

Abstract

Huntington’s disease (HD) is a lethal autosomal dominant neurodegenerative disorder resulting from a CAG repeat expansion in the huntingtin (HTT) gene. The product of translation of this gene is a highly aggregation-prone protein containing a polyglutamine tract >35 repeats (mHTT) that has been shown to colocalize with histone deacetylase 4 (HDAC4) in cytoplasmic inclusions in HD mouse models. Genetic reduction of HDAC4 in an HD mouse model resulted in delayed aggregation of mHTT, along with amelioration of neurological phenotypes and extended lifespan. To further investigate the role of HDAC4 in cellular models of HD, we have developed bifunctional degraders of the protein and report the first potent and selective degraders of HDAC4 that show an effect in multiple cell lines, including HD mouse model-derived cortical neurons. These degraders act via the ubiquitin-proteasomal pathway and selectively degrade HDAC4 over other class IIa HDAC isoforms (HDAC5, HDAC7, and HDAC9).
Introduction
The histone deacetylase (HDAC) enzyme family plays a key role in the epigenetic mechanisms that regulate transcription through deacetylation of lysine residues within the N-terminus of histone proteins. HDACs are capable of shuttling between the cytoplasm and nucleus and are directly linked to cellular signaling networks.1−3 Recently, the pharmacologic inhibition of HDACs has emerged as a promising strategy to treat neurodegenerative disease, with protective effects in various animal models of Alzheimer’s and Parkinson’s diseases, amyotrophic lateral sclerosis, and Huntington’s disease (HD).4–6
A hallmark of HD is the translation of the mutant HTT gene to an aggregation-prone, polyglutamine-rich (>35 Q) mutant huntingtin protein (mHTT). In transgenic mouse models of HD (R6/2 and HdhQ150), the HDAC class IIa isoform HDAC4 associates with mHTT in the cytoplasm and colocalizes with cytoplasmic aggregates.7 Genetic suppression of HDAC4 in R6/2 mice delayed aggregation of mHTT in the central nervous system (CNS) and resulted in elevated expression of brain-derived neurotrophic factor (BDNF) with a concomitant amelioration of disease phenotype and an increased life span. In a precedent study, a reduction in the level of HDAC4 protein in the cortex and brain stem of R6/2 mice was observed after chronic administration of a HDAC pan-inhibitor (suberoylanilide hydroxamic acid: SAHA) with a decrease in aggregate load observed in the same brain regions.8 Based on this precedent, we have developed class IIa-selective HDAC inhibitors to investigate the effect of HDAC4 inhibition in preclinical HD models.9−12 Specifically, two chemical classes were pursued that resulted in the development of potent, orally available, and brain-penetrant class IIa HDAC inhibitors. As a companion approach, we also tethered our class IIa HDAC inhibitors to a binding moiety to the von Hippel–Landau (VHL) E3 ubiquitin ligase to produce intracellular bifunctional degraders of HDAC4 protein.
Bifunctional protein degraders, often referred to as proteolysis-targeting chimeras (PROTACs),13−15 catalyze degradation of the target protein of interest (PoI) by recruiting E3 ubiquitin ligases to specifically catalyze the ubiquitination of the target protein, resulting in subsequent degradation by the 26S proteasome. The degrader is comprised of a ligand to the PoI (aka “warhead”) tethered through a linker to an E3 ubiquitin ligase-recruiting ligand. Due to the catalytic nature of the degradation mechanism, a sustained effect can be observed at sub-stoichiometric compound concentrations. Furthermore, degrader efficiency and selectivity are not solely a function of the binding affinity and specific to the target PoI, but rather on the ternary complex formed between PoI, degrader, and E3 ligase such that a bifunctional degrader may exhibit high isoform selectivity despite a relatively poor target-binding selectivity profile.16
While several bifunctional degraders of HDAC proteins have been developed,17−27 none target HDAC4. Here we describe the design, synthesis, and in vitro characterization of the first generation of highly isoform-selective HDAC4 protein degraders.
Results and Discussion
Design and Synthesis of Bifunctional HDAC4 Degraders
As one of our starting points, we selected the hydroxamic acid (HA) 1, a potent, cell-permeable, and CNS-penetrant class IIa HDAC pan-selective inhibitor previously reported by Luckhurst et al. (Figure 1).9 Based on the co-crystal X-ray structure of the closely related analogue 3 (Figure 2) in HDAC4, it was envisaged that the 5-position of the pyrimidine ring would accommodate a linker to the E3 ligase ligand with little impact on binding, as this vector points toward solvent in the co-crystal structure (Figure 2A).
Figure 1.

Class IIa HDAC inhibitors: hydroxamic acid 1 and trifluoromethyloxadiazole 2.
Figure 2.
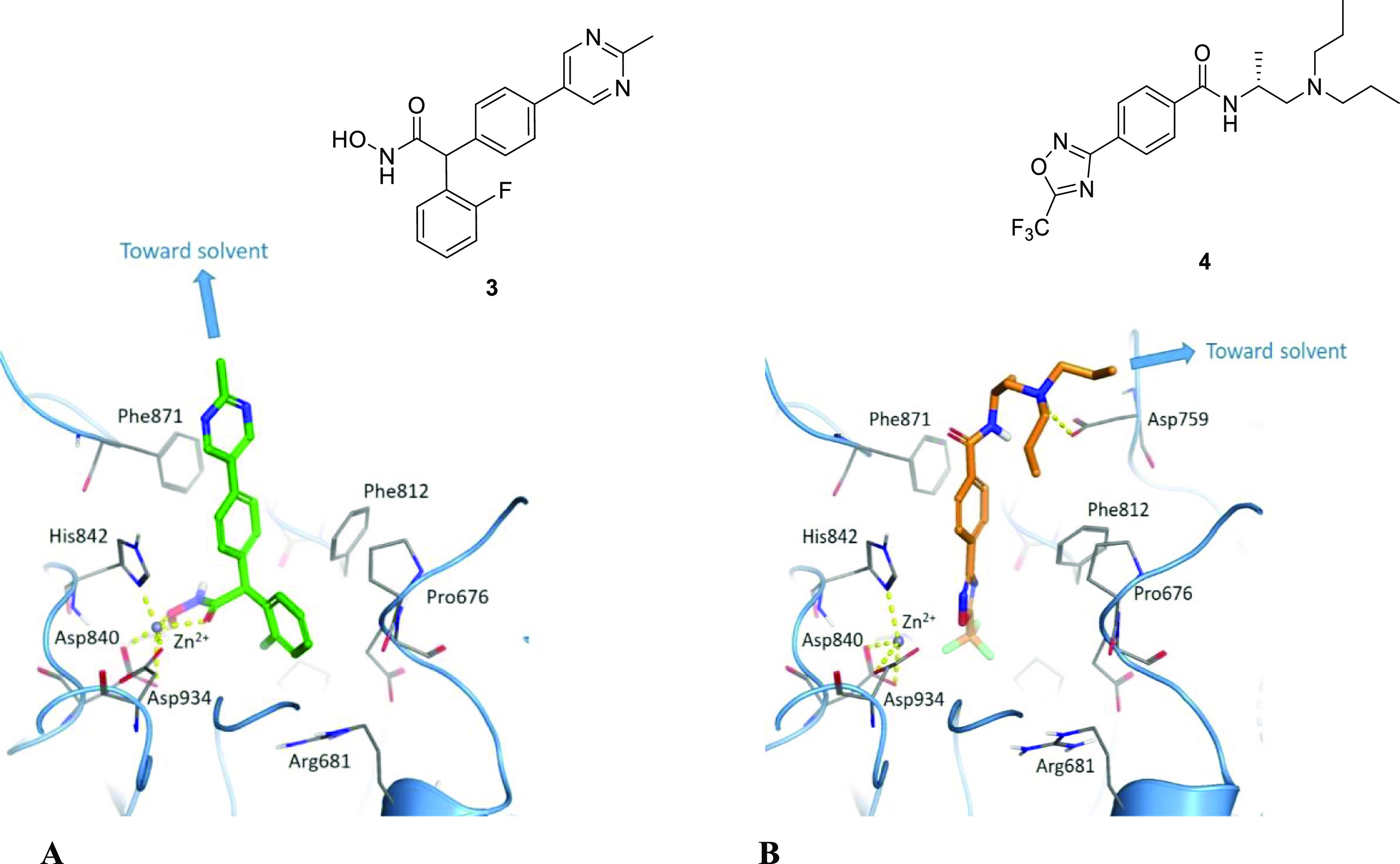
(A) Co-crystal X-ray structure showing the (S)-enantiomer of HA compound 3 at 2.1 Å resolution (PDB: 6FYZ), and (B) TFMO compound 4 docked into the HDAC4 active site, each illustrating a suitable site of attachment for a tether to the E3 ligase ligand.
As well as the HA degraders, we also explored degraders derived from the trifluoromethyloxadiazole (TFMO) series exemplified for compound 2, a potent, cell permeable and CNS-penetrant class IIa HDAC pan-selective inhibitor previously reported (Figure 1).11 It is well established in the degrader literature that increasing the H-bond donor count negatively correlates with degrader potency, partly due to the effect on cell permeability and also reduces the probability of achieving high oral bioavailability.28−31 As the target is located in the CNS, it was anticipated that incorporating a nonacidic functionality into our compounds would increase the likelihood of achieving acceptable brain penetration.32 On the basis of docking of the TFMO compound 4 in HDAC4 (Figure 2B), we envisaged that the terminus of the propyl group would provide a suitable attachment point for a linker to the E3 ligase ligand, as this position is directed toward the solvent.
Based on the early reports that the VHL E3 ligase had been successfully applied toward degradation of oncology targets33,34 we chose the VHL ligand for our preliminary studies.35 It has since been well documented that linker composition and length can affect degrader efficiency and, in the absence of structural information, that optimal linker composition must be empirically derived.36 We therefore designed HDAC4 degraders based on tethering compounds 1 and 4 to VHL E3 ubiquitin ligase ligands using PEGylated linkers of varying lengths (n = 2–4 ethoxy chains) (Figure 3).
Figure 3.
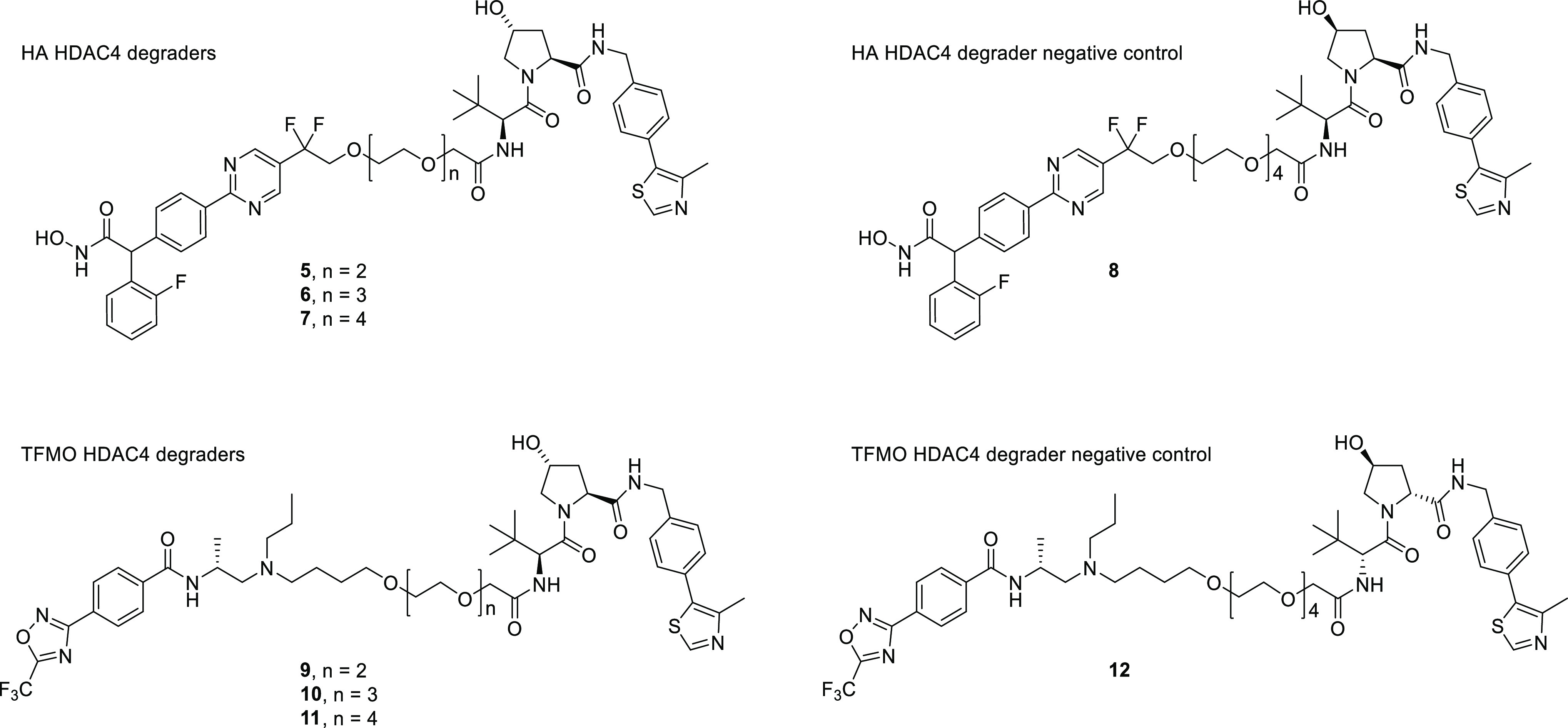
Structures for HDAC4 degraders and negative controls.
