 5 after the 5 min at 95°C ramp down to 25 °C at a rate of 5 °C/min.
- This step is not required if the CIP treatment is also omitted (Step 3).
- Set up the ligation reaction. Add the following together (we also recommend doing a ‘no-insert’ control to check for vector background ligation)
- X μl cut vector from step one (add 50 ng)
- 1 μl 1:200 dilution of the phosphorylated and annealed oligos from step three.
- 1 μl 10X T4 ligase buffer (NEB)
- 1 μl T4 DNA ligase (NEB)
- MiliQ water up to 10 μl
- 10 μl total
Incubate for 30 min at RT
- Transform into competent MachOne or DH5 alpha E. coli as per lab instructions (or any other cloning strain of choice can be used)
- These vectors are AmpR
- For the Cas13-expressing plasmids, which use a lentiviral backbone, it’s advisable to use cloning strains that have been optimized for maintaining repeat elements (e.g. NEB Stable or Invitrogen Stbl3 cell lines).
Miniprep from individual clones, determine concentration by nanodrop spectrophotometry and send for Sanger sequencing to confirm proper insertion of the spacer sequence. A standard U6 sequencing primer is often available from sequencing providers.
Table 1.
Expression Plasmids Used
| Plasmid Name/Addgene code | Gene Expressed |
|---|---|
| pC0044-EF1a-RanCas13b-NES-mapk (Addgene cat. #103855) | RanCas13b-NLS |
| pC0046-EF1a-PspCas13b-NES-HIV (Addgene cat. #103862) | PspCas13b-NLS |
| pXR001-EF1a-CasRX-2A-EGFP (Addgene cat. #109049) | RfxCas13d-NLS |
| pC0041-RanCas13b crRNA backbone (Addgene cat. # 103852) | RanCas13b gRNA expression plasmid |
| pC0043-PspCas13b crRNA backbone (Addgene cat. # 103854) | PspCas13b gRNA expression plasmid |
| pXR003-RfxCas13d gRNA cloning backbone (Addgene cat. # 109053) | RfxCas13d gRNA expression plasmid |
Table 2.
gRNA expression plasmids, sequences, and genes targeted
| gRNA expression plasmid | gRNA Sequence (5’-3’) | Gene targeted | gRNA | Figure |
|---|---|---|---|---|
| pC0041 | GTATTATTTATGGCAAATACACAAAGAAAG | KRAS | 9 | 1B |
| pC0041 | GCAGGGTTTTCCCAGTCACGACGTTGTAAAA | NT | NT | 1B |
| pC0043 | GTATTATTTATGGCAAATACACAAAGAAAG | KRAS | 9 | 1B |
| pC0043 | GCAGGGTTTTCCCAGTCACGACGTTGTAAAA | NT | NT | 1B |
| pXR003 | CTATAATGGTGAATATCTTCAAATGATTTA | KRAS | K7 | 4A |
| pXR003 | TCAGAGTCCTTAACTCTTTTAATTTGTTCT | KRAS | K8 | 1B, 2B, 2C, 4A, S1C, S2C |
| pXR003 | TAATTTGTTCTCTATAATGGTGAATATCTT | KRAS | K9 | 4A |
| pXR003 | TCACCAGAAGCGTACCATACTCACGAACAG | NT30 | NT | 1B, 2B, 2C, 3B, 4B, 4C, S1A, S1B, S2A, S2B |
| pXR003 | GGTAATGCCTGGCTTGTCGACGCATAGTCT | NT57 | NT | 4B,4C |
| pXR003 | CAGACAATTGTGTCGCTGCCATC | CD46 | 1 | 3A, 3B, 3C, 3D |
| pXR003 | ACAATTGTGTCGCTGCCATCGAG | CD46 | 2 | 3A, 3B, 3C, 3D |
| pXR003 | TCATTACAAATAAAGTGCATCTG | CD46 | 3 | 3A, 3C, 3D |
| pXR003 | CCAGAAGCAACCCAGGAAAGCGC | CD46 | 4 | 3A, 3C, 3D |
| pXR003 | TGATCCTTTAAGTTCACAATATA | CD46 | 5 | 3A, 3C, 3D |
| pXR003 | CTGAATTGTCACCACAATAAATC | CD46 | 6 | 3A, 3C, 3D |
| pXR003 | TCAAAAGATGAACTGGCAAACCA | CD46 | 7 | 3A, 3C, 3D |
| pXR003 | TTGCAATATTAGCTAAGCCACAG | CD46 | 8 | 3A, 3C, 3D |
| pXR003 | TTAGAGAATTTTCCAATAATATT | CD46 | 9 | 3A, 3C, 3D |
| pXR003 | TAATTTTGCCAATTTTATAAAAT | CD46 | 10 | 3A, 3C, 3D |
| pXR003 | AGACAATTGTGTCGCTGCCATCG | CD46 | 11 | 3A, 3B, 3C, 3D |
| pXR003 | ATAACAATCACAGCAATGACCCA | CD46 | 12 | 3A, 3C, 3D |
| pXR003 | TCACAAATAGTATGGGTGGCAAG | CD46 | 13 | 3A, 3C, 3D |
| pXR003 | GCGGCGGCCGGGAGGCTCCATGC | CD46 | 14 | 3A, 3C, 3D |
| pXR003 | AACTCGTAAGTCCCATTTGCAGG | CD46 | 15 | 3A, 3C, 3D |
| pXR003 | ATGTGCCTTTCTTCTTCCTCCTT | CD46 | 16 | 3A, 3C, 3D |
2.2. Cell culture and co-transfection
2.2.1. Determining how much DNA transfected impacts Cas13 KD efficiency
- Make DMEM Complete medium. Mix in a 500 ml bottle of DMEM (Gibco or Lonza):
- 50 ml of BD Hyclone FBS
- 5 ml of 100 mM Sodium Pyruvate (from 100X stock Gibco)
- 5 ml of 100 mM Penicillin-Streptomycin (from 100X stock Gibco)
- 5 ml of 100 mM L-Glutamine (from 100X stock Gibco)
Aliquot medium, store in 4 °C or warm media to 37 °C in water bath for immediate use.
In a 96-well tissue cultured treated dish (Thermofisher scientific) seed 10,000 Human Embryonic Kidney 293T (HEK293T) cells (ATCC CRL-3216) with 200 μl DMEM per well [50,000 cells/ml].
- After one doubling (16 hours), transfect the cells with either 100 ng, 200 ng, 300 ng, or 400 ng of total DNA per well. Cells are to be co-transfected with the same amount of RfxCas13d-EGFP expressing plasmid (pXR001) and gRNA expressing plasmid (pXR003).
- Use JetPrime Transfection Reagent and prepare samples according to manufacturer’s protocol.
