

Abstract
Inosine-5’-monophosphate dehydrogenase (IMPDH) is a potential target for microorganisms. However, identifying inhibitor design determinants for IMPDH orthologs continues to evolve. Herein, a series of mycophenolic anilide inhibitors of Cryptosporidium parvum and human IMPDHs are reported. Furthermore, molecular docking of 12 (e.g. SH-19; CpIMPDH Ki,app = 0.042 ± 0.015 μM, HsIMPDH2 Ki,app = 0.13 ± 0.05 μM) supports different binding modes with the two enzymes. For CpIMPDH the inhibitor extends into a pocket in an adjacent subunit. In contrast, the inhibitor interacts with Ser276 in the NAD binding site in HsIMPDH2, as well as an adjacent pocket within the same subunit. These results provide further guidance for generating IMPDH inhibitors for enzymes found in an array of pathogenic microorganisms, including Mycobacterium tuberculosis.
Keywords: Cryptosporidium parvum, IMPDH, Inosine-5’-monophosphate dehydrogenase, Inhibitor, Binding mode
Graphical Abstract
Inosine-5’-monophosphate dehydrogenase (IMPDH) catalyzes the nicotinamide adenine dinucleotide (NAD)-dependent oxidation of inosine 5’-monophosphate (IMP) to xanthosine 5’-monophosphate (XMP) as the rate-limiting step in the biosynthesis of guanine nucleotides.1 Therefore, IMPDH regulates intracellular guanine nucleotide pools and is critical for cell proliferation in both eukaryotes and prokaryotes.2
IMPDH inhibition has recently gained momentum as a potential treatment of microbial infections. For example, blocking prokaryotic IMPDH could provide a strategy for growth inhibition of bacteria such as Mycobacterium tuberculosis (Mtb) and Staphylococcus aureus (Sa).3, 4 In addition, this enzyme has been targeted in the protozoan Cryptosporidium parvum (Cp), which has a similar IMPDH to some prokaryotes, likely resulting from lateral gene transfer from bacteria.5, 6 These prokaryotic IMPDHs are structurally distinct from their human counterparts.2 The most dramatic difference is in the adenosine subsite (A-site) of the NAD binding site7, 8, and several inhibitor scaffolds (e.g. 1 – 6 in Figure 1A) exploit this divergence.9–17
Figure. 1.
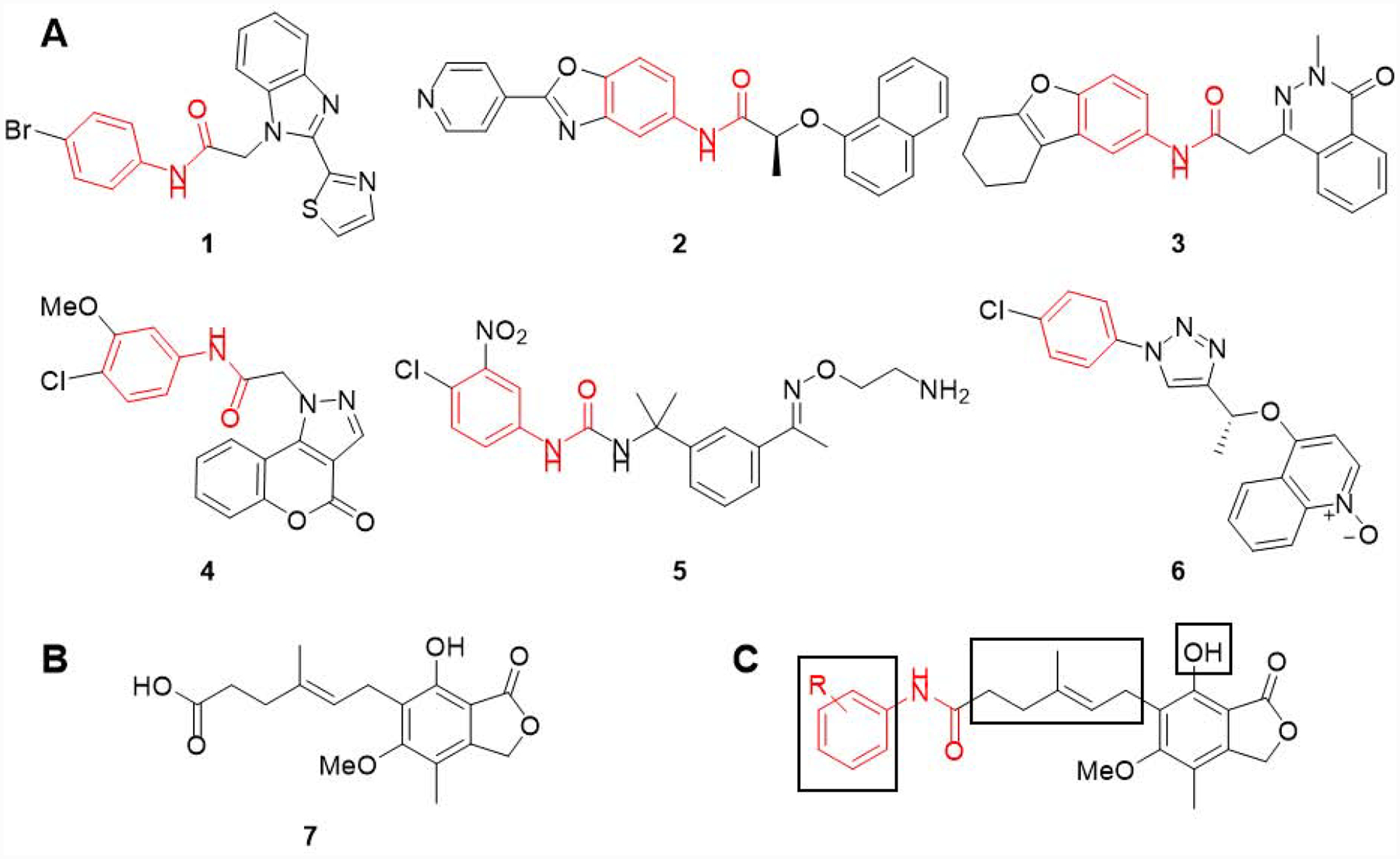
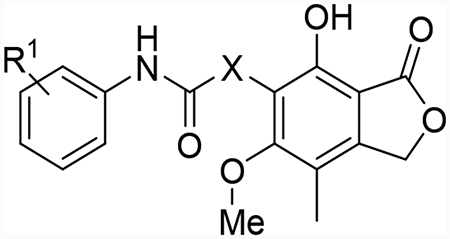
(A) Six structurally distinct CpIMPDH inhibitors (CpIMPDH IC50 = 12 – 64 nM).27 The fragments found in these inhibitors that interact with the adenosine subsite (A-site) of the NAD binding site are highlighted in red. This is based on co-crystal structures of 1, 2, 4 and 5 with CpIMPDH and 6 with Clostridium perfringens IMPDH (ClpIMPDH). The interactions of inhibitor 3 are assumed based on structural similarity since it has not been co-crystalized with an IMPDH. (B) Structure of mycophenolic acid (MPA, 7). (C) Mycophenolic anilides with three regions explored herein shown in boxes.
Mycophenolic acid (MPA, 7, Figure 1B) is a prototypic human (Hs) IMPDH inhibitor18, 19 (e.g. HsIMPDH1 Ki = 33 nM and HsIMPDH2 Ki = 7 nM)20 used clinically as an ester prodrug (e.g. mycophenolate mofetil) for immunosuppression in preventing rejection following organ transplantation.21 Interestingly, MPA (7) binds in the nicotinamide subsite, but is a poor inhibitor of prokaryotic and C. parvum IMPDHs (e.g. CpIMPDH Ki = 9.3 μM).22 Mycophenolic anilides have also been found to inhibit HsIMPDH2,23–25 but their activities against CpIMPDH and bacterial orthologs have not been described.