Synthesis of the HA series of HDAC4 degraders (5–7) and negative control 8 is shown in Scheme 1. The synthesis started by the treatment of 1-bromo-2-fluorobenzene (13) with n-butyl lithium, followed by the addition of 4-bromobenzaldehyde to afford an alcohol that was subsequently converted into nitrile 14 by InBr3-catalyzed cyanation. The nitrile was hydrolyzed to a carboxylic acid and coupled to O-(tetrahydro-2H-pyran-2-yl)hydroxylamine with HATU to provide the THP-protected hydroxamic acid intermediate 15. Palladium-mediated borylation of 15 with bis(pinacolato)diboron provided the Suzuki precursor 16. The other coupling partners for the Suzuki reaction were synthesized starting from 5-iodo-2-methoxy-pyrimidine (17). Treatment of 17 with ethyl 2-bromo-2,2-difluoroacetate in the presence of copper afforded an ester, which was reduced to alcohol 18. Heating 18 with cyanomethylenetributylphosphorane and t-butyl ester PEG linkers (19, 20, 21) provided a series of PEG-linked esters of varying length that were converted into the acid stable methyl esters and prepared for Suzuki cross-coupling by chlorination with POCl3 to produce the chloropyrimidines 22. These chloropyrimidines were coupled to boronate 16 under palladium-mediated conditions, under which conditions the methyl esters were saponified directly to the acids (23). HATU-mediated coupling of the acids (23) to VHL ligand 24 followed by removal of the THP protecting group provided the HA series HDAC4 degraders 5, 6, and 7. Following the same method, intermediate 23 and epimeric VHL ligand 25 afforded the HA HDAC degrader negative control 8.
Scheme 1. Synthesis of HA Series HDAC4 Degraders 5–7 and Negative Control 8.

Reagents and conditions: (a) 1.6 M nBuLi in hexanes, 4-bromobenzaldehyde −78 °C, 3 h, 99%; (b) InBr3, TMSCN, 4 Å molecular sieves, DCM, RT, 18 h, 60%; (c) 6 N NaOH, EtOH, 84 °C, 18 h, 96%; (d) O-(tetrahydro-2H-pyran-2-yl)hydroxylamine, HATU, iPr2NEt, DMF, RT, 0.5 h, 82%; (e) bis(pinacolato)diboron, CsF, Pd(PPh3)4, DME, MeOH, 110 °C, 1.5 h, 52%; (f) ethyl 2-bromo-2,2-difluoroacetate, Cu, DMSO, 50 °C, 18 h, 92%; (g) LiBH4, THF/EtOH, 0 °C for 2 h, then 6 N HCl, MeOH, RT, 1.5 h, 37%; (h) cyanomethylenetributylphosphorane, toluene, 90 or 100 °C, 18 h, 80% (n = 2); (i) 2 N HCl, 1,4-dioxane, 100 °C, 2 h; (j) 12 N HCl, MeOH, RT, 18 h, 47% over 2 steps (n = 2); (k) POCl3, reflux, 2 h, 78% (n = 2); (l) Pd(PPh3)4, K2CO3, 1,4-dioxane, H2O, 100 °C, 1 h, then 110 °C, 1.5 h, 39% (n = 2); (m) HATU, iPr2NEt, DMF, RT, 0.5 h, 67% (n = 2); (n) TFA, MeOH, RT, 18 h, 62% (n = 2).
Scheme 2 highlights the sequence used to synthesize TFMO HDAC4 degraders 9–11 and the VHL epimer negative control 12. Mono-alkylation of the commercially available diols 26–28 with tert-butyl 2-bromoacetate gave the hydroxy intermediates 29 which were mesylated, reacted with butane-1,4-diol, and trans-esterified to afford the methyl esters 30. Oxidation of 30 to aldehydes 31 followed by reductive amination and N-deprotection afforded the primary amine intermediates 32. Introduction of the amido functionality followed by ester hydrolysis provided the carboxylic acids 33, which were subsequently coupled with the VHL ligand 24 to afford the desired targets 9–11, or, in the case of coupling with the VHL epimer 34, the negative control 12.
Scheme 2. Synthesis of the TFMO-Series HDAC4 Degraders 9–11 and Negative Control 12.

Reagents and conditions: (a) KOtBu, THF, 45 °C for 0.5 h then tert-butyl 2-bromoacetate, 0 °C to RT, 24 h, 47% (n = 2); (b) MsCl, Et3N, DCM, 0 °C to RT, 20 h, 99.6% (n = 2); (c) butane-1,4-diol, KOtBu, THF, RT, 22 h; (d) H2SO4, MeOH, RT, 3 days, 54% over 2 steps (n = 2); (e) (COCl)2, DMSO, Et3N, DCM, −78 °C to RT, 3.5 h, 99% (n = 2); (f) tert-butyl (R)-(1-(propylamino)propan-2-yl)carbamate, NaBH(OAc)3, DCM, RT, 20 h, 65% (n = 2); (g) 4 M HCl in 1,4-dioxane, MeOH, 0 °C to RT, 2 h, 99% (n = 2); (h) 4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)benzoic acid, EDC, HOBt, iPr2NEt, DCM, RT, 15 h, 70% (n = 2); (i) LiOH, MeOH, H2O, RT, 5 h, 91% (n = 2); (j) HATU, iPr2NEt, DMF, RT, 0.5 h, 45% (n = 2).
Confirmation of Target Engagement
The HDAC4 degraders were profiled in comparison to the unmodified HDAC inhibitors 1 and 4 in recombinant HDAC enzymatic inhibition assays9 to confirm target engagement and selectivity relative to the other class IIa isoforms (Table 1). With respect to the HA degraders, these were produced based on the SAR reported by Luckhurst et al.,9 in which all the analogues are racemic, and a comparison between (R)- and (S)-activity was not made. For the sake of comparison to the previous studies, we continued with the racemate. For the HA series we observed a 5- to 20-fold reduction in HDAC4 enzymatic inhibition for the degraders 5–7, and negative control (8) relative to the parent compound (1). Presumably the reduction in HDAC4 inhibition is due to steric interference introduced by the PEG-linked VHL ligand.
Table 1. Recombinant HDAC Activity, Cellular HDAC Functional Activity and Aqueous Solubility of the HDAC4 Inhibitors and Degraders.
| compound |
HDAC enzymatic IC50 (μM)a |
HDAC cellular IC50 (μM)b |
solubility (μM)e | |||||
|---|---|---|---|---|---|---|---|---|
| HDAC4 | HDAC5 | HDAC7 | HDAC9 | class IIac | class Id | |||
| HA series | 1 | 0.054 | 0.060 | 0.031 | 0.050 | 0.069 | >5 | 142f |
| 5 | 0.41 | 0.23 | 0.21 | 0.29 | 0.15 | >5 | 5.2 | |
| 6 | 0.56 | 0.31 | 0.21 | 0.34 | 0.16 | >5 | 5.0 | |
| 7 | 1.1 | 0.26 | 0.25 | 0.42 | 0.12 | >5 | 4.8 | |
| 8 | 0.23 | n.d.g | n.d. | n.d. | 0.13 | >5 | 4.8 | |
| TFMO series | 4 | 0.003 | 0.005 | 0.013 | 0.013 | 0.024 | 3.0 | 145h |
| 9 | 0.090 | 0.14 | 0.22 | 0.13 | 0.033 | 4.3 | 100 | |
| 10 | 0.10 | 0.25 | 0.23 | 0.22 | 0.002 | 2.8 | 132 | |
| 11 | 0.092 | 0.15 | 0.15 | 0.094 | 0.006 | >5 | 144 | |
| 12 | 0.15 | n.d. | n.d. | n.d. | 0.18 | >5 | n.d. | |
Values are the geomean of ≥2 replicates; standard deviations are <10% of the mean value.
Jurkat E6.1, treated for 24 h; values are the geomean of ≥2 replicates; standard deviations are <10% of the mean value.
HDAC substrate = Boc-Lys(TFA)AMC.
HDAC substrate = Boc-Lys(Ac)AMC.
Kinetic solubility measured in pH 7.4 PBS, 2% v/v DMSO, n = 1 unless otherwise indicated.
Average of two measurements.
n.d. = not determined.
Average of four measurements.
Nevertheless, the HA series degraders showed inhibition comparable to the parent compound in the class IIa Jurkat cell assay [IC50 (cell): 1 = 0.069 μM, 5 = 0.15 μM, 6 = 0.16 μM, 7 = 0.12 μM, 8 = 0.13 μM], thus indicating good cell permeability for these degraders despite a significant reduction in aqueous solubility relative to parent. None of the HA series compounds demonstrated class IIa isoform selectivity in the HDAC enzymatic assay but showed high selectivity over the class I isoforms in the class I Jurkat cell assay. Compared to the HA series, the TFMO-series showed a more pronounced reduction in HDAC4 inhibition upon introduction of the PEG-linked VHL ligand to the parent [HDAC4 IC50: 4 (parent) = 0.003 μM, 9 = 0.09 μM, 10 = 0.10 μM, 11 = 0.092 μM, 12 = 0.15 μM]. Appending the PEG-linked VHL ligand did not have a significant impact on solubility in the TFMO-series. With the exception of the negative control 12, these TFMO-series degraders showed comparable class IIa functional activity in Jurkat compared to the parent [IC50 (cell): 4 = 0.024 μM, 9 = 0.033 μM, 10 = 0.002 μM, 11 = 0.006 μM, 12 = 0.18 μM] and good selectivity over the class I isoforms based on the cell assay.
Assessing Degradation of HDAC4 by MSD and Immunoblotting
To quantify cellular HDAC4 protein levels and compare the potency of the degraders, an ELISA-based electrochemiluminescence immunoassay for HDAC4 was developed using the Meso Scale Discovery (MSD) platform and commercially available HDAC4 antibodies (Abcam, ab12171 for capture and Abcam, ab123513 or Santa Cruz, sc-11418 for detection). The amount of HDAC4 protein remaining based on MSD was determined in response to an eleven-point concentration response from 10 μM to 0.05 nM (Figures 4, S6A and S6B). DC50 values (concentration at 50% maximum degradation) and Dmax (maximum % degradation compared to DMSO) were determined for the selected degraders 5–12 (Table 2).
Figure 4.

Determination of HDAC4 degradation potency by HDAC4 MSD assay in Jurkat cells for (A) HA degraders 5–7 and HA negative control 8; (B) TFMO degraders 9–11 and TFMO negative control 12.
Table 2. HDAC4 Degradation Efficiency for HA HDAC4 Degraders 5–8 and TFMO HDAC4 Degraders 9–12 Based on MSDa.
| HA degraders |
TFMO
degraders |
|||||||
|---|---|---|---|---|---|---|---|---|
| compound | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| linker length | n = 2 | n = 3 | n = 4 | n = 4 | n = 2 | n = 3 | n = 4 | n = 4 |
| DC50 (nM) | 28 | 48 | 37 | >10,000 | 3 | 4 | 4 | >10,000 |
| Dmax (%) | 77 | 82 | 83 | 20 | 72 | 85 | 78 | 50 |
Jurkat E6-1 cells were treated with compounds for 24 h; DC50 and Dmax values are the geometric and arithmetic means, respectively, of 2–10 experiments; standard deviations are <10% of the mean value.
The HA HDAC4 degraders 5–7 gave comparable DC50 values, ranging from 28–48 nM with associated Dmax values in the range of 77–83%. Similarly, for the TFMO degraders 9–11, there was little difference in DC50 values across the series, 3–4 nM, with Dmax range of 72–85%. These results show that varying the PEG linker length from n = 2 to n = 4 for both series had no influence on degradation efficiency. As expected, the negative controls, epimers 8 and 12, did not demonstrate significant degradation activity up to a concentration of 10 μM (20 and 50% HDAC4 reduction at 10 μM, respectively).
To confirm concentration-dependent degradation for a representative degrader from each series, cells were treated with an 11-point titration (10 μM–0.05 nM) of HA derivative 7 or TFMO derivative 11 for 24 h and analyzed using immunoblotting (Figures 5 and S1). A hook effect was observed at 10 μM for compound 7, indicating competition between binary and ternary complexes at this high concentration. In contrast, a hook effect was not observed for compound 11. Negative controls 8 and 12, as expected, did not induce HDAC4 degradation at 10 μM in this assay, confirming that these degraders do indeed mediate HDAC4 degradation through interaction with VHL E3 ligase. In the immunoblot study shown in Figure 5, quantification for HDAC4 was normalized to the housekeeping protein actin. This study revealed that HDAC4 reduction was observed with a DC50 of 11 and 2 nM for compounds 7 and 11 respectively, confirming that 11 is a more potent degrader of HDAC4 compared to 7.
Figure 5.
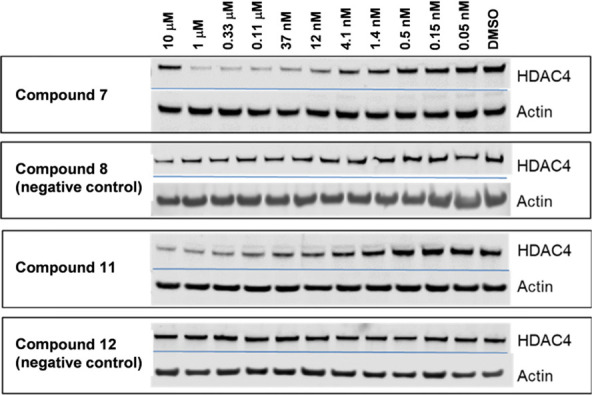
Compounds 7 and 11, but not their negative controls 8 and 12, demonstrate concentration-dependent degradation of HDAC4 by WB in Jurkat E6-1 cells.