- Total amount of DNA varied from 100-400 ng (50-200 ng of each vector)
Exchange medium containing transfection mix with 200 μl of fresh medium in each well 8 hours post transfection.
- 48 hrs post transfection, harvest cells for RNA extraction:
- Aspirate off medium (leave just enough to cover the bottom of the well because cells are loosely adherent and can be damaged from too aspirating too close).
- Lyse cells with 200 μl TRIzol (Ambion) or TRIreagent (Molecular Research Center) and transfer to separate, clean, 1.7 ml Eppendorf tubes. Can either store −80°C or directly proceed to Phenol-Chloroform extraction (see section 2.3).
2.2.2. Determining the optimal transfection time for maximal Cas13 KD Efficiency
- If you have not already done this in the above experiment, prepare DMEM Complete medium. Mix in a 500 ml bottle of DMEM (Gibco or Lonza):
- 50 ml of BD Hyclone FBS
- 5 ml of 100 mM NaPy (from 100x stock Gibco)
- 5 ml of 100 mM Penicillin-Streptomycin (form 100x stock Gibco)
- 5 ml of 100 mM L-Glutamine (from 100x stock Gibco)
Warm medium in 37°C water bath before use
In a 96-well tissue culture treated dish seed 10,000 Human Embryonic Kidney 293T (HEK293T) cells with 200 μl DMEM per well [50,000 cells/ml].
- After one doubling (16 hours), transfect the cells with 200 ng of total DNA per well. Cells are to be co-transfected with the same amount of RfxCas13d-EGFP expressing plasmid (pXR001) and gRNA expressing plasmid (pXR003).
- Use JetPrime Transfection Reagent and prepare samples according to manufacturer’s protocol.
- Total amount of DNA is 200 ng (100 ng of each vector)
Exchange medium containing transfection mix with 200 μl of fresh medium in each well after 4, 8, 16, 20, or 24 hours.
- Harvest cells for RNA extraction:
- Aspirate off medium (leave just enough to cover the bottom of the well because cells are loosely adherent and can be damaged from too aspirating too close).
- Lyse cells with 200 ul TRIzol (Ambion) or TRIreagent (Molecular Research Center) and transfer to separate, clean, 1.7 ml Eppendorf tubes. Can either store −80°C or proceed to Phenol-Chloroform extraction (see section 2.3).
2.2.3. Testing gRNA efficiency in RfxCas13d mediated KD of CD46 cell surface marker mRNA
- If you have not already done this in the above experiment, prepare DMEM Complete medium. Mix in a 500 ml bottle of DMEM (Gibco or Lonza):
- 50 ml of BD Hyclone FBS
- 5 ml of 100 mM NaPy (from 100x stock Gibco)
- 5 ml of 100 mM Penicillin-Streptomycin (form 100x stock Gibco)
- 5 ml of 100 mM L-Glutamine (from 100x stock Gibco)
Warm medium in 37°C water bath for use
In a 96-well tissue culture treated dish (Thermofisher scientific) seed 10,000 Human Embryonic Kidney 293T (HEK293T) cells with 200 ul DMEM per well [50,000 cells/ml].
- After one doubling (16 hours), transfect the cells with 200 ng of total DNA per well. Cells are to be co-transfected with the same amount of RfxCas13d-EGFP expressing plasmid (pXR001) and gRNA expressing plasmid (pXR003).
- Use JetPrime Transfection Reagent and prepare samples according to manufacturer’s protocol.
- Total amount of DNA is 200 ng (100 ng of each vector)
Exchange medium containing transfection mix with 200 μl of fresh medium in each well after 8 hours.
- Harvest cells for flow cytometry:
- Wash cells with 200 μl 1x PBS
- Transfer to clean 1.7 ml Eppendorf tube or U-bottom or V-bottom 96-well tissue culture treated plates (Fisherbrand), spin down at 1200X g in either tabletop microfuge or swinging rotor centrifuge
- Aspirate PBS and keep on ice. Move on to staining the live cells for flow cytometry (see section 2.6)
2.3. RNA Extraction and Purification
- Add chloroform and vortex to mix: 0.2 mL of chloroform per 1 mL of TRIzol® Reagent or TRI-Reagent used for homogenization. Cap the tube securely.
- Let sit at room temp 2-3 min
Spin down in a centrifuge at 4°C at top speed for 5 min
Take off top layer (aqueous layer), move to a fresh tube and add an additional volume of chloroform to this aqueous layer to remove any trace organic layer carry over.
Take aqueous layer and put in a fresh tube.
Add 0.1 volumes of 3M DEPC-treated NaOAc (sodium acetate) pH 5.2 to aqueous layer
Add 1 μl GlycoBlue Coprecipitant to the tube
Mix and add 2.5 volumes of 100 % ice-cold EtOH and mix by pipetting up and down and/or vortexing
Incubate at −20°C for at least 2 hours to overnight
- Spin top speed at 4°C in microfuge for at least 30 min
- Orient all tubes with cap hinge facing out to know where the pellet will form
After spin has completed, remove supernatant without disturbing the pellet
Add 200 μl of ice-cold 70% EtOH (in DEPC-treated water) to wash pellet (carefully resuspend pellet to ensure the RNA is sufficiently washed from contaminating salts)
Spin again top speed and 4°C in microfuge for 5 min to pellet RNA
Careful remove supernatant as to not disrupt the RNA pellet
- Let dry in sterile environment for 5- 10 mins (be careful not to over-dry the pellet) and then resuspend in 10 μl of DEPC-treated water.
- If you only have a several samples, use a Nanodrop spectrophotometer to determine purified RNA concentration (ng/ul)
- If you have a large number of samples, it is advisable to use a Quant-iT RNA Assay kit (ThermoFisher) or equivalent and a fluorescence-capable plate reader to determine RNA concentration (ng/ul)
Store in −80C or proceed directly to cDNA synthesis
2.4. cDNA synthesis
We recommend using the High Capacity cDNA Reverse Transcription Kit with Multiscribe Reverse Transcriptase (Applied Biosystems)
- Prepare the 2X reverse transcription master-mix, per reaction:
Component Volume/Reaction (μl) 10X RT Buffer 0.5 25X dNTP Mix (100 mM) 0.2 10X random hexamers 0.5 Multiscribe Reverse Transcriptase 0.25 Nuclease-free Water 1.05 Total per Reaction 2.5 - It is advisable to make excess master-mix with ~five additional reaction volumes
- NB: To be more economical, we’ve adjusted these final volumes relative to the manufacturer’s recommendation, and we’ve found no loss in RT-qPCR performance.
- NB: Some kits use RNase inhibitor to help prevent RNA degradation by endogenous or environmental RNases. We’ve found it is not necessary in our hands with HEK293T cells and careful technique at the bench, but for other tissue types or environment situations, one might consider adding RNase inhibitor
Add 2X master-mix to the side of each PCR tube or well in a 96-well PCR plate.
