

Abstract
A growing number of G-protein-coupled receptor (GPCR) structures reveal novel transmembrane lipid-exposed allosteric sites. Ligands must first partition into the surrounding membrane and take lipid paths to these sites. Remarkably, a significant part of the bound ligands appears exposed to the membrane lipids. The experimental structures do not usually account for the surrounding lipids, and their apparent contribution to ligand access and binding is often overlooked and poorly understood. Using classical and enhanced molecular dynamics simulations, we show that membrane lipids are critical in the access and binding of ORG27569 and its analogs at the transmembrane site of cannabinoid CB1 receptor. The observed differences in the binding affinity and cooperativity arise from the functional groups that interact primarily with lipids. Our results demonstrate the significance of incorporating membrane lipids as an integral component of transmembrane sites for accurate characterization, binding-affinity calculations, and lead optimization in drug discovery.
Introduction
Membrane proteins such as G-protein-coupled receptors (GPCRs) and ion channels constitute therapeutic targets for more than 50% of the FDA-approved drugs.1−3 Recent developments in the structural biology of integral membrane proteins, such as advances in X-ray crystallography, cryo-electron microscopy (cryo-EM), protein stabilization techniques, and the use of lipid nanodisc approaches, resulted in an increasingly large number of structures in complex with agonists, antagonists, and allosteric modulators.4,5 Remarkably, many structures offer unprecedented details on the existence of transmembrane allosteric sites that can be reached only by lipid paths.6−8 These transmembrane allosteric sites appear to have significantly different characteristics from druggable orthosteric binding sites that are typically accessible from the aqueous phase. The geometrical and physicochemical features of these shallow pockets, such as volume, compactness, solvent-accessible surface area (SASA), and distribution of polar and nonpolar residues, do not entirely conform to the typical characteristics of druggable sites.9−14 For lipid-facing transmembrane sites, binding of ligands often does not involve desolvation or removal of water molecules from the site but is preceded by the relocation of lipids that are directly in contact with the site residues. Most fascinatingly, ligands bound at these sites remain significantly exposed to the membrane lipids. A recent study showed that for a set of allosteric ligands bound at the transmembrane sites of class A and class B GPCRs, approximately 40–55% of their nonpolar SASA was exposed to the membrane lipids, whereas a large part (50–90%) of their polar SASA was buried within the sites.8 These findings raise an important question on the exact contribution of lipids toward the binding affinity of ligands. Lipids appear to actively participate in the ligand-binding process at these sites and thus seem to potentially affect the dissociation rates (koff) and residence time.15 Importantly, to reach transmembrane lipid-facing sites, a ligand must first partition into the membrane and take lipid-mediated paths. Therefore, the local membrane concentration of the ligand, determined by its relative affinity for the membrane versus the aqueous phase, and the actual volume of the membrane containing the ligand would affect the association rate (kon) of the binding process and thus the overall binding kinetics.16−18
Membrane lipids have been shown to actively participate in the access and binding of ligands to transmembrane orthosteric and allosteric sites of many integral membrane proteins. This topic has been extensively discussed in recent reviews.7,8 Lipid pathways have been described for several ligands binding to various GPCRs such as cannabinoid,19−21 sphingosine-1-phosphate,22 rhodopsin,23 protease-activated receptor-1,24 β2 adrenergic receptor,25,26 dopamine D3,27 angiotensin II receptor type 1,28 and P2Y purinoceptor 129 indicating a crucial role of the membrane in enabling access to receptor binding sites. The plasma membrane can act as a “reservoir” or “depot” for lipophilic and amphiphilic drugs, such as salmeterol, a β2 adrenergic receptor agonist, which on dissociation, partitions back into the membrane, enabling rebinding and thereby prolonging the duration of action.26,30 The lipid bilayer has also been shown to “preconfigure” molecules in the orientation and conformation favorable for binding by reducing the dimensionality of the ligands as it takes a two-dimensional lateral diffusion route to access the binding sites.29,31,32
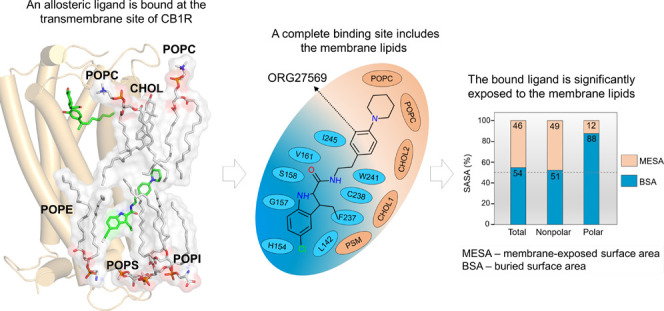
Recently, a crystal structure of cannabinoid CB1 receptor (CB1R) was reported with an agonist, CP55940, bound to the orthosteric site and a negative allosteric modulator (NAM), ORG27569, bound to a lipid-facing extrahelical transmembrane site located near the inner leaflet of the membrane33 (Figure 1 and Supporting Table S1). This extrahelical allosteric binding site is located between transmembrane helices (TMH) 1, 2, and 4 and overlaps with the conserved cholesterol-binding site observed in many GPCRs. Surprisingly, a significant part of the NAM, ORG27569, in its crystal-bound mode, is exposed to the membrane environment and in direct contact with the membrane lipids, suggesting a critical role of the surrounding lipids in stabilizing the ligand at the binding site. Since the discovery of ORG27569 in 2005,34 several ligand-based structure–activity relationship (SAR) studies35−38 have been carried out to identify functional groups that are critical for the allosteric activity and substitutions that affect the receptor binding affinity (Supporting Table S2). It is important to note that these (SAR) studies were conducted without the knowledge that ORG27569 binds to the transmembrane allosteric site. In addition to the binding affinity (the equilibrium dissociation constant, KB), these studies also reported the cooperativity factor (α), a parameter quantifying the effect of allosteric modulators on the binding affinity of the agonist at the orthosteric site. α values greater than 1.0 indicate that the allosteric modulator increases the agonist binding (positive allosteric modulation).
Figure 1.

Orthosteric and allosteric binding sites of CB1R. (A) Extracellular view of the orthosteric and allosteric sites of CB1R (PDB ID 6KQI). Important orthosteric site residues S1732.60, V1963.32, I267ECL2, W3566.48, and S3837.39, are shown in licorice representations (yellow). The CB1R agonist, CP55940 (purple), is shown in the orthosteric site, while the allosteric NAM, ORG27569, is shown in combined licorice and surface representations (light blue). (B) Membrane view of the orthosteric and allosteric binding sites of CB1R. Some of the key allosteric site residues, H1542.41, G1572.44, V1612.48, V2344.43, and W2414.50, are shown in the licorice representation (magenta). ORG27569 is shown in light representation (light blue), while the agonist is shown in combined licorice and surface representations.
In this study, using classical and enhanced molecular dynamics (MD) simulation techniques, we show that the membrane lipids are an integral part of the transmembrane allosteric site of CB1R and significantly contribute to the access and binding of ORG27569 and its structural analogs. The obtained results reveal that the experimentally observed differences in the binding affinity and cooperativity among the studied compounds may, at least in part, arise from the differences in functional groups that largely interact with the lipids. The binding free energies calculated using the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method provide valuable insights into the contribution of individual binding site residues and lipid molecules toward the binding affinity.
Results
Transmembrane Allosteric Site Has Significantly Different Physicochemical Properties and Lower Druggability than the Orthosteric Site
The cannabinoid CB1 receptor (CB1R), like other class A GPCRs, has a well-defined and deeply embedded orthosteric binding site located at the center of the transmembrane helices (Figure 1A). When not occupied, the binding pocket is solvated with water and is generally accessible to ligands from the extracellular aqueous bulk solution. The binding of ligands should be preceded by the desolvation of water molecules from the site and the ligands. In contrast, the lipid-facing extrahelical allosteric site (where ORG27569 binds) is located between and around TMH 1, 2, and 4, near the core and lower leaflet of the bilayer (Figure 1B). In the ligand-free state, this site is occupied by membrane lipids. A ligand must first partition into the membrane and take a lipid path to reach this site. Also, the binding of ligands at this site involves the removal of the lipid molecules that occupy the site. The difference in the locations, geometry, and (de)solvation characteristics of the orthosteric and transmembrane allosteric pockets are intriguing. To quantify the differences, we used fpocket39 to calculate the various properties of these binding sites (Supporting Table S3). The orthosteric site had a high druggability score (0.9) as well as large volume and solvent-accessible surface area (SASA), typical characteristics of druggable binding sites. However, notably, the allosteric site had a low druggability score (0.1) and smaller values for descriptors such as pocket volume and SASA. The druggability score, which assumes any numerical value between 0 and 1, indicates the likelihood of a binding pocket being druggable by small-molecule ligands. This druggability score is calculated by a logistic regression function using the pocket descriptors such as the normalized mean local hydrophobic density, the residue-based mean hydrophobicity score, and the normalized polarity score. Further details on the score can be found elsewhere.11
The motivation and rationale for this study came from the fact that this transmembrane binding site would typically be classified undruggable considering merely the binding site residues, and therefore, membrane-exposed pockets warrant cautious calculation and interpretation of the characteristics to determine their druggability. Importantly, access and binding of a ligand to this transmembrane site involves the partitioning of the ligand into the membrane and gaining accessibility near the site as a first step, followed by eviction of lipids from the site, and finally, occupation of the site by the ligand. Despite eviction from the site, the lipids are in close contact with the bound ligand and potentially contribute to its binding and stability at the site. Using the cannabinoid CB1 receptor–ORG27569 complex as a model system, we attempted to elucidate this complex interplay between the receptor, ligand, and the membrane lipids, providing mechanistic insights into the role of the membrane in the ligand access and binding processes to the transmembrane lipid-facing site. Specifically, we determined the membrane partitioning characteristics, examined the receptor access paths, binding orientations, and molecular interactions of ORG27569, and quantified the contribution of lipids to ligand binding and stability.