Herein, we report a structure-activity relationship study for a series of mycophenolic anilides that incorporate a molecular fragment common to several classes of CpIMPDH inhibitors (Figure 1C). Furthermore, an analysis of these anilides was conducted to elucidate additional structural determinants required for selective inhibition of CpIMPDH and related prokaryotic orthologs versus HsIMPDHs.
An initial set of MPA-anilide derivatives 8 – 16 were prepared via EDC-mediated coupling (Scheme 1). In addition, 10 was further modified by phenol alkylation or alkene reduction to provide derivatives 17 and 18, respectively.28
Scheme 1.
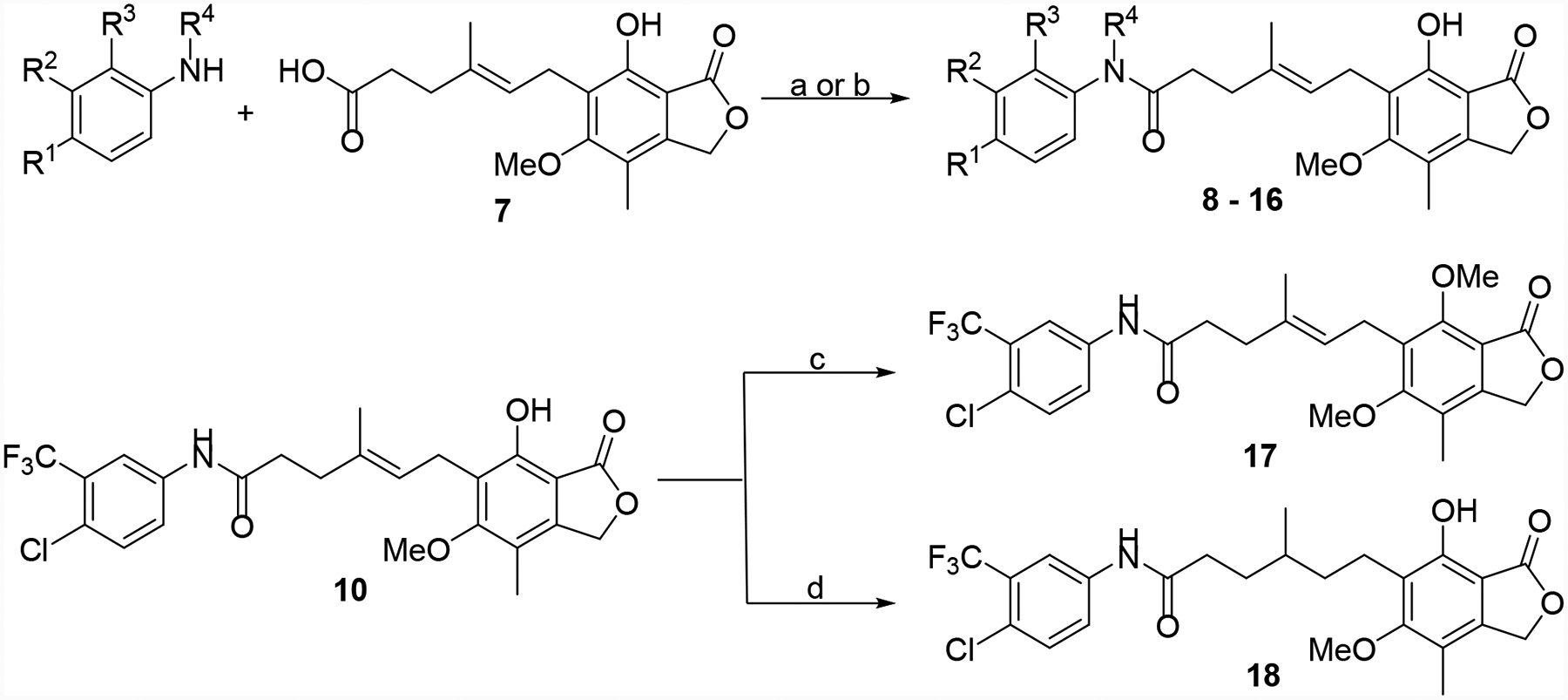
Synthesis of 8 – 18. Reagents and conditions: (a) EDC•HCl, HOBt or HOAt or DMAP, TEA or DIPEA, DMF or DCM, rt or 0 °C to rt, 16 h, 17–80%. (b) i) oxalyl chloride, DMF (cat.), DCM, rt. ii) aniline, TEA, rt, 6 h, 40%. (c) MeI, K2CO3, acetone, rt, 16 h, 64%. (d) 10% Pd/C, H2 (1 atm), EtOAc, rt, 3 h, 34%.
Replacement of the alkene with a cyclopropane bioisostere was also pursued. The synthesis of both enantiomers is illustrated in Schemes 2 and 3. MPA (7) was converted to aldehyde 19,23 which was protected and then reduced to provide alcohol 21 (Scheme 2). A chiral auxiliary was attached generating key intermediate 23a using the methodology of Charette et al.29, 30 Finally, anomerization generated a second crucial intermediate 23b. Both of these intermediates were partially de-protected generating 24a and 24b, which were subjected to cyclopropanation conditions to give 25a and 25b, respectively (Scheme 3).31, 32 These two materials were then subjected to a similar series of transformations (Scheme 4).33, 34 The chiral auxiliary was removed followed by alcohol oxidation, Horner-Wadsworth-Emmons reactions and alkene reduction to produce 29a and 29b. Finally, ester hydrolysis and EDC-mediated aniline coupling provided 31a and 31b.
Scheme 2.
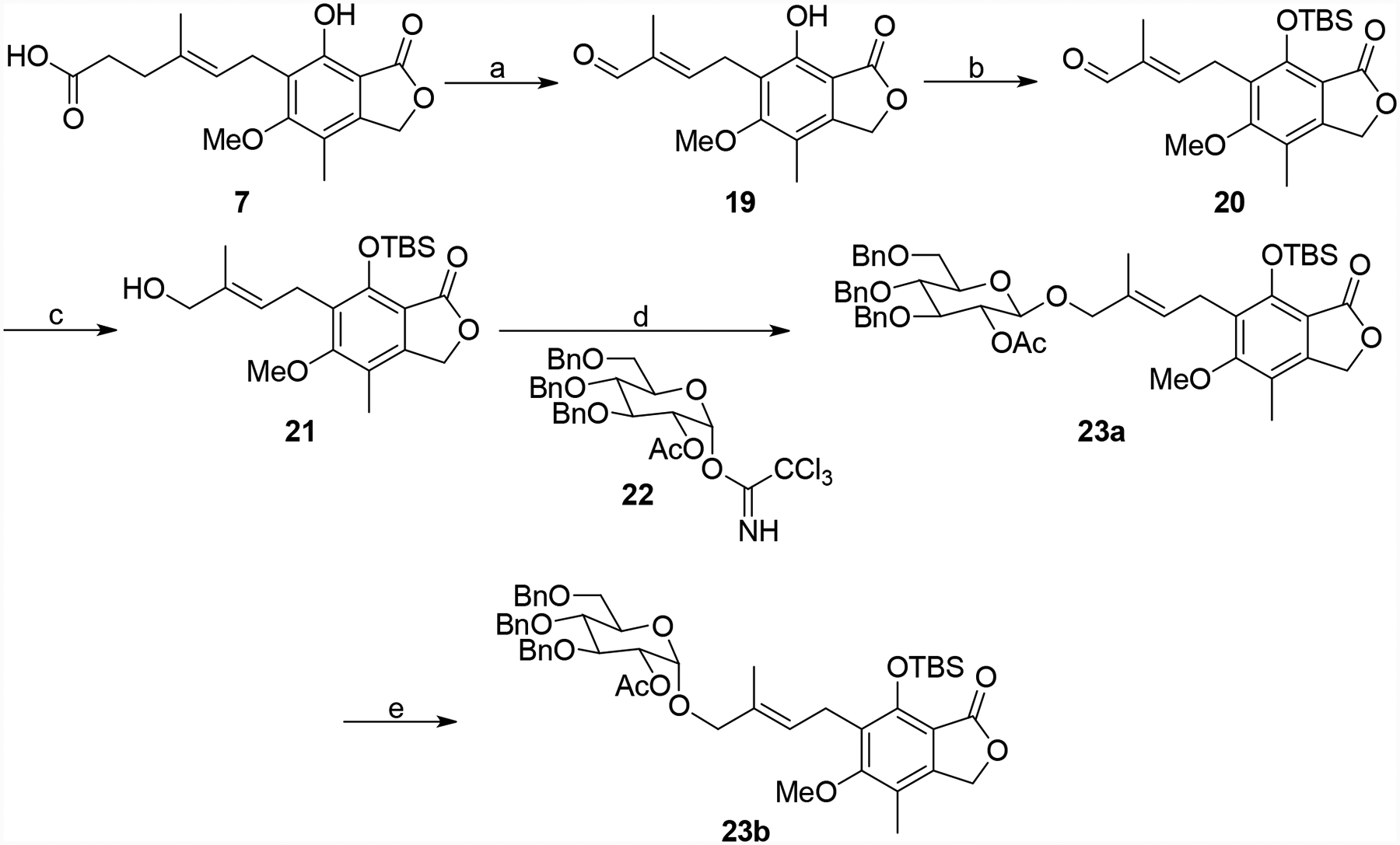
Synthesis of intermediates 23a and 23b. Reagents and conditions: (a) i) OsO4, NMO, NaIO4, THF/H2O, rt, 1.5 h. ii) PPh3=C(CH3)CHO, benzene, reflux, 24 h, 77%. (b) TBSCl, imidazole, DCM, rt, 16 h, 71%. (c) NaBH, MeOH, 0 °4 C to rt, 1 h, 92%. (d) 22, BF3•OEt2, DCM, −78 °C for 0.5 h then 0 °C for 0.5 h, 88%. (e) TiCl4, DCM, −78°C for 10 min then 0 °C for 0.5 h, 99%.