Investigating the Mode of Action
To further confirm the proteasomal-mediated degradation, Jurkat E6-1 cells were treated with compound 7 or 11 at a concentration 25-fold higher than the DC50, and the protein levels analyzed using immunoblotting. Treatment with 1 μM of compound 7 was shown to reduce the cellular HDAC4 level after a 6 h incubation period (Figure 6). In the presence of 1 μM proteosome inhibitors MG132 orcarfilzomib, HDAC4 degradation was prevented, demonstrating that compound 7 promotes HDAC4 degradation via a proteasomal degradation pathway. The same effect was observed for compound 11 when Jurkat E6-1 cells were treated with 100 nM of compound for 6 h with and without proteasome inhibitors MG132 orcarfilzomib (Figure 6).
Figure 6.

HDAC4 degradation in Jurkat E6-1 cells upon treatment with compounds 7 and 11 dosed at 1 μM and 100 nM, respectively, compared to DMSO control in the presence and absence of 1 μM proteasome inhibitors MG132 orcarfilzomib after 6 h.
HDAC4 Degraders Demonstrate Isoform Selectivity
To assess the selectivity of compounds 7 and 11 against other class IIa HDACs, western blot was performed in Jurkat cells for HDAC1, HDAC3, HDAC5, HDAC7, and HDAC9, multiplexed with HDAC4 and actin as controls. No concentration-dependent reduction in immunoreactivity was observed against any other HDACs tested for either compound 7 or 11, while demonstrating potent HDAC4 degradation (Figure 7). This experiment demonstrated that compounds 7 and 11 have good degradation selectivity for HDAC4 against other HDACs, despite 7 having slightly higher (3- to 4-fold) inhibition of HDAC5, HDAC7, and HDAC9 compared to HDAC4, and 11 being equally potent across all four HDAC class IIa isoforms in the HDAC enzymatic activity assays (Table 1). Hence, it is the case that 7 and 11 bind to HDAC4 with no preference over binding to HDAC5, HDAC7, and HDAC9, yet selectively degrade the HDAC4 protein.
Figure 7.

Concentration-dependent degradation over 24 h of HDAC4 in Jurkat E6-1 cells was observed for compounds 7 and 11, but neither compound demonstrated degradation of HDAC1, HDAC5, HDAC7, and HDAC9. Compound 11 showed HDAC3 degradation at the highest dose of 10 μM.
In a related fashion, “PROTAC 3i” has been reported to selectively degrade TBK1 and not IKKε, despite this compound having a similar binding affinity for both proteins.36 Furthermore, apparent degradation selectivity has been observed within the HDAC family proteins for a HDAC6 PROTAC, generated from a nonselective HA HDAC inhibitor linked to pomalidomide.17
A correlation between target-degradation specificity and target-binding affinity has been explored by Bondeson et al. for multiple kinases.37 This study postulated that highly efficient degraders oriented VHL E3 ligase and target proteins in a way that allows a favorable protein–protein interaction to occur and facilitates effective ubiquitination of target proteins. In our case, compounds 7 and 11 may be able to form favorable interactions between HDAC4 and VHL and achieve efficient ubiquitination of lysine residues accessible on HDAC4, but not for other class IIa HDACs. Alternative explanations for the observed HDAC degradation selectivity profile may lie in a slower rate of de-ubiquitination for HDAC4 compared to its class IIa isoforms, or that for the class IIa isoforms ubiquitinylation leads to a biological response different to proteasomal degradation.36,38
Profiling of HDAC4 Degradation in Neuronal Cell Lines
Based on HDAC4 degradation efficiency in Jurkat cells and favorable solubility properties, we selected compound 11 from the TFMO-series HDAC4 degraders to conduct assessment of degradation efficiency in alternate cell lines. By MSD, dose-dependent HDAC4 degradation was measured in the mouse neuroblastoma (Neuro-2a) cell line at 24 h for compound 11, however, with approximately a 20-fold decrease in degradation efficiency relative to the Jurkat cell line (Table 3). Suspecting that the compound may be subject to efflux in the Neuro-2a cell line, we repeated the experiments following pretreatment with elacridar, a known P-glycoprotein (MRP-1, ABCB1) and breast cancer resistance protein (BCRP, ABCG2) inhibitor. In the presence of 10 μM elacridar administered 1 h prior to compound addition, the DC50 value improved significantly (DC50 Neuro-2a: −elacridar = 81 nM, +elacridar = 2.3 nM). A comparable shift in DC50 in the presence of elacridar was not observed in Jurkat cells. The expression levels of P-gp and BCRP in Neuro-2a are not well characterized so we cannot conclude based on these results alone that compound 11 is subject to efflux; however, a similar shift in DC50 was observed in the human neuroblastoma cell line SH-SY5Y (see supplemental WB data in Figure S5 and Table S1). The susceptibility of heterobifunctional degraders to multidrug resistant transporters such as P-gp has previously been reported in SH-SY5Y.39,40 Indeed, compound 11 shows moderate permeability in Madin–Darby canine kidney (MDCK) wild-type cells and in MDCK cells overexpressing multidrug resistance mutation 1 (MDR1) and BCRP (Papp (nm/s): 21, 26, and 14, respectively), but high efflux ratios were demonstrated in each of these cell types (MDCK efflux ratio (B > A/A > B): 13 (wild-type), 7 (MDR1), and 5 (BCRP). Consequently, it is unlikely that compound 11 would efficiently permeate the blood–brain barrier.
Table 3. HDAC4 Degradation Efficiency for Compound 11 in Various Cell Lines witha and withoutb Pretreatment with Elacridar.
| Jurkat
DC50 (nM)c |
Neuro-2a
DC50 (nM)d |
Q175
mouse cortical neuron DC50 (nM)e |
|||
|---|---|---|---|---|---|
| –elacridar | +elacridar | –elacridar | +elacridar | –elacridar | +elacridar |
| 1.7 | 1.6 | 81 | 2.3 | 1.1 | n.d.f |
Cells were treated with 10 μM elacridar 1 h prior to compound addition.
Cells were treated with DMSO 1 h prior to compound addition.
Jurkat DC50 was determined by western blot, −elacridar is based on nine experiments (Figure S1), +elacridar is based on one experiment (Figure S2).
Neuro-2a DC50 was determined by MSD, geometric means of two experiments, standard deviations are <10% of the mean value (Figure S3).
Q175 mouse cortical neuron DC50 was determined by MSD, geometric means of three experiments; standard deviations are <10% of the mean value (Figure S4).
n.d.: not determined.
Finally, to assess compound 11 in a cell type relevant to in vivo proof-of-concept studies in an HD model, we assessed HDAC4 degradation in cultured mouse primary cortical neurons prepared from Q175 knock-in mice carrying a chimeric mouse/human allele containing a Q175 human exon1 sequence within the native murine huntingtin gene. In these primary neurons, compound 11 demonstrated potent degradation efficiency with a DC50 value = 1 nM (Table 3, Figures S4, S6C, S6D, S6E, and S6F).
Conclusions
We report here our discovery of the first potent and selective HDAC4 bifunctional degraders. Starting from pan class IIa HDAC inhibitors in two distinct series and adopting a structure-based approach, we have identified and validated suitable vectors from which to grow the linker units to the E3 ligase ligand that do not disrupt binding to HDAC4. Compounds 7 and 11 are the first reported examples of VHL-based degraders that potently degrade HDAC4 in primary neurons, whereas a peptide-based PROTAC and a cereblon-based PROTAC have demonstrated degradation of tau in mouse primary neuronal cultures and human patient-derived iPSC neurons, respectively.41,42 The potent activity of these degraders, likely to be coupled with slow nascent HDAC4 protein synthesis in these neurons, demonstrated low nanomolar range concentration-dependent reduction in HDAC4 levels. This characteristic of small molecule degraders is potentially advantageous in an in vivo setting, particularly for a CNS application where delivery methods to the brain could be challenging due to poor permeability across the blood–brain barrier.43
Proteasomal inhibition experiments confirmed that these compounds act via the ubiquitin-proteasomal pathway and are selective against HDAC5, HDAC7, and HDAC9, based on our multiplexed western blot studies. Overall, these degraders can be regarded as valuable tools in the armory for examining the role of HDAC4 in HD.
Experimental Section
General Procedures
Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. All reactions involving air or moisture sensitive reagents were performed under a nitrogen atmosphere using dried solvents and glassware. Proton nuclear magnetic resonance spectra were obtained on a Bruker Avance 300 spectrometer at 300 MHz, Bruker Ascend 500 spectrometer at 500 MHz or Bruker AV 400 at 400 MHz. Spectra are given in ppm (δ) and coupling constants, J values, are reported in hertz (Hz). Tetramethylsilane was used as an internal standard for proton nuclear magnetic resonance for samples run in CDCl3 or DMSO-d6. Mass spectra were obtained using a Sciex Qtrap 4500 mass spectrometer, a Varian 1200L single quadrupole mass spectrometer, or a Shimadzu 2020 single quadrupole mass spectrometer. HPLC analyses were obtained using either a Phenomenex Luna C18(2) column, 5 μm (4.6 × 150 mm), Phenomenex Kinetex C18 column, 5 μm (4.6 × 150 mm), or BEH Shield RP18 column eluting with a gradient of 5–100% B (A = 0.1% trifluoroacetic acid in water; B = 0.1% trifluoroacetic acid in acetonitrile) over 10.0 min then 100% B for 3.0 min at a flow rate of 2.0 mL/min. BEH Shield RP18 column detection was by UV at 254 nm or at 254 and 215 nm. The following abbreviations are used: br = broad signal, s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet. All compounds were >95% pure by analytical HPLC.
2-(4-Bromophenyl)-2-(2-fluorophenyl)acetonitrile (14)
To a solution of 1-bromo-2-fluorobenzene (13) (31 mL, 280 mmol) in THF (286 mL) cooled with a dry ice/acetone bath was added a solution of 1.6 M nBuLi in hexanes (170 mL, 272 mmol) over 20 min, with the internal temperature maintained below −60 °C. The mixture was stirred at −78 °C for 1 h, and a solution of 4-bromobenzaldehyde (48.0 g, 259 mmol) in THF (143 mL) was added over 20 min, with the internal temperature maintained below −60 °C. The mixture was stirred at −78 °C for 1 h and at 0 °C for 1 h. After this time, saturated ammonium chloride (285 mL) was added, and the mixture was extracted with ethyl acetate (2 × 250 mL). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated to afford (4-bromophenyl)(2-fluorophenyl)methanol (74.2 g, 99%) as a brown oil: 1H NMR (500 MHz, CDCl3) δ 7.47–7.44 (m, 3H), 7.29–7.24 (m, 3H), 7.15 (td, J = 7.5, 1.0 Hz, 1H), 7.04–7.00 (m, 1H), 6.10 (d, J = 4.0 Hz, 1H), 2.34 (d, J = 4.0 Hz, 1H).
In an oven-dried flask, a mixture of (4-bromophenyl)(2-fluorophenyl)methanol (38.00 g, 135.2 mmol) and 4 Å molecular sieves (7 g) in dichloromethane (270 mL) was stirred at room temperature for 10 min, and the solution was poured into an addition funnel. In another oven-dried 1 L flask, a mixture of indium(III) bromide (4.79 g, 13.5 mmol) and 4 Å molecular sieves (14 g) in dichloromethane (270 mL) was sonicated for 5 min and stirred at room temperature for 10 min. Trimethylsilyl cyanide (43.0 mL, 325 mmol) was added, and the mixture was stirred for 5 min. To the resulting mixture was added the solution of (4-bromophenyl)(2-fluorophenyl)methanol in dichloromethane over 10 min, and stirring continued overnight. After this time, 2 M sodium carbonate (330 mL) was added, and the mixture was stirred at room temperature for 1 h. The layers were separated, and the organic layer was washed with brine (200 mL), dried over sodium sulfate, filtered, and concentrated at reduced pressure. The residue obtained was purified by chromatography (silica gel; heptane to 95:5 heptane/ethyl acetate; gradient elution) to afford impure 2-(4-bromophenyl)-2-(2-fluorophenyl)acetonitrile (27.3 g) as a brown oil. The impure product was further purified by chromatography in two equal portions (silica gel; heptane to 95:5 heptane/ethyl acetate; gradient elution) to afford pure 2-(4-bromophenyl)-2-(2-fluorophenyl)acetonitrile (14) (23.5 g, 60%) as a brown oil: 1H NMR (300 MHz, CDCl3) δ 7.53–7.48 (m, 2H), 7.44 (td, J = 7.5, 1.8 Hz, 1H), 7.39–7.32 (m, 1H), 7.28–7.24 (m, 2H), 7.20 (td, J = 7.5, 1.2 Hz, 1H), 7.14–7.07 (m, 1H), 5.40 (s, 1H); MS (ESI) m/z 290 [M + H]+.