ORG27569 Preferentially Localizes near the Core of the Bilayer
ORG27569 is a highly lipophilic molecule with a calculated log P value of 5.89 (using Bioloom40) and a relatively small topological polar surface area (TPSA = 48.1 Å2) for a ligand with 29 heavy atoms. ORG27569 contains two H-bond donors and two H-bond acceptors and is flexible with six rotatable bonds (Supporting Table S1). We examined the membrane partitioning characteristics, such as the energetically favorable bilayer location, orientation, and conformation of ORG27569 (Figure 2A), using a combination of steered molecular dynamics and umbrella sampling (see the Methods Section for details). The potential of mean force (PMF) curve, which provides the free energy of solvation of the ligand into the membrane, indicates that ORG27569 has a highly favorable free energy of partitioning (∼−8 kcal/mol) from the aqueous phase to the membrane (Figure 2B). Also, the presence of a negligible energy barrier for crossing the membrane core (at Z = 0 Å) indicates that the ligand can be found in both leaflets with equal probability. The free energy minimum for the center-of-mass (COM) of ORG27569 is located at |Zmin| ∼ 2 Å from the bilayer core, which corresponds to the vicinity of the allosteric site. At this preferred location, ORG27569 assumes orientations in which the head part (indole ring) is near the polar headgroups of the bilayer and the tail part (piperidinyl-benzyl rings) is around the membrane core (Figure 2C). As ORG27569 contains six rotatable bonds and is flexible, we wanted to assess the ligand’s most preferred conformation. To account for the ligand flexibility, the internal angle of the ligand was calculated between two vertices formed by the head and tail parts using the amide nitrogen as the intersecting point. In most of the simulation frames analyzed, the internal angle is around 120° indicating a relatively extended conformation (Figure S1). We further investigated the molecular interactions of the ligand with various lipid functional groups as contact occupancy of the ligand’s heavy atoms representing both head and tail parts (Figure 2D). The ligand–lipid contact occupancy calculates the fraction of the simulation time during which a given atom is within 4 Å of the lipid functional groups such as choline, phosphate, and glyceryl oxygens (of the lipid head) and alkyl carbons (of the lipid tail). Consistent with the ligand orientation described above, the tail part of ORG27569 (N-piperidinyl-phenyl fragment) was seen deeply embedded near the lipid bilayer core, likely due to its strong hydrophobic interactions with the lipid alkyl tails. The chlorine (at the 5-position) and nitrogen (N3) atoms of the indole ring were mostly in contact with the polar headgroups, specifically near the phosphate and glyceryl groups. Also, these atoms (Cl and N3) appeared to have negligible contact with the membrane core (alkyl tails). In contrast, the nitrogen of the piperidinyl ring and (N1) nitrogen (N2) and oxygen (O1) of the carboxamide group were in close contact with the lipid alkyl tail. No intramolecular H-bond was observed for ORG27569 during partitioning through the bilayer.
Figure 2.
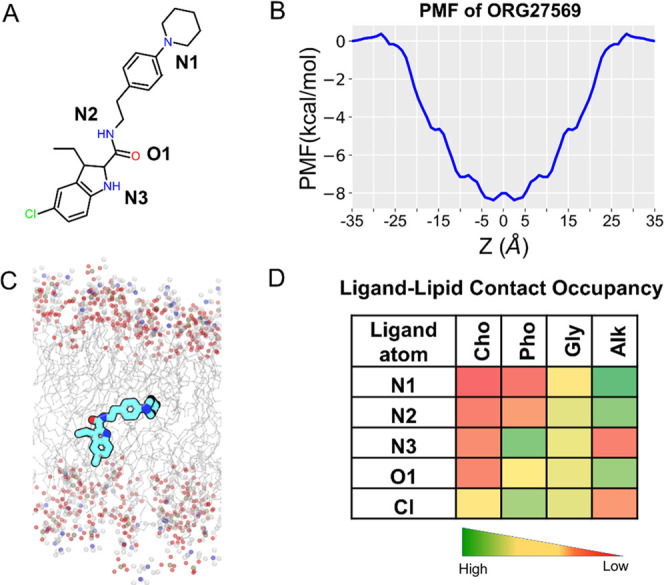
Membrane partitioning characteristics of ORG27569 in a model membrane bilayer made of POPC and cholesterol. (A) Two-dimensional (2D) structure of ORG27569 with the heteroatoms labeled. (B) Potential of mean force (PMF) curve indicates the energetically favorable location for the center-of-mass of ORG27569 around the bilayer core. The favorable location of ORG27569 corresponds to the vicinity of the allosteric binding site. (C) Time-average preferred orientation of ORG27569 within the bilayer. The head part (indole ring) of ORG27569 is mostly located near the bilayer headgroups, whereas the tail part (piperidinyl-benzyl fragment) is deeply embedded within the lipid alkyl tails, reaching out to the membrane core. The various lipid components are shown, with the alkyl lipid tails represented as lines in gray color and the headgroups, including phosphorus atoms of the phosphate, oxygen atoms of the glyceryl carbonyl, and nitrogen atoms of the choline represented as balls in olive green, red, and blue colors, respectively. (D) Extent of ligand atoms’ contact with the various lipid components was calculated as percentage occupancy, indicating the fraction of the simulation time during which an atom is at least within 4 Å of the lipid group. The atoms from the tail and middle parts (N1, N2, and O1) are mostly in contact with the lipid alkyl tail, whereas the atoms from the head part (Cl and N3) are located near the headgroups. Cho—Choline, Pho—Phosphate, Gly—Glycerol, and Alk—Alkyl.
Access and Binding Mechanisms of ORG27569 to the Transmembrane Binding Site
To elucidate the role of membrane lipids in the access and binding of ORG27569 to the transmembrane allosteric site of CB1R, we performed association simulations using an enhanced sampling technique called well-tempered metadynamics (see the Methods Section for details). We embedded CB1R in an asymmetric bilayer of the heterogeneous lipid composition, closely mimicking its native environment41,42 (Supporting Table S4). As shown earlier, ORG27569 preferentially partitions into the membrane and localizes around the membrane core near the allosteric site. Therefore, we conducted multiple association simulations with the ligand randomly placed around the receptor near the bilayer core and ∼20 Å away from the binding site. In addition, we also performed several simulations with the ligand randomly placed in the extracellular aqueous bulk, 10–20 Å away from the receptor. In all simulations, the free energy associated with the ligand access and binding to the receptor was characterized by two collective variables: (1) the distance between the COMs of the ligand and that of the binding site residues, and (2) the internal angle of the ligand, accounting for its conformation (Figure 3).
Figure 3.

Free energy surface (FES) depicts the access and binding of ORG27569 to the cannabinoid CB1 receptor (PDB ID 6KQI) using well-tempered metadynamics (WT-metaD). The free energy in kcal/mol was characterized using two collective variables: (1) distance between the center-of-mass (COM) of the ligand and the COM of the binding site residues in nm (x-axis) and (2) internal angle of ORG275697 in degrees (y-axis). (A) As ORG27569 (licorice representation, magenta color) approaches the receptor from the aqueous phase, the piperidinyl tail makes initial contact with R1863.22, E258ECL2, and K259ECL2 and the chloro-substituted indole ring of the ligand stays in contact with the phosphatidylcholine (POPC) headgroups. (B) ORG27569 undergoes significant conformational changes as it attempts to access the binding pocket but is blocked off by the cholesterol molecule already bound to the pocket. (C) Cholesterol molecule dissociates but remains within 4 Å of the binding site. This allows the ORG27569 molecule to slide into the binding site, replicating interactions and a binding pose similar to the crystal structure pose (PDB ID 6KQI) (Shao et al., 2019). In this pose, ORG27569 forms hydrophobic interactions with H1542.41, V1612.48, V2344.43, and W2414.50. The binding site residues (light blue) are illustrated in the licorice representation. Lipids shown in the licorice representation include cholesterol (orange), POPC (green), and PSM (yellow).
In simulations where ORG27569 started its journey from within the membrane, the ligand stayed near the bilayer core and took a lateral walk mostly by interactions with the alkyl tails of the membrane lipids, maintaining its orientation suitable for binding to the allosteric site. At the beginning of the simulation, the binding site was occupied by lipids such as POPC, POPE, and cholesterol that formed a H-bond with R2304.39 from the allosteric site. All three of these lipid molecules were within 4 Å of the binding site. The ligand’s access to the site was obstructed by these lipids, and thus, the ligand drifted upward where it interacted with membrane-facing protein residues, including T2424.51, I2454.54, V2494.58, and L2524.61. The ligand moved back out into the membrane, extensively interacting with the surrounding lipids before starting to access the site in a horizontal position. It then flipped into the allosteric pocket, indole ring first, displacing the bound POPE lipid. The binding pose of the ligand was finetuned by the alkyl tails of the POPE and POPC, which propelled the ligand into an upright position in the binding pocket where approximately 50% of the NAM was embedded in the binding site, and the rest was in contact with membrane lipids. In its final bound pose, which is almost identical to the crystal structure, the ligand was in close contact with L1384.48, L1424.43, H1542.41, V1612.48, W2414.50, and I2452.44.
In most of the simulations where the ligand was placed in the aqueous phase initially, ORG27569 quickly made its way into the membrane, approaching in a horizontal orientation with the piperidinyl tail making initial contact with R1863.22, E258ECL2, and K259ECL2 (Figure 3A). However, the chloro-substituted indole ring stayed in contact with the nearby phosphatidylcholine (POPC) molecules. The ligand spent some time near the lipid headgroups forming additional contacts with N1873.23, L1903.26, I2454.54, V2494.58, and L2524.61 (Figure 3A). It then proceeded into the membrane with the chloro-substituted indole ring (head) first. The ligand spent a considerable time in the membrane interacting further with I2454.54, V2494.58, and L2524.61, which are membrane-facing receptor residues, while making its way to the binding site. The ligand underwent significant conformational changes, eventually arriving at an orientation suitable for binding. During this time, a cholesterol molecule was already bound at the site and appeared to hinder the ligand’s entrance to the binding site (Figure 3B). On being barred from entry, ORG27569 moved back into the membrane, where it continued to interact extensively with the membrane lipids while trying to renegotiate for a possible entry into the binding site. Upon successful approach by the ligand this time, the cholesterol molecule dissociates from the site creating space for the ligand, which finally makes its way into the binding site (Figure 3C). Two POPC molecules from the upper leaflet appear to propel the ligand with their alkyl tails toward the allosteric site. In its final bound pose, ORG27569 adopts a half-in, half-out orientation identical to the crystal-bound pose with the indole ring making extensive interactions with H1542.41, G1572.44, V1612.48, V2344.43, and W2414.50 (Figure 3C) while buried in the protein. The tail part of ORG27569, the piperidinyl-benzyl ring, however, sticks into the membrane, interacting mostly with the surrounding lipids (Supporting Movie M1).