Scheme 3.
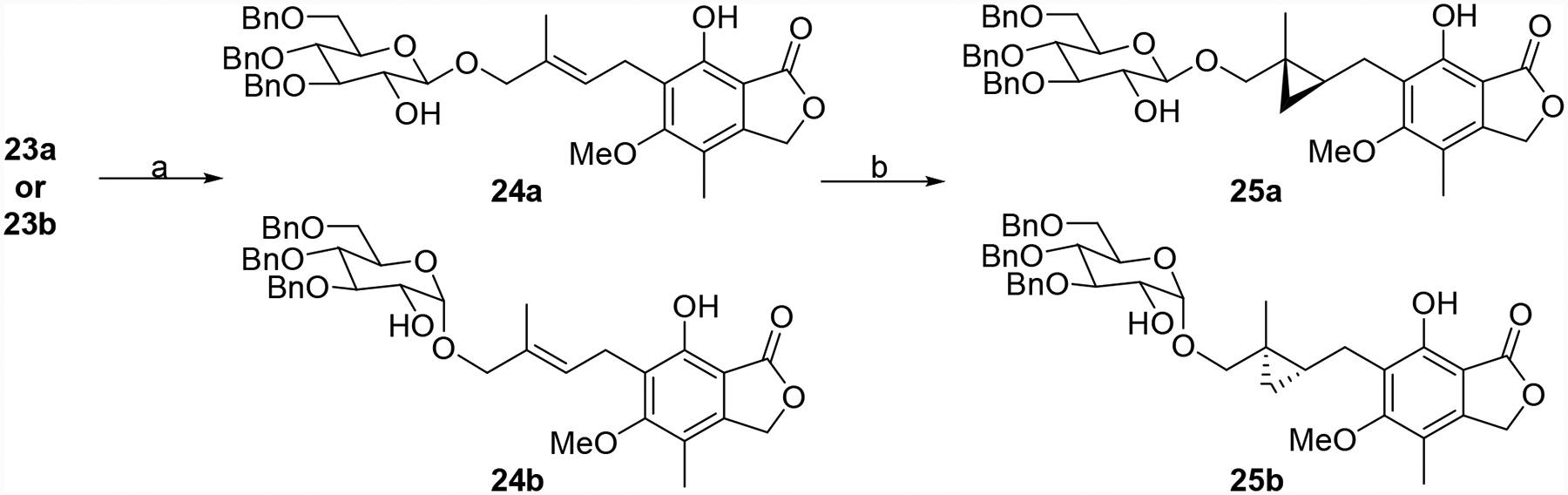
Synthesis of intermediates 25a and 25b. Reagents and conditions: (a) NaOMe, MeOH, 0 °C to rt, 16 h, 55 and 77%. (b) For 25a: Et Zn, CH o2 2I2, toluene, −20 °C, 3 h, 76%. For 25b: Et2Zn, CH2I2, toluene, −78 °C to rt, 16 h (dr 6:1), 98%.
Scheme 4.
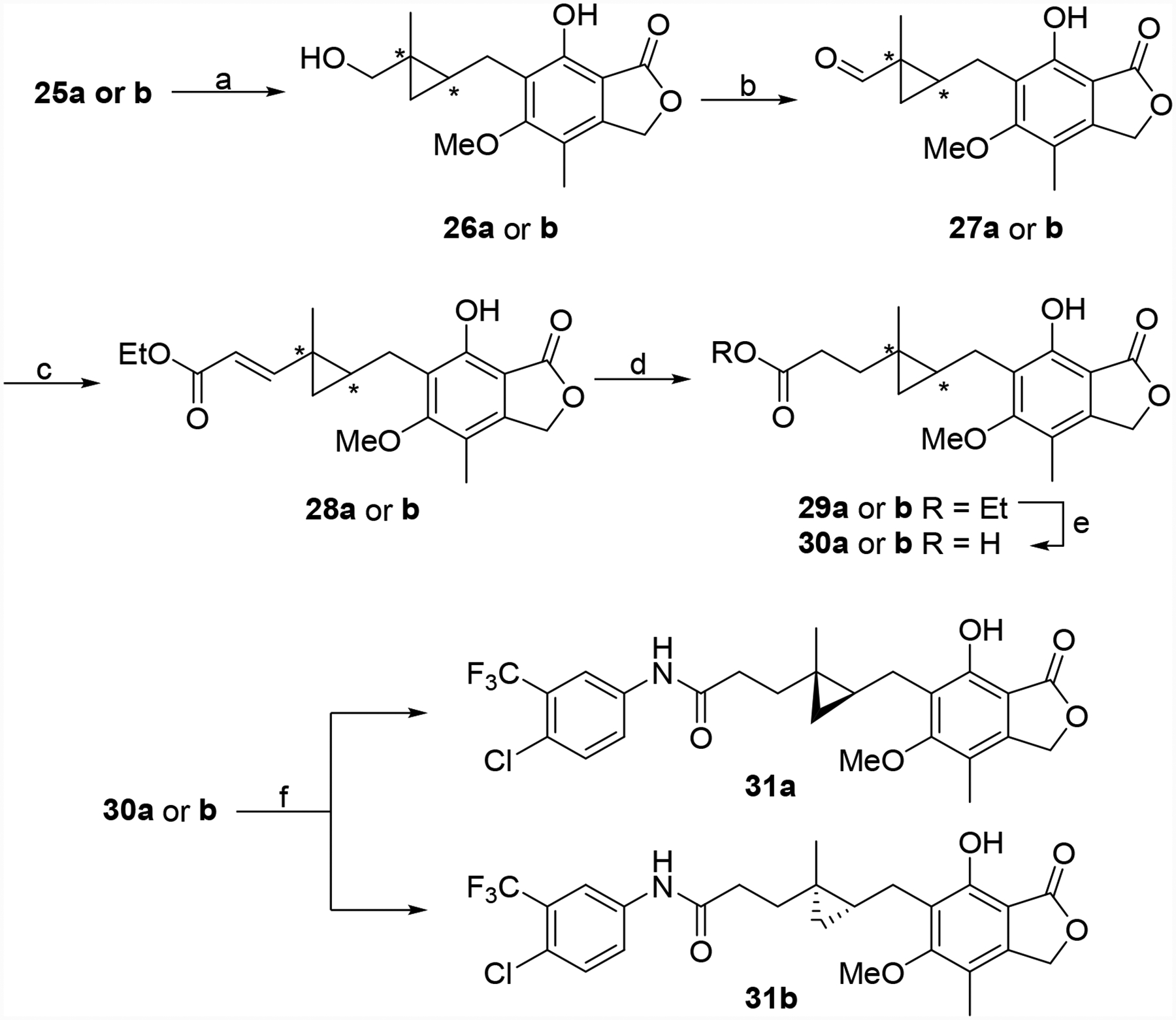
Synthesis of 31a and 31b. Reagents and conditions: (a) i) Tf2O, pyridine, DCM, −20 °C to rt; ii) DMF, pyridine, H2O, 120 °C, 10 min, 81% and 93%. (b) DMP, DCM, rt, 0.5 h, 99%, and 83%. (c) triethyl phosphonoacetate, NaH, benzene, rt, 1 h, 57% and 51% (d) CoCl2, NaBH4, MeOH/DMF, rt, 0.5 h, 62% and 87% (e) LiOH, THF/H2O, rt, 3 h, 86% and 85%. (f) 4-chloro-3-(trifluoromethyl)aniline, EDC•HCl, HOAt, DMF, rt, 16 h, 81% and 71%.