Add 100 ng RNA in equal volume to the 2X master mix
If using a multi-well plate, cover plate with plastic cover, making sure to seal each well tightly.
Spin tubes down in a tabletop centrifuge with PCR tube adaptors to get mastermix and RNA to the bottom of the tube. If using a plate, spin down plate in a swinging plate rotor at 2000 rpm for 5 min. Keep on ice as much as possible.
- Run the following reaction using a thermocycler:
Step 1 Step 2 Step 3 Step 4 Temperature (°C) 25 37 85 4 Time (min) 10 120 5 Infinity
2.5. Analysis of Target Gene Expression by qPCR
- Make Master mix for qPCR reactions:
- If using Taqman probes (See Table 3 for probe sequences):
- You can make one single master mix with all probes if all probes are spectrally compatible (i.e. can be read out by separate channels in the machine and don’t spectrally overlap). If there is overlap you will need separate master mixes.
Reagent Volume for 1 reaction (μl) 2x Mastermix 4 DEPC-treated water 2.5 Probe set 1 0.5 Probe set 2 (optional) 0.5 Housekeeping probe 0.5 cDNA 2 Total 10
- If using SYBR Green Master Mix (See Table 4 for primer sequences):
- Make a separate master mix for each primer set/gene you are targeting:
Reagent Per reaction (μl) 2x SYBR 5 (F) Target primer 0.2 (R) Target primer 0.2 DNA 2 DEPC-treated water 2.6 Total 10 Reagent Per reaction (μl) 2x SYBR 5 (F) Housekeeping primer 0.2 (R) Housekeeping primer 0.2 DNA 2 DEPC-treated water 2.6 Total 10
Make master mix without cDNA with enough for at least five extra reactions to account for carryover. Add 8 μl of master mix to the top inside of each well of a 384-well plate.
To the top inside of each well, add 2 μl of cDNA working concentration
Cover plate with clear plastic plate cover and seal, separating each well to prevent spillover between samples.
Spin down in swinging rotor plate spinner for 5 min at 2000 rpm at 4C
Program qPCR thermocycler according to your plate set up
Run program and analyze data using delta delta Ct method [55]. We generally normalize to mock transfected controls, but there are variations on this in the literature.
Table 3.
Taqman Probes
| Forward Primer (5’ – 3’) | Probe sequence (5’ – 3’) | Reverse Primer (5’ – 3’) | Gene and Probe ID |
|---|---|---|---|
| CCT ACT AGG ACC ATA GGT ACA TCT | /5FAM/AGT CCT TAA /ZEN/CTC TTT TAA TTT GTT CTC TAT AAT GGT GAA T/3IABkFQ/ | AGG GCT TTC TTT GTG TAT TTG C | KRAS (Hs.PT.58.41104810) |
| TGT AGT TGA GGT CAA TGA AGG G | /5Cy5/AAG GTC GGA GTC AAC GGA TTT GGT C/3IAbRQSp/ | ACA TCG CTC AGA CAC CAT G | GAPDH (Hs.PT.39a.22214836) |
Table 4.
qPCR Primers
| Forward primer (5’-3’) | Reverse primer (5’-3’) | Gene |
|---|---|---|
| CTA CAA AGC AAC AGT TAT GTT TGA | GAC ACT TTA AGA CAC TTT GGA AC | CD46 spanning cut site |
| GTG ATC CTG CAC CTG GAC CAG AT | GGA AAT CGA CAT TTG ACC ACT TTA CAC TC | CD46 upstream of cut site |
| TCT CAA ATT ATC CAG GAT ATC CTA | GGA CAA CAC AAA TTA CTG CAA | CD46 downstream of cut site |
| GAGTCAACGGATTTGGTCG | GACAAGCTTCCCGTTCTCAG | GAPDH |
2.6. Staining Live Cells with Labeled Primary Antibody for Flow Cytometry.
After completing the steps in Section 2.2.3, continue here.
Block cells with 1% (w/v) BSA (VWR) in Cold 1X PBS (Lonza) for 10 min at RT on tabletop shaker/mixer.
Spin down 1200X G in swinging rotor centrifuge (Eppendorf) (at 4°C)
Carefully pipette off supernatant
Wash with Cold 1X PBS 3 min on shaker, pipette up and down gently
Spin down 1200X G in swinging rotor centrifuge at 4°C
Carefully pipette off supernatant
Stain for 20 min with 2 μl PE labelled primary antibody (BioLegend) in 1% (w/v) BSA in 1X cold PBS (at 4°C on a plate shaker).
Spin down 1200X G in swinging rotor centrifuge (make sure balanced at 4°C)
Carefully pipette off supernatant
Wash 1X with cold 1X 1% (w/v) BSA in 1X PBS
Spin down 1200X G in swinging rotor centrifuge (make sure balanced at 4°C)
Carefully pipette off supernatant
Resuspend in 100 μl cold 1X PBS and keep on ice
Collect data on BD Accuri flow cytometer using PE channel
Analyze data using FCS express 7 (DeNovo Software) or other flow cytometry software normalizing experimental samples to mock transfected controls
3. Results:
3.1. Evaluating Cas13 ortholog KD efficiencies in our laboratory.
Previous studies evaluating the RNA KD activity of a range of Cas13 orthologs in mammalian cells have revealed that certain Cas13 orthologs appear to be more effective at specific RNA knockdown in mammalian cells. To this point, LbuCas13a, LwaCas13a, PspCas13b, RanCas13b, and RfxCas13d have proven to be the superior Cas13 proteins from each Type VI subtype in achieving optimal target RNA KD. Cox et al. observed that RanCas13b and PspCas13b possessed more robust KD across a range of RNA targets compared to several Cas13a orthologs[7], while Konermann et al. showed that RfxCas13d achieved even greater target KD compared to the Cas13b proteins [8]. With this mind and an interest in developing Cas13 KD targeting approaches in our laboratory, we decided to test the KD activity of each of these Cas13 orthologs is in our own hands.
Cas13 and gRNA expression plasmids targeting KRAS mRNA were co-transfected into HEK293T cells, cultured for 48 hours post transfection, and then harvested to measure KRAS mRNA expression by RT-qPCR (Fig. 1A). Our results were consistent with previously published data, with RanCas13b achieving greater KRAS mRNA KD relative to PspCas13b, and RfxCas13d exhibiting the largest KD of all three orthologs relative to an untreated HEK293T cell control[7, 8](Fig. 1B). With this result, we decided to move forward and see whether we could further optimize RfxCas13d KD efficiency by exploring several transient transfection parameters.
Figure 1. RfxCas13d has more efficient target mRNA KD activity than other Cas13 orthologs.