WT-MetaD Simulations Reproduced the Crystal-Bound Pose of ORG27569
The WT-metaD association simulations recreated the crystal-bound pose of ORG27569 (Figure 4A) with the chloro-substituted indole ring embedded within the receptor and forming mostly hydrophobic interactions with the binding site residues. On the other hand, a majority of the tail part of the ligand (the piperidinyl-benzyl ring) “sticks out” of the binding pocket making extensive interactions with the membrane lipids. While in all of the WT-metaD simulations, ORG27569 adopted binding poses and interactions similar to the crystal-bound pose, the ligand slid further into the pocket than in the crystal structure. Interestingly, the ligand’s carbonyl oxygen of the amide group assumes a different orientation relative to the crystal structure, flipping outward and facing the membrane, where it forms a H-bond with a neighboring cholesterol molecule (Figure 4B).
Figure 4.

Membrane lipids assist and contribute to the stability and binding of ORG27569 to the cannabinoid CB1 receptor (PDB ID 6KQI). (A) Well-tempered metadynamics recreates the crystal-bound pose. The binding pose obtained from well-tempered metadynamics (magenta) was compared to the crystal structure pose (blue). (B) Final bound pose of ORG27569 (magenta) with the amide oxygen flipped outwards, forming a H-bond with the cholesterol hydroxyl group (green). (C) Final pose of ORG27569 (magenta) shows membrane lipids within 4 Å of the ligand, “supporting” its upper region. Lipids shown include cholesterol (orange), phosphatidyl sphingomyelin (purple), and phosphatidylcholine (light green).
Lipids such as Cholesterol and PSM Support the Ligand in the Binding Site
The cholesterol molecule that dissociated upon ORG27569’s entry into the site remained within 4 Å of the ligand. The cholesterol molecule, together with a sphingomyelin (PSM) lipid molecule and a second cholesterol molecule, formed a “lipid support site,” holding up the tail part of the ORG27569, which projects out into the membrane from the binding pocket (Figure 4C). Solvent-accessible surface area (SASA) calculations were carried out using the final bound pose from the WT-metaD association simulations to estimate the extent to which the ligand is buried within the protein (buried surface area, BSA) and in contact with the membrane lipids (membrane-exposed surface area, MESA). As shown in Figure 5A,B and Supporting Table S5, ∼46% of ORG27569 remains exposed to the membrane, with its nonpolar part contributing significantly to this number, while most of the polar regions are embedded in the binding site. This is comparable to the crystal structure where ∼42% of the ligand was exposed to the membrane. Next, we recalculated the druggability score and other binding site characteristics of the transmembrane allosteric site, incorporating the lipids that are directly in contact with ORG27569 (Supporting Table S3). With the inclusion of lipid molecules, there was a significant increase in the values of various pocket descriptors, with the druggability score increasing to 0.9 and the volume and SASA values much higher than those calculated without the lipids.
Figure 5.

Membrane lipids are an integral part of the transmembrane binding site and contribute to ORG27569 binding. (A) 2D depiction of the “complete” allosteric site, including binding site residues and lipids that are within 4 Å of ORG27569 in its final binding orientation at the end of the association simulation. Lipids (orange) shown include phosphatidylcholine (POPC), cholesterol (CHOL), and phosphatidyl sphingomyelin (PSM). (B) Extent (%) of the total, nonpolar, and polar solvent-accessible surface area (SASA) of ORG27569 buried within the protein (buried surface area, BSA), and exposed to the membrane lipids (membrane-exposed surface area, MESA), are given. These values were calculated using the final binding pose from the association simulations. A significant amount of the ligand remains exposed to the membrane lipids in its final bound form (total MESA = 46%). Most of the polar surface area of the ligand is buried within the binding pocket (polar BSA = 88%).
Delipidation and Rearrangement of Lipids by ORG27569’s Entry into the Allosteric Binding Site
In most of the simulations, in the beginning, a cholesterol molecule was observed to be in or around the allosteric site (Figure 6A). Although the direct access of the ligand to the site was initially impeded by this cholesterol, the ligand’s approach led to the perturbation of the H-bonds formed between the hydroxyl group of the same cholesterol and the side-chain amino groups of the residues, H1542.41 and R2304.39 (Figure 6B). The eventual binding of the ligand, as depicted by the distance between the COMs of ORG27569 and the binding site and the internal angle of the ligand (Figure 6C,D), and the distance between the ligand chlorine atom and the side-chain amino groups of the residues, H1542.41 and R2304.39 (Figure 6E) corresponds to the time of the eventual dissociation of the cholesterol molecule (Figure 6B). We also observed rearrangements of several other lipids around the binding site (Figure S2). It was observed that most of the lipids moved away from the binding site to create enough space for the ligand and further reordering and stabilization of the same lipids around the binding site.
Figure 6.

ORG27569 competes with cholesterol for the allosteric binding site of the cannabinoid CB1 receptor (PDB ID 6KQI). (A) Cholesterol molecule was bound to the allosteric site at the beginning of the WT-metaD simulation. The cholesterol (magenta) molecule and binding site residues H1542.41 and R2304.39 (green) are shown in licorice representation. (B) H-bond distance between the cholesterol 3-OH group and the side-chain amino groups of H1542.41 and R2304.39 are shown in blue and orange lines, respectively. Around 10 ns, there were fluctuations in the bond distance as the ligand attempted to make its way into the pocket. This step was followed by the dissociation of the cholesterol molecule, as shown by the red arrow, enabling ligand binding. (C) Distance between the COMs of ORG27569 and the binding site residues during the association process. As shown by the red arrow, the distance comes closer to 4 Å, which indicates its bound state. (D) Fluctuation in the internal angle of ORG27569 during WT-metaD is shown in blue. The ligand mostly favors a relatively extended conformation and ends up in a conformation similar to the crystal-bound one. (E) Distance between the chlorine atom and the side chains of H1542.41 and R2304.39 are shown in magenta and cyan, respectively. Both distances come close to 4 Å, indicating the ligand-bound state.
Assessment of ORG27569’s Orientation and Molecular Interactions Using Unbiased All-Atom MD Simulations
We performed unbiased MD simulations in three replicates to assess the stability of the ligand in the binding site as well as the contributions of the membrane lipids to its binding and interactions. This is important as the crystal structure gives no information about the interaction of ORG27569 with the neighboring lipid molecules. The simulation started with ORG27569 in the crystal structure-bound pose along with the cocrystallized agonist (CC55940), and the total simulation time was 1 microsecond (μs). The ligand was stable in the binding site, as shown by its root-mean-square deviation (RMSD) (Figure S3A) and the distance between the COMs of the ligand and the binding site residues, which include I141, H154, G157, S158, V161, F237, C238, and W241 (Figure S3B). Similar to orientations observed in WT-metaD association simulations, the ligand moved slightly deeper into the binding pocket while still having a significant part outside of the pocket, interacting extensively with the membrane. We also observed a flipping of the carbonyl oxygen of the carboxamide group toward the membrane, and it remained in this orientation for the majority (∼85%) of the simulation (Figure S3C), suggesting an alternate orientation for ORG27569 observed in the crystal structure. However, caution should be exercised in assessing this finding as electronic polarizability, accounting for the response of the charge distribution to variation in the surrounding electric field was not considered in this simulation, given the anisotropic nature of the aqueous-membrane-protein environment.
Membrane Lipids Significantly Contribute to the Binding of ORG27569 at the Transmembrane Allosteric Site
Since ORG27569 was significantly exposed to the membrane and had contact with many lipid molecules, we sought to estimate the contribution of these membrane lipids to its binding using the 1 μs unbiased MD simulation trajectory. The relative contributions of the membrane lipids and protein residues were estimated using the MMPBSA method by including the membrane lipids as part of the binding site (Supporting Tables S6 and S7). The results indicate that the protein residues such as W2414.50, C2384.47, F2374.46, and S1582.45, which are directly in contact with the ligand for most of the simulation time (Figure 7A), contribute significantly to the overall binding of ORG27569, as indicated by the magnitude of the negative energy values (Supporting Table S6). Also, several lipid molecules, including cholesterol, POPC, POPE, POPI, and POPS, which directly interacted with the ligand during the simulation, contributed favorably to the binding of the ligand (Figure 7B,C, and Supporting Table S7). Some of these lipids, such as POPI5 and POPS6, reached the ligand only after 200 and 600 ns, respectively, and stayed in contact for the rest of the simulation time.
Figure 7.

Contribution of the binding site residues and lipids to the overall binding of ORG27569 to CB1R (PDB ID 6KQI). (A) Contact frequency (%) of ORG27569 accounts for the extent of molecular interactions with the binding site residues. The contact frequency represents the fraction of the total simulation time during which a given residue is within 4 Å of the ligand. For example, the binding site residues, C2384.47, W2414.50, and F2374.46, are in close contact with ORG27569 for >95% of the simulation time. (B) Several membrane lipids such as cholesterol, POPC, POPE, POPI, and POPS are directly in contact with ORG27569 likely stabilizing the ligand within the binding site, as observed during the 1 μs long unbiased all-atom MD simulation. (C) Distances between ORG27569 and several lipids were monitored during the entire simulation. The dotted lines represent an arbitrary 4 Å distance in the graphs. The distances are shown for cholesterol (CHOL1, deep sky blue), phosphatidylcholine (POPC2, black), phosphatidylethanolamine (POPE4, blue), phosphatidylinositol (POPI5, purple), and phosphatidylserine (POPS6, red).