Several other derivatives were prepared that incorporated additional changes to the linker region of the hybrid molecules. MPA (7) was cleaved to aldehyde 32, which was reduced and protected to give 34 (Scheme 5). Alkylation of the primary alcohol, ester hydrolysis, aniline coupling and deprotection produced the ether linked derivative 38.35 MPA (7) was also converted to aldehyde 40a, which via a Wittig reaction with 4-ClPhNH(C=O)CH2P+Ph3Cl− gave 41a (Scheme 6).36, 37 Similarly, 34 was oxidized to aldehyde 40b, which was converted into 41b. These two intermediates were de-protected or reduced/de-protected to provide 42a and 42b, respectively. Finally, intermediate 32 was oxidized to carboxylic acid 43, which was coupled to 4-chloroaniline to give the truncated derivative 44 (Scheme 7).
Scheme 5.
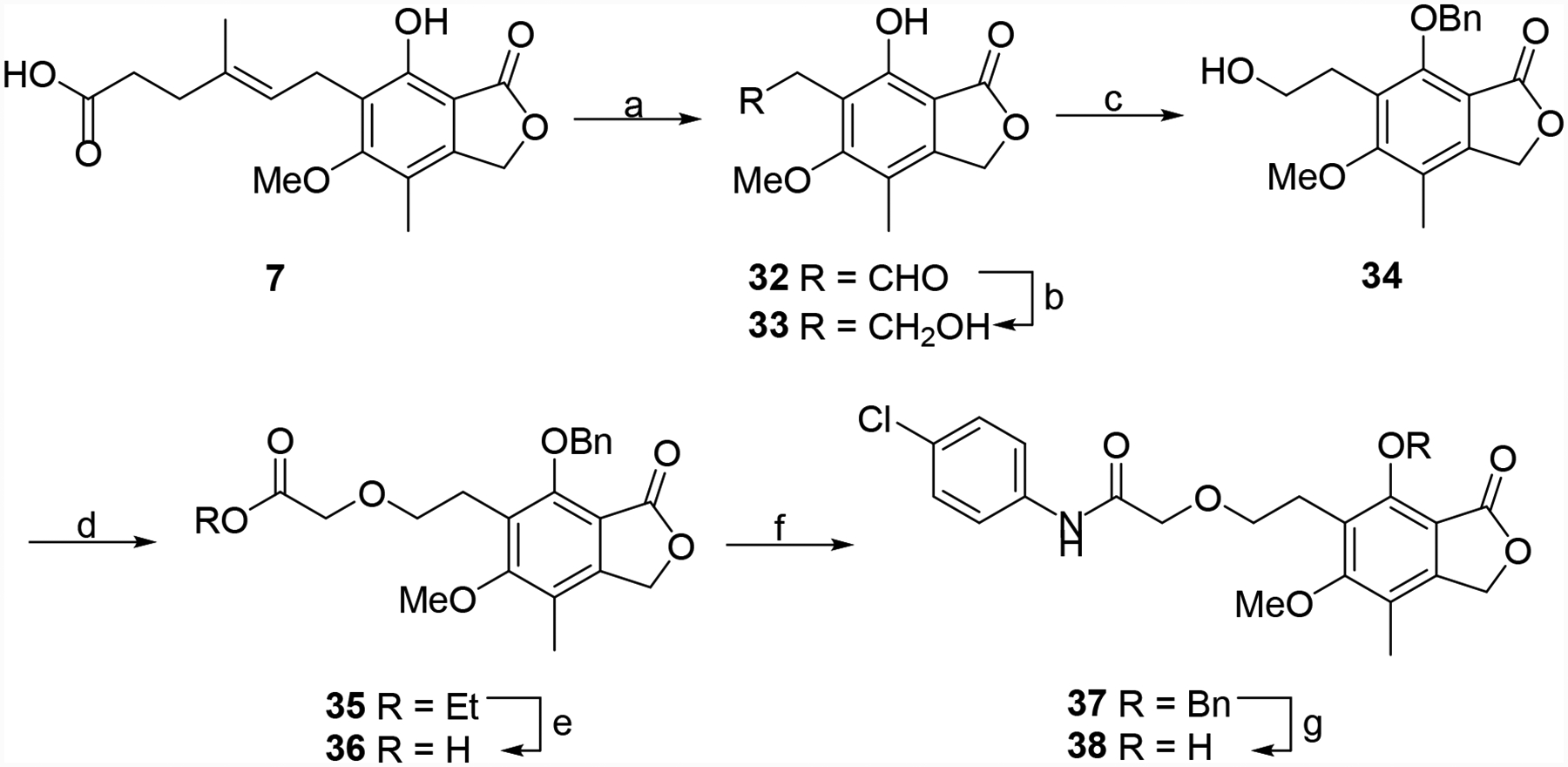
Synthesis of 38. Reagents and conditions: (a) OsO4, NMO, NaIO4, THF/H2O, rt, 1.5 h. (b) NaBH4, EtOH, rt, 3 h, 70–81%. (c) benzyl bromide, TBAF, rt, 6.5 h, 81–89%. (d) InCl3, ethyl diazoacetate, DCM, rt, 16 h, 57%. (e) LiOH, MeOH/H2O, rt, 3 h, 76–95%. (f) EDC•HCl, HOAt, 4-chloroaniline, DMF, rt, 16 h, 65–80%. (g) 10% Pd/C, H2 (1 atm), MeOH, rt, 1 h, 27–56%.
Scheme 6.