2-(4-Bromophenyl)-2-(2-fluorophenyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)acetamide (15)
A mixture of 2-(4-bromophenyl)-2-(2-fluorophenyl)acetonitrile (14) (52.30 g, 180.3 mmol) and 6 N sodium hydroxide (300 mL, 1.80 mol) in ethanol (600 mL) was stirred at 84 °C overnight. After this time, the mixture was cooled to room temperature and concentrated to remove ethanol. The remaining suspension was treated with methyl tert-butyl ether (1.5 L) and extracted with water (2.5 and 0.5 L). The combined aqueous layers were washed with methyl tert-butyl ether (1 L), acidified with 12 N HCl (180 mL), and extracted with dichloromethane (4 × 300 mL). The combined dichloromethane layers were washed with brine (400 mL), dried over sodium sulfate, filtered, and concentrated to afford 2-(4-bromophenyl)-2-(2-fluorophenyl)acetic acid (53.6 g, 96%) as an off-white solid: 1H NMR (500 MHz, d6-DMSO) δ 12.93 (s, 1H), 7.55 (d, J = 8.5 Hz, 2H), 7.36–7.31 (m, 1H), 7.27 (d, J = 8.5 Hz, 2H), 7.22–7.15 (m, 3H), 5.25 (s, 1H).
HATU (0.676 g, 1.78 mmol) was added to a solution of 2-(4-bromophenyl)-2-(2-fluorophenyl)acetic acid (0.50 g, 1.6 mmol), O-(tetrahydro-2H-pyran-2-yl)hydroxylamine (0.186 g, 1.58 mmol) and N,N-diisopropylethylamine (0.85 mL, 4.9 mmol) in N,N-dimethylformamide (16 mL) at room temperature, and the reaction mixture was stirred for 30 min. After this time, water (200 mL) was added, and the mixture extracted with ethyl acetate (3 × 100 mL). The organic layers were combined, washed with brine (3 × 200 mL), dried over sodium sulfate, filtered and concentrated in vacuo. The residue obtained was purified by chromatography (silica gel; heptane to 60:40 heptane/ethyl acetate; gradient elution) to afford 2-(4-bromophenyl)-2-(2-fluorophenyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)acetamide (15) (0.540 g, 82%) as a white solid: 1H NMR (500 MHz, d6-DMSO) δ 11.58 (d, J = 6.5 Hz, 1H), 7.54 (d, J = 1.5 Hz, 2H), 7.53–7.31 (m, 2H), 7.26–7.15 (m, 4H), 5.06 (d, J = 2.5 Hz, 1H), 4.82 (d, J = 11.5 Hz, 1H), 3.93–3.86 (m, 1H), 3.51–3.48 (m, 1H), 1.66–1.64 (m, 3H), 1.50–1.42 (m, 3H); MS (ESI) m/z 409 [M + H]+.
2-(2-Fluorophenyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)-2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acetamide (16)
Tetrakis(triphenylphosphine)palladium(0) (0.057 g, 0.049 mmol) was added to a sparged solution of 2-(4-bromophenyl)-2-(2-fluorophenyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)acetamide (15) (0.200 g, 0.500 mmol), bis(pinacolato)diboron (0.137 g, 0.539 mmol) and cesium fluoride (0.153 g, 1.00 mmol) in 1,2-dimethoxyethane (13 mL)/methanol (3 mL), and the mixture was heated at 110 °C for 1.5 h. After this time, the mixture was cooled to room temperature, quenched with saturated ammonium chloride (10 mL) and water (10 mL), and extracted with dichloromethane (3 × 15 mL). The organic layers were combined, dried over sodium sulfate, filtered, and concentrated at reduced pressure. The residue obtained was purified by chromatography (silica gel; heptane to 70:30 heptane/ethyl acetate; gradient elution) to afford 2-(2-fluorophenyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)-2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl) acetamide (16) (0.117 g, 52%) as a yellow, tacky solid: 1H NMR (500 MHz, d6-DMSO) δ 11.58 (d, J = 7.0 Hz, 1H), 7.64 (d, J = 8.0 Hz, 2H), 7.34–7.26 (m, 4H), 7.19–7.14 (m, 2H), 5.06 (d, J = 1.5 Hz, 1H), 4.82 (d, J = 16.0 Hz, 1H), 3.92–3.87 (m, 1H), 3.51–3.47 (m, 1H), 1.69–1.60 (m, 3H), 1.57–1.46 (m, 3H), 1.28 (m, 12H).
2,2-Difluoro-2-(2-methoxypyrimidin-5-yl)ethan-1-ol (18)
A mixture of ethyl 2-bromo-2,2-difluoroacetate (4.87 g, 24.0 mmol), 5-iodo-2-methoxypyrimidine (17) (2.83 g, 12.0 mmol), dimethyl sulfoxide (48 mL), and copper powder (1.98 g, 31.2 mmol) was stirred at 50 °C overnight. After this time, the reaction mixture was cooled to room temperature, and treated with saturated ammonium chloride (200 mL), water (50 mL) and ethyl acetate (100 mL). The suspension was filtered through diatomaceous earth, the filtrate layers were separated, and the aqueous layer was extracted with ethyl acetate (2 × 50 mL). The combined organic layers were washed with brine (3 × 75 mL), dried over magnesium sulfate, filtered and concentrated at reduced pressure. The residue obtained was purified by chromatography (silica gel; heptane to 50:50 heptane/ethyl acetate; gradient elution) to afford ethyl 2,2-difluoro-2-(2-methoxypyrimidin-5-yl) acetate (2.57 g, 92%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 8.74 (s, 2H), 4.37–4.33 (m, 2H), 4.08 (s, 3H), 1.39–1.33 (m, 3H); MS (ESI) m/z 233 [M + H]+.
To a solution of ethyl 2,2-difluoro-2-(2-methoxypyrimidin-5-yl) acetate (1.80 g, 7.75 mmol) in ethanol (150 mL) at 0 °C was added a solution of 2 M lithium borohydride in tetrahydrofuran (2.71 mL, 5.43 mmol), and the reaction mixture was stirred at 0 °C for 2 h. After this time, methanol (150 mL) was added, followed by 6 N hydrochloric acid (7.7 mL). The reaction mixture was warmed to room temperature and stirred for 80 min. Solid sodium bicarbonate (20 g) was added, and the mixture was sonicated for 20 min, adsorbed onto silica gel, and chromatographed (silica gel; heptane to 60:40 heptane/ethyl acetate; gradient elution) to afford 2,2-difluoro-2-(2-methoxypyrimidin-5-yl)ethanol (18) (0.548 g, 37%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 8.68 (s, 2H), 4.06–3.99 (m, 5H), 2.10 (br s, 1H); MS (ESI) m/z 191 [M + H]+.
Methyl 2-(2-(2-(2-(2-Chloropyrimidin-5-yl)-2,2-difluoroethoxy)ethoxy)ethoxy) Acetate (22; n = 2)
To a solution of tert-butyl 2-(2-(2-hydroxyethoxy)ethoxy) acetate (19) (0.834 g, 3.79 mmol) and 2,2-difluoro-2-(2-methoxypyrimidin-5-yl)ethan-1-ol (18) (0.360 g, 1.89 mmol) in toluene (7.2 mL), was added cyanomethylenetributylphosphorane (1.5 mL, 5.7 mmol), and the mixture was stirred at 90 °C overnight. After this time, the reaction mixture was cooled to room temperature, diluted with dichloromethane (80 mL), adsorbed onto silica gel, and purified by chromatography (silica gel; heptane to ethyl acetate; gradient elution) to afford tert-butyl 2-(2-(2-(2,2-difluoro-2-(2-methoxypyrimidin-5-yl)ethoxy)ethoxy)ethoxy) acetate (0.597 g, 80%) as a brown oil: 1H NMR (500 MHz, CDCl3) δ 8.68 (s, 2H), 4.06 (s, 3H), 4.02 (s, 2H), 3.97 (t, J = 12.0 Hz, 2H), 3.75–3.73 (m, 2H), 3.71–3.68 (m, 2H), 3.66–3.63 (m, 4H), 1.47 (s, 9H); MS (ESI) m/z 393 [M + H]+.
To a solution of tert-butyl 2-(2-(2-(2,2-difluoro-2-(2-methoxypyrimidin-5-yl)ethoxy)ethoxy)ethoxy)acetate (0.755 g, 1.92 mmol) in 1,4-dioxane (36 mL) was added 2 N hydrochloric acid (24 mL, 48 mmol), and the reaction mixture stirred at 100 °C for 2 h. After this time, the reaction mixture was cooled to room temperature and concentrated at reduced pressure. Azeotropic concentration of the residue with methanol (2 × 20 mL) followed by drying in vacuo for 30 min afforded crude 2-(2-(2-(2,2-difluoro-2-(2-hydroxypyrimidin-5-yl)ethoxy)ethoxy)ethoxy)acetic acid (0.75 g) as a brown gum, which was used in the next step without purification: MS (ESI) m/z 323 [M + H]+.
To a solution of crude 2-(2-(2-(2,2-difluoro-2-(2-hydroxypyrimidin-5-yl)ethoxy)ethoxy)ethoxy)acetic acid (estimated 1.92 mmol) in methanol (80 mL) was added 12 N hydrochloric acid (0.40 mL, 4.8 mmol), and the reaction mixture was stirred at room temperature overnight. After this time, the mixture was concentrated at reduced pressure. The residue obtained was treated with saturated sodium bicarbonate (30 mL) and extracted with dichloromethane (3 × 50 mL). The combined dichloromethane layers were dried over magnesium sulfate, filtered and concentrated at reduced pressure to afford crude methyl 2-(2-(2-(2,2-difluoro-2-(2-hydroxypyrimidin-5-yl)ethoxy)ethoxy)ethoxy) acetate (0.300 g, 47%) as a brown oil, which was used in the next step without further purification: MS (ESI) m/z 337 [M + H]+.
A solution of crude methyl 2-(2-(2-(2,2-difluoro-2-(2-hydroxypyrimidin-5-yl)ethoxy)ethoxy)ethoxy) acetate (0.150 g, 0.446 mmol) in phosphorus oxychloride (5.0 mL, 53 mmol) was stirred at reflux for 2 h. After this time, the reaction mixture was cooled to room temperature, diluted with dichloromethane (10 mL) and heptane (10 mL), and concentrated at reduced pressure. The residue obtained was cooled with an ice water bath and treated with dichloromethane (10 mL) and saturated sodium bicarbonate (40 mL). The mixture was stirred at 0 °C for 5 min and extracted with dichloromethane (3 × 50 mL). The combined dichloromethane layers were washed with brine (50 mL), dried over magnesium sulfate, filtered, and concentrated at reduced pressure to afford impure methyl 2-(2-(2-(2-(2-chloropyrimidin-5-yl)-2,2-difluoroethoxy)-ethoxy)ethoxy) acetate (22; n = 2) (0.123 g, 78%) as a brown oil, which was used in the subsequent Suzuki coupling reaction without further purification: 1H NMR (500 MHz, CDCl3) δ 8.92 (d, J = 2.5 Hz, 2H), 8.42 (3, J = 8.0, Hz, 2H), 7.49–7.42 (m, 2H), 7.41–7.39 (m, 1H), 7.14–7.03 (m, 3H), 5.17 (d, J = 4.5 Hz, 1H), 5.00 (s, 1H), 4.12–3.87 (m, 5H), 3.78–3.66 (m, 3H), 3.62–3.48 (m, 7H), 1.88–1.73 (m 3H), 1.63–1.48 (m, 3H);MS (ESI) m/z 634 [M + H]+.
(2S,4R)-1-((2S)-2-(tert-Butyl)-14,14-difluoro-14-(2-(4-(1-(2-fluorophenyl)-2-(hydroxyamino)-2-oxoethyl)phenyl)pyrimidin-5-yl)-4-oxo-6,9,12-trioxa-3-azatetradecan-1-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (5)
To a suspension of 2-(2-fluorophenyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)-2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acetamide (16) (0.221 g, 0.485 mmol) and methyl 2-(2-(2-(2-(2-chloropyrimidin-5-yl)-2,2-difluoroethoxy)ethoxy)ethoxy) acetate (22; n = 2) (0.123 g, 0.347 mmol) in 1,4-dioxane/water (4:1, 10 mL) was added potassium carbonate (0.105 g, 0.763 mmol) followed by tetrakis(triphenylphosphine)palladium(0) (0.060 g, 0.052 mmol), and the reaction mixture was stirred at 100 °C for 1 h and at 110 °C for 1.5 h. After this time, the mixture was cooled to room temperature, treated with 50% saturated ammonium chloride (40 mL), and extracted with dichloromethane (2 × 60 mL). The combined dichloromethane layers were washed with brine (20 mL), dried over magnesium sulfate, filtered, and adsorbed onto silica gel. The residue obtained was chromatographed (silica gel; dichloromethane to 80:20 dichloromethane/methanol; gradient elution) to afford impure 2-(2-(2-(2,2-difluoro-2-(2-(4-(1-(2-fluorophenyl)-2-oxo-2-(((tetrahydro-2H-pyran-2-yl)oxy)amino)ethyl)phenyl)pyrimidin-5-yl)ethoxy)ethoxy)ethoxy)acetic acid (23; n = 2) (0.085 g, 39%) as an off-white solid, which was used in the next step without further purification: MS (ESI) m/z 634 [M + H]+.