Differences in the Binding Affinities of ORG27569 Analogs May Be Attributed to Functional Groups That Interact Largely with the Membrane Lipids
To delineate the structure–membrane interaction relationships of the allosteric ligands at the transmembrane site, we extended our study by investigating a few structural analogs of ORG27569 (Figure 8). The analogs resulted from several ligand-based structure–activity relationship studies to optimize the binding affinity and cooperativity of ORG27569 (Supporting Table S2). From these studies, structural variation at the C-3 position of the indole ring was demonstrated to affect the activity of ORG27569, with an increase in the alkyl chain length at this position resulting in a corresponding increase in the binding affinity or cooperativity in general.36−38,43 We, therefore, investigated five analogs of ORG27569 with varying alkyl chain lengths and/or tail substitutions using unbiased MD simulations (Figure 8A). All of the studied analogs were docked at the site using ORG27569 as a template, and the protein–ligand complexes embedded in their native heterogeneous membrane environment were subjected to 200 ns long MD simulations. The resulting trajectories were analyzed for the stability of the ligands at the site, binding orientations, and molecular interactions with the site residues and the surrounding lipids. The analogs assumed orientations that are similar to that of ORG27569 in the allosteric binding pocket and remained within the site for the entire simulation time (Figures S4 and S5). The contact heatmap generated from the MD simulation trajectory shows the extent of contacts made by the head, tail, and alkyl chain parts of ORG27569 and the analogs with the binding site residues and the membrane lipids (Figure 8B and Supporting Table S8). All of the analogs have significant contact with the lipid alkyl tails indicating that they remain in the site and interact with the core of the bilayer. The indole head group (H) of all of the ligands except Compound 21 is entirely in contact with the protein residues and the lipid alkyl tails. In the case of Compound 21, the ligand moves down toward the lower leaflet and appears to make notable contact with the choline functional groups and, to a smaller extent, with the glyceryl atoms. The tail part (T) of all of the ligands except ICAM-b interact mostly with the lipid alkyl region. In the case of ICAM-b, the ligand moves up toward the upper leaflet and appears to make some contact with the choline and glyceryl functional groups. In the crystal structure, the alkyl chain at the C-3 position of the indole ring in ORG27569 was positioned away from the site and appeared to project toward the membrane lipids. In simulations of all of the analogs, the alkyl chain remained positioned toward the membrane and engaged in interactions with the lipids indicating that the differences in the substitution at the C-3 position mainly alter interactions with the membrane lipids and not the binding site residues (Supporting Movie M2). The extent of membrane interactions was also quantified by the fraction of the ligands’ solvent-accessible surface area that is in contact with the membrane lipids, MESA, and the fraction that is buried within the binding site, BSA (Supporting Table S5). The MESA values for ICAM-b and Compound 21 were higher than that of ORG27569 (57, 51, and 46%, respectively), likely due to differences in their relative binding orientations within the site. As expected, the BSA value was much higher for ORG27569 as compared to that of Compound 21 and ICAM-b (53.9, 49, and 43%, respectively). In the case of analogs with the dimethylamino tail, both Compound 13 and Compound 11j have much lower MESA values compared to Compound 12f (35, 37, and 54%, respectively). This difference is mainly attributed to the smaller dimethylamino tail as compared to the N-piperidinyl ring. However, the presence of the n-hexyl chain in Compound 12f seems to pull the molecule up and contribute to a higher MESA value. The number of lipid molecules surrounding the ligand (within 4 Å) calculated for all of the analogs confirm the above observations (Figure S7).
Figure 8.

ORG27569’s analogs show distinct contacts with the membrane with varying alkyl chain lengths. (A) 2D structures of ORG27569 and its analogs. (B) Binding affinity of the ligands to the allosteric site (as either the equilibrium dissociation constant KB or the EC50 value), the binding cooperativity factor (α), and the extent of contact with the protein and lipids of the studied ORG27569 analogs. The heatmap shows the percentage occupancy (the fraction of the simulation time during which the given ligand is within 4 Å of the protein residue or membrane lipids) was calculated as the extent of contact the ligand atoms make with the various lipid components throughout the simulation time. The longer alkyl chains at C-3 positions of the analogs such as Compounds 11j, Compound 12f, and ICAM-b appear to be mostly in contact with the membrane lipids. Ligand parts: T—tail (piperidinyl-benzyl rings), A—alkyl chain at the C-3 position, and H—head (indole ring); P—protein residues; and Membrane: C—Choline, P—Phosphate, G—Glyceryl, and A—Alkyl.
The alkyl chain at the C-3 position seems to cause notable differences in the orientation of the analogs at the site (Figure 9A). These differences were quantified as the orientational angle of the ligands with respect to the bilayer normal (z-axis) (Figure 9B). Interestingly, the analogs with longer alkyl chains (ICAM-b, Compound 11j, and Compound 12f) and one without the alkyl substitution at the C-3 position (Compound 21) appear to make smaller orientational angles from the bilayer normal (z-axis) compared to ORG27569 and Compound 13, both with an ethyl substitution. The orientational angle and MESA of the ligands seem to be affected by several factors, including the alkyl chain length at the C-3 position, polar interactions of the carbonyl functional group, and the aryl–aryl interactions between the indole ring of the ligands and the side-chain imidazole ring of the binding site residue H1542.41.
Figure 9.

Preferred orientations of ORG27569 and its analogs at the transmembrane allosteric binding site. (A) Orientation of ligands with respect to three critical binding site residues, H1542.41, V1612.48, and W2414.50, reveals the differences in the molecular interactions with protein residues and the surrounding membrane lipids. Most importantly, compounds with long alkyl substitutions at position C-3 are oriented such that their alkyl groups point toward the membrane lipids, affecting the overall orientation of the ligands. (B) Boxplot shows the orientational angle of each ligand through the entire simulation time that was quantified as a tilt angle between the bilayer normal (z-axis) and the vector, connecting the two ends of the ligand as depicted in Figure S5B (chlorine atom at one end and the farthest aromatic carbon atom on the piperidine ring or the nitrogen of the dimethylamino group at the other end). The N-piperidinyl and dimethylamino analogs are shown in blue and red colors, respectively.
For all of the analogs, the orientation and polar interaction of the carbonyl group were monitored by calculating the distance between the carbonyl oxygen and the side-chain hydroxyl group of S1582.45 in the allosteric binding site (Figure S5A). In addition, the relative bilayer depth of the bound ligands was quantified by the distance between the COMs of the indole ring of the ligands and the side-chain imidazole ring of the binding site residue H1542.41 (Figure S5B). Interestingly, the analog with the longest alkyl chain and dimethylamino-benzyl tail (Compound 12f) moved above the imidazole ring within the first 5 ns and maintained a distance of ∼10 Å for the rest of the simulation time. However, other analogs with smaller alkyl chain lengths in that group (Compound 11j and Compound 13) maintained the indole ring much closer to the imidazole ring (average distance of ∼5 Å). Among the analogs with the piperidinyl-benzyl tail, Compound 21 and ICAM-b had similar profiles with distances ranging between 5 and 8 Å. Compound 21, which has no alkyl chain at the C-3 position, showed a relatively distinct lipid interaction profile such that its head part appears to make more contact with the choline headgroups (Figure 8B), suggesting that the C-3 alkyl chain might serve as an anchor to the membrane, stabilizing the position of the ligand in the membrane core.
Using the MMPBSA method, we calculated the individual contributions of the binding site residues and lipids within 4 Å of the bound ligand toward the binding free energies of the analogs (Supporting Tables S6 and S7, respectively). Specifically, the contributions by 3–4 lipid molecules from each leaflet (U1, U2, and U3 from the upper leaflet and L1, L2, L3, and L4 from the lower leaflet), as illustrated in Figure S6 are given. The analogs with the longer alkyl chains seem to have higher relative free energies of binding. The relative contributions by many critical residues such as V1612.48, F2374.46, C2384.47, and W2414.50 seem to have notable differences among the analogs. Also, the relative contributions of various lipid molecules have considerable variations, raising the possibility that the differences in the binding affinity and cooperativity observed in the experiments among the analogs may be attributed to interactions not only with binding site residues but also with the lipids. The experimental binding affinity values along with the confidence intervals reported in the original articles are given in Supporting Table S2. As can be seen, the binding affinity (KB) values for several compounds, including ICAM-b, have quite large confidence intervals. Therefore, it is critical not to entirely rule out a potential trend between the experimental data and the calculated MMPBSA energies. Importantly, for ORG27569 and Compound 12f, the reported confidence interval is relatively narrow, and the calculated MMPBSA energies seem to capture the observed trend (Supporting Table S6). It should be noted that all of these compounds have been reported by the same research group using an identical assay procedure. Unfortunately, only the EC50 value was found for Compound 21, generated by a different group. To further highlight the contribution of lipids that are directly in contact with the ligands, we employed the heterogeneous dielectric implicit membrane model to estimate the binding energies (Supporting Table S9). Although this membrane model allows the dielectric constant to vary along the bilayer normal to represent the anisotropic nature, the calculated binding affinities show poor correlation with the experimental data, even among compounds with tight confidence intervals.
The molecular interactions of the CB1R agonist, CP55940, at the orthosteric site were also assessed in the presence of ORG27569 and its analogs using the same simulation trajectories. The molecular interactions, specifically, the H-bonds formed between the agonist and the orthosteric binding residues, S1732.60, I267ECL2, and S3837.39 (Figure S8A), were monitored. Except for transient disruptions, as seen with ORG27569, ICAM-b, and Compound 13, the H-bonds remain relatively stable for all analogs. Contacts between the agonist and other binding site residues were also evaluated using a 4 Å cutoff (Figure S8B). Although there is a high degree of similarity in the agonist contacts (F1702.57, S1732.60, L1933.29, I267ECL2, F268ECL2, F3797.35, and S3837.39) in the presence of ORG27569 and other analogs, subtle differences are observed for several residues (F108Nterm, F1772.64, F1893.25, T1973.33, Y2755.39, L2765.40, and L3596.51).
Discussion and Conclusions
A growing number of experimental structures reveal the existence of transmembrane extrahelical binding sites in integral membrane proteins such as GPCRs, ion channels, and transporters.7,44 These relatively less conserved sites are attractive targets for allosteric modulation and offer immense advantages in terms of selectivity for ligands compared to orthosteric sites. However, the transmembrane sites pose unique challenges in that a ligand must first partition into the membrane and navigate through the surrounding lipid environment before it can access and bind to the site.8 Earlier studies have suggested that interactions of ligands with the membrane lipids, driven by their distinct lipophilic and amphiphilic properties, can result in a difference in properties such as binding kinetics and rebinding to the receptors affecting their overall pharmacology, including the onset and duration of action.17,25,45 According to the microkinetic model, specific interactions of ligands with the membrane lipids can determine their local concentrations surrounding the target site and preorganize them in orientations and conformations suitable for binding. Importantly, while bound, a significant part of these ligands remains directly in contact with the membrane lipids, revealing an intriguing and often overlooked situation where the surrounding lipids actively contribute to the binding and stability at the site.