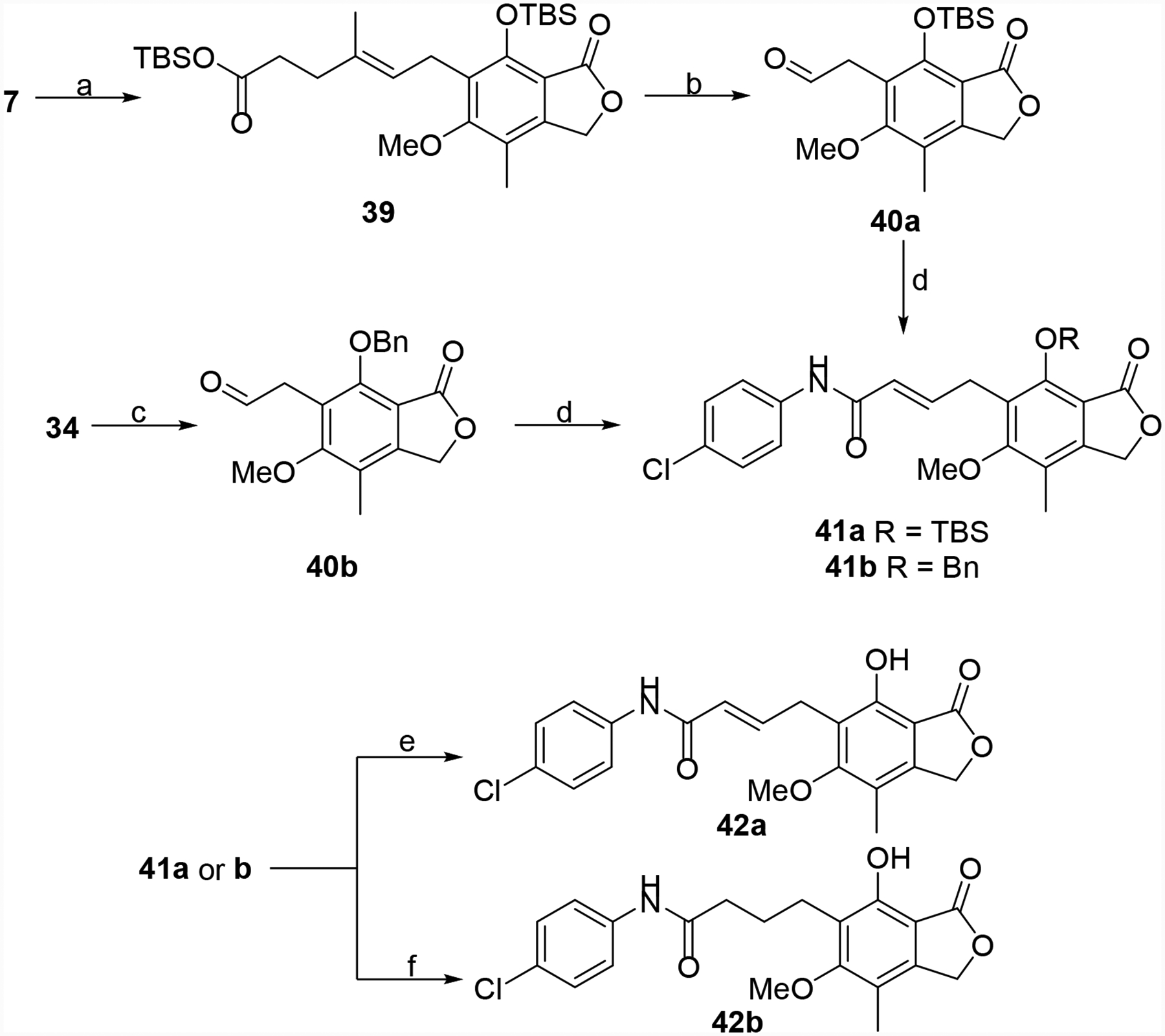
Synthesis of 42a and 42b. Reagents and conditions: (a) TBSCl, imidazole, DMF, rt, 6 h, 66%. (b) OsO4, NMO, NaIO4, THF/H2O, rt, 1.5 h, 70%. (c) DMP, DCM, rt, 3 h, 80–88%. (d) LDA, 4-ClPhNH(C=O)CH2P+Ph3Cl− (see Supporting Information compound S2), THF/toluene, 0°C to rt, 3 h, 73% and 51% (e) TBAF, THF, 0 °C to rt, 0.5 h, 22% (f) 10% Pd/C, H2 (1 atm), MeOH, rt, 1 h, 46%.
Scheme 7.
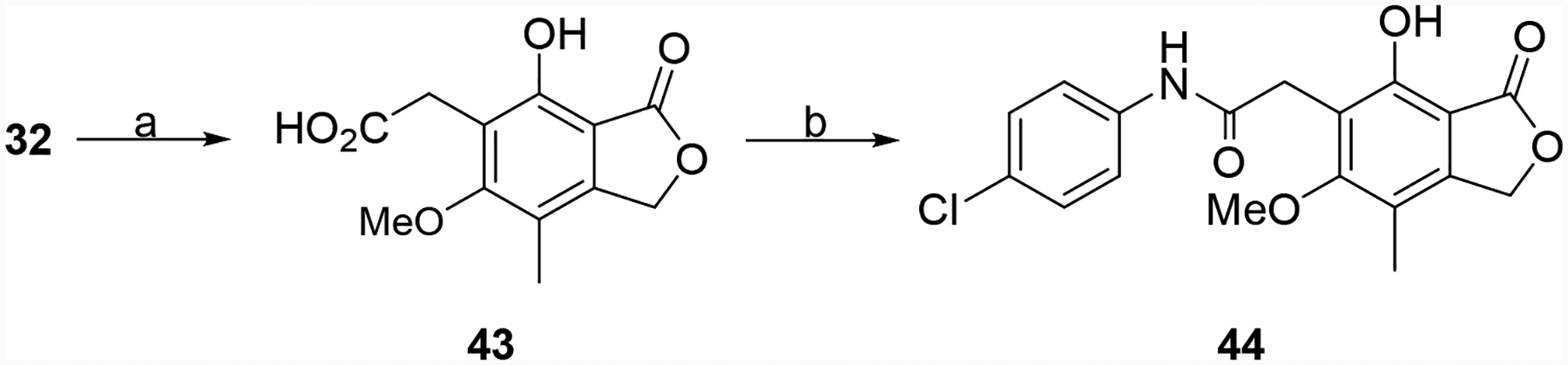
Synthesis of 44. Reagents and conditions: (a) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH/H2O, rt, 4 h, 40 mg (b) EDC, DIPEA, 4-chloroaniline, THF/DMF, rt, 16 h, 38%.
Recombinant CpIMPDH and HsIMPDH2 were purified from E. coli.38 Enzyme activity was determined by monitoring the production of NADH.39 For CpIMPDH, enzyme (10 nM) and inhibitor (1 nM to 5 μM) were incubated in the presence of 50 mM Tris-HCl, pH 8.0, 100 mM KCl, 3 mM EDTA, 1 mM dithiothreitol at 25 °C for 5 min prior to addition of substrates NAD (300 μM) and IMP (250 μM). For HsIMPDH2, enzyme (20 nM) and inhibitor (1 nM to 5 μM) were incubated in the presence of 50 mM Tris-HCl, pH 8.0, 100 mM KCl, 3 mM EDTA, 1 mM dithiothreitol at 25 °C for 5 min prior to addition of substrates NAD (60 μM) and IMP (250 μM).41 Production of NADH was monitored by fluorescence. Ki,app values were determined for each inhibitor against CpIMPDH and HsIMPDH2 using Dynafit.40
Introducing an anilide as the common fragment to MPA improved CpIMPDH inhibition. For example, 8, which incorporated the anilide from 4 with MPA, potently inhibited CpIMPDH with Ki,app value of 0.046 μM (Table 1). Interestingly, the compound also inhibited HsIMPDH2 (Ki,app = 0.35 μM), in contrast to all prior reported CpIMPDH inhibitors. Since electron withdrawing groups were preferred in previously developed CpIMPDH inhibitors, the methoxy group in 8 was replaced with a chloro (9). This compound showed similar inhibitory potency as 8 for both enzymes. However, replacing with a trifluoromethyl (10) increased potency against both enzymes. Transposing the para-chloro to the ortho-position (11) resulted in loss of CpIMPDH inhibitory activity. This was consistent with the SAR of other CpIMPDH inhibitors, possibly resulting from a clash with Y358’ and loss of a halogen bond with G357’.9 A survey of various electron withdrawing groups at the 4-position demonstrated that chloro (12) provided potent and balanced CpIMPDH and HsIMPDH2 inhibition. Reminiscent of other CpIMPDH inhibitors, methylation of the anilide (16) resulted in loss of activity. Similarly, methylation of the phenol (17), known to eliminate MPA inhibition of HsIMPDH,41 also resulted in loss of CpIMPDH inhibition.