To a solution of crude 2-(2-(2-(2,2-difluoro-2-(2-(4-(1-(2-fluorophenyl)-2-oxo-2-(((tetrahydro-2H-pyran-2-yl)oxy)amino)ethyl)phenyl)pyrimidin-5-yl)ethoxy)ethoxy)ethoxy)acetic acid (23; n = 2) (0.0850 g, 0.134 mmol) and (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (24) (0.066 g, 0.15 mmol)13 in N,N-dimethylformamide (5.37 mL) was added N,N-diisopropylethylamine (0.187 mL, 1.07 mmol) and the mixture was stirred at room temperature for 5 min. To the resulting mixture was added HATU (0.059 g, 0.15 mmol), and the reaction mixture was stirred at room temperature for 28 min. After this time, the mixture was quenched with 50% saturated sodium bicarbonate (100 mL) and stirred for 5 min. The resulting precipitate was collected by filtration and washed with water (3 × 30 mL). The wet solid was dissolved in ethyl acetate (80 mL), and the solution was washed with brine (25 mL), dried over magnesium sulfate, filtered, and concentrated at reduced pressure. The residue obtained was chromatographed (silica gel; dichloromethane to 80:20 dichloromethane/methanol; gradient elution) to afford (2S,4R)-1-((2S)-2-(tert-butyl)-14,14-difluoro-14-(2-(4-(1-(2-fluorophenyl)-2-oxo-2-(((tetrahydro-2H-pyran-2-yl)oxy)-amino)ethyl)phenyl)pyrimidin-5-yl)-4-oxo-6,9,12-trioxa-3-azatetradecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (0.094 g, 67%) as a white solid: 1H NMR (500 MHz, d6-DMSO) δ 11.64 (d, J = 7.5 Hz, 1H), 9.07 (s, 2H), 8.97 (s, 1H), 8.58 (t, J = 6.0 Hz, 1H), 8.39 (d, J = 8.5 Hz, 2H), 7.46–7.32 (m, 9H), 7.22–7.17 (m, 2H), 5.19–5.14 (m, 2H), 4.90–4.80 (m, 1H), 4.55 (d, J = 9.5 Hz, 1H), 4.45–4.36 (m, 3H), 4.27–4.23 (m, 1H), 4.11 (t, J = 13.0 Hz, 2H), 4.00–3.85 (m, 3H), 3.69–3.65 (m, 3H), 3.61–3.49 (m, 8H), 2.43 (s, 3H), 2.10–2.00 (m, 1H), 1.95–1.85 (m, 1H), 1.70–1.61 (m, 3H), 1.57–1.46 (m, 3H), 0.91 (s, 9H).
To a solution of (2S,4R)-1-((2S)-2-(tert-butyl)-14,14-difluoro-14-(2-(4-(1-(2-fluorophenyl)-2-oxo-2-(((tetrahydro-2H-pyran-2-yl)oxy)amino)ethyl)-phenyl)pyrimidin-5-yl)-4-oxo-6,9,12-trioxa-3-azatetradecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (0.093 g, 0.089 mmol) in methanol (20 mL) was added trifluoroacetic acid (1 mL), and the reaction mixture stirred at room temperature overnight. After this time, the volatiles were removed at reduced pressure, and the residue obtained was purified by chromatography (silica gel; dichloromethane to 80:20 dichloromethane/methanol; gradient elution). The product obtained was repurified by reverse phase chromatography (C18; 95:5 water with 0.1% trifluoroacetic acid/acetonitrile to acetonitrile; gradient elution). The product obtained was purified again by chromatography (silica gel; dichloromethane to 80:20 dichloromethane/methanol; gradient elution), followed by lyophilization from acetonitrile/water (1:1, 6 mL) to afford (2S,4R)-1-((2S)-2-(tert-butyl)-14,14-difluoro-14-(2-(4-(1-(2-fluorophenyl)-2-(hydroxyamino)-2-oxoethyl)phenyl)-pyrimidin-5-yl)-4-oxo-6,9,12-trioxa-3-azatetradecan-1-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (5) (0.053 g, 62%) as a white solid: no clear melt observed; 1H NMR (500 MHz, d6-DMSO, observed as a 93:7 mixture of rotational isomers; only shifts for the major rotamer are reported) δ 11.05 (s, 1H), 9.06 (s, 3H), 8.97 (s, 1H), 8.58 (t, J = 6.0 Hz, 1H), 8.39 (dd, J = 6.5, 2.0 Hz, 2H), 7.53–7.31 (m, 9H), 7.21–7.17 (m, 2H), 5.14 (d, J = 3.5 Hz, 1H), 5.12 (s, 1H), 4.55 (d, J = 9.5 Hz, 1H), 4.45–4.35 (m, 3H), 4.27–4.23 (m, 1H), 4.11 (t, J = 13.5 Hz, 2H), 3.95 (s, 2H), 3.71–3.65 (m, 3H), 3.61–3.51 (m, 7H), 2.43 (s, 3H), 2.10–2.00 (m, 1H), 1.92–1.87 (m, 1H), 0.92 (s, 9H); MS (ESI) m/z 962 [M + H]+; HPLC: Phenomenex Kinetex C18 column, tR = 6.42 min, 99% (AUC) at both 254 and 215 nm.
(2S,4R)-1-((2S)-2-(tert-Butyl)-17,17-difluoro-17-(2-(4-(1-(2-fluorophenyl)-2-(hydroxyamino)-2-oxoethyl)phenyl)pyrimidin-5-yl)-4-oxo-6,9,12,15-tetraoxa-3-azaheptadecan-1-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (6)
Compound 6 was prepared according to the procedure detailed for compound 5, starting from tert-butyl 2-(2-(2-2-(hydroxyethoxy)ethoxy)ethoxy) acetate (20). white solid: no clear melt observed; 1H NMR (500 MHz, d6-DMSO, observed as a 94:6 mixture of rotational isomers; only shifts for the major rotamer are reported) δ 11.04 (dd, J = 8.5, 1.5 Hz, 1H), 9.06 (s, 3H), 8.96 (s, 1H), 8.58 (t, J = 11.0 Hz, 1H), 8.39 (d, J = 8.5 Hz, 2H), 7.51 (t, J = 8.0 Hz, 1H), 7.47–7.31 (m, 8H), 7.20–7.17 (m, 2H), 5.14 (d, J = 3.5 Hz, 1H), 5.11 (s, 1H), 4.55 (d, J = 9.5 Hz, 1H), 4.45–4.34 (m, 3H), 4.24 (dd, J = 16.0, 4.5 Hz, 1H), 4.11 (t, J = 13.0 Hz, 2H), 3.93 (s, 2H), 3.67–3.46 (m, 14H), 2.42 (s, 3H), 2.07–2.03 (m, 1H), 1.92–1.87 (m, 1H), 0.92 (s, 9H); MS (ESI) m/z 1006 [M + H]+; HPLC: Phenomenex Kinetex C18 column, tR = 6.44 min, 99% (AUC) at both 254 and 215 nm.
(2S,4R)-1-((2S)-2-(tert-Butyl)-20,20-difluoro-20-(2-(4-(1-(2-fluorophenyl)-2-(hydroxyamino)-2-oxoethyl)phenyl)pyrimidin-5-yl)-4-oxo-6,9,12,15,18-pentaoxa-3-azaicosan-1-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (7)
Compound 7 was prepared according to the procedure detailed for compound 5, starting from tert-butyl 14-hydroxy-3,6,9,12-tetraoxatetradeconate (21). white solid: no clear melt observed; 1H NMR (500 MHz, d6-DMSO, observed as a 94:6 mixture of rotational isomers; only shifts for the major rotamer are reported) δ 11.05 (s, 1H), 9.07–9.06 (m, 3H), 8.97 (s, 1H), 8.59 (t, J = 6.0 Hz, 1H), 8.40 (d, J = 8.5 Hz, 2H), 7.53–7.31 (m, 9H), 7.21–7.17 (m, 2H), 5.14–5.12 (m, 2H), 4.56 (d, J = 9.5 Hz, 1H), 4.45–4.35 (m, 3H), 4.28–4.22 (m, 1H), 4.13 (t, J = 13.0 Hz, 2H), 3.95 (s, 2H), 3.70–3.55 (m, 6H), 3.55–3.40 (m, 12H), 2.43 (s, 3H), 2.10–2.00 (m, 1H), 1.92–1.87 (m, 1H), 0.93 (s, 9H); MS (ESI) m/z 1050 [M + H]+; HPLC: Phenomenex Kinetex C18 column, tR = 6.44 min, 98.1% (AUC) at 254 nm and tR = 6.43 min, 97.0% (AUC) at 215 nm.
(2S,4S)-1-((2S)-2-(tert-Butyl)-20,20-difluoro-20-(2-(4-(1-(2-fluorophenyl)-2-(hydroxyamino)-2-oxoethyl)phenyl)pyrimidin-5-yl)-4-oxo-6,9,12,15,18-pentaoxa-3-azaicosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (8)
Compound 8 was prepared according to the procedure for 7, using the VHL epimer (2S,4S)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (25) (0.066 g, 0.15 mmol).13 off-white solid: no clear melt observed; 1H NMR (400 MHz, CDCl3) 8.92 (d, J = 3.4 Hz, 2H), 8.66 (s, 1H), 8.45 (dd, J = 8.2, 8.2 Hz, 2H), 8.22–8.04 (m, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.38–7.28 (m, 6H), 7.20 (d, J = 9.0 Hz, 1H), 7.11–7.01 (m, 2H), 5.92 (s, 1H), 5.18 (d, J = 27.4 Hz, 1H), 4.64–4.55 (m, 3H), 4.42 (s, 1H), 4.35–4.28 (m, 1H), 4.01–3.87 (m, 6H), 3.69–3.33 (m, 18H), 2.49 (s, 3H), 2.21–2.05 (m, 2H), 0.96 (s, 9H); one exchangeable proton was not observed; MS (ESI) m/z 1050 [M + H]+; HPLC: Acquity UPLC BEH Shield RP18 column, tR = 4.58 min, 96.6% (AUC) at 210–400 nm (DAD).
(2S,4R)-1-((3R,20S)-20-(tert-Butyl)-3-methyl-1,18-dioxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16-trioxa-2,5,19-triazahenicosan-21-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidin-2-carboxamide (9)
Diethylene glycol (26) (9.70 mL, 102 mmol) was added to a solution of potassium tert-butoxide (1.08 g, 9.62 mmol) in tetrahydrofuran (10 mL), and the mixture was placed in a preheated aluminum block at 45 °C for 30 min. After this time, the resulting mixture was cooled to 0 °C, and tert-butyl 2-bromoacetate (1.51 mL, 10.3 mmol) was added in one portion. The cooling bath was removed, and the reaction mixture stirred at room temperature for 24 h. After this time, brine (100 mL) was added, and the mixture was extracted with dichloromethane (3 × 80 mL). The combined organic layers were washed with water (2 × 50 mL), dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure to afford tert-butyl 2-(2-(2-hydroxyethoxy)ethoxy) acetate (29; n = 2) (1.06 g, 47%) as a light-yellow oil: 1H NMR (500 MHz, CDCl3) δ 4.02 (s, 2H), 3.76–3.73 (m, 2H), 3.72–3.71 (m, 4H), 3.64–3.62 (m, 2H), 2.41 (br s, 1H), 1.48 (s, 9H).
A solution of methanesulfonyl chloride (0.667 g, 5.82 mmol) in dichloromethane (10 mL) was added to a mixture of tert-butyl 2-(2-(2-hydroxyethoxy)ethoxy) acetate (29; n = 2) (1.06 g, 4.81 mmol) and triethylamine (1.0 mL, 7.2 mmol) in dichloromethane (30 mL) at 0 °C, and the mixture was stirred at 0 °C for 30 min and at room temperature for 20 h. After this time, the reaction mixture was treated with saturated aqueous sodium bicarbonate (100 mL), stirred at room temperature for 5 min, and the biphasic mixture was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure to afford tert-butyl 2-(2-(2-((methylsulfonyl)oxy)ethoxy)ethoxy) acetate (1.43 g, 99.6%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 4.40–4.38 (m, 2H), 4.00 (s, 2H), 3.80–3.78 (m, 2H), 3.72–3.70 (m, 4H), 3.08 (s, 3H), 1.48 (s, 9H). A 1.0 M solution of potassium tert-butoxide in tetrahydrofuran (7.2 mL, 7.2 mmol) was added to a solution of butane-1,4-diol (4.3 mL, 49 mmol) in tetrahydrofuran (16 mL), and the mixture was stirred at room temperature for 30 min. A solution of tert-butyl 2-(2-(2-((methylsulfonyl)oxy)ethoxy)ethoxy) acetate (1.43 g, 4.79 mmol) in tetrahydrofuran (65 mL) was then added dropwise over 10 min, and the mixture stirred at room temperature for 22 h. After this time, volatiles were removed under reduced pressure, and the residue obtained was dissolved in methanol (250 mL). Sulfuric acid (1.8 mL, 34 mmol) was added dropwise, and the mixture was stirred at room temperature for 3 days. After this time, saturated aqueous sodium bicarbonate (200 mL) and water (100 mL) were added, and the mixture was extracted with dichloromethane (4 × 100 mL). The combined organic layers were washed with brine (100 mL), dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure. The residue obtained was purified by chromatography (silica gel; dichloromethane to 80:20 dichloromethane/methanol; gradient elution) to afford methyl 2-(2-(2-(4-hydroxybutoxy)ethoxy)ethoxy) acetate (30; n = 2) (0.649 g, 54% yield over two steps) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 4.18 (s, 2H), 3.75–3.73 (m, 5H), 3.70–3.68 (m, 2H), 3.67–3.63 (m, 4H), 3.62–3.60 (m, 2H), 3.52 (t, J = 6.0 Hz, 2H), 2.36 (t, J = 6.0 Hz, 1H), 1.73–1.63 (m, 4H); MS (ESI) m/z 251 [M + H]+.