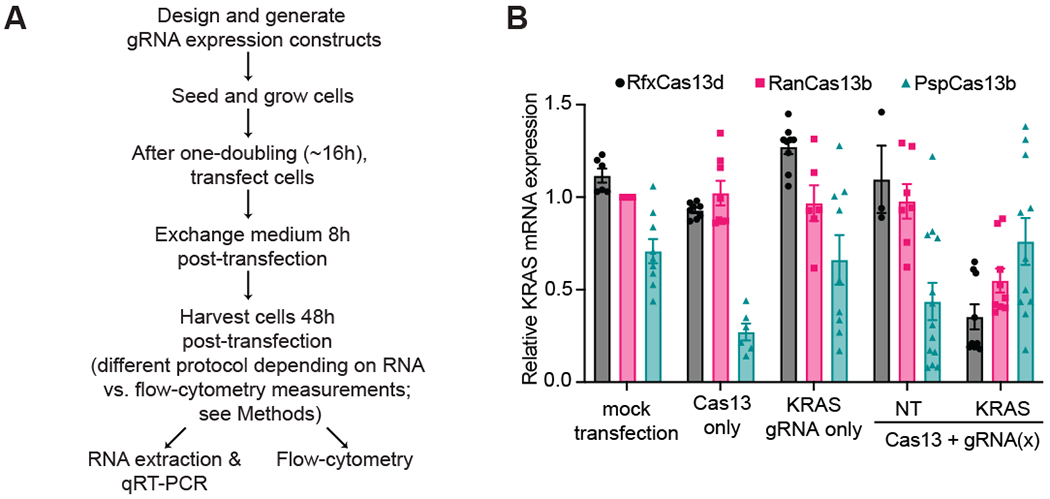
(A) A flowchart for the workflow of a typical Cas13 RNA KD experiment in mammalian cell cultures. (B) Comparison of KD efficiencies of different Cas13 proteins targeting the same region of KRAS mRNA. 100 ng of Cas13 expressing vector were co-transfected into HEK293T cells with 100 ng of corresponding gRNA expressing vector for 8 hours before media change and harvested to measure KRAS mRNA expression levels by RT-qPCR (n>3 replicates) with GAPDH used as a housekeeping control. Data were normalized to a mock transfection control. NT: non-targeting control gRNA. The error bars represent the mean ± SEM, respectively.
3.2. Optimal Transfection Conditions for RfxCas13 target mRNA KD
Initial Cas13 RNA KD studies have used saturating amounts of transfection reagent and DNA which could prove toxic to cells and potentially alter gene expression. Abbudayyeh et al. and Cox et al. transfected 300ng of total DNA in HEK293T cells in 96-well plates when targeting RNA with Cas13a and Cas13b orthologs[6, 7]. Konermann et al. transfected 400ng of total DNA in HEK293T cells in 96-well plates when targeting RNA with Cas13d[8]. This amount of DNA is 1.5-2x the maximum recommended amount for transfecting HEK293T cells in 96-well plates, according to most manufacturer’s instructions.
To this end, we decided to explore whether altering the total amount of DNA transfected into HEK293T cells seeded in a 96-well plate would affect RfxCas13d’s KD efficiency. Specifically, we transfected 100-400 ng of total DNA (50-200 ng of the Cas13 expressing plasmid and the gRNA expressing plasmid, respectively) using JetPrime, a transfection reagent we and others find milder (in terms of cell toxicity) than other commercially available alternatives[56]. We find that although transfecting a greater amount of total DNA yields greater KRAS mRNA KD (Fig. 2A), it becomes increasingly toxic for the cells with observable toxicity and loss of cell density (via light microscopy) apparent when transfecting greater than 200-300 ng of total DNA (Sup. Fig. 1). Therefore, in our hands, transfecting 200-300 ng of total DNA is the best balance achieve the sufficient KD while mitigating cellular toxicity.
Figure 2. Determining the optimal conditions for co-transfecting RfxCas13d and gRNA expression plasmids in HEK293T cells.
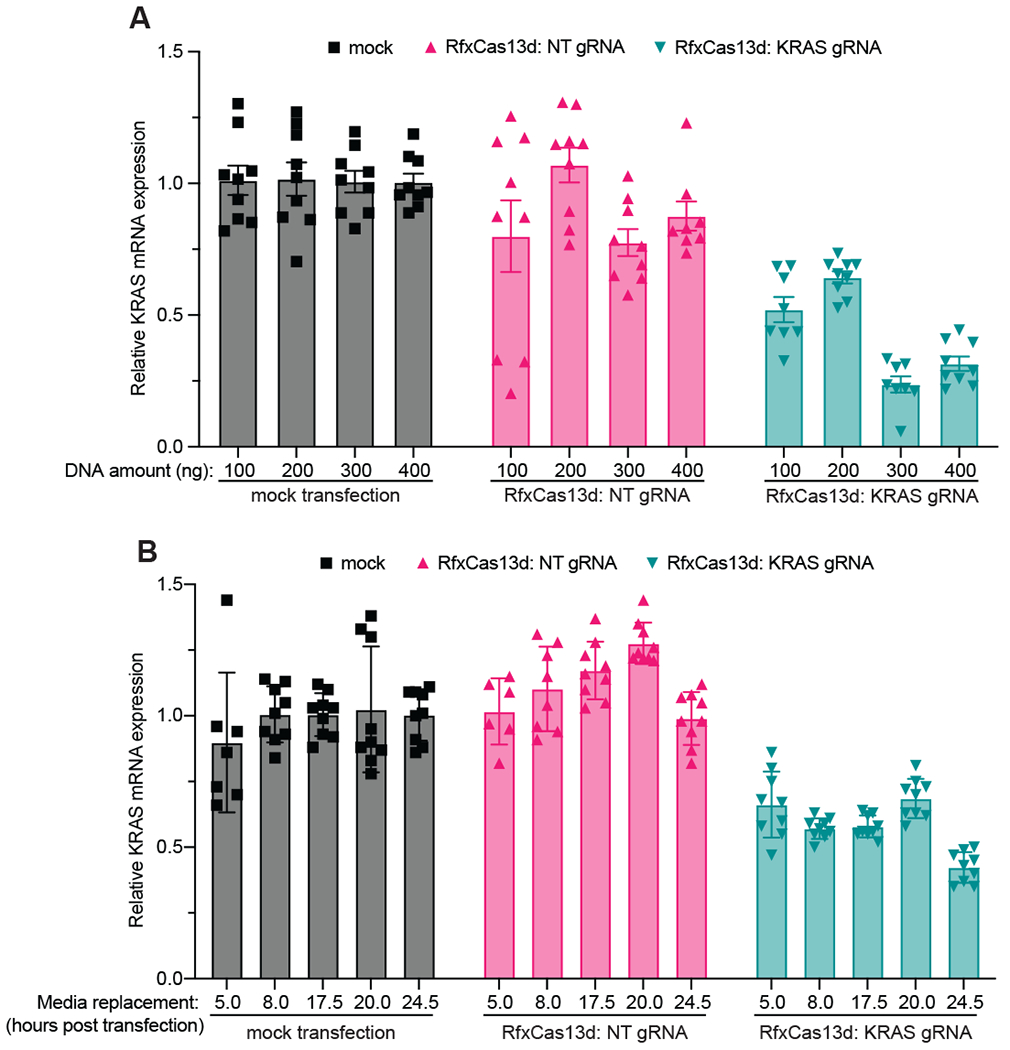
(A) The magnitude of target gene KD targeting the same region of KRAS mRNA as determined by RT-qPCR, as a function of total DNA transfected (n>3 replicates). GAPDH was used as a housekeeping control. Data were normalized to a mock transfection control. The error bars represent the mean ± SEM, respectively. (B) The magnitude of target gene KD targeting the same region of KRAS mRNA as determined by RT-qPCR, as a function of the time the cells were exposed to transfection reagent (n>3 replicates). Data were normalized to a mock transfection control. NT: non-targeting control gRNA. The error bars represent the mean ± SEM, respectively.