Table 1.
CpIMPDH and HsIMPDH inhibitory activities of 8 – 17.
| Compound | R1 | R2 | R3 |
CpIMPDH Ki,app (μM) |
HsIMPDH Ki,app (μM) |
|---|---|---|---|---|---|
| 8 | 3-OMe, 4-Cl | H | H | 0.046 (±0.013)σ | 0.35 (±0.09)σ |
| 9 | 3,4-di-Cl | H | H | 0.041 (±0.010)σ | 0.34 (± 0.11)σ |
| 10 | 3-CF3, 4-Cl | H | H | 0.016 (±0.007)σ | 0.23 (±0.08)σ |
| 11 | 2,3-di-Cl | H | H | 0.681 (0.143)r | ND* |
| 12 | 4-Cl | H | H | 0.042 (±0.02)σ | 0.13 (±0.05)σ |
| 13 | 4-F | H | H | 0.180 (±0.06)r | 0.27 |
| 14 | 4-CF3 | H | H | 0.151 (±0.04)r | 0.40 |
| 15 | 4-CN | H | H | 0.11 | 0.35 |
| 16 | 4-Cl | Me | H | 0.87 (±0.13)r | ND* |
| 17 | 3-CF3 4-Cl | H | Me | 1.7 | ND* |
ND: Not Determined
Standard deviation
Range
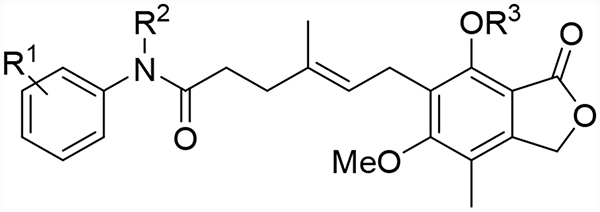
Next, the central linker was explored using various bioisosteres and by truncation (Table 2). 3-Trifluoromethyl-4-chloro or 4-chloro substituted anilides were chosen based on 10 and 12. Reduction of the alkene (18) resulted in some loss of potency for CpIMPDH inhibition, which was more dramatic for HsIMPDH2. Bioisosteric replacement of the alkene with a cyclopropane revealed that one enantiomer (31b) retained potent CpIMPDH inhibitory activity (Ki,app = 0.066 μM), although HsIMPDH2 inhibition was reduced (Ki,app = 0.46 μM). The eudysmic ratios of 31b and 31a for CpIMPDH and HsIMPDH inhibitions were 8.6 and 3.1, respectively. Several additional changes to the linker connecting the two aryl groups resulted in reduced CpIMPDH and HsIMPDH2 inhibitory activities.
Table 2.
CpIMPDH and HsIMPDH inhibitory activities of 18, 31a, 31b, 38, 42a, 42b and 44.
| Compound | R1 | X |
CpIMPDH Ki,app (μM) |
HsIMPDH Ki,app (μM) |
|---|---|---|---|---|
| 18 | 3-CF3, 4-Cl |
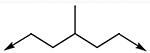
|
0.060 (±0.01)r | ND* |
| 31a | 3-CF3, 4-Cl |
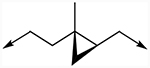
|
0.57 | 1.40 |
| 31b | 3-CF3, 4-Cl |
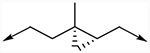
|
0.066 (±0.023)σ | 0.46 (±0.28)σ |
| 38 | 4-Cl |
|
0.405 (±0.176)r | 0.87 |
| 42a | 4-Cl |
|
0.48 | 0.55 |
| 42b | 4-Cl |
|
0.45 (±0.13)r | 0.29 |
| 44 | 4-Cl |
|
2.8 | 0.8 |
ND: Not Determined
Standard deviation
Range
Two critical differences between HsIMPDH2 and CpIMPDH are in a loop structure of the NAD binding region and a proximal site in the adjacent subunit. The A-site of HsIMPDH2 is largely within the same monomer as the IMP site, but for CpIMPDH and related prokaryotic IMPDHs, a portion of the A-site is located in the adjacent subunit.7, 8 This hydrophobic pocket is absent in HsIMPDH.42 As illustrated in the sequence alignments of HsIMPDH2 and CpIMPDH (Figure 2), the NAD loop of HsIMPDH contains a hydrophilic serine at residue 276, while CpIMPDH has a hydrophobic alanine at the equivalent position (residue 165’; note that CpIMPDH lacks an approximately 100 residue regulatory domain present in most IMPDHs). For the A-site, there are also distinct residue differences with aspartic acid (e.g. 470’) in HsIMPDH corresponding to tyrosine (e.g. Y358’) in CpIMPDH.42 Interaction with this tyrosine residue has previously been shown to be critical for achieving selective CpIMPDH inhibitors.9–17, 27 Interestingly, a number of other microorganisms, including several pathogenic Gram-(+) and Gram-(−) bacteria listed in Figure 2, also have alanine and tyrosine residues in these two positions and are inhibited by CpIMPDH inhibitors.27
Figure. 2.

Sequence alignment of IMPDH enzymes from human, C. parvum and several bacteria highlighting the NAD loop and A-site. aY358’ is based on numbering for CpIMPDH and the ‘ denotes the adjacent subunit.
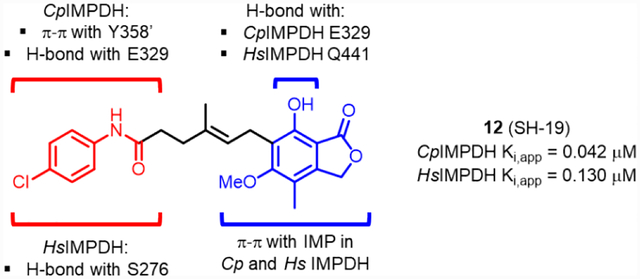
In order to understand potential binding modes of MPA-anilides to CpIMPDH and HsIMPDH, molecular docking studies were conducted using Autodock Tools. Docking of 12 into CpIMPDH (PDB: 3KHJ) provided a low energy pose that satisfied the three key interactions of previously co-crystalized selective CpIMPDH inhibitors. Specifically, the phenol portion of MPA had a π-π interaction with the hypoxanthine of IMP, the anilide NH formed a hydrogen bond with Glu329 and the anilide extended into the adjacent subunit forming a π-π interaction with Tyr358’ (Figure 3A). Additionally, the phenol OH formed an ionic-dipole interaction with Glu329. This binding mode is also consistent with several of the observed structure-activity relationship features, such as alkylation of the anilide or phenol disrupting interactions with Glu329. In addition, truncation of the linker would result in an inability of the inhibitor to extend into the other subunit to provide the critical π-π interaction with Tyr358’. Overlay of this docking model with the co-crystal structure of CpIMPDH•IMP•5 (PDB: 4RVB) further illustrates that 12 can readily be accommodated in a binding mode similar to selective CpIMPDH inhibitors (see Supporting Information, Figure S1A).10
Figure 3.