Dimethyl sulfoxide (0.37 mL, 5.2 mmol) was added dropwise to a solution of oxalyl chloride (0.34 mL, 3.9 mmol) in dichloromethane (20 mL) at −78 °C, and the mixture was stirred at −78 °C for 25 min. A solution of methyl 2-(2-(2-(4-hydroxybutoxy)ethoxy)ethoxy) acetate (30; n = 2) (0.648 g, 2.59 mmol) in dichloromethane (20 mL) was added dropwise over 5 min, and the reaction mixture was stirred at −78 °C for 20 min. Triethylamine (1.5 mL, 11 mmol) was then added, and stirring was continued at −78 °C for 2 min. The cooling bath was then removed, and the mixture was stirred at room temperature for 3 h. After this time, the reaction mixture was poured into saturated aqueous sodium bicarbonate (50 mL), the layers were separated, and the aqueous phase was extracted with dichloromethane (2 × 50 mL). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure to afford methyl 2-(2-(2-(4-oxobutoxy)ethoxy)ethoxy) acetate (31; n = 2) (0.664 g, 99%) as a light-orange semi-solid: 1H NMR (500 MHz, CDCl3) δ 9.78 (t, J = 1.5 Hz, 1H), 4.18 (s, 2H), 3.75 (s, 3H), 3.74–3.72 (m, 2H), 3.70–3.68 (m, 2H), 3.64–3.62 (m, 2H), 3.59–3.57 (m, 2H), 3.50 (t, J = 6.5 Hz, 2H), 2.53 (td, J = 7.0, 1.5 Hz, 2H), 1.95–1.89 (m, 2H).
In a 10 mL microwave vial (R)-2-((tert-butoxycarbonyl)amino)propyl methanesulfonate (5.054 g, 19.95 mmol)10 was diluted with propan-1-amine (15.0 mL, 182 mmol), and the reaction vial was sealed. The mixture was placed in a preheated aluminum block and stirred at 50 °C. After 2.5 days, the mixture was cooled to room temperature, diluted with saturated aqueous sodium bicarbonate (150 mL), and extracted with ethyl acetate (3 × 75 mL). The combined organic layers were washed with brine (75 mL), dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure to afford tert-butyl (R)-(1-(propylamino)propan-2-yl)carbamate (3.73 g, 86%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 4.71 (br s, 1H), 3.78–3.69 (m, 1H), 2.62–2.53 (m, 5H), 1.53–1.46 (m, 2H), 1.45 (s, 9H), 1.14 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 7.4 Hz, 3H). Sodium triacetoxyborohydride (0.878 g, 4.14 mmol) was added to a solution of tert-butyl (R)-(1-(propylamino)propan-2-yl)carbamate (0.564 g, 2.61 mmol) and methyl 2-(2-(2-(4-oxobutoxy)ethoxy)ethoxy) acetate (31; n = 2) (0.643 g, 2.59 mmol) in dichloromethane (38 mL), and the mixture stirred at room temperature for 20 h. After this time, the reaction mixture was diluted with saturated aqueous sodium bicarbonate (50 mL), and the layers were separated. The aqueous layer was extracted with dichloromethane (2 × 50 mL), and the combined organic layers were dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure. The residue obtained was purified by chromatography (silica gel; dichloromethane to 90:10 dichloromethane/methanol; gradient elution) to afford methyl (R)-2,2,6-trimethyl-4-oxo-8-propyl-3,13,16,19-tetraoxa-5,8-diazahenicosan-21-oate (0.754 g, 65%) as a clear oil: 1H NMR (500 MHz, CDCl3) δ 4.77 (br s, 1H), 4.18 (s, 2H), 3.75 (s, 3H), 3.75–3.73 (m, 2H), 3.71–3.69 (m, 2H), 3.66–3.64 (m, 2H), 3.59–3.54 (m, 3H), 3.46 (t, J = 6.5 Hz, 2H), 2.46–2.30 (m, 5H), 2.27 (dd, J = 13.0, 6.5 Hz, 1H), 1.63–1.57 (m, 2H), 1.48–1.37 (m, 13H), 1.13 (d, J = 6.5 Hz, 3H), 0.86 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 449 [M + H]+.
4.0 M HCl in 1,4-dioxane (2.0 mL, 8.0 mmol) was added to a solution of methyl (R)-2,2,6-trimethyl-4-oxo-8-propyl-3,13,16,19-tetraoxa-5,8-diazahenicosan-21-oate (0.350 g, 0.780 mmol) in methanol (2.0 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 5 min and at room temperature for 2 h. After this time, the solvent was removed under reduced pressure, and the residue obtained was azeotroped with dichloromethane (2 × 10 mL) to afford methyl (R)-16-amino-14-propyl-3,6,9-trioxa-14-azahepta-decanoate dihydrochloride (32; n = 2) (0.335 g, 99%) as a viscous, yellow oil: 1H NMR (500 MHz, d6-DMSO) δ 10.62 (br s, 1H), 8.53 (br s, 3H), 4.14 (s, 2H), 3.88–3.77 (m, 1H), 3.65 (s, 3H), 3.61–3.58 (m, 2H), 3.56–3.52 (m, 4H), 3.50–3.48 (m, 2H), 3.44–3.40 (t, J = 6.5 Hz, 3H), 3.27–2.98 (m, 5H), 1.80–1.68 (m, 4H), 1.60–1.51 (m, 2H), 1.30 (d, J = 6.5 Hz, 3H), 0.91 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 349 [M + H]+.
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 0.181 g, 0.944 mmol) and hydroxybenzotriazole monohydrate (0.146 g, 0.953 mmol) were added to a solution of N,N-diisopropylethylamine (0.55 mL, 3.2 mmol) and 4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)benzoic acid (0.201 g, 0.780 mmol) in dichloromethane (4 mL), and the mixture was stirred at room temperature for 5 min. A solution of methyl (R)-16-amino-14-propyl-3,6,9-trioxa-14-azaheptadecanoate dihydrochloride (32; n = 2) (0.329 g, 0.780 mmol) in dichloromethane (3 mL) was added and stirring continued at room temperature for 15 h. After this time, the mixture was poured into saturated aqueous sodium bicarbonate (75 mL), the layers were separated, and the aqueous layer was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure. The residue obtained was purified by chromatography (silica gel; dichloromethane to 90:10 dichloromethane//90:10 methanol/conc. ammonium hydroxide; gradient elution) to afford methyl (R)-3-methyl-1-oxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16-trioxa-2,5-diazaoctadecan-18-oate (0.323 g, 70%) as a clear oil: 1H NMR (500 MHz, CDCl3) δ 8.20–8.18 (m, 2H), 7.94–7.92 (m, 2H), 6.87 (br s, 1H), 4.15 (s, 2H), 4.06–4.00 (m, 1H), 3.74 (s, 3H), 3.72–3.70 (m, 2H), 3.68–3.66 (m, 2H), 3.63–3.61 (m, 2H), 3.56–3.54 (m, 2H), 3.46–3.42 (m, 2H), 2.55–2.34 (m, 6H), 1.63–1.38 (m, 6H), 1.32 (d, J = 6.0 Hz, 3H), 0.85 (t, J = 7.0 Hz, 3H); MS (ESI) m/z 589 [M + H]+.
A 1 M aqueous lithium hydroxide solution (1.06 mL, 1.06 mmol) was added to a solution of methyl (R)-3-methyl-1-oxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16-trioxa-2,5-diazaoctadecan-18-oate (0.312 g, 0.530 mmol) in methanol (42 mL) and water (9.5 mL), and the mixture was stirred at room temperature for 5 h. After this time, the mixture was poured into saturated aqueous sodium bicarbonate (75 mL) and extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate, and filtered, and the filtrate concentrated under reduced pressure. The residue obtained was purified by reverse phase chromatography (C18; water w/0.1% TFA to acetonitrile w/0.1% TFA; gradient elution) and the product fractions were concentrated under reduced pressure. The residue obtained was azeotroped with 1:1 water/conc. ammonium hydroxide (2 × 5 mL) to remove TFA, followed by azeotroping twice with a mixture of N,N-diisopropylethylamine (2 mL)/toluene (3 mL)/dichloromethane (10 mL) to afford N-ethyl-N-isopropylpropan-2-amine hemi((R)-3-methyl-1-oxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16-trioxa-2,5-diazaoctadecan-18-oate) (33; n = 2) (0.401 g, 91%) as a clear, colorless oil: 1H NMR (500 MHz, d6-DMSO) δ 8.40 (d, J = 8.5 Hz, 1H), 8.16–8.14 (m, 2H), 8.06–8.04 (m, 2H), 4.19–4.10 (m, 1H), 3.97 (s, 2H), 3.57–3.55 (m, 2H), 3.52–3.43 (m, 10H), 3.38–3.33 (m, 3H), 3.02–2.91 (m, 4H), 2.55–2.35 (m, 6H), 1.51–1.38 (m, 6H), 1.23–1.14 (m, 33H), 0.83 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 575 [M + H]+.
(2S,4R)-1-((S)-2-Amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (24), 0.101 g, 0.235 mmol)13 was added to a solution of N-ethyl-N-isopropylpropan-2-amine hemi((R)-3-methyl-1-oxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16-trioxa-2,5-diazaoctadecan-18-oate (33; n = 2) (0.194 g, 0.233 mmol) and N,N-diisopropylethylamine (0.10 mL, 0.57 mmol) in N,N-dimethylformamide (5 mL) at room temperature. 1-Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU, 0.095 g, 0.25 mmol) was added, and the mixture stirred at room temperature for 30 min. After this time, saturated aqueous sodium bicarbonate (40 mL) was added, and the mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with brine (20 mL), dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure. The residue obtained was purified by chromatography (silica gel; dichloromethane to 80:20 dichloromethane/methanol; gradient elution) followed by reverse phase chromatography (C18; water w/0.1% TFA to acetonitrile w/0.1% TFA; gradient elution). The product fractions were diluted with saturated aqueous sodium bicarbonate (100 mL) and extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure. The residue obtained was lyophilized from 1:1 acetonitrile/water to afford (2S,4R)-1-((3R,20S)-20-(tert-butyl)-3-methyl-1,18-dioxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16-trioxa-2,5,19-triazahenicosan-21-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidin-2-carboxamide (9) (0.106 g, 45%) as a white solid: no clear melt observed; 1H NMR (500 MHz, d6-DMSO, observed as an 89:11 mixture of rotational isomers; chemical shifts are given for the major rotamer) δ 8.98 (s, 1H), 8.59 (t, J = 5.5 Hz, 1H), 8.32 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 8.5 Hz, 2H), 8.04 (d, J = 8.0 Hz, 2H), 7.46–7.33 (m, 5H), 5.14 (d, J = 3.5 Hz, 1H), 4.56 (d, J = 10.0 Hz, 1H), 4.46–4.32 (m, 3H), 4.24 (dd, J = 16.0, 5.5 Hz, 1H), 4.14–4.07 (m, 1H), 3.96 (s, 2H), 3.68–3.49 (m, 10H), 3.45 (m, 2H), 2.47–2.28 (m, 9H), 2.09–2.01 (m, 1H), 1.90 (ddd, J = 12.5, 9.0, 4.5 Hz, 1H), 1.51–1.32 (m, 6H), 1.15 (d, J = 6.5 Hz, 3H), 0.94 (s, 9H), 0.81 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 987 [M + H]+; HPLC: Phenomenex Luna C18(2) column, tR = 6.65 min, 99.0% (AUC); elemental analysis for C48H65F3N8O9S·H2O: calcd: C, 57.36; H, 6.72; N, 11.15; found: C, 57.10; H, 6.68; N, 11.11.
(2S,4R)-1-((3R,23S)-23-(tert-Butyl)-3-methyl-1,21-dioxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16,19-tetraoxa-2,5,22-triazatetracosan-24-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (10)
Compound 10 was prepared according to the procedure detailed for compound 9, using triethylene glycol (27). clear, colorless gum: 1H NMR (500 MHz, CD3OD, observed as an 87:13 mixture of rotational isomers; chemical shifts are given for the major rotamer) δ 8.87 (s, 1H), 8.21–8.19 (m, 2H), 8.00–7.98 (m, 2H), 7.46–7.44 (m, 2H), 7.42–7.40 (m, 2H), 4.69 (s, 1H), 4.58–4.49 (m, 3H), 4.35 (d, J = 15.5 Hz, 1H), 4.29–4.23 (m, 1H), 4.07–3.99 (m, 2H), 3.87–3.85 (m, 1H), 3.79 (dt, J = 11.0, 4.0 Hz, 1H), 3.72–3.60 (m, 8H), 3.59–3.57 (m, 2H), 3.53–3.51 (m, 2H), 3.44 (t, J = 6.0 Hz, 2H), 2.65–2.43 (m, 9H), 2.22 (ddt, J = 13.5, 7.5, 2.0 Hz, 1H), 2.08 (ddd, J = 13.5, 9.5, 4.5 Hz, 1H), 1.59–1.45 (m, 6H), 1.25 (d, J = 6.5 Hz, 3H), 1.03 (s, 9H), 0.88 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 1031 [M + H]+; HPLC: Phenomenex Luna C18(2) column, tR = 6.66 min, 99% (AUC); elemental analysis for C50H69F3N8O10S·H2O: calcd: C, 57.24; H, 6.82; N, 10.68; found: C, 57.23; H, 6.58; N, 10.59.