Next, we wanted to determine the effect of altering the amount of time the cells were exposed to transfection reagent before a media change. In this case, we transfected 100 ng each of RfxCas13d and KRAS mRNA targeting gRNA expression plasmids for different amounts of time ranging from 5 to 24.5 hours before the media was changed, allowing the cells to grow for 48h in total post initial transfection. We see that in as little as 5 hours we observe a consistent KD and this generally remains consistent across all time points, with a slightly improved KD at the 24.5-hour time point (Fig. 2B). However, we observe a similar toxicity phenomenon as above when the cells are exposed to transfection reagent for longer than 8 hours (Sup. Fig. 2). Taken together, we find that transfecting 200 ng of total DNA for 8 hours before changing the medium yields the optimal conditions to transfect HEK293T cells for RfxCas13d mediated RNA KD.
3.3. Where you and how you measure matters: considerations for the measurement of Cas13 target RNA KD
After some issues obtaining sufficient and reproducible RNA knockdown for several new guide designs and/or target RNA we were using in our lab, we eventually noticed that the level of KD measured using RT-qPCR is highly dependent on whether the amplicon spans the Cas13:gRNA target site or is upstream or downstream of the target site (Fig. 3A). For example, we noticed that for several CD46 gRNAs we tested for RNA KD in HEK293T cells, we observed different magnitudes of RNA KD depending on whether we measured upstream, downstream, or spanning the target site (Fig 3B). In particular, we noticed that amplicons that span the target site result in a larger apparent KD than when using either upstream or downstream amplicons. We hypothesize that this might be the reason that other researchers in the community have observes various levels of KD when trying to apply it to new targets. Here, we suggest always measuring across the intended gRNA target site when using RT-qPCR to measure RNA KD.
Figure 3. Considerations for measurement of Cas13 target RNA KD using RT-qPCR or flow-cytometry.
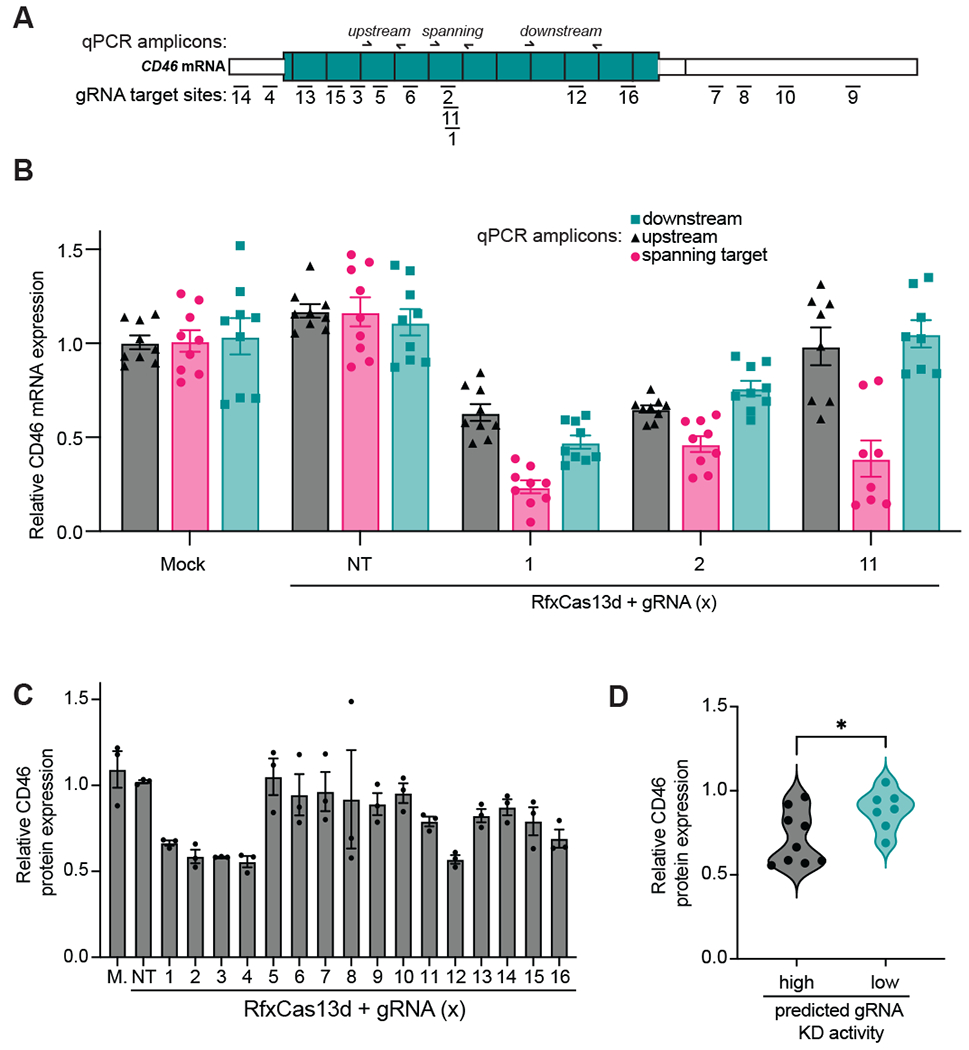
(A) A Schematic of CD46 mRNA and the positions targeted by each gRNA (used in panels B-D) and the positions of the RT-qPCR amplicons used in panel B. (B) The magnitude of target gene KD targeting CD46 mRNA and with the specified gRNAs and primer pairs as determined by RT-qPCR (n>3 replicates). GAPDH was used as a housekeeping control. Data were normalized to a mock transfection control. The error bars represent the mean ± SEM, respectively. (C) The magnitude of protein KD after targeting CD46 mRNA as determined by flow cytometry using a PE-labeled anti-CD46 antibody (n>3 replicates). Data were normalized to a mock (M.) transfection control. NT: non-targeting control gRNA. The error bars represent the mean ± SEM, respectively. (D) A correlation plot comparing a binned predicted gRNA score from Wessels et al. gRNA design algorithm (‘high’ and ‘low’ scoring gRNAs with corresponding scores above and below 0.5, respectively) with CD46 protein flow cytometry measurements obtained in panel C. * indicates statistical significance (p value: 0.0298 as determined by a two-tailed unpaired Welch’s t test).