In this study, the solvation free energy profile obtained from the membrane partitioning simulations revealed that ORG27569 has a much higher affinity for the membrane than the aqueous bulk. Intriguingly, the energetically favorable bilayer location of ORG27569, which is near the membrane core, corresponds to the location of the allosteric site, ensuring high local concentrations of the ligand near the site (Figures 1B and 2B). Surprisingly, ORG27569’s preferred orientations and conformations within the membrane observed in the simulations are comparable to its crystal-bound pose,33 indicating that the membrane lipids likely facilitate the binding process by preorganizing the ligand favorable for binding.
Our WT-metaD association simulations offer useful insights into how ORG27569 accesses the binding site deeply embedded within the lipid bilayer. The association simulations revealed that the membrane lipids play an important role in facilitating ligand partitioning, preconfiguration, and eventual binding. The ligand first partitions into the lipid bilayer assumes orientations that are optimal for binding, and then binds to the target site (Figure 3A–C). These correspond to the earlier suggested mechanisms of binding of ligands to membrane-embedded sites.8,26,46 Interestingly, the ligand made initial contact with several residues near the extracellular surface. These interactions were seen consistently in multiple simulations and appear to be critical in drawing the molecule around the transmembrane allosteric site. Multiple sequence alignment performed with several other class A GPCRs, including CB2 receptor; histamine 1 receptor; β-1 and β-2 adrenergic receptors; dopamine 1 and 2 receptors; δ, μ, and κ opioid receptors; angiotensin II type 1 receptor; chemokine receptor 6; cholecystokinin-1 receptor; orexin-1 receptor; adenosine A2A receptor; and sphingosine-1-phosphate receptor, reveals negligible conservation among these residues (Supporting Figure S9). The sequence alignment also revealed a moderate to high sequence similarity among certain allosteric site residues (Supporting Figure S10). This specificity at the extracellular surface residues may likely play a critical role in imparting the receptor selectivity for these ligands, potentially reducing off-target interactions. In addition to these apparent fundamental steps, desolvation of the site residues and the ligand must precede the binding event. Desolvation prior to the ligand binding is a thermodynamically important process by which solvent molecules within the binding site as well as those surrounding the ligand get freed (gain of entropy), facilitating the receptor–ligand complex formation.47−50 In the case of the CB1R transmembrane allosteric site, lipids molecules that are in close contact with the site residues must be evacuated, as well as the lipids from the membrane bulk surrounding the ligand, must be stripped off prior to binding. Notably, the binding pocket of the allosteric site has structural features similar to Cholesterol Consensus Motif (CCM), which has been described for other GPCRs51 (Supporting Table S10). Although this site’s primary amino acid sequence does not entirely meet the CCM requirement, a cholesterol molecule has previously been crystallized at this site.52,53 Surprisingly, in all of the association simulations, a cholesterol molecule was seen in and around the site, likely competing with the ligand.52 However, once the ligand reaches the binding site, these cholesterol molecules stay near the site and appear to stabilize the ligand even by forming a H-bond (Figure 6). We also observed the rearrangement of several lipids around the binding site during the association process (Figure S2). This rearrangement process involved the selective recruitment of certain lipids near the site as well as the displacement of others from the vicinity of the site. These point to an interesting and complex interplay between membrane lipids and ligands that bind to the transmembrane region and suggests an integral role of the membrane lipids in promoting ligand stability. Also, these complex phenomena might significantly impact the overall thermodynamics of the binding process, affecting the binding kinetics and affinity of ligands. Intriguingly, incorporating the surrounding lipids as part of the transmembrane allosteric site seems to significantly improve the binding site characteristics (Supporting Table S3), suggesting that transmembrane lipid-facing binding sites need careful consideration and renewed interpretation of the typical characteristics sought after druggable binding sites. We believe that this strategy might result in the rediscovery of potential transmembrane sites, previously considered undruggable.
While bound to the allosteric site, most of the tail part of ORG27569 and its analogs were directly in contact with the lipids. Specifically, most of the piperidinyl-benzyl ring, representing up to 46% of the solvent-accessible surface area of ORG27569, was exposed to the membrane lipids (MESA in Figure 5B and Supporting Table S5). The other studied analogs bind in orientations similar to ORG27569 and expose a significant part of their tails (piperidinyl-benzyl ring for Compound 21 and ICAM-b, and dimethylamino-benzyl ring for Compound 13, Compound 11j, and Compound 12f), which varies from ∼35 to 57% of MESA (Supporting Table S5). A similar range in the MESA values has been reported for the ligands bound to transmembrane allosteric sites of class A and class B GPCRs before.8 For all of the studied ligands, we monitored the trajectory of every single lipid that came in contact (within 4 Å) with the ligands (Supporting Figures S11–S15). Figure 7C features the distance plots for lipids with a percentage occupancy of at least 50 (%) and above for ORG27569. As can be seen from the graphs, these lipids seem to stay in contact with the ligands for the majority of the simulation time. Interestingly, in a few simulations, lipid molecules (for example, cholesterol surrounding Compound 12f, Supporting Figure S15), diffused farther from the ligand but stayed within ∼10 Å. Furthermore, the binding free energies calculated by the MMPBSA method revealed that the lipids surrounding the ligands make a significant energetic contribution to ligand binding (Figure 7B,C, and Supporting Table S7). Among the studied analogs, the major structural differences are (1) the length of the alkyl chain at the C-3 position of the indole ring and (2) the substitution at the para position of the phenyl ring, either the N-piperidinyl ring or dimethylamino group. Importantly, the crystal-bound orientation of ORG27569 and our simulations show that both these functional groups interact mostly with the membrane lipids. Also, changes in these positions seem to affect the binding affinity (KB) and the cooperativity factor in a more complex and nontrivial fashion. For example, ICAM-b and Compound 11j have the same length alkyl chain (n-pentyl group) at the C-3 position but differ in the tail part. The replacement of the N-piperidinyl group (in ICAM-b) with the dimethylamino group (in Compound 11j) seems to produce a ∼3-fold increase in the binding affinity (KB) with no effect on the cooperativity factor. However, an identical replacement of the tail part among ORG27569 and Compound 13 did not affect the binding affinity but produced a ∼3-fold increase in the cooperativity factor. Furthermore, among the two compounds containing the dimethylamino substitution at the phenyl ring, Compound 11j and Compound 12f, increasing the alkyl chain length at the C-3 position (from n-pentyl to n-hexyl) seems to affect both the binding affinity (a 2-fold increase) and the cooperativity factor (a 3-fold decrease) in opposite ways.36,54 Apparently, the differences in the structural features among ORG27569 and its analogs (the alkyl chain length at the C-3 position and the para- substitution on the phenyl ring) seem to dictate varied interactions with the membrane lipids, resulting in distinct binding orientations, molecular interactions with the binding site residues, and the extent to which a given ligand is buried within the binding site (BSA) or exposed to the membrane lipids (MESA). The nontrivial structure–activity relationships (both KB and α) among these ligands can be attributed, at least in part, to the differences in their membrane lipid interactions. It is essential to recognize that only a limited number of compounds were evaluated here. Studying more compounds from additional series is necessary to fully understand the role of ligand–lipid interactions on the binding affinity and cooperativity.
The insights obtained from this study suggest that while seeking ligands for transmembrane lipid-facing sites, a rational structural design strategy should include consideration for optimizing membrane interactions along with receptor interactions toward desirable pharmacodynamic outcomes. This idea indicates a paradigm shift from the strategy of uncoupling the membrane interactions and improving structure–activity relationships of ligands through interactions with binding site residues alone. We presume that the proposed strategy, incorporating membrane lipids as an integral component of the binding site, might require efforts toward meeting one or more of the following objectives.7,55,56 First, ligands must have optimal lipophilic and amphiphilic properties resulting in favorable membrane partitioning characteristics to achieve adequate local concentrations surrounding the target site. Second, ideal membrane lipid interactions of ligands may facilitate the access and binding processes by preorganizing the ligands in suitable orientations and conformations near the site. Third, the bound ligands should engage in the most favorable interactions with the surrounding lipids such that their binding orientations and residue interactions at the site result in desirable efficacy. It should be noted that these requirements may differ significantly depending upon the location of the site with respect to the bilayer depth. For example, ligands binding to a site near the bilayer headgroups might engage mostly in polar interactions in contrast to ligands binding to a site near the bilayer core where hydrophobic interactions may dominate. A recent study57 offered a valuable retrospective analysis of TRPA1 antagonists from two different chemical series. These molecules target two distinct binding sites: (1) proline sulfonamides that bind to an intrahelical transmembrane binding site, and (2) hypoxanthine analogs that bind to an intracellular binding site located near the lower leaflet of the bilayer. Intriguingly, the physicochemical and pharmacokinetic properties of the studied molecules seem to be dictated by the location and characteristics of the sites and the extent to which the ligands were exposed to the lipids.
In summary, our work provides valuable insights into the active role of membrane lipids in orchestrating the access and binding process for ligands targeting the lipid-facing transmembrane allosteric site of CB1R. As shown in this study, the membrane lipids play a critical role in not only facilitating the ligand partitioning and access but also actively contributing to the binding affinity and thus the stability of ligands at the binding site. As we witness staggering progress in the structural biology of integral membrane proteins, including GPCRs58−60 and ion channels, transmembrane allosteric sites offer attractive alternatives to the more conserved orthosteric sites to gain better selectivity and efficacy, safety, and prospect for biased signaling.8,57 Factors such as the asymmetry among the upper- and lower leaflets, organ-, tissue- and subcellular organelle-specific heterogeneity in the lipid composition of the bilayer, both in normal and disease states, would warrant additional consideration while incorporating the membrane into the drug discovery workflow.61−63 It is certain that with the growing knowledge of the structure–membrane interaction relationships for small-molecule ligands, additional tools and techniques to rationally account for the role of membrane lipids would facilitate the effective design of drug candidates for transmembrane extrahelical target sites.
Methods
Calculation of Pocket Descriptors Using fPocket
The pocket descriptors for the orthosteric and allosteric pockets were calculated using the default parameters in fPocket.39 For the protein–ligand complex without lipids, calculations were done using the crystal structure of CB1R bound to the NAM, ORG27569, and the agonist, CP55940 (PDB ID 6KQI). Later calculations with lipids were done using a frame extracted from the WT-metaD simulation, as described elsewhere in this section.