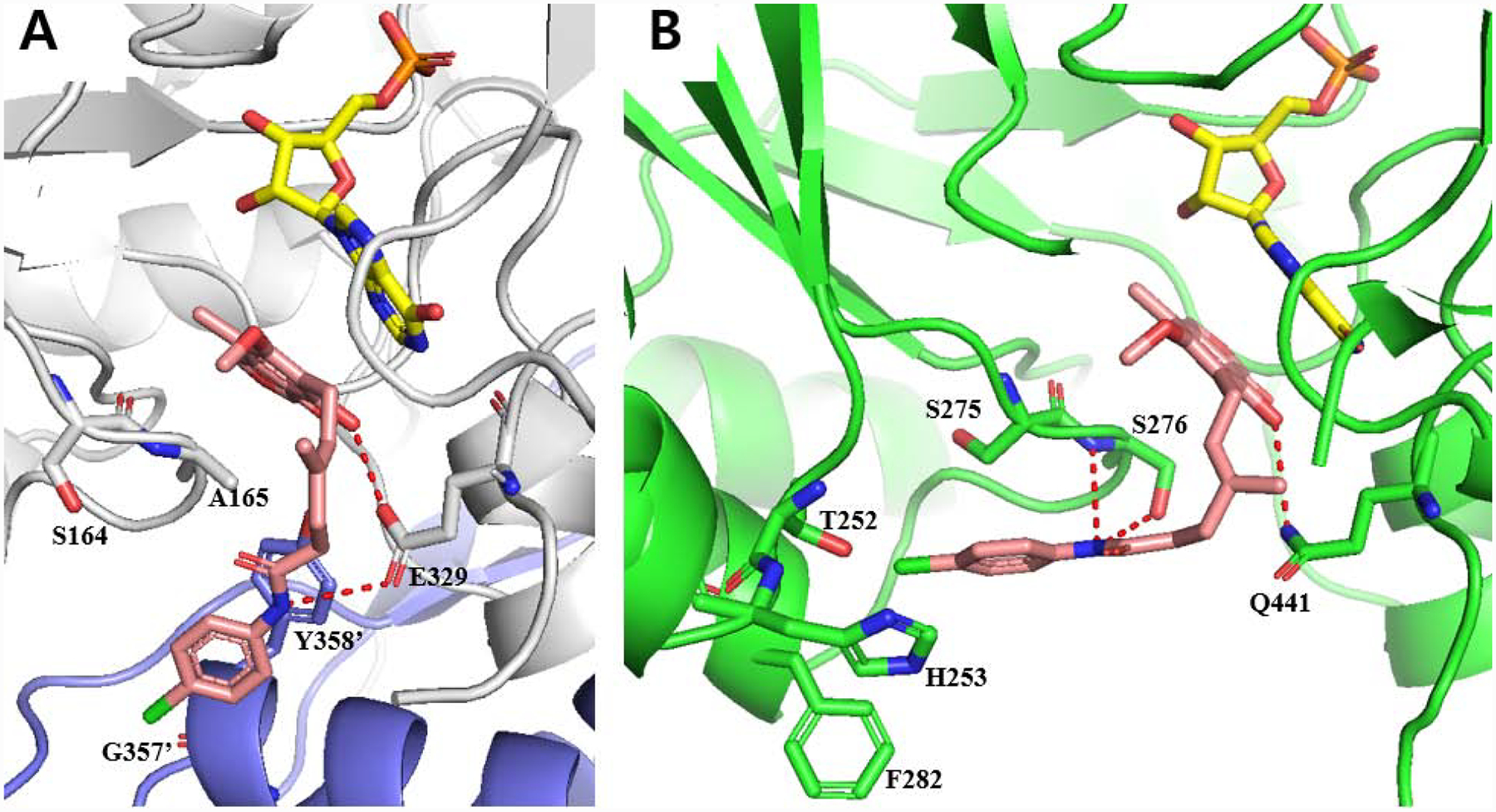
(A) Docked model of 12 (pink) with CpIMPDH (gray, PDB: 3KHJ) and IMP (yellow). Adjacent monomer protein is shown as purple and residue numbers are differentiate by prime (‘). Hydrogen bonds are shown as red dashes (< 3.0 Å). Docking score of 12 with CpIMPDH•IMP was −9.86. (B) Docked structure of 12 (pink) with hamster IMPDH2 (green, PDB: 1JR1) and IMP (yellow), which has only eight non-binding site amino acid differences compared to HsIMPDH243. Hydrogen bonds are shown as red dashes (< 3.5 Å). Docking score of 12 with hamster IMPDH2•IMP was −8.83. Water, K+ and other protein subunits were deleted for docking and presentation for clarity.
Docking of 12 into hamster IMPDH2 (PDB : 1JR1), which has only eight non-binding site amino acid differences43 compared to HsIMPDH2, provided a low energy pose with similar interactions as MPA (7), including H-bonding of the anilide NH and phenol to Ser276 in the NAD binding site and Gln441, respectively (Figure 3B; for an overlay of the docked structure of 12 with hamster IMPDH2•IMP•7 see Supporting Information, Figure S1B). The parachlorophenyl occupies a modestly large pocket created by Thr252, His253, Phe282 and Ser275, as well as being within 3.2 Å of the methylene portion of this later residue’s side-chain. Interestingly, attempts to dock a CpIMPDH selective inhibitor (e.g. 5) into hamster IMPDH2 did not produce reasonable binding modes with low binding energies (data not shown). This could result from the compound being less flexible and not being able to form a productive interaction with Ser276 in the NAD site. Collectively, these data elucidated an additional structural criterion for achieving CpIMPDH inhibitor selectivity: the inability to form interactions with Ser276.
Since MPA anilides inhibit CpIMPDH, we also assessed inhibition of Mycobacterium tuberculosis (Mtb) IMPDH that likewise has alanine in the NAD loop and tyrosine in the A-site (Figure 2). Similar to other active CpIMPDH inhibitors, compound 10 potently blocked MtbIMPDH (Ki,app = 0.060 μM).
In conclusion, mycophenolic anilides were found that inhibit both CpIMPDH and HsIMPDH by incorporation of a molecular fragment from previously reported CpIMPDH inhibitors. Prior studies combined with molecular docking assessments revealed that selectivity for microorganism IMPDHs (e.g. those with alanine in the NAD loop and Y358’ in the adjacent subunit) requires two distinct design elements: 1) interaction with the adjacent subunit via π-π interactions with Y358’ and 2) lack of interactions with Ser276 in the NAD binding site of HsIMPDH. These two features provide further guidance for generating selective IMPDH inhibitors for a subset of susceptible microorganisms.
Supplementary Material
Acknowledgements
This work was supported in part by a grant from the National Institutes of Health (R01AI125362).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at.