An alternative to needing to design multiple amplicons to measure the performance of several gRNAs across a gene of interest, in some cases we wondered whether flow cytometry could allow for a more direct measurement of KD performance in our lab. To test out this idea, we used the Wessels et al. gRNA design tool to design a set of 16 gRNAs targeting CD46 (Fig. 3A), a well-expressed cell surface marker amenable to flow cytometry analysis[47] and carried out an RNA KD experiment followed by flow cytometry and observed a range of gRNA KD efficiencies (Fig. 3C). We specifically designed these gRNAs to span across the whole range of gRNA efficiency scores to test out performance of the gRNA design algorithm in our hands. To visualize the relationship between gRNA score and KD efficiency, we correlated the experimental KD level we observed via flow cytometry with a binned predicted gRNA score (‘high’ and ‘low’ scoring gRNAs with corresponding scores above and below 0.5, respectively) and found evidence of a statistically significant binary correlation between high scoring gRNAs and better KD performance (Fig. 3D), indicating that one has a higher chance of finding a well-performing gRNA if one selects multiple high-scoring gRNAs using this algorithm. Finally, it should be noted that flow-cytometry may not always be the best choice for rapid Cas13 KD measurements, especially if good quality conjugated antibodies validated for flow cytometry are not available and/or significant optimization is required (this is generally the case for intracellular targets).
3.4. Evidence that increasing the length of the gRNA spacer can rescue poorly performing gRNAs
Finally, we have evidence supporting a rescue effect when increasing the length of a gRNA spacer from 22 to 30-nt. (Fig. 4). Here, we selected three gRNAs targeting KRAS (denoted K7, K8, K9 originally from Konermann et al.), generated 22-nt. and 30-nt. spacers for these gRNAs and measured KD efficiency using RT-qPCR. We observe that for a poorer performing 22-nt. gRNA (e.g. K7) we’re able to restore better performance with a longer 30-nt. gRNA. This is also somewhat observed with gRNA K9. We hypothesize that this effect is most likely due to increasing the thermodynamic stability of the gRNA-target RNA duplex, or increasing the stability of the gRNA. Additional studies are required to understand this phenomenon and whether it can be generalized across many gRNAs and targets. One could imagine this approach could prove useful to achieve controlled target RNA KD by manipulating the structure and composition of a single gRNA. This could enable researchers to obtain a range of KD activity using different lengths/versions of a single gRNA, leading to the possibility of titratable RfxCas13d mediated RNA KD technology.
Figure 4. Increasing length of gRNA increases KD performance.
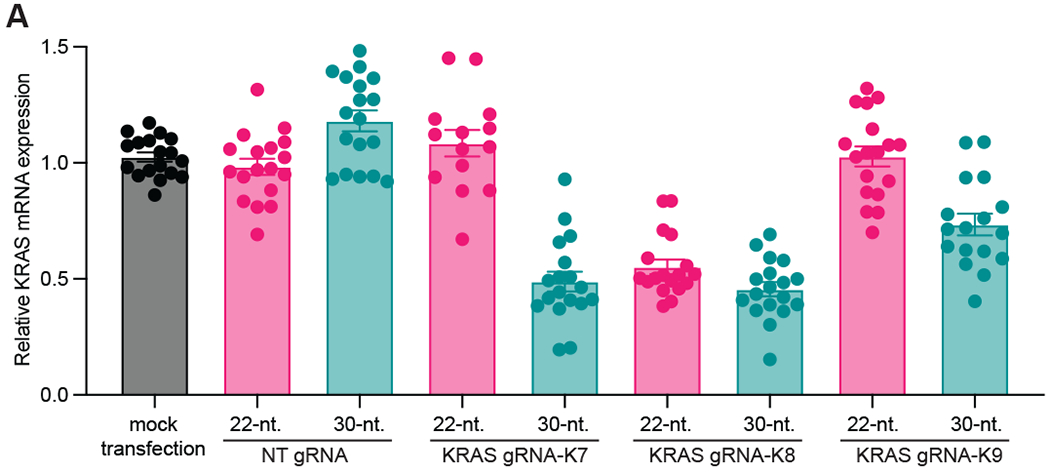
(A) The magnitude of target gene KD targeting KRAS mRNA and with the specified gRNAs containing either 22-nt. or 30-nt. gRNA-spacers, as determined by RT-qPCR (n>3 replicates). GAPDH was used as a housekeeping control. Data were normalized to a mock transfection control. The error bars represent the mean ± SEM, respectively.
4. Troubleshooting tips
- Issues with transfection and cellular toxicity? In our hands, transfecting more than 200 ng of Cas13 and gRNA plasmids in wells of a 96 well plate can be cytotoxic. Likewise, leaving the transfection reagent incubating with the cells for increasing amounts of time can also be cytotoxic. To determine the cytotoxic thresholds of transfecting DNA into your cells of interest, you can try testing a couple of different conditions:
- Transfect your cells using a range of DNA amounts to determine lower and upper limits for cytotoxicity. Transfect for a standard 4 hours before changing media (keeping the time consistent). Pay attention to rate of growth and cell morphology to evaluate cytotoxicity. Measure RNA KD with RT-qPCR and pay attention to consistency in control samples and KD in your target samples.
- Once you have optimized the amount of DNA to transfect, transfect samples for variable amounts of time before exchanging transfection containing media for fresh media (4-24 hours). Pay attention to rate of growth and cell morphology to evaluate cytotoxicity. Measure RNA KD with RT-qPCR and pay attention to consistency in control samples and KD in your target samples.
Interest in optimizing gRNA design? If measuring gRNA performance to learn more about gRNA design, choose mRNAs that encode cell surface proteins. By using this method, you can rapidly readout target KD for a large number of gRNAs by flow cytometry. This is particularly useful because it is a direct readout of gene expression.
Issues with RT-qPCR readout of RNA KD? Be sure to design your probe/primer sets spanning the Cas13: gRNA target site. Cas13 mediated cleavage of target RNA yields metastable RNA fragments in the cell, therefore yielding differential levels of RNA expression based on where your qPCR amplicon is relative to the target site. We have observed that designing the probe/primer set over the cleavage junction is a good proxy for RNA KD and gRNA performance.