Unbiased MD Simulations Protocol
In this study, we used the crystal structure of CB1R bound to the NAM, ORG27569, and the agonist, CP55940 (PDB ID 6KQI). The protein structure was prepared for simulations using Molecular Operating Environment (MOE).64 The T4 lysozyme fused to the intracellular loop (ICL) 3 was removed, and the ICL3 was modeled using Modeller.65 The mutations T210A, S203K, E373K, T238V, and R340E were reverted to the wild-type sequence. Structure preparation was carried out in MOE to fill in missing atoms, assign partial charges, and assign protonation states at pH 7. The analogs of ORG27569 used included Compound 21,35 ICAM-b,36 Compound 13,35 Compound 11j,37 and Compound 12f.38 The ligands, ORG27569 and CP55940, and the five ORG27569 analogs were then parameterized using the CHARMM-GUI ligand reader and modeler,66 and charges were assigned using CGenFF.67 Further, a short energy minimization step to remove any steric clashes in the protein–ligand complexes was carried out using MOE, and the resulting structures were used to build the systems utilizing CHARMM-GUI.68 The membrane builder module in CHARMM-GUI69 was used to build a heterogeneous membrane consisting of 16:0/18:1 phosphatidylcholine (POPC), cholesterol, 18:1/16:0 phosphatidyl sphingomyelin (PSM), 16:0/18:1 phosphatidylserine (POPS), 16:0/18:1 phosphatidylinositol (POPI), and 16:0/18:1 phosphatidylethanolamine (POPE) distributed asymmetrically. Further details on lipid distribution between the leaflets are contained in the supplementary information (Supporting Table S4). The system was solvated with TIP3P70 water molecules up to about 23 Å padding on both sides, and a 0.15 M concentration of NaCl was used to neutralize the system charge. The final system size was 90 × 90 × 115 Å3 and contained 247 lipid molecules. The resulting protein–ligand complex in its native membrane environment surrounded by solvent and ion molecules was subjected to 5000 steps of steepest descent minimization and then equilibrated using the CHARMM-GUI recommended six-step equilibration protocol.71 All MD simulations were carried out using GROMACS 5.1.4.72
For the production run, a cutoff distance of 12 Å was used for the van der Waals and short-range electrostatic interactions. The long-range electrostatic interactions were computed with the particle-mesh Ewald summation method. A 2-fs integration time step was used for all MD simulations. The system was then simulated using NPT conditions at 1 atm pressure and a temperature of 310 K for a total simulation time of 1 μs with frames saved every 10 ps. For the analogs, the total simulation time was 200 ns for each system.
Association Simulations Using Well-Tempered Metadynamics (WT-MetaD) to Characterize Ligand Access Path(s)
To bind to the transmembrane allosteric site, a ligand must partition into the membrane and access the site only through lipid path(s). Also, the binding of a ligand at this site involves the removal of lipids that are directly in contact with the binding site residues. These complex access and binding events were investigated using an enhanced simulation technique called well-tempered metadynamics.73 Well-tempered metadynamics is an adaption of metadynamics and has been used extensively to investigate rare and interesting events within reasonable timescales by adding a bias that constrains the system to explore only relevant physical regions of conformational space.73 Following equilibration, WT-MetaD was conducted in triplicate using PLUMED v2.374 with the ligand placed in random positions in the aqueous bulk and another triplicate of ligands placed in the membrane. The distance between the center-of-mass (COM) of the binding site residues, H1542.41, V1612.48, V2344.43, and W2414.50, and the COM of ligands was used as the first collective variable (CV) while the internal angle of the ligand was utilized as the second CV. The distance between the chlorine atom and H1542.41 was used as a committor, which essentially turned off the bias once the distance was at least 4 Å. For all WT-metaD, simulations were carried out at 310 K with an upper wall of 3.0 nm, a bias factor of 15, sigma of 0.05, height of 1.5, and a force of 200 kJ/mol. The corresponding free energy surface was then created using MEPSA.75
Membrane Partitioning Simulations
We determined the membrane partitioning characteristics of ORG27569 using a combination of steered molecular dynamics (SMD) and umbrella sampling, as previously published.26,76 The ligand was parameterized using the CHARMM-GUI ligand reader and modeler,66 and charges were assigned using CGenFF.67 The CHARMM-GUI69 membrane builder was used to generate a bilayer consisting of 65 lipids per leaflet, of which 59 of them were POPC lipids and 6 cholesterol molecules, adding up to a total of 130 lipid molecules. The system was solvated with TIP3P70 water molecules, and the ionic concentration was kept at 0.15 M using adequate NaCl. The system was then subjected to equilibration using the CHARMM-GUI six-step equilibration71 process, which uses an NVT ensemble for the first two equilibration steps and an NPT ensemble for the last four steps. The equilibration started with a harmonic force constant of 5 kcal/mol restraints on the heavy atoms of the protein, planar restraints to restrain the headgroups of the lipid components along the z-axis, and dihedral restraints to restrain chirality of lipid headgroups and double bonds for 250 ps. This step was followed by two steps that ran for 250 ps with a scaling factor on the restraint, slowly releasing the restraints. The final three steps of the equilibration continued with a scaling restraint, but for 500 ps with the force totally removed in the last step. An additional 50 ns equilibration was carried out before starting the SMD.
The ligand’s initial position was in the bulk water, 35 Å from the center of the bilayer. During the SMD, the ligand was pulled through the membrane bilayer from its initial position. The pulling was carried out at a speed of 1 Å per ns with a 1 fs time step along the bilayer normal (z-axis) with a harmonic restraint of 5 kcal/mol/Å2 on the ligand. A total of 35 windows, extending from the bulk solvent to the middle of the bilayer, each 1 Å apart, were extracted from the simulation trajectories and used as starting points for the umbrella sampling simulations. Each window was equilibrated for 10 ns, and umbrella sampling was carried out for another 40 ns. A biasing harmonic constraint of 1.5 kcal/mol/Å2 was used to restrain the z-component of the distance between the center-of-mass of the lipid atoms and the heavy atoms of the ligand during the umbrella sampling simulations. All simulations were carried out using the CUDA version of NAMD 2.12.77 The potential of mean force (PMF) was constructed using the weighted histogram analysis method (WHAM).78
Solvent-Accessible Surface Area (SASA) calculations
Studies have shown that the standard free energy of transfer of a solute from water to the membrane is empirically related to the solute’s solvent-accessible surface area (SASA).79−81 Also, the extent of the ligand’s SASA that was buried during the receptor binding has been shown to directly correlate with the binding affinity. Apparently, for ligands binding to the lipid-exposed sites, the extent of ligand’s SASA exposed to the membrane would have similar effects. Therefore, we calculated the solvent-accessible surface area (SASA) of protein–ligand complexes and the unbound species (protein and ligands) of CB1R, ORG27569, and its analogs using NACCESS.82 The NACCESS program calculates the atomic accessible surface by rolling a probe (size = 1.4 Å) around the given molecule’s van der Waals surface. Specifically, the solvent-accessible surface area of a ligand in its receptor-bound form is defined as the membrane-exposed surface area (MESA), while the difference between the SASA of an unbound ligand and its MESA value represents the buried surface area (BSA).
Binding Free Energy Calculations Using the MMPBSA Method
We used the Molecular Mechanics Poisson–Boltzmann Surface Area (MMPBSA) method to calculate the binding free energy of the studied compounds as well as the contribution of the binding site residues of the receptor and the membrane lipids. The MMPBSA method83 combines both molecular mechanics and continuum solvation models. EMM, the average molecular mechanical energy, includes the energy of both bonded (bond, angle, dihedral, and improper interactions) and nonbonded interactions (van der Waals and electrostatic interactions) and is calculated using molecular mechanics (MM) force-field parameters.84 We applied the MMPBSA method using the latest implementation of the g_mmpbsa tool.85−87 Lipids within a 4 Å distance from the ligand were included as part of the binding site in the MMPBSA free energy calculations. The energy contribution of the protein residues and lipid components was then calculated using the following equation88
where ΔRXBE is the binding energy of residue x, n is the total number of atoms in the residue, Ai and Aifree are the energy of the ith atom from the x residue in bound and unbound forms, respectively. We further employed the heterogeneous dielectric implicit membrane model89 implemented in gmx_MMPBSA90 to estimate the MMPBSA binding free energies. In contrast to the implicit solvent model with a single dielectric constant used earlier, this membrane model allows the dielectric constant to vary along the bilayer depth in the membrane. Memopt = 2 and memopt = 3 were tested and gave similar results, so memopt = 2 was used for all calculations.
Glossary
Abbreviations
- BSA
buried surface area
- CB1R
cannabinoid receptor 1
- COM
center-of-mass
- Cryo-EM
cryo-electron microscopy
- ECL
extracellular
- H-bond
hydrogen bond
- ICL
intracellular loop
- MESA
membrane-exposed surface area
- MMPBSA
molecular mechanics Poisson–Boltzmann surface area
- MOE
molecular operating environment
- NaCl
sodium chloride
- NAM
negative allosteric modulator
- ns
nanosecond
- PMF
potential of mean force
- POPC
phosphatidylcholine
- POPE
phosphatidylethanolamine
- POPI
phosphatidylinositol
- POPS
phosphatidylserine
- PSM
phosphatidyl sphingomyelin
- ps
picosecond
- SASA
solvent-accessible surface area
- TPSA
topological polar surface area
- WT-MetaD
well-tempered metadynamics
- μs
microsecond
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00946.
Structural and physicochemical properties of ORG27569; total, nonpolar, and polar BSA and MESA; amino acid details for the CCM in GPCRs; internal angle of ORG27569; binding orientations of ORG7569 and its analogs; and critical residues at the transmembrane allosteric binding site of the CB1 receptor (PDF)
This movie captured the access and binding mechanism of ORG27569 to CB1R from an extracellular aqueous environment, investigated by the association simulations using well-tempered metadynamics (Movie M1) (MP4)
This movie captured the binding and molecular interactions of Compound 12f, an analog of ORG27569 to CB1R. The n-hexyl alkyl chain at the C-3 position of the indole ring is entirely in contact with the surrounding membrane environment, investigated by all-atom classical MD simulations (Movie M2) (MP4)
Author Contributions
P.O. and S.N. conceived and designed the experiments; P.O. carried out the experiments (modeling and simulations); and P.O. and S.N. performed data processing and analysis, and wrote the paper.