References
- 1.Hedstrom L IMP dehydrogenase: structure, mechanism, and inhibition. Chem Rev. 2009;109(7): 2903–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hedstrom L, Liechti G, Goldberg JB, Gollapalli DR. The antibiotic potential of prokaryotic IMP dehydrogenase inhibitors. Curr Med Chem. 2011;18(13): 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makowska-Grzyska M, Kim Y, Gorla SK, et al. Mycobacterium tuberculosis IMPDH in Complexes with Substrates, Products and Antitubercular Compounds. PLoS One. 2015;10(10): e0138976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeswanth S, Kumar CL, Swarupa V, et al. Characterization of inosine monophosphate dehydrogenase from Staphylococcus aureus ATCC12600 and its involvement in biofilm formation. 2013;11: 12. [Google Scholar]
- 5.Striepen B, Pruijssers AJ, Huang J, et al. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc Natl Acad Sci U S A. 2004;101(9): 3154–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Striepen B, White MW, Li C, et al. Genetic complementation in apicomplexan parasites. Proc Natl Acad Sci U S A. 2002;99(9): 6304–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makowska-Grzyska M, Kim Y, Maltseva N, et al. A novel cofactor-binding mode in bacterial IMP dehydrogenases explains inhibitor selectivity. J Biol Chem. 2015;290(9): 5893–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colby TD, Vanderveen K, Strickler MD, Markham GD, Goldstein BM. Crystal structure of human type II inosine monophosphate dehydrogenase: implications for ligand binding and drug design. Proc Natl Acad Sci U S A. 1999;96(7): 3531–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Z, Khan J, Makowska-Grzyska M, et al. Synthesis, in vitro evaluation and cocrystal structure of 4-oxo-[1]benzopyrano[4,3-c]pyrazole Cryptosporidium parvum inosine 5’-monophosphate dehydrogenase (CpIMPDH) inhibitors. J Med Chem. 2014;57(24): 10544–10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim Y, Makowska-Grzyska M, Gorla SK, et al. Structure of Cryptosporidium IMP dehydrogenase bound to an inhibitor with in vivo antiparasitic activity. Acta Crystallogr F Struct Biol Commun. 2015;71(Pt 5): 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson CR, Gorla SK, Kavitha M, et al. Phthalazinone inhibitors of inosine-5’-monophosphate dehydrogenase from Cryptosporidium parvum. Bioorg Med Chem Lett. 2013;23(4): 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorla SK, Kavitha M, Zhang M, et al. Optimization of benzoxazole-based inhibitors of Cryptosporidium parvum inosine 5’-monophosphate dehydrogenase. J Med Chem. 2013;56(10): 4028–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirubakaran S, Gorla SK, Sharling L, et al. Structure-activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg Med Chem Lett. 2012;22(5): 1985–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorla SK, Kavitha M, Zhang M, et al. Selective and potent urea inhibitors of cryptosporidium parvum inosine 5’-monophosphate dehydrogenase. J Med Chem. 2012;55(17): 7759–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharling L, Liu X, Gollapalli DR, Maurya SK, Hedstrom L, Striepen B. A screening pipeline for antiparasitic agents targeting cryptosporidium inosine monophosphate dehydrogenase. PLoS Negl Trop Dis. 2010;4(8): e794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gollapalli DR, Macpherson IS, Liechti G, Gorla SK, Goldberg JB, Hedstrom L. Structural determinants of inhibitor selectivity in prokaryotic IMP dehydrogenases. Chem Biol. 2010;17(10): 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maurya SK, Gollapalli DR, Kirubakaran S, et al. Triazole inhibitors of Cryptosporidium parvum inosine 5’-monophosphate dehydrogenase. J Med Chem. 2009;52(15): 4623–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hager PW, Collart FR, Huberman E, Mitchell BS. Recombinant human inosine monophosphate dehydrogenase type I and type II proteins. Purification and characterization of inhibitor binding. Biochem Pharmacol. 1995;49(9): 1323–1329. [DOI] [PubMed] [Google Scholar]
- 19.Franklin TJ, Cook JM. The inhibition of nucleic acid synthesis by mycophenolic acid. Biochem J. 1969;113(3): 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carr SF, Papp E, Wu JC, Natsumeda Y. Characterization of human type I and type II IMP dehydrogenases. J Biol Chem. 1993;268(36): 27286–27290. [PubMed] [Google Scholar]
- 21.van Gelder T, Hesselink DA. Mycophenolate revisited. Transpl Int. 2015;28(5): 508–515. [DOI] [PubMed] [Google Scholar]
- 22.Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. Cryptosporidium parvum IMP dehydrogenase: identification of functional, structural, and dynamic properties that can be exploited for drug design. J Biol Chem. 2004;279(39): 40320–40327. [DOI] [PubMed] [Google Scholar]
- 23.Pankiewicz KW, Lesiak-Watanabe KB, Watanabe KA, et al. Novel mycophenolic adenine bis(phosphonate) analogues as potential differentiation agents against human leukemia. J Med Chem. 2002;45(3): 703–712. [DOI] [PubMed] [Google Scholar]
- 24.Chen L, Wilson D, Jayaram HN, Pankiewicz KW. Dual inhibitors of inosine monophosphate dehydrogenase and histone deacetylases for cancer treatment. J Med Chem. 2007;50(26): 6685–6691. [DOI] [PubMed] [Google Scholar]
- 25.Shah CP, Kharkar PS. Newer human inosine 5’-monophosphate dehydrogenase 2 (hIMPDH2) inhibitors as potential anticancer agents. J Enzyme Inhib Med Chem. 2018;33(1): 972–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuny GD, Suebsuwong C, Ray SS. Inosine-5’-monophosphate dehydrogenase (IMPDH) inhibitors: a patent and scientific literature review (2002–2016). Expert Opin Ther Pat. 2017;27(6): 677–690. [DOI] [PubMed] [Google Scholar]
- 27.Mandapati K, Gorla SK, House AL, et al. Repurposing cryptosporidium inosine 5’-monophosphate dehydrogenase inhibitors as potential antibacterial agents. ACS Med Chem Lett. 2014;5(8): 846–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Wang S, Crispino GA, Tenhuisen K, Singh A, Grosso JAJTl. Selective removal of a benzyl protecting group in the presence of an aryl chloride under gaseous and transfer hydrogenolysis conditions. Tetrahedron Lett. 2003;44(21): 4041–4043. [Google Scholar]
- 29.Charette AB, Turcotte N, Côté BJJocc. One-pot synthesis of substituted allyl-α-D-glucopyranosides by an in situ anomerization protocol. J Carbohydr Chem. 1994;13(3): 421–432. [Google Scholar]
- 30.Pilgrim W, Murphy PV. SnCl(4)- and TiCl(4)-catalyzed anomerization of acylated O- and S-glycosides: analysis of factors that lead to higher alpha:beta anomer ratios and reaction rates. J Org Chem. 2010;75(20): 6747–6755. [DOI] [PubMed] [Google Scholar]
- 31.Charette AB, Turcotte N, Marcoux J-FJTl. The use of α-d-glucopyranosides as surrogates for the β-l-glucopyranosides in the stereoselective cyclopropanation reaction. Tetrahedron Lett. 1994;35(4): 513–516. [Google Scholar]
- 32.Charette AB, Cote BJJotACS. Stereoselective synthesis of all four isomers of coronamic acid: a general approach to 3-methanoamino acids. J Am Chem Soc. 1995;117(51): 12721–12732. [Google Scholar]
- 33.Charette AB, Cote BJTJoOC. Asymmetric cyclopropanation of allylic ethers: cleavage and regeneration of the chiral auxiliary. J Org Chem. 1993;58(4): 933–936. [Google Scholar]
- 34.Vega-Pérez JM, Periñán I, Palo-Nieto C, Vega-Holm M, Iglesias-Guerra FJTA. Alkenyl β-dgalactopyranoside derivatives as efficient chiral templates in stereoselective cyclopropanation and epoxidation reactions. Tetrahedron-Asymmetry. 2010;21(1): 81–95. [Google Scholar]
- 35.Krishna PR, Prapurna YL, Alivelu MJTl. InCl3 catalyzed carbene insertion into O–H bonds: efficient synthesis of ethers. Tetrahedron Lett. 2011;52(27): 3460–3462. [Google Scholar]
- 36.Zhaowen L, Li Z, Chunfen X, Yong Y, Fanbo Z, Kaixun HJMCR. Anticancer activities of some arylcarbamoylalkyltriphenylphosphonium chlorides. Med Chem Res. 2007;16(7–9): 380–391. [Google Scholar]
- 37.Soli ED, Braun MPJJoLC, Society RTOJotII. Synthesis of [phenyl-U-14C] aryl and [8–14C] carboxy labeled tracers of vorinostat. J Labelled Compd Rad. 2006;49(5): 437–443. [Google Scholar]
- 38.Umejiego NN, Gollapalli D, Sharling L, et al. Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against cryptosporidiosis. Chem Biol. 2008;15(1): 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farazi T, Leichman J, Harris T, Cahoon M, Hedstrom L. Isolation and characterization of mycophenolic acid-resistant mutants of inosine-5’-monophosphate dehydrogenase. J Biol Chem. 1997;272(2): 961–965. [DOI] [PubMed] [Google Scholar]
- 40.Kuzmic P Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237(2): 260–273. [DOI] [PubMed] [Google Scholar]
- 41.Mitsuhashi S, Takenaka J, Iwamori K, Nakajima N, Ubukata M. Structure-activity relationships for inhibition of inosine monophosphate dehydrogenase and differentiation induction of K562 cells among the mycophenolic acid derivatives. Bioorg Med Chem. 2010;18(22): 8106–8111. [DOI] [PubMed] [Google Scholar]
- 42.Macpherson IS, Kirubakaran S, Gorla SK, et al. The structural basis of Cryptosporidium -specific IMP dehydrogenase inhibitor selectivity. J Am Chem Soc. 2010;132(4): 1230–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collart FR, Huberman E. Cloning and sequence analysis of the human and Chinese hamster inosine-5’-monophosphate dehydrogenase cDNAs. J Biol Chem. 1988;263(30): 15769–15772. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.