Struggling to find high performing gRNAs for your RNA of interest? Try increasing the length of a gRNA-spacer as this may improve the KD.
5. Conclusions
There is a growing body of evidence showing the need for optimization in utilizing Cas13 when targeting RNA in mammalian cell culture. Transfecting DNA can be cytotoxic for live cells, decreasing reproducibility and reliability of RNA KD experiments. We found by optimizing the amount and duration of transfection conditions for human cells, we can study the changes in target gene expression without causing significant cellular toxicity.
Choosing the optimal techniques to measure the experimental perturbations is also critical in generating interpretable results from Cas13 mediated RNA KD experiments. Quantitative measures of protein expression using for example flow cytometry provide a direct readout of changes in mRNA expression but is easiest when measuring expression of more accessible proteins, such as cell surface proteins. Measuring RNA expression via RT-qPCR can also be reliable, but the probe/primer sets must be designed over the Cas13:gRNA target site for the most accurate expression readout.
Lastly, we found that the performance of a particular gRNA to KD target RNA expression can be increased by increasing the length of the gRNA-spacer. In our case, increasing the length of several KRAS-targeting gRNA-spacers from 22-nt. to 30-nt., increases the level of KD achieved with each gRNA, providing another means to improve the performance of Cas13-mediated RNA KD in mammalian cells.
Supplementary Material
Supplementary Figure 1. Determining DNA amounts for transfection and maintenance of cell health for RfxCas13d mediated RNA KD experiments. Brightfield Microscopy images determining how DNA transfection amounts impacts cell proliferation for (A) mock transfected, (B) Cas13:gRNA-NT transfected and (C) Cas13:gRNA-KRAS transfected cells. Images were taken at 48 hours post transfection. Cell morphology images were taken under incandescent light to show the physical shape of the transfected cells. Healthy cells appear flat and star-shaped and stressed cells appear rounded. All samples were transfected for 8 hours before the media was exchanged for fresh media.
Supplementary Figure 2. Determining the length of time exposed to transfection reagent and maintenance of cell health for RfxCas13d mediated RNA KD experiments. Brightfield Microscopy images determining exposing cells to transfection reagents for varying lengths of time impacts cell proliferation for (A) mock transfected, (B) Cas13:gRNA-NT transfected and (C) Cas13:gRNA-KRAS transfected cells. Images were taken at 48 hours post transfection. Cell morphology images were taken under incandescent light to show the physical shape of the transfected cells. Healthy cells appear flat and star-shaped and stressed cells appear rounded.
All samples transfected with 200 ng total DNA.
Highlights.
In our laboratory, compared to PspCas13b and RanCas13b, RfxCas13d yields the largest RNA knockdown in HEK293T cells
By studying a range of transfection conditions, we report optimal conditions that balance KD efficiency and cell toxicity
The position of the qPCR amplicon relative to the Cas13 target site matters for accurate RNA knockdown readout
Increasing the length of gRNA spacers can help rescue inefficient gRNAs
Acknowledgements
This study and authors were supported by the National Institutes of Health National Institutes of Health grant R35GM133462, National Institutes of Health Training Grant in Cellular, Biochemical, and Molecular Sciences (T32 GM068411), University of Rochester Sproull Fellowship (B.J.D), and URMC O’Connell Lab Startup Funds. Competing interests: M.R.O is an inventor on patent applications related to CRISPR-Cas systems and uses thereof. M.R.O is a member of the scientific advisory boards for Dahlia Biosciences and LocanaBio, and an equity holder in Dahlia Biosciences and LocanaBio. The authors would like to thank Mary Pulvino, Ning Tong, Bogdan Polevoda, Xenia Schaffer, and Matthew Raymonda for their insight and advice regarding tissue culture and measuring changes in gene expression.
References
- 1.Horvath P and Barrangou R, CRISPR/Cas, the immune system of bacteria and archaea. Science, 2010. 327(5962): p. 167–70. [DOI] [PubMed] [Google Scholar]
- 2.Koonin EV, Makarova KS, and Zhang F, Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol, 2017. 37: p. 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohanraju P, et al. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science, 2016. 353(6299): p. aad5147. [DOI] [PubMed] [Google Scholar]
- 4.Abudayyeh OO, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science, 2016. 353(6299): p. aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.East-Seletsky A, et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature, 2016. 538(7624): p. 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abudayyeh OO, et al. RNA targeting with CRISPR-Cas13. Nature, 2017. 550(7675): p. 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox DBT, et al. RNA editing with CRISPR-Cas13. Science, 2017. 358(6366): p. 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konermann S, et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell, 2018. 173(3): p. 665–676 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ai Y, Liang D, and Wilusz JE, CRISPR/Cas13 effectors have differing extents of off-target effects that limit their utility in eukaryotic cells. BioRxiv, 2021: p. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelley CP, Maja C, and Wang ET, Negative autoregulation mitigates collateral RNase activity of repeat-targeting CRISPR-Cas13d in mammalian cells. BioRxiv, 2021: p. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi P, Murphy MR, Aparicio AO, Kesner J. s., Fang Z, Chen Z, Trehan A, and Wu X, RNA-guided cell targeting with CRISPR/RfxCas13d collateral activity in human cells. BioRxiv, 2021: p. 20. [Google Scholar]
- 12.Vialetto E, Yu Y, Collins SP, Wandera KG, Barquist L, and Beisel CL, A target expression threshold dictates invader defense and autoimmunity by CRISPR-Cas13. BioRxiv, 2021: p. 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Doval C and Jinek M, Molecular architectures and mechanisms of Class 2 CRISPR-associated nucleases. Curr Opin Struct Biol, 2017. 47: p. 157–166. [DOI] [PubMed] [Google Scholar]
- 14.Corey DR, Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat Neurosci, 2017. 20(4): p. 497–499. [DOI] [PubMed] [Google Scholar]
- 15.Smith I, et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol, 2017. 15(11): p. e2003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson AL, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol, 2003. 21(6): p. 635–7. [DOI] [PubMed] [Google Scholar]
- 17.Jackson AL, et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA, 2006. 12(7): p. 1179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berkhout B and Jeang KT, RISCy business: MicroRNAs, pathogenesis, and viruses. J Biol Chem, 2007. 282(37): p. 26641–26645. [DOI] [PubMed] [Google Scholar]
- 19.Sanson KR, et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun, 2018. 9(1): p. 5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burmistrz M, Krakowski K, and Krawczyk-Balska A, RNA-Targeting CRISPR-Cas Systems and Their Applications. Int J Mol Sci, 2020. 21(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelles DA, et al. Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell, 2016. 165(2): p. 488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tambe A, et al. RNA Binding and HEPN-Nuclease Activation Are Decoupled in CRISPR-Cas13a. Cell Rep, 2018. 24(4): p. 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu L, et al. Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell, 2017. 168(1–2): p. 121–134 e12. [DOI] [PubMed] [Google Scholar]