This work was supported by the National Institutes of Health/National Institute of General Medical Sciences [Grants R15 GM131293-01 and R01 GM137022].
The authors declare no competing financial interest.
Supplementary Material
References
- Sriram K.; Insel P. A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs?. Mol. Pharmacol. 2018, 93, 251. 10.1124/mol.117.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. How many drug targets are there?. Nat. Rev. Drug Discovery 2006, 5, 993–996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Santos R.; Ursu O.; Gaulton A.; Bento A. P.; Donadi R. S.; Bologa C. G.; Karlsson A.; Al-Lazikani B.; Hersey A.; Oprea T. I.; Overington J. P. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery 2017, 16, 19–34. 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J. P. Recent innovations in membrane-protein structural biology. F1000Research 2019, 8, 211. 10.12688/f1000research.16234.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch J.; Cheruvara H.; Gamage N.; Harrison P. J.; Lithgo R.; Quigley A. Changes in membrane protein structural biology. Biology 2020, 9, 401. 10.3390/biology9110401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Zhang J. Small molecule allosteric modulators of G protein-coupled receptors: Drug–target interactions. J. Med. Chem. 2019, 62, 24–45. 10.1021/acs.jmedchem.7b01844. [DOI] [PubMed] [Google Scholar]
- Payandeh J.; Volgraf M. Ligand binding at the protein–lipid interface: Strategic considerations for drug design. Nat. Rev. Drug Discovery 2021, 20, 710–722. 10.1038/s41573-021-00240-2. [DOI] [PubMed] [Google Scholar]
- Szlenk C. T.; Gc J. B.; Natesan S. Does the lipid bilayer orchestrate access and binding of ligands to transmembrane orthosteric/allosteric sites of G protein-coupled receptors?. Mol. Pharmacol. 2019, 96, 527–541. 10.1124/mol.118.115113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduk P. J.; Huth J. R.; Tse C. Predicting protein druggability. Drug Discovery Today 2005, 10, 1675–1682. 10.1016/S1359-6446(05)03624-X. [DOI] [PubMed] [Google Scholar]
- Cheng A. C.; Coleman R. G.; Smyth K. T.; Cao Q.; Soulard P.; Caffrey D. R.; Salzberg A. C.; Huang E. S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. 10.1038/nbt1273. [DOI] [PubMed] [Google Scholar]
- Schmidtke P.; Barril X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 2010, 53, 5858–5867. 10.1021/jm100574m. [DOI] [PubMed] [Google Scholar]
- Mason J. S.; Bortolato A.; Congreve M.; Marshall F. H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Wagner J. R.; Lee C. T.; Durrant J. D.; Malmstrom R. D.; Feher V. A.; Amaro R. E. Emerging computational methods for the rational discovery of allosteric drugs. Chem. Rev. 2016, 116, 6370–6390. 10.1021/acs.chemrev.5b00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J. J.; Orban Z. J.; Carlson H. A. Predicting binding sites from unbound versus bound protein structures. Sci. Rep. 2020, 10, 15856 10.1038/s41598-020-72906-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney D. C.; Haubrich B. A.; Van Liefde I.; Vauquelin G. The role of binding kinetics in GPCR drug discovery. Curr. Top Med. Chem. 2015, 15, 2504–2522. 10.2174/1568026615666150701113054. [DOI] [PubMed] [Google Scholar]
- Sykes D. A.; Parry C.; Reilly J.; Wright P.; Fairhurst R. A.; Charlton S. J. Observed drug-receptor association rates are governed by membrane affinity: The importance of establishing “micro-pharmacokinetic/pharmacodynamic relationships” at the β2-adrenoceptor. Mol. Pharmacol. 2014, 85, 608–617. 10.1124/mol.113.090209. [DOI] [PubMed] [Google Scholar]
- Dickson C. J.; Hornak V.; Velez-Vega C.; McKay D. J. J.; Reilly J.; Sandham D. A.; Shaw D.; Fairhurst R. A.; Charlton S. J.; Sykes D. A.; Pearlstein R. A.; Duca J. S. Uncoupling the structure–activity relationships of β2 adrenergic receptor ligands from membrane binding. J. Med. Chem. 2016, 59, 5780–5789. 10.1021/acs.jmedchem.6b00358. [DOI] [PubMed] [Google Scholar]
- Sykes D. A.; Stoddart L. A.; Kilpatrick L. E.; Hill S. J. Binding kinetics of ligands acting at GPCRs. Mol. Cell. Endocrinol. 2019, 485, 9–19. 10.1016/j.mce.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst D. P.; Grossfield A.; Lynch D. L.; Feller S.; Romo T. D.; Gawrisch K.; Pitman M. C.; Reggio P. H. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 2010, 285, 17954–17964. 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y.; Mercier R. W.; Anday J. K.; Thakur G. A.; Zvonok A. M.; Hurst D.; Reggio P. H.; Janero D. R.; Makriyannis A. Ligand-binding architecture of human CB2 cannabinoid receptor: Evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem. Biol. 2008, 15, 1207–1219. 10.1016/j.chembiol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakowiecki J.; Filipek S. Hydrophobic ligand entry and exit pathways of the CB1 cannabinoid receptor. J. Chem. Inf. Model. 2016, 56, 2457–2466. 10.1021/acs.jcim.6b00499. [DOI] [PubMed] [Google Scholar]
- Stanley N.; Pardo L.; Fabritiis G. D. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016, 6, 22639 10.1038/srep22639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand P. W.; Scheerer P.; Park J. H.; Choe H. W.; Piechnick R.; Ernst O. P.; Hofmann K. P.; Heck M. A ligand channel through the G protein-coupled receptor opsin. PLoS One 2009, 4, e4382 10.1371/journal.pone.0004382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch M. P.; Jo H.; Valcourt J. R.; Srinivasan Y.; Pan A. C.; Capponi S.; Grabe M.; Dror R. O.; Shaw D. E.; DeGrado W. F.; Coughlin S. R. Entry from the lipid bilayer: A possible pathway for inhibition of a peptide G protein-coupled receptor by a lipophilic small molecule. Biochemistry 2018, 57, 5748–5758. 10.1021/acs.biochem.8b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes D. A.; Parry C.; Reilly J.; Wright P.; Fairhurst R. A.; Charlton S. J. Observed drug-receptor association rates are governed by membrane affinity: The importance of establishing ““micro-pharmacokinetic/pharmacodynamic relationships” at the β2-adrenoceptor. Mol. Pharmacol. 2014, 85, 608–617. 10.1124/mol.113.090209. [DOI] [PubMed] [Google Scholar]
- Szlenk C. T.; Gc J. B.; Natesan S. Membrane-facilitated receptor access and binding mechanisms of long-acting β2-adrenergic receptor (β2-AR) agonists. Mol. Pharmacol. 2021, 100, 406–427. 10.1124/molpharm.121.000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato F.; Juhola H.; Zak A.; Postila P. A.; Saukko A.; Rissanen S.; Enkavi G.; Vattulainen I.; Kepczynski M.; Róg T. Membrane-dependent binding and entry mechanism of dopamine into its receptor. ACS Chem. Neurosci. 2020, 11, 1914–1924. 10.1021/acschemneuro.9b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriakidi S.; Chatzigiannis C.; Papaemmanouil C.; Tzakos A. G.; Mavromoustakos T. Exploring the role of the membrane bilayer in the recognition of candesartan by its GPCR AT1 receptor. Biochim. Biophys. Acta, Biomembr. 2020, 1862, 183142 10.1016/j.bbamem.2019.183142. [DOI] [PubMed] [Google Scholar]
- Yuan X.; Raniolo S.; Limongelli V.; Xu Y. The molecular mechanism underlying ligand binding to the membrane-embedded site of a G protein-coupled receptor. J. Chem. Theory Comput. 2018, 14, 2761–2770. 10.1021/acs.jctc.8b00046. [DOI] [PubMed] [Google Scholar]
- Vauquelin G. On the ‘micro’-pharmacodynamic and pharmacokinetic mechanisms that contribute to long-lasting drug action. Expert Opin. Drug Discovery 2015, 10, 1085–1098. 10.1517/17460441.2015.1067196. [DOI] [PubMed] [Google Scholar]
- Sargent D. F.; Schwyzer R. Membrane lipid phase as catalyst for peptide--receptor interactions. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 5774–5778. 10.1073/pnas.83.16.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwyzer R. In search of the ‘bio-active conformation’: Is it induced by the target cell membrane?. J. Mol. Recognit. 1995, 8, 3–8. 10.1002/jmr.300080103. [DOI] [PubMed] [Google Scholar]
- Shao Z.; Yan W.; Chapman K.; Ramesh K.; Ferrell A. J.; Yin J.; Wang X.; Xu Q.; Rosenbaum D. M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. 10.1038/s41589-019-0387-2. [DOI] [PubMed] [Google Scholar]
- Price M. R.; Baillie G. L.; Thomas A.; Stevenson L. A.; Easson M.; Goodwin R.; McLean A.; McIntosh L.; Goodwin G.; Walker G.; Westwood P.; Marrs J.; Thomson F.; Cowley P.; Christopoulos A.; Pertwee R. G.; Ross R. A. Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol. 2005, 68, 1484–1495. 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Piscitelli F.; Ligresti A.; La Regina G.; Coluccia A.; Morera L.; Allarà M.; Novellino E.; Di Marzo V.; Silvestri R. Indole-2-carboxamides as allosteric modulators of the cannabinoid CB1 receptor. J. Med. Chem. 2012, 55, 5627–5631. 10.1021/jm201485c. [DOI] [PubMed] [Google Scholar]
- Ahn K. H.; Mahmoud M. M.; Samala S.; Lu D.; Kendall D. A. Profiling two indole-2-carboxamides for allosteric modulation of the CB1 receptor. J. Neurochem. 2013, 124, 584–589. 10.1111/jnc.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud M. M.; Ali H. I.; Ahn K. H.; Damaraju A.; Samala S.; Pulipati V. K.; Kolluru S.; Kendall D. A.; Lu D. Structure-activity relationship study of indole-2-carboxamides identifies a potent allosteric modulator for the cannabinoid receptor 1 (CB1). J. Med. Chem. 2013, 56, 7965–7975. 10.1021/jm4009828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L.; Ali H. I.; Olszewska T.; Ahn K. H.; Damaraju A.; Kendall D. A.; Lu D. Optimization of chemical functionalities of indole-2-carboxamides to improve allosteric parameters for the cannabinoid receptor 1 (CB1). J. Med. Chem. 2014, 57, 3040–3052. 10.1021/jm5000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: An open-source platform for ligand pocket detection. BMC Bioinf. 2009, 10, 168 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bio-Loom, BioByte Corp, 212 E Rowland St, DPT#55021, Covina, CA 91723. 2021.