(2S,4R)-1-((3R,26S)-26-(tert-Butyl)-3-methyl-1,24-dioxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16,19,22-pentaoxa-2,5,25-triazaheptacosan-27-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (11)
Compound 11 was prepared according to the procedure detailed for compound 9, using tetraethylene glycol (28). off-white solid: 1H NMR (500 MHz, d6-DMSO, observed as an 86:14 mixture of rotational isomers; chemical shifts are given for the major rotamer) δ 8.98 (s, 1H), 8.59 (t, J = 6.0 Hz, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 8.0 Hz, 2H), 8.04 (d, J = 8.0 Hz, 2H), 7.46–7.35 (m, 5H), 5.15 (d, J = 8.5 Hz, 1H), 4.56 (d, J = 8.5 Hz, 1H), 4.46–4.32 (m, 3H), 4.24 (dd, J = 10.5, 5.5 Hz, 1H), 4.16–4.05 (m, 1H), 3.96 (s, 2H), 3.68–3.39 (m, 18H), 3.36–3.31 (m, 4H), 2.44 (s, 3H), 2.41–2.28 (m, 4H), 2.09–2.02 (m, 1H), 1.93–1.86 (m, 1H), 1.52–1.33 (m, 6H), 1.16 (d, J = 6.5 Hz, 3H), 0.94 (s, 9H), 0.82 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 1075 [M + H]+; HPLC: Phenomenex Luna C18(2) column, tR = 6.62 min, 97.7% (AUC); elemental analysis for C52H73F3N8O11S·2H2O: calcd: C, 56.20; H, 6.98; N, 10.08; found: C, 55.95; H, 7.07; N, 9.85.
(2R,4S)-1-((3R,26R)-26-(tert-Butyl)-3-methyl-1,24-dioxo-5-propyl-1-(4-(5-trifluoromethyl-1,2,4-oxadiazol-3-yl)phenyl)-10,13,16,19,22-pentaoxa-2,5,25-triazaheptacosan-27-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (12)
Compound 12 was prepared according to the procedure detailed for 11, using the epimer (2R,4S)-1-((R)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (34)13 to afford the product as an off-white solid: no clear melt observed; 1H NMR (500 MHz, d6-DMSO, observed as a 92:8 mixture of rotational isomers; only shifts for the major rotamer are reported) δ 8.97 (s, 1H), 8.56 (t, J = 6.0 Hz, 1H), 8.32 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 8.0 Hz, 2H), 8.04 (d, J = 8.0 Hz, 2H), 7.45–7.37 (m, 5H), 5.14 (d, J = 8.5 Hz, 1H), 4.56 (d, J = 8.5 Hz, 1H), 4.45–4.34 (m, 3H), 4.24 (dd, J = 10.5, 5.5 Hz, 1H), 4.12–4.10 (m, 1H), 3.96 (s, 2H), 3.67–3.41 (m, 18H), 3.36–3.31 (m, 4H), 2.43 (s, 3H), 2.36–2.30 (m, 4H), 2.07–2.03 (m, 1H), 1.92–1.87 (m, 1H), 1.50–1.38 (m, 6H), 1.14 (d, J = 6.5 Hz, 3H), 0.94 (s, 9H), 0.81 (t, J = 7.5 Hz, 3H); MS (ESI) m/z 1075 [M + H]+; HPLC: Phenomenex Luna C18(2) column, tR = 6.32 min, 99% (AUC); elemental analysis for C52H73F3N8O11S·0.5H2O: calcd: C, 57.60; H, 6.88; N, 10.33; found: C, 57.67; H, 6.97; N, 10.26.
Cell Cultures
Jurkat E6-1 cells (obtained from the European Collection of Cell Cultures (ECACC)) were cultured in RPMI 1640 growth medium (Invitrogen, 11835063) supplemented with 10 mM HEPES (Invitrogen 15630056), 2 mM l-glutamine (Invitrogen 25030081) and 10% fetal bovine serum (FBS) (Invitrogen 16140063).
Mouse cortical neuron cultures were prepared using Q175 neo minus knock-in mice (zQ175DN on a C57BL6 background, carrying a chimeric mouse/human allele containing human exon1 with an expanded Q sequence within the native murine huntingtin gene.44 Briefly, cortices of E18 mouse brains were micro-dissected, and genotypes were determined by HTRF-based genotyping (mouse monoclonal 2B7-Terbium and D2 labeled MW1 of the hind brain tissue, allowing for rapid and reliable identification of the knock-in mice in mixed genotype litters. Single cell suspensions were prepared from the pooled zQ175 heterozygous cortices by dissociation with papain (Worthington Biochemical, LK003176) and DNase (Worthington Biochemical, LK003170) for 10 min at 37 °C. The tissue was triturated using a P1000 pipette and after the tissue was allowed to settle for 2 min, the supernatant was collected, and the remaining tissue triturated. Following a centrifugation step, the neuronal cells were cultured at a seeding density of 62,500 cells/well on a WT embryonic rat glial feeder layer at PN04 (3000 cells/well grown for 3 days) seeded on poly-d-lysine hydrobromide (PDL) precoated (0.5 mg/mL; Sigma, P7280) 96-well plates in culture medium (Neurobasal medium; Gibco, 21103049) with 1X B27-supplement (Invitrogen, 17504044), 9.5 mM KCl (Sigma, P5405) 2 mM l-alanyl-l-glutamine dipeptide (GlutaMAX, Life Technologies, 1735245), 4.75 μg/mL Gentamycin (Invitrogen, 1555919) and incubated at 37 °C/5% CO2. After 4 days in vitro (DIV4), 25% of the medium was refreshed with culture medium, with 3 μM cytosine β-d-arabinofuranoside hydrochloride (AraC; Sigma, C6645) to prevent glia overgrowth.
In Vitro Studies
When conducting in vitro studies for the TFMO compounds, we used fresh aliquots of dry powder for each study, to circumvent any potential issues that could arise from solution instability for this class of compounds.
Recombinant and Cellular HDAC Activity Assays
The HDAC activity of the test compounds against HDACs was determined using the catalytic domains with AMC-conjugated substrate, in 384-well microtiter plates, as described by Beconi et al.45 Briefly, HDAC enzymatic activity against HDAC4, HDAC5, HDAC7 and HDAC9 were determined using Boc-Lys(TFA)-AMC substrate (Bachem, 4060676). Concentrations of substrates and incubation times were used, where each enzymatic reaction was linear. HDAC activities were quantified by the substrate de-acetylation followed by fluorophore AMC release after tryptic digest. Fluorescence was determined (Ex. 355 nm, Em. 460 nm) using an EnVision plate reader (Perkin Elmer).
Cellular potency of the test compounds was determined in Jurkat E6-1 cells, as described by Beconi et al.,45 using Boc-Lys(Ac)-AMC substrate for class I activity and Boc-Lys(TFA)-AMC substrate class IIa activity. Cellular HDAC activities were quantified using an EnVision plate reader, following overnight incubation with lysis buffer (50 mM Tris–HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1% Nonidet P40 Substitute, pH 8.0) supplemented with 0.06% trypsin at 37 °C.
Western Blot Assay
Cell lysates were prepared by adding NP-40 lysis buffer ((20 mM Tris–HCl, 137 mM NaCl, 10% glycerol, 1% IGEPAL CA630 (NP40 substitute), pH 8.0, 1 × Halt protease inhibitor cocktail (Thermo Scientific)) to cell pellets. Cells were titrated, vortexed and incubated on ice for 30 min before clarifying by centrifugation (13,000 rpm for 15 min at 4 °C). Cell lysate protein concentration was determined using a Pierce BCA protein assay kit (ThermoFisher, 23225). Lysates were mixed with NuPAGE LDS sample buffer (ThermoFisher, NP0008), boiled at 95 °C for 5 min, and an equal amount of protein was loaded onto NuPAGE 4–12% bis-tris protein gels (ThermoFisher, 23225 NP0323PK2). Electrophoresis was performed at 200 V for 40 min, using NuPAGE MES SDS running buffer (ThermoFisher, NP0002) and proteins transferred at 30 V for 70 min to Immobilon-FL PVDF membrane (Merck, IPFL00010) using NuPAGE transfer buffer (ThermoFisher, NP0006). The membranes were blocked for 1 h at room temperature with Odyssey blocking buffer (PBS) (LI-COR, 927-40000) and incubated overnight at 4 °C with β-actin (Cell Signaling Technology, 4970) and HDAC4 antibodies (Santa Cruz Biotechnology, sc-365093) diluted 1:10,000 and 1:200, respectively in Odyssey blocking buffer (PBS). The membranes were then washed three times with TBS-T and incubated for 1 h at room temperature with IRDye secondary antibodies (IRDye 800CW Donkey anti-Mouse 925-32212 and IRDye 680RD Donkey anti-Rabbit 925-68073, LI-COR) diluted 1:10,000 in Odyssey PBS blocking buffer. The membranes were washed and imaged on Odyssey (LI-COR) and analyzed on ImageStudio software (LI-COR). Other antibodies used in multiplex HDAC selectivity western blots were: HDAC1 (Sigma, H3284), HDAC3 (Cell Signaling Technology, 3949), HDAC5 (CHDI-90000155, rabbit polyclonal antibody raised to a synthetic peptide corresponding to residues 474–493 of HDAC5 produced at Thermo Scientific Pierce for CHDI), HDAC7 (Abcam, ab12174), and HDAC9 (Abcam, ab109446).
HDAC4 MSD Assay
Cell lysates were prepared by adding MSD lysis buffer (Tris lysis buffer; Meso Scale Discovery, R60TX-3, Phosphatase inhibitor II; Sigma, P5726, Phosphatase inhibitor III; Sigma, P0044), 2 mM PMSF; Sigma, P7626, Complete protease inhibitors (EDTA-free); Roche Diagnostics, 04693159001, 10 mM NaF; Sigma, S7920) to 96-well cell culture plates (50 μL per well for 100 μL per well Jurkat in cell suspension or 100 μL per well for Neuro-2a after media removal and PBS wash), and shaking on an orbital shaker at 1000 rpm for 5 min. An uncoated 96-well MSD MULTI-ARRAY plate (Meso Scale Discovery, L15XA) was coated with 25 μL/well capture HDAC4 antibody (Abcam, ab12171) diluted 1:2000 in capture antibody dilution buffer (15 mM NaHCO3, 35 mM Na2CO3, pH 9.6 adjusted with 5% HCl) and incubated overnight at 4 °C. The MSD capture plate was washed three times with 100 μL/well PBS-T (0.2% Tween-20), blocked with 150 μL/well 2% Probumin (Merck, 810033) PBS-T block buffer for 1 h at room temperature, and incubated with 50 μL/well cell lysate for 1 h at room temperature. The MSD capture plate was washed and incubated for 1 h at room temperature with 25 μL/well HDAC4 detection antibody (Abcam, ab123513 or Santa Cruz, sc-11418) diluted 1:1500 in 2% Probumin block buffer. The MSD capture plate was washed and incubated for 1 h at room temperature with 25 μL/well SULFO-TAG labeled antibody (Meso Scale Discovery, R32AB-1) diluted 1:2000 in 2% Probumin block buffer. The plate was washed, 150 μL/well MSD read buffer (Meso Scale Discovery, R92TC-1) was added and electrochemiluminescence emission was read on Sector Imager 600 or 6000 (Meso Scale Discovery). In parallel 96-well plates, total protein concentrations in lysate were determined using Pierce BCA Protein Assay Kit (ThermoFisher, 23225). MSD counts was normalized to the protein amount (μg) per well. These normalized values were further normalized to the average signal of DMSO control wells and expressed as % HDAC4 (of DMSO control) and analyzed on GraphPad Prism version 8.
Q175 Cortical Neuron Experiments
The test compounds were administered during a partial refresh in DMSO (0.1% final concentration) at DIV7. After 4 to 72 h of compound treatment, Hoechst (10 μg/mL; Invitrogen, H3570) and DRAQ7 (0.6 μM; Biostatus, DR71000) in culture medium were added to the cells for 15 min at 37 °C/5% CO2 followed by live-imaging using InCell 2200 automated high content imaging and analysis platform (GE Healthcare) to quantify changes in neuronal viability by scoring viable cells according to their level of nuclear condensation using live-cell nuclear staining with Hoechst (1:1000) and image segmentation in InCell developer software. Cultures were subsequently washed with PBS, harvested and lysed in 100 μL MSD assay buffer and stored at −80 °C until HDAC4 levels were quantified using the MSD immunoassay. The HDAC4 MSD studies were performed as described above.
Data are expressed as the percentage of vehicle conditions after quantitation on a per well basis and data points on the graph represent means ± standard errors of the mean. Significance was calculated using Dunnett’s post hoc test following ANOVA. Four-parameter nonlinear regression was used to fit the concentration-response curve and only in case of incomplete curves, top constraints were used. Significance and curve fitting were performed using GraphPad Prism version 8.