- 24.Gootenberg JS, et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science, 2018. 360(6387): p. 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horlbeck MA, et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res, 2015. 25(8): p. 1147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batra R, et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell, 2017. 170(5): p. 899–912 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Connell MR, Molecular Mechanisms of RNA Targeting by Cas13-containing Type VI CRISPR-Cas Systems. J Mol Biol, 2019. 431(1): p. 66–87. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Xu J, Guo X, Li Z, Cao L, Liu S, Guo Y, Wang G, Luo Y, Zhang Z, Wei X, Zhao Y, Liu T, Wang X, Xia H, Kuang M, Guo Q, Li J, Chen L, Wang Y, Wang F, Liu Q, You F, Collateral Cleavage of 28s rRNA by RfxCas13d causes death of mice. BioRxiv, 2022: p. 39. [Google Scholar]
- 30.Buchman AB, et al. Programmable RNA Targeting Using CasRx in Flies. CRISPR J, 2020. 3(3): p. 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vialetto E, et al. A target expression threshold dictates invader defense and prevents autoimmunity by CRISPR-Cas13. Cell Host Microbe, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozcan A, et al. Programmable RNA targeting with the single-protein CRISPR effector Cas7–11. Nature, 2021. 597(7878): p. 720–725. [DOI] [PubMed] [Google Scholar]
- 33.Kellner MJ, et al. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat Protoc, 2019. 14(10): p. 2986–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gootenberg JS, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science, 2017. 356(6336): p. 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ackerman CM, et al. Massively multiplexed nucleic acid detection with Cas13. Nature, 2020. 582(7811): p. 277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granados-Riveron JT and Aquino-Jarquin G, CRISPR-Cas13 Precision Transcriptome Engineering in Cancer. Cancer Res, 2018. 78(15): p. 4107–4113. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Rauch S, and Dickinson BC, Programmable technologies to manipulate gene expression at the RNA level. Curr Opin Chem Biol, 2021. 64: p. 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pei Y and Lu M, Programmable RNA manipulation in living cells. Cell Mol Life Sci, 2019. 76(24): p. 4861–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pickar-Oliver A and Gersbach CA, The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol, 2019. 20(8): p. 490–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kannan S, et al. Compact RNA editors with small Cas13 proteins. Nat Biotechnol, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu C, et al. Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nat Methods, 2021. 18(5): p. 499–506. [DOI] [PubMed] [Google Scholar]
- 42.Powell JE, et al. Targeted gene silencing in the nervous system with CRISPR-Cas13. Sci Adv, 2022. 8(3): p. eabk2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou C, et al. CasRx-mediated RNA targeting prevents choroidal neovascularization in a mouse model of age-related macular degeneration. Natl Sci Rev, 2020. 7(5): p. 835–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang LZ, et al. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol Cell, 2019. 76(6): p. 981–997 e7. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, et al. CRISPR-mediated live imaging of genome editing and transcription. Science, 2019. 365(6459): p. 1301–1305. [DOI] [PubMed] [Google Scholar]
- 46.Wu QW and Kapfhammer JP, The Bacterial Enzyme RfxCas13d Is Less Neurotoxic Than PspCas13b and Could Be a Promising RNA Editing and Interference Tool in the Nervous System. Brain Sci, 2021. 11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wessels HH, et al. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat Biotechnol, 2020. 38(6): p. 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bandaru S, et al. Structure-based design of gRNA for Cas13. Sci Rep, 2020. 10(1): p. 11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendez-Mancilla A, et al. Chemically modified guide RNAs enhance CRISPR-Cas13 knockdown in human cells. Cell Chem Biol, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo X, Wessels HH, Mendez-Mancilla A, Haro D, and Sanjana NE, Transcriptome-wide Cas13 guide RNA design for model organisms and viral RNA pathogens. BioRxiv, 2020: p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wessels HH, Mendez-Mancila A, Papalexi E, Mauck WM, Morris JA, Mimitou E, Smibert P, Sanjana NE, and Sajita R, Efficient combinatorial targeting of RNA transcripts in single cells with Cas13 RNA Perturb-seq. BioRxiv, 2022: p. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei J, Lofty P, Faizi K, Kitano H, Hsu PD, and Konermann S, Deep learning of Cas13 guide activity from high-throughput gene essentiality screening. BioRxiv, 2021: p. 40. [Google Scholar]
- 53.Krohannon A, et al. CASowary: CRISPR-Cas13 guide RNA predictor for transcript depletion. BMC Genomics, 2022. 23(1): p. 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tong H, Huang J, Xiao Q, He B, Dong X, Liu Y, Yang X, Han D, Wang Z, Ying W, Zhang R, Wei Y, Wang X, Xu C, Zhou Y, Li Y, Cai M, Wang Q, Xue M, Li G, Fang K, Zhang H, ad Yang H, High-fidelity Cas13 variants for targeted RNA degradation with minimal collateral effect. BioRxiv, 2021: p. 40. [DOI] [PubMed] [Google Scholar]
- 55.Riedel G, et al. An extended DeltaCT-method facilitating normalisation with multiple reference genes suited for quantitative RT-PCR analyses of human hepatocyte-like cells. PLoS One, 2014. 9(3): p. e93031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sandbichler AM, Aschberger T, and Pelster B, A method to evaluate the efficiency of transfection reagents in an adherent zebrafish cell line. Biores Open Access, 2013. 2(1): p. 20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Determining DNA amounts for transfection and maintenance of cell health for RfxCas13d mediated RNA KD experiments. Brightfield Microscopy images determining how DNA transfection amounts impacts cell proliferation for (A) mock transfected, (B) Cas13:gRNA-NT transfected and (C) Cas13:gRNA-KRAS transfected cells. Images were taken at 48 hours post transfection. Cell morphology images were taken under incandescent light to show the physical shape of the transfected cells. Healthy cells appear flat and star-shaped and stressed cells appear rounded. All samples were transfected for 8 hours before the media was exchanged for fresh media.
Supplementary Figure 2. Determining the length of time exposed to transfection reagent and maintenance of cell health for RfxCas13d mediated RNA KD experiments. Brightfield Microscopy images determining exposing cells to transfection reagents for varying lengths of time impacts cell proliferation for (A) mock transfected, (B) Cas13:gRNA-NT transfected and (C) Cas13:gRNA-KRAS transfected cells. Images were taken at 48 hours post transfection. Cell morphology images were taken under incandescent light to show the physical shape of the transfected cells. Healthy cells appear flat and star-shaped and stressed cells appear rounded.
All samples transfected with 200 ng total DNA.