- Ingólfsson H. I.; Melo M. N.; van Eerden F. J.; Arnarez C.; Lopez C. A.; Wassenaar T. A.; Periole X.; de Vries A. H.; Tieleman D. P.; Marrink S. J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- Van Meer G.; Voelker D. R.; Feigenson G. W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopart R.; Lu D.; Lichtman A. H.; Kendall D. A. Allosteric modulators of cannabinoid receptor 1: Developing compounds for improved specificity. Drug Metab. Rev. 2018, 50, 3–13. 10.1080/03602532.2018.1428342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Zhang J. Small molecule allosteric modulators of G protein-coupled receptors: Drug-target interactions. J. Med. Chem. 2019, 62, 24–45. 10.1021/acs.jmedchem.7b01844. [DOI] [PubMed] [Google Scholar]
- Sykes D. A.; Charlton S. J. Slow receptor dissociation is not a key factor in the duration of action of inhaled long-acting β2-adrenoceptor agonists. Br. J. Pharmacol. 2012, 165, 2672–2683. 10.1111/j.1476-5381.2011.01639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrini A.; Joseph V.; Dourado M.; Reese R. M.; Shields S. D.; Rougé L.; Bravo D. D.; Chernov-Rogan T.; Austin C. D.; Chen H.; Wang L.; Villemure E.; Shore D. G. M.; Verma V. A.; Hu B.; Chen Y.; Leong L.; Bjornson C.; Hötzel K.; Gogineni A.; Lee W. P.; Suto E.; Wu X.; Liu J.; Zhang J.; Gandham V.; Wang J.; Payandeh J.; Ciferri C.; Estevez A.; Arthur C. P.; Kortmann J.; Wong R. L.; Heredia J. E.; Doerr J.; Jung M.; Vander Heiden J. A.; Roose-Girma M.; Tam L.; Barck K. H.; Carano R. A. D.; Ding H. T.; Brillantes B.; Tam C.; Yang X.; Gao S. S.; Ly J. Q.; Liu L.; Chen L.; Liederer B. M.; Lin J. H.; Magnuson S.; Chen J.; Hackos D. H.; Elstrott J.; Rohou A.; Safina B. S.; Volgraf M.; Bauer R. N.; Riol-Blanco L. A TRPA1 inhibitor suppresses neurogenic inflammation and airway contraction for asthma treatment. J. Exp. Med. 2021, 218, e20201637 10.1084/jem.20201637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Dewdney T. G.; Liu Z.; Reiter S. J.; Brunzelle J. S.; Kovari I. A.; Kovari L. C. Higher desolvation energy reduces molecular recognition in multi-drug resistant HIV-1 protease. Biology 2012, 1, 81–93. 10.3390/biology1010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T.; Abel R.; Kim B.; Berne B. J.; Friesner R. A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 808–813. 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal J.; Friesner R. A.; Berne B. J. Role of desolvation in thermodynamics and kinetics of ligand binding to a kinase. J. Chem. Theory Comput. 2014, 10, 5696–5705. 10.1021/ct500584n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deganutti G.; Zhukov A.; Deflorian F.; Federico S.; Spalluto G.; Cooke R. M.; Moro S.; Mason J. S.; Bortolato A. Impact of protein-ligand solvation and desolvation on transition state thermodynamic properties of adenosine A(2A) ligand binding kinetics. In Silico Pharmacol. 2017, 5, 16 10.1007/s40203-017-0037-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M. A.; Cherezov V.; Griffith M. T.; Roth C. B.; Jaakola V. P.; Chien E. Y.; Velasquez J.; Kuhn P.; Stevens R. C. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008, 16, 897–905. 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornaiuolo M.; Bruno A.; Botta L.; Regina G. L.; Cosconati S.; Silvestri R.; Marinelli L.; Novellino E. Endogenous vs exogenous allosteric modulators in GPCRs: A dispute for shuttling CB1 among different membrane microenvironments. Sci. Rep. 2015, 5, 15453 10.1038/srep15453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T.; Vemuri K.; Nikas S. P.; Laprairie R. B.; Wu Y.; Qu L.; Pu M.; Korde A.; Jiang S.; Ho J.-H.; Han G. W.; Ding K.; Li X.; Liu H.; Hanson M. A.; Zhao S.; Bohn L. M.; Makriyannis A.; Stevens R. C.; Liu Z.-J. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. 10.1038/nature23272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K. H.; Mahmoud M. M.; Kendall D. A. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein independent ERK1/2 kinase activation. J. Biol. Chem. 2012, 287, 12070–12082. 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccatelli F.; Aliagas I.; Zheng H. Why decreasing lipophilicity alone is often not a reliable strategy for extending IV half-life. ACS Med. Chem. Lett. 2018, 9, 522–527. 10.1021/acsmedchemlett.8b00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin H.; Altman M. D.; Ellis J. M.; Fuller P.; Johnson S. A.; Lahue B.; Lapointe B. Strategy for extending half-life in drug design and its significance. ACS Med. Chem. Lett. 2018, 9, 528–533. 10.1021/acsmedchemlett.8b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemure E.; Terrett J. A.; Larouche-Gauthier R.; Déry M.; Chen H.; Reese R. M.; Shields S. D.; Chen J.; Magnuson S.; Volgraf M. A retrospective look at the impact of binding site environment on the optimization of TRPA1 antagonists. ACS Med. Chem. Lett. 2021, 12, 1230–1237. 10.1021/acsmedchemlett.1c00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster D. J.; Conn P. J. Allosteric modulation of GPCRs: New insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron 2017, 94, 431–446. 10.1016/j.neuron.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey Conn P.; Christopoulos A.; Lindsley C. W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discovery 2009, 8, 41–54. 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Lindsley C. W.; Meiler J.; Niswender C. M. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discovery 2014, 13, 692–708. 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casares D.; Escribá P. V.; Rosselló C. A. Membrane lipid composition: Effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int. J. Mol. Sci. 2019, 20, 2167. 10.3390/ijms20092167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harayama T.; Riezman H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- Fadeel B.; Xue D. The ins and outs of phospholipid asymmetry in the plasma membrane: Roles in health and disease. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 264–277. 10.1080/10409230903193307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2021, 2019.
- Webb B.; Sali A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Lee J.; Jo S.; Brooks C. L. 3rd; Lee H. S.; Im W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. 10.1002/jcc.24829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. Jr. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Cheng X.; Swails J. M.; Yeom M. S.; Eastman P. K.; Lemkul J. A.; Wei S.; Buckner J.; Jeong J. C.; Qi Y.; Jo S.; Pande V. S.; Case D. A.; Brooks C. L.; MacKerell A. D.; Klauda J. B.; Im W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Lim J. B.; Klauda J. B.; Im W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Jo S.; Kim T.; Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One 2007, 2, e880 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Barducci A.; Bussi G.; Parrinello M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Marcos-Alcalde I.; Setoain J.; Mendieta-Moreno J. I.; Mendieta J.; Gómez-Puertas P. MEPSA: Minimum energy pathway analysis for energy landscapes. Bioinformatics 2015, 31, 3853–3855. 10.1093/bioinformatics/btv453. [DOI] [PubMed] [Google Scholar]
- GC J. B.; Szlenk C. T.; Gao J.; Dong X.; Wang Z.; Natesan S. Molecular dynamics simulations provide insight into the loading efficiency of proresolving lipid mediators Resolvin D1 and D2 in cell membrane-derived nanovesicles. Mol. Pharmaceutics 2020, 17, 2155–2164. 10.1021/acs.molpharmaceut.0c00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Rosenberg J. M.; Bouzida D.; Swendsen R. H.; Kollman P. A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. 10.1002/jcc.540130812. [DOI] [Google Scholar]
- Juffer A. H.; Eisenhaber F.; Hubbard S. J.; Walther D.; Argos P. Comparison of atomic solvation parametric sets: Applicability and limitations in protein folding and binding. Protein Sci. 1995, 4, 2499–2509. 10.1002/pro.5560041206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds J. A.; Gilbert D. B.; Tanford C. Empirical correlation between hydrophobic free energy and aqueous cavity surface area. Proc. Natl. Acad. Sci. U.S.A. 1974, 71, 2925–2927. 10.1073/pnas.71.8.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C. Hydrophobic bonding and accessible surface area in proteins. Nature 1974, 248, 338–339. 10.1038/248338a0. [DOI] [PubMed] [Google Scholar]
- Hubbard SJ T. J.NACCESS. Computer program, Department of Biochemistry and Molecular Biology, University College London,1993. [Google Scholar]
- Srinivasan J.; Cheatham T. E.; Cieplak P.; Kollman P. A.; Case D. A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate–DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. 10.1021/ja981844+. [DOI] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Kollman P. A. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. 10.1021/jm000241h. [DOI] [PubMed] [Google Scholar]
- Ferrari A. M.; Degliesposti G.; Sgobba M.; Rastelli G. Validation of an automated procedure for the prediction of relative free energies of binding on a set of aldose reductase inhibitors. Bioorg. Med. Chem. 2007, 15, 7865–7877. 10.1016/j.bmc.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Kumari R.; Kumar R.; Lynn A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Miller B. R.; McGee T. D.; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Greene D. A.; Qi R.; Nguyen R.; Qiu T.; Luo R. Heterogeneous dielectric implicit membrane model for the calculation of MMPBSA binding free energies. J. Chem. Inf. Model. 2019, 59, 3041–3056. 10.1021/acs.jcim.9b00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés-Tresanco M. S.; Valdés-Tresanco M. E.; Valiente P. A.; Moreno E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.