In Vitro ADME Assay
Kinetic Solubility Assay
Using a 10 mM stock solution of each test and control compound (hydrocortisone, reserpine) in 100% DMSO, dilutions were prepared to a theoretical concentration of 200 μM in both 0.1 M potassium phosphate buffer containing 0.8% NaCl (PBS), pH 7.4 (2% DMSO final), and in 100% DMSO. An aliquot of the 200 μM DMSO solution was then further diluted to 10 μM, and all dilutions (n = 2, in 96-well plates) allowed to equilibrate at room temperature on an orbital shaker for 2 h. The PBS dilutions were filtered using a MultiScreen HTS solubility filter plate (Millipore), and the filtrate was analyzed by LC-UV with confirmation of the peak of interest by mass spectrometry. The concentration of the compound in PBS filtrate was determined by comparing the UV absorbance peak with that of the two DMSO dilutions as calibration standards.
Acknowledgments
The authors would like to acknowledge Dr. Mark Chambers and Dr. Phil Mitchell for useful discussions in the preparation of this manuscript.
Glossary
Abbreviations
- BCRP
breast cancer resistance protein
- BET
bromodomain and extra-terminal
- Boc
tert-butyloxycarbonyl
- CNS
central nervous system
- DCM
dichloromethane
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- ELISA
enzyme-linked immunosorbent assay
- HA
hydroxamic acid
- HATU
O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphate
- HD
Huntington’s disease
- HDAC
histone deacetylase
- HOBT
1-hydroxybenzotriazole
- HTRF
homogeneous time resolved fluorescence
- HTT
huntingtin
- iPSC
induced pluripotent stem cells
- mHtt
mutant huntingtin
- MSD
Meso Scale Discovery
- PBS
phosphate-buffered saline
- PBST
phosphate-buffered saline/tween 20
- PEG
polyethylene glycol
- P-gp
P-glycoprotein
- POI
protein of interest
- PROTAC
proteolysis-targeting chimera
- RT
room temperature
- TBST
tris-buffered saline/tween 20
- TFMO
trifluoromethyloxadiazole
- THF
tetrahydrofuran
- THP
tetrahydropyran
- TFA
trifluoroacetic acid
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01149.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Lahm A.; Paolini C.; Pallaoro M.; Nardi M. C.; Jones P.; Neddermann P.; Sambucini S.; Bottomley M. J.; Lo Surdo P.; Carfí A.; Koch U.; De Francesco R.; Steinkühler C.; Gallinari P. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17335–17340. 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.; Altamura S.; De Francesco R.; Gallinari P.; Lahm A.; Neddermann P.; Rowley M.; Serafini S.; Steinkühler C. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg. Med. Chem. Lett. 2008, 18, 1814–1819. 10.1016/j.bmcl.2008.02.025. [DOI] [PubMed] [Google Scholar]
- Gu H.; Liang Y.; Mandel G.; Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 7571–7576. 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sando R. III; Gounko N.; Pieraut S.; Liao L.; Yates J. III; Maximov A. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 2012, 151, 821–834. 10.1016/j.cell.2012.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ziemka-Nalecz M.; Jaworska J.; Sypecka J.; Zalewska T. Histone deacetylase inhibitors: A therapeutic key in neurological disorders?. J. Neuropathol. Exp. Neurol. 2018, 77, 855–870. 10.1093/jnen/nly073. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Tekwani B. L. Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front. Pharmacol. 2020, 11, 537. 10.3389/fphar.2020.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M.; Landles C.; Weiss A.; Bradaia A.; Seredenina T.; Inuabasi L.; Osborne G.; Wadel K.; Touller C.; Butler R.; Robertson J.; Franklin S. A.; Smith D. L.; Park L.; Marks P. A.; Wanker E. E.; Olson E. N.; Luthi-Carter R.; van der Putten H.; Beaumont V.; Bates G. P. HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 2013, 11, e1001717 10.1371/journal.pbio.1001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M.; Benn C. L.; Franklin S. A.; Smith D. L.; Woodman B.; Marks P. A.; Bates G. P. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS One 2011, 6, e27746 10.1371/journal.pone.0027746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckhurst C. A.; Aziz O.; Beaumont V.; Bürli R. W.; Breccia P.; Maillard M. C.; Haughan A. F.; Lamers M.; Leonard P.; Matthews K. L.; Raphy G.; Stott A. J.; Munoz-Sanjuan I.; Thomas B.; Wall M.; Wishart G.; Yates D.; Dominguez C. Development and characterization of a CNS-penetrant benzhydryl hydroxamic acid class IIa histone deacetylase inhibitor. Bioorg. Med. Chem. Lett. 2019, 29, 83–88. 10.1016/j.bmcl.2018.11.009. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Munoz-Sanjuan I.; Beaumont V.; Beconi M.; Buerli R. W.; Haughan A. F.; Luckhurst C. A.; Wall M.; Raphy G.; Thomas B.. Histone deacetylase inhibitors and compositions and methods of use. WO 2014159210, 2014.
- Stott A. J.; Maillard M. C.; Beaumont V.; Allcock D.; Aziz O.; Borchers A. H.; Blackaby W.; Breccia P.; Creighton-Gutteridge G.; Haughan A. F.; Jarvis R. E.; Luckhurst C. A.; Matthews K. L.; McAllister G.; Pollack S.; Saville-Stones E.; Van de Poël A. J.; Vater H. D.; Vann J.; Williams R.; Yates D.; Muñoz-Sanjuán I.; Dominguez C. Evaluation of 5-(trifluoromethyl)-1,2,4-oxadiazole-based class IIa HDAC inhibitors for Huntington’s disease. ACS Med. Chem. Lett. 2021, 12, 380–388. 10.1021/acsmedchemlett.0c00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C., Maillard M. C., Haughan A. F., Van de Poel A., Stott A. J., Luckhurst C. A., Saville-Stones E. A., Wishart G., Wall M., Breccia P., Jarvis R. E.. Preparation of N-substituted oxadiazolylbenzamides as histone deacetylase inhibitors and compositions thereof, for treatment of neurodegenerative diseases. WO 2016179554, 2016.
- Neklesa T. K.; Winkler J. D.; Crews C. M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- Wang P.; Zhou J. Proteolysis targeting chimera (PROTAC): A paradigm-shifting approach in small molecule drug discovery. Curr. Top. Med. Chem. 2018, 18, 1354–1356. 10.2174/1568026618666181010101922. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Crews C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K. H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.; Song Y.; Xie H.; Wu H.; Wu Y. T.; Leisten E. D.; Tang W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. 10.1016/j.bmcl.2018.05.057. [DOI] [PubMed] [Google Scholar]
- An Z.; Lv W.; Su S.; Wu W.; Rao Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. 10.1007/s13238-018-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Yang K.; Zhang Z.; Leisten E. D.; Li Z.; Xie H.; Liu J.; Smith K. A.; Novakova Z.; Barinka C.; Tang W. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J. Med. Chem. 2019, 62, 7042–7057. 10.1021/acs.jmedchem.9b00516. [DOI] [PubMed] [Google Scholar]
- Cao F.; de Weerd S.; Chen D.; Zwinderman M. R. H.; van der Wouden P. E.; Dekker F. J. Induced protein degradation of histone deacetylases 3 (HDAC3) by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 208, 112800. 10.1016/j.ejmech.2020.112800. [DOI] [PubMed] [Google Scholar]
- Smalley J. P.; Adams G. E.; Millard C. J.; Song Y.; Norris J. K. S.; Schwabe J. W. R.; Cowley S. M.; Hodgkinson J. T. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun. 2020, 56, 4476–4479. 10.1039/D0CC01485K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Wang J.; Zhao L. Y.; Chen X.; Zheng G.; Zhang X.; Liao D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. 10.1039/D0CC03243C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y.; Donovan K. A.; Eleuteri N. A.; Kirmani N.; Yue H.; Razov A.; Krupnick N. M.; Nowak R. P.; Fischer E. S. Chemo-proteomics exploration of HDAC degradability by small molecule degraders. Cell Chem. Biol. 2021, 28, 1514–1527.e4. 10.1016/j.chembiol.2021.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez-Cantudo L.; Ramos A.; Coderch C.; de Pascual-Teresa B. Proteasomal degradation of Zn-dependent Hdacs: The E3-ligases implicated and the designed protacs that enable degradation. Molecules 2021, 26, 5606. 10.3390/molecules26185606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues D. A.; Roe A.; Griffith D.; Chonghaile T. N. Advances in the Design and Development of PROTAC-mediated HDAC degradation. Curr. Top. Med. Chem. 2022, 22, 408–424. 10.2174/1568026621666211015092047. [DOI] [PubMed] [Google Scholar]
- Fischer F.; Alves Avelar L. A.; Murray L.; Kurz T. Designing HDAC-PROTACs: lessons learned so far. Future Med. Chem. 2022, 14, 143–166. 10.4155/fmc-2021-0206. [DOI] [PubMed] [Google Scholar]
- Smalley J. P.; Baker I. M.; Pytel W. A.; Lin L. Y.; Bowman K. J.; Schwabe J. W. R.; Cowley S. M.; Hodgkinson J. T. Optimization of class I histone deacetylase PROTACs reveals that HDAC1/2 degradation is critical to induce apoptosis and cell arrest in cancer cells. J. Med. Chem. 2022, 65, 5642–5659. 10.1021/acs.jmedchem.1c02179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maple H. J.; Clayden N.; Baron A.; Stacey C.; Felix R. Developing degraders: principles and perspectives on design and chemical space. MedChemComm 2019, 10, 1755–1764. 10.1039/C9MD00272C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike A.; Williamson B.; Harlfinger S.; Martin S.; McGinnity D. F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discovery Today 2020, 25, 1793–1800. 10.1016/j.drudis.2020.07.013. [DOI] [PubMed] [Google Scholar]
- Edmondson S. D.; Yang B.; Fallan C. Proteolysis targeting chimeras (PROTACs) in ’beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- Atilaw Y.; Poongavanam V.; Svensson Nilsson C.; Nguyen D.; Giese A.; Meibom D.; Erdelyi M.; Kihlberg J. Solution conformations shed light on PROTAC cell permeability. ACS Med. Chem. Lett. 2021, 12, 107–114. 10.1021/acsmedchemlett.0c00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman B. B. 3rd; Yang L.; Rankovic Z. Practical approaches to evaluating and optimizing brain exposure in early drug discovery. Eur. J. Med. Chem. 2019, 182, 111643. 10.1016/j.ejmech.2019.111643. [DOI] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel–Lindau (VHL) E3 ubiquitin ligase and the Hypoxia Inducible Factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57, 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crew A. P.; Raina K.; Dong H.; Qian Y.; Wang J.; Vigil D.; Serebrenik Y. V.; Hamman B. D.; Morgan A.; Ferrano C.; Siu K.; Neklesa T. K.; Winkler J. D.; Coleman K. G.; Crews C. M. Identification and characterization of Von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase 1. J. Med. Chem. 2018, 61, 583–598. 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018, 25, 78–87.e5. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Shao X.; Zhong T.; Wu Y.; Xu A.; Sun X.; Gao H.; Liu H.; Lan T.; Tong Y.; Tao X.; Du W.; Wang W.; Chen Y.; Li T.; Meng X.; Deng H.; Yang B.; He Q.; Ying M.; Rao Y. Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat. Chem. Biol. 2021, 17, 567–575. 10.1038/s41589-021-00742-5. [DOI] [PubMed] [Google Scholar]
- Powell C. E.; Gao Y.; Tan L.; Donovan K. A.; Nowak R. P.; Loehr A.; Bahcall M.; Fischer E. S.; Jänne P. A.; George R. E.; Gray N. S. Chemically induced degradation of anaplastic lymphoma linase (ALK). J. Med. Chem. 2018, 61, 4249–4255. 10.1021/acs.jmedchem.7b01655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva I. V.; Pande P.; Patton W. F. Sensitive and specific fluorescent probes for functional analysis of the three major types of mammalian ABC transporters. PLoS One 2011, 6, e22429 10.1371/journal.pone.0022429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu T. T.; Gao N.; Li Q. Q.; Chen P. G.; Yang X. F.; Chen Y. X.; Zhao Y. F.; Li Y. M. Specific knockdown of endogenous tau protein by peptide-directed ubiquitin-proteasome degradation. Cell Chem. Biol. 2016, 23, 453–461. 10.1016/j.chembiol.2016.02.016. [DOI] [PubMed] [Google Scholar]
- Silva M. C.; Ferguson F. M.; Cai Q.; Donovan K. A.; Nandi G.; Patnaik D.; Zhang T.; Huang H. T.; Lucente D. E.; Dickerson B. C.; Mitchison T. J.; Fischer E. S.; Gray N. S.; Haggarty S. J. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. eLife 2019, 8, e45457 10.7554/eLife.45457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Southwell A. L.; Smith-Dijak A.; Kay C.; Sepers M.; Villanueva E. B.; Parsons M. P.; Xie Y.; Anderson L.; Felczak B.; Waltl S.; Seunghyun K.; Cheung D.; Dal Cengio L.; Slama R.; Petoukhov E.; Raymonds L. A.; Hayden M. R. An enhanced Q175 knock-in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum. Mol. Genet. 2016, 25, 3654–3675. 10.1093/hmg/ddw212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beconi M.; Aziz O.; Matthews K.; Moumné L.; O’Connell C.; Yates D.; Clifton S.; Pett H.; Vann J.; Crowley L.; Haughan A. F.; Smith D. L.; Woodman B.; Bates G. P.; Brookfield F.; Bürli R. W.; McAllister G.; Dominguez C.; Munoz-Sanjuan I.; Beaumont V. Oral administration of the pimelic diphenylamide HDAC inhibitor HDACi 4b is unsuitable for chronic inhibition of HDAC activity in the CNS in vivo. PLoS One 2012, 7, e44498 10.1371/journal.pone.0044498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.